
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of novel 1,2,4-triazole tethered β-hydroxy sulfides as bacterial tyrosinase inhibitors: synthesis and biophysical evaluation through in vitro and in silico approaches†
Sadaf Saeeda,
Muhammad Jawwad Saif *b,
Ameer Fawad Zahoor*a,
Hina Tabassumc,
Shagufta Kamald,
Shah Faisale,
Rabia Ashrafa,
Samreen Gul Khana,
Usman Nazeerf,
Ali Irfana and
Mashooq Ahmad Bhat*g
*b,
Ameer Fawad Zahoor*a,
Hina Tabassumc,
Shagufta Kamald,
Shah Faisale,
Rabia Ashrafa,
Samreen Gul Khana,
Usman Nazeerf,
Ali Irfana and
Mashooq Ahmad Bhat*g
aDepartment of Chemistry, Government College University Faisalabad, 38000-Faisalabad, Pakistan. E-mail: fawad.zahoor@gcuf.edu.pk
bDepartment of Applied Chemistry, Government College University Faisalabad, 38000-Faisalabad, Pakistan. E-mail: jawwadsaif@gmail.com
cLondon Metropolitan University, 166-220 Holloway Road, London N7 8DB, UK
dDepartment of Biochemistry, Government College University Faisalabad, 38000-Faisalabad, Pakistan
eDepartment of Chemistry, Islamia College University Peshawar, Peshawar 25120, Pakistan
fDepartment of Chemistry, University of Houston, 3585 Cullen Boulevard, Texas 77204-5003, USA
gDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia. E-mail: mabhat@ksu.edu.sa
First published on 13th May 2024
Abstract
In this study, a series of 1,2,4-triazole-tethered β-hydroxy sulfide scaffolds 11a–h was synthesized in good to remarkable yields (69–90%) through the thiolysis of oxiranes by the thiols in aqueous basic catalytic conditions. The synthesized 1,2,4-triazole-tethered β-hydroxy sulfides were screened against bacterial tyrosinase enzyme, and Gram-positive and Gram-negative bacterial cultures i.e., (S. aureus) Staphylococcus aureus & (E. coli) Escherichia coli. Among the synthesized derivatives, the molecules 11a (IC50 = 7.67 ± 1.00 μM), 11c (IC50 = 4.52 ± 0.09 μM), 11d (IC50 = 6.60 ± 1.25 μM), and 11f (IC50 = 5.93 ± 0.50 μM) displayed the better tyrosinase inhibitory activity in comparison to reference drugs ascorbic acid (IC50 = 11.5 ± 1.00 μM) and kojic acid (IC50 = 30.34 ± 0.75 μM). The molecule benzofuran-triazol-propan-2-ol 11c proved to be the most potent bacterial tyrosinase inhibitory agent with a minimum IC50 of 4.52 ± 0.09 μM, as compared to other synthesized counterparts and both standards (kojic acid and ascorbic acid). The compound diphenyl-triazol-propan-2-ol 11a and benzofuran-triazole-propan-2-ol 11c showed comparable anti-bacterial chemotherapeutic efficacy with minimum inhibitory concentrations (MIC = 2.0 ± 2.25 mg mL−1 and 2.5 ± 0.00 mg mL−1, respectively) against S. aureus bacterial strain in comparison with standard antibiotic penicillin (MIC = 2.2 ± 1.15 mg mL−1). Furthermore, among the synthesized derivatives, only compound 11c demonstrated better anti-bacterial activity (MIC = 10 ± 0.40 mg mL−1) against E. coli, which was slightly less than the standard antibiotic i.e., penicillin (MIC = 2.4 ± 1.00 mg mL−1). The compound 11c demonstrated a better binding score (−7.08 kcal mol−1) than ascorbic acid (−5.59 kcal mol−1) and kojic acid (−5.78 kcal mol−1). Molecular docking studies also validate the in vitro anti-tyrosinase assay results; therefore, the molecule 11c can be the lead bacterial tyrosinase inhibitor as well as the antibacterial agent against both types of bacterial strains after suitable structural modifications.
1. Introduction
Tyrosinase is a Cu-containing polyphenol oxidase that is ubiquitously distributed in insects, plants, animals, and microorganisms.1–4 It catalyzes the initial two steps required for melanin biosynthesis, which is responsible for the pigmentation of eyes, skin and hair.5,6 Unregulated stimulation of tyrosinase can lead to severe dermatological afflictions (i.e., scarring with hyperpigmentation and ephelides) and neurodegenerative diseases (i.e., Parkinson's disease).7–9 Tyrosinase inhibitors have become vital in the agriculture, cosmetics, food business, and pharmaceutical industries to prevent hyperpigmentation.10,11 Lately, surplus natural and laboratory-generated tyrosinase inhibitors have been documented, including kojic acid, aromatic acids, ascorbic acid, polyphenols, arbutin, hydroquinone, aromatic aldehydes, tropolone, and aromatic alcohols.12 However, several widely recognized brightening substances i.e., kojic acid and hydroquinone, are regarded as dangerous agents attributable to the undesired adverse effects i.e., neurological disorders, dermatological cancer, skin irritation, and cytotoxicity.13–16 The exploration of novel, manageable and harm-less tyrosinase inhibitors has become a matter of grave concern.Triazole derivatives are recognized for their remarkable heterocyclic structures, which offer a diverse array of therapeutic and biological effects.17–19 These compounds have demonstrated significant roles in combating viruses, cancers, bacteria, seizures, pain, inflammation, diabetes, fungal infections, oxidative stress, and hypertension. Their versatility in addressing such wide-ranging health concerns underscores their importance in pharmaceutical research and development.20–22
Previous research has identified various heterocyclic compounds such as benzofuran-oxadiazole as bacterial tyrosinase inhibitors,23 oxadiazoles as human tyrosinase inhibitors,24 and piperazines as bacterial tyrosinase inhibitors.25 Nitrogenous triazole scaffolds have been found to exhibit mushroom tyrosinase inhibition, such as 1,2,4-triazole hydrazones and 1,2,4-triazole clubbed N-phenylacetamides (Fig. 1).26,27 The anti-tyrosinase inhibition of 1,2,4-triazole derivatives lies in the fact that the N-atoms of the triazole functionality can coordinate with the two Cu ions constituting the catalytic hub (active site) of enzyme. Furthermore, van der Waals interactions between the residues covering the hydrophobic void and phenyl group, augment the binding capability of enzyme.
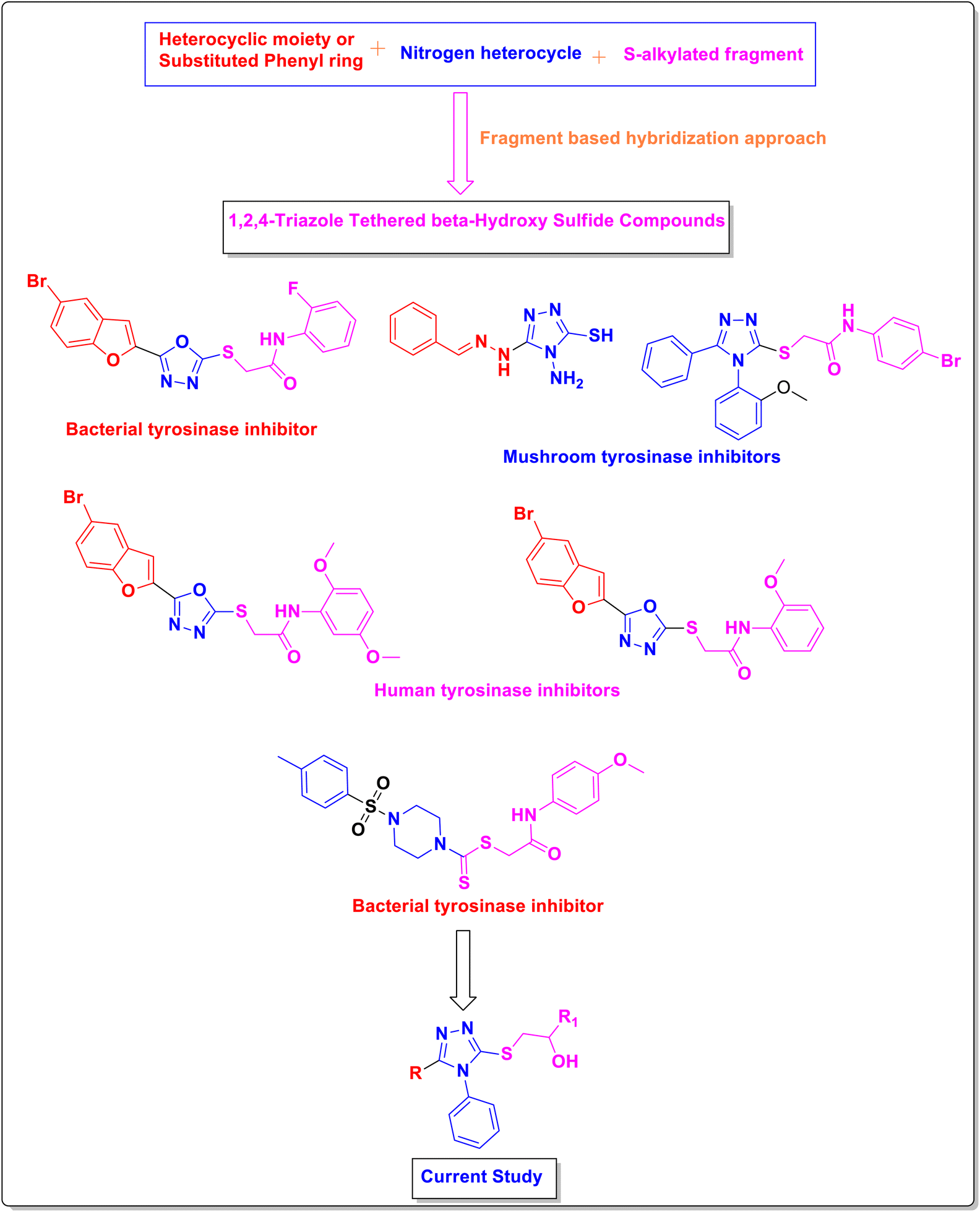 | ||
| Fig. 1 Rational design of fragment-based hybridization approach. | ||
In organic synthesis, epoxides facilitate the generation of several organic frameworks by acting as pivotal forerunners. They undergo treatment with a variety of nucleophiles including thiols, alcohols and amines to afford several organic functionalities.28,29 The ring cleavage of epoxides with –SH (thiols) lead to the generation of β-hydroxy sulfides which act as fascinating precursors in synthetic organic chemistry and can be employed to achieve the natural products, biologically potent molecules and other products including leukotrienes such as LTC4 and LTD4.30–32
Here, a fragment-based hybridization approach was applied to develop rationale of the current study by the incorporation of various heterocyclic moieties such as benzofuran, bromobenzofuran, naphthofuran, and acefylline along with substituted phenyl ring around the 1,2,4-triazole core. The thiol functionality of triazole core was modified by the incorporation of β-hydroxy sulfides as depicted in Fig. 1.
Encouraged by the biological importance and applications of the above compounds in various fields, the aspiration of the current study involves the functional layout and preparation of some novel anti-bacterial and anti-tyrosinase agents through the coupling of 1,2,4-triazole derivatives with different epoxides e.g. 2-methyloxirane, 2-((naphthalen-2-yloxy)methyl)oxirane, and 2-(phenoxymethyl)oxirane via thiolysis. Molecular docking studies were utilized to validate the tyrosinase inhibition activity of the potent compound and finally, a detailed structure–activity relationship was inferred.
2. Results and discussion
2.1. Chemistry
The designed framework to access β-hydroxy sulfides 11a–h has been depicted in Scheme 1. Various substituted aromatic aldehydes 1 and phenol 2 were reacted with ethyl chloroacetate 3 using potassium carbonate in DMF to obtain corresponding esters in 76–87% yield.33,37 Besides this, various carboxylic acids were transformed into respective esters (68–80% yield) via Fischer esterification, by using a catalytic amount of sulfuric acid.38 Corresponding hydrazides 6a–h were synthesized in 47–90% yield by treating various esters 5a–h with hydrazine monohydrate in ethanol.39 These synthesized compounds were refluxed with aryl isothiocyanate 7 in ethanol to get key intermediates (thiosemicarbazide) 8a–h. These intermediates, after refluxing with KOH solution, were further transformed into the desired 1,2,4-triazoles 9a–h (68–83%).40 Targeted 1,2,4-triazole-based β-hydroxy sulfides 11a–h (Table 1) were prepared in excellent yield (69–90%) by ring opening of epoxides 10a–c with triazoles 9a–h, in the presence of potassium carbonate.41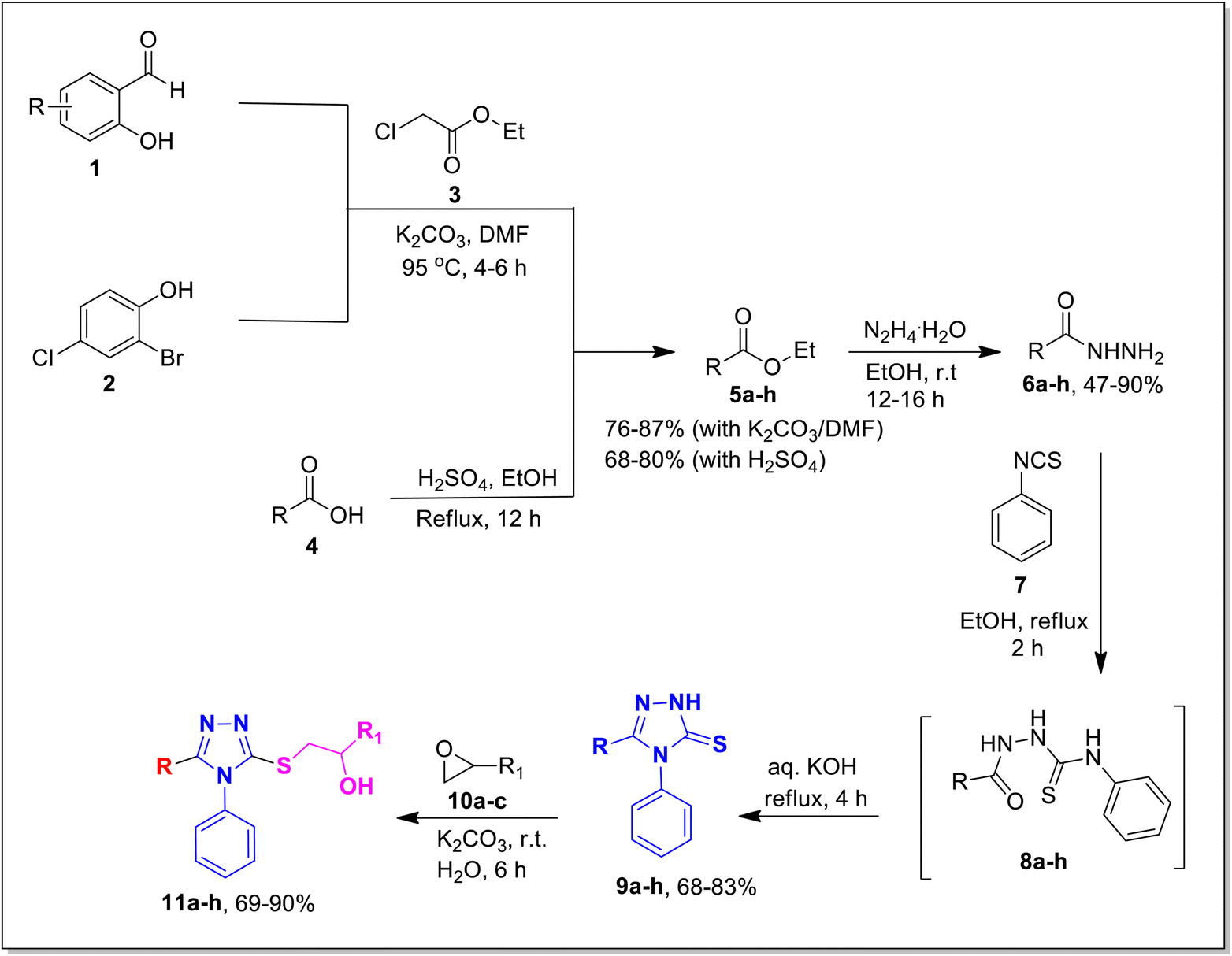 | ||
| Scheme 1 Schematic layout of the synthesis of 1,2,4-triazoles tethered β-hydroxy sulfides (11a–h). | ||
| Compound | R | R1 | Product | Yield (%) |
|---|---|---|---|---|
| 11a | 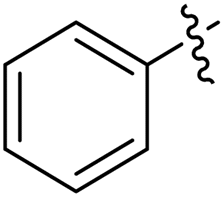 |
CH3– | 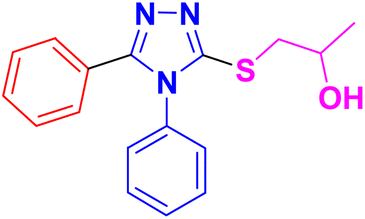 |
79 |
| 11b |  |
CH3– | 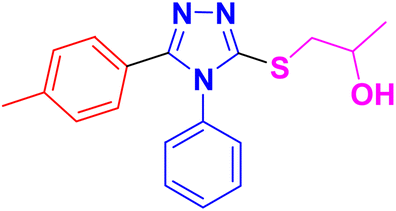 |
79 |
| 11c |  |
CH3– | 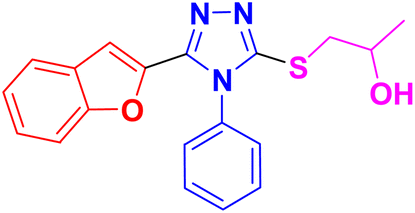 |
85 |
| 11d | 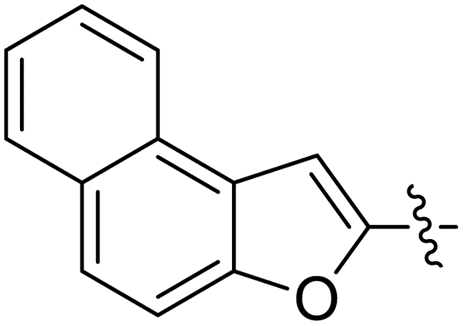 |
CH3– | 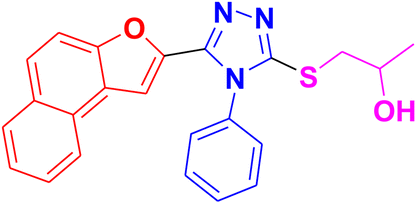 |
86 |
| 11e | 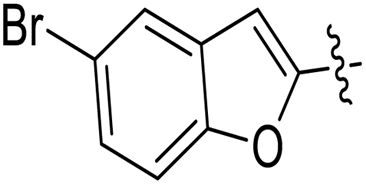 |
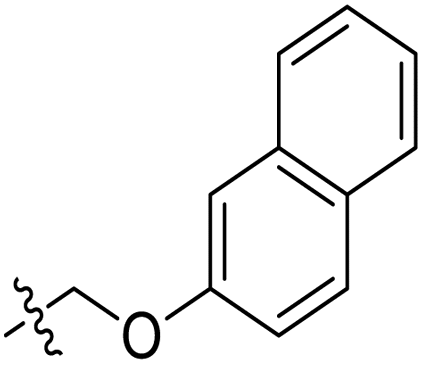 |
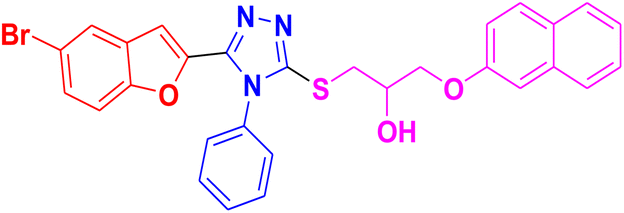 |
90 |
| 11f | 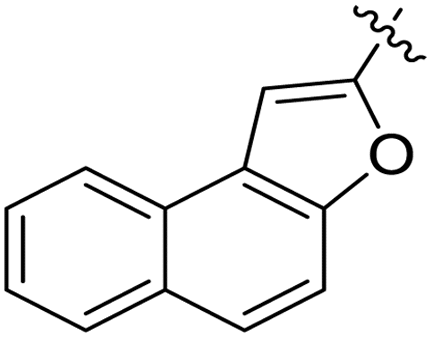 |
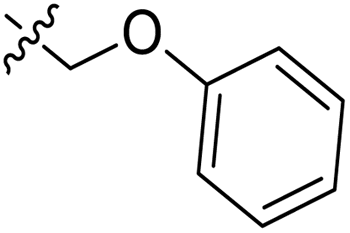 |
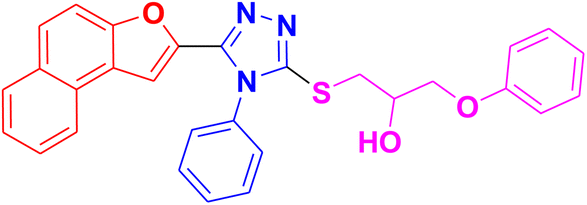 |
81 |
| 11g | 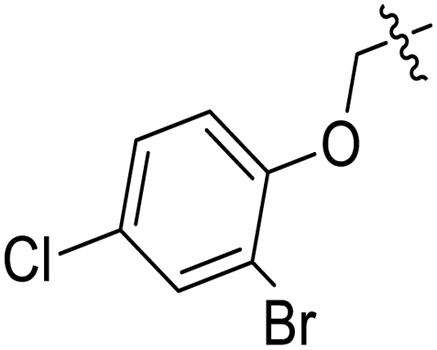 |
CH3– | 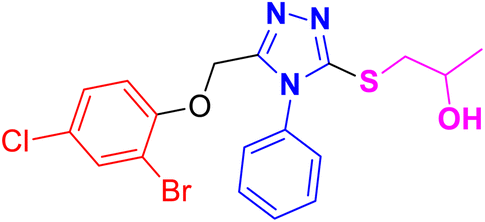 |
69 |
| 11h | 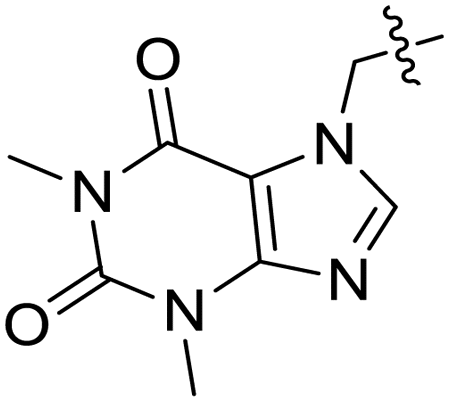 |
CH3– | 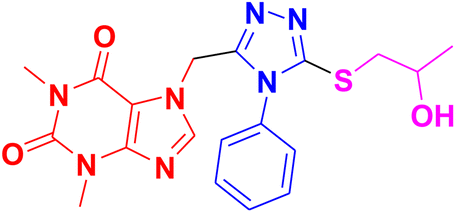 |
71 |
2.2. Bacterial tyrosinase inhibition activity of 1,2,4-triazole-tethered β-hydroxy sulfides (11a–h)
The in vitro anti-tyrosinase activity of the β-hydroxy sulfides 11a–h containing the 1,2,4-triazole moiety was examined and compared with that of ascorbic acid and kojic acid. Table 2 depicts the inhibitory effects of the novel compounds against bacterial tyrosinase and their IC50 values ranged from 4.52 ± 0.09 to 51.40 ± 0.16 μM. Interestingly, the tested compounds 11a, 11c, 11d and 11f were found to be more potent inhibitors of bacterial tyrosinase than the reference inhibitors, ascorbic acid and kojic acid. Over all, compound 11c exhibited the most robust anti-tyrosinase activity, demonstrating IC50 = 4.52 ± 0.09 μM, whereas, compound 11e displayed poor anti-tyrosinase effect with 51.40 ± 0.16 μM IC50 value. The remaining tested compounds displayed moderate inhibitory activity.| Compounds | Tyrosinase inhibition IC50 (μM) |
|---|---|
| 11a | 7.67 ± 1.00 |
| 11b | 14.43 ± 1.50 |
| 11c | 4.52 ± 0.09 |
| 11d | 6.60 ± 1.25 |
| 11e | 51.40 ± 0.16 |
| 11f | 5.93 ± 0.50 |
| 11g | 13.50 ± 0.50 |
| 11h | 13.41 ± 1.15 |
| Ascorbic acid | 11.5 ± 1.00 |
| Kojic acid | 30.34 ± 0.75 |
2.3. Structure–activity relationship
The anti-tyrosinase efficacy of prepared compounds 11a–h relied significantly on the key parameters, including the position of substituents, the nature of substituents as electron withdrawing or donating, and the electronegativity of attached moieties to the triazole ring. These factors play crucial roles in establishing the structure activity relationship (SAR).Regarding the nature of the substituent, the benzofuran moiety conferred a remarkable tyrosinase inhibition potential over other functionalities. Interestingly, compound 11c bearing a benzofuran moiety was found to be the most promising and potent bacterial tyrosinase inhibitory agent, (which) exhibited the minimum inhibitory concentration (IC50 = 4.52 ± 0.09 μM) as displayed in Fig. 2. The replacement of benzofuran moiety in compound 11c with aryl (11a, IC50 = 7.67 ± 1.00 μM), naphthofuran (11d, IC50 = 6.60 ± 1.25 μM), and benzofuran and propan-2-ol of compound 11c with naphthofuran and 3-phenoxypropan-2-ol (11f, IC50 = 5.93 ± 0.50 μM) led to a dramatic decline in (the) tyrosinase inhibitory activity (Fig. 2). Additionally, compound 11e (IC50 = 51.40 ± 0.16) with 5-bromobenzofuran and 3-(naphthalen-2-yloxy)propan-2-ol substituents was found to be less active as compared to other synthesized compounds as displayed in Fig. 2. The placement of electron-donating groups on the aryl ring, i.e., 4-CH3 (11b, IC50 = 14.43 ± 1.50 μM), also resulted in decreased inhibitory activity. Whereas, the introduction of halogens (bromo and chloro) at the 2- and 4-position of the phenyl ring 11g (attached to the 1,2,4-triazole ring), respectively, diminished the inhibitory effect against the tested enzyme with an IC50 of 13.50 ± 0.50 μM as in compound 11g. The anti-tyrosinase effect of 11h (IC50 = 13.41 ± 1.15 μM) bearing acefylline scaffold was found to be almost similar in activity to that of the compound 11g, that is, 13.50 ± 0.50 μM.
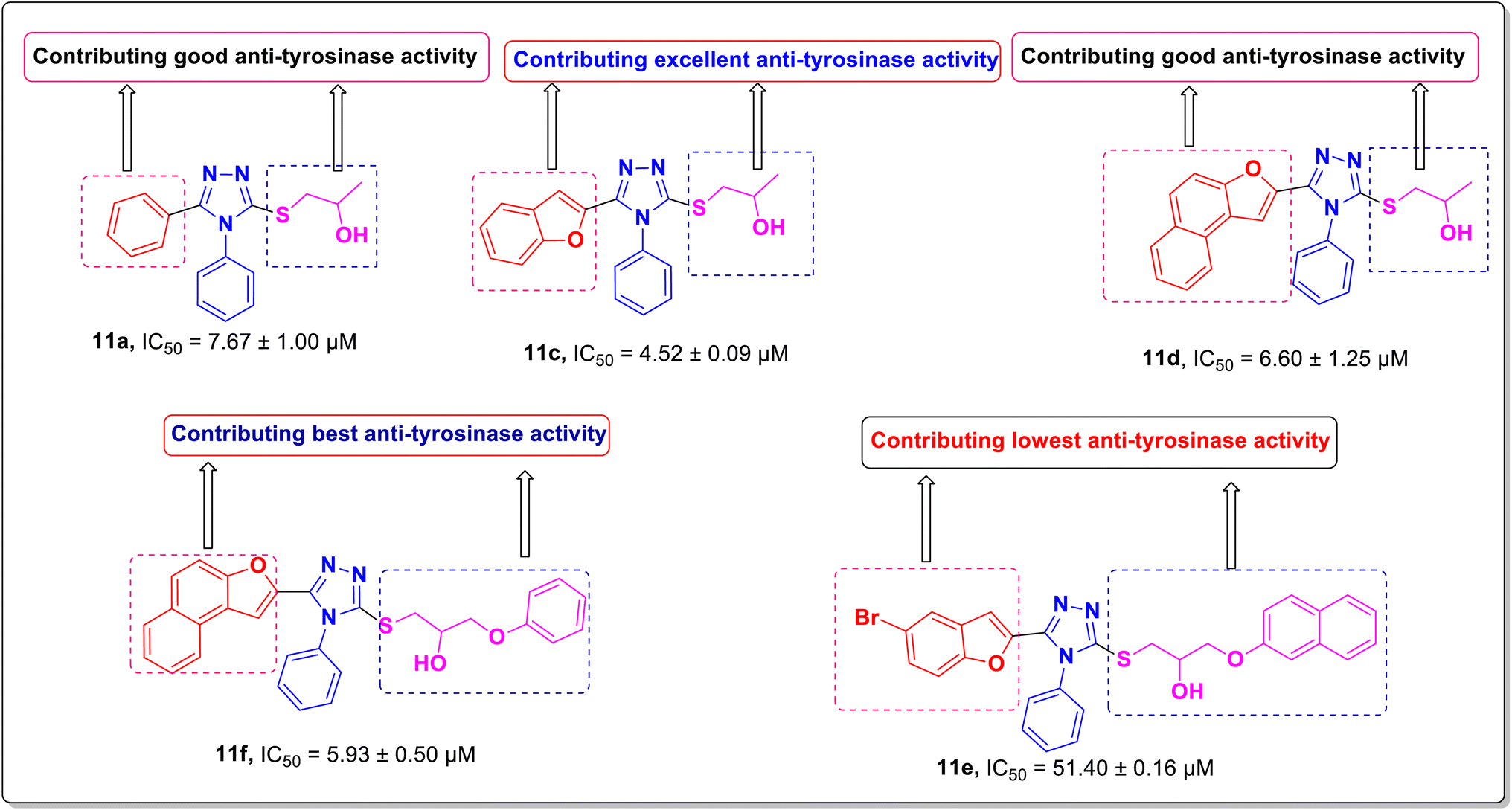 | ||
| Fig. 2 Structure–activity relationship of the excellent to least potent tyrosinase inhibitors. | ||
2.4. Molecular docking studies of the most bioactive 1,2,4-triazole-tethered β-hydroxy sulfide 11c
The in vitro investigations of the prepared compounds reflected that the compound 11c showed a significantly good inhibition profile against the tyrosinase enzyme. Encouraged by the results, we further explored the binding modes and the molecular interactions of compound 11c with the target enzyme utilizing the computation molecular docking investigations. The results of molecular docking investigation revealed that the compound 11c did bind to the catalytic hub (active site) of the tyrosinase enzyme with an anchoring affinity score of −7.08 kcal mol−1 in comparison to the reference standard tyrosinase inhibitors (i.e.) kojic acid (−5.78 kcal mol−1) and ascorbic acid (−5.59 kcal mol−1), respectively. The binding affinities showed that compound 11c has a significantly better affinity towards the tyrosinase enzyme in comparison with the control compounds.The conformational analysis of the compound 11c inside the substrate-binding site of the target enzyme showed that it binds strongly and makes robust molecular interactions of diverse types with the catalytic hub residues of the tyrosinase enzyme. It was noted that during the conformational analysis, the Ser360 active site residue was engaged by the –OH group of this compound with a hydrogen bond contact. Moreover, the nearby sulfur atom of compound 11c was able to interact via a pi-sulfur contact with the Phe347 residues of the target enzyme. The triazole moiety of compound 11c exhibited several interactions within the tyrosinase active site and formed notable molecular contacts of π–π, T-shaped coordination with Phe347 and a π-alkyl water-repellant contact (hydrophobic) with Ile368. Additionally, the triazole-linked phenyl ring also participated in a similar type of interaction with Val377. Furthermore, the benzofuran moiety, the main pharmacophore of this compound, significantly contributed to binding through dual π-alkyl water-repellant interactions (hydrophobic) with Val377. The benzofuran moiety of compound 11c further engaged with His367, a critical active-site histidine in the tyrosinase enzyme. This residue gets coordinated to a copper ion, which is essential for the enzyme's catalytic activity. Moreover, the benzofuran moiety established a dual interaction with His367 i.e., π–π, T-figured and a π–π stacked type interactions. Other weak types of van der Waals (interactions) were also noted (observed) with the copper ions that are located in the substrate-binding site of tyrosinase enzyme along with the other amino acid vestiges of the active site. Further, preliminary computational SAR studies showed that the substitution of bulky groups on the benzofuran ring lowers the binding affinities of the derivatives towards the catalytic hub (active site) of the tyrosinase while the introduction of a small polar group like –OH (hydroxyl) on the benzofuran moiety enhances the binding of these compounds with the tyrosinase enzyme. The binding energies of the evaluated compounds are presented in Table 1, while the graphical representation of the compound 11c inside the tyrosinase active site can be viewed in Fig. 3.
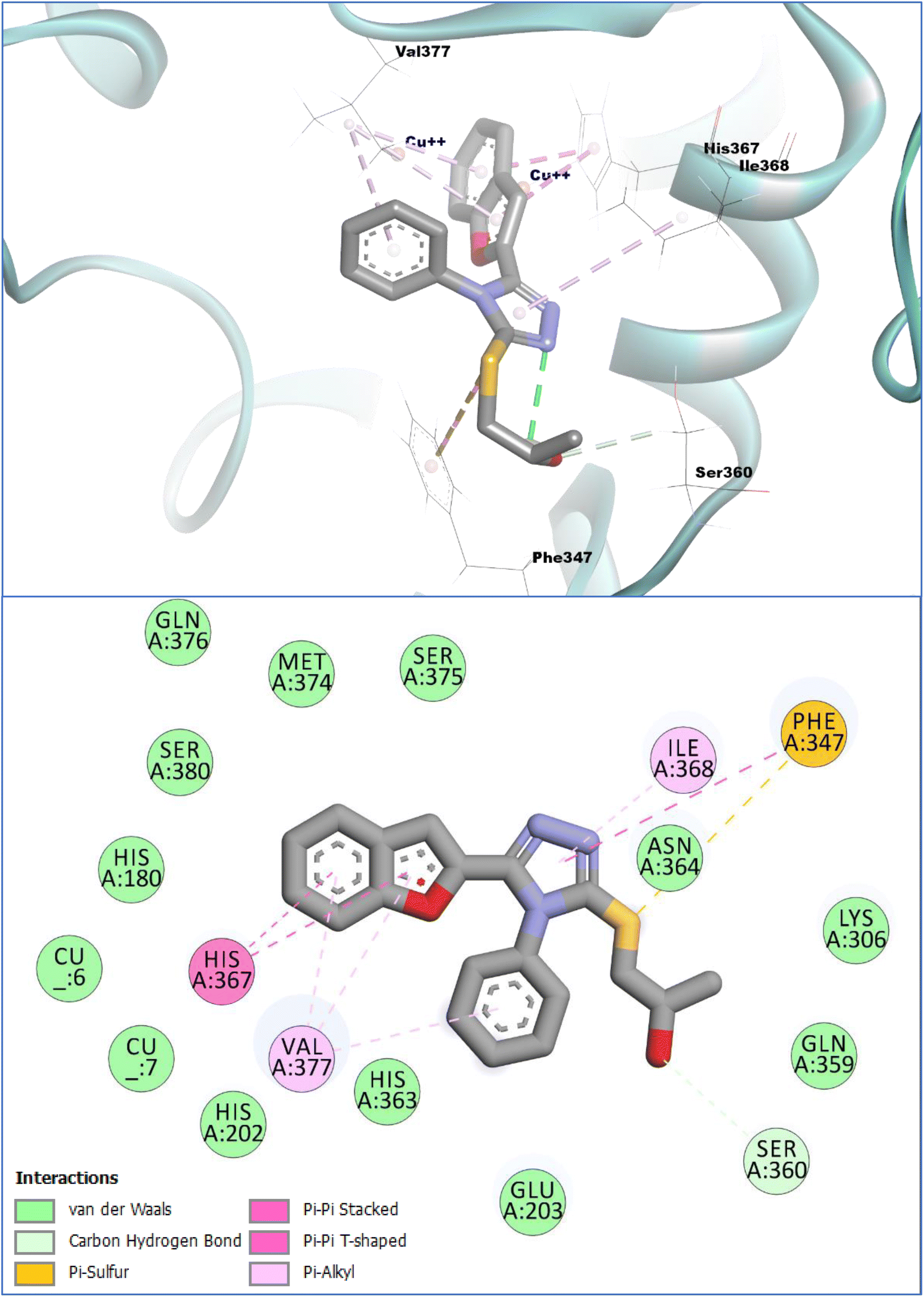 | ||
| Fig. 3 Binding conformation and interaction profile of compound 11c with the tyrosinase enzyme. Molecular docking was performed using Webina (an online version of Autodock Vina). | ||
The probable mechanism of action of compound 11c against the tyrosinase enzyme can be explained by the direct contact of the benzofuran moiety with the important histidine residue and other stronger interactions with the active site residues as seen in the molecular docking studies. This compound may obstruct the association of the catalytic Cu ions inside the catalytic hub of tyrosinase enzyme by interfering with the co-ordination process of these metal ions with the coordinating histidine residues. This may result in altering the oxidation states of these ions, resulting in the loss of activity of the tyrosinase enzyme.
2.5. Anti-bacterial activity
The outcomes of the anti-bacterial action of newly synthesized compounds presented as MIC values in comparison to the control (penicillin) are illustrated in Table 3. Among the evaluated compounds, most of the derivatives showed strong to moderate inhibition in defiance of gram +ve and −ve negative bacterial cultures i.e., S. aureus and E. coli respectively. All-encompassing, compound 11a demonstrated remarkable effectiveness in inhibiting the growth of the Gram-positive bacterium S. aureus, thereby exhibiting minimum inhibitory concentration (MIC) = 2.0 ± 2.25 mg mL−1. However, against Gram-negative bacteria, compound 11c exhibited notable potency, displaying an MIC of 10 ± 0.40 mg mL−1. These findings underscore the diverse anti-bacterial properties of these compounds, with 11a showing specificity towards Gram-positive strains and 11c demonstrating efficacy against Gram-negative bacteria as depicted in Table 4.| Sr. No. | Compounds | Binding affinities |
|---|---|---|
| 1 | Compound 11c | −7.08 kcal mol−1 |
| 2 | Ascorbic acid | −5.59 kcal mol−1 |
| 3 | Kojic acid | −5.78 kcal mol−1 |
| Compound | S. aureus | E. coli | ||
|---|---|---|---|---|
| ZI (mm) | MIC (mg mL−1) | ZI (mm) | MIC (mg mL−1) | |
| 11a | 10 ± 0.0 | 2.0 ± 2.25 | 12 ± 1.0 | 11.0 ± 1.00 |
| 11b | 10 ± 1.0 | 3.5 ± 0.25 | 11.33 ± 1.52 | 12.5 ± 1.25 |
| 11c | 11.33 ± 1.52 | 2.5 ± 0.00 | 10.33 ± 0.57 | 10 ± 0.40 |
| 11d | 9.33 ± 0.57 | 10 ± 1.00 | 15.66 ± 2.08 | 16 ± 1.00 |
| 11e | ND | ND | ND | ND |
| 11f | ND | ND | ND | ND |
| 11g | 19.33 ± 2.08 | 12 ± 1.00 | 14.33 ± 2.51 | 20.5 ± 1.00 |
| 11h | 12.66 ± 1.52 | 9.0 ± 1.50 | 11 ± 1.0 | 15.0 ± 0.75 |
| Penicillin | 26 ± 0.75 | 2.2 ± 1.15 | 24 ± 0.50 | 2.4 ± 1.00 |
SAR for β-hydroxy sulfides (11a–h) regarding antibacterial activity demonstrated that tested compounds exhibited desirable activity, in which compound 11a (MIC = 2.0 ± 2.25 mg mL−1) having an aryl group at the triazole (ring), showed the most potent activity against S. aureus (Gram-positive) comparable to penicillin (MIC = 2.2 ± 1.15 mg mL−1). The p-tolyl moiety, when introduced in place of aryl(ring), decreased the activity from 2.2 ± 1.15 mg mL−1 to 3.5 ± 0.25 mg mL−1. The association of a benzofuran heterocycle at the 5th locale of the triazole ring in compound 11c (MIC = 2.5 ± 0.00 mg mL−1) revealed promising anti-bacterial activity. The introduction of the naphthofuran moiety in compound 11d has resulted in a decreased activity (MIC = 10 ± 1.00 mg mL−1). Similarly, the incorporation of 2-bromo-4-chlorophenoxymethyl 11g (MIC = 12 ± 1.00 mg mL−1) and acefylline moiety 11h (MIC = 9.0 ± 1.50 mg mL−1) did not significantly enhance the activity. All β-hydroxy sulfides (11a–h) were found to be less potent against Gram-negative bacteria than the standard drug. Among the synthesized derivatives, however, compound 11c was interpreted to be functional with 10 ± 0.40 mg mL−1 MIC value. Other hybrids with p-tolyl 11b, benzofuran 11c, naphthofuran 11d, 2-bromo-4-chlorophenoxy)methyl 11g, and acefylline 11h showed moderate activity with MIC values ranging between 11.0 ± 1.00 to 20.5 ± 1.00 mg mL−1 towards similar strain, whereas, compounds 5-bromobenzofuran 11e and naphthofuran 11f were found inactive against both strains.
3. Experimental
Reagents and starting materials used in this research were obtained from Merck and Sigma-Aldrich. All the reactions were conducted in a flask (round bottom) with magnetic stirring. All experiments were kept under surveillance by thin-layer chromatographic analysis on silica gel (60 F254) precoated plates. TLC spots were detected under ultraviolet (UV) light after elution. The melting points (m.p.) were determined with the help of the Gallenkamp equipment. 1H and 13C-NMR spectra of prepared derivatives were chronicled on a Bruker spectrophotometer functioning at 400 MHz and 100 MHz for proton and carbon correspondingly. Chemical shifts values were measured in ppm (parts per million), which were obtained with respect to the internal reference i.e., tetramethylsilane (TMS). 1H-NMR signal multiplicities were expressed in “s”, “d”, “t”, and “m” symbols for singlet, doublet, triplet, and multiplet correspondingly.3.1. Synthesis of compounds
3.1.6.1. Synthesis of 1-((4,5-diphenyl-4H-1,2,4-triazol-3-yl)thio)propan-2-ol 11a. According to the general procedure in Section 3.1.6, 3,4-diphenyl-1H-1,2,4-triazole-5(4H)-thione 9a (0.1 g, 0.39 mmol), 2-methyloxirane 10a (0.028 g, 0.47 mmol) and K2CO3 (0.05 g, 0.39 mmol) in water (2 mL) were reacted to obtain 11a in 79% yield.
3.1.6.2. Synthesis of 1-((4-%phenyl-5-(p-tolyl)-4H-1,2,4-triazol-3-yl)thio)propan-2-ol 11b. According to the general procedure in Section 3.1.6, 4-phenyl-3-(p-tolyl)-1H-1,2,4-triazole-5(4H)-thione 9b (0.1 g, 0.37 mmol) was made to react with 2-methyloxirane 10a (0.026 g, 0.45 mol) & K2CO3 (0.05 g, 0.37 mmol) in water (2 mL) to attain 11b in 79% yield.
3.1.6.3. Synthesis of 1-((5-(benzofuran-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)propan-2-ol 11c. In accordance with the general procedure Section 3.1.6, benzofuran-based triazole 9c (0.1 g, 0.34 mmol), 2- methyloxirane 10a (0.024 g, 0.41 mol) and potassium carbonate (0.047 g, 0.34 mmol) were reacted in water (2 mL) to obtain 11c in 85% yield.
3.1.6.4. Synthesis of 1-((5-(naphtho[2,1-b]furan-2-yl)-4-phenyl-4H-1,2,4-triazol-3- yl)thio)propan-2-ol 11d. In accordance with the general procedure Section 3.1.6, naphthofuran-based triazole 9d (0.1 g, 0.29 mmol) was reacted with 2-methyloxirane 10a (0.02 g, 0.35 mmol) and K2CO3 (0.04 g, 0.29 mmol) in water (2 mL) to afford 11d in 86% yield.
3.1.6.5. Synthesis of 1-((5-(5-bromobenzofuran-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-3-(naphthalen-2-yloxy)propan-2-ol 11e. In accordance with the general procedure Section 3.1.6, the reaction of benzofuran-based triazole 9e (0.1 g, 0.26 mmol) upon treatment with 2-((naphthalen-2-yloxy)methyl)oxirane 10b (0.065 g, 0.32 mmol) and K2CO3 (0.037 g, 0.26 mmol) in water (2 mL) yielded 11e in 90% yield.
3.1.6.6. Synthesis of 1-((5-(naphtho[2,1-b]furan-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-3-phenoxypropan-2-ol 11f. In accordance with the general procedure Section 3.1.6, the reaction of naphthofuran-linked triazole 9d (0.1 g, 0.29 mmol) was carried out with 2-(phenoxymethyl)oxirane 10c (0.052 g, 0.35 mmol) and potassium carbonate (0.04 g, 0.29 mmol) in water (2 mL). Resultantly, targeted compound 11f was formed in 81% yield.
3.1.6.7. Synthesis of 1-((5-((2-bromo-4-chlorophenoxy)methyl)-4-phenyl-4H-1,2,4-triazol- 3-yl)thio)propan-2-ol 11g. According to the general procedure in Section 3.1.6, triazole derivative 9g (0.1 g, 0.25 mmol) was subjected to react with 2-methyloxirane 10a (0.018 g, 0.30 mmol), and K2CO3 (0.035 g, 0.25 mmol) in water (2 mL) to yield desired compound 11g (69% yield).
3.1.6.8. Synthesis of 7-((5-((2-hydroxypropyl)thio)-4-phenyl-4H-1,2,4-triazol-3-yl)methyl)-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione 11h. According to the general procedure in Section 3.1.6, acefylline bearing triazole 9h (0.1 g, 0.27 mmol) upon treatment with 2-methyloxirane 10a (0.019 g, 0.32 mmol) and K2CO3 (0.037 g, 0.27 mmol) in water (2 mL) afforded 11h in 71% yield.
3.2. Characterization data
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH). 13C-NMR (100 MHz, CDCl3) δ 22.14, 27.82, 29.80, 40.54, 40.76, 66.92, 106.50, 126.92, 130.21, 130.79, 131.52, 141.68, 148.29, 150.93, 151.42, 154.70, 154.84. MS (ESI) m/z: 428.14 [M + 1]+; anal. calcd. For C19H21N7O3S: C, 53.38; H, 4.95; N, 22.94; found; C, 53.39; H, 4.96; N, 22.92.
CH). 13C-NMR (100 MHz, CDCl3) δ 22.14, 27.82, 29.80, 40.54, 40.76, 66.92, 106.50, 126.92, 130.21, 130.79, 131.52, 141.68, 148.29, 150.93, 151.42, 154.70, 154.84. MS (ESI) m/z: 428.14 [M + 1]+; anal. calcd. For C19H21N7O3S: C, 53.38; H, 4.95; N, 22.94; found; C, 53.39; H, 4.96; N, 22.92.3.3. Tyrosinase inhibition assay
A novel series of naphthofuran based triazole scaffolds was synthesized and assessed for their ability to inhibit tyrosinase activity, an enzyme involved in melanin production. The details about the isolation, purification, characterization, and kinetic constants (Km and Vmax) etc. of bacterial tyrosinase enzyme are) provided as ESI† (Section 2: Bacterial tyrosinase enzyme data). The evaluation of tyrosinase inhibition followed a modified protocol based on methods as outlined by Kim.50 In this protocol, a reaction mixture comprising of L-tyrosine (700 μL; 2 mM), phosphate buffer solution (0.05 M), and synthesized molecules (100 μL) was prepared and subjected to incubation for ten minutes at room temperature. Afterwards, the 200 μL of bacterial tyrosinase (approximately 48 U mL−1) was introduced to the solution, which was then subjected to incubation at 37 °C for 20 minutes. The dopachrome formation was observed after 2 minutes incubation by gauging the escalation in optical density (OD) at λmax = 275 nm. The prepared derivatives (1 mM) were solubilized in DMSO to furnish stock solution, from which further five different dilutions were prepared. Kojic acid (1 mM), a known tyrosinase inhibitor, was taken as standard for comparison. The effectiveness of inhibition was expressed as IC50 values, representing the concentration at which 50% inhibition occurred. To quantify the inhibitory activity, the inhibition percentage was evaluated by using the below formula:| % Inhibition = [(A − B)/A] × 100 |
3.4. Molecular docking study
The protein structure of the target enzyme human Tyrosinase (hTYR) has no resolved X-ray crystallographic or cryo-EM structure available in the protein databank yet. Therefore, the amino acid sequence of this enzyme was taken from the UniProt database with sequence ID-P14679 and a new protein structure of this enzyme was homology modelled by utilizing the SWISS-MODEL51 server. The template (used) for modelling this structure, was hTYRP1 (human tyrosinase-related protein-1) with PDB ID-5M8L52 as this enzyme showed the highest homology with the human tyrosinase. As the hTYRP1 contains the Zn++ as active catalytic atoms and the hTYR contains the Cu++ ions utilizing the MOE v2022 software, the copper ions were modelled into the prepared homology modelled hTYR enzyme active site. The protein structure was further optimized via the protein preparation function in the MOE v2022. The structures of ligands were prepared in ChemDraw professional v16.0 and then it was imported to MOE for further optimization. After that, the ligands were docked using the online version of AutodockVina (v1.2.3) known as Webina53–55 by creating a box with XYZ dimensions of 22 Å and around the catalytic hub of the modelled tyrosinase enzyme with the grid coordinates of the docking site centered at X = 32.154, Y = 140.176, Z = 215.585. The molecular interaction and conformational analysis of the ligand–protein complexes were performed in Biovia DS v2017 software.3.5. Antibacterial assay
The antibacterial efficacy of designated synthesized structural motifs was assessed by using disc diffusion technique.56,57 A suspension of hundred microliter constituting 108 cfu per milliliter of bacteria was spread onto nutrient agar using a sterilized loop. Filter paper discs with 5.6 mm diameter, impregnated with the compound solution at a specified concentration of 25 μg/100 μL in CHCl3, and were carefully positioned on the agar surface. Penicillin was served as a reference drug (25 μg per disc). Following the one hour incubation at 4 °C, it was proceeded at 27 °C for 24 hours. The diameter of the inhibition zone, including the disc's 5.6 mm diameter, was measured in millimeters using a reference control.4. Conclusion
A mild and efficient synthetic protocol has been utilized to achieve 1,2,4-triazole-tethered β-hydroxy sulfide structural motifs 11a–h in good to remarkable yields (69–90%) by the thiolysis of oxiranes with substituted 1,2,4-triazole-3-thiol using (basic conditions) and characterized by various spectroscopic techniques. These novel 1,2,4-triazole-tethered β-hydroxy sulfides 11a–h were screened against bacterial tyrosinases. Among these synthesized triazole derivatives, molecules 11a (IC50 = 7.67 ± 1.00 μM), 11c (IC50 = 4.52 ± 0.09 μM), 11d (IC50 = 6.60 ± 1.25 μM), and 11f (IC50 = 5.93 ± 0.50 μM) demonstrated the higher tyrosinase inhibition efficacy in comparison to the reference standards ascorbic acid (IC50 = 11.5 ± 1.00 μM) and kojic acid (IC50 = 30.34 ± 0.75 μM). Specifically, the benzofuran (ring) containing compound 11c unveiled the most significant inhibition activity (IC50 = 4.52 ± 0.09 μM), surpassing both kojic acid and ascorbic acid. Moreover, triazole compounds 11a and 11c demonstrated comparable anti-bacterial efficacy with MIC of 2.0 ± 2.25 mg mL−1 and 2.5 ± 0.00 mg mL−1, respectively, against the S. aureus strain, comparable to the standard antibiotic penicillin (MIC = 2.2 ± 1.15 mg mL−1). Notably, compound 11c exhibited slightly less anti-bacterial activity (MIC = 10 ± 0.40 mg mL−1) against E. coli, and was evidently less potent as compared to penicillin (MIC = 2.4 ± 1.00 mg mL−1). Furthermore, the benzofuran-moiety based compound 11c demonstrated a better binding score (−7.08 kcal mol−1) than both ascorbic acid (−5.59 kcal mol−1) and kojic acid (−5.78 kcal mol−1), indicating the strong molecular interactions. Molecular docking studies corroborate the in vitro findings, suggesting the compound 11c as promising candidate for bacterial tyrosinase inhibition. Thus, it could be inferred that compound 11c shows dual functionality, offering a means to combat melanogenesis and bacterial infections simultaneously. Its versatility underscores its significance in pharmaceutical research, suggesting avenues for novel therapeutic interventions against microbial threats.Author contributions
S. S.: methodology, investigation, data curation, writing—original draft preparation. M. J. S.; visualization; writing—review and editing, validation, project administration. A. F. Z.: conceptualization, resources, writing—review and editing, supervision, project administration; H. T.; writing—review and editing, formal analysis, data curation; S. K.: biological activity, writing—review and editing; S. F.: molecular docking software, writing—original draft, writing—review and editing; R. A.; methodology, writing—review and editing. S. G. K.: resources, data curation, writing—review and editing, funding acquisition; U. N.: analysis, writing—original draft preparation, resources, data curation; A. I.: writing—review and editing, visualization, funding acquisition; M. A. B.: project administration, writing—review and editing, and funding acquisition.Conflicts of interest
Authors have no conflict of interest for this research work.Acknowledgements
We are thankful for the Researchers supporting project number (RSPD2024R740), King Saud University, Riyadh, Saudi Arabia.References
- Y. M. Ha, J. Y. Park, Y. J. Park, D. Park, Y. J. Choi, J. M. Kim and H. Y. Chung, Bioorg. Med. Chem. Lett., 2011, 21, 2445 CrossRef CAS PubMed.
- N. A. El-Zawawy and S. S. Ali, Microb. Pathog., 2016, 100, 213 CrossRef CAS PubMed.
- S. Parvez, M. Kang, H. S. Chung and H. Bae, Phytother. Res., 2007, 21, 805 CrossRef CAS PubMed.
- S. Y. Lee, N. Baek and T. G. Nam, J. Enzyme Inhib. Med. Chem., 2016, 31, 1 CrossRef CAS PubMed.
- J. Liu, F. Wu and C. Chen, Bioorg. Med. Chem. Lett., 2015, 25, 5142 CrossRef CAS PubMed.
- T. Pillaiyar, M. Manickam and V. Namasivayam, J. Enzyme Inhib. Med. Chem., 2017, 32, 403 CrossRef CAS PubMed.
- H. Dong, J. Liu, X. Liu, Y. Yu and S. Cao, J. Mol. Struct., 2018, 1151, 353 CrossRef CAS.
- S. Ullah, Y. Park, C. Park, S. Lee, D. Kang, J. Yang and H. R. Moon, Bioorg. Med. Chem., 2019, 27, 2192 CrossRef CAS PubMed.
- M. Fan, G. Zhang, X. Hu, X. Xu and D. Gong, Food Res. Int., 2017, 100, 226 CrossRef CAS PubMed.
- S. Chortani, V. D. Nimbarte, M. Horchani, H. B. Jannet and A. Romdhane, Bioorg. Chem., 2019, 92, 103270 CrossRef PubMed.
- M. T. H Khan, Curr. Med. Chem., 2012, 19, 2262 CrossRef PubMed.
- T. H. Zhu, S. W. Cao and Y. Y. Yu, Int. J. Biol. Macromol., 2013, 62, 589 CrossRef CAS PubMed.
- L. Xia, A. Idhayadhulla, Y. R. Lee, Y. J. Wee and S. H. Kim, Eur. J. Med. Chem., 2014, 86, 605 CrossRef CAS PubMed.
- Y. Yuan, W. Jin, Y. Nazir, C. Fercher, M. A. Blaskovich, M. A. Cooper, R. T. Barnard and Z. M. Ziora, Eur. J. Med. Chem., 2020, 187, 111892 CrossRef CAS PubMed.
- M. Talebi, K. Majidi, K. Bassam, M. Abdi, M. Daneshvar, S. S. Moayedi and M. Amanlou, J. Mol. Struct., 2022, 1268, 133707 CrossRef CAS.
- G. Karakaya, A. Türe, A. Ercan, S. Öncül and M. D. Aytemir, Bioorg. Chem., 2019, 88, 102950 CrossRef PubMed.
- W. Xie, J. Zhang, X. Ma, W. Yang, Y. Zhou, X. Tang, Y. Zou, H. Li, J. He, S. Xie, Y. Zhao and F. Liu, Chem. Biol. Drug Des., 2015, 86, 1087 CrossRef CAS PubMed.
- H. M. A. Abdullah, S. Kamal, A. U. Rehman, S. Rasool and M. S. A. Hamid, ACS Omega, 2022, 7, 32360–32368 CrossRef PubMed.
- M. Hassan, B. D. Vanjare, K. Y. Sim, H. Raza, K. H. Lee, S. Shahzadi and A. Kloczkowski, Molecules, 2022, 27, 1731 CrossRef CAS PubMed.
- M. Özil, H. T. Balaydın and M. Şentürk, Bioorg. Chem., 2019, 86, 705 CrossRef PubMed.
- H. Kumar, S. A. Javed, S. A. Khan and M. Amir, Eur. J. Med. Chem., 2008, 43, 2688 CrossRef CAS PubMed.
- S. Kumar, S. L. Khokra and A. Yadav, Futur. J. Pharm. Sci., 2021, 7, 106 CrossRef PubMed.
- A. Irfan, A. F. Zahoor, S. Kamal, M. Hassan and A. Kloczkowski, Int. J. Mol. Sci., 2022, 23, 10979 CrossRef CAS PubMed.
- A. Irfan, S. Faisal, S. Ahmad, S. A. Al-Hussain, S. Javed, A. F. Zahoor, B. Parveen and M. E. A. Zaki, pharmaceuticals, 2023, 16, 344 CrossRef CAS PubMed.
- A. F. Zahoor, F. Hafeez, A. Mansha, S. Kamal, M. N. Anjum, Z. Raza, S. G. Khan, J. Javid, A. Irfan and M. A. Bhat, Biomedicines, 2023, 11, 2739 CrossRef CAS PubMed.
- Z. Peng, G. Wang, Q. H. Zeng, Y. Li, Y. Wu, H. Liu and Y. Zhao, Food Chem., 2021, 341, 128265 CrossRef CAS PubMed.
- B. D. Vanjare, P. G. Mahajan, N. C. Dige, H. Raza, M. Hassan, Y. Han and K. H. Lee, Mol. Diversity, 2021, 25, 2089 CrossRef CAS PubMed.
- W. Guo, J. Chen, D. Wu, J. Ding, F. Chen and H. Wu, Tetrahedron, 2009, 65, 5240 CrossRef CAS.
- V. Kesavan, D. Bonnet-Delpon and J. P. Bégué, Tetrahedron Lett., 2000, 41, 2895 CrossRef CAS.
- V. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Tetrahedron Lett., 2015, 56, 2892 CrossRef CAS.
- F. Fringuelli, F. Pizzo, S. Tortoioli and L. Vaccaro, Green Chem., 2003, 5, 436 RSC.
- H. Hosseinpoor, S. M. Farid, A. Iraji, M. S. Asgari, N. Edraki, S. Hosseini and M. Khoshneviszadeh, Bioorg. Chem., 2021, 114, 104979 CrossRef CAS PubMed.
- Z. A. Kaplancikli, Molecules, 2011, 16, 7662 CrossRef CAS PubMed.
- S. Faiz, A. F. Zahoor, M. Ajmal, S. Kamal, S. Ahmad, A. M. Abdelgawad and M. E. Elnaggar, J. Heterocycl. Chem., 2019, 56, 2839 CrossRef CAS.
- M. Kowalewska, H. Kwiecień, M. Śmist and A. Wrześniewska, J. Chem., 2013, 52, 22 Search PubMed.
- D. N. Rao, A. R. G. Prasad, Y. N. Spoorthy, D. R. Rao, L. K. Ravindranath and J. Taibah Uni, Med. Sci., 2014, 9, 293 Search PubMed.
- S. Jupudi, M. A. Azam and A. Wadhwani, Chem. Pap., 2021, 75, 1221 CrossRef CAS.
- M. Nazir, M. A. Abbasi, S. Z. Siddiqui, K. M. Khan, U. Salar, M. Shahid and F. A. Khan, Bioorg. Chem., 2018, 81, 253 CrossRef CAS PubMed.
- I. Shahzadi, A. F. Zahoor, A. Rasul, N. Rasool, Z. Raza, S. Faisal, B. Parveen, S. Kamal, M. Zia-ur-Rehman and F. M. Zahid, J. Heterocycl. Chem., 2020, 57, 2782 CrossRef CAS.
- I. Shahzadi, A. F. Zahoor, A. Rasul, A. Mansha, S. Ahmad and Z. Raza, ACS Omega, 2021, 6, 11943 CrossRef CAS PubMed.
- S. M. Abaee, M. M. Mojtahedi, H. Abbasi and E. R. Fatemi, Synth. Commun., 2008, 38, 282–289 CrossRef CAS.
- G. Fancellu, K. Chand, D. Tomás, E. Orlandini, L. Piemontese, D. F. Silva, S. M. Cardoso, S. Chaves and M. A. Santos, J. Enzyme Inhib. Med. Chem., 2020, 35, 211 CrossRef CAS PubMed.
- A. H. F. Abdelwahab and S. A. H. Fekry, Eur. J. Chem., 2021, 12, 340 CrossRef CAS.
- P. L. Baccon-Sollier, Y. Malki, M. Maye, L. M. A. Ali, L. Lichon, P. Cuq, L. A. Vincent and N. Masurier, J. Enzyme Inhib. Med. Chem., 2020, 35, 935–949 CrossRef CAS PubMed.
- Mehr-un-Nisa, M. A. Munawar, F. A. Chattha, S. Kousar, J. Munir, T. Ismail, M. Ashraf and M. A. Khan, Bioorg. Med. Chem., 2015, 23, 6014 CrossRef CAS PubMed.
- P. K. Sahoo, R. Sharma and P. Pattanayak, Med. Chem. Res., 2010, 19, 127 CrossRef CAS.
- M. B. Halli, R. B. Sumathi and M. Kinni, Spectrochim. Acta, Part A, 2012, 99, 46 CrossRef CAS PubMed.
- E. S. H. El-Ashry, N. Rashed, L. F. Awad, E. S. Ramadan, S. M. Abdel-Maggeed and N. Rezki, J. Carbohydr. Chem., 2008, 27, 70 CrossRef CAS.
- A. A. Aly, A. A. Hassan, M. M. Makhlouf and S. Brase, Molecules, 2020, 25, 3036 CrossRef CAS PubMed.
- J. H. Kim, J. Y. Yoon, S. Y. Yang, S. K. Choi, S. J. Kwon, I. S. Cho, M. H. Jeong, Y. H. Kim and G. S. Choi, J. Enzyme Inhib. Med. Chem., 2017, 32, 78 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. De Beer, C. Rempfer and L. Bordoli, et al, Nucleic Acids Res., 2018, 46, W296 CrossRef CAS PubMed.
- X. Lai, H. J. Wichers, M. Soler-Lopez and B. W. Dijkstra, Angew. Chem., 2017, 129, 9944 CrossRef.
- Y. Kochnev, E. Hellemann, K. C. Cassidy and J. D. Durrant, Bioinformatics, 2020, 36, 4513 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455 CrossRef CAS PubMed.
- S. D. Sarker, L. Nahar and Y. Kumarasamy, Methods, 2007, 42, 321 CrossRef CAS PubMed.
- S. Saeed, I. Shahzadi, A. F. Zahoor, A. A. Al-Mutairi, S. Kamal, S. Faisal, A. Irfan, S. A. Al-Hussain, M. T. Muhammed and M. E. A. Zaki, Front. Chem., 2024, 12, 1372378 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01252f |
| This journal is © The Royal Society of Chemistry 2024 |