
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of epinephrine using a K2Fe4O7 modified glassy carbon electrode†
Mingcheng Jiang,
Decheng Zeng,
Xinxin Zheng and
Hongming Yuan *
*
State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun, 130012, PR China. E-mail: hmyuan@jlu.edu.cn
First published on 13th May 2024
Abstract
Iron-based electrochemical catalysts used to modify electrodes for biosensing have received more attention from biosensor manufacturers because of their excellent biocompatibility and low cost. In this work, a fast-ion conductor potassium ferrite (K2Fe4O7) modified glassy carbon electrode (GCE) was prepared for detecting epinephrine (EP) by electrochemical techniques. The obtained K2Fe4O7/GCE electrode exhibited not only a wide linear range over EP concentration from 2 μM to 260 μM with a detection limit of 0.27 μM (S/N = 3) but also high selectivity toward EP in the presence of common interferents ascorbic acid (AA) and uric acid (UA), as well as good reproducibility and stability.
Introduction
Epinephrine (EP), C9H13NO3 [(R)-4-(1-hydroxy-2-[methylamin]ethyl) benzene-1,2-diol], is a vital neurotransmitter belonging to the catecholamine group in the central nervous system of mammals.1 In pharmaceutical applications, EP has been utilized to treat various conditions such as myocardial infarction, bronchial asthma, Parkinson's disease, and hypertension.2,3 The multifaceted effects of EP include cardiovascular and cerebrovascular constriction, augmentation of heart activity, and elevation of blood pressure.4 These ailments are closely associated with the concentration levels of EP in the human body. Accurate detection of EP in biological fluids like blood and urine holds immense significance.5Several quantitative analytical methods have been established to determine EP concentrations, such as high-performance liquid chromatography (HPLC),6 fluorimetry,7 and electrochemiluminescence.8 Since EP is an electroactive molecule, electrochemical techniques have emerged as practical and cost-effective approaches.9 Electrochemical sensors utilizing glassy carbon electrodes (GCE) and carbon paste electrodes have been commonly employed for EP detection. To enhance their performance, various materials, such as organic molecules,10 polymers11, carbon nanotubes12, graphene13, and precious metals14, have been explored as catalysts to modify the bare electrodes, resulting in improved electrocatalytic activity for EP.
Iron, the fourth most abundant element in the Earth's crust, has garnered considerable attention for its catalytic properties, particularly iron oxides.15 Iron is also integral to biomolecules such as hemoglobin and myoglobin. Consequently, iron-containing materials have shown promise in biosensor applications. Recent studies have successfully detected dopamine (DA), another catecholamine neurotransmitter, using electrodes modified with iron oxide nanoparticles,16–18 Fe-based metal–organic frameworks (MOFs),19 and cobalt-doped FePS3.20 However, no reported electrode is modified by iron-based materials that demonstrate electrocatalytic activity for the oxidation of EP.
K2Fe4O7, a compound displaying superionic conducting properties, is particularly favorable for enhancing the transfer ability of electrons. Our research group has previously reported successfully applying a K2Fe4O7-modified GCE electrode to determine DA.21 Building on this, we explore the electrocatalytic activity of the K2Fe4O7/GCE electrode towards the oxidation of EP in this work. Furthermore, we investigate the performance of the electrode in detecting EP in the presence of interfering substances, such as ascorbic acid (AA) and uric acid (UA). The reproducibility and stability of the EP sensor are also evaluated.
Materials and methods
Materials
Epinephrine (EP) (99%), uric acid (UA) (99%), and ascorbic acid (AA) (99%) were bought from Sigma-Aldrich (Shanghai, China). Fe (NO3)3·9H2O, KOH, KH2PO4, HCl, and K2HPO4, were purchased from Sinopharm Chemical Reagent. Co. (China). All chemicals were analytical grade and used without any treatment. The phosphate buffer solution (PBS, 0.1 M) was prepared with K2HPO4 and KH2PO4, and the pH value ranging from 6.0 to 7.8 was adjusted by changing the radio of K2HPO4 and KH2PO4. Aqueous solutions used in experiments were prepared using deionized water with a resistance of 18.5 MΩ cm−1 obtained from an ultrapure water system (Wotepu, China).Instruments and measurements
Powder X-ray diffraction (PXRD) experiments were measured on a Rigaku D-Max 2550 diffractometer (Rigaku, Japan) with Cu-Kα radiation (λ = 1.5418 Å). UV-Vis absorption spectra were collected with a UV-2450 spectrometer (Hitachi, Japan). All electrochemical measurements were tested at room temperature by CHI660C electrochemical workstation (Chenhua Co., Shanghai, China). Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were performed in a standard three-electrode system, which included bare and modified GCE as the working electrodes, Ag/AgCl/KCl (0.1 M) as the reference electrode, and platinum as the counter electrode.Preparation of K2Fe4O7
We acquired the single-phase crystalline K2Fe4O7 through the hydrothermal method previously reported.22 To ensure purity, the crystal surface was cleaned with 2.0 M HCl and washed several times with distilled water. After washing, the crystal was dried in a vacuum oven at 60 °C. The crystal was finally ground into a powder using a mortar for about one hour.Construction of the modified electrode
The GCE (diameter = 3 mm) was polished with 0.05 μm alumina slurry. Afterward, the electrodes were ultrasonically cleaned in distilled water for 5 min and left to air dry at room temperature. The K2Fe4O7 dispersion was prepared by adding 3 mg K2Fe4O7 powder to a mixture of 5 mL dimethylformamide and 1 μL Nafion and then sonicating for 30 min. Take 10 μL of the dispersion at a time and drop it on the surface of the electrode, then let it dry naturally at room temperature and repeat three times until the K2Fe4O7 is spread evenly over the surface of the electrode.Results and discussion
Characterization of K2Fe4O7
The crystal phase and purity of K2Fe4O7 were confirmed through X-ray Diffraction analysis, as shown in Fig. 1. The results indicate that K2Fe4O7 was successfully synthesized with a high purity level (Detailed crystal information is given in ESI S1†). Moreover, the samples obtained after grinding exhibited similarity to those identified through single crystal structure simulation. This observation suggests that the crystal structure of K2Fe4O7 remained unchanged even after the grinding process.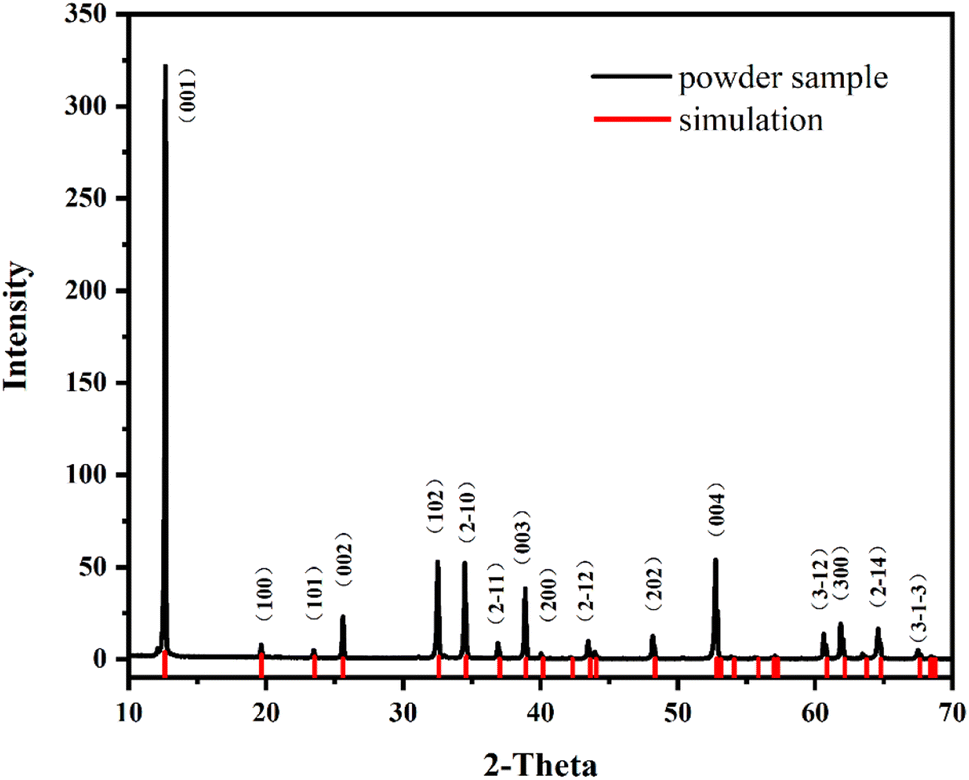 | ||
| Fig. 1 The simulation and powder XRD patterns of K2Fe4O7. | ||
The electrochemical response of EP
The electrochemical response of EP on the electrode surface and the electrocatalytic behavior of K2Fe4O7 were investigated through the CV method. Initially, the bare electrode and K2Fe4O7/GCE electrode were separately tested in PBS containing 100 μM EP. The CV results, as illustrated in Fig. 2(a), indicate that the response current of the K2Fe4O7/GCE electrode was notably higher as compared to the bare electrode, thus indicating a considerable electrocatalytic effect of K2Fe4O7 on EP. It is important to note that the CV plot clearly shows two sets of redox peaks. Specifically, A1/B1 corresponds to the redox process between adrenaline and adrenoquinone, and the relevant calculations in the testing process are based on the data from this oxidation peak unless otherwise specified. The other redox peaks represent the redox process between adrenochrome and 5,6-dihydroxy-N-methylindole23 (Fig. S2†). The reason for the appearance of this specific pair of redox peaks is that K2Fe4O7 has previously been demonstrated to be used as a catalyst for the oxygen evolution reaction. During the test, oxygen generation accompanies the oxidation of epinephrine to adrenochrome, resulting in a second pair of redox peaks. Additionally, the change in response current with scan rate can reflect the kinetic process of EP on the electrode surface. With a scan rate increase from 10 mV s−1 to 100 mV s−1, there was a noticeable linear relationship between the response current and the square root of the scan rate, as shown in Fig. 2(b and c). This linearity proves that the redox process of EP is diffusion-controlled24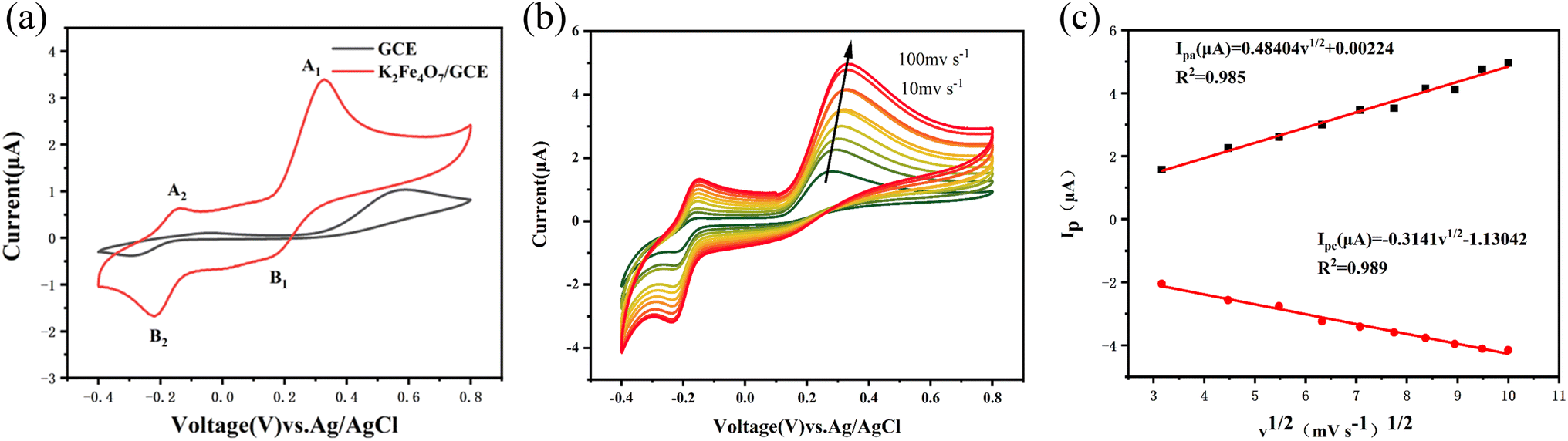 | ||
| Fig. 2 (a) CV of bare GCE and K2Fe4O7/GCE in 0.1 M PBS (pH = 7.4) containing 100 μM EP. (b) CVs of K2Fe4O7/GCE in 0.1 M PBS (pH = 7.4) containing 100 μM EP at scan rates of 10–100 mV s−1. (c) The plots of response currents and the square root of scan rate. | ||
The effect of pH
To investigate the effect of pH on the performance of the electrochemical sensor for EP, PBS solutions with various pH values containing 100 μM EP were prepared and tested (Fig. 3(a)). The results showed that the response potential of the sensor displayed a linear decrease with the increase of pH, with a slope of approximately 0.052 mV pH−1, indicating the transfer of two protons during the reaction.25 The response current of the sensor increased with the pH value until it reached its maximum value at pH 7.4, after which it decreased with further increases in pH. As a result, pH 7.4 was determined to be the optimal pH value for the sensor (Fig. 3(a)). Subsequent experiments were conducted at this pH value unless otherwise specified.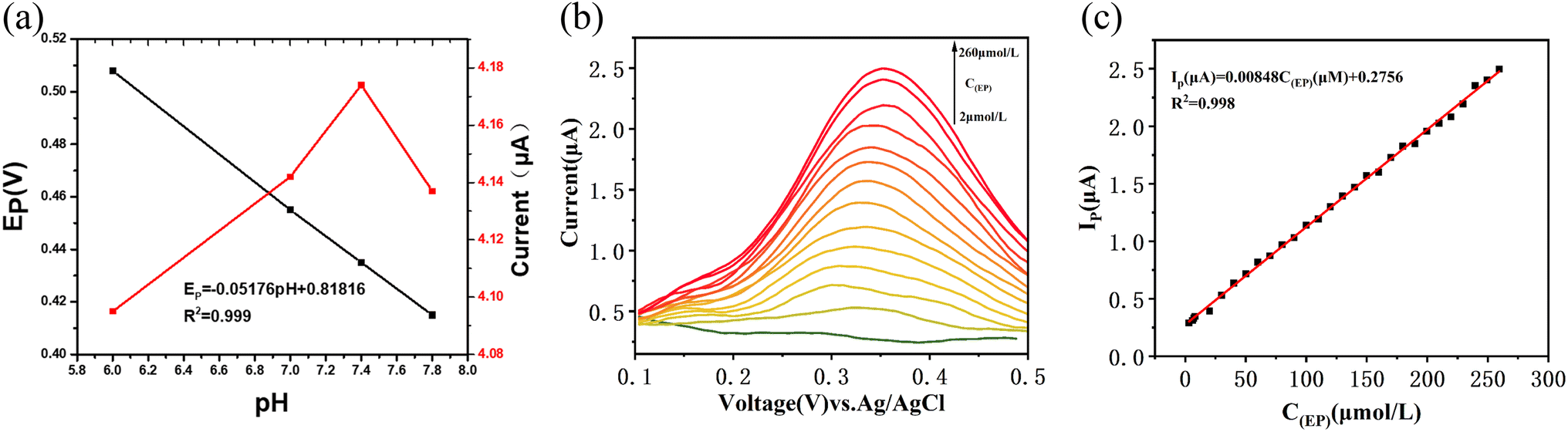 | ||
| Fig. 3 (a) The plots of pH with response current and response potential in 0.1 M PBS containing 100 μM EP. (b) DPV of K2Fe4O7/GCE for different EP concentrations in 0.1 M PBS (pH = 7.4). (c) Plots of the response current and EP concentrations (2–260 μM). | ||
Calibration curve
Differential pulse voltammetry (DPV) was employed as a highly responsive and rapid detection technique for adrenaline (EP). To establish the relationship between the response current and the concentration of EP, DPV scans were performed in phosphate-buffered saline (PBS) with varying concentrations of EP (Fig. 3(b and c)). The results revealed a robust linear correlation between the response current and EP concentration within the range of 2 μM to 260 μM. The equation for this relationship is Ip (μA) = 0.00848C(EP) (μM) + 0.2756, with an impressive coefficient of determination (R2 = 0.998). Additionally, the limit of detection for this sensor was determined to be 0.27 μM (See S3† for specific LOD calculations). The comparative analysis presented in Table 1 demonstrates the successful development of an EP sensor compared with other previously published studies.Selectivity of the sensor
During the electrochemical quantification of EP using biosensors, interfering substances such as ascorbic acid (AA) and uric acid (UA) can affect the accuracy of the results.32 The DPV method was employed to assess the selectivity of the K2Fe4O7/GCE electrode towards EP. The electrochemical activity of AA and UA on the surface of the K2Fe4O7/GCE electrode was investigated, as depicted in Fig. 4(a). Interestingly, it was observed that AA exhibited no detectable electrochemical response, and although urea showed a weak electrochemical response around 0.32 V, it did not affect the detection results. The results indicate the sensor's exceptional selectivity towards EP.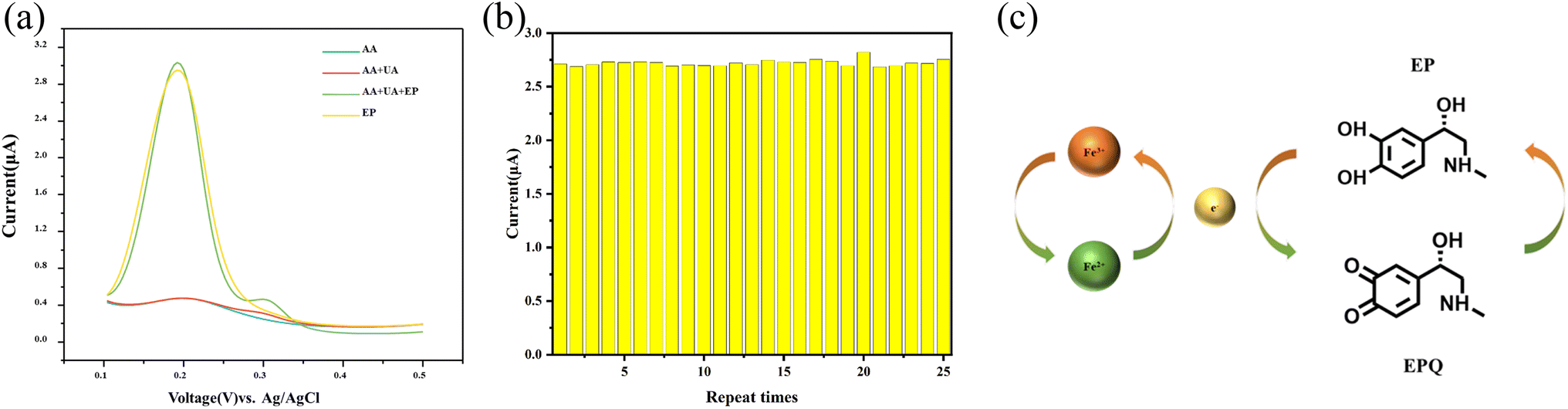 | ||
| Fig. 4 (a) DPV response of K2Fe4O7/GCE in 0.1 M PBS (pH = 7.4) containing 1 mM AA; 1 mM AA and 100 μM UA; 100 μM UA and 100 μM EP; 100 μM EP respectively. (b) Results of eight independent tests of K2Fe4O7/GCE. (c) Brief detection mechanism. | ||
Repeatability and stability
K2Fe4O7 has been demonstrated to be a viable material for electrochemical analysis. Maintaining good repeatability and stability is crucial for electrochemical sensors in the detection process. The modified electrode was subjected to 25 independent measurements in a buffering solution containing 100 μM EP to further validate its repeatability and stability as an electrochemical sensor. As shown in Fig. 4(b), the results exhibited a relative standard deviation of 2.92%. The electrochemical signal remained stable, showing the sensor's suitability for EP detection.Detection mechanism
K2Fe4O7 has been demonstrated to be a viable material for electrochemical analysis. However, according to our previous research, the molecule cannot directly gain electrons on the electrode surface.21 But K2Fe4O7 has been shown to be a solid electrolyte,22 which further illustrates that the valence state of elemental iron can undergo a reversible transition. Thus, we proposed a brief detection mechanism (Fig. 4(c)).Conclusions
In summary, K2Fe4O7 was investigated as an electrode modification material to detect EP efficiently. The response currents of the electrode display a linear increase proportional to the concentrations of EP from 2 μM to 260 μM, with a detection limit of 0.27 μM (S/N = 3). Additionally, K2Fe4O7/GCE performs a high selectivity and stability. This electrochemical sensor provides another application of iron-based catalyst and offers a new choice in detecting EP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported financially by the State Key Laboratory of Inorganic Synthesis and Preparative Chemistry.References
- S. Immanuel and R. Sivasubramanian, Bull. Mater. Sci., 2020, 43, 79 CrossRef CAS.
- T. Tavana, M. A. Khalilzadeh, H. Karimi-Maleh, A. A. Ensafi, H. Beitollahi and D. Zareyee, J. Mol. Liq., 2012, 168, 69–74 CrossRef CAS.
- T. Thomas, R. J. Mascarenhas, O. J. D. Souza, S. Detriche, Z. Mekhalif and P. Martis, Talanta, 2014, 125, 352–360 CrossRef CAS PubMed.
- D. M. Fouad and W. A. El-Said, J. Nanomater., 2016, 2016, 6194230 Search PubMed.
- C. Wang, F. X. Hu, X. C. Zou, Y. Q. Wang, Y. R. Ren and J. Tan, Talanta, 2022, 248, 123621 CrossRef CAS.
- M. A. Fotopoulou and P. C. Ioannou, Anal. Chim. Acta, 2002, 462, 179–185 CrossRef CAS.
- X. L. Zhu, P. N. Shaw and D. A. Barrett, Anal. Chim. Acta, 2003, 478, 259–269 CrossRef CAS.
- Y. Y. Su, J. A. Wang and G. N. Chen, Talanta, 2005, 65, 531–536 CrossRef CAS PubMed.
- T. Łuczak, Electroanalysis, 2009, 21, 2557–2562 CrossRef.
- M. D. Tezerjani, A. Benvidi, A. Dehghani Firouzabadi, M. Mazloum-Ardakani and A. Akbari, Measurement, 2017, 101, 183–189 CrossRef.
- F. Liu and X. Kan, J. Electroanal. Chem., 2019, 836, 182–189 CrossRef CAS.
- H. Yi, D. Zheng, C. Hu and S. Hu, Electroanalysis, 2008, 20, 1143–1146 CrossRef CAS.
- M. Taleb, R. Ivanov, S. Bereznev, S. H. Kazemi and I. Hussainova, J. Electroanal. Chem., 2018, 823, 184–192 CrossRef CAS.
- L. Zou, Y. Li, S. Cao and B. Ye, Talanta, 2013, 117, 333–337 CrossRef CAS.
- Y. Nishibayashi, Dalton Trans., 2018, 47, 11290–11297 RSC.
- W. Zhang, J. Zheng, J. Shi, Z. Lin, Q. Huang, H. Zhang, C. Wei, J. Chen, S. Hu and A. Hao, Anal. Chim. Acta, 2015, 853, 285–290 CrossRef CAS PubMed.
- Y. Huang, Y. Zhang, D. Liu, M. Li, Y. Yu, W. Yang and H. Li, Talanta, 2019, 201, 511–518 CrossRef CAS.
- Y. Liu, W. Zhu, D. Wu and Q. Wei, Measurement, 2015, 60, 1–5 CrossRef.
- X. Ke, Z. Zhao, J. Huang, C. Liu, G. Huang, J. Tan, H. Zhu, Z. Xiao, X. Liu, Y. Mei and J. Chu, ACS Appl. Mater. Interfaces, 2023, 15, 12005–12016 CrossRef CAS PubMed.
- W. Shan, X. Ma, G. Chen, F. Xu, H. Zhao, L. Dong, X. Yan, Z. Bi, L. Yu and M. Qiu, J. Electrochem. Soc., 2023, 170, 047514 CrossRef CAS.
- X. Sun, L. Zhang, X. Zhang, X. Liu, J. Jian, D. Kong, D. Zeng, H. Yuan and S. Feng, Biosens. Bioelectron., 2020, 153, 112045 CrossRef CAS PubMed.
- H. M. Yuan, H. Li, T. S. Zhang, G. H. Li, T. M. He, F. Du and S. H. Feng, J. Mater. Chem. A, 2018, 6, 8413–8418 RSC.
- M. Rafiee, L. Khalafi, F. Mousavi, F. Babaloii and F. Kalhori, Electroanalysis, 2017, 29, 2004–2007 CrossRef CAS.
- S. Majdi, A. Jabbari, H. Heli and A. A. Moosavi-Movahedi, Electrochim. Acta, 2007, 52, 4622–4629 CrossRef CAS.
- F. Fallah, M. R. Shishehbore and A. Sheibani, Talanta, 2023, 252, 123776 CrossRef CAS PubMed.
- S. Renjini, P. Abraham, S. Aparna and V. A. Kumary, J. Electrochem. Soc., 2019, 166, B1321–B1329 CrossRef CAS.
- P. Shaikshavali, T. M. Reddy, T. V. Gopal, G. Venkataprasad, V. S. Kotakadi, V. N. Palakollu and R. Karpoormath, Colloids Surf., A, 2020, 584, 124038 CrossRef CAS.
- P. Gopal, G. Narasimha and T. M. Reddy, Process Biochem., 2020, 92, 476–485 CrossRef.
- L. V. da Silva, N. D. dos Santos, A. K. A. de Almeida, D. dos Santos, A. C. F. Santos, M. C. Franca, D. J. P. Lima, P. R. Lima and M. O. F. Goulart, J. Electroanal. Chem., 2021, 881, 114919 CrossRef CAS.
- M. F. Chen and X. Y. Ma, Russ. J. Appl. Chem., 2014, 87, 200–206 CrossRef CAS.
- B. Mekassa, M. Tessema, B. S. Chandravanshi, P. G. L. Baker and F. N. Muya, J. Electroanal. Chem., 2017, 807, 145–153 CrossRef CAS.
- M. L. Yola and N. Atar, Composites, Part B, 2019, 175, 107113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00242c |
| This journal is © The Royal Society of Chemistry 2024 |