
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic effect between In2O3 and ZrO2 in the reverse water gas shift reaction†
Jiayu Donga,
Hong Wangd,
Guofeng Zhao *c,
Dong Jiang*b and
Haitao Xu*a
*c,
Dong Jiang*b and
Haitao Xu*a
aSchool of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: xuhaitao@ecust.edu.cn
bSchool of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: jiangdong@ecust.edu.cn
cKey Laboratory of Functional Molecular Solids, Ministry of Education, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241002, China. E-mail: gfzhao@chem.ecnu.edu.cn
dInstitute of Optical Functional Materials for Biomedical Imaging, School of Chemistry and Pharmaceutical Engineering, Shandong First Medical University, Shandong Academy of Medical Sciences, Taian 271016, China
First published on 8th May 2024
Abstract
Efficient activation of CO2 at low temperature was achieved through the interface effect between In2O3 and ZrO2 by their geometric and electronic effects. The results show that 75In2O3–25ZrO2 (In2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ZrO2 molar ratio of 3:1), as a catalyst for the reverse water gas shift reaction, can achieve 28% CO2 conversion with 96% CO selectivity at 400 °C, 0.1 MPa, a H2:CO2 molar ratio of 3:1 and a gas hourly space velocity of 10000 mL g−1 h−1. In situ FTIR experiments provide a basis for clarifying the pivotal role of formate (facilitated at In2O3–ZrO2 interface) in this reaction.
:ZrO2 molar ratio of 3:1), as a catalyst for the reverse water gas shift reaction, can achieve 28% CO2 conversion with 96% CO selectivity at 400 °C, 0.1 MPa, a H2:CO2 molar ratio of 3:1 and a gas hourly space velocity of 10000 mL g−1 h−1. In situ FTIR experiments provide a basis for clarifying the pivotal role of formate (facilitated at In2O3–ZrO2 interface) in this reaction.
1. Introduction
Throughout the course of industrial development, humans have heavily relied on fossil fuels to meet the substantial demand for energy, resulting in a continuous increase in greenhouse gas emissions and exacerbation of climate change.1 Utilizing carbon dioxide, an abundant and economical carbon resource, to produce high-value-added chemicals or liquid fuels is of significant importance for energy conservation, emissions reduction, and the sustainable utilization of carbon resources.2 In recent years, carbon capture and utilization (CCU) technology has attracted much attention and is considered as one of the useable ways to reduce CO2 emissions.3–7 The thermal catalytic reduction of CO2 refers to the process of converting CO2 into hydrocarbons or carbon monoxide (CO) with green hydrogen, typically carried out with the aid of catalysts at elevated temperature.8 The rapid development of renewable energy lowers the cost of green hydrogen production,9 prompting the urgent need for catalysts with high activity, selectivity, and stability.The reverse water gas shift (RWGS) reaction hydrogenates CO2 into CO, which can be further used to synthesize methanol, breaking through the thermodynamic equilibrium limit of direct methanol production from CO2,10,11 and can also be combined with Fischer–Tropsch synthesis (FTS) process to prepare useful chemicals such as olefins.12–15 Whether producing methanol through the CAMERE method (carbon dioxide hydrogenation to form methanol via a RWGS reaction) or preparing low-carbon olefins via the CO2-FTS method, the RWGS reaction with high CO yield is a crucial step. Therefore, the RWGS reaction is considered as the most promising and prospective pathway in re-utilizing CO2.
Catalysts used in the RWGS reaction can be classified into noble metal catalysts, such as Rh,16 Ru,17 and Pt,18 and non-noble metal catalysts, such as Co,19 Fe,20,21 and Mo.22,23 The noble metal catalysts exhibit outstanding performance due to their effective hydrogen dissociation capabilities, but their high costs and instability (nanoparticle agglomeration) limit their industrial application; the non-noble metal catalysts need high temperature to deliver the same performance as noble metal ones.24 Therefore, there is of significant importance in developing low-temperature, high-performance catalysts to address these limitations. In recent years, indium oxide (In2O3) has been found as a proficient catalyst for CO2 hydrogenation, with its pronounced catalytic activity attributed to the abundant oxygen vacancy (Ov) on its surface.25–28 Furthermore, In2O3 can be easily supported and/or modified by promoters to form more Ov sites, thereby activating more CO2 molecules, and stabilizing surface intermediates near Ov.29–33
Moreover, ZrO2 is also used as catalyst support in RWGS reaction, but its role plays in the reaction is still unclear.34 Unfortunately, there are relatively few reports related to the synergistic interfacial effect between In2O3 and ZrO2, hampering the rational design of mixed oxides for the RWGS reaction. For the optimal 75In2O3–25ZrO2 (In2O3:ZrO2 molar ratio of 3:1) with abundant In2O3–ZrO2 interface, 28% CO2 conversion and 96% CO selectivity can be achieved at 400 °C, 0.1 MPa, H2:CO2 molar ratio of 3:1 and GHSV (gas hourly space velocity) of 10000 mL g−1 h−1. Control experiments and characterization results testify that the as-formed oxygen vacancies (Ovs) caused by the reduction of In2O3 to In2O3−x significantly enhance catalytic activity for 75In2O3–25ZrO2. In addition, in situ Fourier transform infrared spectroscopy (FTIR) shows that HCOO* (formate) plays an important role in this reaction. For 75In2O3–25ZrO2 with abundant In2O3–ZrO2 interface, HCOO* is easily hydrogenated into CO. However, for In2O3, the content of HCOO* is relatively lower, thus contributing to its lower catalytic activity. For ZrO2, the CO32− is relatively stable, correlating well with its low catalytic activity. This work elucidates the synergistic effect between mixed In2O3 and ZrO2, paving a way to design industrial catalyst with abundant In2O3–ZrO2 interface to offer excellent catalytic performance for RWGS reaction.
2. Experimental
2.1. Catalyst preparation
The mixed In–Zr oxides were synthesized by a co-precipitation method. For instance, for the 75In2O3–25ZrO2 (In2O3:ZrO2 molar ratio of 3:1), 1.5612 g In(NO3)3·xH2O and 0.3713 g Zr(NO3)4·5H2O were dissolved in 20 mL deionized water, followed by the addition of the mixed solution of NH4OH (10 mL, 25 wt% in H2O, Alfa Aesar) and ethanol (30 mL, Titan) until the pH reaching 9.2. The resulting slurry was heated to 80 °C with vigorous stirring and aged for 30 min. Then the solid was separated by high-pressure filtration, washed with 500 mL deionized water, dried at 60 °C for 12 h, and calcined at 500 °C (heating rate of ca. 2 °C min−1) for 3 h. Other catalysts such as In2O3, ZrO2, and aIn2O3–bZrO2 (a and b represent In2O3 and ZrO2 molar ratio (a = 25%, 50%, and 75%, b = 1 − a)) were prepared using the same method by simply tuning the molar ratio of In(NO3)3·xH2O and Zr(NO3)4·5H2O.
2.2. Catalyst characterization
The N2 sorption was conducted using the ASAP 2020 instrument (Mack, USA). The specific surface area (SBET) was determined by Brunauer–Emmett–Teller (BET) model and the pore size was calculated by Barret–Joyner–Halenda (BJH) model. The In and Zr loadings were detected by an inductively coupled plasma-atomic emission spectrometry (ICP-AES) at 167–785 nm/725 instrument (Agilent Corporation, USA). The power X-ray diffraction (XRD) patterns of catalysts were obtained on a Rigaku D/Max 2550 VB/PC instrument (Rigaku, Japan) using a scanning rate of 10° min−1. The fine structures were observed by a transmission electron microscopy (TEM) at an accelerating voltage of 200 kV on JEM-2100 (JEOL, Japan). The energy dispersive X-ray spectroscopy (EDX) was measured by the JEM-2100 (JEOL, Japan) with an amplification of 8000–300000. X-ray photoelectron spectroscopy (XPS) was measured at ESCALAB 250Xi photoelectron spectrometer (Thermo Fisher Scientific, USA) equipped with an Al-Kα X-ray source. All the binding energies were calibrated on the basis of the internal standard of the binding energy of C 1s (284.8 eV). Electron paramagnetic resonance (EPR) spectroscopy was performed using the CIQTEK EPR200-Plus. Spectra were collected accumulating 1 scan for field sweeps of 3250–3850 G at 298 K with a magnetic field modulation frequency of 100 kHz. The spectrum of an empty tube was subtracted to correct for the background signal.
The experiments of H2-temperature programmed reduction (H2-TPR) and CO2-temperature programmed desorption (CO2-TPD) were carried out on a ChemBET Pulsar automatic adsorption apparatus (Quantachrome Company, USA) equipped with a thermal conductivity detector (TCD), and the efflux were monitored by an on-line mass spectrometer (MS, SHP8400PMS-L, Shanghai Sunny Hengping Scientific Instrument Co. Ltd, China). For H2-TPR, each catalyst (0.1 g) was pretreated in Ar flow (30 mL min−1) at 300 °C for 0.5 h and cooled down to room temperature. Then, the gas was switched to H2/Ar flow (10 vol% H2, 50 mL min−1) and the catalyst was reduced from room temperature to 800 °C at a heating rate of 10 °C min−1. For CO2-TPD, each catalyst (0.1 g) was pretreated in Ar flow (30 mL min−1) at 400 °C for 1 h, and then reacted in mixture gas (the molar ratio of H2:CO2 is 3:1, 50 mL min−1) at 400 °C for 2 h. Then, the catalyst was cooled to 50 °C in the same flow followed by CO2 (50 mL min−1) adsorption at 50 °C for 2 h. After that, the catalyst was flushed in He flow (50 mL min−1) for 0.5 h, followed by heated from 50 to 800 °C at a rate of 10 °C min−1, and signals of CO2 were monitored by MS on line.
The in situ Fourier transform infrared (FT-IR) was conducted on a IRPrestige-21 equipment (Shimadzu, Japan). A resolution of 8 cm−1 and scanning times of 50. 50 mg catalyst and 100 mg KBr were pressed into a wafer and placed in the in situ chamber. All the samples were pretreated at 400 °C in H2 flow (37.5 mL min−1) for 10 min and cooled to the room temperature to obtain the background spectrum. When the adsorption of CO2, the flow was switched to CO2 (12.5 mL min−1, 99.99%) at room temperature for 10 min, after that, CO2 was switched off and the catalyst was maintained at 50 °C for 2 h. Subsequently, catalyst was purged with a He flow (30 mL min−1) for 5 minutes and then raised from 50 to 400 °C, with the spectra were collected. After raising to 400 °C, the flow was switched to H2 for 10 s, H2 was switched off and the spectra was collected at 0.5 MPa. When the co-adsorption of CO2 and H2, the flow was switched to the mixed gas (the molar ratio of H2:CO2 is 3:1, 50 mL min−1). The temperature was raised from 100 to 400 °C and the spectra were collected.
2.3. Catalytic evaluation
In this work, a continuous fixed-bed reactor was used to evaluate the performance of catalysts. Typically, 0.3 g catalyst was loaded into a reactor with an inner diameter of 7 mm and the length of 700 mm. H2 (36 mL min−1), CO2 (12 mL min−1), and Ar (2 mL min−1) was controlled by mass flow controllers, forming a H2/CO2/Ar (molar ratio of 72/24/4) mixture and passing through the catalyst bed. The Ar was used as the internal standard gas. Then, the temperature was successively raised from room temperature to 400 °C and maintained for 2 h. The effluent was analyzed by online gas chromatography (GC7900), equipped with a thermal conductivity detector (TCD) and TDX-1 column. The CO2 conversion (XCO2), CO selectivity (SCO), CO yield (YCO) and CH4 selectivity (SCH4) were calculated as follows:
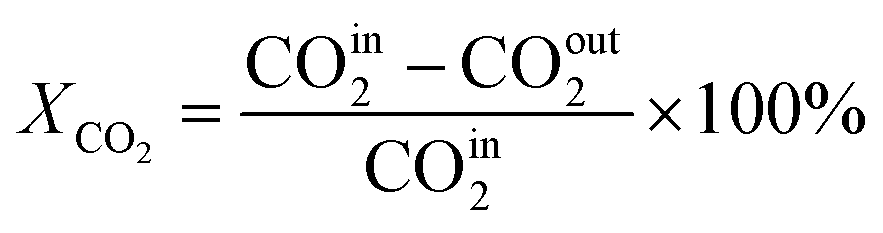 | (1) |
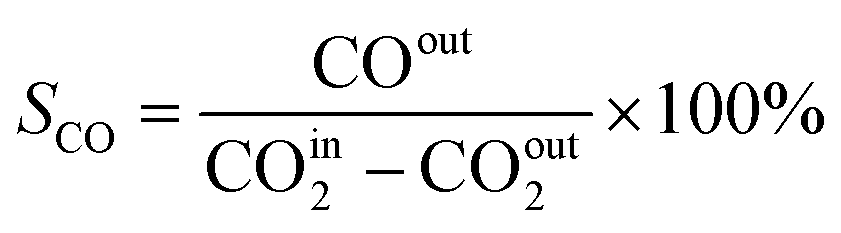 | (2) |
| YCO = XCO2 × SCO | (3) |
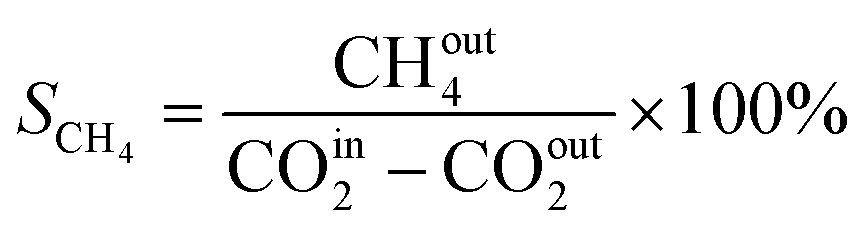 | (4) |
3. Results and discussion
3.1. Structures and chemical states of fresh catalysts
Five In2O3–ZrO2 catalysts were prepared by the co-precipitation method, varying the molar content of In2O3 of 0, 25%, 50%, 75% and 100%. The inductively coupled plasma-atomic emission spectrometry (ICP-OES) measurements confirmed that In and Zr contents were almost identical to the theoretical value. Adding ZrO2 to In2O3 will slightly increase the specific surface area (Table S1†), but the specific surface area of 75In2O3–25ZrO2 is close to that of In2O3, albeit the catalyst area is not the key factor determining catalytic activity.35 Moreover, the type IV hysteresis loop testifies the mesoporous structure of this series of catalysts (Fig. S1†). TEM images of this series of catalysts show the similar morphologies, with the diameter of 8–15 nm (Fig. S2†).For 75In2O3–25ZrO2, the average particle size is 10.0 ± 1.4 nm, and the HRTEM images illustrate the lattice distances of 0.292, 0.275, and 0.297 nm, corresponding to the In2O3(222), In2O3(321), and t-ZrO2(101) planes, respectively (Fig. S3a and b†). The STEM-EDX mapping images show that In and Zr elements are randomly distributed on the catalyst surface (Fig. S3c and d†), forming abundant In2O3–ZrO2 interface and tentatively contributing to excellent catalytic performance.
X-ray diffraction (XRD) was used to figure out the effect of Zr modification on bulk structure. The XRD patterns in Fig. S4a.† display that the pure ZrO2 (i.e., 0In2O3–100ZrO2) prefers to crystallize to its thermodynamically stable monoclinic structure; the presence of In steers the growth of ZrO2 toward metastable tetragonal phase,28 suggesting that partial In is incorporated into the ZrO2 lattice in the form of In–O–Zr bond, as evidenced by the HRTEM image of 75In2O3–25ZrO2 sample.36 The transition from In–O–In bond to In–O–Zr bond should greatly improve the CO2 conversion and CO selectivity of In2O3–ZrO2 catalysts (see the results in Section 3.2). Owing to the fact that the lattice parameters of cubic In2O3 (JCPDS card 06-0416) and t-ZrO2 (JCPDS card 37-1413) are akin, their XRD patterns are virtually identical. However, as shown in Fig. S4b,† the diffraction peak moves from 30.167° (t-ZrO2(111)) to 30.580° (c-In2O3(222)) with the increase of In2O3 content, and such tiny peak shift confirms the generation of In2O3–ZrO2 solid solution.
The surface chemical states of In2O3–ZrO2 catalysts were characterized by XPS (Fig. 1) and EPR (Fig. S6†). The symmetric binding energy peaks at ∼452 and ∼444.3 eV testify that In species exists in the form of In3+.37 With the increase of In2O3 content, the binding energy of In3+ decreases slightly, indicating the electron transfer from Zr to In.36 The symmetric binding energy peaks at ∼184.5 and ∼182.0 eV testify that Zr species exists in the form of Zr4+.38 For 50In2O3–50ZrO2 and 75In2O3–25ZrO2, the binding energies of Zr are higher that of 25In2O3–75ZrO2, also coinciding with the electron transfer. For the O 1s XPS spectra, the major peak at 529.5–531.0 eV corresponds to lattice oxygen, the peak at 531.0–532.0 eV to Ov, and the one at 532.5–533.0 eV to surface OH.39 Obviously, with the increase of In2O3 content, the Ov content increases progressively. Fig. S5† shows that there is a positive correlation between the CO STY (space-time yield) and the oxygen vacancy concentration, which means that the Ov may play an important role in the RWGS reaction. Furthermore, the EPR results in Fig. S6† reveals a signal of g = 1.890 for fresh In2O3, which implies that the surface vacancies exist on In2O3.40 Pure ZrO2 sample exhibits an isotropic EPR signal at g = 1.973, which is assigned to the bulk Zr3+ ions located at axially symmetric sites. The 75In2O3–25ZrO2 demonstrates a prominent signal that can be attributed to unpaired electrons trapped in symmetric site at g = 2.004, which is always typically assigned to oxygen vacancies.41 This means the synergistic effect between In2O3 and ZrO2 in 75In2O3–25ZrO2 solid solution is beneficial to produce new oxygen vacancies at g = 2.004, which is in line with the XPS result.
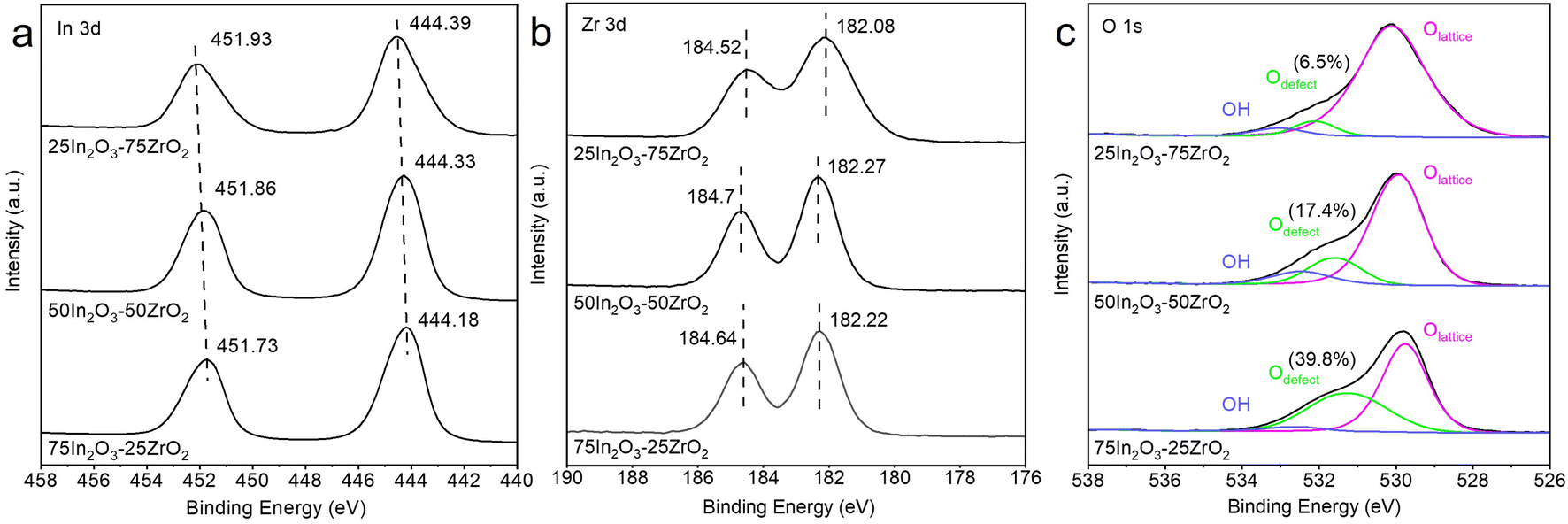 | ||
| Fig. 1 XPS spectra of (a) In 3d, (b) Zr 3d, and (c) O 1s for the 25In2O3–75ZrO2, 50In2O3–50ZrO2 and 75In2O3–25ZrO2 catalyst. | ||
H2 temperature-programmed reduction (H2-TPR) tests were conducted to determine the reactivity of the In2O3–ZrO2 catalyst toward H2 activation in the temperature range of 50–800 °C, as shown in Fig. S7a.† The H2-TPR profiles revealed that reduction temperature of bulk In2O3 in In2O3 and 75In2O3–25ZrO2 are 662 °C and 697 °C respectively, while the reduction temperature of surface In2O3 are 189 °C and 225 °C respectively. However, the H2-TPR of ZrO2 demonstrates no significant H2 consumption, which means the neglectable reducibility of ZrO2. Interestingly, for 75In2O3–25ZrO2, the reduction signals of surface and the bulk In2O3 are located at a higher temperature than that of pure In2O3 catalyst, hinting a stronger interaction between In2O3 and ZrO2.29 This also shows the increasing Ov content over 75In2O3–25ZrO2 catalyst, which is in accordance with the XPS result and the prominent catalytic activity.42
CO2 temperature programmed desorption (CO2-TPD) was conducted to further investigate the CO2 adsorption behaviour on the In2O3–ZrO2 catalyst, as shown in Fig. S7b.† The profiles exhibit several significant CO2 evolution signals from the ZrO2 and 75In2O3–25ZrO2 catalyst in the temperature range of 134–220, 273–315 and 396–477 °C. While the signal of CO2 adsorbed on pure In2O3 are not detectable. The signal peak around 153 °C belongs to the physisorption of CO2. Other signal peaks belong to the chemically absorbed CO2 on the H2-induced oxygen vacancy sites (Ov).42 Additionally, CO2-TPD has been widely used to measure the surface basicity of catalysts, and high desorption temperature promised a strong basic site.43 Compared with ZrO2 catalyst, the CO2 desorption peak of 75In2O3–25ZrO2 catalyst shift to the higher temperatures of 315 °C and 75In2O3–25ZrO2 catalyst have strong site at around 450 °C. Specifically, the addition of In enhances the strength of CO2 adsorption on these sites, owing to the increase in basic intensity.42 The characterization results of H2-TPR and CO2-TPD consistently confirm that In2O3–ZrO2 interface benefits the formation of oxygen vacancies, thus enhancing the ability of 75In2O3–25ZrO2 catalyst to CO2 adsorption and H2 activation.
3.2. Catalytic performance
CO2 hydrogenation mainly involves the following three reactions (5)–(7) to produce three products of CO, CH4 and CH3OH, respectively.| CO2 + H2 ↔ CO + H2O, ΔrHθm = 41.2 kJ mol−1 | (5) |
| CO2 + 4H2 ↔ CH4 + H2O, ΔrHθm = −164.9 kJ mol−1 | (6) |
| CO2 + 3H2 ↔ CH3OH + H2O, ΔrHθm = −49.4 kJ mol−1 | (7) |
Fig. 2a shows the CO2 conversion, CO selectivity, and CO yield over the five catalysts. The catalytic performance of pure ZrO2 (i.e., 0In2O3–100ZrO2) is extremely poor, with CO2 conversion of only 4% and CO selectivity of only 53%, while the pure In2O3 (i.e., 100In2O3–0ZrO2) gives higher CO2 conversion of 23.5% and CO selectivity of 95.8%. Interestingly, the In2O3–ZrO2 catalysts (i.e., 25In2O3–75ZrO2, 50In2O3–50ZrO2, and 75In2O3–25ZrO2) all offers CO selectivity above 92%, with volcano evolution of CO2 conversion. Most notably, the 75In2O3–25ZrO2 offers the highest CO selectivity of 96% and highest CO2 conversion of 28%. Due to a pronounced synergistic effect between ZrO2 and In2O3, the In–Zr interface within the bimetallic oxides augments the density of Ov on the In2O3 surface, thereby significantly enhancing the adsorption and hydrogenation capacities towards CO2. In addition, no methane can be detected, and a small amount of methanol was the only by-product.
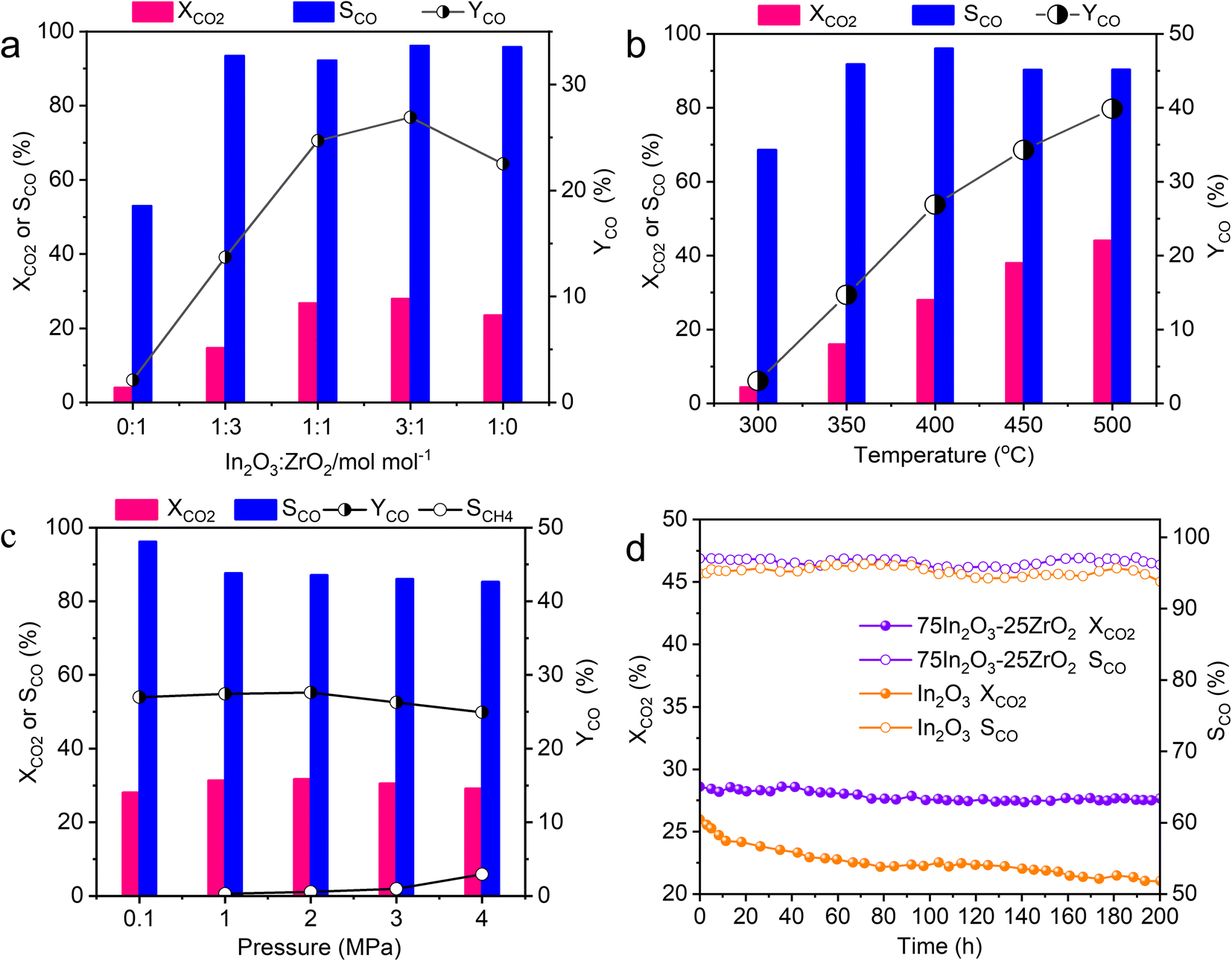 | ||
| Fig. 2 (a) The catalytic performance of the In2O3–ZrO2 catalysts with different In2O3 ratio (reaction conditions: 400 °C, 0.1 MPa, H2:CO2:Ar ratio = 72:24:4, 10000 mL g−1 h−1); (b) influence of reaction temperature on the catalytic performance of 75In2O3–25ZrO2 (reaction conditions: 0.1 MPa, H2:CO2:Ar ratio = 72:24:4, 10000 mL g−1 h−1); (c) influence of pressure on the catalytic performance of 75In2O3–25ZrO2 (reaction conditions: 400 °C, H2:CO2:Ar ratio = 72:24:4, 10000 mL g−1 h−1); (d) the stability test of 75In2O3–25ZrO2 and In2O3 (reaction conditions: 400 °C, 0.1 MPa, H2:CO2:Ar ratio = 72:24:4, 10000 mL g−1 h−1). | ||
The influence of reaction temperature, pressure, and gas hourly space velocity (GHSV) on catalytic performance is exhibited in Fig. 2b, c and S8.† At 0.1 MPa, and GHSV of 10000 mL g−1 h−1, with the temperature rising from 300 to 500 °C, CO2 conversion increases from 4% to 44%, and the highest CO selectivity is 96% at 400 °C. At 400 °C, and GHSV of 10000 mL g−1 h−1, with the pressure increasing from 0.1 to 4 MPa, CO2 conversion slightly increases from 28% to 29%, but CO selectivity decreases from 96% to 85% (with the formation of new by-product CH4), because high reaction pressure is beneficial to CO2 methanation reaction.44 Moreover, at 0.1 MPa, and 400 °C, CO2 conversion decreases from 35% to 27.7% with increasing GSHV from 6000 to 14000 mL g−1 h−1, while the maximum CO selectivity is 94% at the GSHV of 10000 mL g−1 h−1. Hence, the optimized reaction condition is as follows: 0.1 MPa, 400 °C and GHSV of 10000 mL g−1 h−1. For the best catalyst 75In2O3–25ZrO2, under the best reaction conditions, the CO2 conversion and CO selectivity are 28% and 96% in the 200 h-test. However, for In2O3, the conversion decreases from 26% to 21%. Compared with pure In2O3, the stability of mixed oxides is obviously enhanced. Hence, the In2O3–ZrO2 interface is of great importance in improving and maintaining catalytic activity (Fig. 2d). We compared the catalyst 75In2O3–25ZrO2 with other catalysts including non-noble metal and noble metal catalysts in the RWGS reaction in Table S3.† CO2 conversion, CO selectivity and STY of 75In2O3–25ZrO2 are very promising. Notably, the STY of 75In2O3–25ZrO2 is higher than other catalysts (apart from Ag/Al2O3). Furthermore, compared with noble metal catalysts, In-based catalysts have lower cost and more prospects in industry applications.
3.3. Surface intermediates and reaction mechanism
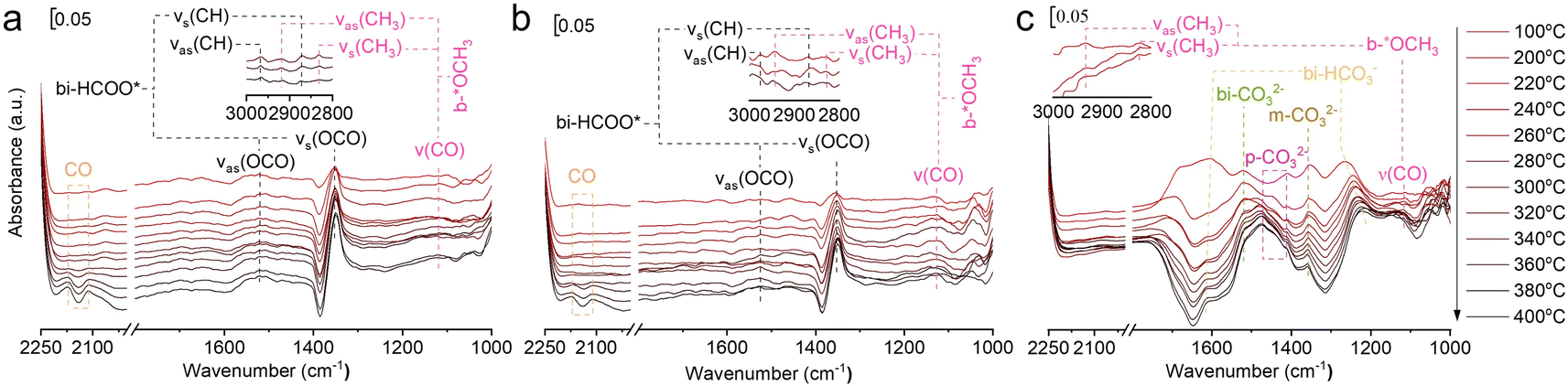
In situ FTIR was used to investigate the evolution of key surface intermediates for RWGS reaction, and the wavenumbers of the intermediates are summarized in Table S4.†33,34,36,37,39,45–55 Firstly, the three catalysts (75In2O3–25ZrO2, In2O3, and ZrO2) were placed into the chamber and reduced with hydrogen at 400 °C. Subsequently, CO2 was introduced into the chamber for adsorption. Finally, the gaseous CO2 was purged by He flow and the spectra were collected from 50 to 400 °C (Fig. 3). For 75In2O3–25ZrO2 (Fig. 3a), the following characteristic bands can be observed: bi-HCOO* (bidentate formate, at 1350, 1589, 2873 and 2967 cm−1);33,36,39,46,48,50,52–55 b-*OCH3 (bridged methoxy, at 1128, 2822 and 2930 cm−1).33,39,50–53 With the increase of temperature, the peak area of bi-HCOO* increases significantly, testifying that CO2 could be transformed into bi-HCOO* easily. For In2O3 (Fig. 3b), similar characteristic bands are also found, but the content of bi-HCOO* is relatively lower, corresponding well with its lower catalytic activity and tentatively showing that bi-HCOO* may play an important role in this reaction. For ZrO2 (Fig. 3c), the following characteristic bands can be observed: bi-HCO3− (bidentate bicarbonate, at 1284 and 1636 cm−1);34,49,54 m-CO32− (monodentate carbonate, at 1355 cm−1);34,49 bi-CO32− (bidentate carbonate, at 1523 cm−1);34,47 p-CO32− (polydentate carbonate, at 1463 and 1411 cm−1);47,48,54 b-*OCH3 (bridged methoxy, at 1126, 2830 and 2925 cm−1).33,39,50–53 With the increase of temperature, bi-HCO3− decomposes rapidly, while bi-CO32− and m-CO32− decompose sluggishly. Because of with strong thermal resistance and a rather low separation between the two C–O stretching modes, polydentate carbonate species is relatively stable (νas(CO3) = 1463 cm−1 and νs(CO3) = 1411 cm−1).54 In addition, the peak area of p-CO32− increases slightly, indicating that the above species may transform into p-CO32−.37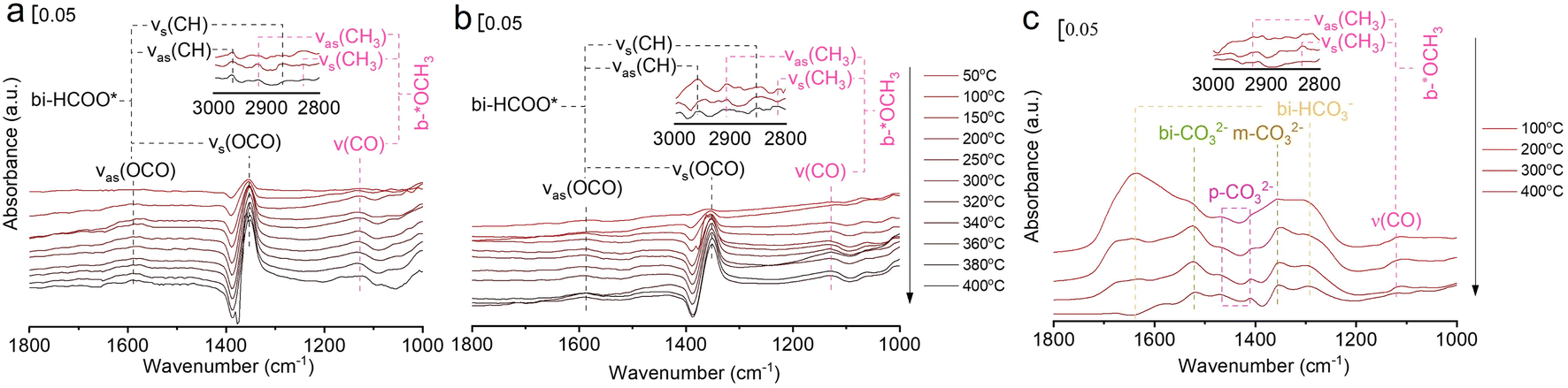 | ||
| Fig. 3 In situ FTIR spectra of CO2 adsorption at different temperatures over (a) 75In2O3–25ZrO2, (b) In2O3, and (c) ZrO2. | ||
In order to testify the pivotal role of bi-HCOO* playing in this reaction, the reaction of H2 and CO2 (molar ratio of H2 and CO2 is 3:1) over these three reduced catalysts were tracked by in situ FTIR (Fig. 4). For 75In2O3–25ZrO2, the characteristic band of CO is observed at 320 °C (ν(CO) = 2111.1 and 2170 cm−1). However, for In2O3, CO starts to appear at 360 °C, corresponding well with its lower catalytic activity. For ZrO2, the characteristic bands of CO are not observed, showing that bi-HCO3−, bi-CO32−, m-CO32−, and p-CO32− can't be hydrogenated easily.48 Lastly, for all the three catalysts, 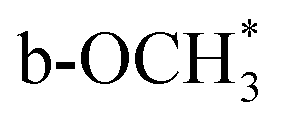 is also observed, but this species could only be hydrogenated to CH4 at relative higher 0.5 MPa, thus excluding the role of
is also observed, but this species could only be hydrogenated to CH4 at relative higher 0.5 MPa, thus excluding the role of 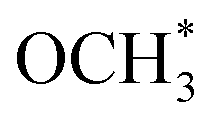 playing under the reaction conditions (Fig. S9†). But the CH4 is not formed in the real fixed-bed reaction process, which is likely caused by the different conditions between in situ FTIR and real reaction process.
playing under the reaction conditions (Fig. S9†). But the CH4 is not formed in the real fixed-bed reaction process, which is likely caused by the different conditions between in situ FTIR and real reaction process.
 | ||
| Fig. 4 In situ FTIR spectra of the reaction of CO2 and H2 over (a) 75In2O3–25ZrO2, (b) In2O3, and (c) ZrO2. | ||
Combined with the above analyses, it can be suggested that CO2 hydrogenation on the In2O3–ZrO2 catalyst through HCOO* intermediates (Scheme S1†). H2 adsorbed on the exposed surface of In2O3 crystal to form 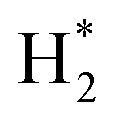 , and then formed
, and then formed 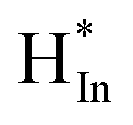 and
and 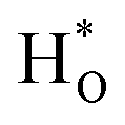 at In site and O site, respectively. At the same time, CO2 is adsorbed on a base on the surface of the composite oxide, activated by oxygen vacancy, and then combined with activated
at In site and O site, respectively. At the same time, CO2 is adsorbed on a base on the surface of the composite oxide, activated by oxygen vacancy, and then combined with activated 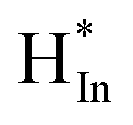 to form formate intermediate (HCOO*). HCOO* interacts with the site of ZrO2, undergoes the cleavage of C–O and C–H bonds, and forms O–H bonds at the same time, producing CO* and OH*, and CO* desorbs to produce CO.33,37 In this case, ZrO2 can not only modify In2O3, but also serve as an active site. In2O3–ZrO2 constitutes a bimetallic In–Zr oxide catalyst system.
to form formate intermediate (HCOO*). HCOO* interacts with the site of ZrO2, undergoes the cleavage of C–O and C–H bonds, and forms O–H bonds at the same time, producing CO* and OH*, and CO* desorbs to produce CO.33,37 In this case, ZrO2 can not only modify In2O3, but also serve as an active site. In2O3–ZrO2 constitutes a bimetallic In–Zr oxide catalyst system.
4. Conclusions
In this work, the optimal 75In2O3–25ZrO2 and the contrastive In2O3, ZrO2 were prepared by the coprecipitation method, and 75In2O3–25ZrO2 exhibits excellent 28% conversion and 96% selectivity in the RWGS reaction under the best reaction conditions (400 °C, 0.1 MPa, H2:CO2 molar ratio of 3:1 and gas hourly space velocity of 10000 mL g−1 h−1). XRD and STEM-EDX show that the In2O3–ZrO2 solid solution is formed, and XPS testifies that the electron transfer effect plays an important role in this reaction. In situ FTIR shows that: for 75In2O3–25ZrO2 with abundant In2O3–ZrO2 interface, HCOO* is easily hydrogenated into CO; however, for In2O3, the content of HCOO* is relatively lower, thus contributing to its lower catalytic activity; for ZrO2, the CO32− is relatively stable, correlating well with its low catalytic activity. This work definitely testifies the pivotal role of HCOO* in the RWGS reaction, but also paves a way to design bimetal oxide catalyst with excellent catalytic performance for RWGS reaction.
Conflicts of interest
The authors declare that there is no conflict of interest.References
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- Z. Zhang, S.-Y. Pan, H. Li, J. Cai, A. G. Olabi, E. J. Anthony and V. Manovic, Renewable Sustainable Energy Rev., 2020, 125, 109799 CrossRef CAS.
- A. Al-Mamoori, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, Energy Technol., 2017, 5, 834–849 CrossRef.
- A. Kaetelhoen, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187–11194 CrossRef PubMed.
- I. Mohsin, T. A. Al-Attas, K. Z. Sumon, J. Bergerson, S. McCoy and M. G. Kibria, Cell Rep. Phys. Sci., 2020, 1, 100104 CrossRef.
- L. J. Mueller, A. Kaetelhoen, M. Bachmann, A. Zimmermann, A. Sternberg and A. Bardow, Front. Energy Res., 2020, 8, 15 CrossRef.
- E. Kawai, A. Ozawa and B. D. Leibowicz, Appl. Energy, 2022, 328, 120183 CrossRef CAS.
- T. Len and R. Luque, Green Chem., 2023, 25, 490–521 RSC.
- Y. H. Zheng, M. Ma and H. Y. Shao, Carbon Neutrality, 2023, 2, 23 CrossRef.
- O. S. Joo, K. D. Jung, I. Moon, A. Y. Rozovskii, G. I. Lin, S. H. Han and S. J. Uhm, Ind. Eng. Chem. Res., 1999, 38, 1808–1812 CrossRef CAS.
- S. W. Park, O. S. Joo, K. D. Jung, H. Kim and S. H. Han, Appl. Catal., A, 2001, 211, 81–90 CrossRef CAS.
- R. Liu, D. Leshchev, E. Stavitski, M. Juneau, J. N. Agwara and M. D. Porosoff, Appl. Catal., B, 2021, 284, 119787 CrossRef CAS.
- D. Wang, Z. Xie, M. D. Porosoff and J. G. Chen, Chem, 2021, 7, 2277–2311 CAS.
- A. D. N. Kamkeng and M. Wang, Chem. Eng. J., 2023, 462, 142048 CrossRef CAS.
- Y. F. Zhu, B. Xie, R. Amal, E. C. Lovell and J. Scott, Small Struct., 2023, 4, 2200285 CrossRef CAS.
- G. Kim, S. Shin, Y. Choi, J. Kim, G. Kim, K.-J. Kim and H. Lee, JACS Au, 2022, 2, 1115–1122 CrossRef CAS PubMed.
- R. Tang, Z. Zhu, C. Li, M. Xiao, Z. Wu, D. Zhang, C. Zhang, Y. Xiao, M. Chu, A. Genest, G. Rupprechter, L. Zhang, X. Zhang and L. He, ACS Mater. Lett., 2021, 3, 1652–1659 CrossRef CAS PubMed.
- L. Chen, L. Kovarik and J. Szanyi, ACS Catal., 2021, 11, 12058–12067 CrossRef CAS.
- D. Zagoraios, S. Tsatsos, S. Kennou, C. G. Vayenas, G. Kyriakou and A. Katsaounis, ACS Catal., 2020, 10, 14916–14927 CrossRef CAS.
- L. Yang, L. Pastor-Perez, J. J. Villora-Pico, A. Sepulveda-Escribano, F. Tian, M. Zhu, Y.-F. Han and T. R. Reina, ACS Sustain. Chem. Eng., 2021, 9, 12155–12166 CrossRef CAS.
- Y. Meng, X. Y. Liu, Y. J. Ma, X. H. Gao and X. D. Wen, Mol. Catal., 2022, 529, 112538 CrossRef CAS.
- A. Pajares, X. Liu, J. R. Busacker, P. Ramirez de la Piscina and N. Homs, Nanomaterials, 2022, 12, 3165 CrossRef CAS PubMed.
- J. J. Xu, X. X. Gong, R. R. Hu, Z. W. Liu and Z. T. Liu, Mol. Catal., 2021, 516, 111954 CrossRef CAS.
- G. Zhou and X. Ai, Journal of Chongqing Technology and Business University, 2023, 40, 8–14 Search PubMed.
- Q. Sun, J. Ye, C.-j. Liu and Q. Ge, Greenhouse Gases, 2014, 4, 140–144 CrossRef CAS.
- A. Tsoukalou, P. M. Abdala, D. Stoian, X. Huang, M.-G. Willinger, A. Fedorov and C. R. Mueller, J. Am. Chem. Soc., 2019, 141, 13497–13505 CrossRef CAS PubMed.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- J. Wang, G. Zhang, J. Zhu, X. Zhang, F. Ding, A. Zhang, X. Guo and C. Song, ACS Catal., 2021, 11, 1406–1423 CrossRef CAS.
- T. P. Araujo, C. Mondelli, M. Agrachev, T. Zou, P. O. Willi, K. M. Engel, R. N. Grass, W. J. Stark, O. V. Safonova, G. Jeschke, S. Mitchell and J. Perez-Ramirez, Nat. Commun., 2022, 13, 5610 CrossRef PubMed.
- P. C. Zonetti, V. L. Bridi, G. G. Gonzalez, C. R. Moreira, O. C. Alves, R. R. de Avillez and L. G. Appel, ChemCatChem, 2019, 11, 4011–4020 CrossRef CAS.
- M. S. Frei, C. Mondelli, A. Cesarini, F. Krumeich, R. Hauert, J. A. Stewar, D. C. Ferre and J. Perez-Ramirez, ACS Catal., 2020, 10, 1133–1145 CrossRef CAS.
- X. Zhang, A. V. Kirilin, S. Rozeveld, J. H. Kang, G. Pollefeyt, D. F. Yancey, A. Chojecki, B. Vanchura and M. Blum, ACS Catal., 2022, 12, 3868–3880 CrossRef CAS.
- T.-y. Chen, C. Cao, T.-b. Chen, X. Ding, H. Huang, L. Shen, X. Cao, M. Zhu, J. Xu, J. Gao and Y.-F. Han, ACS Catal., 2019, 9, 8785–8797 CrossRef CAS.
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17, 4297–4303 CrossRef CAS.
- H. Zhang, Y. Y. Dong, W. P. Fang and Y. X. Lian, Chin. J. Catal., 2013, 34, 330–335 CrossRef CAS.
- C. Yang, C. Pei, R. Luo, S. Liu, Y. Wang, Z. Wang, Z.-J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 19523–19531 CrossRef CAS PubMed.
- C. Y. R. Vera, N. Manavi, Z. Zhou, L.-C. Wang, W. Diao, S. Karakalos, B. Liu, K. J. Stowers, M. Zhou, H. Luo and D. Ding, Chem. Eng. J., 2021, 426, 131767 CrossRef.
- L. Yao, X. Shen, Y. Pan and Z. Peng, J. Catal., 2019, 372, 74–85 CrossRef CAS.
- Y. Wei, F. Liu, J. Ma, C. Yang, X. Wang and J. Cao, Mol. Catal., 2022, 525, 112354 CrossRef CAS.
- N. Siedl, P. Gügel and O. Diwald, J. Phys. Chem., 2013, 117, 20722–20729 CAS.
- C. Gionco, M. C. Paganini, E. Giamello, R. Burgess, C. Di Valentin and G. Pacchioni, Chem. Mater., 2013, 25, 2243–2253 CrossRef CAS.
- Z. Lu, K. H. Sun, J. Wang, Z. T. Zhang and C. J. Liu, Catalysts, 2020, 10, 1360 CrossRef CAS.
- Y. Cui, L. L. Xu, M. D. Chen, X. B. Lian, C. E. Wu, B. Yang, Z. C. Miao, F. G. Wang and X. Hu, Catal. Sci. Technol., 2019, 9, 5605–5625 RSC.
- K. Stangeland, D. Kalai, H. L. Li and Z. X. Yu, 8th International Conference on Applied Energy (ICAE), 2016, vol. 105, pp. 2022–2027 Search PubMed.
- G. C. Cabilla, A. L. Bonivardi and M. A. Baltanás, J. Catal., 2001, 201, 213–220 CrossRef CAS.
- S. S. Dang, B. Qin, Y. Yang, H. Wang, J. Cai, Y. Han, S. G. Li, P. Gao and Y. H. Sun, Sci. Adv., 2020, 6, eaaz2060 CrossRef CAS PubMed.
- L. Z. Gao and C. T. Au, J. Catal., 2000, 189, 1–15 CrossRef CAS.
- L. Lin, S. Yao, Z. Liu, F. Zhang, N. Li, D. Vovchok, A. Martínez-Arias, R. Castañeda, J. Lin, S. D. Senanayake, D. Su, D. Ma and J. A. Rodriguez, J. Phys. Chem. C, 2018, 122, 12934–12943 CrossRef CAS.
- Y. Wang, L. Zhu, Y. Liu, E. I. Vovk, J. Lang, Z. Zhou, P. Gao, S. Li and Y. Yang, Appl. Surf. Sci., 2023, 631, 157534 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- B. Liu, T. Fang and Y. He, Catal. Sci. Technol., 2022, 12, 300–309 RSC.
- W. Wang, Z. Qu, L. Song and Q. Fu, J. Catal., 2020, 382, 129–140 CrossRef CAS.
- W. Wang, Z. Qu, L. Song and Q. Fu, J. Energy Chem., 2020, 40, 22–30 CrossRef.
- S. Collins, M. Baltanas and A. Bonivardi, J. Catal., 2004, 226, 410–421 CrossRef CAS.
- F. Ouyang, J. N. Kondo, K. Maruya and K. Domen, Catal. Lett., 1998, 50, 179–181 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01372g |
| This journal is © The Royal Society of Chemistry 2024 |