
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles study of the effect of oxygen vacancy and iridium doping on formaldehyde adsorption on the La2O3(001) surface
Youhui Xua,
Xiaoying Caoa,
Xiuwu Chena,
Fanting Konga,
Hongbo Lianga,
Hengjiao Gao *b,
Hongxia Caoa and
Jieyu Lia
*b,
Hongxia Caoa and
Jieyu Lia
aSchool of Media Engineering, Lanzhou University of Arts and Science, Lanzhou 730000, Gansu Province, China
bScience and Technology on Vacuum Technology and Physics Laboratory, Lanzhou Institute of Physics, Lanzhou 730000, Gansu Province, China. E-mail: gaohengjiao@163.com
First published on 8th July 2024
Abstract
Formaldehyde adsorption on intrinsic La2O3 surface, four-fold coordinated oxygen vacancy (VO4c), six-fold coordinated oxygen vacancy (VO6c), and iridium-doped La2O3(001) surface was studied by the first-principles method. The results show that formaldehyde adsorption on the Ir-doped La2O3(001) surface with VO6c is the strongest because of the directional movement of electrons caused by the interaction of the Ir-5d orbitals and internal oxygen vacancy, wherein the adsorption energy is 3.23 eV. This model showed a significant increase in adsorption energy, indicating that Ir doping improves the formaldehyde adsorption capacity of the La2O3(001) surface. The energy band analysis shows that iridium doping introduces impurity energy levels into the intrinsic La2O3 energy band, which enhances the interaction between the La2O3(001) surface and formaldehyde molecules. Density of state analysis indicated that the adsorption of formaldehyde molecules on the La2O3(001) surface is mainly due to the interaction between the O-2p, C-2p orbitals of formaldehyde and the Ir-5d orbital of iridium atoms. Furthermore, the existence of VO4c and VO6c defects has no effect on the position and shape of the valence and conduction bands. The effects of oxygen vacancy and iridium doping on the optical properties mainly appeared in the low-energy infrared and visible regions, making the O-2p, C-2p orbitals of formaldehyde and the Ir-5d, O-2p orbitals of the La2O3(001) surface become hybridized near the Fermi level and the electronic transition from the valence band to conduction band more likely to occur. The La2O3 material can be used as an ideal photocatalytic material for formaldehyde degradation.
Introduction
Formaldehyde (HCOH) is a colorless, volatile, and pungent-smelling toxic substance that may have a significant impact on human health and the natural environment. Currently, the International Agency for Research on Cancer (IARC) classifies formaldehyde as a group I human carcinogen, and the U.S. Environmental Protection Agency (EPA) lists it as a toxic and harmful water pollutant.1,2 Long-term exposure to low concentrations of formaldehyde can lead to chronic respiratory diseases, and long-term exposure to high concentrations of formaldehyde may cause severe lung diseases, which are extremely detrimental to human life. Therefore, monitoring and removing indoor formaldehyde is crucial.3–5 The development of simple, fast, and effective methods to detect and remove formaldehyde completely is attractive and urgent for environmental protection and air purification.6 Photocatalytic technology involves the irradiation of a photocatalyst with sunlight at room temperature for increased surface reduction–oxidation ability to achieve pollutant purification, material synthesis, and transformation. Through photocatalytic technology, formaldehyde can be oxidized and decomposed into non-toxic carbon dioxide (CO2) and water (H2O).7,8 The photocatalyst is a critical factor in determining the photocatalytic degradation of formaldehyde. Exploring and developing various potential photocatalysts with high efficiency are important research areas.9,10 The photocatalysts reported thus far involve s-block elements such as sodium (Na), potassium (K), and strontium (Sr), p-block elements such as gallium (Ga), indium (In), germanium (Ge), and bismuth (Bi), d-block elements such as titanium (Ti), nickel (Ni), cobalt (Co), and zinc (Zn), and La-series elements such as lanthanum (La), cerium (Ce), and samarium (Sm). The photocatalytic degradation of formaldehyde begins from its adsorption and activation on the photocatalyst surface. Therefore, the theoretical study of the adsorption process of formaldehyde can determine the adsorption stability and bonding mechanism between formaldehyde and the substrate surface.11–13 This can provide a foundation for subsequent studies on the degradation mechanism of formaldehyde. Wu et al.14 studied the mechanism of formaldehyde oxidation by manganese (Mn)-doped cerium oxide (CeO2). They reported that the oxidation included formaldehyde adsorption, C–H bond breaking, formation and desorption of H2O and CO2 molecules, a decrease in coordinated oxygen atoms on the surface of the catalyst, and more oxygen vacancies. This lowered the potential barrier of C–H bond breaking and was more conducive to the oxidation of formaldehyde. Alvarado et al.15 studied the adsorption process of formaldehyde on the surface of monolayer hydrogenated gallium nitride (GaN). They reported that the adsorption of formaldehyde molecules on the N atom and Ga atom sides of the monolayer were physical and exothermic chemical adsorption processes, respectively. The hydrogen atoms adjacent to the surface were captured, and stable methoxy (CH3O) was formed, resulting in the appearance of a hydrogen vacancy on the surface. Zhang et al.16 reported that the adsorption of formaldehyde molecules was easier on the bridging oxygen (O) atoms and oxygen vacancies of the TiO2(110) surface at low temperatures. Zhou et al.17 studied the adsorption of formaldehyde molecules on Al-doped single-walled carbon nanotubes with vacancies. The interaction between the adsorbed formaldehyde molecules and carbon nanotubes was promoted by the Al doping and the vacancies, resulting in increased adsorption energy and significant charge transfer.Lanthanum oxide (La2O3) has the advantages of a wide bandgap, high dielectric constant, good thermal stability, and low price. It is widely used in the fields of optical glass, catalysts, ceramics, and thermoelectric materials. The wide bandgap semiconductor materials enable the design of photocatalysts with better performance by providing them with various opportunities for modification and a larger space for electronic structure regulation. Since the bandwidth of La2O3 is about 5.8 eV, only ultraviolet light can photogenerate the electron–hole pairs.18 Therefore, the practical applications of La2O3 as a photocatalyst are limited due to the low utilization of light energy. Thus, the bandwidth of La2O3 should be reduced to respond in infrared light and improve its light absorption efficiency. Loading, doping, and compounding are usually used to improve the photocatalytic activity of photocatalysts. Metal ion doping effectively extends the absorption region of wide-bandgap photocatalysts to the visible region.19,20 It has been reported that the La2O3 surface has good adsorption capacity and activation performance.21 It has wide application prospects as a photocatalyst for the degradation of indoor formaldehyde. Studies on the formaldehyde degradation by La2O3 photocatalysts and its mechanism have not been reported. With the explosive growth in computer performance, computing power and the continuous development of quantum mechanics theory,22–26 a first-principles calculation based on density functional theory (DFT) was used to study the modification of La2O3 due to formaldehyde adsorption and oxidation processes. Li et al.27 theoretically studied the oxidative changes in La2O3(001) and (011) surfaces after La atoms were substituted by different doped cations, such as Cu, Zn, Mg, Fe and Al. They reported that the doping of cations reduced the vacancy-formation energy of La2O3 surfaces, effectively improving its catalytic performance. Cong et al.28 studied the activity change on Sr-doped La2O3 due to the catalytic oxidative coupling of methane. The result indicated that Sr-doped La2O3 clusters had higher thermodynamic and kinetic activity due to the catalytic oxidative coupling of methane, wherein the formed Sr–O bond had stronger reactivity with methane and a lower energy barrier to overcome the reaction with methane than the La–O bond. Bannikov et al.29 reported that the bonding of the doped elements had a significant impact on the magnetic properties of La2O3. The nitrogen (N)-doped La2O3 combined with six ligands had magnetic, narrow-band semiconductor characteristics. In contrast, the N-doped La2O3 combined with four ligands had magnetic, semi-metallic characteristics. At present, there are no relevant reports on the adsorption of formaldehyde molecules on different structures of the La2O3 surface. Relevant studies can provide a theoretical basis for the application of La2O3 as an efficient photocatalyst in the removal of indoor formaldehyde by photocatalysis technology.
It has been widely reported that La2O3 has increased activity in chemical reactions. The adsorption behavior of formaldehyde molecules on the La2O3 surface will help to understand the fundamental principles of its increased activity. Herein, the adsorption processes of formaldehyde molecules on the surface of intrinsic La2O3, La2O3 with oxygen vacancy, and iridium-doped La2O3 were studied. The influence of the oxygen vacancy and iridium doping on the adsorption process of formaldehyde molecules was studied. The adsorption energy, density of states, and optical properties of formaldehyde molecules on La2O3 with different surface states were calculated, and the bonding characteristics and adsorption mechanism of formaldehyde molecules on the La2O3 surface were studied.
Modeling and calculating methods
Modeling
La2O3 has a hexagonal crystal structure with the space group of P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1. As shown in Fig. 1, the bulk phase structure of La2O3 is the superposition of two layers of (LaO)nn+ and one layer of O2−. The La atoms are seven coordinated and marked as La7c. The O atoms, which are four- (2/3 share) and six-coordinated (1/3 share), are marked as O4c and O6c, respectively.30 The cell parameters of La2O3 are given as a = b = 3.939 Å, c = 6.136 Å, α = β = 90°, and γ = 120°. The cell parameters of La2O3 were optimized to verify the rationality of the pseudopotential atomic calculations in La2O3.
m1. As shown in Fig. 1, the bulk phase structure of La2O3 is the superposition of two layers of (LaO)nn+ and one layer of O2−. The La atoms are seven coordinated and marked as La7c. The O atoms, which are four- (2/3 share) and six-coordinated (1/3 share), are marked as O4c and O6c, respectively.30 The cell parameters of La2O3 are given as a = b = 3.939 Å, c = 6.136 Å, α = β = 90°, and γ = 120°. The cell parameters of La2O3 were optimized to verify the rationality of the pseudopotential atomic calculations in La2O3.
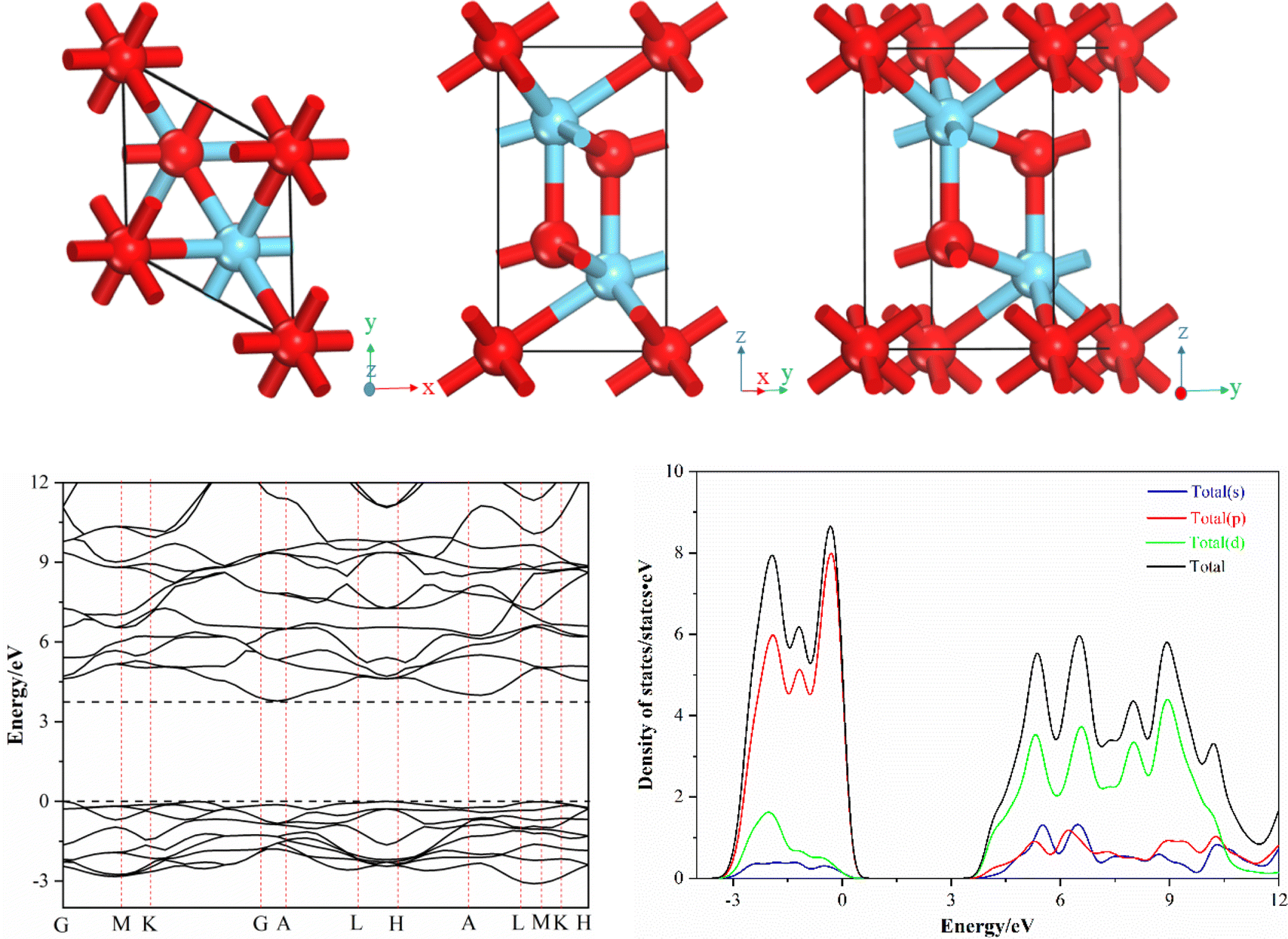 | ||
| Fig. 1 Bulk phase structure, band structure and DOS of bulk La2O3. | ||
The bulk La2O3 is an indirect bandgap semiconductor, wherein the valence band top is mainly composed of the 2p orbital of the O atom and the conduction band is mainly composed of the 5d orbital of the La atom. We validate the effectiveness of the DFT method by calculating the error between the theoretical and experimental values of the lattice parameters. If the error is less than 5%, it indicates that the theoretical simulation method is effective. The optimized cell parameters are a = b = 3.963 Å, c = 6.179 Å, α = β = 90°, and γ = 120°. The errors are less than 1% compared with the initial values, indicating that the pseudopotential atomic calculations and the selection of calculation parameters were reasonable.
In this paper, the most common and stable La2O3(001) surface was selected as the studied object.31–33 The inclusion of more atomic layers in the calculation leads to higher accuracy. However, including too many atomic layers in the analysis will increase its cost and reduce its efficiency. In general, a model will represent the properties of the bulk material when the number of atomic layers exceeds 5. In this study, the number of atomic layers was set as 6, and the procedure of the supercell model of the La2O3(001) surface is as follows. First, the cell model of La2O3 was imported from the software model library. Second, we cut the surface in the (001) direction through the surfaces-cleave surface instruction of the Build menu. Third, according to the symmetry-Supercell instruction in the Build menu bar, we inputted 2 and 2 at the U and V positions, respectively, and an expanded cell structure of the section plane was formed. Four, in order to eliminate the impact of the interaction between periodic units, the Crystals-Build Vacuum Slab Crystal instruction in the Build menu was selected and the value of 15 Å was input in the vacuum layer thickness field. Then, the build button was clicked, and a p(2 × 2 × 2) supercell model of La2O3(001) is finally obtained. The total number of atoms in the surface model is forty, of which sixteen are La atoms, sixteen are four-coordinated oxygen atoms, and eight are six-coordinated oxygen atoms. After replacing one iridium atom or removing one oxygen atom, the corresponding concentrations are 6.25%, 6.25%, and 12.50%, respectively. In order to make the established La2O3 surface model consistent with the actual situation, the bottom three atomic layers of the La2O3 surface model are constrained. They remain stationary during the model optimization and performance simulation process, and are regarded as La2O3 bulk materials. The three upper layers are not constrained and fully relaxed to simulate the active atoms of the La2O3 surface. There are two types of O atoms in the La2O3 surface model, corresponding to the two modes of oxygen vacancies: four-fold coordinated oxygen vacancy (VO4c) and six-fold coordinated oxygen vacancy (VO6c). Consequently, as shown in Fig. 2, there are two models of Ir-doped La2O3(001) denoted as IrLa–VO4c and IrLa–VO6c, respectively.
 | ||
| Fig. 2 p(2 × 2) Ir-doped model of La2O3(001) with different oxygen vacancies. | ||
In the adsorption modeling process of the formaldehyde molecule on the La2O3(001) surface of the (2 × 2 × 2) supercell, the C and O atoms of the formaldehyde molecule bond with the O and La atoms on the La2O3(001) surface, respectively. The O atom can bond with the La atom in three possible ways: adjacent adsorption bonding, diagonal adsorption bonding, and single bond adsorption. In this study, the effects of Ir doping and oxygen vacancies on the adsorption of formaldehyde molecules were compared. There are six possible configurations for formaldehyde molecule adsorption, which are as follows: La2O3(001) surface, La2O3(001) surface containing VO4c, La2O3(001) surface containing VO6c, Ir-doped La2O3(001) surface, Ir-doped La2O3(001) surface containing VO4c, Ir-doped La2O3(001) surface containing VO6c, Ir-doped La2O3 containing VO4c and VO6c in turn. Additionally, the O atom of the formaldehyde molecule can bond with either the La or Ir atom on the Ir-doped La2O3(001) surface, resulting in 33 adsorption configurations. The formaldehyde molecular adsorption configurations are shown in Fig. 3. The distances of La–O, C–O and C–H are set as 3.0 Å, 3.0 Å and 1.14 Å, respectively. The adsorption energy for each adsorption configuration is different, which impacts the adsorption process significantly.
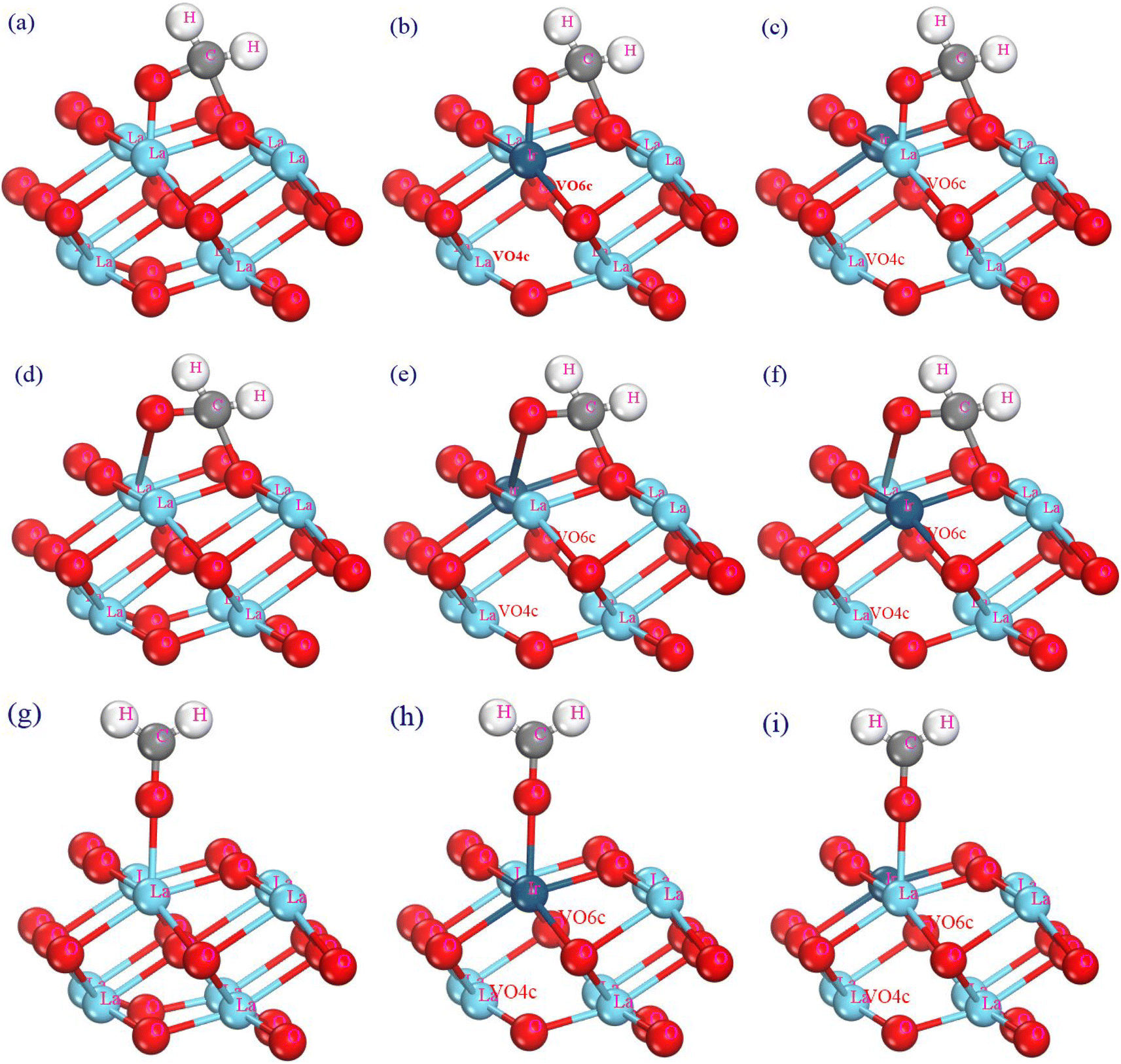 | ||
| Fig. 3 The adsorption configurations of the formaldehyde molecule on the La2O3(001) surface: (a) adjacent adsorption; (b) adjacent adsorption bonding with the Ir atom; (c) adjacent adsorption bonding with the La atom; (d) diagonal adsorption; (e) diagonal adsorption bonding with the Ir atom; (f) diagonal adsorption bonding with the La atom; (g) single bond adsorption; (h) single bond adsorption bonding with the Ir atom; (i) single bond adsorption bonding with the La atom. | ||
Calculating methods
The calculations were performed by the CASTEP module of Materials Studio 2019 software based on density functional theory (DFT). The interaction between ions and valence electrons was described by the projected additive wave (PAW) method, and the exchange–correlation potential between electrons was processed by the Perdew–Burke–Ernzerhof (PBE) functional of generalized gradient approximation (GGA).29,32,33 Through testing, the plane wave truncation kinetic energy was set as 600 eV, the force was distributed to each atom, and the convergence value of the total system energy was less than 3.0 × 10−3 eV nm−1 and 1.0 × 10−5 eV per atom, respectively. The deviations of stress and displacement were less than 0.05 GPa and 1.0 × 10−4 nm, respectively. The size of the k lattice point in the Brillouin zone on the La2O3(001) surface was set as 4 × 4 × 1.34 The valence electron configurations of the La, O, Ir, and C atoms are taken as La 5d1 6s2, O 2s2 2p4, Ir 4f14 5d7 6s2, and C 2s2 2p2, respectively.Results and discussions
Analysis of the adsorption energy calculation results
Adsorption energy (Eads) is defined as the change in total energy of the system before and after adsorption, indicating the stability of the substance in the adsorption system. The sign and calculated value of Eads indicate the possibility and tightness of adsorption, respectively. The equation for Eads is given as follows:35| Eads(AB) = E(CH2O/La2O3(001)) − ECH2O − ELa2O3(001) | (1) |
| Adsorption configuration | Adjacent (eV) | Diagonal (eV) | Single bond (eV) | |||
|---|---|---|---|---|---|---|
| With La | With Ir | With La | With Ir | With La | With Ir | |
| Bulk | −3.07 | −1.25 | −1.01 | |||
| VO4c | −3.12 | −2.12 | −1.04 | |||
| VO6c | −1.98 | −2.47 | −1.04 | |||
| Ir-doped | −0.97 | −1.63 | −2.52 | −1.25 | −1.11 | −0.52 |
| Ir-doped and VO4c | −1.03 | −2.57 | −1.22 | −0.32 | −1.13 | −0.99 |
| Ir-doped and VO6c | −2.54 | −3.23 | −2.21 | 0.46 | −0.95 | −2.02 |
| Ir-doped, VO4c and VO6c | −1.16 | −1.83 | −1.78 | −1.01 | −0.85 | −0.95 |
It can be seen that the Eads of adjacent adsorption is the largest, followed by the Eads of diagonal adsorption. The Eads of single bond adsorption is the smallest. Except for the configuration of diagonal adsorption bonding with the Ir atom of the La2O3(001) surface with the VO6c vacancy, the Eads values of all configurations of formaldehyde molecule adsorption on the La2O3(001) surface are negative, indicating that formaldehyde molecules can be spontaneously adsorbed on different La2O3(001) surfaces. The Eads of single bond adsorption bonding with the Ir atom of the Ir-doped and Ir-doped La2O3(001) surfaces with VO4c vacancy is −0.52 eV and −0.32 eV, respectively, indicating a physical adsorption process. The Eads of other adsorption configurations is less than −0.62 eV, indicating a stable chemical adsorption process. The Eads of formaldehyde gas molecules on the La2O3(001) surface is affected by the type of oxygen vacancy, noble metal doping, and the bonding mode of O atoms in formaldehyde molecules. Because of the existence of VO4c in the La2O3 bulk material, the Eads of different adsorption configurations of formaldehyde molecules on the La2O3(001) surface increases. For an intrinsic La2O3(001) surface with VO6c oxygen vacancy, the Eads of the adjacent adsorption configuration decreases, whereas it increases for the diagonal adsorption and single bond adsorption configurations. The Eads for formaldehyde molecules on different La2O3(001) surfaces decreased after the bulk La2O3 is doped with the Ir element, indicating that the Ir doping in La2O3 has an adverse effect on the formaldehyde molecule adsorption. When the VO4c or VO6c oxygen vacancy existed in the Ir-doped La2O3(001) surface, the Eads for formaldehyde molecules at the adjacent and single bond top sites increased, and the Eads of the diagonal sites decreased, respectively. The largest Eads is the formaldehyde adsorption on the Ir-doped La2O3(001) surface with VO6c vacancy, which is −3.23 eV. The Eads is larger than that for formaldehyde molecules on the La2O3(001) surface containing an oxygen vacancy, indicating that formaldehyde molecule adsorption benefits the most from the existence of a VO6c oxygen vacancy in Ir-doped La2O3 surface structures. The Eads for formaldehyde molecules on the Ir-doped La2O3(001) surface with VO4c and VO6c vacancy is smaller than that on the La2O3(001) surface with VO6c defects.
The mechanism of the photocatalytic technology of La2O3 semiconductor materials involves four stages. This paper mainly focuses on the adsorption mechanism of formaldehyde molecules on the surface of photocatalytic materials in the first stage. The larger the adsorption energy is, the stronger the adsorption of formaldehyde molecules, and the more conducive to improving the efficiency of the photocatalytic oxidation of formaldehyde molecules. The theoretical research of La2O3 mainly focuses on the surface properties of La2O3, the oxidative coupling of methane (OCM), and the adsorption and desorption of the oxygen molecule. At present, studies on the degradation of formaldehyde by La2O3 photocatalysts and its mechanism have not been reported. Compared with other types of catalyst materials, such as BN (2.14 eV),5 ZnO (0.85 eV),12 CeO2 (0.86–1.31 eV),14 TiO2 (1.66 eV),16 aluminum-doped carbon nanotubes (0.65 eV),17 and others, the adsorption energy of formaldehyde adsorbed on the surface of modified La2O3(001) is calculated to reach 3.23 eV. The adsorption energy has been greatly improved, and it can be theoretically used as an ideal photocatalytic material for formaldehyde degradation.
Adsorption mechanism of formaldehyde molecules on the La2O3(001) surface
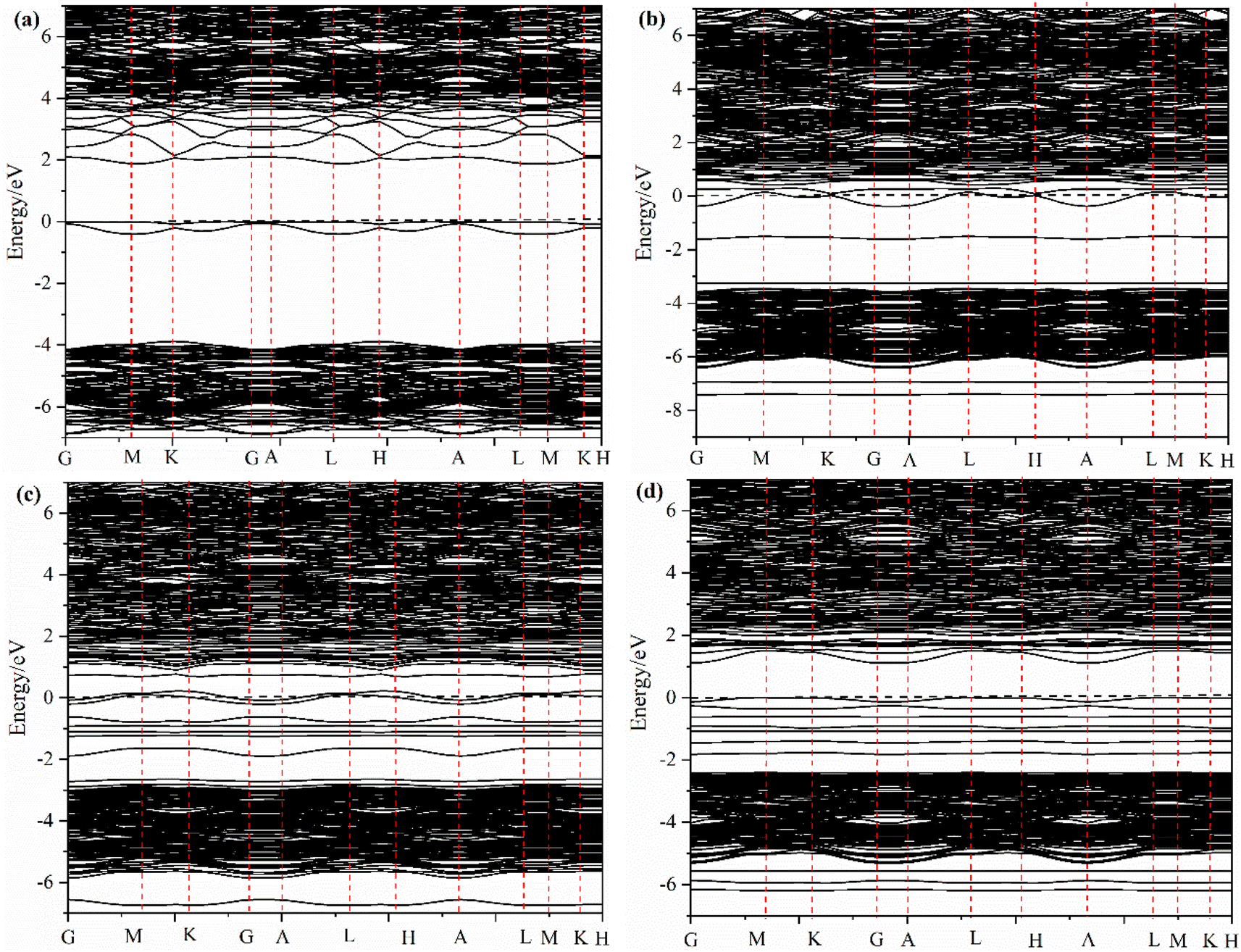 | ||
| Fig. 4 The band structure of different La2O3(001) surface structures and adsorbed formaldehyde molecule: (a) intrinsic La2O3(001) surface; (b) Ir-doped La2O3(001) surface with VO6c oxygen vacancy; (c) formaldehyde molecule adsorbed on the intrinsic La2O3(001) surface; (d) formaldehyde molecule adsorbed on the Ir-doped La2O3(001) surface with the VO6c oxygen vacancy. | ||
The effect of the oxygen vacancy and iridium doping on the energy band structure before and after adsorption of formaldehyde molecules on the La2O3(001) surface can be summarized as follows. Compared with the intrinsic La2O3(001) surface, the new energy levels between the top of the valence band and the bottom of the conduction band enhances the electronic transition process because of the interaction of the 2p orbital of the oxygen vacancy atoms and the 5d orbital of the Ir atom, which acts as a bridge and is conducive to the electronic transition process.
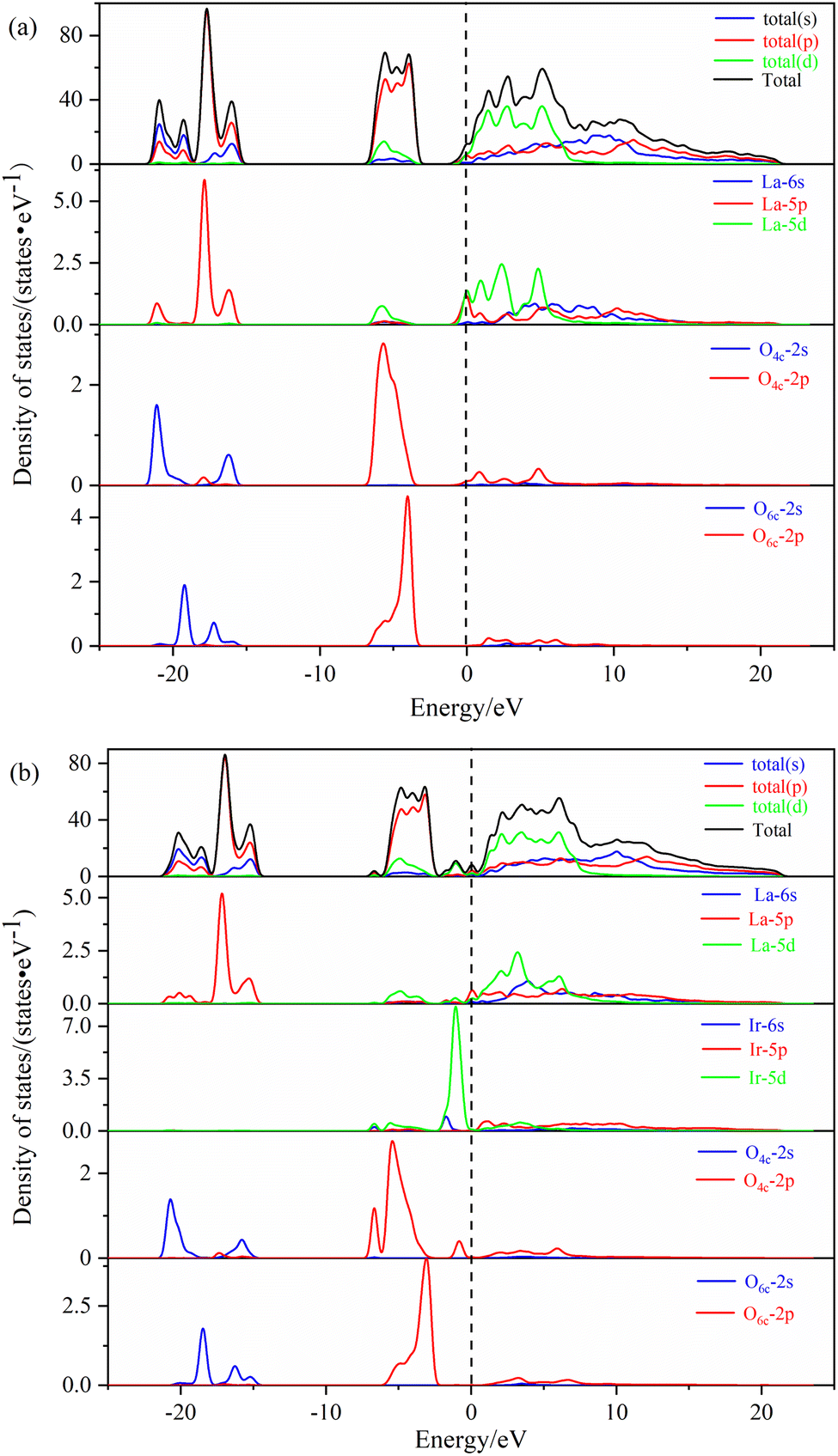 | ||
| Fig. 5 The density of states of different La2O3(001) surface structures: (a) intrinsic La2O3(001) surface; (b) Ir-doped La2O3(001) surface with VO6c oxygen vacancy. | ||
In Fig. 5b, the shapes of the peak and corresponding energy intervals in the high-energy conduction band and low-energy valence band agree with the density of states analysis. The Ir-doped and VO6c oxygen vacancy do not have a significant effect on the valence band. However, the density of state peak of the O4c-2p orbital splits and broadens due to Ir doping, Ir atom bonding with O4c, electron transfer from the Ir atom to O4c, and interaction between the Ir-5d orbital and O4c-2p orbital. Furthermore, Ir doping and O6c enable the La-5p orbital to play an important role near the Fermi level. Therefore, two new peaks of the density of states emerge close to the conduction and valence bands, which is conducive to the electronic transition process.
The density of states of free formaldehyde molecules, intrinsic La2O3(001) surface, and Ir-doped La2O3(001) surface with VO6c oxygen vacancy are shown in Fig. 6. As shown in Fig. 6a, the peaks of the merged and orbital electron density of states for free formaldehyde molecules are sharp and narrow with high localization, which is consistent with the density of states characteristics for general gaseous molecules. After the formaldehyde molecules are adsorbed on the La2O3(001) surface, the orbitals broaden and merge, and the orbital electrons show typical delocalization, which is due to interaction between the formaldehyde molecules and surface atoms. As shown in Fig. 6b, the density of states of the intrinsic La2O3(001) surface changes significantly after adsorption of formaldehyde molecules. The conduction band shifts to a high-energy direction, and two new isolated peaks of the density of states appear between the valence and conduction bands of the intrinsic La2O3(001) surface structure. The peak at the Fermi level is due to the La-5p orbital. The peak in the low-energy direction is due to hybridization of the O-2p and C-2p orbitals in formaldehyde molecules. This indicates that there is a strong interaction between a single formaldehyde molecule and the La2O3(001) surface. Upon adsorption of formaldehyde molecules, impurity levels are generated between the conduction and valence bands. These impurity levels hinder the charge transfer after subsequent adsorption of formaldehyde molecules, which results in the unstable adsorption of more formaldehyde molecules. Fig. 6c shows that the two isolated peaks for the density of states between the conduction band and the valence band disappear. For the Ir-doped La2O3(001) surface structure containing VO6c defects, the peak for the density of states of the valence band widens and extends to a position near the Fermi level. The top of the valence band is due to the Ir-5d and O4c-2p orbitals and the O-2p and C-2p orbitals of the formaldehyde molecules. The doping of Ir atoms in the La2O3(001) surface structure causes the hybridization of the O-2p and C-2p orbitals of formaldehyde molecules and the Ir-5d and O4c-2p orbitals of the surface layer, resulting in an easier transfer of the lone pair of electrons in the formaldehyde molecules to the La2O3(001) surface. Therefore, the VO6c defect and Ir doping reduce the gap between the valence and conduction bands and enhance the interaction between the formaldehyde molecules and La2O3(001) surface by promoting charge transfer. Consequently, the same La2O3(001) surface area adsorbs more formaldehyde molecules.
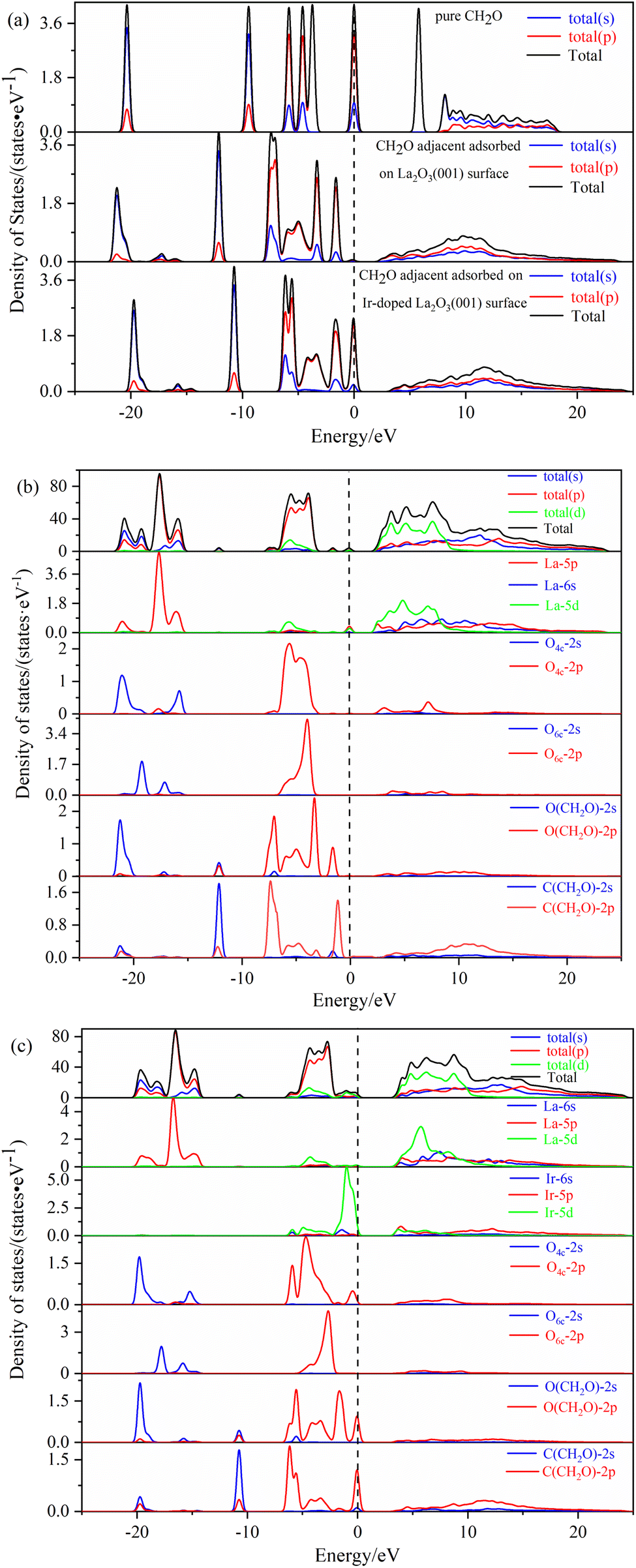 | ||
| Fig. 6 The density of states for: (a) free formaldehyde; (b) formaldehyde adjacent adsorbed on the La2O3(001) surface; (c) formaldehyde adjacent adsorbed on the Ir-doped La2O3(001) surface with VO6c oxygen vacancy. | ||
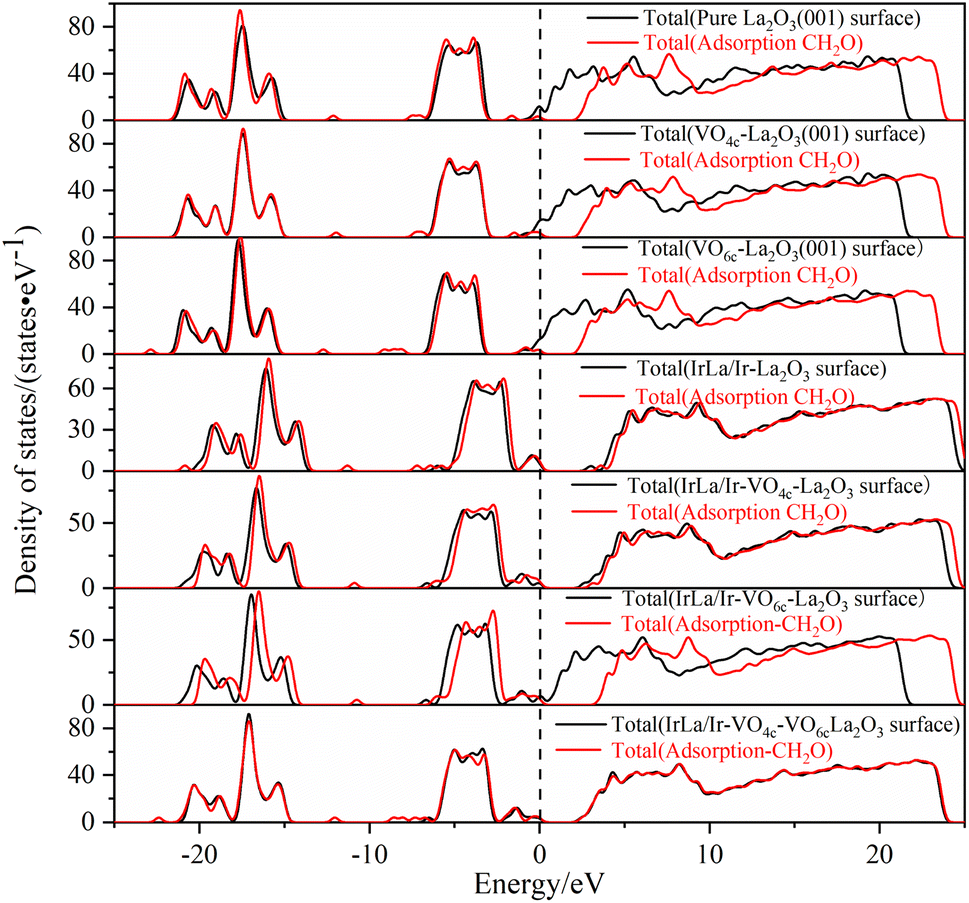 | ||
| Fig. 7 The density of states of different La2O3(001) surface structures before and after the adsorption of a formaldehyde molecule. | ||
Therefore, based on the above research results, we summarize the conclusion as follows. The physical nature of oxygen vacancy and iridium doping on the electronic properties before and after the adsorption of formaldehyde molecules on the La2O3(001) surface is the interaction of the 2p orbital of the oxygen vacancy atoms and 5d orbital of the Ir atom, which forms new energy levels between the top of the valence band and the bottom of the conduction band. These energy levels provide a bridge that facilitates the excited transition of valence band electrons towards the conduction band. After the adsorption of formaldehyde molecules on the La2O3(001) surface, the transition electrons will react with the C-2p and O-2p orbital electrons of the formaldehyde molecules. Subsequently, the oxidative degradation of formaldehyde molecules occurs. Compared with the intrinsic La2O3(001) surface, the Ir-doped La2O3(001) surface with oxygen vacancy significantly enhanced the adsorption of formaldehyde and interaction between the orbital electrons, which is more conducive to the process of formaldehyde oxidative degradation.
Optical properties
| ε(ω) = ε1(ω) + iε2(ω) | (2) |
The first-principles method was used to calculate the optical properties of different La2O3(001) surface structures and the formaldehyde molecules after adsorption. Furthermore, since the calculated value of the bandgap was smaller than the experimental value, the scissor operator was used for the correction. A correction factor of 2.29 eV was used to convert the calculated value of 2.51 eV to the experimental value of 5.8 eV. The curves of the calculated values of the real and imaginary parts of the dielectric function in the selected energy range of 0–10 eV are shown in Fig. 8 and 9. Considering only the electronic contribution to the dielectric constant, it can be seen from Fig. 8 that the static dielectric constants of different La2O3(001) surfaces before formaldehyde molecule adsorption are 3.45, 17.14, 6.10, 3.46, 3.16, 2.99 and 3.86, and the corresponding values after formaldehyde molecule adsorption are 3.09, 3.17, 4.74, 3.14, 3.50, 2.98 and 3.25, respectively. Except for the configuration of the diagonal adsorption bonding with the Ir atom of the La2O3(001) surface with the VO6c oxygen vacancy, the static permittivities of different La2O3(001) surface configurations decrease after the formaldehyde molecules are adsorbed. In the low energy region (<2 eV), the real part ε1(ω) of the dielectric function increases with an increase in energy and peaks at about 2 eV. The density of states indicates that this is mainly caused by the electronic transition from the La-5d or doped Ir-5d orbital to the O-2p orbital on the La2O3(001) surface. As shown in Fig. 9, the peaks of ε2(ω) emerge in two stages with the increase in photon energy. The peak of stage 1 (about 4.74 eV < e < 15.00 eV) is due to an electronic transition from the top of the valence band to the bottom of the conduction band. In addition, the peak of stage 2 (15 eV < e < 40 eV) is due to an electronic transition from the center and top of the valence band to the bottom and center of the conduction band.
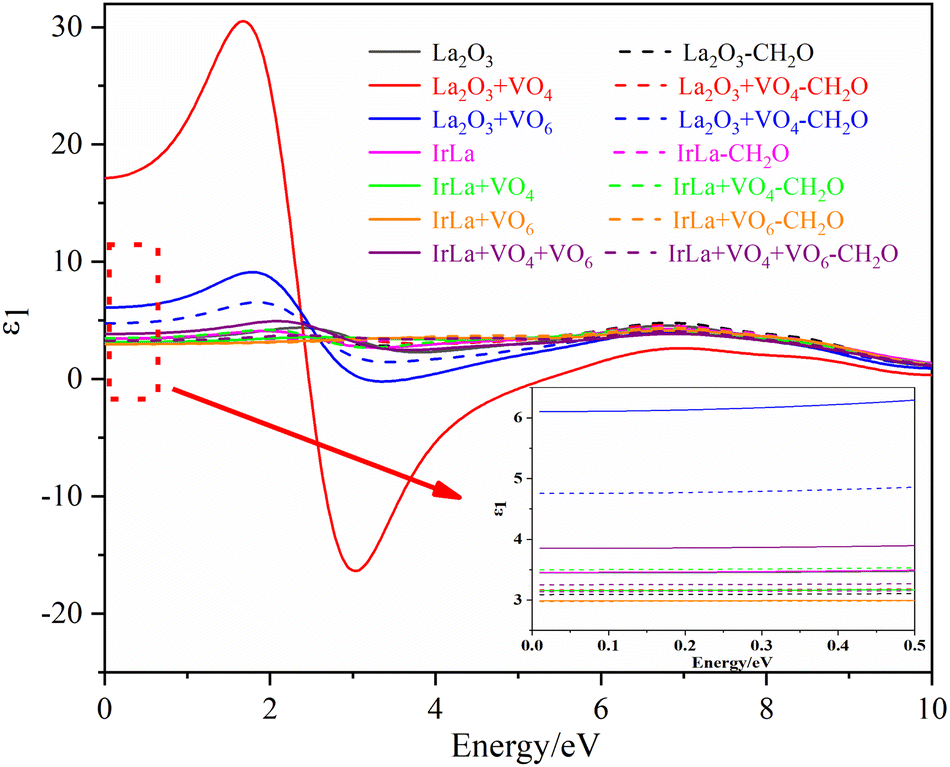 | ||
| Fig. 8 The real part of the permittivity of different La2O3(001) surfaces and adsorbing formaldehyde molecule. | ||
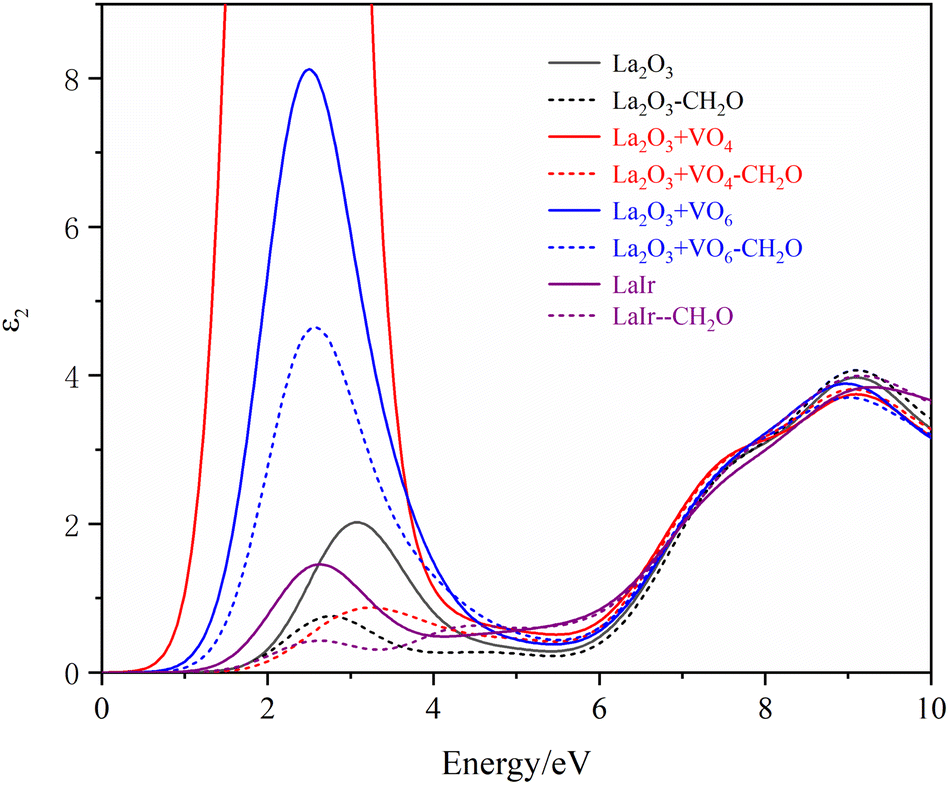 | ||
| Fig. 9 The imaginary part of the permittivity of different La2O3(001) surfaces and the adsorbing formaldehyde molecule. | ||
| α(ω) = 2(ω)[ε12(ω) + ε22(ω) − ε1(ω)]1/2 | (3) |
The calculated absorption spectrum of different La2O3(001) surfaces and adsorbing formaldehyde molecule is shown in Fig. 10. It can be seen that the oxygen vacancy and Ir doping have a significant impact on the absorption spectrum of La2O3(001) surfaces. Both VO4c and VO6c oxygen vacancies can increase the absorption peak of the La2O3(001) surface in the visible region, and Ir doping obviously weakens the absorption intensity in this region. The absorption peaks in the low-power infrared and visible light region disappear when there is a single type of oxygen vacancy or Ir doping on the La2O3(001) surface. However, they reappear when the VO4c oxygen vacancy, CO6c oxygen vacancy, and Ir doping exist at the same time. The absorption peaks of adjacent adsorption configurations of different La2O3(001) surfaces are located at 3.34 eV (371 nm), 3.03 eV (409 nm), 3.07 eV (404 nm), 2.84 eV (437 nm), and 3.15 (394 nm), and the corresponding absorption edges are 1.68 eV, 0.77 eV, 1.11 eV, 1.32 eV, and 1.30 eV, respectively. This indicates that the La2O3(001) surface with the VO4c oxygen vacancy has a stronger long-wave absorption capacity, and the absorption wavelength can extend to the infrared region (1610 nm). The main reason is that the absorption spectrum is determined by the imaginary part ε2(ω) of the dielectric function. Due to the change in the electron distribution near the Fermi level of different La2O3(001) surfaces, the absorption band edge and band gap are different. This eventually leads to various degrees of difficulty, and the process of electronic transition from the top of the valence band to the bottom of the conduction band. After the adsorption of formaldehyde molecules on different La2O3(001) surfaces, the absorption peaks of other configurations disappeared in the visible region. This is due to the hybridization near the Fermi level between the O-2p, C-2p orbitals of the formaldehyde molecule and the Ir-5d, O4c-2p orbitals of the top atomic layer on the La2O3(001) surfaces. The gap between the valence band and conduction band is significantly reduced to form approximately zero band gap materials, such that electronic transitions from the valence band to the conduction band can occur with almost no energy barriers.
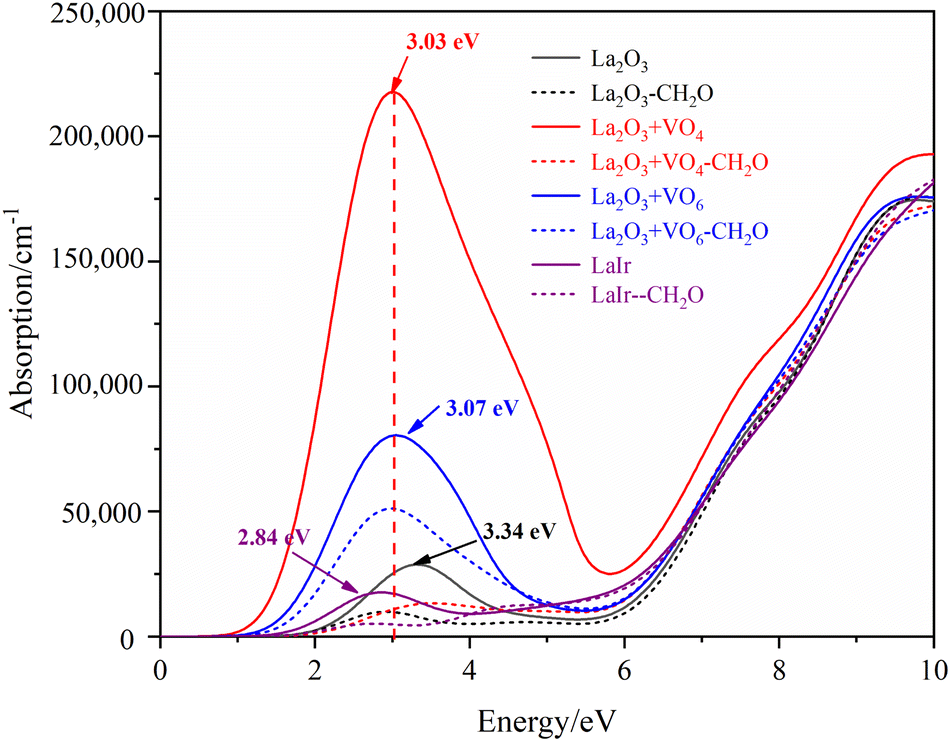 | ||
| Fig. 10 The absorption spectra of different La2O3(001) surfaces and adsorbed formaldehyde molecule. | ||
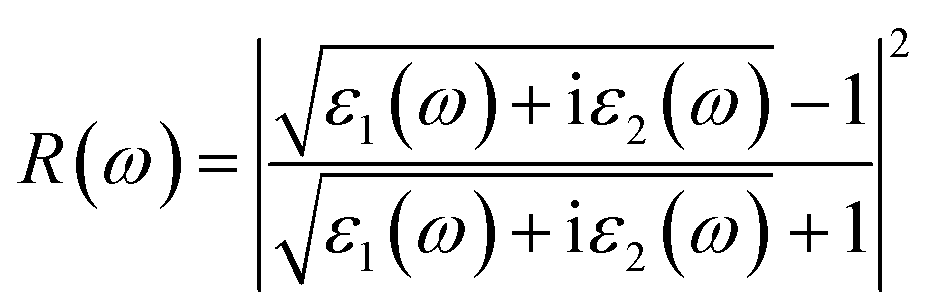 | (4) |
The R(ω) is the macroscopic manifestation of a solid electronic interband transition under the perturbation of an optical electromagnetic field, which is determined by the real part of the dielectric function. Fig. 11 shows the calculated absorption spectra of different La2O3(001) surfaces and adsorbed formaldehyde molecule. It can be seen that the variation trends of the reflection spectrum and absorption spectrum are similar. Furthermore, the reflectivity increases with increasing photon energy in the low-energy infrared and visible regions. Because of the interaction between reflection and absorption, the La2O3(001) surface effectively reflects light in a certain range. It can also absorb light in the same wavelength range. There are three reflection peaks on the adjacent adsorption configurations of different La2O3(001) surfaces, and the corresponding energies are located at around 2.65 eV, 12.70 eV, and 26.45 eV, respectively, whereas the reflection spectra of the La2O3(001) surfaces with the single oxygen vacancy (VO4c or VO6c) and Ir doping only have two peaks at 12.70 eV and 26.45 eV. After adsorption of formaldehyde molecules, the reflection peaks in the low-energy infrared and visible regions near 2.65 eV disappear, and the reflection spectrum contains two peaks near 12.70 eV and 26.45 eV.
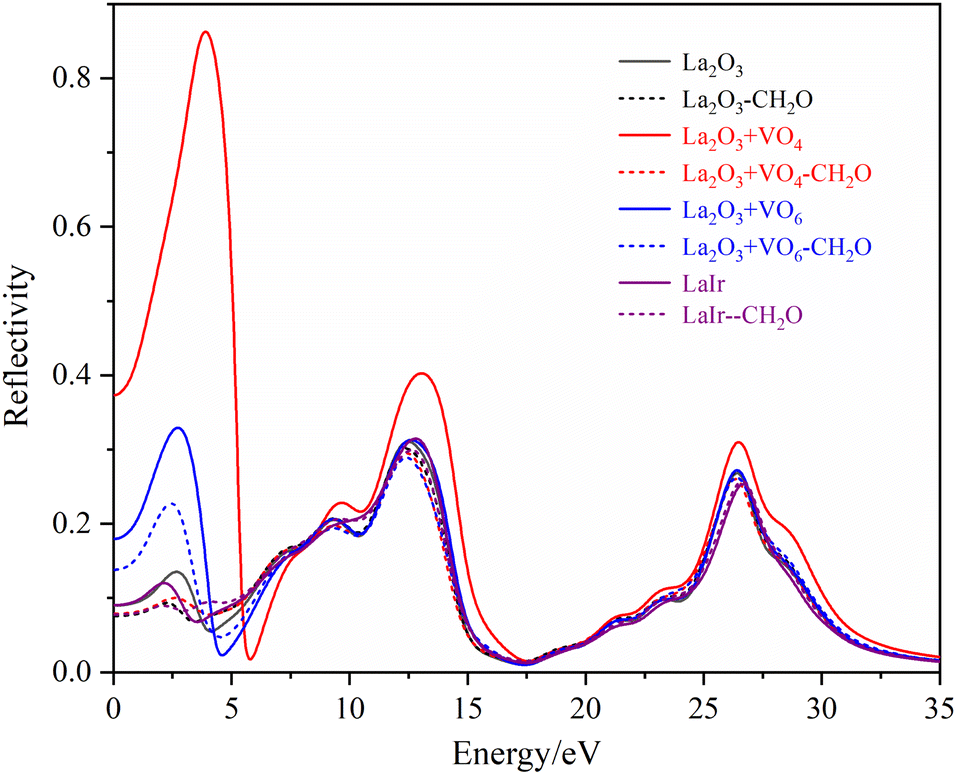 | ||
| Fig. 11 The reflectance spectra of different La2O3(001) surfaces and adsorbed formaldehyde molecule. | ||
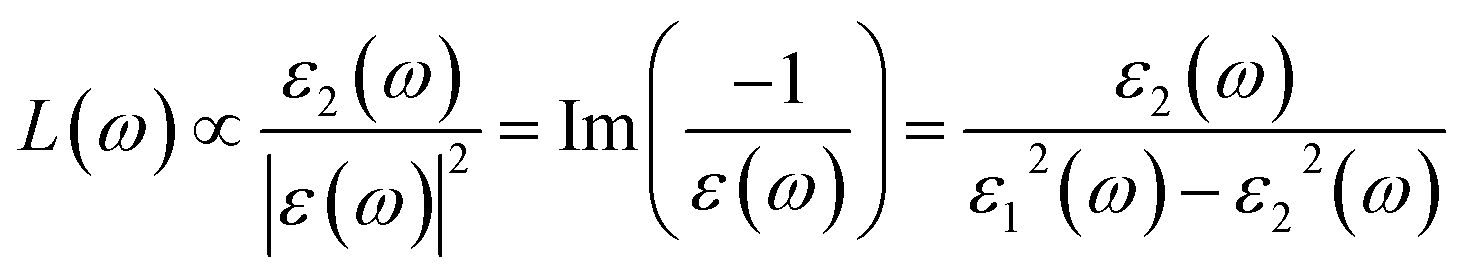 | (5) |
The loss functions of different La2O3(001) surfaces and adsorbed formaldehyde molecule are shown in Fig. 12. There are three main peaks in the energy loss function spectrum. The corresponding energy and change trend are basically consistent with the reflection spectrum. Conversely, the change in the peak value is opposite to the high value of the reflection peak, which corresponds to the low value of the peak in the energy loss function spectrum. The peak position represents the frequency of the excited electron and vibrational frequency of the particles. The peak of the energy loss function corresponds to the sharp drop area of the reflection peak, and all of the peak values are within the energy range of 1–50 eV.
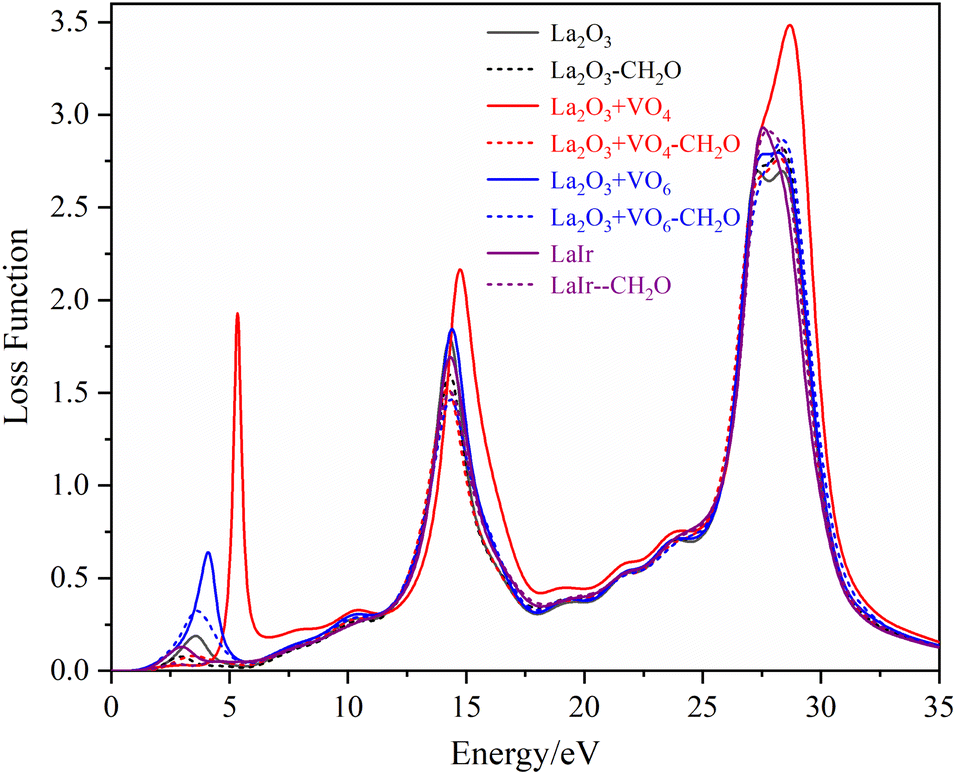 | ||
| Fig. 12 The loss functions of different La2O3(001) surfaces and adsorbed formaldehyde molecule. | ||
Conclusions
The effects of the oxygen vacancy (VO4c and VO6c) and iridium doping on formaldehyde adsorption on the La2O3(001) surface were studied by the first-principles method. From the view of energy, the adjacent adsorption of the iridium-doped La2O3(001) surface containing the VO6c oxygen vacancy is the most stable adsorption site for formaldehyde molecules. Due to the significant doping of the Ir-5d orbital and internal VO6c oxygen vacancies, the effects of the oxygen vacancy and iridium doping on the optical properties of the La2O3(001) surface are mainly shown in the low-energy infrared and visible regions.Compared with the ZnO, CeO2, TiO2, and Al-doped carbon nanotube catalyst materials, the Eads of formaldehyde adsorbed on the modified La2O3(001) surface is 3.23 eV, which is a significant improvement. The La2O3 semiconductor material can be used as an ideal photocatalytic material for formaldehyde degradation and indoor detection of formaldehyde gas. In the subsequent research process, we will start the technical study on the noble metal iridium doping of La2O3 by atomic layer deposition technology and photocatalytic degradation of formaldehyde, and conduct an overall evaluation of the performance and application prospect of the La2O3 photocatalytic material.
Data availability
The data that support the findings of this study can be obtained from the corresponding author. Hengjiao Gao, Email: E-mail: gaohengjiao@163.com.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge support from the Talent Introduction Research Fund Project (Grant No. 2021BSQD005), the Young Doctor of Gansu Province Colleges and Universities (Grant No. 2021QB-130), and the Key Fund Project of Equipment Pre-research Key Laboratory (Grant No. 6142207220102).References
- S. E. Diltemiz and K. Ecevit, High-performance formaldehyde adsorption on CuO/ZnO composite nanofiber coated QCM sensors, J. Alloys Compd., 2019, 783, 608–616 CrossRef CAS
.
- A. Noguerón, H. N. Fernández-Escamilla, J. Guerrero-Sánchez and N. Takeuchi, Formaldehyde adsorption on a hydrogenated aluminum nitride monolayer: a self-propagated reaction, Comput. Theor. Chem., 2019, 1159, 18–22 CrossRef
- M. W. Zheng, Y. T. Lin and S. H. Liu, TiO2@g-C3N4/SiO2 with superior visible-light degradation of formaldehyde for indoor humidity control coatings, Mater. Today Sustain., 2023, 24, 100496 CrossRef
- J. Pei, Z. Bai, Y. Pan and Q. Wu, Long-lasting removal of indoor formaldehyde in continuous airflow by B–TiO2@AC composites, Built Environ., 2024, 256, 111458 CrossRef
- H. Zhang, Z. Zheng, T. Yu, C. Liu, H. Qian and J. Li, Seasonal and diurnal patterns of outdoor formaldehyde and impacts on indoor environments and health, Environ. Res., 2022, 205, 112550 CrossRef CAS PubMed
- L. Lu, T. Xiao, X. Yang, X. Zhou and J. Yan, Refinement and predicting formaldehyde concentrations of indoor fabric: effects of temperature and humidity, Chemosphere, 2023, 342, 140096 CrossRef CAS PubMed
- V. Gupta, S. Sarkar, O. Aftenieva, T. Tsuda, L. Kumar, D. Schletz, J. Schultz, A. Kiriy, A. Fery and N. Vogel, et al., Nanoimprint lithography facilitated plasmonic-photonic coupling for enhanced photoconductivity and photocatalysis, Adv. Funct. Mater., 2021, 31, 2105054 CrossRef CAS
- P. A. F. Cortés, M. Marx, M. Trose and M. Beller, Heteroleptic copper complexes with nitrogen and phosphorus ligands in photocatalysis: overview and perspectives, Chem Catal., 2021, 1, 298–338 CrossRef
- B. Tang, Y. He, Z. Zhang, Z. Wang, L. Ji, T. Ma, S. Li, Y. Dai and G. Zhang, Influence of N doping and the functional groups of graphene on a RGO/TiO2 composite photocatalyst, Sci. China: Technol. Sci., 2020, 63, 1045–1054 CrossRef CAS
- W. Xie, Q. Fu, C. Cheng, G. Zhang, N. Yan and Z. Wang, Effect of La2O3 on the oxidation resistance of SiC ceramic at 1973K: experimental and theoretical study, J. Am. Ceram. Soc., 2020, 103, 614–621 CrossRef CAS
- H. Zhao, K. An, Z. Wang, X. Liu, M. He, X. Yang, Z. Tang, S. Lai, S. Han, Z. Sun and Y. Jiao, The impact of water co-adsorption on the removal of formaldehyde from the indoor air by oxygen-rich activated carbons: a theoretical and experimental study, Appl. Surf. Sci., 2023, 635, 157729 CrossRef CAS
- H. Tran and M. Spencer, Zinc oxide for gas sensing of formaldehyde: density functional theory modeling of the effect of nanostructure morphology and gas concentration on the chemisorption reaction, Mater. Chem. Phys., 2017, 163, 274–284 CrossRef
- J. Xiao, X. Li, H. Lian, M. Pan, C. Xu, C. Xia, S. Lan and W. Li, Deep eutectic solvents for effective removal of indoor formaldehyde: theoretical design, experiment, and adsorption mechanism study, J. Mol. Liq., 2023, 392, 123473 CrossRef CAS
- H. Wu, S. Ma, W. Song and E. J. Hensen, Density functional theory study of the mechanism of formaldehyde oxidation on Mn-doped ceria, J. Phys. Chem., 2016, 120, 13071–13077 CAS
- L. A. Alvarado-Leal, H. N. Fernandez-Escamilla, J. Guerrero-Sanchez, E. Martínez-Guerra and N. Takeuchi, Formaldehyde adsorption on a hydrogenated gallium nitride monolayer: a density functional theory study, Appl. Surf. Sci., 2020, 506, 144944 CrossRef CAS
- Z. Zhang, M. Tang, Z.-T. Wang, Z. Ke, Y. Xia, K. T. Park, I. Lyubinetsky, Z. Dohnalek and Q. Ge, Imaging of formaldehyde adsorption and diffusion on TiO2(110), Top. Catal., 2015, 58, 103–113 CrossRef CAS
- Q. Zhou, C. Wang, Z. Fu, H. Zhang and Y. Tang, Adsorption of formaldehyde molecule on Al-doped vacancy-defected single-walled carbon nanotubes: a theoretical study, Comput. Mater. Sci., 2014, 82, 337–344 CrossRef CAS
- S. Mirza, M. Zulfiqar and S. Azam, Effect of hydrostatic pressure on electronic, elastic, and optical properties of hexagonal lanthanum oxide (La2O3): a first principles calculations, Phys. B, 2024, 676, 415686 CrossRef CAS
- S. Li, Y. Lin, Y. Wu, Y. Wu, X. Li and W. Tian, Ni doping significantly improves dielectric properties of La2O3 films, J. Alloys Compd., 2020, 822, 153469 CrossRef CAS
- D. Salinas, N. Escalona, G. Pecchi and J. L. G. Fierro, Fierro. Lanthanum oxide behavior in La2O3-Al2O3 and La2O3-ZrO2 catalysts with application in FAME production, Fuel, 2019, 253, 400–408 CrossRef CAS
- S. Li, G. Liu, S. Zhang, K. An, Z. Ma, L. Wang and Y. Liu, Cerium-modified Ni-La2O3/ZrO2 for CO2 methanation, J. Energy Chem., 2020, 43, 155–164 CrossRef
- M. Du, J. Zheng, L. Mei, Y. Zhang and C. Hou, Exploring the conversion mechanism of formaldehyde to CO2 and H2 catalyzed by bifunctional ruthenium catalysts: a DFT study, Mol. Catal., 2022, 530, 112630 CrossRef CAS
- C. Parks, K. Hughes and M. Pourkashanian, Furthering the understanding of product formation in monoethanolamine degradation: a mechanistic DFT study, Int. J. Greenhouse Gas Control, 2022, 119, 103732 CrossRef CAS
- J. Sun, W. Du, B. Xiao, Y. Wu, Y. Liu and T. Zhang, First-principles study of multiple-site substitutions of alloying elements in Ni-based single crystal superalloys, Sci. China: Technol. Sci., 2021, 64, 1276–1284 CrossRef CAS
- L. Liu and J. Zhao, Formaldehyde adsorption and decomposition on rutile (110): a first-principles study, Surf. Sci., 2016, 652, 156–162 CrossRef CAS
- V. Nagarajan and R. Chandiramouli, DFT investigation of formaldehyde adsorption characteristics on MgO nanotube, J. Inorg. Organomet. Polym., 2014, 24, 1038–1047 CrossRef CAS
- B. Li and H. Metiu, DFT studies of oxygen vacancies on undoped and doped La2O3 surfaces, J. Phys. Chem. C, 2010, 114, 12234–12244 CrossRef CAS
- L. Cong, Y. Zhao, S. Li and Y. Sun, Sr-doping effects on La2O3 catalyst for oxidative coupling of methane, Chin. J. Catal., 2017, 38, 899–907 CrossRef CAS
- V. Bannikov, I. Shein and A. Ivanovskii, Magnetic and electronic properties of nitrogen-doped lanthanum sesquioxide La2O3 as predicted from first principles, J. Supercond. Novel Magn., 2011, 24, 1693–1696 CrossRef CAS
- C. Liu, X. Fan and S. Ahmed, Structures and electronic properties of four crystal GeO2 and two rare-earth element oxides La2O3 and CeO2: first principles calculation, J. Theor. Comput. Chem., 2013, 5, 1350031 CrossRef
- Z. Wang, D. Wang and X. Gong, Strategies to improve the activity while maintaining the selectivity of oxidative coupling of methane at La2O3: a density functional theory study, ACS Catal., 2020, 10, 586–594 CrossRef CAS
- W. Cheng, W. Xia and H. Wan, Influence of surface reactivity of lanthanum oxide on the activation of methane and oxygen, Chem. J. Chin. Univ., 2019, 40, 940–949 CAS
- M. S. Palmer, M. Neurock and M. M. Olken, Periodic density functional theory study of the dissociative adsorption of molecular oxygen over La2O3, J. Phys. Chem. B, 2022, 106, 6543–6547 CrossRef
- S. Wang, S. Li and D. A. Dixon, Mechanism of selective and complete oxidation in La2O3-catalyzed oxidative coupling of methane reaction, Catal. Sci. Technol., 2020, 10, 2602–2614 RSC
- H. Gao, Y. Xiong, X. Liu, D. Zhao, Y. Feng, L. Wang and J. Wang, A first-principles study of oxygen adsorption on Ir(111) surface, Appl. Surf. Sci., 2016, 389, 211–215 CrossRef CAS
- J. Liu, F. Zhou, Q. Dai and H. Gao, Effect of Ca2+, Mg2+, Ba2+ and Sr2+ cations on calcium carbonate scaling formation in oil-gas well: based on density functional theory study and molecular dynamics simulation, J. Cryst. Growth, 2021, 563, 126089 CrossRef CAS
- M. Irfan, S. Azam and A. Iqbal, Proposal of new stable ABC2 type ternary semiconductor pnictides K3Cu3P2 and K3Ni3P2: first-principles calculations and prospects for thermosphysical and optoelectronic applications, Int. J. Energy Res., 2020, 45, 2980–2996 CrossRef
- A. Nadeem, A. I. Bashir, S. Azam, A. U. Rahman and M. A. Iqbal, First-principles quantum analysis on the role of V-doping on the tuning of electronic and optical properties of spinel oxides MnTi2O4, Mater. Sci. Eng., B, 2022, 278, 115643 CrossRef CAS
- S. Alnujaim, A. Bouhemadou, A. Bedjaoui, S. Bin-Omran, Y. Al-Douri, R. Khenata and S. Maabed, Ab initio prediction of the elastic, electronic and optical properties of a new family of diamond-like semiconductors, Li2HgMS4(M = Si, Ge and Sn), J. Alloys Compd., 2020, 843, 155991 CrossRef CAS
- Y. Zhang and Q. Hou, First-principle study on the effect of point defects on the mechanical properties, thermal conductivity, and optical properties of wurtzite AlN, Vacuum, 2023, 207, 111694 CrossRef CAS
- T. Hieu and T. Nguyen, Energy-loss function including damping and prediction of plasmon lifetime, J. Electron Spectrosc. Relat. Phenom., 2014, 193, 79–85 CrossRef
| This journal is © The Royal Society of Chemistry 2024 |