
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved and ligand-free copper-catalyzed cyclization for an efficient synthesis of benzimidazoles from o-bromoarylamine and nitriles†
Emmanuel Mintah Bonku‡
 ab,
Hongjian Qin‡abc,
Abdullajon Odilovab,
Safomuddin Abduahadiab,
Samuel Desta Gumaab,
Feipu Yanga,
Fuqiang Zhud,
Haji A. Aisa*bc and
Jingshan Shen*ab
ab,
Hongjian Qin‡abc,
Abdullajon Odilovab,
Safomuddin Abduahadiab,
Samuel Desta Gumaab,
Feipu Yanga,
Fuqiang Zhud,
Haji A. Aisa*bc and
Jingshan Shen*ab
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, P. R. China. E-mail: shenjingshan@simm.ac.cn
bUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, P. R. China
cState Key Laboratory Basis of Xinjiang Indigenous Medicinal Plants Resource Utilization, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi, Xinjiang 830011, P. R. China. E-mail: haji@ms.xjb.ac.cn
dTopharman Shanghai Co., Ltd., No. 388 Jialilue Road, Zhangjiang Hitech Park, Shanghai 201203, P.R. China
First published on 26th February 2024
Abstract
We present an improved copper-catalyzed cyclization for an efficient synthesis of benzimidazoles from o-bromoarylamine and nitriles, under mild and ligand-free conditions. The optimal conditions yielded exceptional products of up to 98%, demonstrating the broad applicability of this synthetic strategy in generating a wide range of valuable imidazole derivatives. This methodology enables the efficient synthesis of various substituted benzimidazole derivatives and offers an environmentally friendly alternative to conventional methods. By eliminating the use of harsh reagents and high temperatures associated with traditional synthesis approaches, this method proves to be more efficient and robust. Notably, we successfully applied this synthetic approach to the synthesis of bendazol and thiabendazole, yielding 82% and 78%, respectively, on a 100 gram scale.
Introduction
Benzimidazole (2) and its derivatives are crucial nitrogen-containing heterocycles that serve as privileged scaffolds in the pharmaceutical industry.1 This is primarily due to their prominence in various bioactive compounds and pharmaceutical active ingredients (APIs). These include antihypertensive drugs like telmisartan, bendazol, and candesartan, as well as antihelminthic drugs such as albendazole, thiabendazole, and mebendazole. Additionally, benzimidazoles are utilized in the production of antiviral drugs like enviradine, proton pump inhibitors such as esomeprazole, lansoprazole, and pantoprazole, then as antihistamines like semizole (Fig. 1).2 Due to the significant biological activities, unique structural features, and wide-ranging industrial applications, the synthesis of benzimidazole-based drugs has garnered considerable attention from chemists, resulting in the development of various strategies.2 One of the key focal points has been the efficient construction of the essential benzimidazole skeleton.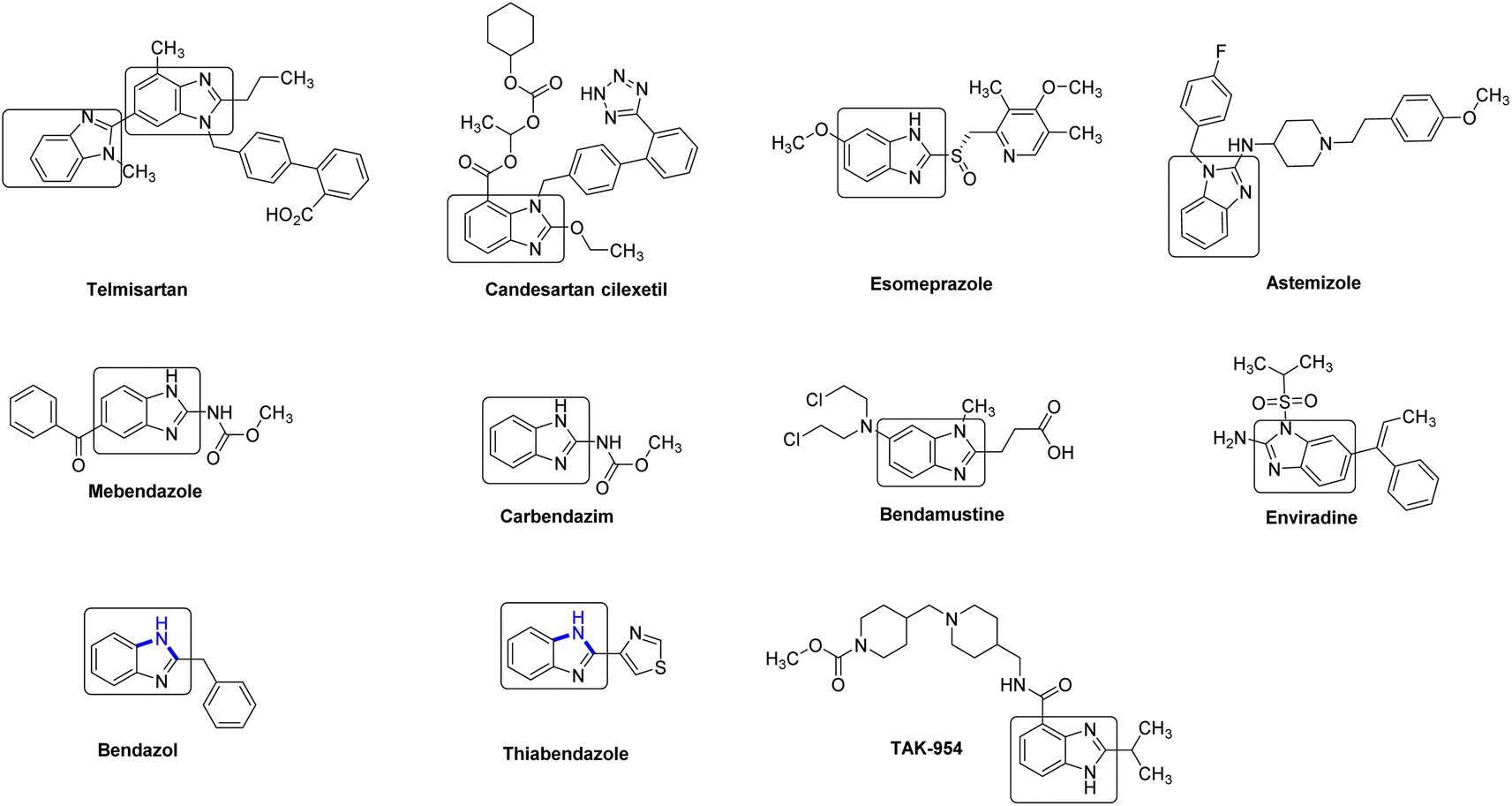 | ||
| Fig. 1 Selected examples of drugs containing benzimidazole ring unit. | ||
The conventional method for synthesizing benzimidazoles involves the condensation and cyclization of substituted o-phenylenediamines with carboxylic acid derivatives and aldehydes under strong acidic conditions, high temperatures, or with equivalent amounts of oxidants. Additionally, the production of the required substituted o-phenylenediamines typically necessitates the incorporation of nitro groups through nitration with nitrosulfuric acid on the substituting aniline, followed by reduction (Scheme 1A). Several alternative approaches have been evaluated to improve the synthesis of benzimidazoles, including the use of aldehydes as substrates and iodine as a catalyst in the presence of hydrogen peroxide,3 employing metal-catalyzed or oxidative cyclization of amidines (Scheme 1B),4 and the use of an iridium photocatalyst to generate benzimidazoles from N-phenylamidoxime esters (Scheme 1C), and a recently reported approach involving Pd-catalyzed carbonylative cyclization.5,6 However, certain underlying issues remain unresolved or new drawbacks have emerged, such as the commercial unavailability of viable raw materials, laborious procedures, the need for homogeneous precious-metal catalysts, and high cost. Moreover, these approaches generate significant amounts of stoichiometric waste due to the presence of leaving groups or additives in the substrate. Furthermore, the original synthetic route for constructing benzimidazole intermediates, which involved nitration and polyphosphoric acid (PPA)-mediated cyclization, resulted in safety issues and the generation of acidic sewage that required treatment.7
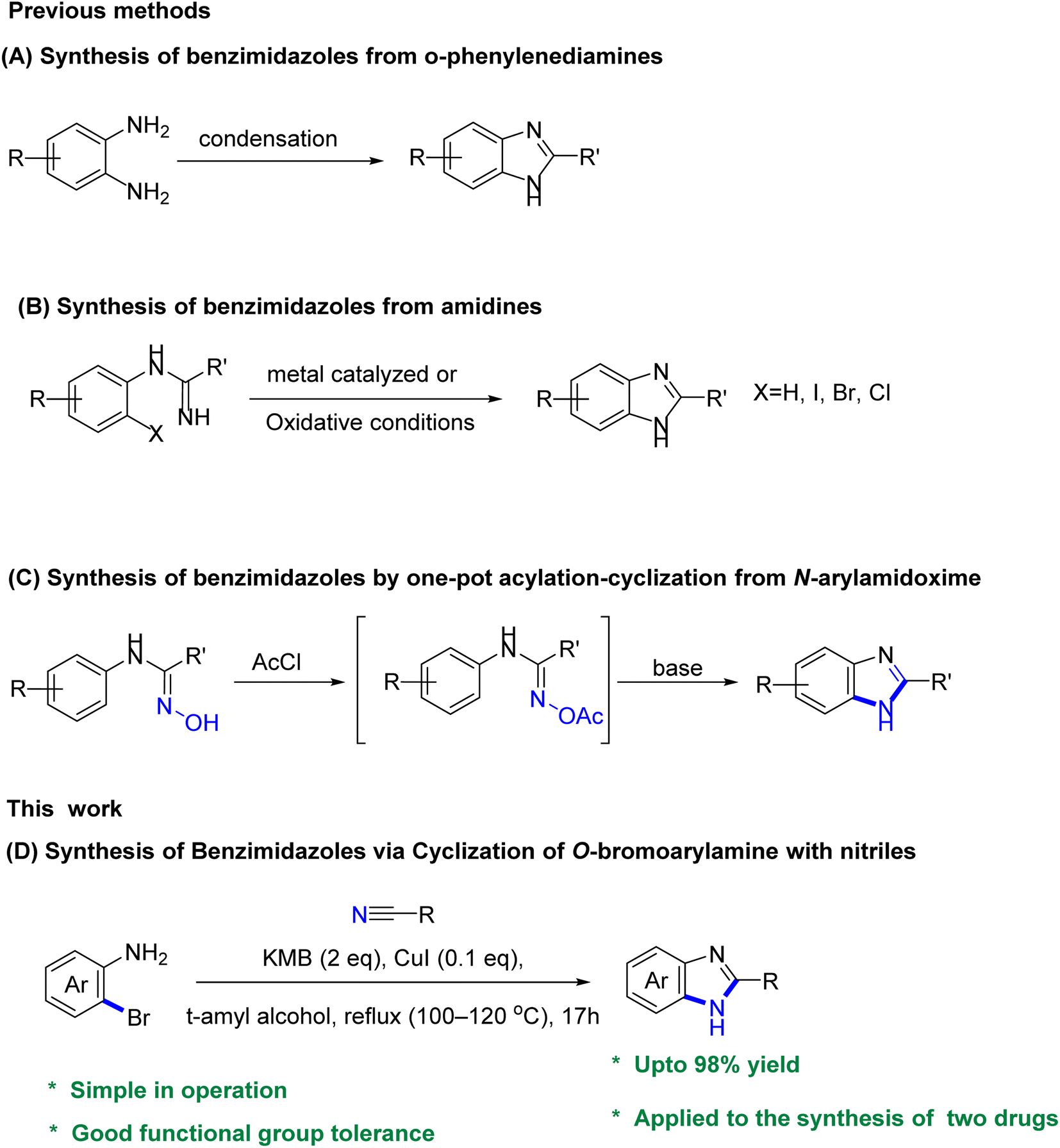 | ||
| Scheme 1 Previous methods (A–C) for producing benzimidazoles, and our method (D) for the synthesis of benzimidazoles via cyclization of o-bromoarylamine with nitriles. | ||
In prior research, various synthesis methods for benzimidazoles have been reported (Scheme 1A–C), including the copper-catalyzed Ullman-type C–N coupling of o-haloanilides with primary amides in a one-pot reaction.8 It's worth noting that Xiang et al.9 and Yu et al.10 demonstrated the synthesis of benzimidazoles through intermolecular cyclization reactions of 2-iodoanilines with aryl nitriles. Although their approaches offers route to synthesize a range of amides and benzoxazole derivatives, its application in terms of catalytic synthesis of heterocycles is limited. More so, using nitriles as nitrogen nucleophiles, the direct reaction with arylamines through a catalytic cyclization system streamlines the synthesis of benzimidazoles compared to that with amides, and even with ligands.
In light of previous studies and existing knowledge, particularly Xiang et al.9 and Yu et al.,10 we were inspired to investigate the feasibility of employing nitriles as an alternative to amides for the synthesis of benzimidazoles, and expand the substrate scope through the cyclization of anilines with nitriles to generate the desired product (Scheme 1D), and further demonstrate the synthetic application of our approach in a number of benzimidazole-based drugs.
Herein, we present an improved approach for the synthesis of benzimidazoles by cyclization of o-bromoarylamine with nitriles. Remarkably, the reaction occurs under mild and ligand-free conditions, using a relatively low catalytic load. This methodology utilizes readily accessible starting materials, thus promoting a safe and efficient synthesis pathway. It is noteworthy that this method exhibits a high tolerance for functional groups on the substrate and enables the efficient construction of structurally diverse benzimidazoles compared to previous approaches. In addition, this method provided an improved approach for making bendazol and thiabenzadole, avoiding the undesired cyclization with polyphosphoric acid, high reaction temperature, and other drawbacks in the existing operations.
Results and discussion
The cyclization reaction conditions for the formation of benzimidazole (2) were investigated in this study, with a focus on the reaction between anilines and n-butyronitrile (3) as our model starting materials. We specifically chose anilines due to their cleaner and more promising alternative to other aromatic amines. Previous reports corroborate that anilines are readily available, cost-effective, and easy to handle.11 Consequently, we anticipated that the cyclization process with anilines under mild conditions would offer significant advantages by reducing the reliance on harsh reagents and conditions typically associated with traditional methods.In our investigation, we initially explored the synthesis of benzimidazoles using the cyclization reaction of 2-iodoaniline (1I) and arylnitrile, as previously reported by Xiang's9 and Yu's10 research group. Building upon this method, we aimed to directly cyclize o-bromoaniline (1Br) or o-chloroaniline (1Cl) with n-butyronitrile in the absence of copper catalyst and ligand. However, our experimental results indicated that the desired product was only obtained in limited quantities (Table 1, entries 1–3).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Starting material | Catalyst | Base | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reactions were conducted by dissolving compound 1Br, 1Cl or 1I (1.0 mmol) in solvent (6 ml), then adding n-butyronitrile 3 (2 equiv.), catalysts (0.1 equiv.) and a base (2.0 equiv.), followed by stirring at 84–140 °C for 17 h.b Isolated yield.c With 1 equivalent of base.d Not detected.e Stirring for 36 h.f Addition of ligand.g With 0.5 equivalent of base. | |||||||
| 1 | 2-Iodoaniline | — | t-BuOK | t-BuOH | 84 | 17 | 14 |
| 2 | 2-Chloroaniline | — | t-BuOK | t-BuOH | 84 | 17 | Trace |
| 3 | o-Bromoaniline | — | t-BuOK | t-BuOH | 84 | 17 | 26 |
| 4 | o-Bromoaniline | CuBr | t-BuOK | t-BuOH | 84 | 17 | 80 |
| 5 | o-Bromoaniline | CuBr | t-BuOK | t-BuOH | 84 | 17 | 39c |
| 6 | o-Bromoaniline | CuCl | t-BuOK | t-BuOH | 84 | 15 | 68 |
| 7 | o-Bromoaniline | Cu2O | t-BuOK | t-BuOH | 84 | 15 | 71 |
| 8 | o-Bromoaniline | CuBr2 | t-BuOK | t-BuOH | 84 | 16 | 41 |
| 9 | o-Bromoaniline | CuCl2 | t-BuOK | t-BuOH | 84 | 16 | 35 |
| 10 | o-Bromoaniline | Cu(OAc)2 | t-BuOK | t-BuOH | 84 | 16 | 38 |
| 11 | o-Bromoaniline | CuI | t-BuOK | t-BuOH | 84 | 16 | 86 |
| 12 | o-Bromoaniline | CuI | KOH | t-BuOH | 84 | 16 | Trace |
| 13 | o-Bromoaniline | CuI | K2CO3 | t-BuOH | 84 | 16 | Ndd |
| 14 | o-Bromoaniline | CuI | K3PO4 | t-BuOH | 84 | 16 | Ndd |
| 15 | o-Bromoaniline | CuI | t-BuONa | t-BuOH | 84 | 16 | 64 |
| 16 | o-Bromoaniline | CuI | (t-BuO)2Mg | t-BuOH | 84 | 16 | 55 |
| 17 | o-Bromoaniline | CuI | t-BuOLi | t-BuOH | 84 | 16 | 74 |
| 18 | o-Bromoaniline | CuI | KMB | t-BuOH | 84 | 18 | 87 |
| 19 | o-Bromoaniline | CuI | KMB | 1,4-Dioxane | 100 | 18 | 56 |
| 20 | o-Bromoaniline | CuI | KMB | Toluene | 110 | 16 | 45 |
| 21 | o-Bromoaniline | CuI | KMB | DMF | 140 | 17 | 50 |
| 22 | o-Bromoaniline | CuI | KMB | NMP | 140 | 18 | 55 |
| 23 | o-Bromoaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | 92 |
| 24 | o-Bromoaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | 93e |
| 25 | 2-Chloroaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | 20 |
| 26 | 2-Iodoaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | 85 |
| 27 | o-Bromoaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | 86f |
| 28 | o-Bromoaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | 85f |
| 29 | o-Bromoaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | Traceg |
| 30 | o-Bromoaniline | CuI | KMB | t-Amyl alcohol | 120 | 17 | 43c |
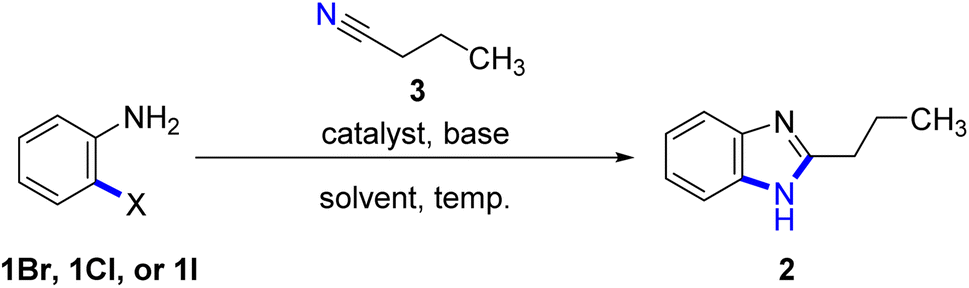
Based on the aforementioned observations, we hypothesized that the desired product could be obtained through the cyclization reaction of o-bromoaniline (1Br) with nitriles under optimized conditions (Table 1, entry 3). Hence, our investigation focused on determining the optimal conditions for this cyclization reaction, starting with an evaluation of the base and catalyst's effect.
When the reaction was conducted in the absence of a base, the desired product was not obtained. Moreover, reducing the equivalent of the base resulted in a decreased yield compared to the other case (entry 5 vs. 4).
Again, our findings revealed that the inclusion of a copper catalyst was essential, as its absence led to a significant decrease in product yield and a notable unconsumed amount of starting material (Table 1, entries 1–3, vs. 4–11). We attempted the addition of different copper catalysts to react in t-BuOH as a solvent to coordinate with o-bromoaniline. We found that copper(I) bromide (CuBr), a univalent copper catalyst, could afford the desired product in 80% yield (Table 1, entry 4). We also examined the catalytic effects of cuprous chloride (CuCl) and cuprous oxide (Cu2O). However, it was observed that these catalysts led to a decreased yield compared to CuBr (Table 1, entries 6–7, vs. 4). In addition to the previously tested copper catalysts, we also investigated divalent copper catalysts such as copper bromide (CuBr2), copper chloride (CuCl2), and copper acetate (Cu(OAc)2). However, in comparison to the monovalent copper catalysts, these divalent copper catalysts showed lower catalytic efficiency (Table 1, entries 8–10 vs. 4, 6 and 7). After screening various copper catalysts, it was determined that copper iodide (CuI) exhibited the best catalytic effect (Table 1, entry 11). The yield of the cyclization product using copper iodide reached 86%.
Several bases were tested as alternatives to potassium tert-butoxide (t-BuOK) in our study, including potassium hydroxide (KOH), potassium carbonate (K2CO3), and potassium phosphate (K3PO4). However, the use of these bases (Table 1, entries 12–14) yielded only trace amounts or no detectable cyclization product. We also explored the use of other bases such as sodium tert-butoxide (t-BuONa), magnesium di-tert-butoxide ((t-BuO)2Mg), and lithium tert-butoxide (t-BuOLi), which were found to promote the reaction and generate the desired products. Nevertheless, their efficiency was comparatively lower than that of potassium tert-amylate (KMB) (Table 1, entries 15–17 vs. 18). We investigated the effect of different solvents on the cyclization reaction. Various solvents, including tert-amyl alcohol, ethylene glycol, dioxane, toluene, dimethylformamide (DMF), and N-methylpyrrolidone (NMP), were examined. Among these screened solvents, tert-amyl alcohol was found to be more effective than other solvents (Table 1, entry 23 vs. 19–22). Under the same reaction conditions, the yield did not increase significantly with the extension of reaction time (Table 1, entry 24). It was observed that, under same conditions, the reaction of 2-chloroaniline and 2-iodoaniline resulted in the desired product with yields of 20% and 85% respectively. Nonetheless, it is evident that 2-bromoaniline proves to be the optimal 2-haloaniline derivative for this reaction (Table 1, entry 23 vs. 25–26).
Interestingly, when attempting to add N,N-dimethylethylenediamine or proline as a ligand, the yield rather decreased (Table 1, entries 27–28). The results clearly indicate that maintaining a reaction time of 16–17 hours without the use of ligands is the optimal approach. Therefore, we selected copper iodide as the catalyst, potassium tert-amylate (KMB) as the base, and a refluxing (120 °C) cyclization reaction in tert-amyl alcohol as the optimal reaction conditions (Table 1, entry 23).
Additional control experiments were conducted under optimized conditions, specifically without a base, resulting in no detectable cyclization product. Using less than 2 equivalent of the base resulted in a trace amount (entry 29) and low yield of the desired product, indicating a lower catalytic efficiency as compared to other yield (entry 30 vs. 23). These results unequivocally demonstrate that copper-catalyzed cyclization reactions require a substantial amount of base, along with a catalytic amount of copper catalyst, to produce the desired benzimidazoles.12
Following the establishment of the optimal cyclization reaction conditions, we proceeded to explore the scope and versatility of aliphatic nitrile substrates and o-bromoaromatic amines (Table 2). Remarkably, our research revealed that when the benzene ring of the o-bromoaromatic amine substrate (1a–1c, 1f, 1k, 1o) was substituted with a methyl group or an ester, the reaction proceeded smoothly, leading to the formation of the desired products (2a–2c, 2f, 2k, 2o) with yields ranging between 86-95%. We further explored the influence of different substituents on the cyclization reaction by investigating the electronic effects of substituents on the benzene ring of o-bromoaromatic amine. Remarkably, the presence of a methoxy group in the phenyl substrates (1d, 1e) resulted in efficient cyclization reactions, yielding benzimidazoles (2d and 2e) with a high yield of 98%. Moreover, when the phenyl o-bromoaromatic amine substrates (1g–1h) contained chlorine or fluorine substituents, the cyclization products (2g and 2h) were obtained in even higher yields of 94% and 88%, respectively.
| a The reactions were performed on the scale of 1.0 mmol of 1Br, 1a–1i, 1k–1o and 1q–1r under the conditions: 2.0 equiv. of nitriles, 2 equiv. of KMB, 0.1 equiv. of CuI catalyst, 6 ml t-amyl alcohol solvent, 120 °C for 17 h. The yields were given as isolated yields. |
|---|
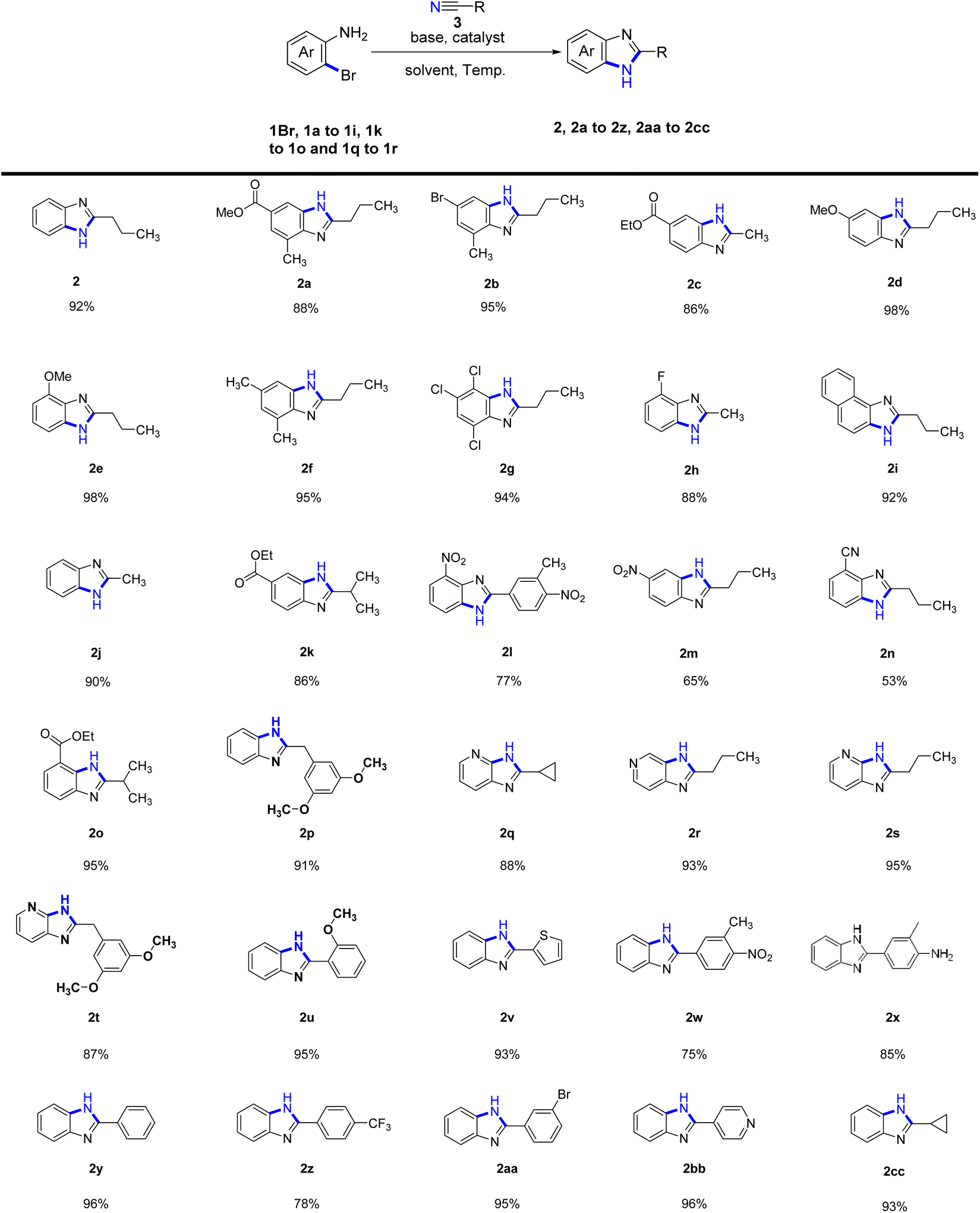 |
Furthermore, we successfully investigated the reactivity of the phenyl unsubstituted o-bromoaniline substrate (1Br) in cyclization reactions with various nitriles, resulting in higher yields of products (2, 2j, 2cc, 2p). However, when o-bromoaniline substrates with electron-withdrawing groups such as nitro (1m) and nitrile (1n) substituents were used, the yields of the corresponding products (2m, 2n) were lower, ranging from 53% to 65%. Interestingly, o-bromoaromatic amines bearing 1-naphthyl (1i) and pyridyl (1q, and 1r) substituents also exhibited reactivity in the intermolecular cyclization reactions, affording products 2i (92% yield), 2q (88% yield), 2r (93% yield), 2s (95% yield) and 2t (87% yield).
Again, an investigation was conducted on the scope of aromatic nitrile substrates and o-bromoaromatic amines. The results showed that most aromatic nitrile substrates and o-bromoarylamine cyclization yielded satisfactory results. However, it was observed that the presence of charge-drawing functional groups, such as 3-methyl-4-nitrobenzonitrile and p-trifluoromethylbenzonitrile, led to lower cyclization yields (2l, 2w, and 2z at 75–78%). Other aromatic nitrile substrates, such as benzonitrile, 2-methoxybenzonitrile, 2-nitrilethiophene, and 3-methyl-4-aminobenzonitrile can all undergo cyclization with o-bromoaniline (1Br) to afford the desired products with higher yields. Specifically, the desired products 2y, 2u, 2v, and 2x were obtained with yields of 96%, 95%, 93%, and 85%, respectively. Remarkably, even when 3-bromobenzonitrile was cyclized with o-bromoaniline (1Br), the reaction exhibited good functional group compatibility as the bromine atom did not participate in the reaction, leading to the formation of the desired product 2aa with a yield of 95%. Similarly, the reaction of aromatic nitriles with heterocyclic rings, such as 4-nitrilepyridine and o-bromoaniline (1Br) successfully gave the desired product 2bb with a yield of 96%.
Post-application for the synthesis of thiabendazole, and bendazol
Subsequently, the applicability of our cyclization approach was demonstrated in a concise synthesis of antihypertensive drug, bendazol, and anthelminthic drug, thiabenzadole.2 Gram-scale cyclization of 2-bromoaniline with thiazole-4-carbonitrile and potassium-2-methylbutan-2-olate in tert-amyl alcohol in the presence of CuI at 120 °C afforded thiabendazole with an impressive isolated yield of 78%, surpassing the reported yield of 64% achieved using a catalyzed-polyphosphoric acid cyclization method.13 Likewise, this succinct approach was utilized to synthesize the antihypertensive drug, bendazol, yielding a light yellow solid of bendazol in a 82% isolated yield through DCM/hexane re-crystallization, showcasing outstanding functional group compatibility for the cyclization of the benzonitrile substrate as compared to previous report.14Based on the experimental results and relevant literature,11,13 we propose a reaction mechanism pathway for the synthesis of benzimidazole compounds via the cyclization of o-bromoaromatic amines and nitrile substrates, as depicted in Fig. 2. Initially, under basic conditions, the amino group of the o-bromoaromatic amine undergoes deprotonation, resulting in the formation of intermediate C with a negatively charged nitrogen atom. Subsequently, it reacts with the cyano group of the nitrile substrate to produce intermediate amidine D. Introducing a copper iodide catalyst leads to the formation of a metal intermediate E. Subsequently, intramolecular bromine transmetalation occurs, resulting in the formation of intermediate F. Ultimately, through reduction and elimination reactions, the final product, benzimidazole compound, is generated, and the catalyst is released, thereby completing the catalytic cycle. These results demonstrate the efficiency of this synthetic pathway for the synthesis of nitrile-based compounds. The findings further demonstrate the broad applicability of the present synthetic strategy in producing valuable imidazole derivatives.
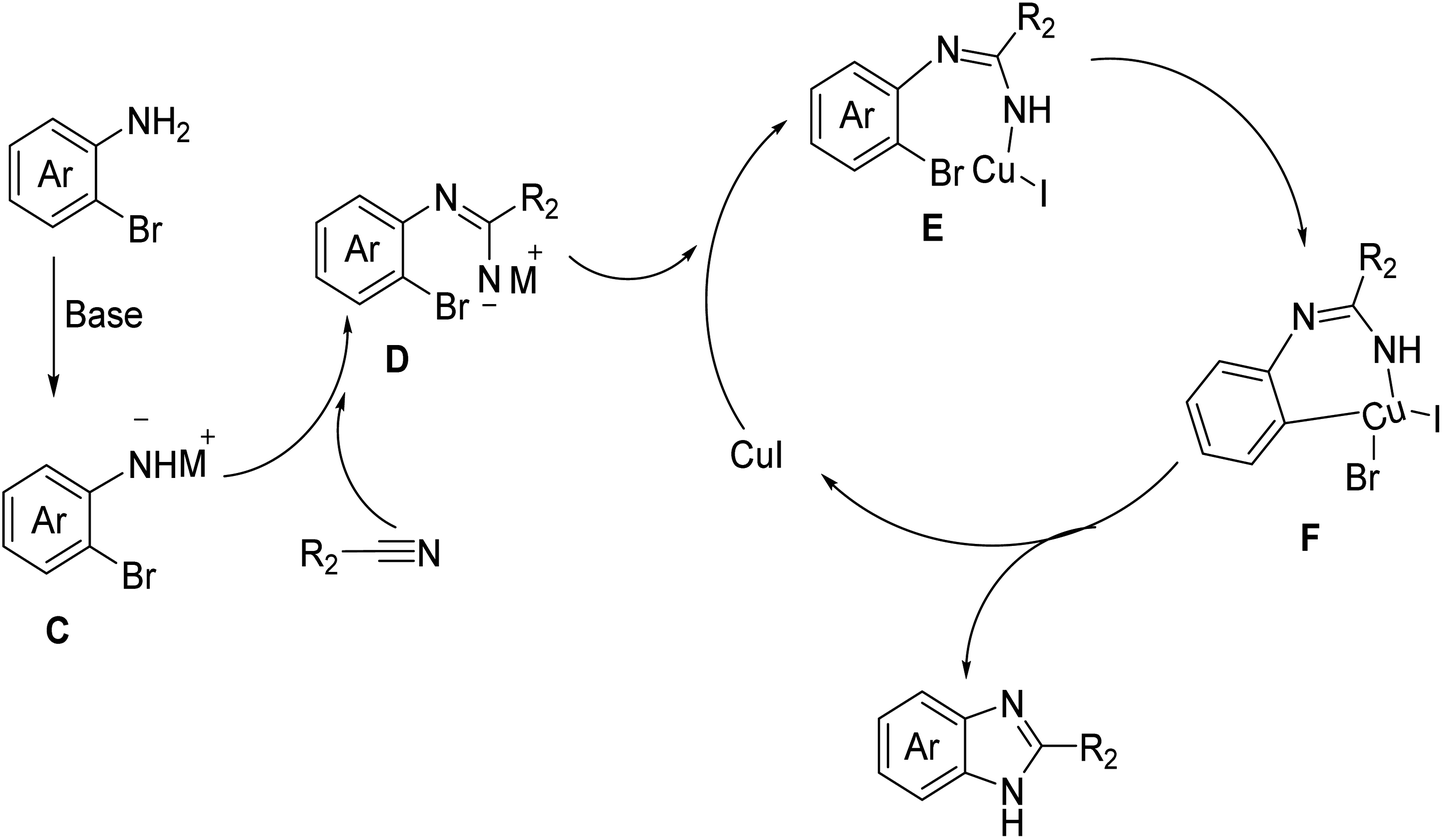 | ||
| Fig. 2 Proposed mechanism of the cyclization reaction between o-bromoaromatic amines and nitrile substrates. | ||
Conclusions
In this study, we have successfully developed an improved and ligand-free copper-catalyzed cyclization method for the synthesis of benzimidazoles. By utilizing the cyclization of o-bromoarylamine with nitriles, we have demonstrated remarkable yields of benzimidazoles, with optimization leading to 30 examples with yields of up to 98%. Our approach builds upon the previous work of Xiang et al.9 and Yu et al.,10 offering significant advantages in terms of simplicity, compatibility with various functional groups, substrate scope, and overall efficiency compared to their methods and other existing methodologies. Additionally, we have validated the applicability of our method in the synthesis of two benzimidazole-based drugs, namely bendazol and thiabenzadole, yielding 82% and 78% respectively on a 100 gram scale. Notably, our method eliminates the need for ligands, nitric acid, sulfuric acid, and polyphosphoric acid, emphasizing its versatility and environmentally friendly nature. An ongoing research at our laboratory explores additional applications of this approach.Experimental section
General information
1H, 19F and 13C NMR spectra were recorded on a Brucker 500 Hz or 600 Hz instrument. Chemical shifts for 1H NMR are reported in parts per million (ppm), and multiplicities are denoted as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. Chemical shifts for 13C NMR are reported as the chemical shift. Electrospray ionization (ESI) mass spectra were measured using a Thermo Fisher FINNIGAN LTQ instrument. All high-resolution mass spectroscopy (HRMS) results were obtained on an Agilent 1290-6545 UHPLC-QTOF LC/MS spectrometer. TLC inspections were performed on silica gel plates (GF-254). All commercially available chemicals and solvents were used without further purification unless otherwise noted.General procedure
O-Bromoarylamine (1.0 mmol, 1.0 eq.), potassium-2-methylbutan-2-olate (2.0 mmol, 2 eq.), CuI (0.1 mmol, 0.1 eq.), nitriles 3 (2 mmol, 2 eq.), and tert-amyl alcohol (6.0 ml) were charged to a Schleck tube that had been purged with nitrogen. After addition, the mixture was then warmed to 110–120 °C and held for 17 h. The reaction was considered complete when the conversion of o-bromoarylamine was ≥99%. The reaction mixture was concentrated in vacuum at 40–45 °C to remove most of the solvents to give the residue. The residue was purified by silica-gel column chromatography to give benzimidazoles.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) were observed as indicative of the presence of tautomerism; 1H NMR (400 MHz, DMSO-d6, major isomer) δ 12.30 (s, 1H), 7.51 (s, 1H), 7.08 (s, 1H), 2.77 (t, J = 7.1 Hz, 2H), 2.45 (s, 3H), 1.78 (dt, J = 14.3, 7.2 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, major isomer) δ 156.78, 144.90, 130.26, 124.78, 118.41, 113.43, 111.35, 30.90, 21.44, 17.09, 14.16; 1HNMR (400 MHz, DMSO-d6, minor isomer) δ 12.24 (s, 1H), 7.40 (s, 1H), 7.08 (s, 1H), 2.77 (t, J = 7.1 Hz, 2H), 2.48 (s, 3H), 1.78 (dt, J = 14.3, 7.2 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, minor isomer) δ 156.8, 155.6, 144.9, 142.3, 135.4, 133.6, 130.3, 124.9, 124.2, 123.2, 118.4, 113.8, 113.4, 111.3, 30.1, 30.9, 21.4, 17.1, 16.7, 14.1. HRMS (ESI): m/z [M + H]+ calcd for C11H14BrN2 253.0335, found: 253.0332.
:9) were observed as indicative of the presence of tautomerism; 1H NMR (400 MHz, DMSO-d6, major isomer) δ 12.30 (s, 1H), 7.51 (s, 1H), 7.08 (s, 1H), 2.77 (t, J = 7.1 Hz, 2H), 2.45 (s, 3H), 1.78 (dt, J = 14.3, 7.2 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (100 MHz, DMSO-d6, major isomer) δ 156.78, 144.90, 130.26, 124.78, 118.41, 113.43, 111.35, 30.90, 21.44, 17.09, 14.16; 1HNMR (400 MHz, DMSO-d6, minor isomer) δ 12.24 (s, 1H), 7.40 (s, 1H), 7.08 (s, 1H), 2.77 (t, J = 7.1 Hz, 2H), 2.48 (s, 3H), 1.78 (dt, J = 14.3, 7.2 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, minor isomer) δ 156.8, 155.6, 144.9, 142.3, 135.4, 133.6, 130.3, 124.9, 124.2, 123.2, 118.4, 113.8, 113.4, 111.3, 30.1, 30.9, 21.4, 17.1, 16.7, 14.1. HRMS (ESI): m/z [M + H]+ calcd for C11H14BrN2 253.0335, found: 253.0332.13C NMR (100 MHz, CD3OD) δ 159.6, 132.6, 131.2, 130.7, 128.1, 119.3, 117.7, 29.65, 22.6, 13.9. HRMS (ESI): m/z [M + H]+ calcd for C10H10Cl3N2 262.9904, found: 262.9898.
:1) were observed as indicative of the presence of tautomerism; 1H NMR (400 MHz, DMSO-d6, major isomer) δ 12.47 (s, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.13–7.03 (m, 1H), 6.99–6.84 (m, 1H), 2.49 (s, 3H). 1HNMR (400 MHz, DMSO-d6, minor isomer) δ 12.71 (s, 1H), 7.34 (d, J = 7.6 Hz, 1H), 7.13–7.03 (m, 1H), 7.01–6.89 (m, 1H), 2.49 (s, 3H); 13C NMR (101 MHz, DMSO) δ 154.2, 152.3, 151.7, 138.1, 138.0, 132.0, 122.3, 122.2, 107.5, 106.80, 106.6, 15.0; 19F NMR (377 MHz, DMSO) δ −135.23. HRMS (ESI): m/z [M + H]+ calcd for C8H8FN2 151.0666, found: 151.0662.Synthesis of bendazol. The reaction mixture, consisting of 1 (1 eq.), potassium-2-methylbutan-2-olate (2 eq.), and 2-phenylacetonitrile 3 (2 eq.) in the presence of CuI (0.1 eq.), was vigorously stirred in a reaction vessel for 17 hours at 110–120 °C. Upon completion, the reaction was cooled to 25 °C and quenched with water. The resulting mixture was then extracted with ethyl acetate, and the organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was subsequently purified by re-crystallization from DCM/hexane, yielding bendazol as a light yellow solid (80.9 g, 82%). 1H NMR (400 MHz, DMSO-d6) δ 15.53 (s, 1H), 7.77 (dt, J = 6.7, 3.4 Hz, 2H), 7.62–7.46 (m, 4H), 7.44–6.86 (m, 3H), 4.57 (s, 2H); 13C NMR (101 MHz, DMSO) δ 152.5, 134.0, 130.7, 129.1, 128.9, 127.6, 125.5, 113.8, 31.8.
Synthesis of thiabendazole. Following the general procedure, this material was obtained as white solid in a yield 93.2 g (78%). 1H NMR (400 MHz, DMSO-d6) δ 13.00 (s, 1H), 9.34 (d, J = 2.0 Hz, 1H), 8.47 (d, J = 1.9 Hz, 1H), 7.61 (dd, J = 55.4, 7.1 Hz, 2H), 7.23 (dt, J = 8.6, 4.9 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 155.5, 147.0, 146.9, 143.7, 134.3, 122.5, 121.7, 119.3, 118.7, 111.7.
Conflicts of interest
The authors declare that they have no apparent conflicting financial interests or personal connections that could have appeared to impact the findings presented in this manuscript.Acknowledgements
This work was supported by the Institute of Drug Innovation of the Chinese Academy of Sciences for research on antiviral protease inhibitors (Grant No. CASIMM120234003), the China-Uzbekistan New Drug, Belt and Road Joint Laboratory Construction and Innovative Drug Research for National Key R&D Program of China (Grant No. 2020YFE0205600), and the University of Chinese Academy of Sciences (UCAS), for the UCAS Scholarship for International students.References
- (a) R. S. Keri, A. Hiremathad, S. Budagumpi and B. Nagaraja, Comprehensive Review in Current Developments of Benzimidazole-Based Medicinal Chemistry, Chem. Biol. Drug Des., 2015, 86, 19–65 CrossRef PubMed; (b) X. Liu, Y. Han, X. Ge and Z. Liu, Imidazole and Benzimidazole Modified Half-Sandwich IridiumIII N-Heterocyclic Carbene Complexes: Synthesis, Anticancer Application, and Organelle Targeting, Front. Chem., 2020, 8, 182 CrossRef CAS PubMed; (c) G. Singh and H. Sahota, Impact of benzimidazole and dithiocarbamate fungicides on the photosynthetic machinery, sugar content and various antioxidative enzymes in chickpea, Plant Physiol. Biochem., 2018, 132, 166–173 CrossRef CAS PubMed.
- (a) M. Sharpe, B. Jarvis and K. L. Goa, Telmisartan: a review of its use in hypertension, Drugs, 2001, 61, 1501–1529 CrossRef CAS PubMed; (b) S. C. Benson, H. A. Pershadsingh, C. I. Ho, A. Chittiboyina and T. W. Kurtz, Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity, Hypertension, 2004, 43, 993–1002 CrossRef CAS PubMed; (c) K. P. Barot, S. Nikolova, I. Ivanov and M. D. Ghate, Novel Research Strategies of Benzimidazole Derivatives: A Review, Mini-Rev. Med. Chem., 2013, 13, 1421–1447 CrossRef CAS PubMed; (d) S. I. Alaqeel, Synthetic Approaches to Benzimidazoles from O-Phenylenediamine: A Literature Review, J. Saudi Chem. Soc., 2017, 21, 229–237 CrossRef CAS; (e) S. R. Brishty, M. J. Hossain, M. U. Khandaker, M. R. I. Faruque, H. Osman and S. M. Rahman, A comprehensive account on recent progress in pharmacological activities of benzimidazole derivatives, Front. Pharmacol., 2021, 12, 762807 CrossRef CAS PubMed.
- M. Vaithiyalingam, K. R. Mohan, C. Kamaraj, V. Sugumar, N. Manivannan, S. Kadaikunnan and G. Ghodake, Facile Synthesis of Benzimidazoles via Oxidative Cyclization of Acyclic Monoterpene Aldehyde with Diamines: Studies on Antimicrobial and in Vivo Evaluation of Zebrafish, Chem. Biodiversity, 2023, 20, 6, DOI:10.1002/cbdv.202300315.
- Q. Liu, L. Wang, J. Liu, S. Ruan and P. Li, Facile synthesis of carbamoylated benzimidazo[2,1-a]isoquinolin-6(5H)-ones via radical cascade cyclization under metal-free conditions, Org. Biomol. Chem., 2021, 19, 3489–3496 RSC.
- (a) H. Qin, A. Odilov, E. M. Bonku, F. Zhu, T. Hu, H. Liu, H. A. Aisa and J. Shen, Facile Synthesis of Benzimidazoles via N-Arylamidoxime Cyclization, ACS Omega, 2022, 7, 45678–45687, DOI:10.1021/acsomega.2c06554; (b) A. Odilov, H. Qin, E. M. Bonku, F. Zhu, F. Yang and J. Shen, An efficient synthesis of the last step key intermediate for telmisartan via Pd-catalyzed carbonylative cyclization, Tetrahedron, 2023, 148, 133702 CrossRef CAS.
- C. Chen, C. Chen, B. Li, J. Tao and J. Peng, Aqueous Synthesis of 1-H-2-Substituted Benzimidazoles via Transition-Metal-Free Intramolecular Amination of Aryl Iodides, Molecules, 2012, 17, 12506–12520, DOI:10.3390/molecules171112506.
- P. Li, Y. Ji, W. Chen, X. Zhang and L. Wang, The facile synthesis of 2-bromoindoles via Cs2CO3-promoted intramolecular cyclization of 2-(gem-dibromovinyl)anilines under transition-metal-free conditions, RSC Adv., 2013, 3, 73–78 RSC.
- H. M. Refaat, Synthesis and anticancer activity of some novel 2-substituted benzimidazole derivatives, Eur. J. Med. Chem., 2010, 45, 2949–2956 CrossRef CAS PubMed.
- S. Xiang, W. Tan, D. Zhang, X. Tian, C. Feng, B. Wang, K. Zhao, P. Hu and H. Yang, Synthesis of benzimidazoles by potassium tert-butoxide-promoted intermolecular cyclization reaction of 2-iodoanilines with nitriles, Org. Biomol. Chem., 2013, 11, 7271–7275 RSC.
- J. Yu, Y. Xia and M. L.-P. Lu, an efficient N,O-bidentate ligand for copper-catalyzed intramolecular cyclization reaction of 2-iodoanilines with nitriles for the synthesis of benzimidazoles, Appl. Organomet. Chem., 2014, 28, 764–767 CrossRef CAS.
- H. Göker, C. Kuş, D. W. Boykin, S. Yildiz and N. Altanlar, Synthesis of some new 2-substituted-phenyl-1H-benzimidazole-5-carbonitriles and their potent activity against Candida species, Bioorg. Med. Chem., 2002, 10, 2589–2596 CrossRef PubMed.
- (a) T. Aneeja, M. Neetha, C. M. A. Afsina and G. Anilkumar, Progress and prospects in copper-catalyzed C–H functionalization, RSC Adv., 2020, 10(57), 34429–34458 RSC; (b) X. D. Wang, L. H. Zhu, P. Liu, X. Y. Wang, H. Y. Yuan and Y. L. Zhao, Copper-catalyzed cascade cyclization reactions of diazo compounds with tert-butyl nitrite and alkynes: one-pot synthesis of isoxazoles, J. Org. Chem., 2019, 84(24), 16214–16221 CrossRef CAS PubMed.
- H. D. Brown, A. R. Matzuk, I. Ilves, L. H. Peterson, S. A. Harris, L. H. Sarett and A. C. Cuckler, Antiparasitic drugs. IV. 2-(4'-thiazolyl)-benzimidazole, a new anthelmintic, J. Am. Chem. Soc., 1961, 83(7), 1764–1765 CrossRef CAS.
- A. J. Blacker, M. M. Farah, M. I. Hall, S. P. Marsden, O. Saidi and J. M. Williams, Synthesis of benzazoles by hydrogen-transfer catalysis, Org. Lett., 2009, 11(9), 2039–2042 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Including copies of 1H NMR, 19F NMR, 13C NMR, and mass spectra for all compounds. See DOI: https://doi.org/10.1039/d4ra00245h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |