
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-spin s-triazine linked fluorenyl radical polymer†
Hoa Phan*a,
Tun Seng Herngb,
Hou Xudongc,
Linh Khanh Nguyena,
Vinh The Laa,
Chinh Dang Huynha,
Jun Dingb and
Jishan Wu *c
*c
aSchool of Chemistry and Life Science, Hanoi University of Science and Technology, Hanoi, Vietnam. E-mail: hoa.phanvan@hust.edu.vn
bDepartment of Materials Science and Engineering, National University of Singapore, 119260 Singapore, Singapore
cDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore, Singapore. E-mail: chmwuj@nus.edu.sg
First published on 24th May 2024
Abstract
The syntheses of high-spin organic polymers have been a daunting task due to the highly reactive nature of organic radicals, especially when they are ferromagnetically coupled. In this paper, we report our approach to obtain high-spin organic polymers, in which a reasonably stable fluorenyl radical was employed as the primary radical unit, and s-triazine serves as the connector that facilitates ferromagnetic coupling between them. Initially, the diamagnetic polymer precursor was synthesized by cyclotrimerization of a cyano-monomer. Subsequently, the high-spin polymers were obtained by oxidizing corresponding anionic polymers using O2 (6) or I2 (7). The temperature-dependent magnetic moments, and field-dependent magnetization data obtained from SQUID measurements revealed ferromagnetic couplings between primary radical units, with coupling J = 7.5 cm−1 and 38.6 cm−1. The percentages of primary unit in the radical form are 29%, and 47% for 6 and 7, respectively. Notably, this marks the first reported instance of a high-spin fluorenyl radical polymer exhibiting ferromagnetic coupling.
Introduction
Stable organic radicals are fascinating entities that have been extensively studied. These species possess electrons occupying relatively high energy SOMO orbitals, resulting in rich chemistry. Moreover, the unpaired electrons impart interesting, unique properties, giving rise to numerous potential applications in magnetism,1,7 rechargeable batteries,2 conductivity,3 spintronics,4 MRI, or switches.5 For these potential applications, organic radicals often are in clusters or 2D/3D structures, which serve to enhance the properties of the isolated radicals or introduce new properties due to the coupling between them. Amongst these, achieving ferromagnetic coupling between radicals remains one of the most challenging synthesis tasks, yet it promises to yield intriguing properties applicable to materials, such as high-spin (HS) clusters for magnetic resonance imaging contract agents, organic magnets, organic conductors, molecular switches, etc. These materials also contribute to understanding the nature of magnetic coupling between unpaired electrons within conjugated systems. One widely employed approach to obtain HS organic compounds involves connecting radicals at positions 1, 3, and 5 of benzene or positions 2, 4, and 6 of triazine, as ferromagnetic couplings through these units can be notably strong. Moreover, the chemistry for preparing compounds with radicals connecting through these positions offers a range of variable structures, from small molecules to highly complex.1,6–8 Various radical centers have been used, predominantly being arylmethyl radicals; however, a common issue with these radicals is the disruption of the magnetic coupling path due to defects, which are often present at the radical centers. Various strategies have been devised to address this issue, such as creating multiple magnetic coupling paths by preparing HS macrocycles and connecting them with more than one monoradical bridge.8–10 This, of cause, increases structural complexity and requires significant synthetic efforts. Alternatively, more stable radicals like nitrosyl radicals have been explored; however, their magnetic coupling through π-conjugated systems tends to be weak.11–13In this paper, we present our efforts to synthesize high-spin polymers utilizing fluorenyl radicals. Compared to arylmethyl radicals, fluorenyl radicals offer greater stability. Additionally, the magnetic coupling achieved through π-conjugated systems is expected to be significantly stronger compared to nitroxyl radicals.
The synthesis of the polymers involved several steps, beginning with the polymerization reactions of the –CN containing monomer, as outlined in Scheme 1.14 Compound 1 was synthesized by reacting 2,6-dibromofluoreone with the Grignard reagent, 2-mesitylmagnesium bromide, in THF at room temperature. Compound 2 was obtained through the reduction of 1 using triethylsilane in the presence of BF3·Et2O. The conversion of –Br to –CN to yield compound 3 was performed using microwave irradiation. Polymer 4 was prepared by utilizing the nitrile trimerization reaction to form s-triazine, with triflic acid catalyst added slowly through vapor diffusion at 60 °C under N2 for 18 hours. The resulting material was insoluble in common organic solvents or water. After neutralization with 2,6-lutidine, washing with acetonitrile, and thorough mechanical grinding followed by Soxhlet extraction with acetonitrile as the solvent for 12 hours, the final solid compound 4 was obtained, appearing pale blue.
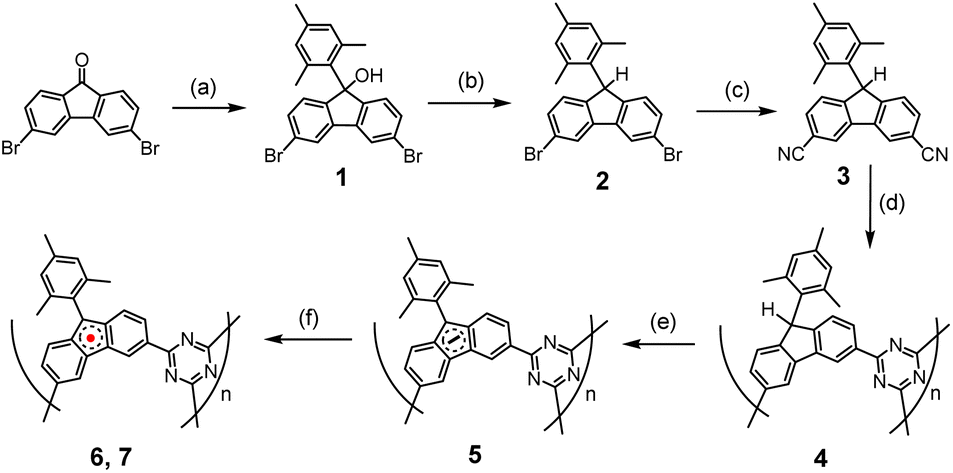 | ||
| Scheme 1 Synthetic route for preparation of 6 and 7.15 (a) n-BuLi, THF, −78 °C, 1 h; MesBr, THF, −78 °C to RT, 12 h. (b) Triethylsilane, BF3·Et2O, DCM, 0 °C, 30 min. (c) CuCN, Pd(OAc)2, Na2CO3, dimethylacetamide, 140 °C, 3.5 min, microwave. (d) Triflic acid vapor, 60 °C, 3 days. (e) t-BuOK, 55 °C, THF, 24 h. (f) Oxidizing agents (O2 or I2). | ||
Polymer 5 was formed by deprotonation at the 9-position of the fluorenyl moiety, using four equivalents of t-BuOK in dry THF heated at 60 °C for 36 hours under N2. The resulting solid was washed twice with dry THF, dried at 60 °C for 6 hours, then at 80 °C for two hours under high vacuum, yielding a black solid.
The final radical polymers, 6 and 7, were prepared using two different methods. Polymer 6 was obtained by adding excess O2 gas at room temperature to degassed 5 for 8 minutes, followed by removal of the remaining O2 under vacuum. Polymer 7 was formed by adding an I2 solution in THF (three equivalents of I2 to one equivalent fluorenyl anion monomer) to the suspension of 5 in THF at −78 °C, then stirring for 1 hour. The solvent with excessive I2 was removed at −78 °C, and the solid was washed with dry THF five times at −78 °C before warming up, drying under vacuum, and storing under N2 in a glovebox.
The formation of compounds 1, 2, and 3 was confirmed by 1H and 13C NMR spectra, as well as MALDI-TOF mass spectrometry (Fig. S1–S4 in the ESI†). The formation of 4 was characterized by IR spectra and X-ray photoelectron spectroscopy (XPS).
The IR spectra (Fig. 1) provide conclusive evidence of the formation of triazine from nitrile during the polymerization process. In 4, the characteristic vibration of the CN stretch of nitrile at 2225 cm−1 is significantly reduced in intensity compared to that of the monomer 3. Additionally, two new intense peaks appear at 1512 cm−1 and 1350 cm−1, corresponding to the CN in-plane and out-of-plane stretches, respectively, of the triazine moiety. The other peaks remain essentially unchanged, indicating the preservation of other molecular components from the monomer.
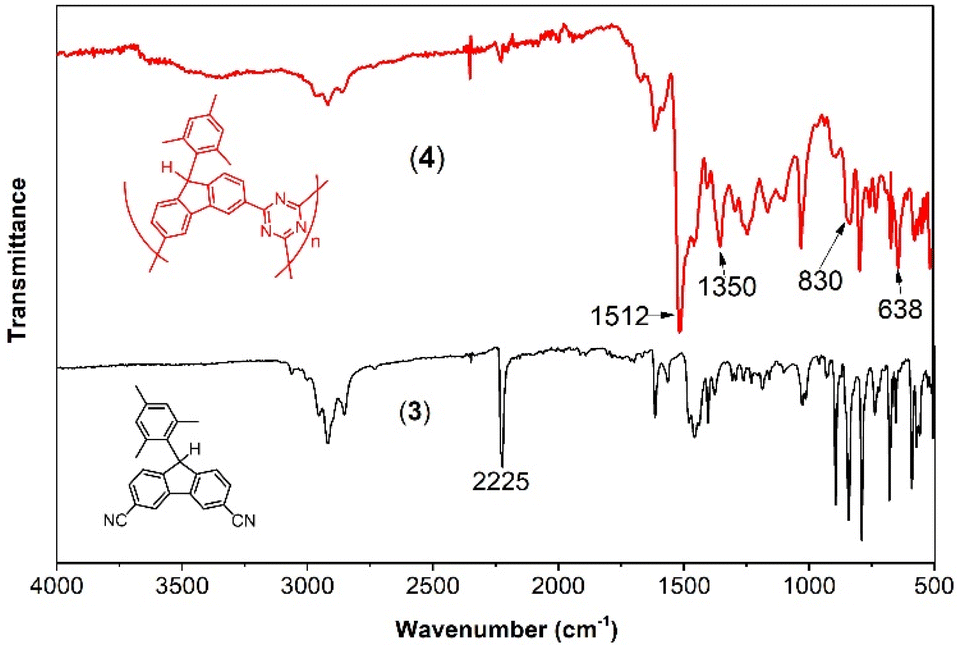 | ||
| Fig. 1 IR spectra of the monomer 3 (solid black bottom spectrum), and its polymerization product, 4 (solid red top spectrum). | ||
XPS curves for monomer 3, and the final products 6 and 7 are depicted in Fig. 2. The C 1s binding energies (in eV) for Csp3 (285.6, 285.5, and 285.7), Csp2–C (286.8, 286.8, and 286.8), and Csp1,2–N (288.2, 287.9, and 288.0) for monomer 3, 6, and 7, respectively, show no significant change. However, notable changes are observed in the N 1s binding energies. This binding energy for monomer 3 is 399.5 eV, while for 6 and 7, it is similar, at 400.2 eV, indicating the conversion of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N to s-triazine.7,8
N to s-triazine.7,8
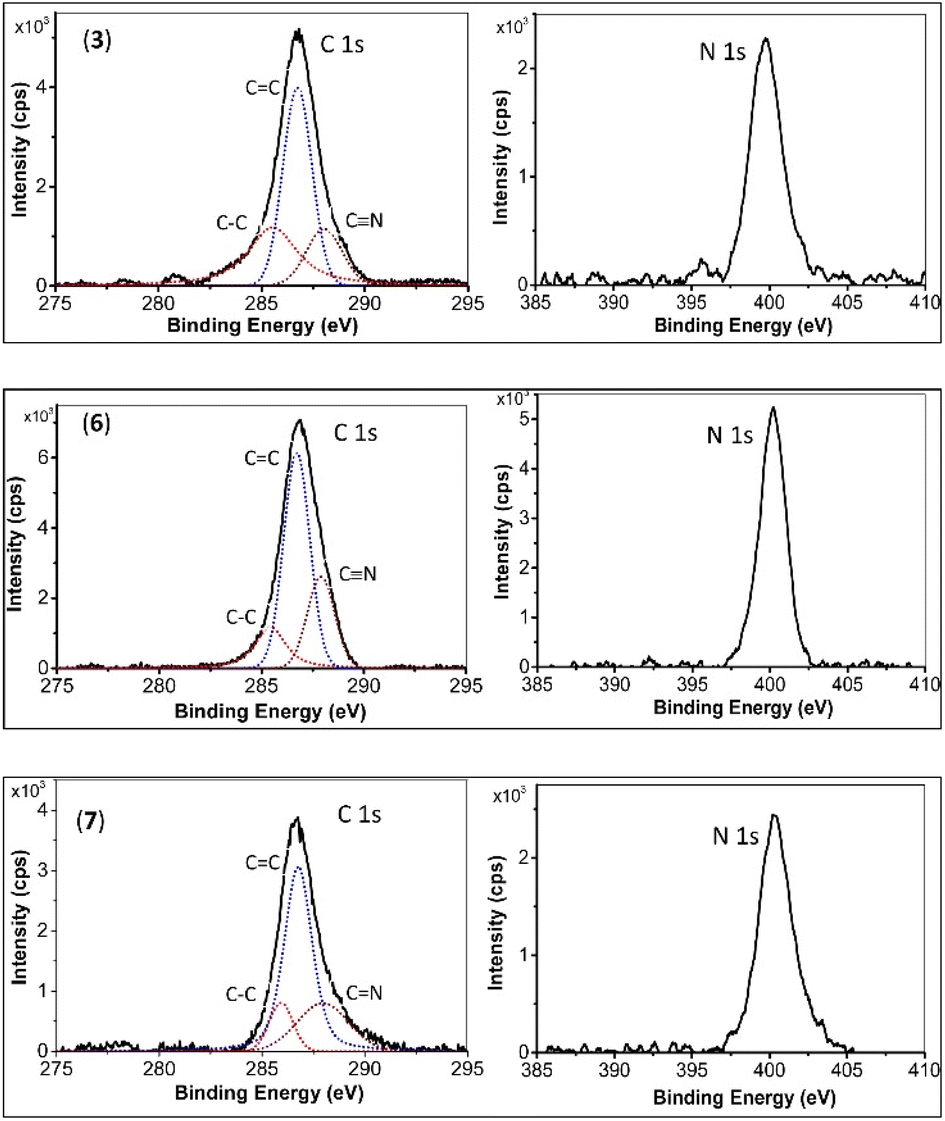 | ||
| Fig. 2 C 1s, N 1s binding energies for 3 (top spectra), 6 (middle spectra) and 7 (bottom spectra). | ||
To comprehend the nature and strength of magnetic coupling between two neighboring radicals connecting through the s-triazine moiety, quantum chemical calculations were executed. Initially, geometrical optimization of an oligomer comprising 21 monomers (depicted in Fig. 3) was performed using the Orca program, employing the B3LYP function and the 6-31G basis set. The rationale behind using such a large oligomer was to simulate similar steric effects experienced by monomers in a larger polymer, particularly focusing on the innermost layer highlighted in purple. These steric effects significantly influence the dihedral angles between s-triazine and radical monomers, thereby affecting the magnetic coupling strength between neighboring radicals. The dihedral angles observed were 17.50°, 13.73°, 13.67°, 13.25°, 12.68°, and 15.73°, with an average angle of 14.43°.
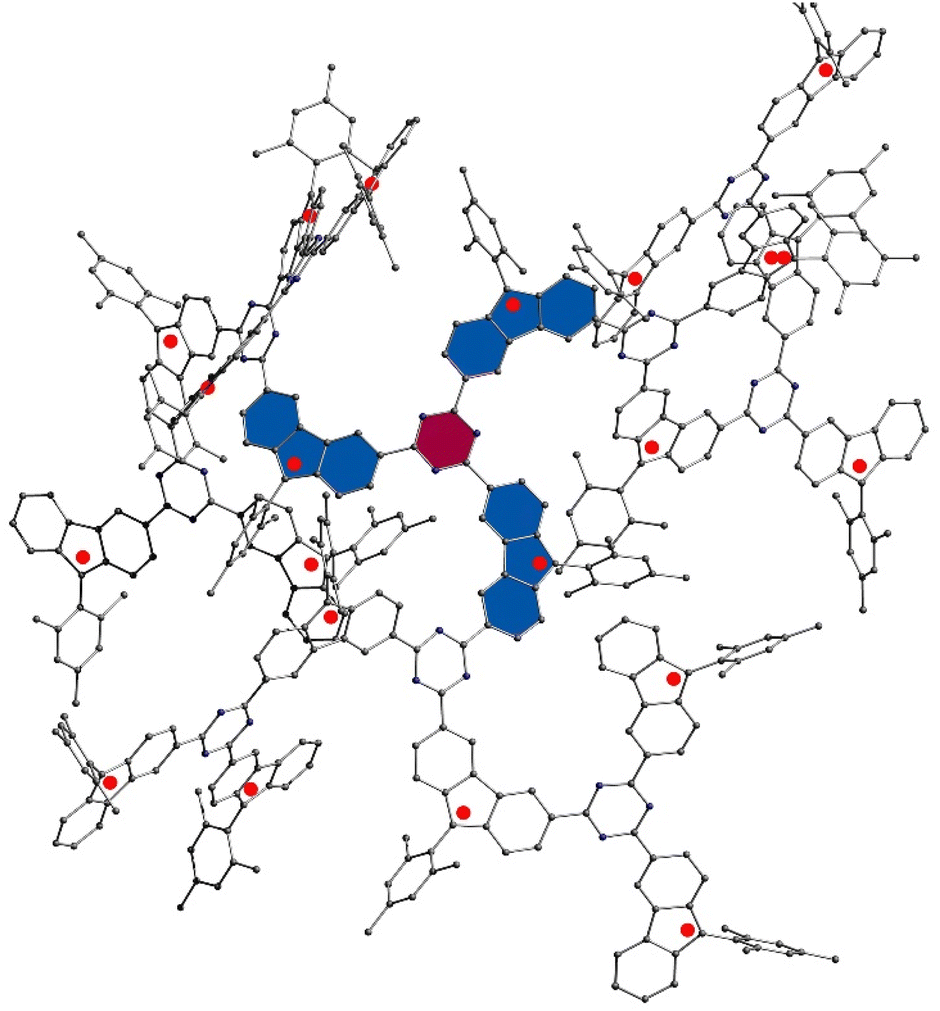 | ||
| Fig. 3 Optimized a structure with 21 primary radical units using B3LYP function and 6-31G basis set, on Orca program. | ||
Subsequently, this average dihedral angle was applied to a biradical model consisting of two fluorenyl radicals linked through the 2 and 4 positions of s-triazine. The singlet–triplet gap (ΔES–T) was determined using the Gaussian 09 package, employing the UCAM-B3LYP level of theory with the Pople basis set 6-31G*. The calculated ΔES–T gap value was found to be 0.15 kcal mol−1, corresponding to a magnetic coupling constant (J) of 26 cm−1 between the two radicals. The triplet state was identified as more stable, indicating a ferromagnetic coupling between the radicals. The calculated spin density, dihedral angles and bond length of some C–C bonds of the dimer in the triplet state is illustrated in Fig. S7 and S8.†
Even after thorough grinding and refluxing under boiling acetonitrile, the particle diameter remains in the range of hundreds of micrometers. To gain insight into the pore size for post-modification purposes, we conducted structural optimization on an oligomer consisting of 45 monomers in the radical form. The resulting pore size was approximately 9 Å (refer to Fig. S5†). Hence, t-BuOK is deemed a suitable deprotonation agent.
The ESR technique was employed to characterize the formation of organic radicals upon oxidation. To obtain compound 6, O2 gas was used as the oxidizing agent. Upon addition of O2 gas at room temperature, the ESR signal substantially jumped up (Fig. 4A). The intensities gradually peaked after 10 minutes before gradually decreasing. After 12 hours, the ESR signal almost disappeared. A g-value of 2.0026 obtained, indicating that the spin density predominantly located on carbon atoms (Fig. 4B). This finding is in good agreement with quantum chemical calculations (Fig. S6†). Notably, no reaction occurred at temperatures below −40 °C. At −20 °C or 0 °C, the reaction proceeded at a slower rate, with ESR intensities at their maxima also lower compared to those at room temperature. Compound 7 was obtained by oxidizing compound 5 with I2 in THF at −78 °C, and the nature of the ESR spectrum was essentially the same as that of compound 6 (Fig. S8†).
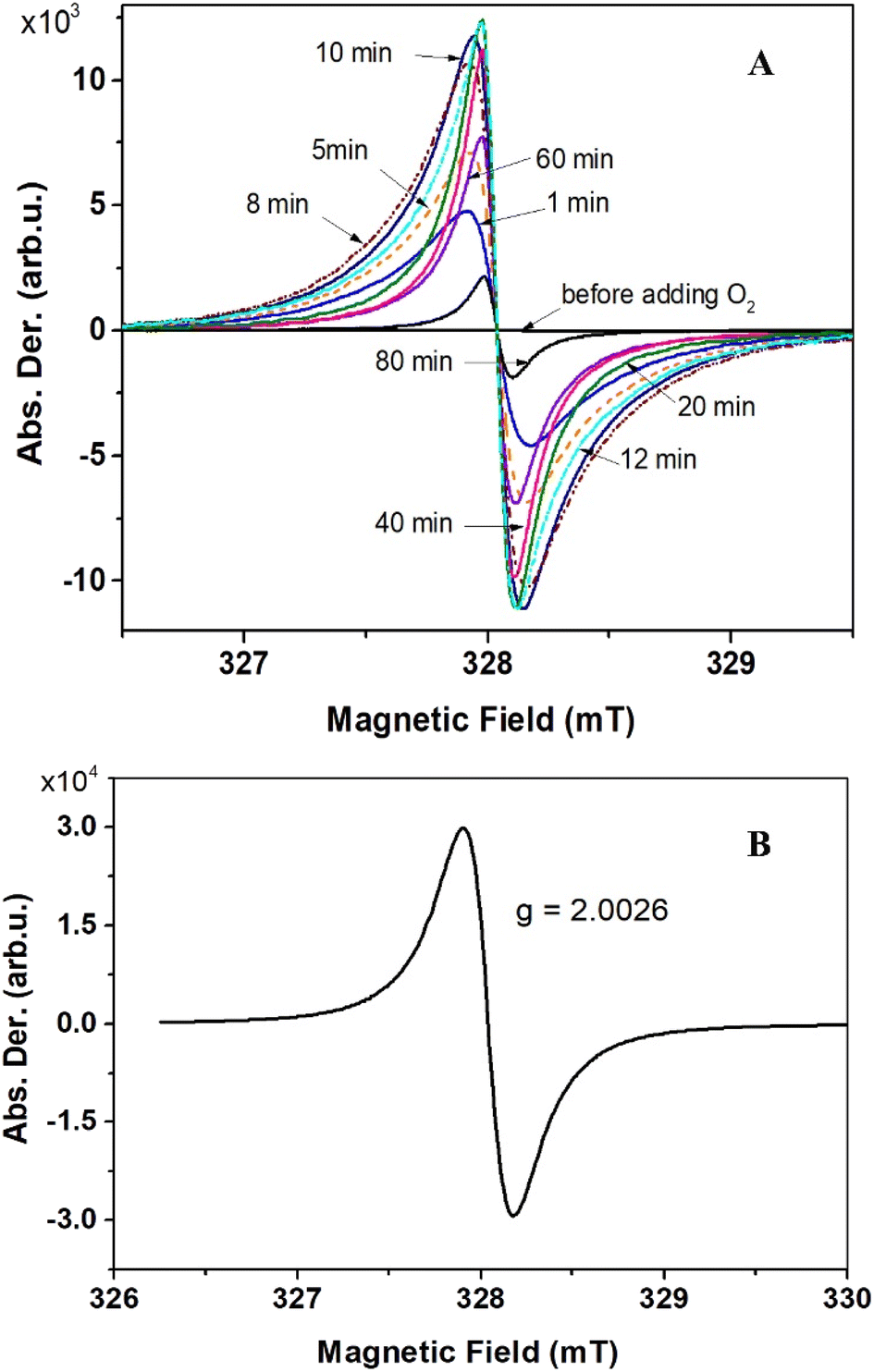 | ||
| Fig. 4 Evolution of the ESR spectra upon adding O2 to compound 5 (A). Solid state ESR spectrum of compound 6, recorded at ambient conditions (B). | ||
The magnetic properties of compounds 6 and 7 were measured using a Superconducting Quantum Interference Device (SQUID) magnetometer. In the case of compound 6, the values of the products of molar magnetic susceptibilities (calculated for one unit or equivalent of one monomer) and temperatures (χunitT) (as shown in Fig. 5A) increased as the temperature decreased. This behavior suggests the presence of ferromagnetic couplings between units (or monomers). However, at room temperature, the χunitT value was approximately 0.13 emu K mol−1, significantly smaller than the value expected for a spin-only uncoupled monoradical (0.375 emu K mol−1). This discrepancy indicates that a significant number of units (or monomers) may not be in the radical form, possibly due to decomposition processes.
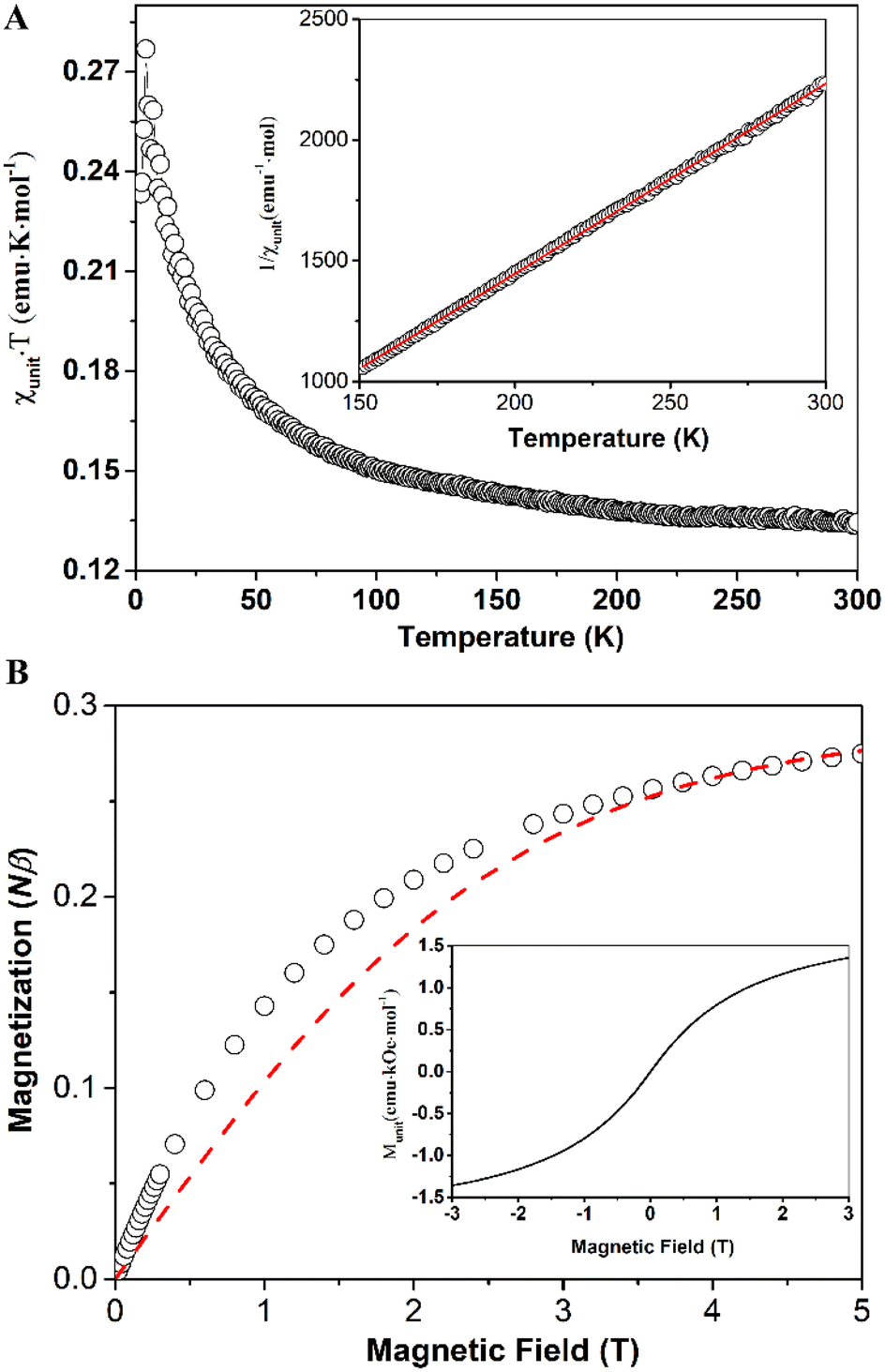 | ||
| Fig. 5 Temperature-dependent of the product of molar magnetic susceptibility and temperature of compound 6 (black open circles), the inset shows 1/χunit versus T (black open triangle) and the best fit using Curie–Weiss law (red solid line) (A); field-dependent magnetization of compound 6 measured at 2 K (open black circles) and fitting curve (red dash line) using Brillouin function for uncoupled, spin-only monoradical, S = 0.5, primary unit in radical form 29% (B), and full M–H curve at 2 K (the inset) (B). | ||
The χunitT reached its maximum value of 0.280 emu K mol−1 at 4.2 K, then decreased at lower temperatures due to zero-field splitting (ZFS) effects or weak antiferromagnetic couplings between radical units through spaces. The best fit using the Curie–Weiss law for the linear high-temperature part of the 1/χunit versus T curve (Fig. 5A, inset) yielded a magnetic coupling constant between two adjacent spins, J = 7.5 cm−1, with Curie constant, C = 0.127 emu K mol−1, and Weiss constant, θ = 16.19 K (see ESI† for details about the fitting). The magnetization curve, M–H, at 2 K is presented in Fig. 5B (open circles) and is fitted (red dashed line) using the Brillouin function for uncoupled, spin-only monoradical with S = 0.5. The magnetization of the sample rises faster at lower magnetic fields than the fitting curve indicates, suggesting ferromagnetic coupling between radical centers in the sample, consistent with our findings in the χunitT versus T curve. The percentage of units in the radical form is determined to be 29%, which agrees with the temperature-variable magnetic susceptibility measurements. The full M–H curve (Fig. 5B, inset) reveals no hysteresis, spontaneous magnetization, or soft permanent magnet behaviors, even at 2 K.
Compound 7 exhibits similar magnetic behaviors to compound 6. The χunitT products increase upon lowering temperature, indicating ferromagnetic couplings between radical centers. The χunitT value at room temperature is 0.23 emu K mol−1 (Fig. 6A), still smaller than that of an uncoupled, spin-only monoradical, but larger than that of compound 6. The χunitT values increase as the temperature decreases, reaching a maximum of 0.5 emu K mol−1 at 14 K, before decreasing at lower temperatures due to antiferromagnetic coupling or zero-field splitting effects. The maximum χunitT value is significantly higher than that of an uncoupled, spin-only monoradical. The field-dependent magnetization data at 2 K for compound 7 are similar to those of compound 6. The higher magnetization compared to the monoradical, S = 0.5, in the low magnetic field range implies ferromagnetic couplings between the radical centers. However, there is no spontaneous magnetization or magnetic order even at 2 K, as indicated by the field-dependent magnetic moment curve (Fig. 6B, inset).
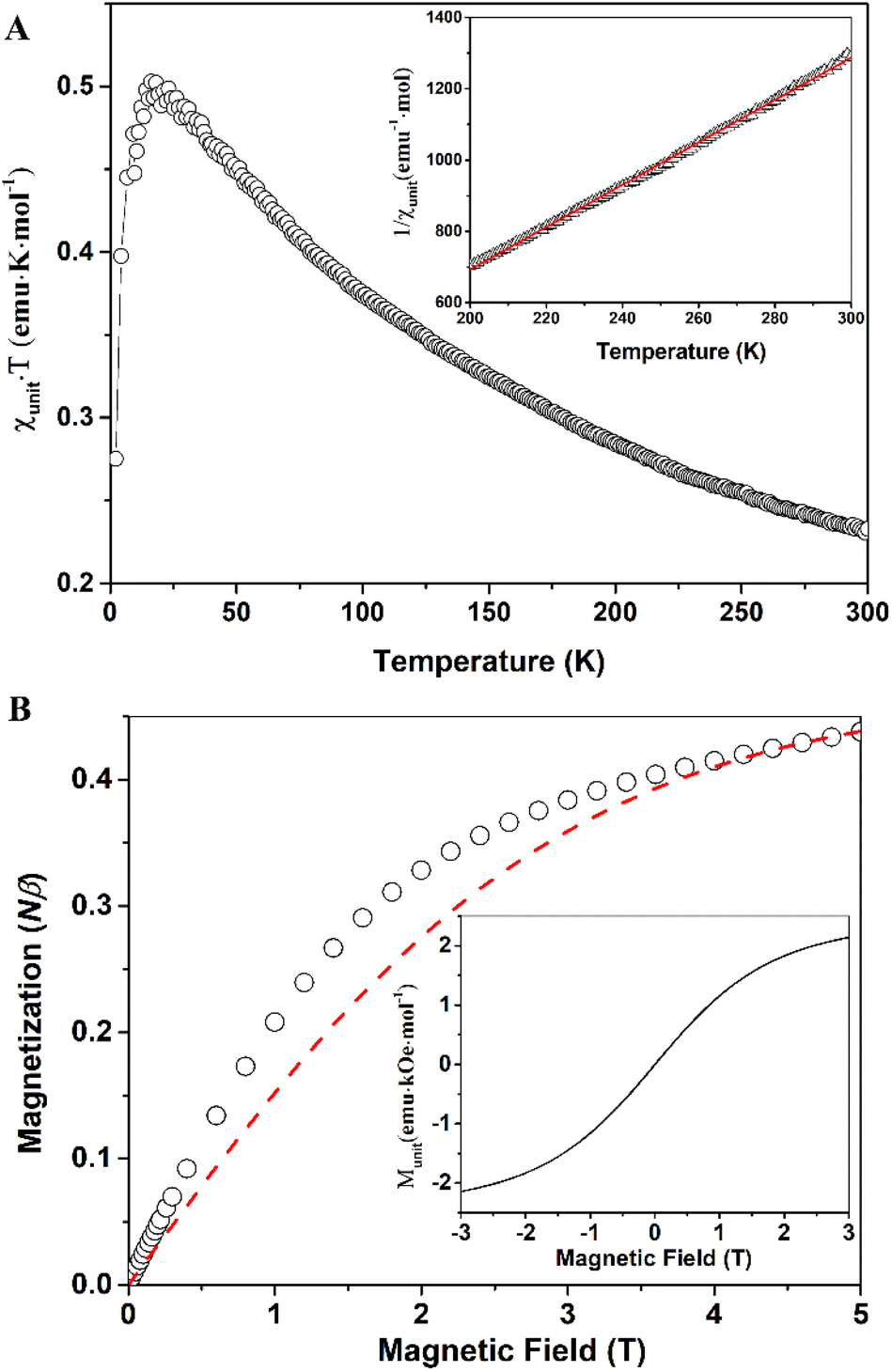 | ||
| Fig. 6 Temperature-dependent of the χunitT products of sample 7 (black open circles), the inset shows 1/χunit versus T (black open triangle), and the best fit using Curie–Weiss law (red solid line) (A); field-dependent magnetization of compound 7 measured at 2 K (open black circles) and fitting curve (red dash line) using Brillouin function for uncoupled, spin-only monoradical, S = 0.5, primary unit in radical form 47% (B), and full M–H curve at 2 K (the inset) (B). | ||
The higher room temperature χunitT value of compound 7 suggests that it has a higher percentage of monomers in the radical form than that of compound 6. Indeed, the percentage of units in the radical form is estimated to be 47% from fitting the field-dependent magnetization of compound 7 at 2 K using Brillouin function for uncoupled, spin-only monoradical, S = 0.5 (Fig. 6B). The magnetic coupling constant between two adjacent spins, J = 38.6 cm−1, was determined by fitting using the Curie–Weiss law for the linear high-temperature part of the 1/χunit versus T curve (Fig. 6A, inset), with a Curie constant, C = 0.167 emu K mol−1, and Weiss constant, θ = 83.53 K. This magnetic coupling value is in good agreement with the DFT calculation data. The larger magnetic coupling J in compound 7 compared to that in 6 can be attributed to the experimental J being an average value. In the ideal scenario, where two radicals connect through only an s-triazine moiety, the largest J value is obtained. However, if two adjacent radicals are chemically bridged through one or more monomer units that are not in the radical form (along with s-triazine), then the J value is significantly reduced. Therefore, the higher the percentage of monomer units in the radical form, the larger the J value becomes.
Conclusions
In summary, we have successfully synthesized high-spin organic polymers based on fluorenyl radicals linked by s-triazine. The trimerization reaction of –CN to form s-triazine was utilized to achieve polymers with a high degree of polymerization. The final radical forms of these polymers were obtained by oxidizing their respective anion forms using O2 gas (6) or I2 in THF (7). Notably, the radical form of compound 7 exhibits a higher percentage of fluorenyl units (47%) compared to compound 6 (29%). The magnetic measurements revealed ferromagnetic couplings between adjacent radicals, with magnetic coupling values of J = 7.5 cm−1 for 6 and 38.6 cm−1 for 7, respectively. These values align well with calculated data regarding the nature and magnitude of the magnetic coupling. The field-dependent magnetization at 2 K for both samples 6 and 7 shows similar behavior, with the increase in magnetization surpassing the expected curves fitted with S = 0.5 in the lower field range, further suggesting a ferromagnetic coupling between radical centers. To the best of our knowledge, these are the first reported high-spin fluorenyl-based radical polymers.Data availability
Data supporting this article has been provided as ESI.†Author contributions
The ideas were initiated by H. P. and J. W. Experiments were conducted and processed by H. P. with contributions from J. W. SQUID data were collected by T. S. H., J. D. and processed by H. P. Computations in the manuscript were performed by H. P. and H. X. The manuscript was written by H. P., J. W., with assistance from L. K. N., and with contributions from all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. W. acknowledges financial support from NRF Investigatorship (NRF-NRFI05-2019-0005), and H. P. acknowledges financial support from Vingroup Innovation Foundation (VINIF) (VINIF.2022.DA00137).References
- (a) J. S. Miller, Mater. Today, 2014, 17, 224–235 CrossRef CAS; (b) J. S. Miller, A. J. Epstein and W. M. Reiff, Science, 1988, 240, 40–47 CrossRef CAS PubMed; (c) J. S. Miller, A. J. Epstein and W. M. Reiff, Chem. Rev., 1988, 88, 201–220 CrossRef CAS; (d) B. Kahr, D. V. Van Engen and P. Gopalan, Chem. Mater., 1993, 5, 729–732 CrossRef CAS; (e) R. Feng, L. Zhang, H. Ruan, Y. Zhao, G. Tan and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6084–6088 CrossRef CAS PubMed; (f) J. Mahmood, J. Park, D. Shin, H.-J. Choi, J.-M. Seo, J.-W. Yoo and J.-B. Baek, Chem, 2018, 4, 2357–2369 CrossRef CAS; (g) Y. Jiang, I. Oh, S. H. Joo, O. Buyukcakir, X. Chen, S. H. Lee, M. Huang, W. K. Seong, J. H. Kim and J. U. Rohde, ACS Nano, 2019, 13, 5251–5258 CrossRef CAS PubMed.
- (a) E. P. Tomlinson, M. E. Hay and B. W. Boudouris, Macromolecules, 2014, 47, 6145–6158 CrossRef CAS; (b) C. Friebe and U. S. Schubert, Top. Curr. Chem., 2019, 375, 19 CrossRef PubMed; (c) T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed; (d) Y. Liu, M. A. Goulet, L. Tong, Y. Liu, Y. Ji, L. Wu, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, Chem, 2019, 5, 1861–1870 CrossRef CAS.
- (a) S. Dongmin Kang and G. Jeffrey Snyder, Nat. Mater., 2017, 16, 252–257 CrossRef CAS PubMed; (b) S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed; (c) T. L. D. Dexter Tam, C. K. Ng, S. L. Lim, E. Yildirim, J. Ko, W. L. Leong, S.-W. Yang and J. Xu, Chem. Mater., 2019, 31, 8543–8550 CrossRef CAS; (d) H. Gao, C. Ge, B. Hou, H. Xin and X. Gao, ACS Macro Lett., 2019, 8, 1360–1364 CrossRef CAS PubMed.
- (a) T. Quintes, M. Mayländer and S. Richert, Nat. Rev. Chem, 2023, 7, 75–90 CrossRef PubMed; (b) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 303–349 RSC; (c) S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC; (d) T. Sugawara, H. Komatsu and K. Suzuki, Chem. Soc. Rev., 2011, 40, 3105–3118 RSC; (e) M. Murrie and D. J. Price, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2007, 103, 20–38 RSC; (f) D. Pinkowicz, S. Chorąży and O. Stefańczyk, Sci. Prog., 2011, 94, 139–183 CrossRef CAS PubMed.
- (a) M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443 CrossRef CAS PubMed; (b) W. Fujita and K. Awaga, Science, 1999, 286, 261 CrossRef CAS PubMed; (c) H. Phan, K. Lekin, S. M. Winter, R. T. Oakley and M. Shatruk, J. Am. Chem. Soc., 2013, 135(42), 15674–15677 CrossRef CAS PubMed; (d) K. Lekin, H. Phan, S. M. Winter, J. WL Wong, A. A. Leitch, D. Laniel, W. Yong, R. A. Secco, J. S. Tse, S. Desgreniers, P. A. Dube, M. Shatruk and R. T. Oakley, J. Am. Chem. Soc., 2014, 136(22), 8050–8062 CrossRef CAS PubMed.
- H. Iwamura, Polyhedron, 2013, 66, 3–14 CrossRef CAS.
- H. Phan, T. S. Herng, D. Wang, X. Li, W. Zeng, J. Ding, K. P. Loh, A. T. Wee and J. Wu, Chem, 2019, 5, 1223–1234 CAS.
- (a) Y. Teki, T. Takui, K. Itoh, H. Iwamura and K. Kobayashi, J. Am. Chem. Soc., 1983, 105, 3722–3723 CrossRef CAS; (b) Y. Teki, T. Takui, K. Itoh, H. Iwamura and K. Kobayashi, J. Am. Chem. Soc., 1986, 108, 2147–2156 CrossRef CAS PubMed.
- (a) A. Rajca, J. Wongsriratanakul and S. Rajca, J. Am. Chem. Soc., 1997, 199, 11674–11686 CrossRef; (b) A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed; (c) A. E. London, H. Chen, M. A. Sabuj, J. Tropp, M. Saghayezhian, N. Eedugurala, B. A. Zhang, Y. Liu, X. Gu, B. M. Wong, N. Rai, M. K. Bowman and J. D. Azoulay, Sci. Adv., 2019, 5, eaav2336 CrossRef CAS PubMed; (d) X. X. Chen, J. T. Li, Y. H. Fang, X. Y. Deng, X. Q. Wang, G. Liu, Y. Wang, X. Gu, S. D. Jiang and T. Lei, Nat. Commun., 2022, 13, 2258 CrossRef CAS PubMed; (e) L. Skorka, P. Kurzep, T. Chauviré, L. Dubois, J. M. Mouesca, V. Maurel and I. Kulszewicz-Bajer, J. Phys. Chem. B, 2017, 121(16), 4293–4298 CrossRef CAS PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- S. Wu, M. Li, H. Phan, D. Wang, T. S. Herng, J. Ding, Z. Lu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8007–8011 CrossRef CAS PubMed.
- (a) K. Irie, K. Shibayama, M. Mito, S. Takagi, M. Ishizuka, K. Lekin and R. T. Oakley, Phys. Rev. B, 2019, 99, 014417 CrossRef CAS; (b) G. Li, H. Phan, T. S. Herng, T. Y Gopalakrishna, C. Liu, W. Zeng, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 129(18), 5094–5098 CrossRef.
- M. Tamura, Y. Nakazawa, D. Shiomi, K. Nozawa, Y. Hosokoshi, M. Ishikawa, M. Takahashi and M. Kinoshita, Chem. Phys. Lett., 1991, 186, 401–404 CrossRef CAS.
- (a) X. Lu, S. Lee, J. Kim, T. Gopalakrishna, H. Phan, T. Herng, Z. Lim, Z. Zeng, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2016, 138, 13048–13058 CrossRef CAS PubMed; (b) H. Zhang, H. Phan, T. S. Herng, T. Y. Gopalakrishna, W. Zeng, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 13484–13488 CrossRef CAS PubMed.
- (a) S. A. Weissman, D. Zewge and C. Chen, J. Org. Chem., 2005, 70, 1508–1510 CrossRef CAS PubMed; (b) D. A. Safin, J. M. Frost and M. Murugesu, Dalton Trans., 2015, 44, 20287–20294 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of ESR, SQUID measurements, and relevant 1H, 13C NMR, MALDI-TOP, DFT calculation data. See DOI: https://doi.org/10.1039/d4ra03034f |
| This journal is © The Royal Society of Chemistry 2024 |