
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in situ phase transformation induced mesoporous heterointerface for the alkaline hydrogen evolution reaction†
Zhiping Lin‡
abd,
Zongpeng Wang‡c,
Tianchen Jina,
Ting Jianga,
Longfei Dinga,
Shijie Shen a,
Jitang Zhang*ade and
Wenwu Zhong*a
a,
Jitang Zhang*ade and
Wenwu Zhong*a
aSchool of Material Science and Engineering, Taizhou University, No. 1139, Shifu Road, Taizhou, 318000, China. E-mail: zhangjt@tzc.edu.cn; zhongww@tzc.edu.cn
bSchool of Chemical Engineering and Technology, China University of Mining and Technology, No. 1, Daxue Road, Xuzhou, 221116, China
cSchool of Material Science and Engineering, Central South University, No. 932, Lushan South Road, Changsha, 410083, China
dERA Co, Ltd, No. 1118, Huangjiao Road, Taizhou, 318020, China
eCollege of Chemical and Biological Engineering, Zhejiang University, No. 866, Yuhangtang Road, Hangzhou, 310058, China
First published on 7th May 2024
Abstract
The phase structure of a catalyst plays a crucial role in determining the catalytic activity. In this study, a facile phosphorization process is employed to achieve the in situ phase transformation from single-phase Co3O4 to CoO/CoP hybrid phases. Characterization techniques, including XRD, BET, SEM, and TEM, confirm the retention of the mesoporous nature during the phase transformation, forming porous CoO/CoP heterointerfaces. Strong charge transfer is observed across the CoO/CoP heterointerface, indicating a robust interaction between the hybrid phases. The CoO/CoP hybrid exhibits significantly enhanced catalytic activity for the alkaline hydrogen evolution reaction (HER) compared to pristine Co3O4. Density Functional Theory (DFT) calculations reveal that the elimination of the band gap in the spin-down band of Co in CoO/CoP contributes to the observed high HER activity. The findings highlight the potential of CoO/CoP hybrids as efficient catalysts for HER, and contribute to the advancement of catalyst design for sustainable energy applications.
1. Introduction
Hydrogen is considered a clean and versatile energy carrier,1–5 and its production through water electrolysis, driven by renewable energy sources, offers a promising solution to reduce our reliance on fossil fuels and mitigate environmental impact.6–14 The hydrogen evolution reaction (HER) is a crucial process in the field of hydrogen production, which takes place at the cathode, where protons form hydrogen gas. HER can occur in both acidic and alkaline environments. Alkaline conditions are considered more environmentally friendly due to the avoidance of corrosive acidic electrolytes.15–22 Furthermore, the other half reaction of water splitting, the oxygen evolution reaction (OER) exhibits more favorable kinetics in alkali than in acid.23–29 Thus, alkaline HER can enable efficient, cost-effective, and environmentally friendly hydrogen production. Efficient and cost-effective catalysts are crucial for accelerating the HER and improving the overall efficiency of hydrogen production. Noble metals such as platinum are effective catalysts but are expensive.30–34 Considerable efforts have been dedicated to working on developing alternative, non-precious metal catalysts, including various metal oxides,35–37 sulfides,38–40 phosphides41–43 and so on.Cobalt oxides have recently garnered attention for their potential applications in water splitting.44–48 Several factors contribute to their attractiveness for large-scale use: firstly, cobalt is abundant on Earth, enhancing the feasibility of widespread applications. Secondly, the variable oxidation states of cobalt in cobalt oxides allow for the acceptance and donation of electrons during the reaction, resulting in a unique electronic structure that can be finely tuned. Strategies such as the incorporation of nitrogen or phosphorus have been explored to enhance overall catalytic activity. Thirdly, cobalt oxides are usually effective involving the participation of hydroxide ions, which is essential in alkaline HER. However, despite their catalytic properties being extensively studied for OER, the catalytic activity of cobalt oxides for the HER remains relatively low and less explored. Considering the mentioned advantages, the development of highly efficient HER catalysts based on cobalt oxides is of paramount importance. Nevertheless, this pursuit faces challenges in achieving the high efficiency of cobalt-oxides-based catalysts, understanding the underlying reaction mechanisms, and ensuring sustained long-term performance. Addressing these challenges will be crucial for advancing the practical applications of cobalt oxides in the field of water splitting. Meanwhile, cobalt phosphide (CoP) has also emerged as a promising catalyst, due to its favorable catalytic activity, abundance, and versatility of modulation. CoP samples with various morphologies such as nanorod,49 nanoflower,50 nanotube51 and others have been demonstrated with excellent HER activities. CoP has also been doped with N,52 Cu,53 W54 and other elements to achieve better performance. Besides those strategies, forming heterointerface has been recognized as a promising method to boost the catalytic activity by introducing interaction between the two phases side by side.55–58 Normally, strong charge transfer and build-in field can be observed across the heterointerface, which will regulate the electron properties and optimize the adsorptive features of the interface, thus realizing advanced catalytic activity. However, the construction of heterointerfaces by sequentially overlaying one phase onto another often results in insufficient contact and mechanical stability. The conversion of one individual phase into hybrid phases simultaneously to fabricate in situ heterointerfaces remains a challenge.
In this work, we have successfully utilized a facile phosphorization process to achieve the in situ transformation of a single-phase Co3O4 material into CoO/CoP hybrid phases. Notably, the mesoporous nature of Co3O4 is preserved throughout the phase transformation, leading to the formation of porous CoO/CoP heterointerfaces. The observed strong charge transfer across these heterointerfaces indicates a robust interaction between the hybrid phases. Importantly, CoO/CoP exhibits significantly enhanced catalytic activity for the hydrogen evolution reaction (HER) compared to pristine Co3O4. DFT calculations provide valuable insights into the observed high HER activity, revealing that the elimination of the band gap in the spin-down band of Co in CoO/CoP plays a crucial role in the improved catalytic performance. This work highlights the potential of CoO/CoP hybrids as efficient catalysts for HER.
2. Experiments
2.1 Synthesis of Co3O4
Mesoporous Co3O4 was synthesized utilizing the two-solvent method,59 employing SBA-15 as template. The typical fabrication procedure is outlined as follows: 40 mL of n-hexane was introduced into a beaker, followed by the addition of 0.8 g of purchased SBA-15. After thorough stirring at room temperature, a measured quantity of Co(NO3)2 solution was incrementally added. The resulting mixture was stirred continuously for 20 hours, followed by ambient temperature washing and drying. Subsequently, the obtained powder was heated in a muffle furnace to 300 °C, maintaining this temperature for 5 hours. Multiple etching steps with NaOH solution, were employed to remove the silica template. After thorough cleaning and drying, mesoporous Co3O4 was successfully obtained.2.2 Synthesis of CoO/CoP
The CoO/CoP was prepared following a phosphorization procedure as shown in Fig. S1.† In an exemplary process, 30 mg mesoporous Co3O4 were placed into a quartz boat within a tubular furnace, while another quartz boat containing 600 mg NaH2PO2·H2O was positioned in the same furnace. In the tube, Co3O4 was put at the downstream side, while NaH2PO2·H2O at the upstream side. The tubular furnace was gradually heated to 350 °C at a rate of 5 °C min−1, and after maintaining this temperature for one hour, it was slowly cooled to room temperature. This controlled thermal treatment resulted in the formation of CoO/CoP.2.3 Characterization methods
X-ray diffraction (XRD) analysis was undertaken utilizing a Bruker-D8 Advance instrument equipped with Cu Kα radiation. Scanning electron microscopy (SEM) images were acquired using a Hitachi S-4800 apparatus. Transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and selected area electron diffraction (SAED) measurements were carried out employing the FEI Tecnai G2 F20. Energy-dispersive X-ray spectroscopy (EDS) analysis was conducted using the Super-X EDS system. X-ray photoelectron spectroscopy (XPS) spectra were obtained utilizing the Thermo Scientific K-Alpha, with an Al Kα radiation source (hν = 1486.6 eV). Brunauer–Emmett–Teller (BET) measurements were performed on the Micromeritics ASAP 2460 3.01.2.4 Electrochemical measurements
The electrochemical assessments were executed employing a CHI 660E electrochemical workstation, employing a standard three-electrode configuration at room temperature. Specifically, a glass carbon electrode served as the substrate for the catalysts and functioned as the working electrode. A graphite rod was utilized as the counter electrode, while the Ag/AgCl electrode was employed as the reference electrode in a 1 M KOH electrolyte. To fabricate the working electrode, a composite mixture of 5 mg catalyst, 750 μL alcohol, 250 μL isopropanol, and 50 μL Nafion solution (5 wt%) was thoroughly homogenized through ultrasonic treatment for a duration of half an hour. Subsequently, 5 μL of the thus-prepared suspension was deposited onto the meticulously cleaned surface of the glass carbon electrode. Following natural drying, the resulting working electrode was rendered suitable for implementation in subsequent electrochemical tests.The hydrogen evolution reaction (HER) efficacy of the respective catalysts was assessed in 1 M KOH solutions. Linear sweep voltammetry (LSV) curves were recorded at a sweep rate of 5 mV s−1 with iR correction. Tafel slopes were determined from the acquired LSV curves. Cyclic voltammetry (CV) measurements spanned the range of 0.11–0.21 V relative to the reversible hydrogen electrode, with the sweep speed incrementally adjusted from 20 to 200 mV s−1. Electrochemical impedance spectroscopy was conducted under a constant overpotential of 120 mV, covering a frequency range from 100 kHz to 0.1 Hz.
3. Results and discussion
The crystal structure of the samples was investigated through X-ray diffraction (XRD), as depicted in Fig. 1a. The Co3O4 sample exhibits diffraction peaks at 31.3, 36.9, 38.5, 44.8, 55.7, 59.4, and 65.2°, aligning with the standard PDF card of Co3O4 (No. 42-1467). No other diffraction peaks are observable, attesting to the high phase purity of the as-prepared Co3O4. Specifically, the identified diffraction peaks correspond to the (220), (311), (222), (400), (422), (511), and (440) crystal facets of Co3O4, respectively. In the case of the CoO/CoP sample, two distinct sets of diffraction peaks were discerned. The set at 36.5, 42.4, and 61.5° aligns with the standard PDF card of CoO (No. 48-1719), corresponding to the (111), (200), and (220) crystal facets, respectively. Conversely, the second set at 31.6, 32, 38.9, 46.2, 48.1, 48.4, 52.3, 56.4, and 56.8° matches with the standard PDF card of CoP (No. 29-0497), corresponding to the (011), (002), (200), (112), (211), (202), (103), (020), and (301) facets of CoP, respectively. Notably, the absence of prominent Co3O4 peaks indicates the thoroughness of the phosphorization process. The XRD results suggest an in situ transformation of Co3O4 into CoO/CoP compounds following phosphorization.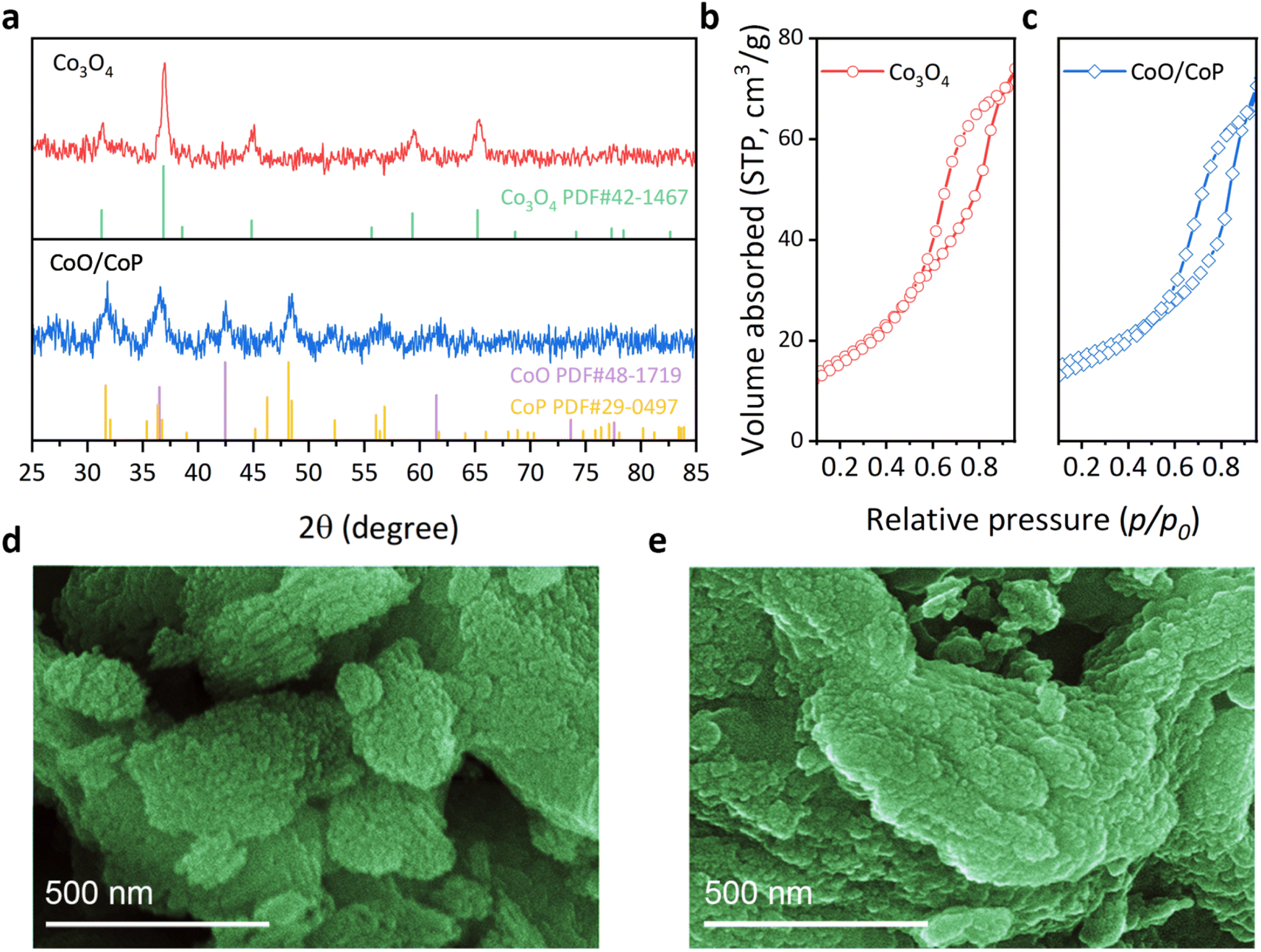 | ||
| Fig. 1 (a) XRD patterns of Co3O4 (red) and CoO/CoP (blue), respectively. (b and c) BET measurements of Co3O4 (red) and CoO/CoP (blue), respectively. (d and e) SEM photos of Co3O4 and CoO/CoP, respectively. | ||
The porous nature of the as-prepared Co3O4 samples were revealed through the Brunauer–Emmett–Teller (BET) method, as depicted in Fig. 1b and c. Co3O4 exhibits a specific surface area of 59 m2 g−1, while 42.2 m2 g−1 for CoO/CoP, suggesting only some portion of porous channels are destructed after phosphorization. The morphology of the synthesized samples was scrutinized through scanning electron microscopy (SEM) images, as depicted in Fig. 1d, e and S2.† The pristine Co3O4 manifests a morphology comprised of stacked flakes, with an outline size ranging from 1 to 4 μm. A congruent morphology has been previously documented in studies utilizing the SBA-15 as templates.60,61 Upon phosphorization, CoO/CoP retains a similar morphology, albeit with more dispersed exfoliated small flakes and softer edges. These observations suggest the preservation of the structural framework during the in situ phase transformation. It is noteworthy that the SEM images do not reveal mesoporous channels, attributable to the diminutive pore size of ∼7 nm.
To further explore the mesoporous channels and the crystal structure of the synthesized CoO/CoP, transmission electron microscopy (TEM) characterizations were conducted. The scanning transmission electron microscopy (STEM) image of CoO/CoP is presented in Fig. 2a, while TEM images at various magnifications are displayed in Fig. 2b and c. Both STEM and TEM images distinctly exhibit mesoporous channel structures, originated from the removal of the SBA-15 template. Analogous mesoporous structures have been documented in prior studies.62–65 To investigate the phase transformation resulted from phosphorization, high-resolution TEM images were obtained. As depicted in Fig. 2d, a well-defined CoO/CoP heterointerface is formed in CoO/CoP. The identified lattice spacings of 2.54 and 2.44 Å (indicated by blue and red lines, respectively) correspond to the (200) and (111) crystal planes of CoP and CoO, respectively. To provide additional evidence for the existence of the CoO/CoP heterointerface, selected area electron diffraction (SAED) was performed. The SAED rings in Fig. 2e reveal the coexistence and polycrystalline nature of CoO/CoP. The specified distances of 2.85, 1.87, and 1.66 Å align with the spacings of (011), (211), and (020) crystal facets in CoP, while the 2.44 Å distance matches the (111) facet spacing in CoO. Energy-dispersive X-ray spectroscopy (EDS) element mapping was also employed to investigate the element distribution in CoO/CoP, as shown in Fig. 2f. It is evident that the Co and O elements are homogeneously distributed throughout the sample. However, the distribution of the P element exhibits noticeable segregation, serving as additional compelling evidence for the CoO/CoP heterointerface. One may speculate that O should distribute separately from P, by simply examining the chemical denotation of “CoO/CoP”. The absence of O segregation in the EDS mapping is attributed to the well-known adsorption tendency of O species on CoP, which is evidenced below. These analyses, in conjunction with the XRD, BET, SEM, and TEM results, unequivocally affirm the transformation of mesoporous Co3O4 into mesoporous CoO/CoP heterointerface material following phosphorization.
 | ||
| Fig. 2 (a) STEM photo of CoO/CoP. (b and c) TEM photos of CoO/CoP at different magnifications. (d) HRTEM photo of CoO/CoP. (e) SAED pattern of CoO/CoP. (f) Element mapping of CoO/CoP. | ||
The chemical state is a crucial parameter in determining the catalytic activity of an electrocatalyst. The full X-ray photoelectron spectroscopy (XPS) survey of the synthesized samples is presented in Fig. S4.† All XPS spectra are aligned using the C 1s peak at 284.8 eV (Fig. S3†). For Co3O4, discernible peaks are observed only for Co, O, and C elements. In the case of CoO/CoP, in addition to the aforementioned elements, an anticipated P element is also evident. The absence of other elemental peaks attests to the high purity of the samples. The high-resolution XPS spectra of the Co 2p orbital for Co3O4 and CoO/CoP are illustrated in Fig. 3a and b, respectively. In the case of Co3O4, two sets of doublets are deconvoluted. The peaks at 780.0 and 795.2 eV pertain to the Co3+ 2p3/2 and 2p1/2 orbitals, respectively.66,67 A second set of peaks at 781.2 and 796.6 eV corresponds to the Co2+ 2p3/2 and 2p1/2 orbitals, respectively. The intensity ratio of Co3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co2+ is determined to be 1.92:1, closely mirroring the nominal ratio for Co3O4. Conversely, in the case of CoO/CoP, the Co3+ peaks are absent, implying a thorough transformation of Co3O4 into other phases following phosphorization. The intensity of the Co3+ peaks increases significantly, indicating that a portion of Co3O4 has transformed into CoO. This inference is further corroborated by the examination of satellite peaks, where the satellite peaks in CoO are more observable compared to those in Co3O4. This observation aligns with the known phenomenon that the 2+ oxidation state of Co exhibits more prominent satellite peaks than other oxidation states.68 Furthermore, a new set of peaks at 778.5 and 793.4 eV emerges, aligning with the Co0 2p3/2 and 2p1/2 orbitals, respectively. The presence of Co0 unequivocally signifies the transformation of some portion of Co3O4 into CoP. The XPS analysis thus provides further confirmation of the existence of the CoO/CoP heterointerface. Meanwhile, a chemical shift of +0.6 eV is observed for the Co2+ 2p3/2 orbital after phosphorization, which signifies a downshift of the valence state of Co, indicating electron transfer from CoP to CoO across the heterointerface. The high-resolution O 1s XPS spectra of Co3O4 and CoO/CoP are depicted in Fig. 3c and d, respectively. The O 1s spectrum is resolved into two distinct peaks. The peak at 530 eV is indicative of O–metal bonds, while the peak at 531.2 eV corresponds to O–H bonds arising from adsorbed surface hydroxyls.69,70 Fig. 3e and f present the high-resolution P 2p XPS spectra. As expected, P signal is absent in Co3O4. On the contrary, two prominent peaks can be identified for CoO/CoP. The peak at 130 eV is attributed to the P–Co bond in CoP, while the peak at 134 eV is ascribed to the P–O bond.71,72
:Co2+ is determined to be 1.92:1, closely mirroring the nominal ratio for Co3O4. Conversely, in the case of CoO/CoP, the Co3+ peaks are absent, implying a thorough transformation of Co3O4 into other phases following phosphorization. The intensity of the Co3+ peaks increases significantly, indicating that a portion of Co3O4 has transformed into CoO. This inference is further corroborated by the examination of satellite peaks, where the satellite peaks in CoO are more observable compared to those in Co3O4. This observation aligns with the known phenomenon that the 2+ oxidation state of Co exhibits more prominent satellite peaks than other oxidation states.68 Furthermore, a new set of peaks at 778.5 and 793.4 eV emerges, aligning with the Co0 2p3/2 and 2p1/2 orbitals, respectively. The presence of Co0 unequivocally signifies the transformation of some portion of Co3O4 into CoP. The XPS analysis thus provides further confirmation of the existence of the CoO/CoP heterointerface. Meanwhile, a chemical shift of +0.6 eV is observed for the Co2+ 2p3/2 orbital after phosphorization, which signifies a downshift of the valence state of Co, indicating electron transfer from CoP to CoO across the heterointerface. The high-resolution O 1s XPS spectra of Co3O4 and CoO/CoP are depicted in Fig. 3c and d, respectively. The O 1s spectrum is resolved into two distinct peaks. The peak at 530 eV is indicative of O–metal bonds, while the peak at 531.2 eV corresponds to O–H bonds arising from adsorbed surface hydroxyls.69,70 Fig. 3e and f present the high-resolution P 2p XPS spectra. As expected, P signal is absent in Co3O4. On the contrary, two prominent peaks can be identified for CoO/CoP. The peak at 130 eV is attributed to the P–Co bond in CoP, while the peak at 134 eV is ascribed to the P–O bond.71,72
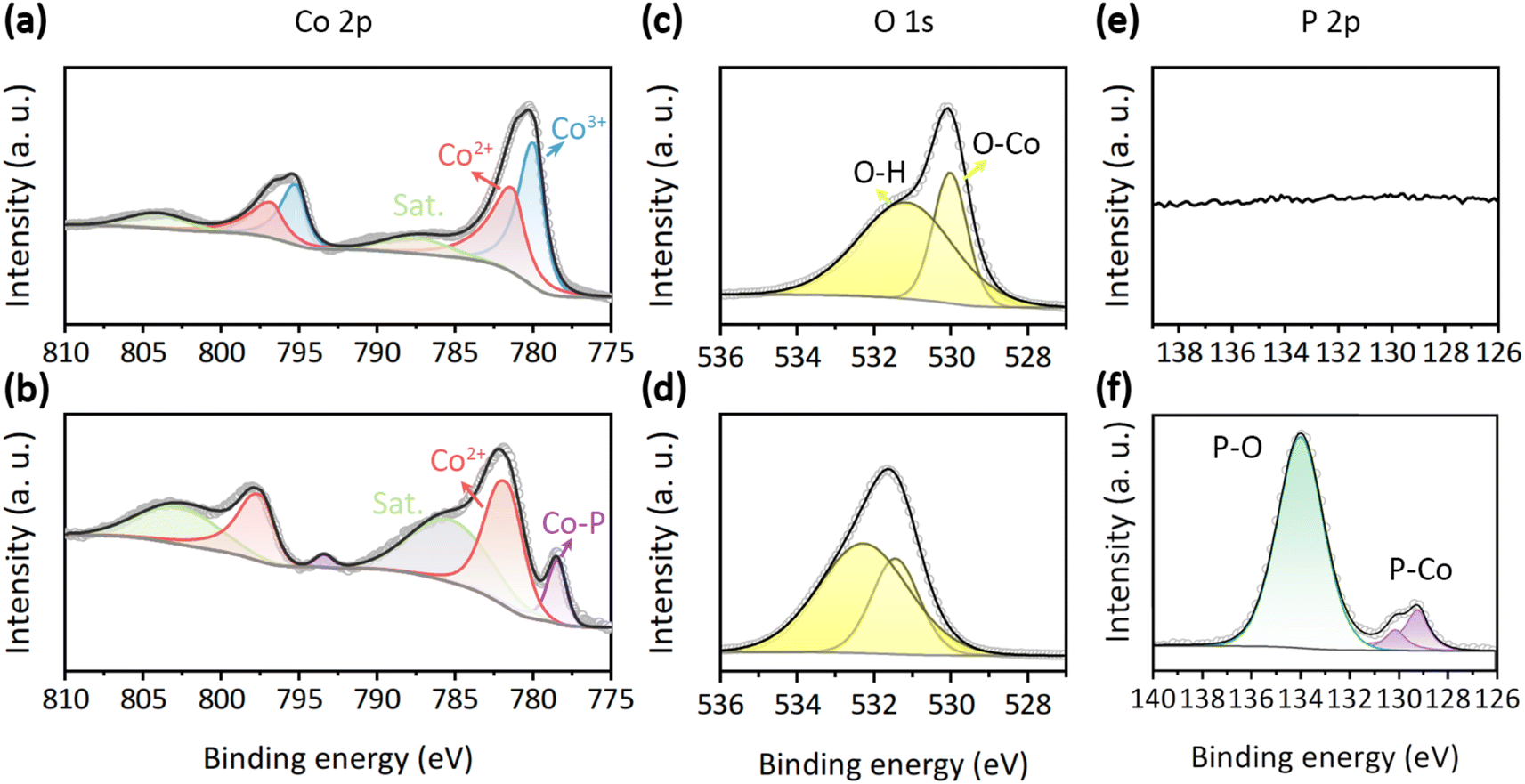 | ||
| Fig. 3 (a and b) Co 2p XPS spectra of Co3O4 and CoO/CoP, respectively. (c and d) O 1s XPS spectra of Co3O4 and CoO/CoP, respectively. (e and f) P 2p XPS spectra of Co3O4 and CoO/CoP, respectively. | ||
The electrocatalytic activity for the hydrogen evolution reaction (HER) of the synthesized samples was assessed using an electrochemical workstation. As illustrated in Fig. 4a, the linear sweep voltammetry (LSV) curves depict the Faraday current as a function of overpotential for Co3O4, CoO/CoP, and Pt/C (wt. 20%) powders. Notably, Co3O4 requires an overpotential of 220 mV to achieve a current density of 10 mA cm−2, whereas CoO/CoP demonstrates a significantly reduced overpotential of 148 mV. This substantial reduction indicates that the formation of the CoO/CoP heterointerface greatly enhances the HER activity. On one hand, the formation of CoO/CoP can facilitate efficient electron transfer between CoO and CoP, which is essential for the electrochemical reduction of protons to hydrogen. This improved electron transfer can accelerate the kinetics of HER and lower the overpotential. On the other hand, the CoO/CoP heterointerface can lead to changes in the local coordination environment of Co ions, which can influence their intrinsic catalytic activity by altering their electronic structure. The Tafel slopes for the respective materials, derived from the LSV curves, are presented in Fig. 4b. The Tafel slope for Co3O4 is measured at 137 mV dec−1, whereas that for CoO/CoP is 84 mV dec−1. These results suggest that the formation of the CoO/CoP heterointerface considerably reduces the kinetic barrier in the HER process. To delve into the underlying physics of the enhanced HER activity of CoO/CoP, the double layer capacity (Cdl) was measured using cyclic voltage methods within the non-Faraday range, as presented in Fig. 4c and S5.† Specifically, CoO/CoP exhibits a Cdl of 4 mF cm−2, surpassing that of Co3O4 (2.5 mF cm−2). This indicates that CoO/CoP possesses a higher electrochemically active area than Co3O4 after phosphorization. Electrochemical impedance spectra were also recorded and are depicted in Fig. 4d. CoO/CoP exhibits significantly lower impedance compared to Co3O4, signifying faster charge transfer at the reaction surface. The measured electrochemical properties are summarized in Table S1.† The stability of a catalyst is also a crucial parameter for evaluating its performance. The long-term stability test of CoO/CoP was conducted at a current density of 10 mA cm−2 for over 65 hours (Fig. 4e).
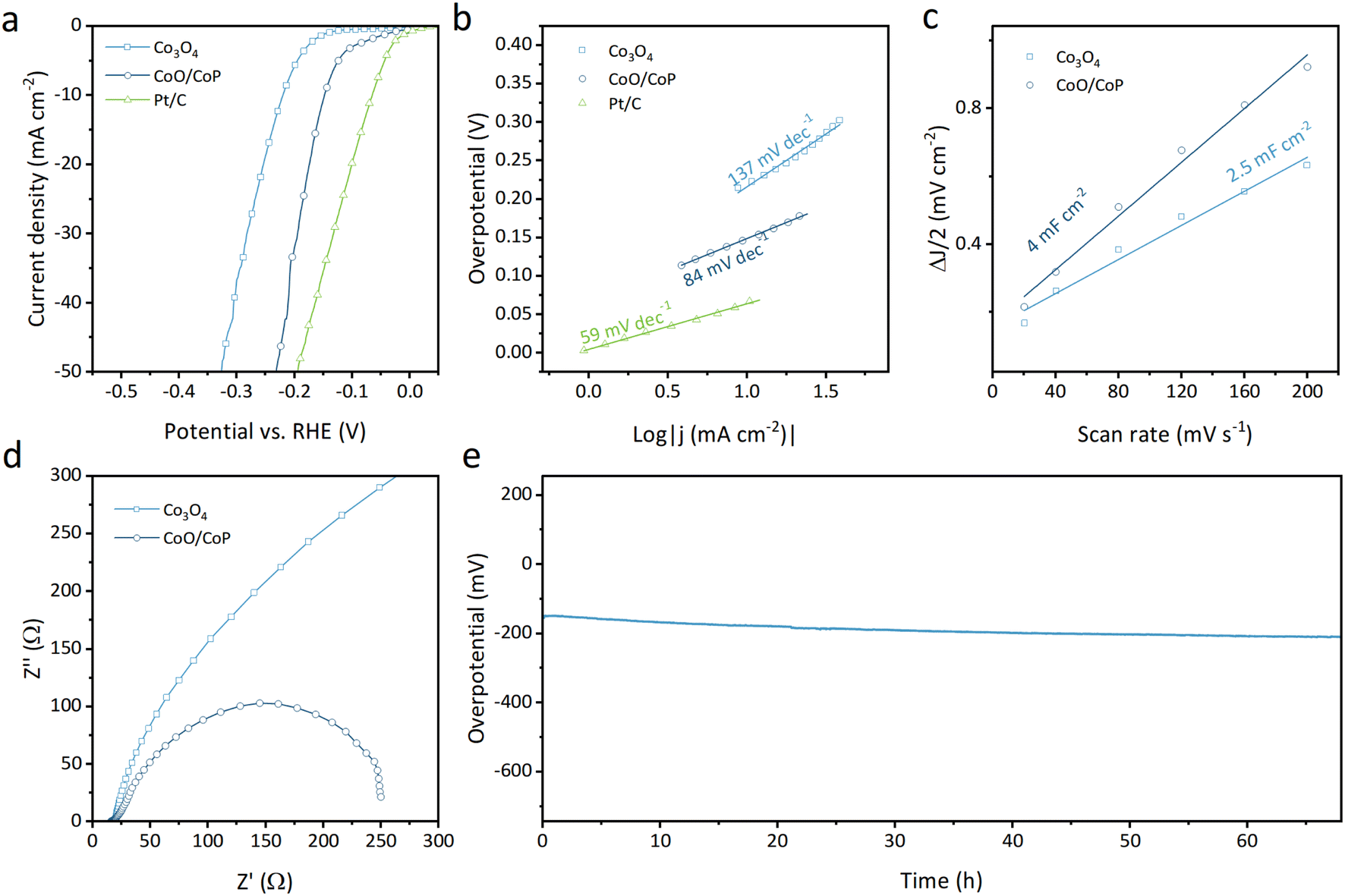 | ||
| Fig. 4 (a) LSV curves of Co3O4 and CoO/CoP, respectively. (b) Tafel slopes of Co3O4 and CoO/CoP, respectively. (c) Cdl of Co3O4 and CoO/CoP, respectively. (d) EIS of Co3O4 and CoO/CoP, respectively. (e) Stability test. | ||
To explore the underlying reasons behind the improved hydrogen evolution reaction (HER) activity of CoO/CoP, theoretical calculations based on the density functional theory (DFT) were conducted. The CoO/CoP heterointerface was constructed and relaxed, in comparison with individual CoO and CoP, as depicted in Fig. S5 and S6.† In alkaline environments, the HER process involves two main steps: the initial dissociation of water and the subsequent desorption of hydrogen from the catalyst surface. The optimized structures for water dissociation and hydrogen desorption for the three considered models are shown in Fig. 5a–c, respectively. The reaction barrier, represented by the Gibbs free energy change, for these models is illustrated in Fig. 5d. It is observed that CoO exhibits excessively strong adsorption for water, leading to a substantial barrier for water dissociation. In contrast, the CoO/CoP heterostructure exhibits a minimal reaction barrier for both water dissociation and hydrogen desorption. The energy barrier of CoP falls between the barriers observed for CoO and CoO/CoP. To elucidate the optimized reaction barrier of the CoO/CoP heterostructure, the partial density of states of the active Co site in the three models was calculated, as illustrated in Fig. 5e. The analysis reveals a large band gap in CoO. The band gap disappears for the up-spin states in CoP, while it persists in the down-spin band. In the case of the CoO/CoP heterostructure, the band gap disappears for both up-spin and down-spin bands. Therefore, the interaction between CoO and CoP leads to the elimination of the bandgap, enhancing the interaction between the catalyst and reaction radicals.
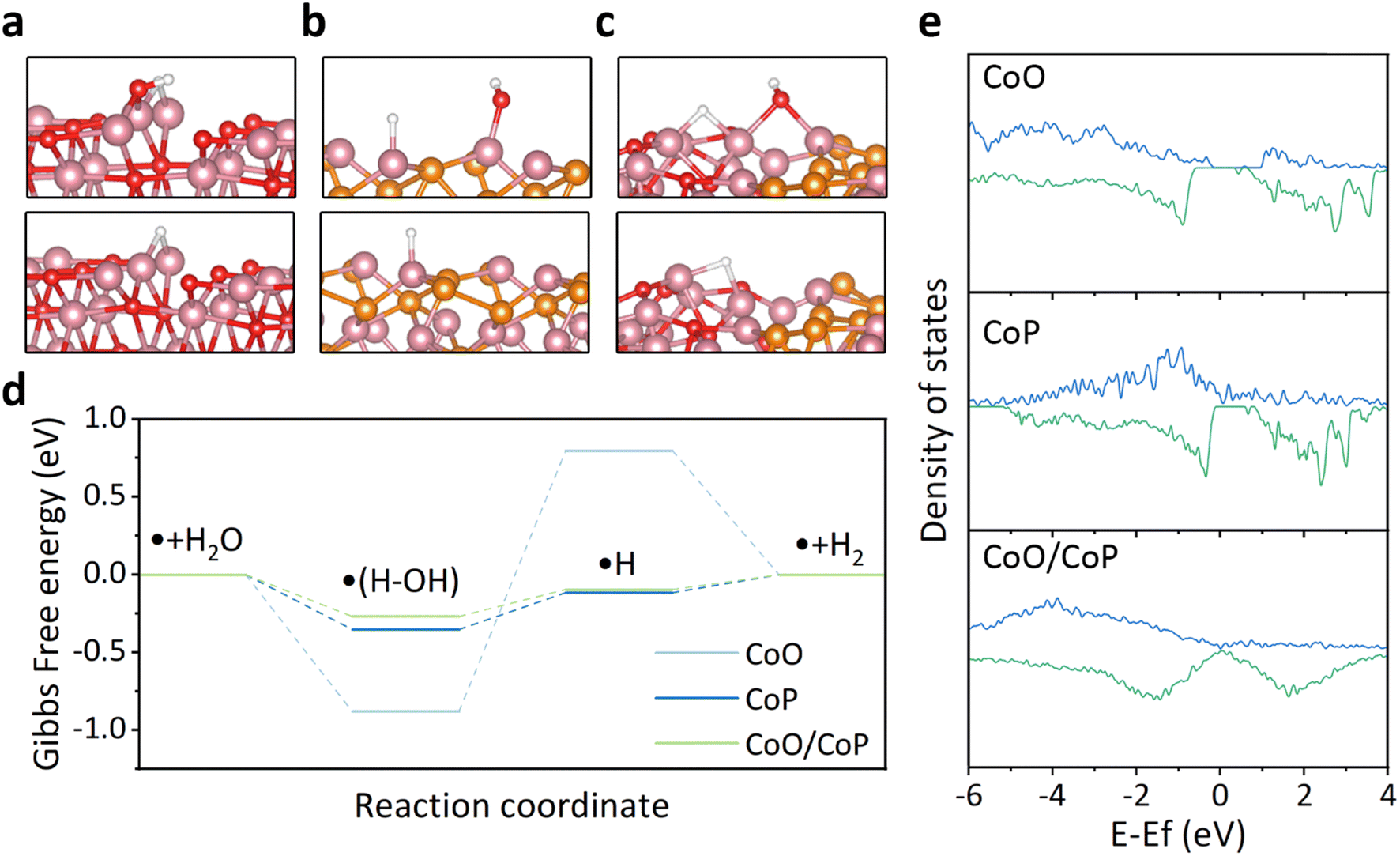 | ||
| Fig. 5 (a–c) DFT calculation structure model for CoO, CoP and CoO/CoP, respectively. (d) Corresponding Gibbs free energy change during HER in alkali. (e) Partial density of states of Co in CoO, CoP and CoO/CoP, respectively. The bule lines indicate spin-up states while the green lines indicate spin-down states. | ||
4. Conclusion
In conclusion, a facile phosphorization process has been successfully employed to achieve the in situ transformation from a single-phase Co3O4 to CoO/CoP hybrid phases. The synthesized samples have undergone comprehensive characterization using various techniques, including XRD, BET, SEM, TEM, among others. Throughout the phase transformation, the mesoporous nature of Co3O4 has been retained, resulting in the formation of porous CoO/CoP heterointerfaces. The presence of strong charge transfer across the CoO/CoP heterointerface suggests a robust interaction between the hybrid phases. Importantly, CoO/CoP demonstrates significantly enhanced catalytic activity for HER compared to pristine Co3O4. DFT calculations offer insights into the observed high HER activity, revealing that the elimination of the band gap in the spin-down band of Co in CoO/CoP contributes to the improved catalytic performance.Conflicts of interest
The authors declare no conflict of interests regarding the publication of this article.Acknowledgements
National Natural Science Foundation of China (22379104), Natural Science Foundation of Zhejiang Province (LY24E020004), and China Postdoctoral Science Foundation (2022M723546).References
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- C. Tarhan and M. A. Çil, J. Energy Storage, 2021, 40, 102676 CrossRef.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- J. Zhang, G. Chen, K. Müllen and X. Feng, Adv. Mater., 2018, 30, 1800528 CrossRef PubMed.
- Z. Wang, Z. Lin, S. Shen, W. Zhong and S. Cao, Chin. J. Catal., 2021, 42, 710–730 CrossRef CAS.
- Z. Chen, J. Cao, X. Wu, D. Cai, M. Luo, S. Xing, X. Wen, Y. Chen, Y. Jin, D. Chen, Y. Cao, L. Wang, X. Xiong and B. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 12223–12233 CrossRef CAS PubMed.
- Y. He, P. Tang, Z. Hu, Q. He, C. Zhu, L. Wang, Q. Zeng, P. Golani, G. Gao, W. Fu, Z. Huang, C. Gao, J. Xia, X. Wang, X. Wang, C. Zhu, Q. M. Ramasse, A. Zhang, B. An, Y. Zhang, S. Martí-Sánchez, J. R. Morante, L. Wang, B. K. Tay, B. I. Yakobson, A. Trampert, H. Zhang, M. Wu, Q. J. Wang, J. Arbiol and Z. Liu, Nat. Commun., 2020, 11, 57 CrossRef CAS PubMed.
- K. Hu, T. Ohto, Y. Nagata, M. Wakisaka, Y. Aoki, J.-i. Fujita and Y. Ito, Nat. Commun., 2021, 12, 203 CrossRef CAS PubMed.
- Z. Lin, C. Wang, Z. Wang, Q. Liu, C. Le, B. Lin and S. Chen, Electrochim. Acta, 2019, 294, 142–147 CrossRef CAS.
- J. Mahmood, F. Li, S.-M. Jung, M. S. Okyay, I. Ahmad, S.-J. Kim, N. Park, H. Y. Jeong and J.-B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- C. Wan, Z. Zhang, J. Dong, M. Xu, H. Pu, D. Baumann, Z. Lin, S. Wang, J. Huang, A. H. Shah, X. Pan, T. Hu, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Mater., 2023, 22, 1022–1029 CrossRef CAS PubMed.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- Z. Wang, Z. Lin, Y. Wang, S. Shen, Q. Zhang, J. Wang and W. Zhong, Adv. Mater., 2023, 35, 2302007 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- Q. He, D. Tian, H. Jiang, D. Cao, S. Wei, D. Liu, P. Song, Y. Lin and L. Song, Adv. Mater., 2020, 32, 1906972 CrossRef CAS PubMed.
- X. Chen, J. Wan, J. Wang, Q. Zhang, L. Gu, L. Zheng, N. Wang and R. Yu, Adv. Mater., 2021, 33, 2104764 CrossRef CAS PubMed.
- J. Kim, S.-M. Jung, N. Lee, K.-S. Kim, Y.-T. Kim and J. K. Kim, Adv. Mater., 2023, 35, 2305844 CrossRef CAS PubMed.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Advanced Science, 2018, 5, 1700464 CrossRef PubMed.
- H. Tan, B. Tang, Y. Lu, Q. Ji, L. Lv, H. Duan, N. Li, Y. Wang, S. Feng, Z. Li, C. Wang, F. Hu, Z. Sun and W. Yan, Nat. Commun., 2022, 13, 2024 CrossRef CAS PubMed.
- Z. Lin, B. Xiao, Z. Wang, W. Tao, S. Shen, L. Huang, J. Zhang, F. Meng, Q. Zhang, L. Gu and W. Zhong, Adv. Funct. Mater., 2021, 31, 2102321 CrossRef CAS.
- X. Chen, X.-T. Wang, J.-B. Le, S.-M. Li, X. Wang, Y.-J. Zhang, P. Radjenovic, Y. Zhao, Y.-H. Wang, X.-M. Lin, J.-C. Dong and J.-F. Li, Nat. Commun., 2023, 14, 5289 CrossRef CAS PubMed.
- A. H. Shah, Z. Zhang, Z. Huang, S. Wang, G. Zhong, C. Wan, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Catal., 2022, 5, 923–933 CrossRef CAS.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed.
- Z. Wang, Z. Lin, J. Deng, S. Shen, F. Meng, J. Zhang, Q. Zhang, W. Zhong and L. Gu, Adv. Energy Mater., 2021, 11, 2003023 CrossRef CAS.
- Z. Wang, B. Xiao, Z. Lin, S. Shen, A. Xu, Z. Du, Y. Chen and W. Zhong, J. Energy Chem., 2021, 54, 510–518 CrossRef CAS.
- T. Guo, L. Li and Z. Wang, Adv. Energy Mater., 2022, 12, 2200827 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- P. Liu, B. Chen, C. Liang, W. Yao, Y. Cui, S. Hu, P. Zou, H. Zhang, H. J. Fan and C. Yang, Adv. Mater., 2021, 33, 2007377 CrossRef CAS PubMed.
- J. Park, S. Lee, H.-E. Kim, A. Cho, S. Kim, Y. Ye, J. W. Han, H. Lee, J. H. Jang and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CrossRef CAS PubMed.
- F.-Y. Yu, Z.-L. Lang, L.-Y. Yin, K. Feng, Y.-J. Xia, H.-Q. Tan, H.-T. Zhu, J. Zhong, Z.-H. Kang and Y.-G. Li, Nat. Commun., 2020, 11, 490 CrossRef PubMed.
- Z. Wang, B. Xiao, Z. Lin, Y. Xu, Y. Lin, F. Meng, Q. Zhang, L. Gu, B. Fang, S. Guo and W. Zhong, Angew. Chem., Int. Ed., 2021, 60, 23388–23393 CrossRef CAS PubMed.
- Z. Li, R. Ge, J. Su and L. Chen, Adv. Mater. Interfaces, 2020, 7, 2000396 CrossRef CAS.
- C. Cui, R. Cheng, H. Zhang, C. Zhang, Y. Ma, C. Shi, B. Fan, H. Wang and X. Wang, Adv. Funct. Mater., 2020, 30, 2000693 CrossRef CAS.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS PubMed.
- Y. Zhu, Q. Lin, Y. Zhong, H. A. Tahini, Z. Shao and H. Wang, Energy Environ. Sci., 2020, 13, 3361–3392 RSC.
- T. Ling, T. Zhang, B. Ge, L. Han, L. Zheng, F. Lin, Z. Xu, W.-B. Hu, X.-W. Du, K. Davey and S.-Z. Qiao, Adv. Mater., 2019, 31, 1807771 CrossRef PubMed.
- C. Huang, L. Yu, W. Zhang, Q. Xiao, J. Zhou, Y. Zhang, P. An, J. Zhang and Y. Yu, Appl. Catal., B, 2020, 276, 119137 CrossRef CAS.
- Z.-Z. Liu, X. Shang, B. Dong and Y.-M. Chai, J. Catal., 2018, 361, 204–213 CrossRef CAS.
- Z. Lin, B. Lin, Z. Wang, S. Chen, C. Wang, M. Dong, Q. Gao, Q. Shao, T. Ding, H. Liu, S. Wu and Z. Guo, ChemCatChem, 2019, 11, 2217–2222 CrossRef CAS.
- T.-W. Wang, T.-L. Wang, W.-J. Chou, L.-F. Wu and S.-H. Lin, Phys. Chem. Chem. Phys., 2021, 23, 2305–2312 RSC.
- S. M. El-Refaei, P. A. Russo and N. Pinna, ACS Appl. Mater. Interfaces, 2021, 13, 22077–22097 CrossRef CAS PubMed.
- S. Xu, H. Zhao, T. Li, J. Liang, S. Lu, G. Chen, S. Gao, A. M. Asiri, Q. Wu and X. Sun, J. Mater. Chem. A, 2020, 8, 19729–19745 RSC.
- X. Yu, Z.-Y. Yu, X.-L. Zhang, P. Li, B. Sun, X. Gao, K. Yan, H. Liu, Y. Duan, M.-R. Gao, G. Wang and S.-H. Yu, Nano Energy, 2020, 71, 104652 CrossRef CAS.
- J. Fester, A. Makoveev, D. Grumelli, R. Gutzler, Z. Sun, J. Rodríguez-Fernández, K. Kern and J. V. Lauritsen, Angew. Chem., Int. Ed., 2018, 57, 11893–11897 CrossRef CAS PubMed.
- C. Wang and L. Qi, Angew. Chem., Int. Ed., 2020, 59, 17219–17224 CrossRef CAS PubMed.
- W. Li, J. Liu, P. Guo, H. Li, B. Fei, Y. Guo, H. Pan, D. Sun, F. Fang and R. Wu, Adv. Energy Mater., 2021, 11, 2102134 CrossRef CAS.
- P. Shi, Y. Zhang, G. Zhang, X. Zhu, S. Wang and A.-L. Wang, J. Mater. Chem. A, 2021, 9, 19719–19724 RSC.
- T. Liu, P. Li, N. Yao, G. Cheng, S. Chen, W. Luo and Y. Yin, Angew. Chem., Int. Ed., 2019, 58, 4679–4684 CrossRef CAS PubMed.
- H. Yang, Y. Zhang, F. Hu and Q. Wang, Nano Lett., 2015, 15, 7616–7620 CrossRef CAS PubMed.
- C. Wu, Y. Yang, D. Dong, Y. Zhang and J. Li, Small, 2017, 13, 1602873 CrossRef PubMed.
- Y. Men, P. Li, F. Yang, G. Cheng, S. Chen and W. Luo, Appl. Catal., B, 2019, 253, 21–27 CrossRef CAS.
- L. Wen, Y. Sun, C. Zhang, J. Yu, X. Li, X. Lyu, W. Cai and Y. Li, ACS Appl. Energy Mater., 2018, 1, 3835–3842 CrossRef CAS.
- J. Wu, N. Han, S. Ning, T. Chen, C. Zhu, C. Pan, H. Wu, S. J. Pennycook and C. Guan, ACS Sustain. Chem. Eng., 2020, 8, 14825–14832 CrossRef CAS.
- H. Wang, W. Fu, X. Yang, Z. Huang, J. Li, H. Zhang and Y. Wang, J. Mater. Chem. A, 2020, 8, 6926–6956 RSC.
- K. Song, H. Zhang, Z. Lin, Z. Wang, L. Zhang, X. Shi, S. Shen, S. Chen and W. Zhong, Adv. Funct. Mater., 2024, 34, 2312672 CrossRef CAS.
- Z. Lin, Z. Wang, J. Gong, T. Jin, S. Shen, Q. Zhang, J. Wang and W. Zhong, Adv. Funct. Mater., 2023, 33, 2307510 CrossRef CAS.
- L. Zhang, X. Shi, A. Xu, W. Zhong, J. Zhang and S. Shen, Nano Res., 2024, 17, 3693–3699 CrossRef CAS.
- K. Jiao, B. Zhang, B. Yue, Y. Ren, S. Liu, S. Yan, C. Dickinson, W. Zhou and H. He, Chem. Commun., 2005, 5618–5620, 10.1039/B512080B.
- T. Liu, T. Jiang, J. Zhu, T. Rui, S. Zheng and Z. Hu, New J. Chem., 2023, 47, 22125–22133 RSC.
- S. Sun, X. Zhao, M. Yang, L. Wu, Z. Wen and X. Shen, Sci. Rep., 2016, 6, 19564 CrossRef CAS PubMed.
- J. Gonzalez-Prior, J. I. Gutierrez-Ortiz, R. Lopez-Fonseca, G. Busca, E. Finocchio and B. de Rivas, Catal. Sci. Technol., 2016, 6, 5618–5630 RSC.
- Q. Li, Y. Du, X. Li, G. Lu, W. Wang, Y. Geng, Z. Liang and X. Tian, Sens. Actuators, B, 2016, 235, 39–45 CrossRef CAS.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608–2613 CrossRef CAS PubMed.
- S. Todorova, J. L. Blin, A. Naydenov, B. Lebeau, H. Kolev, P. Gaudin, A. Dotzeva, R. Velinova, D. Filkova, I. Ivanova, L. Vidal, L. Michelin, L. Josien and K. Tenchev, Catal. Today, 2020, 357, 602–612 CrossRef CAS.
- M. Naushad, T. Ahamad, M. Ubaidullah, J. Ahmed, A. A. Ghafar, K. M. Al-Sheetan and P. Arunachalam, J. King Saud Univ., Sci., 2021, 33, 101252 CrossRef.
- K. Zhu, J. Liu, S. Li, L. Liu, L. Yang, S. Liu, H. Wang and T. Xie, Adv. Mater. Interfaces, 2017, 4, 1700377 CrossRef.
- D. He, X. Song, W. Li, C. Tang, J. Liu, Z. Ke, C. Jiang and X. Xiao, Angew. Chem., Int. Ed., 2020, 59, 6929–6935 CrossRef CAS PubMed.
- L. Jia-bing, W. Xing-zhao, Z. Jun and Z. Yan, J. Fuel Chem. Technol, 2023, 51, 571 CrossRef.
- S.-Y. Zhang, T.-T. Li, H.-L. Zhu and Y.-Q. Zheng, J. Mater. Sci., 2018, 53, 4323–4333 CrossRef CAS.
- Y. Huang, D. Lv, G. Zhang, Y. Cai, Q. Li, H. Wang and Z. Ma, Energy Fuels, 2022, 36, 3339–3346 CrossRef CAS.
- Z. Liu, X. Yu, H. Xue and L. Feng, J. Mater. Chem. A, 2019, 7, 13242–13248 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02063d |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |