
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of transition metals in coinage metal nanoclusters for the remediation of toxic dyes in aqueous systems
Priyanka Sharma
,
Mainak Ganguly
 * and
Mamta Sahu
* and
Mamta Sahu
Department of Chemistry, Manipal University Jaipur, Dehmi Kalan, Jaipur 303007, India. E-mail: humansense2009@gmail.com
First published on 9th April 2024
Abstract
A difficult issue in chemistry and materials science is to create metal compounds with well-defined components. Metal nanoclusters, particularly those of coinage groups (Cu, Ag, and Au), have received considerable research interest in recent years owing to the availability of atomic-level precision via joint experimental and theoretical methods, thus revealing the mechanisms in diverse nano-catalysts and functional materials. The textile sector significantly contributes to wastewater containing pollutants such as dyes and chemical substances. Textile and fabric manufacturing account for about 7 × 105 tons of wastewater annually. Approximately one thousand tons of dyes used in textile processing and finishing has been recorded as being discharged into natural streams and water bodies. Owing to the widespread environmental concerns, research has been conducted to develop absorbents that are capable of removing contaminants and heavy metals from water bodies using low-cost technology. Considering this idea, we reviewed coinage metal nanoclusters for azo and cationic dye degradation. Fluorometric and colorimetric techniques are used for dye degradation using coinage metal nanoclusters. Few reports are available on dye degradation using silver nanoclusters; and some of them are discussed in detailed herein to demonstrate the synergistic effect of gold and silver in dye degradation. Mostly, the Rhodamine B dye is degraded using coinage metals. Silver nanoclusters take less time for degradation than gold and copper nanoclusters. Mostly, H2O2 is used for degradation in gold nanoclusters. Still, all coinage metal nanoclusters have been used for the degradation due to suitable HOMO–LUMO gap, and the adsorption of a dye onto the surface of the catalyst results in the exchange of electrons and holes, which leads to the oxidation and reduction of the adsorbed dye molecule. Compared to other coinage metal nanoclusters, Ag/g-C3N4 nanoclusters displayed an excellent degradation rate constant with the dye Rhodamine B (0.0332 min−1). The behavior of doping transition metals in coinage metal nanoclusters is also reviewed herein. In addition, we discuss the mechanistic grounds for degradation, the fate of metal nanoclusters, anti-bacterial activity of nanoclusters, toxicity of dyes, and sensing of dyes.
 Priyanka Sharma | Priyanka Sharma obtained her BSc and MSc degrees from Maharaja Brij University in India. She is now pursuing her PhD from Manipal University Jaipur, India, under the guidance of Dr Mainak Ganguly. Her area of interest is materials science and environmental science. |
 Mainak Ganguly | Dr Mainak Ganguly received his PhD from Indian Institute of Technology, Kharagpur, India, in 2014. He had acted as postdoctoral researcher up to 2019 in Furman University (USA) and McGill University (Canada). He is currently working as an Asst. Professor in the Department of Chemistry, Manipal University Jaipur (India). His research interests include nanoparticles, clusters, biophysical chemistry, environmental remediation, etc. He has published more than 55 papers and two book chapters. |
 Mamta Sahu | Mamta Sahu received her BSc from Subodh Girls PG collage (Jaipur, India) and MSc degree from RK PG collage (Jaipur, India). She is currently pursuing her PhD under the supervision of Dr Mainak Ganguly from Manipal University Jaipur, India. Her area of interest is environmental nanoscience. |
1. Motivation
The widespread presence of organic dyes in industrial wastewater from the paper, textile, and garment industries pollutes the environment significantly.1 According to multiple investigations, 10–12% of dyes used in the textile industry are lost during synthesis and processing activities and end up in wastewater.2–6 These dye-polluted effluents contain colors that are non-biodegradable, extremely toxic, and detrimental to living organisms.7,8 Dyes are vividly visible in water even at extremely low concentrations (<1 ppm) and can damage aquatic habitats.9–11 As a result, color removal from wastewaters is critical.12 Contamination by different dyes poses serious threats to mankind. A handful of review articles have been published on sensing using coinage metal nanoparticles and nanoclusters. Kanelidis et al.13 reviewed the role of ligands in coinage metal nanoparticles for electronics. Chinta et al.14 summarized colorimetric tests for natural amino acids using coinage metal nanoparticles. Sharma et al.15 reviewed coinage metal-enhanced fluorescence using diiminic Schiff bases for the selective and sensitive detection of aqueous pollutants. Similarly, metal nanocluster-based luminescent biosensors via etching chemistry were reviewed by Si et al.16Nanostructures are classified into nanoparticles (PNls) and nanoclusters (CNs) depending on their size. Size alteration is also related to various physico-chemical properties. CNs exhibit fluorescence due to d–d transition. Moreover, their ultra-small size is related to a very high surface area. Dye degradation with nanoparticles has already been reviewed. Metal nanoclusters of Cu, Ag, and Au for dye degradation is a very important area for environmental remediation and needs to be reviewed in detail. In the present manuscript, a comparative study of coinage metal nanoclusters for dye degradation was reviewed along with dye sensing. This review article may be an important for young researchers to venture into the field of sensing based on dye degradation.
2. Introduction
Significant efforts have been undertaken in recent years to investigate the photophysical and photochemical behavior of multicomponent nanostructured assemblies made up of metals, semiconductors, and photoactive dyes.17–29 Metal nanoclusters (CNsM) are molecularly precise structures made up of a certain number of metal atoms plus capping ligands. Their emergence bridges the gap between tiny molecules and nanomaterials, allowing for a better grasp of many fundamental elements of science.30 CNsM's physical and chemical characteristics are heavily influenced by their core structure as well as the arrangement of their surface ligands. Atomically defined gold, silver and copper CNs are a special type of CNsM that are used in catalysis, optics, biology, and sensing.31–34 Coinage CNsM with atomically precise structures, such as Au, Ag, and Cu, can be regarded as extreme-trivial metal nanoparticles (PNlsM), aggregated from dozens to hundreds of metal atoms protected by single-layer organic ligands. They are metal atom aggregates with sizes generally less than 3 nm, occupying the transition scale among metal-atom and plasmonic PNlsM.35–37 Because CNsM is much smaller than PNslM, the initial surface plasmon resonance effect vanishes. As the particle size is close to the Fermi wavelength, significant alterations take place in the particle's energy levels. This is the result of the quantum size effect, which causes the electron levels of CNsM the transition from a quasi-continuous to a discrete state. This leads to molecular-like behaviors (intense fluorescence due to energy gaps between the LUMO and HOMO).38Due to the presence of electrons with incomplete d-orbitals in their atomic structures, transition metals are categorized as d-block elements.39 They differ from the core group elements in both their physical and chemical properties. Because the energy levels of the d and ns shells are so close together, these elements may show a variety of oxidation states, making them relevant for further study. The range of applications for transition metals is widened as a result as there are an enormous number of oxide combinations possible.40,41
Because of its stability, nontoxicity, and high electronegativity, gold (Au) is the most appealing choice for noble metal-based photocatalysts.42 Fluorescent gold CNs (CNsAu), aggregation of numerous to tens of metal-atoms, have piqued the interest of scientists in recent years due to their exceptional optical, electro-magnetic, and catalytic properties resulting from their very tiny size (2 nm).43–46 Few-atom CNsAu are difficult to fabricate due to their strong aggregation tendency. However, several synthetic approaches have been successfully developed to produce CNsAu with unique properties, showing great and broad applicability in diverse fields like bio-imaging, chemo-sensing, and catalysis.47–51 The formation of CNsAu and CNsAg has also received a lot of attention in investigations on noble metal CNs. In comparison to other precious metals, copper is comparatively plentiful and cheap.52–57 CNsCu, on the other hand, are less stable because of their high oxidation susceptibility, and their fluorescence quantum yield (QY) is lower.58 In recent years, efforts have been undertaken to enhance the physicochemical and optical characteristics of CNsCu to widen their usefulness in a variety of domains, including sensing and nanodrugs.59 Because of their strong photo-stability, high QY emitters, and less toxicity, ligand-protected CNsAg have attracted a lot of interest as new fluorophores. These characteristics render them suitable for use in microscopy settings when DNA is used as a template, with potential biocompatibility and applications in bio-labeling and sensing.
In this review article, we concentrated on the coinage metal CNs for the elimination of hazardous dye both via adsorption and degradation with plausible mechanisms. Moreover, the role of transition metals in such removal processes was also reviewed. The sensing of the dye is discussed here with coinage metal clusters.
3. Dye
Unlike pigments, which chemically bind to the substrate they color, dyes are colored substances that link chemically to the surface (they are applied to). To make dye faster on fiber, a mordant may need to be added. Dye is frequently applied in an aqueous solution.60 Textile dyes, like a wide range of industrial contaminants, are exceedingly poisonous and potentially carcinogenic and have been connected to several diseases in people and animals as well as environmental damage.61 Colors in sediments and soil bioaccumulate are transferred to public water supply systems using their refractory nature under aerobic circumstances, especially in conventional treatment facilities. As the reduction of azo-type compounds leads to risky aromatic amines, they can be partially destroyed or transformed in the presence of anoxic sediments despite the majority's resilience to the environment. Another potential is that dyes will be combined with intermediate synthetic chemicals or their breakdown products to produce new mutagenic and carcinogenic substances. Dyes have many applications and can be found in the effluents of numerous manufacturing and processing units; hence, their discharge has a significant environmental effect. As waste from several industries produces significant pollution, protecting the environment becomes a difficult task. The economy of Asia and other nations is significantly influenced by the textile sector. The textile industry often uses dyes to color fabrics.62 A very little amount of dye in the water is extremely noticeable and can be hazardous to aquatic life. In the textile company, substantial volumes of aqueous wastes and dye effluents with strong persistent color and high biological oxygen demand (BOD) loading are released from the dyeing process, which are both aesthetically and ecologically unsatisfactory.63 Dyes are organic compounds with a complex aromatic molecular structure that may give other things a brilliant and vivid color. However, dyes' complex aromatic molecular structures make them exceedingly durable, making biodegradation more challenging.64 Dyes are generally found in dye house effluent at concentrations ranging from 10 to 50 mg L−1; nevertheless, color has been observed at concentrations greater than 1 mg L−1. Except for the dye, most contaminants may be eliminated using chemical and physical processes. Metals are sequestered by synthetic dyes, producing microtoxicity in fish and other aquatic creatures.65–67Fig. 1 displays the toxicity of dyes in wastewater, the treatment of the dye-contaminated wastewater, and the dye degradation mechanism in a schematic form.
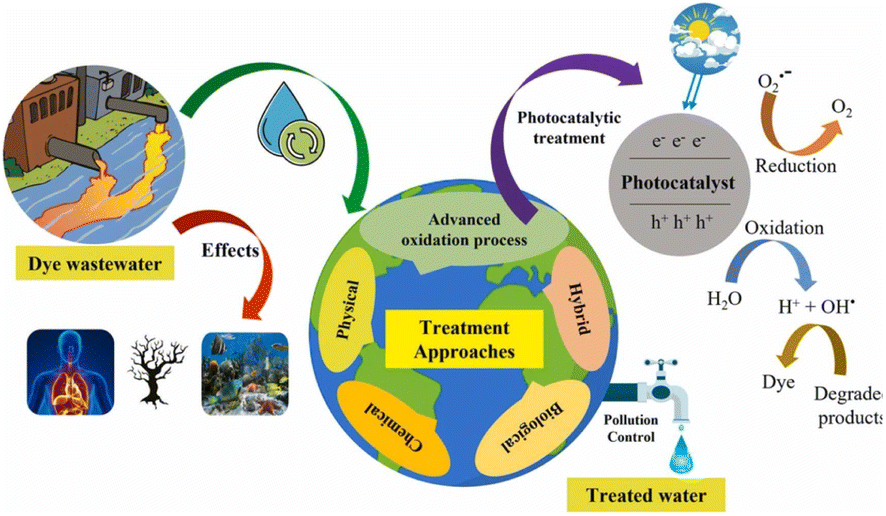 | ||
| Fig. 1 Toxicity of dyes in wastewater. Reproduced with permission from ref. 67, copyright 2024, J. Environ. Chem. Eng. | ||
3.1 Classification of dyes
Dyes can be classified into two classes based on their chemical structure and application techniques [Fig. 2]. However, dyes such as methylene blue, Rhodamine B, methyl orange, congo red, amido black 10b, and coumarin are utilized extensively in textile and natural life. These colors are also poisonous and detrimental to human health. These dyes are so far reported to be degraded using coinage CNsM.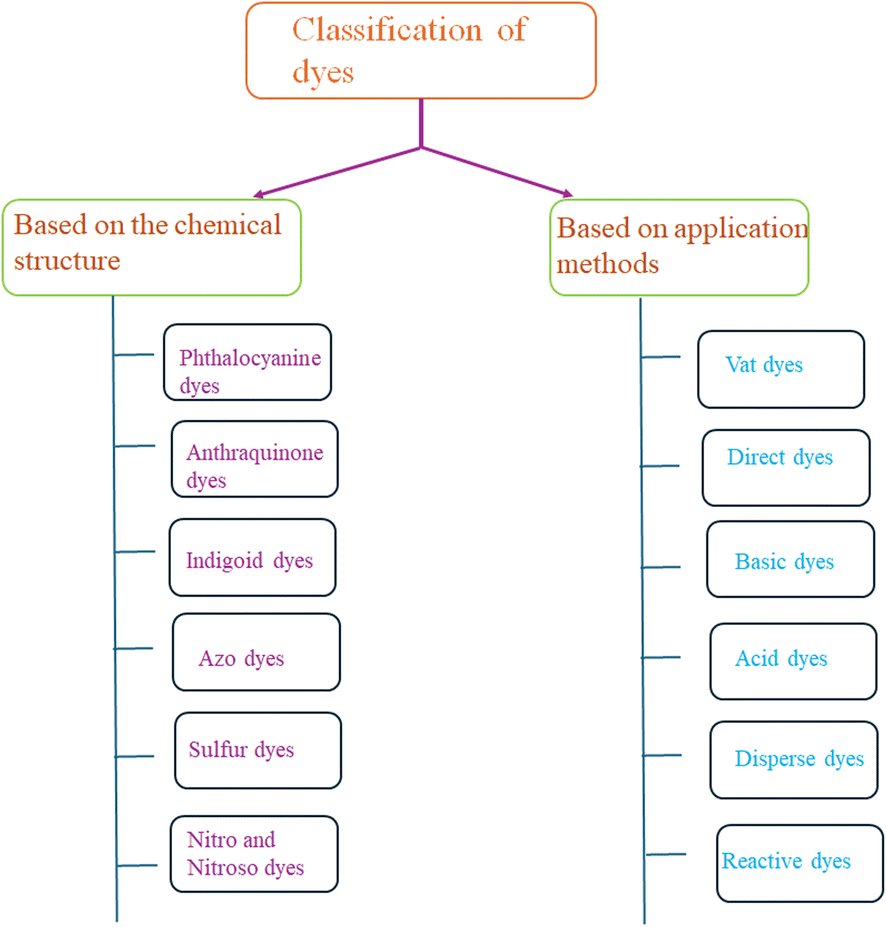 | ||
| Fig. 2 Classification of dye based on chemical structure and applications. | ||
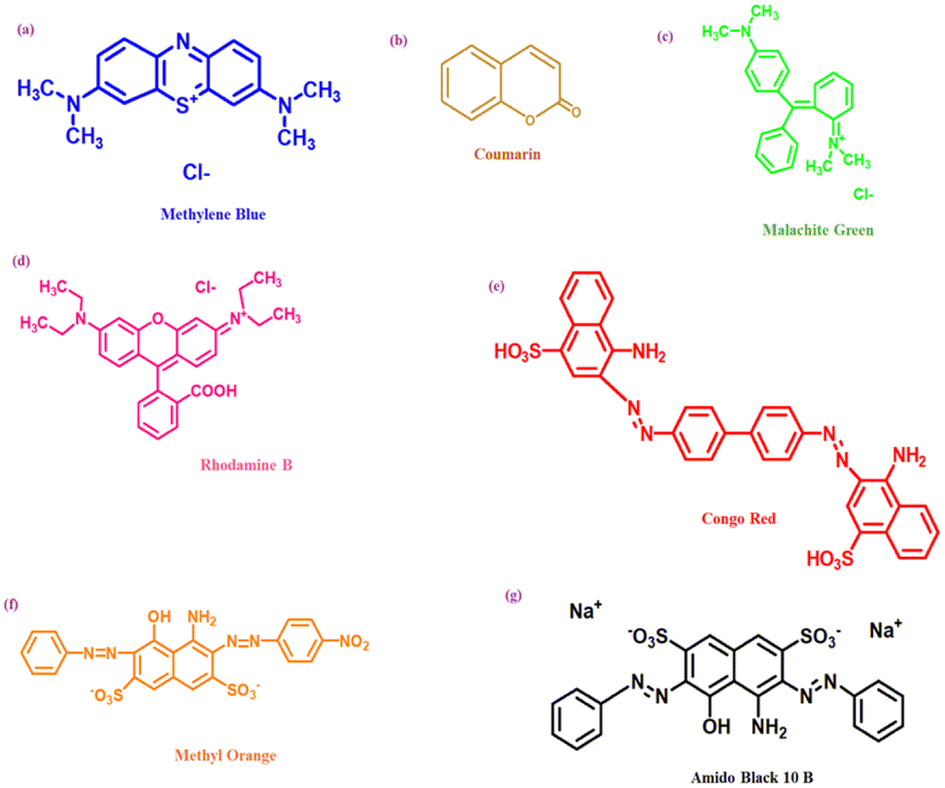 | ||
| Fig. 3 Structure of different dyes [(a) methylene blue, (b) coumarin, (c) malachite green, (d) rhodamine B, (e) congo red, (f) methyl orange and (g) amido black 10B] degradable via CNsM. | ||
4. Nanocluster
Specific formulas can describe nanoclusters, much like well-characterized organic or organometallic materials. These CNs-based molecules represent inorganic–organic hybrid substances that act as a crucial link in communicating between atoms and PNls. Their ultrasmall size, ranging from sub-nanometer to two/three nanometers in size for the metal core,90–93 positions them as a “missing link.” These CNs exhibit robust quantum size effects (characterized by distinct electronic energy levels), numerous absorption bands resulting from one-electron transitions, enhanced emission, magnetic behavior, remarkable catalytic reactivity, and nonlinear optical characteristics.94–96 A scale function of atomicity within the cluster, derived from the energy variations between the HOMO & LUMO, indicates that CNsM with dimensions akin to the electron's Fermi wavelength (approximately 0.7 nm) and size-dependent fluorescence displays molecule-like characteristics with quantifiable electronic properties and luminescence.97–100 Because of the quantum size effect, the size of the CNs is critical for their catalytic activity. Furthermore, the unsaturated coordination site with increased d-band energy improves the catalytic reactivity [Fig. 4].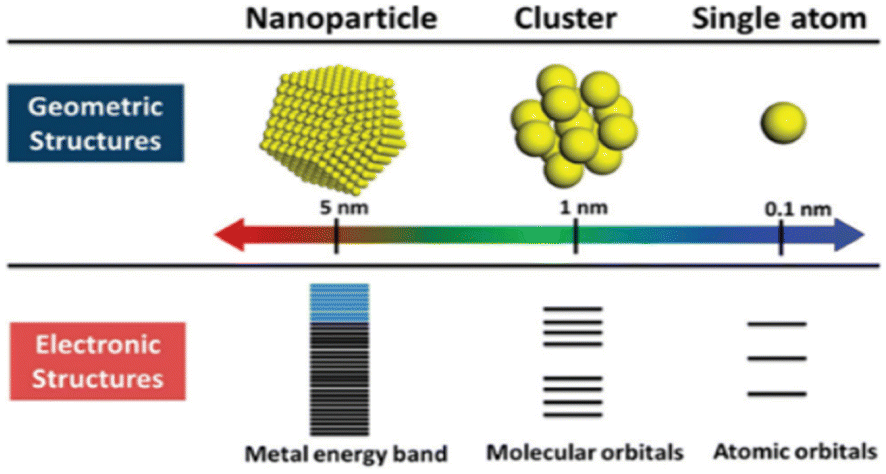 | ||
| Fig. 4 Comparison between geometric and electronic structures of the nanoparticle, cluster, and single atom. Reproduced with permission from ref. 100, copyright 2024, Nanoscale. | ||
5. Synergistic effect
The synergistic effect refers to the combined influence of more than one species when the total of their impacts exceeds the total of their combined effects.101,102 This phenomenon is akin to epistasis, where the function of one gene is dependent on more than one modifier gene in the genetic contextual. In the context of catalysis, synergism plays a crucial role. Certain metals, including silver, copper, nickel, palladium, and cobalt, exert a significant influence on oxygen activation in gold-catalyzed oxidation processes. Notably, Ag and Au, despite being in the same group (11) in the periodic table, commonly exhibit synergistic effects. This highlights the unique interactions and enhanced catalytic outcomes that can arise from the combination of specific metal elements.103 Au–Ag synergism in fluorescence is a very promising phenomenon, and many researchers have lately entered this area. Silver and gold bimetallic clusters have been shown to be a viable approach to circumvent the normally poor λem (in comparison to other popular fluorophores) of CNsAu since the bimetallic CNs display considerably higher QY.104,105Doping is the deliberate insertion of contaminants into a pure material to modify its qualities. Doping commonly converts semiconductors to conductors, resulting in a substantial shift in band-gap energy. Significant changes are witnessed for coinage CNsM with transition metal doping owing to the shift in band-gap energy.106,107 Doping is one kind of synergism of the d-block metal (mostly) with coinage CNsM to increase the CNs's sensitivity and surface volume [Fig. 5].108 It has been observed that coinage CNsM cannot degrade dyes. However, doping with transition metals coinage CNsM can degrade dyes.
 | ||
| Fig. 5 Baldness genes were epistatic to blond or red hair genes. Reproduced with permission from ref. 103, copyright 2024, and RSC Advances. | ||
6. Degradation
In recent years, the elimination of dyes and other contaminants from the aquatic atmosphere has become a critical need.109 To address it, several techniques have been developed, such as ozonation,110 membrane filtration,111 bioadsorption,112,113 ion exchange removal,114–116 adsorption,117–120 photocatalytic degradation,121,122 catalytic reduction,123,124 biological/aerobic therapy,125 and coagulation.126 The absorption technique is often employed in prerelease removal operations because of its simplicity and low cost.127 However, limitations such as low absorption efficacy, incomplete pollutant removal, and poor mechanical stability of adsorbents make efficient pollutant removal challenging.128–130 Over the past several years, the photodegradation of contaminants has gained more attention. Through the process of photocatalysis, a semiconductor photocatalyst uses sunlight absorption to break down a variety of environmental pollutants, including organic pollutants in the air and water.131–134 Photodegradation has benefits over conventional wastewater treatment processes. Numerous operational factors, such as the starting [dye], pH of the solution, exposure intensity, and temperature of the reaction, impact the whole photodegradation method from dye molecule adsorption on the photocatalyst to dye molecule destruction by reactive radicals.135–140 For e.g., Xie et al.87 demonstrated that the decolorization of amino 10 B dye followed pseudo-first-order kinetics, where raising the initial dye concentration reduces the photodegradation efficiency [Fig. 6].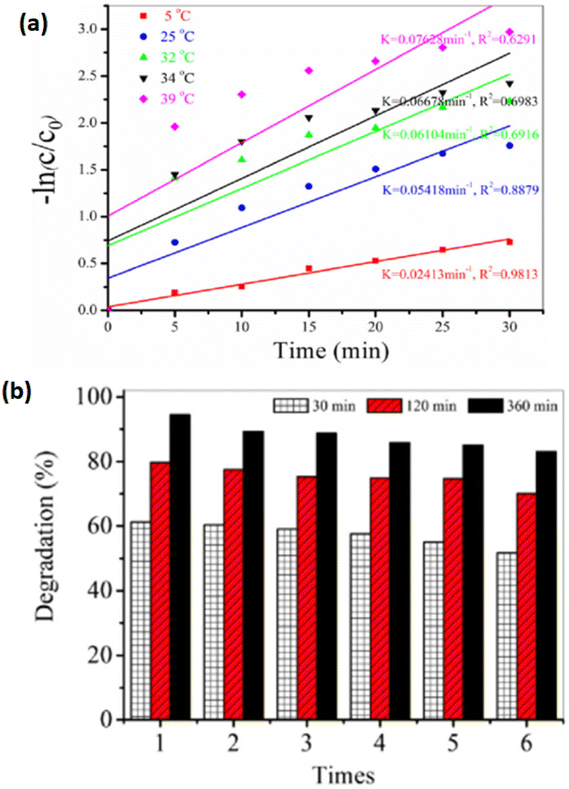 | ||
| Fig. 6 Dye degradation. (a) pseudo first order; (b) reusability of dye. Reproduced with permission from ref. 87, copyright 2024, Water Air Soil Pollut. | ||
Table 1 demonstrated the formation of various coinage CNsM/transition metal hybrid particles. Such hybrids were utilized for dye degradation in the following sections.
| CNs | Capping | UV (nm) | λem/λex (nm) | Hybrid | UV (nm) | λem/λex (nm) | Size (nm) | Oxidation number |
|---|---|---|---|---|---|---|---|---|
| Ag | Perylenetetracarboxylic acid | 530 | — | Ag–TiO2/Nb2O5 | 470 | — | ∼2–3 | 0 |
| Histidine | 254 | 465/373 | Ag:Al2O3:TiO2 | — | — | ∼1.6 | 0 | |
| Mercaptosuccinic acid | 390 | — | AgQCs-GCN | — | 400/350 | >500 | — | |
| Perylenetetracarboxylic acid | 254 | — | ZnO–Ag | — | — | 1.5–2 | 1 | |
| 3-Aminopropyl-trimethoxysilane | ∼370 | 380/325 | ZnO@Ag | 373 | 380/325 | 2.5 | 0 | |
| Glutathione & NaBH4 | 530 | — | Ag–TiO2 hybrid | 454 | — | <3 | 0 | |
| Hydrazine hydrate | — | — | Ag/CN-sr | 400 | 425/316 | 10 | 1 | |
| C4H6O6 | 250 | — | Ag/CeO2 | 320 | — | 5–7 | 0 | |
| BSA & NaBH4 | 305 | 670/420 | Ag@AuCNs/f-MoS2 | 305 | 705/420 | 2–3 | 0 | |
| GSH | — | — | Ag–TiO2 | — | — | — | — | |
| Cu | L-Cysteine | — | 480/400 | CuCNs/C–H2O2 | — | 480/400 | — | 0 |
| C2H2O4 | — | — | (NH4)2CuMo8O26(C10H8N2)2 | — | — | — | 0 | |
| Glutathione | 330 | 450/373 | CuCNS | 330 | 370/330 | 1.7 | 0 | |
| Alcoholic solution | 370 | — | CuNa–K2Ti6O13 | 370 | — | 5 | 2 | |
| Au | Bovine serum albumin & NaOH | — | 440/375 | C-dot-C153-AuCNs | — | 440/375 | 2 ± 1 | 0 |
| GSH | 395 | — | GNCF-300 | 350 | — | 1 | 0 & 1 | |
| 5,5-Dimethyl-1-pyrroline-N-oxide | 500 | — | Au CNs@glass | 655 | — | ∼1.3 | 0 |
6.1 CNsAu for dye degradation
The HAuCl4/THF solution was combined with 10 μL of trimethoxysilane (MPTS) and subjected to UV light irradiation at 365 nm for 8 h. Over this period, the solution's color gradually transformed from nearly colorless to a pale yellow following the mixture of the two reagents. Eventually, CNsAu sol was produced. A photoreduction approach was followed to generate luminescent silane-stabilized CNsAu. The λem of the resulting CNsAu could be manipulated from 538 nm to 580 nm by varying the mole ratio of the stabilizer to the gold precursor. In the absence of light, the MB solution with CNsAu showed no discernible change, indicating the reaction was light-driven. Under continuous visible irradiation for 60 min, the [MB] only marginally reduced by 4% in the absence of CNsAu. This slight decrease could be attributed to the self-photo-bleaching action of MB, when exposed to high energy density irradiation. However, in the existence of CNsAu, the absorption of dye was significantly reduced by 95.6%, demonstrating a strong promoting impact of CNsAu on the photocatalytic degradation of MB. The intensity of the specific charge peak at 284 nm, attributed to [MB + H+], was notably elevated, but after an hour of light irradiation, the intensity dropped dramatically and nearly vanished. No discernible intensity rise was observed for peaks at about 286 nm, 304 nm, & 308 nm, which were attributed to [MBL + H+], [MBL + NH4+], & [MBL + Na+], respectively, indicating the absence of MBL (a photoproduct of MB). The photocatalytic efficacy of CNsAu placed on substrates (CNsAu@glass) was more advantageous for catalyst recycling compared to standalone CNsAu. Interestingly, the catalytic activity of standalone CNsAu was greater than that of CNsAu attached to glass substrates [Fig. 7]. This study highlights the potential of these CNsAu as efficient photocatalysts for the degradation of MB and suggests their applicability in catalyst recycling processes.148 The LUMO boundary orbital of CNsAu is approximately 0.17 V concerning the SHE. The fluorescence emission energy (2.2 eV) indicates that the highly occupied molecular orbital frontier orbit was positioned at 2.3 V against SHE, significantly below the redox potential of MB/LMB (−0.4 V). According to the absorption edge of CNsAu (2.9 eV) and the position of the HOMO frontier orbital (0.17 V), an empty level at −0.50 V ought to produce photo-excited electrons.149,150
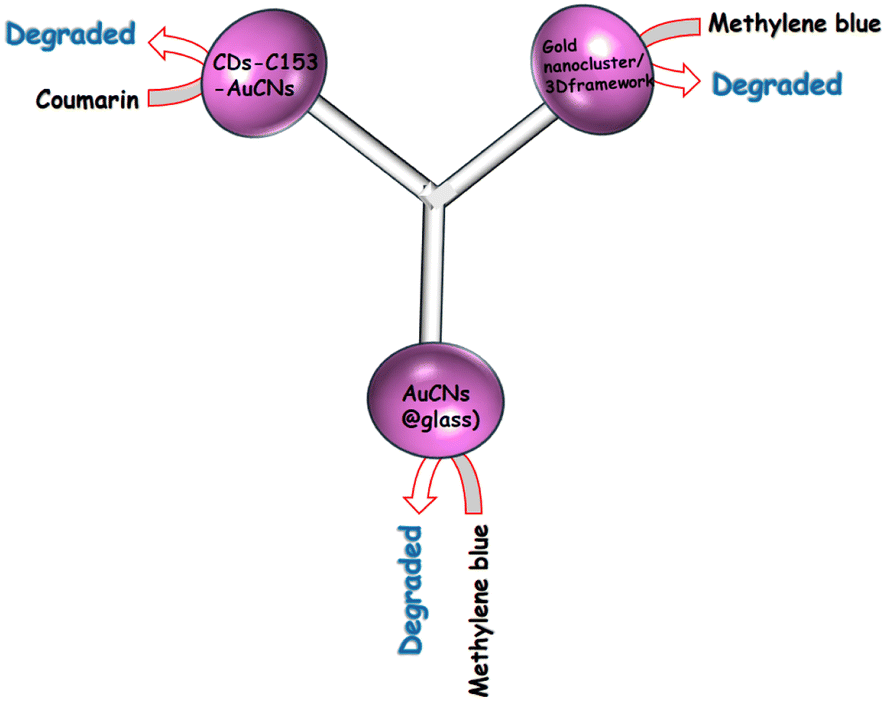 | ||
| Fig. 7 Degradation of various dyes using CNsAu. | ||
| CNsAu + hν → (CNsAu)* |
| (CNsAu)* + O2 → CNsAu(h*) + ˙O2− |
| ˙O2− + H2O + (CNsAu)* → H2O2 + CNsAu(h+) |
| CNsAu(h+) + MB → CNsAu + oxidative products |
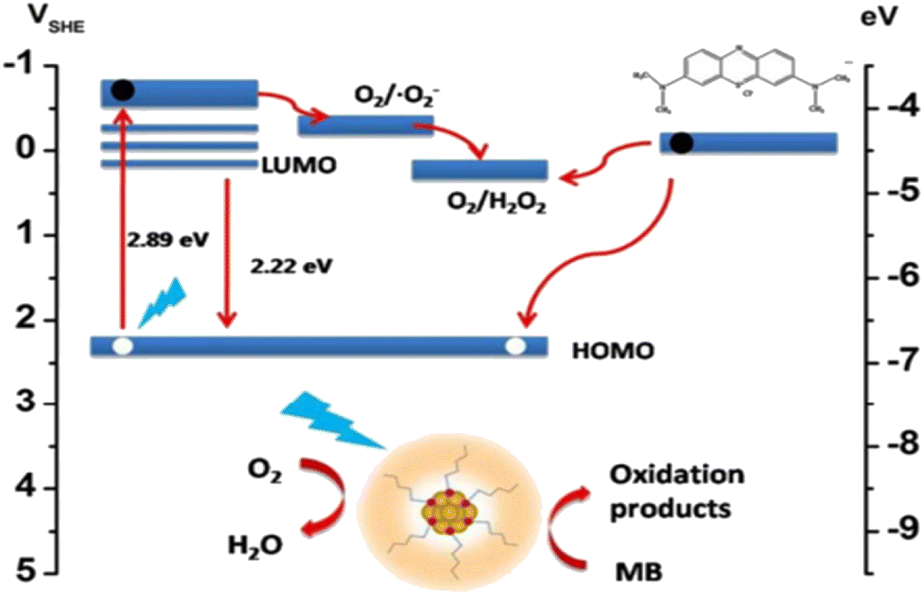 | ||
| Fig. 8 Mechanism of the degradation of MB dye. Reproduced with permission from ref. 150, copyright 2024, Nanoscale. | ||
6.2 Copper nanocluster for dye degradation
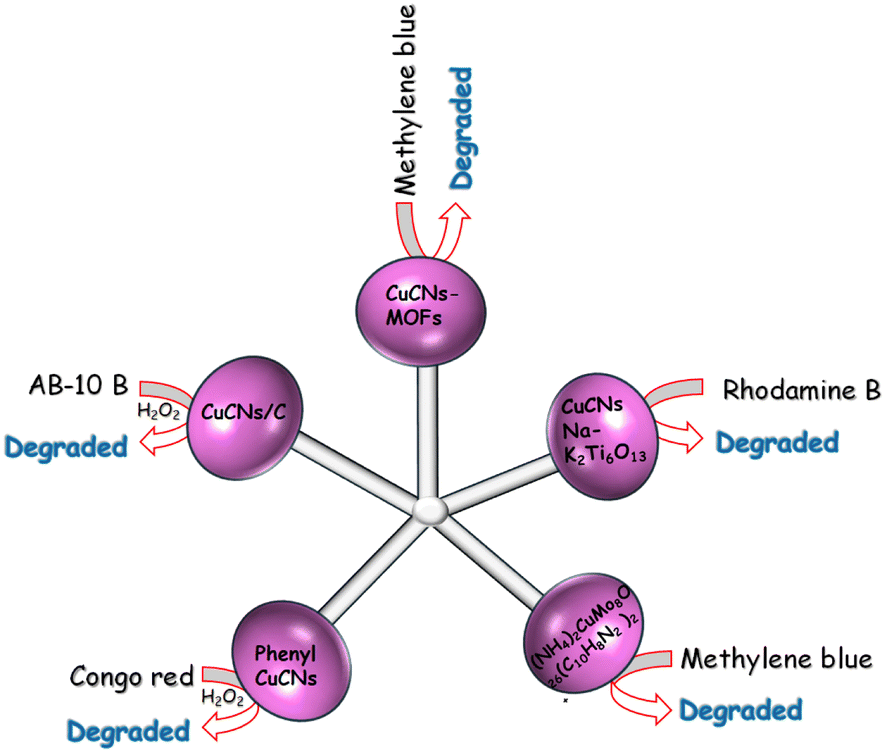 | ||
| Fig. 9 Degradation of various dyes using CNsCu. | ||
CNsCu were synthesized with MAT (3-amino-1,2,4-triazole-5-thiol) ligands, coupled with zinc(II) to produce a nano-sheet CNsCu-MOFs structure, resulting in AIE and improved PL intensity. Water molecules were introduced into S-CNsCu by the H-bond during aging in H2O to enable 3D microflower structures, and weak phosphorescence was converted to strong fluorescence (30%) concurrently.162 After drying, CNsCu released H2O molecules, which were labeled with V-CNsCu and readily coupled with H2O2, which might be converted to radical ˙OH by copper(I). CNsCu-MOFs produced photogenerated e− and h+ when exposed to UV light. The as-obtained electrons reacted with H2O2 to produce additional radical OH, which was subsequently used to catalyze the dyes. The dyes also provided electrons to the remaining holes. The typical three dyes, MB, MO, and RB, as well as the antibiotic tetracycline, might be destroyed in 30 min, when exposed to an XL. Furthermore, in sunlight, the dye-antibiotic mixture might be destroyed after 100 min. V-CNsCu exhibited outstanding photo-Fenton catalytic activity and has promising photocatalytic potential in wastewater.
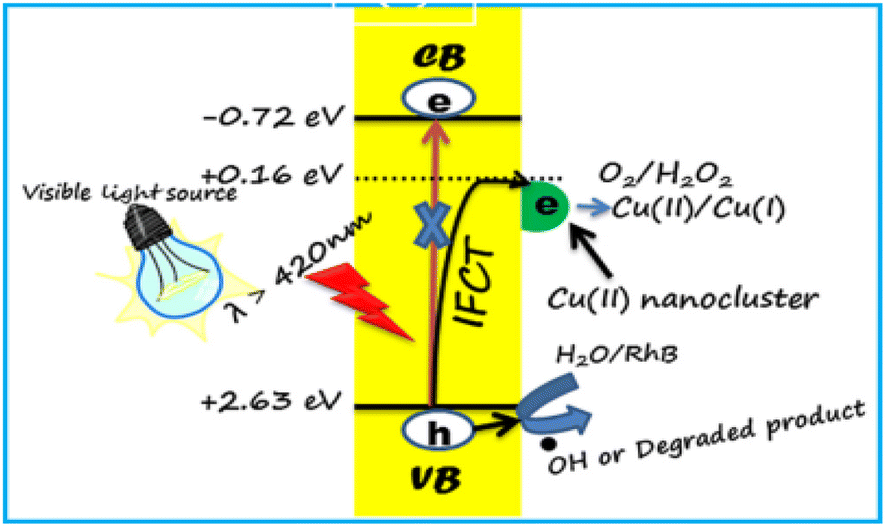 | ||
| Fig. 10 Mechanism of dye degradation using CNsCu. Reproduced with permission from ref. 160, copyright 2024, Mater. Lett. | ||
6.3 Silver nanocluster for dye degradation
A class of hybrid nanomaterials was developed by attaching CNsAg with TiO2/Nb2O5 (Ag/T/N) nanocomposite. It was found that a two-step synthetic technique successfully decorated T/N with CNsAg, whereas a one-step synthesis generated T/N CNs that had minimal Nb2O5 content. When CNsAg were attached to TiO2/Nb2O5, the photo-catalytic activity of the nanocomposite for the elimination of MG was increased as compared to the same materials with insignificant Nb2O5 content. The addition of Nb2O5 to a hybrid nanocomposite allowed for the selective improvement of MG photocatalytic degradation. It indicated the importance of transition metal ions in the removal of MG. The dye was eliminated in half an hour in the dark using Ag/T/N as a catalyst. It suggested dye adsorption with the catalyst, which might be related to the dye's strong intermolecular interaction with the hybrid nanocomposite. The excessive quantity of Nb2O5 loading in the hybrid nanocomposite catalyst might increase the surface acidity of the catalyst, favoring the degradation of cationic dyes like Malachite green. The suspension was exposed to visible light after adsorption and desorption, and fast elimination of the dye was observed. The usage of Ag/T/N resulted in increased visible light absorption. Exposure to visible light produced more e−–h+ pairs. Recombination rates in hybrid nanocomposites were lowered due to the existence of ternary junctions and a larger range of band-gaps. This process resulted in enhanced photocatalytic degradation. The photocatalytic activities of the catalyst were determined by estimating the dye absorbance after photodegradation.169
AgNO3 was thoroughly ground with mercaptosuccinic acid (H2MSA), resulting in an orange-peel-colored powder (silver thiolate), which was further ground with NaBH4, producing a brownish powder. After the addition of distilled water to this suspension, the dark-colored solution was the final product. The embedment of [Ag9(H2MSA)7] on graphitic carbon nitride nanosheets (GCN) produced extended visible-light absorption via multiple single–electron transitions in the silver quantum cluster (AgQCs) and an effective electronic structure for OH˙ radical generation, allowing for enhanced efficacy in the photocatalytic elimination of MB and MO in comparison to pristine GCNs and AgPNls@GCN. Bootharaju and Pradeep171 recently showed that ligand-protected AgQCs might exclusively eliminate cationic dyes due to a favorable electrostatic interaction (between dye molecules and CNs). Due to significant electrostatic repulsion between anionic dye molecules and AgQCs, the removal of anionic dye molecules was a failure. Both types of dyes were utilized as model contaminants to isolate the adsorptive behavior of AgQCs from the photocatalytic activity. The photoelimination of blue dyes by AgQCs-GCN was roughly three times quicker than that of orange dyes owing to electrostatic interaction among sulfur atoms in blue dye molecules and AgQCsCNs. MO and AgQCs exhibited substantial electrostatic repulsion. The sulfonic group of MO had a high electronegative core and was more electron-withdrawing than the carboxylic groups found in AgQCs. The adsorption value was low, which significantly lowered the photo-elimination rate. AgQCs-GCN was a linked semiconductor composite that created a semiconductor heterojunction due to differences in the CB & VB redox energy levels, which enhanced charge carrier separation. The substantial quantum confinement effect in AgQCs allowed it to display many molecular-like single–electron transitions.172,173
The catalytic properties of MOFs could be improved by including metallic species in their porosity post-synthetically. MOFs could be employed to stabilize PNlsM of variable size inside their porosity due to their unusually large surface area and well-defined porous structure via the photo-reduction approach for the CNsAg on a photoactive porous Ti-carboxylate MIL-125-NH2 MOF. The robust MOF interaction among the amino group & silver atoms promoted CNsAg evolution, resulting in the efficient surfactant-free catalyst CNsAg@MIL-125-NH2 with an increment of visible light absorption. The photo-elimination of the emerging organic contaminants (OECs) MB and sulfamethazine (MST) in water treatment, as well as the catalytic hydrogenation of 4-NA to p-phenylenediamine (PDP), were examined. It was worth noting that as compared to the MIL-125-NH2, the composite exhibited enhanced catalytic activity and stability, being able to photodegrade 92% of MB in 1 h, 96% of MST in half an hour, and 100% of 4-NA to PDP in half an hour. CNsAg@MIL-125-NH2 photodegraded up to 72 ± 4 and 92 ± 3% of the dye in the solution in only 10 min and 1 h, respectively. The rapid dye elimination kinetics was most likely attributed to (i) the lower energy band gap induced by the CNsAg plasmon resonance (2.5 and 2.6 eV for CNsAg@MIL-125-NH2 and MIL-125-NH2, respectively);174 (ii) the reduction of the e−–h+ recombination as CNsAg worked as the e− trapping agent and (iii) the chemical affinity of Ag to the sulfur and nitrogen atoms of the MB structure.175 After 1 h in the dark, CNsAg@MIL-125-NH2 absorbed 70% of the dye whereas the pure MOF only removed 40%.176
The strong electrostatic adsorption (SAE) approach was used to deposit monodispersed bare CNsAg with a particle size of 1.3 nm inside the pores of a TiO2-modified mesoporous Mobile Composition of Matter No. 41 (MCM-41). The photocatalytic degradation of MB dye under visible light irradiation was used to assess the performance of the photocatalysts. After 2 h of exposure to TiO2/Ag/MCM-41 (SAE) photocatalyst, about 100% decolorization of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppb MB solution was observed. The SAE catalyst's exceptional performance was ascribed to CNsAg that were uniformly scattered throughout the mesopores of MCM-41, resulting in more silver surface sites accessible for the catalytic reaction.177
000 ppb MB solution was observed. The SAE catalyst's exceptional performance was ascribed to CNsAg that were uniformly scattered throughout the mesopores of MCM-41, resulting in more silver surface sites accessible for the catalytic reaction.177
A low-temperature approach was employed for the production of titanium dioxide-functionalized CNsAg ligated with GSH. CNsAg alone exhibited little visible light photocatalysis, whereas the addition of extreme-trivial titania decreased the band gap of the hybrid CNs and facilitated solar activation, resulting in increased catalytic activity. The elimination of Rh-B revealed the visible light-driven catalytic capability of hybrid CNs. Density Functional Theory (DFT) simulations were utilized to investigate the interaction of surface ligands with titanium dioxide molecules using simple structural models Ag(SCH3)n–TiO2. The absorption spectra of Rh-B solution complied with visible light exposure utilizing TiO2–Ag hybrid CNs as a nano-catalyst at regular time intervals.180 There was a gradual decrease in the absorbance of the peak centered at 550 nm. When photo-catalyzed utilizing CNsAg as a catalyst, there was no discernible change in the absorption spectra of the dye solution. CNsAg were crucial for the solar activation of TiO2 CNs molecules. The introduction of TiO2 (10.8% Ti) significantly improved the visible light-induced photocatalytic activity of CNsAg. The hybrid nanocluster's increased photocatalytic activity could be attributed to decreased e−-hole pair recombination. Metal–metal and metal–ligand interactions shift the absorption band to the visible range, enabling visible light photocatalysis. The photocatalytic activity of PNls was determined by their size, shape, and crystal structure.181,182
The physical characteristics and photocatalytic activities of CNsAg-encumbered graphitic carbon nitride (C3N4-g) nanosheets were investigated. The accumulation of CNsAg on the surface of C3N4-g nanosheets changes the crystal structure and reduces the band-gap energy of C3N4-g. The dramatic drop in intensity indicates that C3N4-g@Ag heterojunctions efficiently avoid photo-generated e−–h+ recombination. The elimination of Rh-B under XL was used to expose the photocatalytic activity of photocatalysts. The increased absorbance in CNsAg occurred due to the SPR effect,183,184 suggesting that it will contribute to RhB degradation. The photocatalytic capabilities of C3N4-g and GCN/Ag samples degraded RhB under XL exposure. The elimination rate of dye was indirectly by varying the intensity of the 554 nm absorption peak of RhB solution using a standard curve linking [RhB] to absorbance. After 50 min of XL irradiation, the 554 nm absorption peak of Rh-B not only vanished but also changed from 554 nm to 530 nm, revealing the disintegration of RhB's conjugated structure. The increase in the photocatalytic activity in photogenerated electrons on C3N4-g nanosheets could be effectively transferred to CNsAg, which decreases the rate of e− and h+ recombination. The intensity of PL was also reduced. UV-vis analysis and the interpretation of CTSPR may have contributed to this significant rise in photocatalytic activity.185
The Ag/g-C3N4 composite (CN-sr@Ag) was enhanced by chemical reduction using ultrasonic spray technology. This technique minimized the aggregation of C3N4-g nanosheets and induced a micron-scale reaction, resulting in uniform distribution and size reduction of CNsAg. The reaction rate constant for RhB degradation was 0.0322 min−1, which was approximately 3 and 7 times greater than that of the Ag/CN composite made using the standard chemical reduction technique (Ag/CN-cr) and bare C3N4-g nanosheets. In 100 min, the [RhB] was reduced by up to 96.47% over a CN-sr@Ag catalyst, whereas it was eliminated by 65% and 43% in the presence of CN-cr@Ag and CN, respectively. The absorption spectra of RhB were at 554 nm. The distinctive absorption spectra of Rh-B at 554 nm decreased rapidly, suggesting that RhB was steadily destroyed as the reaction time increased. The highest absorption site displayed minor hypsochromic changes, which were ascribed to the generation of N-demethylated intermediates of Rh-B during photo elimination. The TEM images of CN-sr@Ag before and after deterioration show that the average size of CNsAg on the surface of CN did not change much after usage (from 15 to 17 nm), but there was aggregation and a wider size distribution range. The Ag content in the composite increased from 3.5 to 3%. The photocatalytic performance of CN-sr@Ag was lowered due to the inevitable agglomeration and loss of silver species. Except for a slightly larger primary peak, the XRD patterns of the CN-sr@Ag samples before and after degradation demonstrate that the peak shape and position were nearly intact.186
A CeO2/Ag nano-structured material with photocatalytic and antibacterial properties was developed utilizing a simple solution combustion synthesis (SCS) technique with tartaric acid as a fuel. Under UV irradiation, CeO2–Ag materials were used as photocatalysts for the catalytic elimination of Rh-B dye in 150 min. RhB showed an absorbance peak at 554 nm. The photodegradation of Rh-B versus the quantity of CeO2/Ag clearly showed that the elimination of Rh-B dye increases with an increase in the amount of catalyst from 100 to 150 mg and subsequently decreases with an increase in the quantity of CeO2/Ag of 200 mg. As a result of the intense redox interaction,187 dye molecules might get adsorbed on the active sites of the CeO2/Ag nanostructured materials. The decrease in catalytic performance with increasing catalyst quantity was thus thought to be attributable to the coarsening of nanoparticles. Kumar and Pandey188 also discovered light scattering, which damages the photocatalyst's catalytic capabilities. It was discovered that CeO2 nano-flakes demonstrated 60% dye degradation in 150 min. The antibacterial activity of Ag/CeO2 nanostructured materials against Gram-positive (S. aureus) and Gram-negative (P. aeruginosa) bacterial strains was also obtained [Fig. 11].189
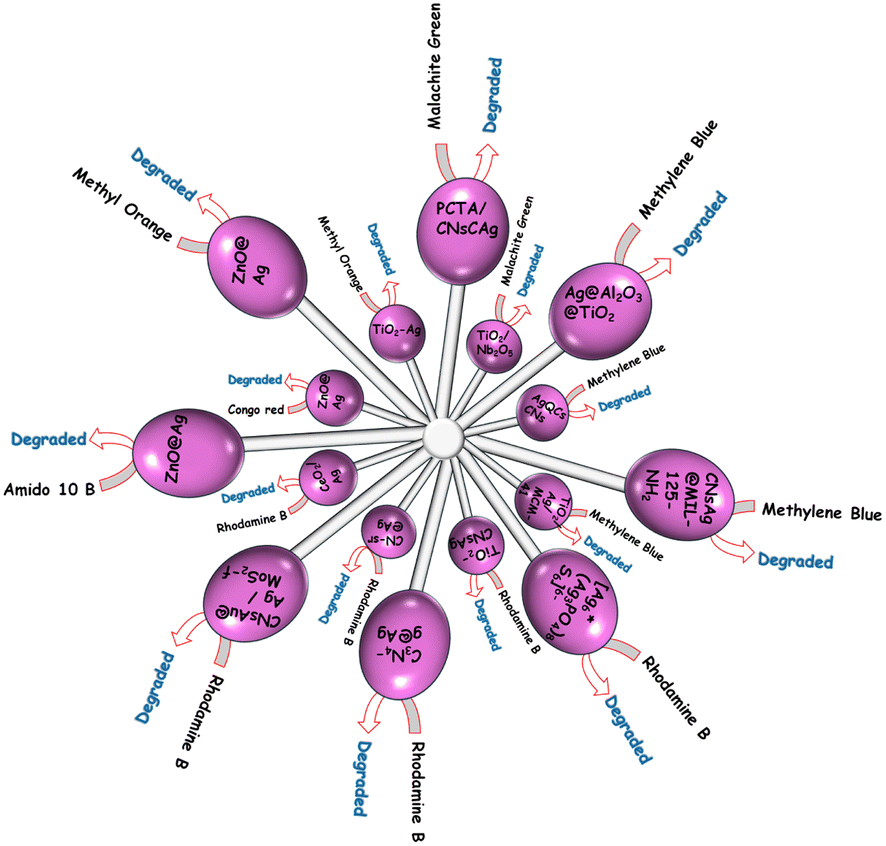 | ||
| Fig. 11 Degradation of various dyes with CNsAg. | ||
CNsM based on Au as core and Ag as shell, named CNsAu@Ag, were electro-statically associated with functionalized semiconducting MoS2 nanosheets named MoS2-f. The Rh-B photo-degradation catalyst CNsAu@Ag/MoS2-f was used for wastewater cleansing from organic contaminant dyes. CNsAu@Ag/f-MoS2 produces extremely reactive hydroxyl radicals by reducing oxygen from charge-separated photogenerated electrons, resulting in superoxide radical ions. The h+ on CNsAu@Ag degraded Rh-B molecules, resulting in a decolorized magenta solution and loss of the distinctive absorbance at 554 nm. Without a catalyst, no changes were shown in RhB, demonstrating that light irradiation alone was insufficient to destroy Rh-B. Similarly, the UV-vis spectra of Rh-B remained virtually unchanged in the existence of solely f-MoS2 (i.e., without any CNsAu@Ag). When a catalytic quantity of bare CNsAu@Ag was applied, the absorption band of Rh-B gradually diminished as time passed, suggesting RhB breakdown. After 180 min of irradiation, the absorption band blue-shifted by 12 nm, resulting in about 70% degradation of Rh-B. The addition of CNsAu@Ag/MoS2-f resulted in virtually quantitative elimination of Rh-B (>85%) in 100 min, which was somewhat longer than the half-time recorded for CNsAu@Ag. Furthermore, after 210 min of light irradiation, RhB degradation was found in CNsAu and CNsAg at about 60 and 50%, respectively. It showed that coinage metal itself was not sufficient for the degradation of dye. The synergism of coinage metal nanocluster was also not able to degrade the dye without the transition metal. Transition metal-capped coinage metal nanoclusters for dye degradation play a vital role.190
Photo Fenton Process (FPP) for UV-light elimination of Methyl Orange (MO) utilizing UV/H2O2/Fe2+ & TiO2-modified photocatalyst with Ag deposition (TiO2-Ag). The rate of kinetics of TiO2-Ag demonstrated greater catalytic performance than FPP since the elimination of the dye took just an hour using TiO2-Ag but it took 2 h for TiO2 using FPP. Because of the plasmon resonance effect of e− transfer from the shallow of TiO2 over metallic Ag at nanoscale particulate sizes, Ag-metalized titanium dioxide photocatalyst had a high surface area and particles were greatly impacted electronically under UV light.194
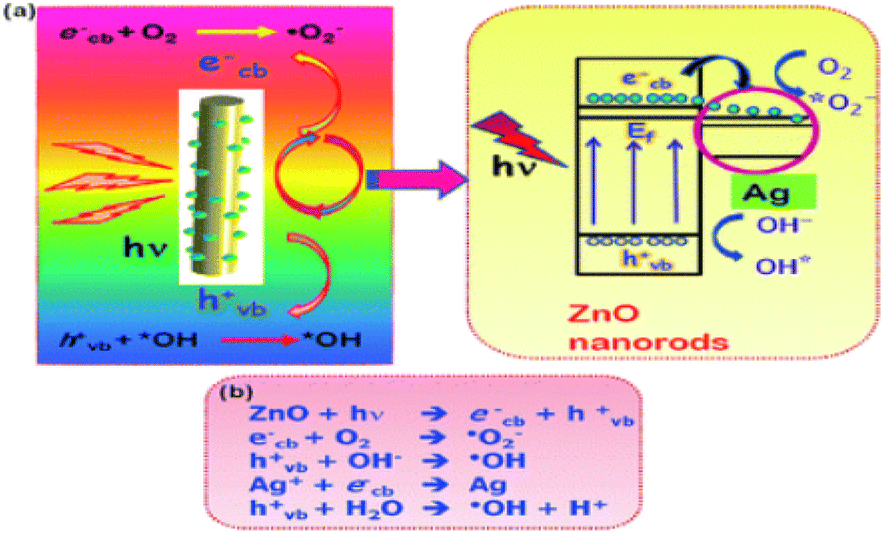 | ||
| Fig. 12 Mechanism of the degradation of dyes. Reproduced with permission from ref. 192, copyright 2024, RSC Advances. | ||
7. Adsorption of dye
Adsorption is a well-established technology for dye removal from wastewater due to its simplicity and cheaper cost when compared to other methods. The development of adsorbents from diverse biomass wastes as a replacement for commercial activated carbons improves the economic effectiveness of the process.1977.1 Rhodamine B
A modified solid-state approach was employed to synthesize an atomically accurate, dithiol-coated CNsAg known as (DMSA)4Ag7 (DMSA: meso-2,3-dimercaptosuccinic acid). The CNs exhibited molecular visual absorption characteristics, featuring a significant peak at 500 nm. In comparison to PNls coated with ligands such as citrate and mercaptosuccinic acid (MSA) and possessing similar chemical structures to DMSA, these clusters displayed significantly higher efficacy for dye removal. The superior dye elimination efficiency of CNs could be attributed to their trivial size, larger shallow area, and favorable electrostatic interactions with cationic dyes. Clusters demonstrated remarkable dye removal performance, absorbing over 80% in 10 min and 99.7% in 15 min, whereas bare alumina adsorbed only about 23% in the same duration. This underscored the highly favourable capability of supported CNs for the elimination of dyes like R-6G. The change in the intensity of dyes corroborated the adsorption of R-6G on CNs. Such CNs, featuring RCOO– groups, exhibited a destructive zeta-potential (PZ) of 75.7 mV, while R-6G maintained a nearly neutral charge (0.1 mV). Upon adding R-6G to the CNs, a reduction in the PZ of the CNs to 57 mV occurred at room temperature, indicating charge neutralization due to electrostatic attraction among oppositely charged ions. The XPS analysis of R-6G-treated supported clusters demonstrated a consistent Ag 3d5/2 peak at 368.1 eV associated with Ag0 in supported clusters, confirming that R-6G uptake was primarily due to adsorption. R6G, MB, MR, and MO had uptake capabilities of 17.2, 16.5, 3.1, and 0.2 mg g−1 of the cluster, respectively.198The SCN− reduction process in colloidal gold solutions results in CNs with a diameter of 2–3 nm. These particles were too tiny to show any surface plasmon absorption characteristics. The introduction of R-6G into colloidal Au solution altered the absorption spectrum significantly. Intermolecular and intercluster interactions were induced by the dye molecules on the Au surface. Furthermore, dye adsorption on AuNPI caused charge neutralisation on the surface, leading the dye/Au assembly to coalesce. The absorbance at 525 nm decreased when the [dye/Au] ratio increased. The monomeric absorption band at 525 nm faded at a [dye/Au] ratio of 11.7, and two additional bands at 507 and 537 nm arose. While a similar amount of SCN− was introduced into an R-6G solution, no similar absorption changes were detected. The robust electrostatic interaction among the dye molecule and the negatively charged SCN@Au was responsible for these absorption variations. These aggregation effects were dominant because the dye molecule was tightly packed around the metal body causing intermolecular interactions [Fig. 13].
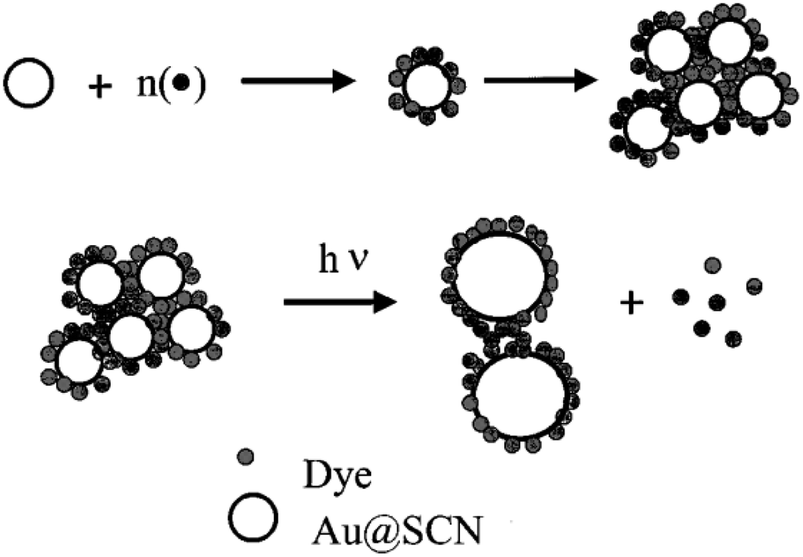 | ||
| Fig. 13 Systematic diagram of dye adsorption. Reproduced with permission from ref. 200, copyright 2024, J. Phys Chem. B. | ||
A herringbone-type aggregation of dyes could not be ruled out. H-type aggregation represented a sandwich-type stacking of dye molecules around AuNPls. The Au colloids formed via thiocyanate reduction were exceedingly tiny, with particle diameters ranging from 2 to 3 nm.199 AuNPls linked to R-6G had comparable particle diameters but existed as dense CNs. The laser-irradiated particles had a comparatively wide diameter of 5–20 nm. During the laser irradiation in Rh-6G/Au CNs' fabrication, larger-sized particles were obtained when AuNPls transformed into tiny aggregated CNs (the laser-induced fusion process).200
8. Sensing of dye with coinage metal nanocluster
Nanosensors utilize PNls recognition sites for selection and signal transduction, indicating the presence of contaminants. Sensing devices are designed to detect a single analyte or a group of analytes at the same time, a concept known as multiplexity [Fig. 14].201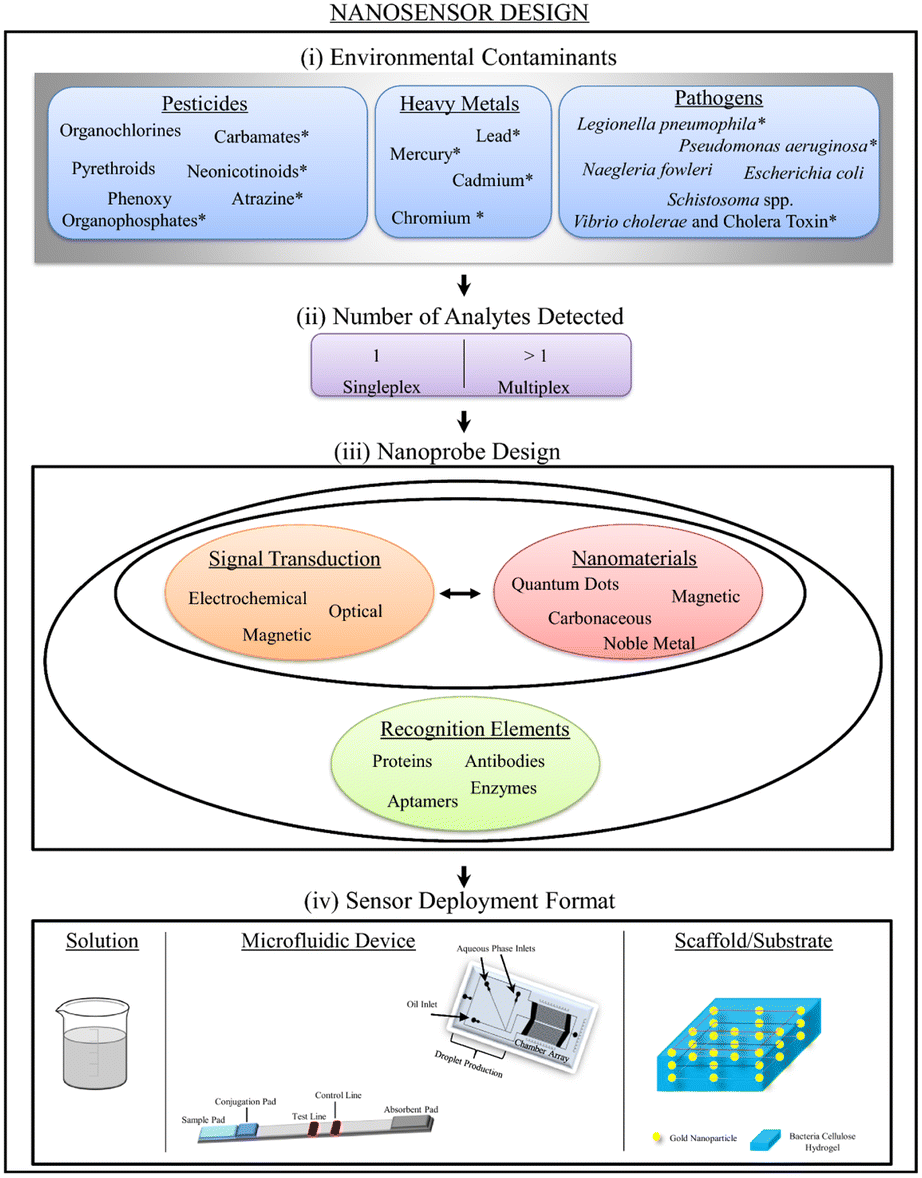 | ||
| Fig. 14 Schematic illustration of nanosensor design, including ambient contaminants, detected analytes, probe design, and deployment format. Reproduced with permission from ref. 201, copyright 2024, J. Nanobiotechnol. | ||
Fluorescence resonance energy (FRE) transfer is a nonradiative energy transfer procedure that uses long-range dipole–dipole coupling to transmit energy from an excited state donor to a nearby ground state acceptor.202 Organic fluorophores,203 fluorescent proteins,204 and quantum dots205 have all been used to build the FRE transfer assay [Table 2].
| Nanocluster | hν | λex/λem | UV (nm) | Size | Oxidation state |
|---|---|---|---|---|---|
| Silver nanocluster | Hyperbranched polyethyleneimine | 375/450 | 365 | ∼2 nm | 0 |
| Copper nanocluster | L-Cysteine | 375/450 | 375 | 3 nm | 0 |
Chen et al.206 demonstrated that FRE transfer among Sudan 1 to 4 & polyethyleneimine-capped (EIP-CNsAg) that were lesser than the critical size for electric resources quantisation (2 nm) were studied and then used for Sudan 1 to 4 sensing in ethanol. When stimulated at 375 nm, the PEI-AgCNs showed significant blue fluorescence, which was substantially amplified when the CNs were dissolved in specified organic solvents, particularly n-propanol and ethanol. The fluorescence of PEI-CNsAg could be effectively diminished by the addition of Sudan dyes. The solvatofluorochromic properties of EIP-capped CNsAg caused the centered wavelength of 455 nm in water to shift to 450 nm in ethanol. In the concentration range of 5–50 μL mL−1, a good linear connection between emissive intensity and [EIP-CNsAg] was established. However, the fluorescence response deviated from linearity. In the presence of Sudan I–IV at a concentration of 10 μL mL−1, a lower concentration fluorescence probe was shown to have greater quenching efficacy. Sudan I and II quenching efficiencies nearly remained constant with rising temperatures; however, Sudan III and IV quenching efficiencies at higher temperatures were larger than those at lower temperatures [Fig. 15].
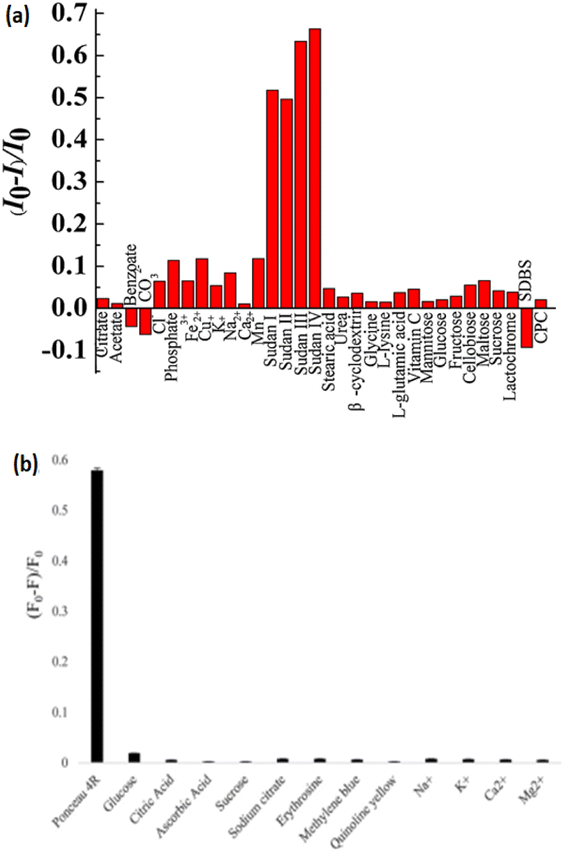 | ||
| Fig. 15 Bar diagram of sensing of dyes. Reproduced with permission from ref. 206 copyright 2024, Sens. Actuators, B; Reproduced with permission from ref. 207 copyright 2024, Journal of Photochemistry and Photobiology A: Chemistry. | ||
CNsCu were developed using a one-pot hydrothermal reaction between Cu and L-Cy (reductant and capping agent). The influence of pH, incubation duration, and temperature on CNsCu emission signals was observed. The introduction of Ponceau-4R to the CNs solution caused fluorescence quenching, and the probable quenching mechanism was static quenching. In the existence and absence of Ponceau-4R, λem & λex were 470 & 375 nm, respectively. The increased addition of L-Cy could increase the rate of Cu2+ reduction, resulting in the production of massive CNs. When the amount of L-Cy was too low, the degree of reduction was inadequate, resulting in poor stability and CNsCu agglomeration. CNsCu had a carboxyl group on their surface, whereas Ponceau-4R contained three RSO3H. Because both RSO3H and O2 atoms on the surface of –COOH group were electronegative, these interactions led to the formation of a complex among Ponceau-4R and CNsCu. The interaction of these two groups and the formation of H-bonds effectively suppressed the fluorescence intensity of CNsCu. Complex molecules generated between the emitter and quencher altered the luminescence property. The blue shift in the λem of CNsCu suggested the altered size. The size of generated CNsCu was 3.3 nm, but after reaction with the dye, it was 17.4 nm, according to dynamic light scattering.207
9. Future prospective & conclusion
Copper nanoclusters have brilliant colors and nearly constant energy levels. Quantum confinement results in a distinct photoluminescence property in CNs including CNsAu, CNsAg, CNsCu, and CNsPt. Coinage metal nanoclusters with transition metals play a vital role in dye degradation because the addition of transition metal with the coinage metal nanocluster increases the surface-to-volume ratio of the nanoclusters. Increasing the surface volume is responsible for the photocatalytic degradation and adsorption behavior of the dye. Colored discharge, particularly dyes from industry, endangers the environment and harms aquatic and human life. Organic dyes are widely utilized as colored chemicals in a variety of sectors, including textiles, paper, cosmetics, leather, and food. For the formation of copper nanocluster, different capping agents were used. Copper nanocluster with transition metal shows an interestingly synergistic effect. In the presence of synergism behavior, the change in nanoclusters' behavior for dye degradation was shown in this review article. Many literature showed the effect of H2O2 on dye degradation. In the overall review article, we showed that in the presence of gold nanoclusters, the time consumption for degradation is lower than that of other metals. Few studies are available on the degradation in copper and gold nanoclusters in comparison to silver nanoclusters. Coinage metal nanoclusters do not show any degradation without doping/alloying with the transition metals. It shows the vital role of synergism behavior on dye degradation. Dye degradation, adsorption, mechanism, and sensing are shown in this review article. It will hopefully be an asset to young researchers, venturing into the field of environmental nanoscience to cope with recent challenges in the environment.Conflicts of interest
There is no conflict of interest.Notes and references
- Y.-H. Chiu, T.-F. M. Chang, C.-Y. Chen, M. Sone and Y.-J. Hsu, Catalysts, 2019, 9, 430 CrossRef CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1 CrossRef CAS.
- F. Deng, L. Min, X. Luo, S. Wu and S. Luo, Nanoscale, 2013, 5, 8703 RSC.
- S. Xu, Y. Zhu, L. Jiang and Y. Dan, Water, Air, Soil Pollut., 2010, 213, 151 CrossRef CAS.
- D. Wang, Y. Wang, X. Li, Q. Luo, J. An and J. Yue, Catal. Commun., 2008, 9, 1162 CrossRef CAS.
- A. Kumar and G. Pandey, Mater. Sci. Eng. Int. J., 2017, 1, 1 Search PubMed.
- M. A. Brown and S. C. De Vito, Environ. Sci. Technol., 1993, 23, 249 CAS.
- S. Chen, J. Zhang, C. Zhang, Q. Yue, Y. Li and C. Li, Desalination, 2010, 252, 149 CrossRef CAS.
- N. Daneshvar, D. Salari and A. Khataee, J. Photochem. Photobiol., A, 2003, 157, 111 CrossRef CAS.
- S. Ledakowicz and M. Gonera, Water Res., 1999, 33, 2511 CrossRef CAS.
- J. Grzechulska and A. W. Morawski, Appl. Catal., B, 2002, 36, 45 CrossRef CAS.
- A. Wahab, Appl. Nanosci., 2019, 9, 1823 CrossRef CAS.
- I. Kanelidis and T. Kraus, Beilstein J. Nanotechnol., 2017, 8, 2625–2639 CrossRef PubMed.
- J. P. Chinta, Sens. Actuators, B, 2017, 248, 733–752 CrossRef CAS.
- P. Sharma and M. Ganguly, IJEST, 2023, 1–20, DOI:10.1007/s13762-023-05171-6.
- H. Si, T. Shu, X. Du, L. Su and X. Zhang, Biosensors, 2022, 12, 511 CrossRef CAS PubMed.
- A. Henglein, J. Phys. Chem., 1993, 97, 5457 CrossRef CAS.
- P. Mulvaney, M. Giersig and A. J. Henglein, Phys. Chem., 1993, 97, 7061 CrossRef CAS.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329 CrossRef.
- A. Haesselbarth, A. Eychmueller, R. Eichberger, M. Giersig, A. Mews and H. Weller, J. Phys. Chem., 1993, 97, 5333 CrossRef CAS.
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, Berlin, 1995 Search PubMed.
- R. G. Freeman, M. B. Hommer, K. C. Grabar, M. A. Jackson and M. J. Natan, J. Phys. Chem., 1996, 100, 718 CrossRef CAS.
- A. Martino, S. A. Yamanaka, J. S. Kawola and D. A. Loy, Chem. Mater., 1997, 9, 423 CrossRef CAS.
- G. L. Hornyak, C. J. Patrissi and C. R. Martin, J. Phys. Chem. B, 1997, 101, 1548 CrossRef CAS.
- B. Shanghavi and P. V. Kamat, J. Phys. Chem. B, 1997, 101, 7675 CrossRef.
- P. V. Kamat, Composite Semiconductor Nanoclusters, in Semiconductor NanoclusterssPhysical, Chemical and Catalytic Aspects, ed. Kamat, P. V. and Meisel, D., Elsevier Science, Amsterdam, 1997, vol. 237 Search PubMed.
- A. Henglein, Chem. Mater., 1998, 10, 444 CrossRef CAS.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- W. T. Wallace, R. B. Wyrwas, R. L. Whetten, R. Mitric and V. Bonacic-Koutecky, J. Am. Chem. Soc., 2003, 125, 8408–8414 CrossRef CAS PubMed.
- S. Kumar and R. Jin, Nanoscale, 2012, 4, 4222–4227 RSC.
- G. Schmid, Chem. Soc. Rev., 2008, 37, 1909–1930 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- Y. Li, M. Zhou and R. Jin, Adv. Mater., 2021, 33, 2006591 CrossRef CAS PubMed.
- Z. Lin, N. Goswami, T. Xue, O. J. H. Chai, H. Xu, Y. Liu, Y. Su and J. Xie, Adv. Funct. Mater., 2021, 31, 2105662 CrossRef CAS.
- Y. Li, M. Zhou, Y. Song, T. Higaki, H. Wang and R. Jin, Nature, 2021, 594, 380–384 CrossRef CAS PubMed.
- X.-M. Luo, C.-H. Gong, F. Pan, Y. Si, J.-W. Yuan, M. Asad, X.-Y. Dong, S.-Q. Zang and T. C. Mak, Nat. Commun., 2022, 13, 1177 CrossRef CAS PubMed.
- W. D. Liu, J. Q. Wang, S. F. Yuan, X. Chen and Q. M. Wang, Angew. Chem., 2021, 60, 11430–11435 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- K. Sridharan, The Electromagnetic Spectrum, 2016, pp. 1–12 Search PubMed.
- A. Manchon and A. Belabbes, Solid State Phys., 2017, 68, 1–89 Search PubMed.
- S. Drummer, T. Madzimbamuto and M. Chowdhury, Materials, 2021, 14, 2700 CrossRef CAS PubMed.
- D. A. Panayotov, A. I. Frenkel and J. R. Morris, ACS Energy Lett., 2017, 2, 1223–1231 CrossRef CAS.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 409–431 CrossRef CAS PubMed.
- R. Jin, Y. Zhu and H. Qian, Chem.–Eur. J., 2011, 17, 6584–6593 CrossRef CAS PubMed.
- C. Zhou, C. Sun, M. Yu, Y. Qin, J. Wang, M. Kim and J. Zheng, J. Phys. Chem. C, 2010, 114, 7727–7732 CrossRef CAS PubMed.
- X. Qu, Y. Li, L. Li, Y. Wang, J. Liang and J. Liang, J. Nanomater., 2015, 2015, 1–23 Search PubMed.
- L. Zhang and E. Wang, Nano Today, 2014, 9, 132–157 CrossRef CAS.
- A. Mathew and T. Pradeep, Part. Part. Syst. Charact., 2014, 31, 1017–1053 CrossRef CAS.
- Y. C. Shiang, C. C. Huang, W. Y. Chen, P. C. Chen and H. T. Chang, J. Mater. Chem., 2012, 22, 12972–12982 RSC.
- W. Jahn and J. Stru, Biol., 1999, 127, 106–112 CAS.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- Y. An, Y. Ren, M. Bick, A. Dudek, E. H.-W. Waworuntu, J. Tang, J. Chen and B. S. Chang, Biosens. Bioelectron., 2020, 154, 112078 CrossRef CAS PubMed.
- X. Hu, T. Liu, Y. Zhuang, W. Wang, Y. Li, W. Fan and Y. Huang, TrAC, Trends Anal. Chem., 2016, 77, 66–75 CrossRef CAS.
- Z. Wang, B. Chen and A. L. Rogach, Nanoscale Horiz., 2017, 2, 135–146 RSC.
- T. Qing, K. Zhang, Z. Qing, X. Wang, C. Long, P. Zhang, H. Hu and B. Feng, Microchim. Acta, 2019, 186, 670 CrossRef CAS PubMed.
- Z. Yuan, Y.-C. Chen, H.-W. Li and H.-T. Chang, Chem. Commun., 2014, 50, 9800–9815 RSC.
- M. Zhang, Y.-Q. Liu, C.-Y. Yu, B.-C. Yin and B.-C. Ye, Analyst, 2013, 138, 4812–4817 RSC.
- E. Thyrhaug, S. A. Bogh, M. R. Carro-Temboury, C. S. Madsen, T. Vosch and D. Zigmantas, Nat. Commun., 2017, 8, 15577 CrossRef CAS PubMed.
- J. M. J. Santillan, D. M. Arboleda, D. Muraca, D. C. Schinca and L. B. Scaffardi, Sci. Rep., 2020, 10, 8217 CrossRef CAS PubMed.
- G. Booth, Dyes, General Survey, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000, ISBN 3527306730, DOI:10.1002/14356007.a09_073.
- B. Lellis, C. Z. Fávaro-Polonio, J. Alencar Pamphile and J. C. Polonio, Biotechnol. Res. Int., 2019, 3, 275–290 Search PubMed.
- M. Benjelloun, Y. Miyah, G. A. Evrendilek, F. Zerrouq and S. Lairini, Arabian J. Chem., 2021, 4, 103031 CrossRef.
- S. B. Wang and H. T. LI, Dyes Pigm., 2007, 72, 308 CrossRef CAS.
- Y. Wang and J. YU, Water Res., 1999, 33, 3512 CrossRef.
- A. K. Mittal and C. Venkobachar, J. Environ. Eng., 1993, 119, 366 CrossRef CAS.
- S. Mondal, Environ. Eng. Sci., 2008, 25, 1–10 CrossRef.
- H. M. Solayman, M. A. Hossen, A. A. Aziz and N. Y. Yahya, J. Environ. Chem. Eng., 2023, 11, 109610 CrossRef CAS.
- X. Liu, W. Li, N. Chen, X. Xing, C. Dong and Y. Wang, RSC Adv., 2015, 5, 34456–34465 RSC.
- Y. Liu, H. Yu, Z. Lv, S. Zhan, J. Yang, X. Peng, Y. Ren and X. Wu, J. Environ. Sci., 2012, 24, 1867–1875 CrossRef CAS PubMed.
- Z. Aksu, Process Biochem., 2005, 40, 997–1026 CrossRef CAS.
- B. Appavu, S. Thiripuranthagan, S. Ranganathan, E. Erusappan and K. Kannan, Ecotoxicol. Environ. Saf., 2018, 151, 118–126 CrossRef CAS PubMed.
- D. Pathania, S. Sharma and P. Singh, Arabian J. Chem., 2017, 10, S1445–S1451 CrossRef CAS.
- E. Haque, J. W. Jun and S. H. Jhung, J. Hazard. Mater., 2011, 185, 507–511 CrossRef CAS PubMed.
- W. Deligeer, Y. W. Gao and Y. Asuha, Appl. Surf. Sci., 2011, 257, 3524–3528 CrossRef CAS.
- V. Ponnusami, S. Vikram and S. N. Srivastava, J. Hazard. Mater., 2008, 152, 276–286 CrossRef CAS PubMed.
- T. Chen, Y. Zheng, J. Lin and G. Chen, J. Am. Soc. Mass Spectrom., 2008, 19, 997–1003 CrossRef CAS PubMed.
- P. C. Dey and R. Das, Spectrochim. Acta, Part A, 2020, 118122 CrossRef CAS PubMed.
- K. V. K. Rao, Toxicol. Lett., 1995, 81, 107–113 CrossRef CAS PubMed.
- Y. Chen, Y. Zhang, C. Liu, A. Lu and W. Zhang, Int. J. Photoenergy, 2011, 2012, 1–6 CrossRef.
- S. D. Richardson, C. S. Wilson and K. A. Rusch, Ground Water, 2004, 42, 678 CrossRef CAS PubMed.
- R. Nagaraja, N. Kottam, C. R. Girija and B. M. Nagabhushana, Powder Technol., 2012, 91, 215–216 Search PubMed.
- M. S. Samuel, K. V. Savunthari and S. Ethiraj, Environ. Sci. Pollut. Res., 2021, 28, 40835–40843 CrossRef CAS PubMed.
- K. Iqbal, A. Iqbal, A. M. Kirillov, W. Liu and Y. Tang, Inorg. Chem., 2018, 57, 13270–13278 CrossRef CAS.
- K. Iqbal, A. Iqbal, A. M. Kirillov, C. Shan, W. Liu and Y. Tang, J. Mater. Chem. A, 2018, 6, 4515–4524 RSC.
- X. Luo, R. Abazari, M. Tahir, W. K. Fan, A. Kumar, T. Kalhorizadeh, A. M. Kirillov, A. R. Amani-Ghadim, J. Chen and Y. Zhou, Coord. Chem. Rev., 2022, 461, 214505 CrossRef CAS.
- X. H. Li, Y. Liu, H. Y. Lin, N. Xu, Z. Zhang, G. C. Liu and X. L. Wang, Cryst. Growth Des., 2022, 22, 3845–3852 CrossRef CAS.
- C. Xie, X. Wen, C. Xiao and S. Wei, Water, Air, Soil Pollut., 2020, 231, 280 CrossRef CAS.
- J. D. Vassallo, Toxicol. Sci., 2004, 80, 249–257 CrossRef CAS PubMed.
- S. L. Born, Food Chem. Toxicol., 2003, 41, 247–258 CrossRef CAS PubMed.
- S. A. Abouelmagd, F. Meng, B.-K. Kim, H. Hyun and Y. Yeo, ACS Biomater. Sci. Eng., 2016, 2, 2294–2303 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- P. Maity, S. Xie, M. Yamauchi and T. Tsukuda, Nanoscale, 2012, 4, 4027–4037 RSC.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- I. Russier-Antoine, F. Bertorelle, M. Vojkovic, D. Rayane, E. Salmon, C. Jonin, P. Dugourd, R. Antoine and P.-F. Breveta, Nanoscale, 2014, 6, 13572–13578 RSC.
- R. Philip, P. Chantharasupawong, H. Qian, R. Jin and J. Thomas, Nano Lett., 2012, 12, 4661–4667 CrossRef CAS.
- G. Ramakrishna, O. Varnavski, J. Kim, D. Lee and T. Goodson, J. Am. Chem. Soc., 2008, 130, 5032–5033 CrossRef CAS PubMed.
- M. S. Devadas, S. Bairu, H. Qian, E. Sinn, R. Jin and G. Ramakrishna, J. Phys. Chem. Lett., 2011, 2, 2752–2758 CrossRef CAS.
- P. Apell, R. Monreal and S. Lundqvist, Phys. Scr., 1988, 38, 174–179 CrossRef CAS.
- D. Lee, R. L. Donkers, G. L. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS PubMed.
- O. J. H. Chai, Z. Liu, T. Chen and J. Xie, Nanoscale, 2019, 11, 20437–20448 RSC.
- A. Wang, X. Y. Liu, C.-Y. Mou and T. Zhang, J. Catal., 2013, 308, 258–271 CrossRef CAS.
- M. Ganguly, J. Jana, A. Pal and T. Pal, RSC Adv., 2015, 6, 17683–17703 RSC.
- M. Ganguly, C. Mondal, J. Pal, A. Pal, Y. Negishi and T. Pal, Dalton Trans., 2014, 43, 11557–11565 RSC.
- T. Y. Zhou, L. P. Lin, M. C. Rong, Y. Q. Jiang and X. Chen, Anal. Chem., 2013, 85, 9839–9844 CrossRef CAS PubMed.
- S. Kumar, M. D. Bolan and T. P. Bigioni, J. Am. Chem. Soc., 2010, 132, 13141–13143 CrossRef CAS PubMed.
- T. U. B. Rao and T. Pradeep, Angew. Chem., 2010, 49, 4218 CrossRef PubMed.
- C. I. Richards, S. Choi, J. C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y. L. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef CAS PubMed.
- J. Sharma, H. C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2010, 46, 3280–3282 RSC.
- S. Aslanzadeh, J. Mol. Model., 2016, 7, 160 CrossRef PubMed.
- K. Naseem, Z. H. Farooqi, R. Begum, W. Wu, A. Irfan and A. G. Al-Sehemi, Macromol. Chem. Phys., 2018, 219, 1800211 CrossRef.
- K. H. H. Aziz, et al., J. Hazard. Mater., 2018, 343, 107 CrossRef PubMed.
- M. Anari-Anaraki and A. Nezamzadeh-Ejhieh, J. Colloid Interface Sci., 2015, 440, 272 CrossRef CAS PubMed.
- M. Borandegi and A. Nezamzadeh-Ejhieh, Colloids Surf., A, 2015, 479, 35 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and M. Kabiri-Samani, J. Hazard. Mater., 2013, 260, 339 CrossRef CAS PubMed.
- A. Nezamzadeh-Ejhieh and M. Karimi-Shamsabadi, Chem. Eng. J., 2013, 228, 631 CrossRef CAS.
- H. Shirzadi and A. Nezamzadeh-Ejhieh, J. Mol. Liq., 2017, 230, 221 CrossRef CAS.
- A. Hernandez-Martínez, React. Funct. Polym., 2018, 122, 75 CrossRef.
- F. Salimi, S. S. Emami and C. Karami, Inorg. Nano-Met. Chem., 2018, 48, 31 CrossRef CAS.
- F. Salimia, M. Eskandaria and C. Karamib, Desalin. Water Treat., 2017, 85, 206 CrossRef.
- F. Salimi, K. Tahmasobi, C. Karami and A. Jahangiri, J. Mex. Chem. Soc., 2017, 61, 250 CAS.
- H. Derikvandi and A. Nezamzadeh-Ejhieh, J. Colloid Interface Sci., 2017, 490, 628 CrossRef CAS PubMed.
- Y. Yu, C. Zhao, X. Liu, M. Sui and Y. Meng, J. Environ. Chem. Eng., 2017, 5, 5406 CrossRef CAS.
- M. Nasrollahzadeh, Z. Issaabadi and S. M. Sajadi, RSC Adv., 2018, 8, 3723 RSC.
- M. Nasrollahzadeh, M. Sajjadi and S. M. Sajadi, Chin. J. Catal., 2018, 39, 109 CrossRef CAS.
- H. Zhang, G. Xue, H. Chen and X. Li, Chemosphere, 2018, 191, 64 CrossRef CAS PubMed.
- Y. Li, S. Guo, H. Yang, Y. Chao, S. Jiang and C. J. R. Wang, RSC Adv., 2018, 8, 8057 RSC.
- N. Demirkıran, G. T. Özdemir, M. Saraç and M. Dardagan, Mong. J. Chem., 2017, 18, 5 CrossRef.
- J. Najeeb, G. Ahmad, S. Nazir, K. Naseem and A. Kanwal, Korean J. Chem. Eng., 2017, 1 Search PubMed.
- M. Ikram, RSC Adv., 2020, 10, 20559 RSC.
- A. Raza, Appl. Nanosci., 2020, 10, 3875 CrossRef CAS.
- A. N. Ejhieh and M. Khorsandi, J. Hazard. Mater., 2010, 176, 629 CrossRef CAS PubMed.
- M. Bahrami and A. Nezamzadeh-Ejhieh, Mater. Sci. Semicond. Process., 2015, 30, 275 CrossRef CAS.
- A. Raza, Appl. Nanosci., 2020, 10, 1535 CrossRef CAS.
- M. Ikram, A. Raza, M. Imran, A. Ul-Hamid, A. Shahbaz and S. Ali, Nanosci. Res. Lett., 2020, 15, 95 CrossRef CAS PubMed.
- M. Ikram, M. I. Khan, A. Raza, M. Imran, A. Ul-Hamid and S. Ali, Phys. E, 2020, 124, 114246 CrossRef CAS.
- T. Shahwan, Chem. Eng. J., 2011, 172, 258 CrossRef CAS.
- B. Neppolian, H. Choi, S. Sakthivel, B. Arabindoo and V. Murugesan, J. Hazard. Mater., 2002, 89, 303 CrossRef CAS PubMed.
- Z. Shams-Ghahfarokhi and A. Nezamzadeh-Ejhieh, Mater. Sci. Semicond. Process., 2015, 39, 265 CrossRef CAS.
- C. Guillard, H. Lachheb, A. Houas, M. Ksibi, E. Elaloui and J.-M. Herrmann, J. Photochem. Photobiol., A, 2003, 158, 27 CrossRef CAS.
- M. Ikram, RSC Adv., 2020, 10, 30007 RSC.
- M. Ikram, RSC Adv., 2020, 10, 24215 RSC.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662 CrossRef CAS PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., 2016, 55, 2230 CrossRef CAS PubMed.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669 CrossRef CAS.
- R. Ullah and J. Dutta, J. Hazard. Mater., 2008, 156, 194 CrossRef CAS PubMed.
- S. Chandra, G. Beaune, N. Shirahata and F. M. Winnik, J. Mater. Chem. B, 2017, 5, 1363 RSC.
- S. Chandra, Nonappa, G. Beaune, A. Som, S. Zhou, J. Lahtinen, H. Jiang, J. V. I. Timonen, O. Ikkala and R. H. A. Ras, Adv. Opt. Mater., 2019, 7, 1900620 CrossRef CAS.
- N. Vilar-Vidal, J. Rivas and M. A. López-Quintela, ACS Catal., 2012, 2, 1693–1697 CrossRef CAS.
- C. Yu, G. Li, S. Kumar, H. Kawasaki and R. Jin, J. Phys. Chem. Lett., 2013, 4, 2847–2852 CrossRef CAS.
- S. Zhou, Y. Duan, F. Wang and C. Wang, Nanoscale, 2017, 9, 4981–4988 RSC.
- S. Bhattacharyya, B. Jana and A. Patra, ChemPhysChem, 2015, 16, 796–804 CrossRef CAS PubMed.
- M. K. Barma, B. Paramanik, D. Bain and A. Patra, Chemistry, 2016, 22, 11699–11705 CrossRef PubMed.
- X. Yang, A. Wolcott, G. Wang, A. Sobo, R. C. Fitzmorris, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2009, 9, 2331–2336 CrossRef CAS PubMed.
- Y. Hou, Z. Wen, S. Cui, X. Feng and J. Chen, Nano Lett., 2016, 16, 2268–2277 CrossRef CAS PubMed.
- Q. Zhang, Y. Huang, L. Xu, J. Cao, W. Ho and S. C. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 4165–4174 CrossRef CAS PubMed.
- B. Wu, Y. Xiong and Y. Ge, Chem. Eng. J., 2018, 331, 343–354 CrossRef CAS.
- U. Bali and B. Karagözoğlu, Dyes Pigm., 2007, 74, 73–80 CrossRef CAS.
- M. Cheng, G. M. Zeng, D. L. Huang, C. Lai, Y. Liu, C. Zhang, J. Wan, L. Hu, C. Y. Zhou and W. P. Xiong, Water Res., 2018, 138, 7–18 CrossRef CAS PubMed.
- H. Zhang, L. H. Guo, D. Wang, L. Zhao and B. Wan, ACS Appl. Mater. Interfaces, 2015, 7, 1816–1823 CrossRef CAS PubMed.
- A. Khan, U. Alam, D. Ali, D. Bahnemann and M. Muneer, Mater. Lett., 2018, 220, 50–53 CrossRef CAS.
- M. Najafi, A. Abbasi, M. Masteri-Farahani and J. Janczak, Dalton Trans., 2015, 44, 6089–6097 RSC.
- Q. Liu, D. Li, X. Lei, Y. Chen, J. Wang, A. Liu, B. Han and G. He, Sep. Purif. Technol., 2023, 310, 122938 CrossRef CAS.
- Y. Wang, Y. Tan, Y. Ding, L. Fu and W. Qing, Colloids Surf., A, 2022, 654, 130072 CrossRef CAS.
- H. Zhang, L. H. Guo, D. Wang, L. Zhao and B. Wan, ACS Appl. Mater. Interfaces, 2015, 7, 1816–1823 CrossRef CAS PubMed.
- T. Goswami, A. Bheemaraju, A. K. Sharma and S. Bhandari, Colloid Polym. Sci., 2021, 299, 925–936 CrossRef CAS.
- Y. Liu, Q. Yao, X. Wu, T. Chen, Y. Ma, C. N. Ong and J. Xie, Nanoscale, 2016, 8, 10145–10151 RSC.
- T. Goswami, M. Singh and K. M. Reddy, Mater. Res. Bull., 2018, 107, 286–294 CrossRef CAS.
- N. K. Chaki, J. Sharma, A. B. Mandle, I. S. Mulla, R. Pasricha and K. Vijayamohanan, Phys. Chem. Chem. Phys., 2004, 1304–1309 RSC.
- A. Bheemaraju, T. Goswami and M. Reddy, ChemistrySelect, 2019, 4, 6790–6799 CrossRef.
- N. K. Pal and C. Kryschi, J. Mol. Catal. A: Chem., 2015, 404 Search PubMed.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2013, 29, 8125–8132 CrossRef CAS PubMed.
- C. Yu, G. Li, S. Kumar, H. Kawasaki and R. Jin, J. Phys. Chem. Lett., 2013, 4, 2847–2852 CrossRef CAS.
- K. Sridharan, E. Jang, J. H. Park, J.-H. Kim, J.-H. Lee and T. J. Park, Chem. –Eur. J., 2015, 21, 9126–9132 CrossRef CAS PubMed.
- L. Collado, A. Reynal, F. Fresno, M. Barawi, C. Escudero, V. Perez-Dieste, J. M. Coronado, D. P. Serrano, J. R. Durrant and V. A. de la Pena O'Shea, Nat. Commun., 2018, 9, 1–10 CrossRef CAS PubMed.
- S. Nair and B. J. Tatarchuk, Fuel, 2010, 89, 3218–3225 CrossRef CAS.
- A. Arenas-Vivo, S. Rojas, I. Ocana, A. Torres, M. Liras, F. Salles, D. Arenas- Esteban, S. Bals, D. Avila and P. Horcajada, J. Mater. Chem. A, 2021, 9, 15704–15713 RSC.
- M. Farrang, J. Photochem. Photobiol., A, 2021, 422, 113551 CrossRef.
- C. Liu, X.-Q. Ren, F. Lin, X.-M. Fu, X.-Z. Lin, T. Li, K.-J. Sun and J.-H. Huang, Angew. Chem., Int. Ed., 2019, 58, 11335 CrossRef CAS PubMed.
- C. Deng, C. Sun, Z. Wang, Y. Tao, Y. Chen, J. Lin, G. Luo, B. Lin, D. Sun and L. Zheng, Angew. Chem., 2020, 59, 12659–12663 CrossRef CAS PubMed.
- T. Goswami, M. Reddy, A. K. Mishra and M. Singh, ChemistrySelect, 2018, 3, 10892–10899 CrossRef CAS.
- N. Balázs, K. Mogyorósi, D. F. Srankó, A. Pallagi, T. Alapi, A. Oszkó, A. Dombi and P. Sipos, Appl. Catal., B, 2008, 84, 356–362 CrossRef.
- P. V. Kamat, Chem. Rev., 1993, 93, 267–300 CrossRef CAS.
- D. Mott, N. T. Thuy, Y. Aoki and S. Maenosono, Philos. Trans. R. Soc., A, 2010, 368, 4275–4292 CrossRef CAS PubMed.
- K. P. Charle, L. Konig, S. Nepijko, I. Rabin and W. Schulze, Cryst. Res. Technol., 1998, 33, 1085–1096 CrossRef CAS.
- T. M. O. Le, T. H. Lam, T. N. Pham, T. C. Ngo, N. D. Lai, D. B. Do and V. M. Nguyen, Polymers, 2018, 10, 633 CrossRef PubMed.
- S. Mao, S. Ning, X. Zhang, M. Xia and F. Wang, Adv. Powder Technol., 2021, 5, 1641–1652 CrossRef.
- R. Nagaraja, N. Kottam, C. R. Girija and B. M. Nagabhushana, Powder Technol., 2012, 215, 91–97 CrossRef.
- A. Kumar and G. Pandey, Mater. Sci. Eng. Int. J., 2017, 1, 00018 Search PubMed.
- K. Negi, A. Umar, M. S. Chauhan and M. S. Akhtar, Ceram. Int., 2019, 45, 20509–20517 CrossRef CAS.
- M. Koklioti, I. Saucedo- Orozco, M. Quintana and N. Tagmatarchis, Mater. Res. Bull., 2019, 114 Search PubMed.
- A. V. Salker and S. D. Gokakakar, Int. J. Phys. Sci., 2005, 4, 377–384 Search PubMed.
- V. P. Dinesh, P. Bijli, A. Ashok, S. K. Dhara, M. Kamruddin, A. K. Tyagi and B. Raj, RSC Adv., 2014, 4, 58930–58940 RSC.
- J. Gonzalez-Rodriguez, L. Fernandez, Y. B. Bava, D. Buceta, C. Vazquez, M. A. Lopez-Quintela, G. Feijoo and M. T. Moreira, Catalysts, 2020, 10, 31 CrossRef CAS.
- M. Reddy and J. Devaraju, Mater. Today: Proc., 2019, 17, 235–238 Search PubMed.
- M. Jakob, H. Levanon and P. V. Kamat, Nano Lett., 2003, 3, 353 CrossRef CAS.
- Z. Yuanhui, Z. Lirong, Z. Yingying, L. Xingyi, L. Z. Qi and W. Kemei, Inorg. Chem., 2007, 46, 6980–7986 CrossRef PubMed.
- R. Agrawal and L. Mulky, ChemBioEng, 2023, 10, 326–335 CrossRef.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2013, 29, 812532 CrossRef PubMed.
- W. Baschong, J. M. Lucocq and J. Roth, Histochemistry, 1985, 83, 409 CAS.
- N. Chandrasekharan, P. V. Kamat, J. Hu and G. Jones, J. Phys. Chem. B, 2000, 47, 11103–11109 CrossRef.
- M. R. Willner and P. J. Vikesland, J. Nanobiotechnol., 2018, 16, 1–16 CrossRef PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., 2006, 45, 4562–4588 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Quantum dots versus organic dyes as fluorescent labels, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- R. D. Mitra, C. M. Silva and D. C. Youvan, Gene, 1996, 173, 13–17 CrossRef CAS PubMed.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- N. Y. chen, H. F. Li, Z. F. Gao and F. Qu, Sens. Actuators, B, 2014, 193, 730–736 CrossRef CAS.
- B. E. Demirhah, H. E. S. Kara and B. Dehmirahan, J. Photochem. Photobiol., A, 2021, 417, 113356 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |