DOI:
10.1039/D4RA02344G
(Paper)
RSC Adv., 2024,
14, 17202-17212
Electrochemical water splitting enhancement by introducing mesoporous NiCoFe-trimetallic phosphide nanosheets as catalysts for the oxygen evolution reaction†
Received
27th March 2024
, Accepted 13th May 2024
First published on 28th May 2024
Abstract
Transition metal-based catalysts are widely used in electrocatalysis, especially in the field of water splitting, due to their excellent electrochemical performance, which focuses on improving the efficiency of the complex oxygen evolution reaction (OER) that occurs at the anode. Transition metal-based catalysts will undergo electrochemical surface reconstruction and form (oxy)hydroxide-based hybrids, which consider the actual active sites for OER. So many efforts have been made to know the origin of the effect of electrochemical surface reconstruction on the performance of the OER. Herein, NiCoFe-phosphide catalyst nanosheets were constructed by a simple one-step hydrothermal reaction by adding oleylamine and ethanol to water solvent during the preparation of the catalyst precursor and high-temperature gas-phase phosphating and significantly showed high effectiveness catalytic activity and conductivity in comparison to normal and traditional preparation methods. Electrochemical analysis, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and high-resolution transmission electron microscopy (HRTEM) demonstrate that the surface was constructed during the electrochemical reaction and formed an amorphous layer of MOx(OH)y active sites, which increased the electrochemical surface area and promoted charge transfer. As well, the synthesized NiCoFePx-PNSs catalyst nanosheets exhibit excellent catalytic activity with a low overpotential equal to 259 mV to achieve the OER at a current density of 10 mA cm−2 and a low Tafel slope of 50.47 mV dec−1 which is better than for most reported transition metal-based electrocatalysts. This work provides a new design for a transition metal-based catalyst for OER as well as further insights into the effect of electrochemical surface reconstruction on intrinsic activity and OER performance.
1. Introduction
Clean and renewable energy technologies, such as water splitting, fuel cells and metal–air batteries, are crucial for mitigating environmental pollution and the energy crisis.1 As an advanced energy conversion technology, water splitting is a prerequisite for the development of reversible fuel cells and rechargeable metal–air batteries.2 It is also considered an appealing solution to address energy-related issues without generating carbon dioxide. Electrochemical water splitting involves two half reactions: the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER).3 Because of the slow four-electron transfer kinetics process of OER, the application of water electrolysis is largely limited, and hence, highly effective electrocatalysts are urgently needed.4 At present, noble metal materials like IrO2 and RuO2 show excellent OER performance under alkaline conditions, but their high cost limits their large-scale application.5 To achieve highly efficient and economic electrolysis of water, it is critical to develop active, stable and cost-effective electrocatalysts for the OER, which is a thermodynamically unfavorable process.6 The first-row transition metals, typically Ni, Co and Fe, are highly attractive due to their low cost, relatively high conductivity, and excellent OER performance.7 Currently, a variety of Ni, Co and Fe transition metal-based nanomaterials (alloys, oxides, hydroxides, sulfides, phosphides and nitrides) have been reported as catalysts with extraordinary catalytic behavior of OER under alkaline conditions.8 Transition metal phosphides (TMPs) are particularly interesting and have attracted considerable attention, given that they exhibit intrinsically high catalytic activity and possess superior electrical conductivity.9 Early studies on TMPs were mainly concentrated on their applications in catalyzing the HER. Later on, TMPs with metal sites and tunable oxygen vacancies (VO) were found to be able to catalyze the OER under alkaline conditions.10 In addition, the later results showed that the starting solvents had an effect on the morphology and properties of the LDH precursors, and the properties and electrochemical activity of oxides and phosphides were closely related to the morphology and properties of their LDH precursors. The LDH precursor's structures and surface morphology with high surface area and abundant lattice defects lead to abundant active sites and confer on these materials unique properties for catalytic reactions by enhancing the kinetics of the conversion between the OER intermediates and increasing their total conductivity, which is the interest of many researchers. It was found that the LDH precursor's mesoporous nanosheet morphology can't only provide greater surface area and swifter electrolyte proximity but also a more rapid gas release rate.11 The starting solvents used to obtain such unique properties are of great interest and the TMPs prepared from LDH precursors with such unique properties must show superior catalytic activity toward OER. Among the TMPs, the nickel cobalt iron phosphide mesoporous nanosheets (NiCoFePx PNSs) have a lot of interest. One of the key advantages of NiCoFePx PNSs as an OER catalyst is their multifunctional nature. Each metal component contributes unique properties that enhance the overall catalytic performance of the material. For example, nickel has high electrical conductivity, which facilitates electron transfer during the OER.12 Cobalt has high catalytic activity and can stabilize high oxidation states, which are both important for efficient OER catalysis.13 Iron provides stability and corrosion resistance to the material, which are important for long-term durability.14 Phosphorus serves as a ligand that helps stabilize the crystal structure and enhance the electronic properties of the material.15 The specific composition and crystal structure of NiCoFePx can significantly impact its electrocatalytic activity and stability in OER. For example, several studies have shown that increasing the Co content in NiCoFePx can enhance its OER activity and reduce its overpotential.16 However, high Co content can also decrease the stability of NiCoFePx PNSs due to the formation of CoOx species during OER. In contrast, increasing the Fe content can improve the stability of NiCoFePx but may also decrease its activity due to the lower catalytic activity of Fe compared to Co.17 So many efforts have been made by researchers to find a suitable structure with a suitable composition and morphology for OER catalysts. Recently, transition metal-based catalysts undergo electrochemical surface reconstruction during the OER and form oxyhydroxide-based hybrid active sites, which consider the actual-active sites and the true-catalysts for the OER. The electrochemical-based catalysts that have this ability are known as pre-catalysts.18 However, the mechanism for enhancing the OER performance and the correlation between the pre-catalysts and the oxyhydroxide active sites are not clear. So, many efforts have been made to understand the origin of the enhancement of the OER performance corresponding to the electrochemical surface reconstruction and to offer a design principle for the structure of these pre-catalysts.19 Compared with the heterostructural catalysts prepared by general methods, the heterostructures formed by the electrochemical surface reconstruction of the pre-catalysts combined with the substrate could have better electrocatalytic performance, which may be attributed to the exposure of more active sites and the optimization of the electronic structure.20
Herein, we create surfaces with abundant lattice defects and active sites of NiCoFe-based catalyst porous nanosheets (PNSs) as OER catalysts for water splitting and provide an insight into their electrochemical surface reconstruction during electrochemical reactions. We will discuss the synthesis approaches and structural characteristics using two different kinds of solvent mixtures, as well as their electrocatalytic performance and durability in alkaline media. For comparison, the catalysts were synthesized via a modification method using starting solvent mixture 1 (OAM, ethanol and water) and showed better catalytic activity than the catalysts prepared using starting solvent mixture 2 (urea, ammonium fluoride and water). The creation procedure started with the hydrothermal method to create layered double hydroxides (LDH), which undergo an oxidation process in the air to obtain the oxide, and ended by subjecting the oxide to NaH2PO2 in a nitrogen (N2) atmosphere, resulting in the formation of metal-based phosphide. The electrochemical analysis, XRD, XPS and HRTEM reveal that the surface was constructed during the electrochemical reaction and formed an amorphous layer of MOx(OH)y active sites, which increased the exposing active sites, the electrochemical surface area, and promoted the charge transfer. As well, the synthesized NiCoFePx-PNSs catalyst nanosheets exhibit excellent catalytic activity with a low overpotential equal to 259 mV in alkaline conditions (1 M KOH) to achieve the OER at a current density of 10 mA cm−2. These findings signify the potential of NiCoFePx as a practical way to enhance the activity and stability of water electrolysis, thereby optimizing the OER performance of NiCoFe-based catalysts.
2. Experimental section
2.1. Materials and chemicals
Nickel(II) nitrate hexahydrate [Ni(NO3)2·6H2O, 98.0%], cobalt(II) nitrate hexahydrate [Co(NO3)2·6H2O, 99.0%], iron(II) nitrate nonahydrate [Fe(NO3)2·9H2O, 98.0%], ammonium fluoride [NH4F, ≥98.0%] and urea [CO(NH2)2, ≥98.0%] were procured from Sinopharm Chemical Reagent Co., Ltd, while oleylamine [C18H37N, 99.0%] and ethanol (C2H5OH; 99.9 wt%) were purchased from Aladdin Reagent Co. Ltd. Potassium hydroxide [KOH, ≥95%] was obtained from Alfa Aesar. The deionized (DI) water used in this study was obtained from a Millipore Autopure system with a resistivity of 18.25 MΩ cm−1. All other materials were of analytical grade and did not require further purification before use.
2.2. Synthesis of NiCoFeLDH and NiCoFeLDH-W PNSs
NiCoFeLDH PNSs were prepared by dissolving Ni(NO3)2·6H2O (0.15 mmol, 0.043 g), Co(NO3)2·6H2O (0.12 mmol, 0.035 g) and Fe(NO3)2·9H2O (0.03 mmol, 0.012 g) in 20 mL of DI water. Solvent mixture 1 of oleylamine (0.5 mL) with ethanol (20 mL) was added to the above solution, which was then stirred for over 30 minutes to ensure uniformity. The resulting solution was transferred to a teflon-lined stainless-steel autoclave with a capacity of 50 mL, which was sealed and heated gradually to 180 °C at a rate of 2 °C per minute. The solution was held at this temperature for 12 hours before cooling to room temperature. The resulting product was then centrifuged, washed multiple times with ethanol and ultrapure (UP) water, and dried under vacuum at 80 °C overnight. The NiCoFeLDH-W PNSs were synthesized in the same way: just replace (solvent mixture 1) by (solvent mixture 2). A solution of urea and ammonium fluoride with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 was dissolved in 20 mL of DI water and stirred until the urea was completely dissolved.
:2 was dissolved in 20 mL of DI water and stirred until the urea was completely dissolved.
2.3. Synthesis of NiCoFeOx and NiCoFeOx-W PNSs
In a typical procedure, the NiCoFeLDH PNSs and NiCoFeLDH-W PNSs were placed in ceramic boats inside a tube furnace. Then, the temperature was raised gradually (5 °C min−1) until it reached 350 °C, maintained there for two hours, and then lowered to room temperature. The final black products of NiCoFeOx and NiCoFeOx-W PNSs were collected.
2.4. Synthesis of NiCoFePx and NiCoFePx-W PNSs
In a standard process, NiCoFeOx PNSs or NiCoFeOx-W PNSs and NaH2PO2 were acquired and subjected to the opposite ends of the ceramic boats within a tube furnace at a 1:10 proportion upstream ratio, all while being exposed to nitrogen gas (N2 flow rate of 5 sccm). The temperature was raised gradually (5 °C min−1) until it reached 350 °C, maintained there for two hours, and then lowered to room temperature. The final black products of NiCoFePx and NiCoFePx-W PNSs were collected.
2.5. Electrochemical measurements
The electrochemical measurements were carried out at room temperature (30 °C) using an electrochemical workstation (CHI-760E, CH Instruments, Inc.). The electrolytic cell, consisting of a three-electrode configuration, has a glassy carbon electrode used as the working electrode; the counter electrode is made of platinum; and the reference electrode is a Hg/HgO electrode filled with 1 M KOH. The catalyst ink was prepared by dispersing 5 mg catalyst and 1 mL Nafion solution (0.1 wt%), then sonicating for 30 minutes to obtain homogenized ink. Before loading the catalyst, the surface of the glassy carbon electrode (GCE, surface area = 0.2 cm2) was polished with 500 nm and 50 nm α-Al2O3 successively. After being washed with ultrapure water and ethanol, 12 μL of ink was dripped on the clean surface (mass loading is 0.3 mg cm−2) and dried at room temperature. Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were performed to investigate the electrocatalytic performance. All voltage readings were converted to voltages versus the reversible hydrogen electrode (RHE) by comparing them to the corresponding reference electrode: E (vs. RHE) = E (Hg/HgO) + 0.92, the overpotential (η) was obtained from: η = E (vs. RHE) − 1.23 V. All the curves were recorded without iR correction. The Tafel plots of catalysts were depicted from their corresponding LSV data, which can be fitted based on the equation below:η = a + b × log(j), where η is the overpotential, j is the steady-state current density to the geometric area of the working electrode, and b is the Tafel slope. The durability test was performed using chronopotentiometry measurements at a current density of 10 mA cm−2. The electrochemical surface area (ECSA) was evaluated by the CV technique in the range of 1.17 to 1.27 V (vs. RHE) under different scan rates from 10 to 50 mV s−1 to avoid the generation of faradaic current. The double-layer capacitance (Cdl) is linearly proportional to the ECSA, and it was estimated by plotting half of the capacitive currents at 1.22 V (vs. RHE) (ΔJ/2, ΔJ is Janode − Jcathode) against scan rate. The overall water splitting performance was assessed in a 1 M KOH solution using a NiCoFe phosphide catalyst working electrode.
2.6. Physical characterization
To examine the material's surface morphology and fine structure, characterization was conducted using a field emission scanning electron microscope (FESEM, Zeiss, SIGMA). The high-resolution transmission electron microscopy (HRTEM), the corresponding selected area electron diffraction (SAED) and energy dispersive X-ray spectroscopy (EDX) results were measured on the FEI Tecnai G2 F20 Field Emission Transmission Microscope. The crystal structure of the samples was revealed through X-ray diffraction (XRD, Bruker D8 Advance) X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å) from 10 to 70°at a scan rate of 5° min−1. N2 adsorption–desorption analysis was conducted on a Micrometrics ASAP 3020 instrument at 77 K. Thermo Fischer's ESCALAB Xi+ was utilized for X-ray photoelectron spectroscopy (XPS) measurement. The calibration of C 1s binding energy was set at 284.6 eV, and the data was analyzed by Casa XPS Software version 2.3.19.
3. Results and discussion
3.1. Construction and characterization of NiCoFePx PNSs
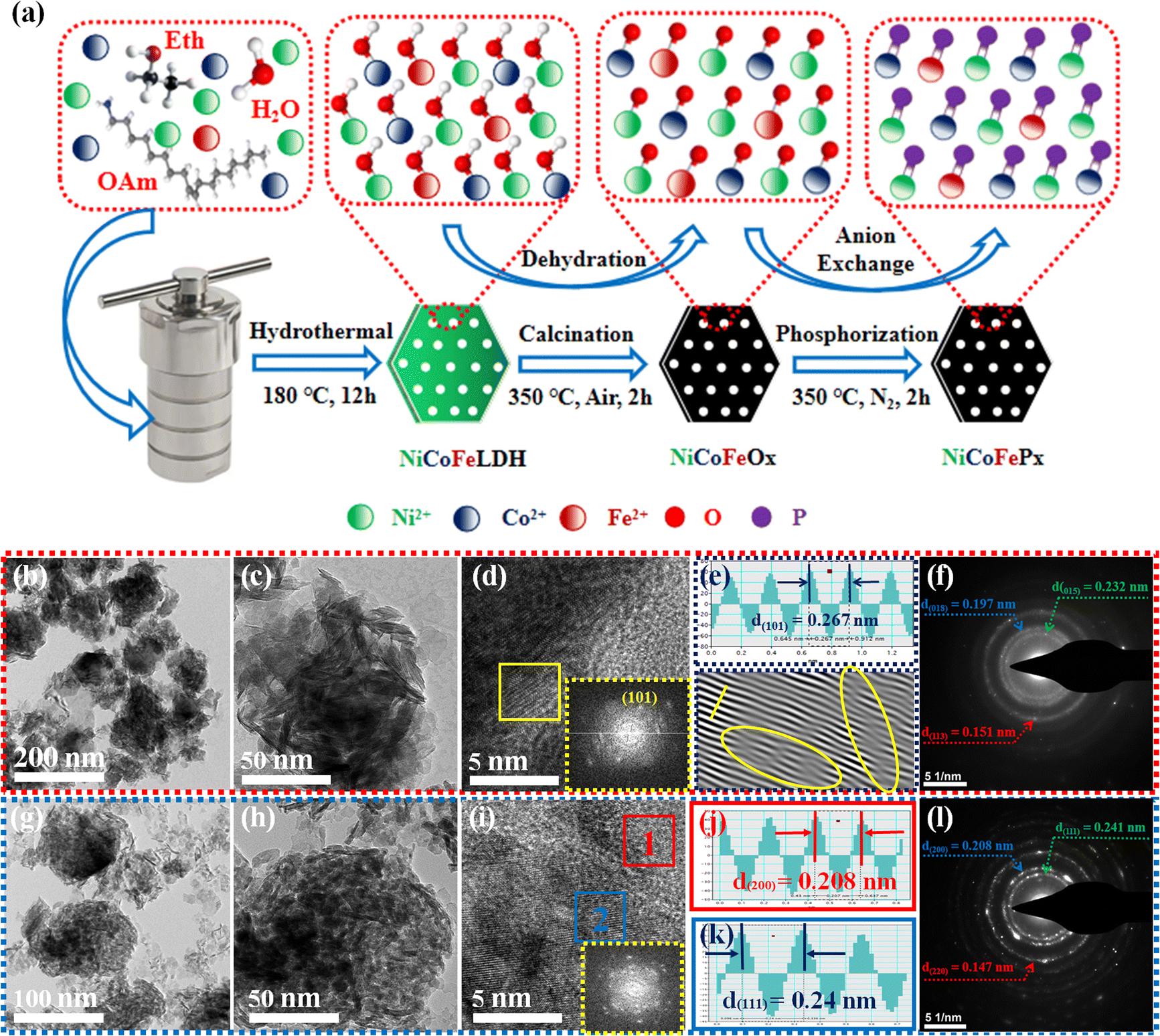
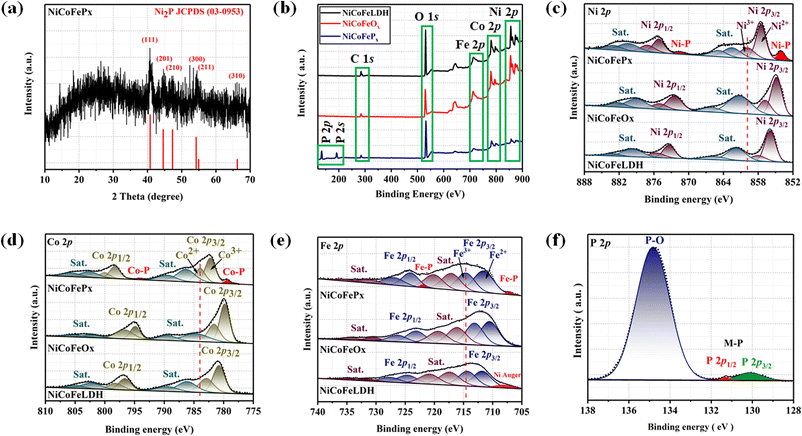
Fig. 1a illustrates the visual path of the synthesis of the NiCoFePx PNSs catalyst. Initially, NiCoFeLDH PNSs are prepared by subjecting a solution of Ni, Co and Fe salts with a solvent mixture 1 (oleylamine, ethanol and water) to a hydrothermal process at 180 °C for 12 h. Subsequently, the synthesized NiCoFeLDH PNSs undergo an oxidation process at 350 °C for 2 h in the air. The last step is the phosphorization process in a nitrogen (N2) environment at 350 °C for 2 h, leading to the formation of NiCoFePx PNSs, which facilitates the development of a surface with abundant lattice defects and active sites, thus improving the catalyst's conductivity and electrical structure. We investigated the morphologies and microstructure of the obtained samples using scanning electron microscopy (SEM) and transmission electron microscopy (TEM) imaging. The SEM images (Fig. S1, ESI†) and TEMimages (Fig. 1b, c and S2, ESI†) of the NiCoFeLDH PNSs revealed a two-dimensional (2D) morphology with ultrathin nanosheets, which indicates that the materials have a layered structure.21 The high-resolution transmission electron microscopy (HRTEM) imaging of one sheet of NiCoFeLDH PNSs (Fig. 1d) revealed that there are some lattice fringes in specific regions with an interplanar spacing of 0.267 nm corresponding to the crystal planes (101) of Ni(OH)2·0.75H2O (JCPDS No. 38-0715) in Fig. 1e, and we can find some of the lattice defects present on the surface. The inset FFT image (Fig. 1e) and the selected area electron diffraction (SAED) image (Fig. 1f) refer to a polycrystalline structure with ring patterns with low intensity diffraction spots, which indicates that the materials possess a low-crystallinity structure.22 SAED rings reveal distances of 0.151 nm, 0.197 nm and 0.232 nm, corresponding to the crystal planes (113), (018), and (015) of Ni(OH)2·0.75H2O (JCPDS No. 38-0715), respectively. The SEM (Fig. S3, ESI†) and TEM images of the NiCoFeOx PNSs (Fig. 1g, h and S4, ESI†) revealed the same morphology as its precursor. The HRTEM image (Fig. 1i) revealed the presence of lattice fringes in some regions that have an interplanar spacing of 0.207 nm and 0.24 nm, respectively, corresponding to the crystal planes (200) and (111) of NiO (JCPDS No. 47-1049), respectively, in Fig. 1j and k. The SAED image (Fig. 1l) refers to polycrystalline materials that possess a high-crystalline structure.23 Some of these spots reveal distances of 0.147 nm, 0.208 nm and 0.241 nm, corresponding to the crystal planes (220), (200) and (111) of NiO (JCPDS No. 47-1049), respectively. The surface morphology of the NiCoFePx PNSs was studied using SEM and TEM images (Fig. 2a–c and S5 ESI†). The images confirmed 2D morphology with ultrathin nanosheets, which indicates that the materials have a layered structure, which is consistent with their precursor as layered double hydroxides. Furthermore, the nanosheets were found to have a mesoporous structure and the BET adsorption theory was used to confirm the mesoporous structure of the catalysts. The hysteresis loop in the BET curve indicated that the catalysts are a typical type IV isotherm, which reveals that the material contains a significant number of mesoporous structures.24 (Fig. S6–S8 and Table S1 ESI†) revealed that the BET surface areas of the NiCoFeLDH, NiCoFeOx and NiCoFePx PNSs were 89.016, 122.28 and 6.51 m2 g−1, with BJH pore volumes of 0.43, 0.37 and 0.017 cm3 g−1 and small pore sizes of 14.75, 10.99 and 10.67 nm, respectively. The transformation from NiCoFeLDH to NiCoFeOx increased the surface area exposed to PH3 gas, which creates the most abundant active sites on the surface of NiCoFePx, but the addition of phosphorus can cause the metal ions to bond more strongly with each other, resulting in a more compact crystal lattice and a reduction in the surface area of NiCoFePx. Furthermore, the pores within the material may be blocked or filled with phosphorous atoms, reducing the available surface area for adsorption. Additionally, the mesoporous structure of the nanosheets facilitates faster mass transportation during the OER process, enhancing the efficiency of the reaction.25 The SAED image (Fig. 2d) and the FFT image inset in Fig. 2e refer to a polycrystalline structure with low-intensity diffraction spots, which indicates that the materials possess a low-crystallinity structure,26 and there are no complete diffraction rings but scattered light spots, which refer to many plane phases on the surface, and there are abundant lattice defects. The HRTEM imaging of one sheet (Fig. 2e) revealed the presence of lattice fringes in certain areas (areas 1 and 2), which revealed an interplanar spacing of 0.221 and 0.203 nm corresponding to the (111) and (201) crystal planes of Ni2P (JCPDS No. 03-0953) in Fig. 2f–i, respectively. The two lattice images are clear evidence that the surface morphology of NiCoFePx PNSs has abundant lattice defects, which form from the reconstruction of atoms during the phosphorization process and facilitate the development of a surface with abundant active sites. The HAADF STEM (Fig. 2j) and elemental EDX mapping (Fig. 2k–q) show the overall distribution of Ni, Co, Fe, P, O and C elements, which prove the homogeneous distribution of the elements through the whole surface of NiCoFePx PNSs. On the other hand, the SEM images are used to study the morphology of the NiCoFeLDH-W, NiCoFeOx-W and NiCoFePx-W prepared using solvent mixture 2 (Fig. S9–S11, ESI†), which revealed a 2D morphology with uniform hexagonal nanosheets. The hexagonal nanosheets observed in the images have a large size, high crystallinity and are very thick in comparison with the above ones. The crystalline structure and chemical composition were characterized by the powder X-ray diffraction (XRD) patterns. The XRD pattern of NiCoFeLDH PNSs (Fig. S12, ESI†) and NiCoFeLDH-W PNSs (Fig. S13, ESI†) showed the same diffraction peaks, which match well with Ni(OH)2·0.75H2O (JCPDS No. 38-0715). The XRD results revealed low intensity and wide peaks of NiCoFeLDH PNSs, referring to their low crystallinity and ultrathin nature in comparison with NiCoFeLDH-W PNSs. The XRD patterns of the NiCoFeOx PNSs (Fig. S14, ESI†) and NiCoFeOx-W PNSs (Fig. S15, ESI†) matched well with NiO (JCPDS No. 47-1049).27 The XRD patterns of the NiCoFePx PNSs (Fig. 3a) and NiCoFePx-W PNSs (Fig. S16, ESI†) show some peaks at 2θ = 40.79°, 44.59°, 54.23° and 66.22° with (111), (201), (300) and (310) plane phases, respectively, matched well with the Ni2P (JCPDS No. 03-0953).28 Inductively coupled plasma-optical emission spectroscopy (ICP-OES) is used to determine the elemental composition of the prepared catalysts. Based on the metal ion ratio obtained from the ICP-OES analysis, the NiCoFe-based samples can be denoted as Ni5Co3.80Fe1LDH, Ni5Co3.84Fe1.02Ox, and Ni5Co4.08Fe1.03Px PNSs, respectively. The results were very close to their feeding ratio, as shown in Table S2, ESI.† Furthermore, the EDX mapping ratio shows that the metal ion ratio of NiCoFePx PNSs is very close to the feeding ratio, as shown in Fig. S17, ESI.†
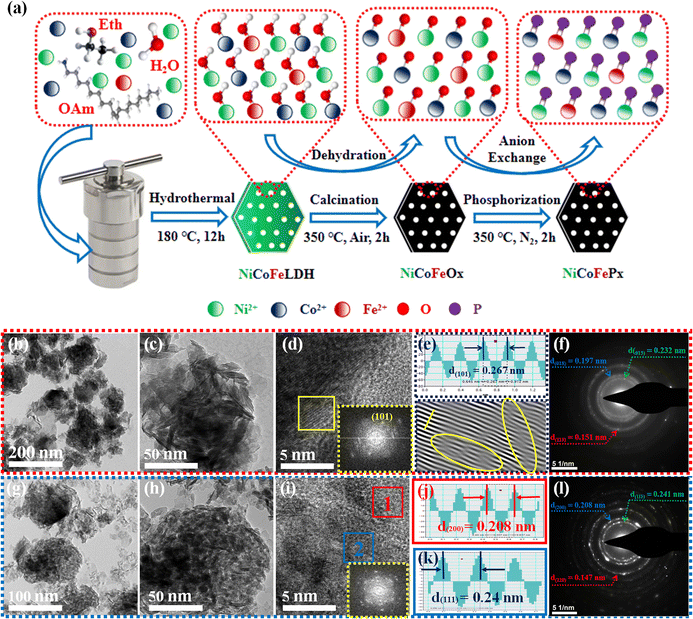 |
| | Fig. 1 (a) Schematic diagram illustrating the synthesis process of NiCoFePx PNSs; morphology characterization of NiCoFeLDH PNSs: (b and c) TEM images; (d) HRTEM image (the inset is FFT image); (e) lattice images of the selected area from HRTEM; (f) SAED pattern; morphology characterization of NiCoFeOx PNSs: (g and.h) TEM images; (i) HRTEM image (the inset is FFT image); (j and k) lattice distances of the selected areas from HRTEM; (l) SAED pattern. | |
 |
| | Fig. 2 Morphology characterization of NiCoFePx PNSs: (a) SEM image; (b and c) TEM images; (d) SAED pattern; (e) HRTEM image (the inset is FFT image); (f and g) lattice images of the two selected areas from HRTEM; (h and i) lattice distance of the two selected areas (j) HADAF image; (k) EDX scan of main metal elements; and (l–q) element mapping. | |
 |
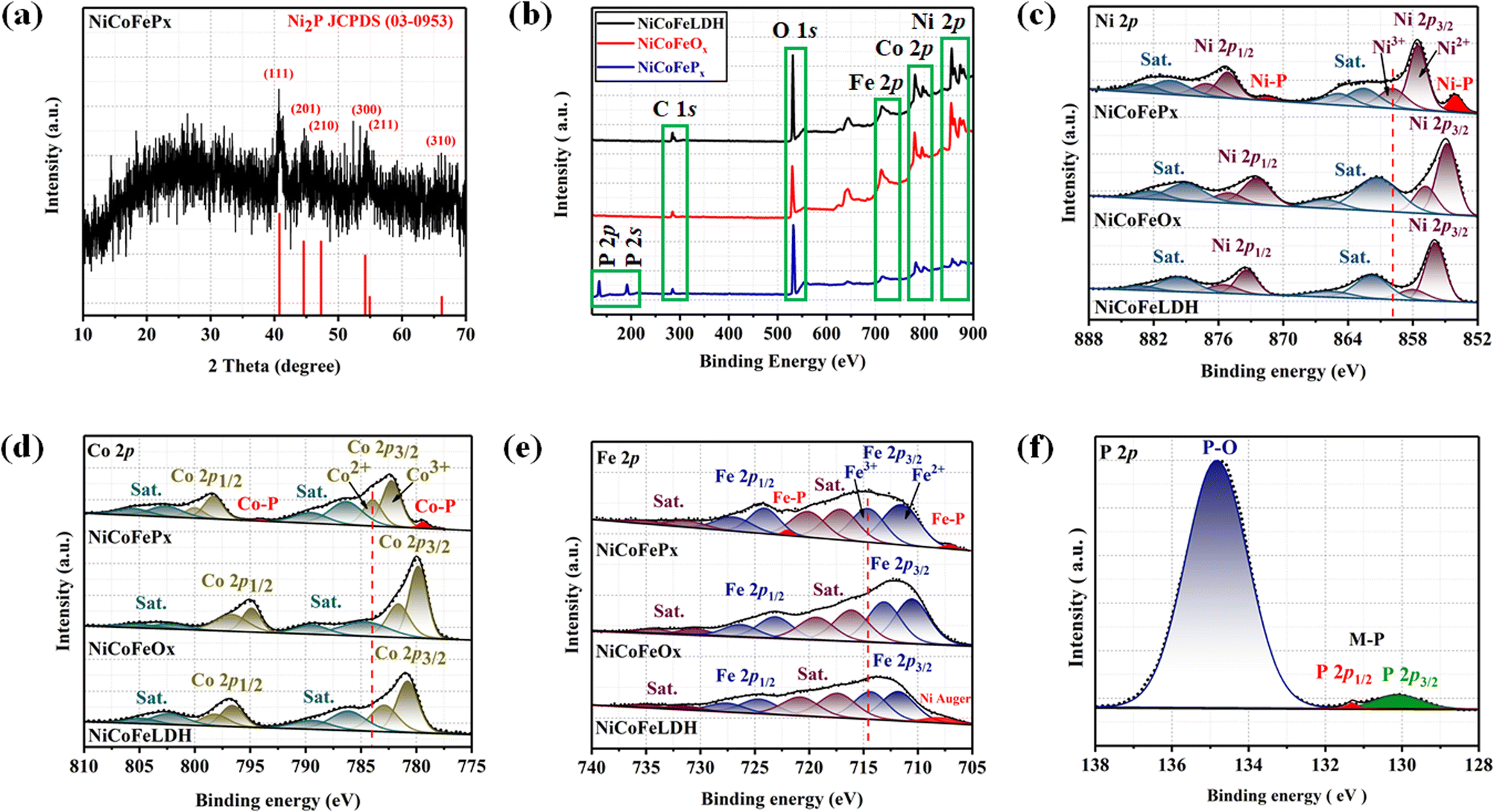
| | Fig. 3 (a) XRD of NiCoFePx; XPS fine spectra of NiCoFeLDH, NiCoFeOx and NiCoFePx PNSs: (b) XPS survey spectra; (c) high resolution Ni 2p; (d) high resolution Co 2p; (e) high resolution Fe 2p; and (f) high resolution P 2p. | |
3.2. Electronic structure and valence state analysis
The electronic structure of the synthesized catalysts was analyzed using X-ray photoelectron spectroscopy (XPS) measurements. The XPS survey spectra (Fig. 3b) confirm the presence of Ni, Co, Fe and O in all three samples. Moreover, two additional peaks of P 2p and P 2s were detected for the NiCoFePx PNSs, indicating the successful formation of NiCoFePx PNSs. The absence of any unexpected peaks in the survey spectra indicates that the synthesized catalysts are pure and free from any impurities or contaminants. The Ni 2p XPS profile (Fig. 3c) shows a spin–orbit doublet of Ni 2p1/2 and Ni 2p3/2, as well as two satellite humps next to the peaks, which refer to the formation of Ni–POx.29 The presence of Ni3+ and Ni2+ indicates that Ni ions are partially oxidized, can participate in redox reactions and allow for the efficient transfer of electrons during the electrocatalytic process, which is important for their catalytic activity. The observation of two small peaks of Ni–P suggests the involvement of phosphorus in the catalyst's formation to form nickel phosphide. The slightly higher binding energy observed in the Ni 2p of NiCoFePx PNSs compared to the other synthesized catalysts indicated that they were beneficial for more Ni3+ active species and promoted the electrocatalytic process. The Co 2p XPS spectrum (Fig. 3d) also shows a two-spin orbit doublet of Co 2p1/2 and Co 2p3/2, along with two satellite humps next to the peaks, referring to the formation of Co–POx.30 The presence of both Co3+ and Co2+ species suggests that the Co ions are partially oxidized and can participate in redox reactions during the electrocatalytic process. Also, two small peaks of Co–P are observed, suggesting the involvement of phosphorus in the catalyst's formation to form cobalt phosphide. The slight shift to high binding energy observed for NiCoFePx PNSs suggests a beneficial formation of Co2+ active species during the electrocatalytic process.31 The Fe 2p XPS spectrum (Fig. 3e) shows a two-spin orbit doublet of Fe 2p1/2 and Fe 2p3/2, along with two satellite humps referring to the formation of Fe–POx.32 These peaks, along with the presence of Fe2+ and Fe3+ species and satellite peaks, suggest that the synthesized catalysts have a complex electronic structure, which can play a crucial role in their electrocatalytic activity. The Fe 2p XPS spectra of NiCoFeLDH PNSs show the auger peak of Ni2+. Additionally, the NiCoFePx PNSs show very small peaks corresponding to the formation of iron phosphide (Fe–P). The P 2p XPS spectrum (Fig. 3f) displays three peaks, which correspond to phosphate and phosphide peaks (P–O and M–P3/2, M–P1/2), respectively.33 The detection of P–O (phosphates PO43− or PO3− or P2O5) peaks suggests that the metal phosphide has undergone oxidation, leading to a higher degree of oxidation probably caused by the exposure of phosphide species to air, which can further enhance the catalytic activity of the synthesized catalysts.
3.3. Electrochemical performance of NiCoFePx PNSs and comparison samples
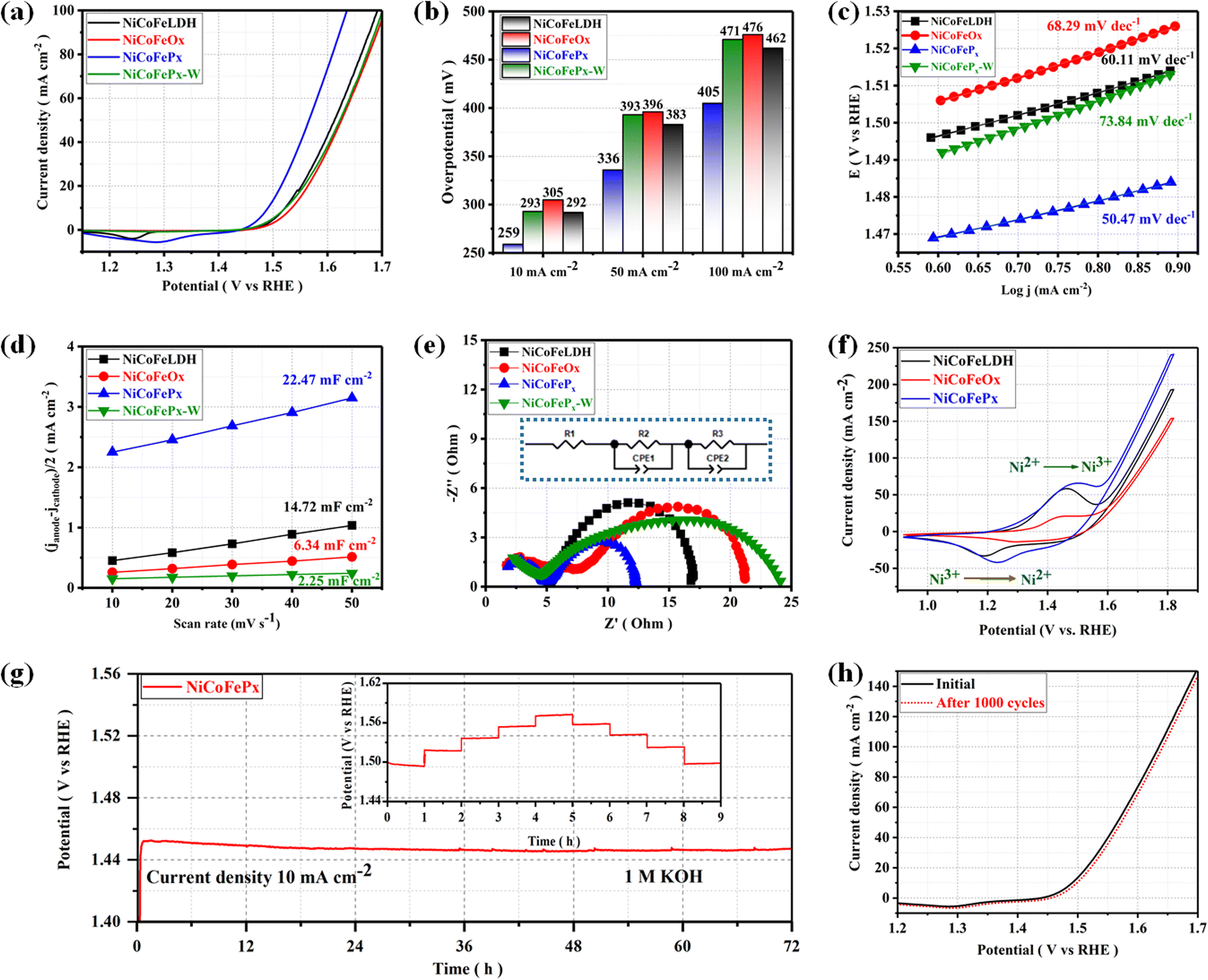
The electrocatalytic performances of the as-prepared samples were investigated in a 1 M KOH solution using a typical three-electrode system. The linear sweep voltammetry (LSV) is used to investigate the electrocatalytic activity of materials for OER at a scan rate of 5 mV s−1 with a working electrode connected to the rotating disk electrode system and rotated at 1600 rpm. The temperature of the entire system was maintained at 30 °C. Starting with the catalyst prepared using solvent mixture 1, the LSV curves (Fig. 4a) show that the NiCoFePx PNSs exhibited the best catalytic activity with the lowest overpotential at 10 mA cm−2 (η10 = 259 mV), which is better than NiCoFeLDH PNSs (η10 = 292 mV) and NiCoFeOx PNSs (η10 = 305 mV). This result indicates that the introduction of a proper amount of P can effectively improve the electrocatalytic OER activities of the catalysts, which may be caused by forming, exposing more active sites, abundant vacancies and defects that accelerate the charge transfer between the intermediates and the active sites and require less energy to drive the reaction compared to the other prepared catalysts. The overpotential plot diagram of the as-synthesized samples at current densities of 10, 50 and 100 mA cm−2 shows that the electrocatalytic behavior of NiCoFePx materials is significantly better than the other samples (Fig. 4b). The OER kinetics were evaluated by the Tafel slope derived from the LSV curves. As shown in Fig. 4c, NiCoFePx PNSs present a Tafel slope (50.47 mV dec−1), which is much smaller than NiCoFeLDH, (60.11 mV dec−1) and NiCoFeOx PNSs (68.29 mV dec−1), indicating that the P doping can improve the reaction kinetics and accelerate the rate of the OER. The intrinsic properties of the electrocatalytic surface of the prepared samples were investigated using electrochemical double-layer capacitance (Cdl). As shown in Fig. 4d, the Cdl value of NiCoFePx PNSs is (22.47 mF cm−2) which is higher than NiCoFeLDH PNSs (14.72 mF cm−2) and NiCoFeOx PNSs (6.34 mF cm−2). The results suggest that the NiCoFePx PNSs have a higher electrochemically active surface area, which is closely related to the density of active sites on the surface, which reflects the reason for the high catalytic activity of NiCoFePx PNSs. Electrochemical impedance spectroscopy (EIS) was utilized to investigate the electrochemical dynamics of charge transfer between the working electrode, catalyst and electrolyte at a potential of 1.52 V (vs. RHE). The Nyquist plots of all the samples were fitted to determine the charge transfer resistance (Rct). The semicircle diameter (Fig. 4e) obtained from the EIS analysis is related to (Rct). The results showed that the NiCoFePx PNSs have the smallest Nyquist fitting semicircle diameter (Rct = 7 Ω), compared to NiCoFeLDH PNSs (Rct = 11.7 Ω), and NiCoFeOx PNSs (Rct = 14 Ω) which refer to low impedance during charge transfer due to the high ECSA and high density of OER active sites on the surface, which can accelerate the rate of the OER and decrease the overpotential. To know the surface reconstruction during the OER and the main metal active sites on the surface of the catalyst, the CV curves in the potential range from 0.9 to 1.8 V were studied. Fig. 4f shows a redox peak around 1.49 V, corresponding to the Ni2+/Ni3+ redox couple. CV curves clearly show that Ni is the main metal active site on the surface, and the NiCoFePx PNSs have a stronger Ni2+/Ni3+ electrochemical response than the other catalyst, indicating that the introduction of the P element during the catalyst preparation could create more electroactive sites on the surface. The long-term electrochemical durability of the NiCoFePx PNSs catalyst was studied by a chronopotentiometric test, as shown in Fig. 4g. The results exhibit excellent long-term stability, with no significant degradation observed during the 72 hour test period. The multi-step chronopotentiometry test is used to further approve the electrocatalytic durability at different current densities from 10 to 50 mA cm−2, with a change of 10 mA cm−2 per hour (inset image, Fig. 4g). The results showed relatively stable current platforms at changing current densities, indicating the great electrochemical universality and high conductivity characteristics of the NiCoFePx PNSs. To further approve the durability, the LSV curves before and after 1000 continuous CV cycles are also compared (Fig. 4h). The LSV curve after the cyclic test remained almost unchanged, suggesting that the NiCoFePx PNSs are a promising candidate for practical applications in various electrochemical processes.
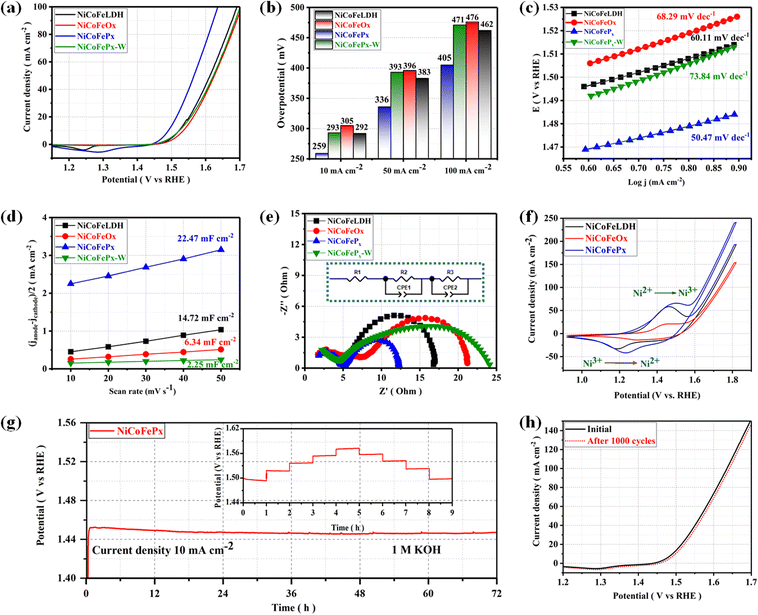 |
| | Fig. 4 Electrochemical measurements result: (a) LSV curves of OER; (b) overpotential histogram at 10, 50 and 100 mA cm−2; (c) Tafel plots; (d) the capacitive current at 1.22 V vs. RHE as a function of the scan rate; (e) Nyquist plots measured at 0.6 V versus Hg/HgO (inset equivalent circuit diagram); (f) CV curves in the potential range from 0.9 to 1.8 V. (g) Chronopotentiometry of the NiCoFePx at a current density of 10 A cm−2 for OER (the inset is durability at different current density); (h) LSV curves of NiCoFePx PNSs before and after multiple cycles stability. | |
On the other hand, the catalytic activity of the catalysts prepared using solvent mixture 2 was studied in Fig. S18, ESI.† The results showed that the properties and electrochemical activity of oxides and phosphides are closely related to the morphology and properties of their LDH precursors. The NiCoFePx-W PNSs exhibited the best catalytic activity, with the lowest overpotential of (η10 = 293 mV), Tafel slope (73.84 mV dec−1), and EIS (Rct = 19 Ω), with stability reaching 24 h at a current density of 10 mA cm−2. The results showed that the catalytic behavior of NiCoFePx PNSs is better than that of NiCoFePx-W PNSs, reflecting the advantage of adding oleylamine and ethanol to the water solvent during catalyst preparation. Some of these advantages include: Firstly, oleylamine is a surfactant that helps in dissolving metal precursors such as nickel, cobalt and iron salts in ethanol and ensures that the metal precursors are uniformly dispersed in the solution, leading to a homogeneous catalyst. Secondly, oleylamine acts as a stabilizing agent, preventing the aggregation or precipitation of metal ions during the synthesis process, which ensures that the resulting catalyst has a high surface area and well-defined morphology, which is crucial for efficient catalytic activity. Thirdly, by controlling the particle size of the NiCoFeLDH catalyst by adjusting the concentration or ratio of these components, it is possible to obtain nanoparticles with the desired sizes, which can significantly influence the catalytic performance.
3.4. Insights into electrochemical surface reconstruction based on HRTEM, XRD and XPS
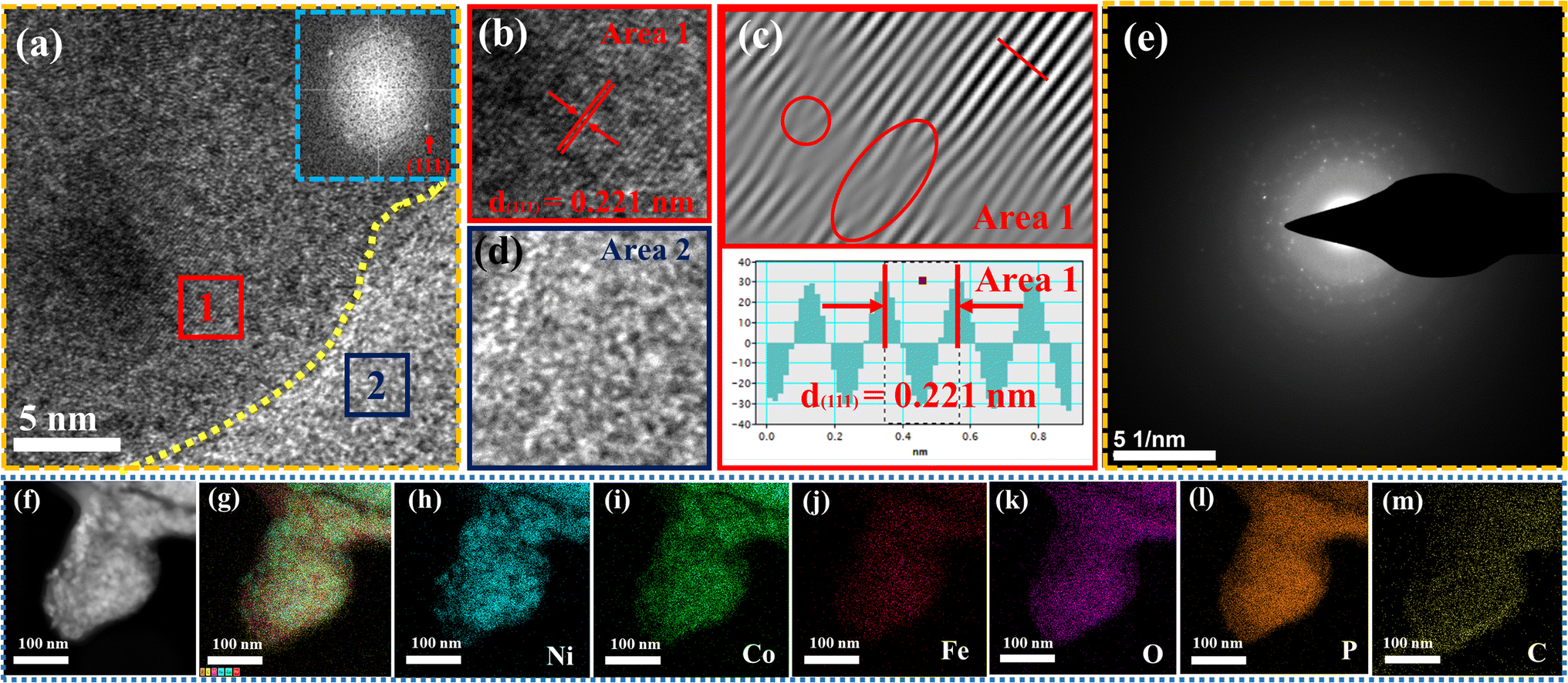
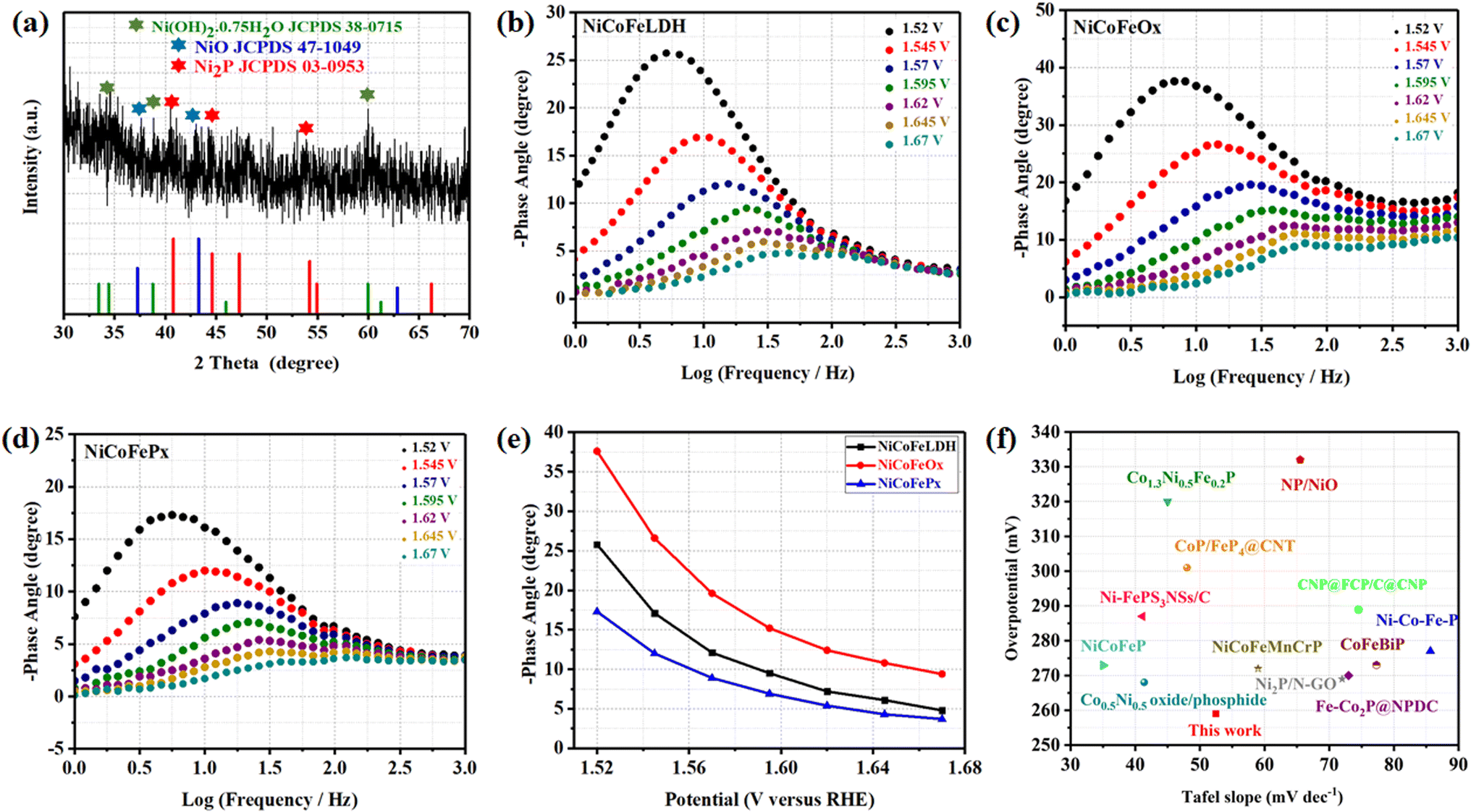
The surface morphology of post OER NiCoFePx PNSs catalyst was investigated using SEM and TEM images (Fig. S19, ESI†), which revealed the surface morphology of nanosheets didn't change during 1000 cycles of CV. Furthermore, the structure of post OER NiCoFePx PNSs catalyst was analyzed using HRTEM imaging (Fig. 5a), which revealed a heterostructure consisting of crystalline and amorphous regions.34 The crystalline region has lattice fringes with an interplanar spacing of 0.221 nm, which corresponds to the (111) crystal plane of Ni2P (JCPDS No. 03-0953) (Fig. 5b–d), and still has abundant lattice defects. The amorphous region corresponds to the formation of MOx(OH)y on the surface. HRTEM confirmed that the internal phosphide structure remains very well and the formation of an amorphous MOx(OH)y layer on the surface. The SAED image (Fig. 5e) and the inset FFT image refer to polycrystalline materials that possess a low-crystalline structure, and there are no complete diffraction rings but scattered light spots. The post OER HAADF STEM (Fig. 5f) and elemental EDX mapping (Fig. 5g–m) showed the homogeneous distribution of Ni, Co, Fe, P, O and C elements through the whole surface of NiCoFePx PNSs. Furthermore, the chemical composition of post OER NiCoFePx PNSs catalyst was investigated by XRD (Fig. 6a). The results showed that the intensity of phosphide peaks decreased with the appearance of some low intensity peaks of NiO and Ni(OH)2, which agree with the formation of an amorphous MOx(OH)y layer on the surface in HRTEM. The results clarify that the catalysts undergo electrochemical surface reconstruction to form oxyhydroxide hybrid active sites, which consider the actual active sites, and Ni2P considers the pre-catalysts for OER.35 The heterostructure formed during the electrochemical activation could have better catalytic activity due to the abundance of active sites and modulation of the electronic structure. Fig. 6b–d exhibits the Bode plots of NiCoFeLDH, NiCoFeOx and NiCoFePx. Significant response signals in the high-frequency region corresponding to the oxidation of Ni2+ to Ni3+ are observed. The potential dependence of the phase angle (Fig. 6e) exhibited that NiCoFePx has the smallest phase angle at all voltages, which refers to the large number of electrons involved in the OER and the ease of adsorption and desorption between the active sites and the intermediates, which may be caused by electrochemical surface reconstruction.36 Also, the NiCoFePx PNSs exhibit not only excellent long-term stability but also superior electrocatalytic activity when compared to other NiCoFe-based OER catalysts reported in recent years (Fig. 6f and Table S3, ESI†).
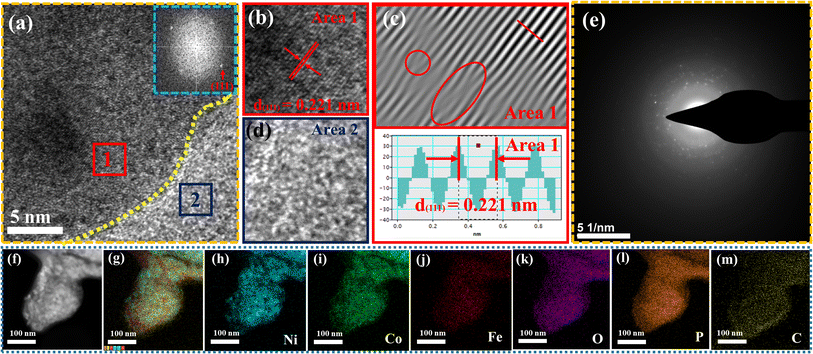 |
| | Fig. 5 Morphology characterization of NiCoFePx PNSs after OER: (a) HRTEM image (the inset is FFT image); (b–d) lattice images of the two selected areas from HRTEM; (e) SAED pattern; (f) HADAF image; (g) EDX scan of main metal elements; and (h–m) element mapping. | |
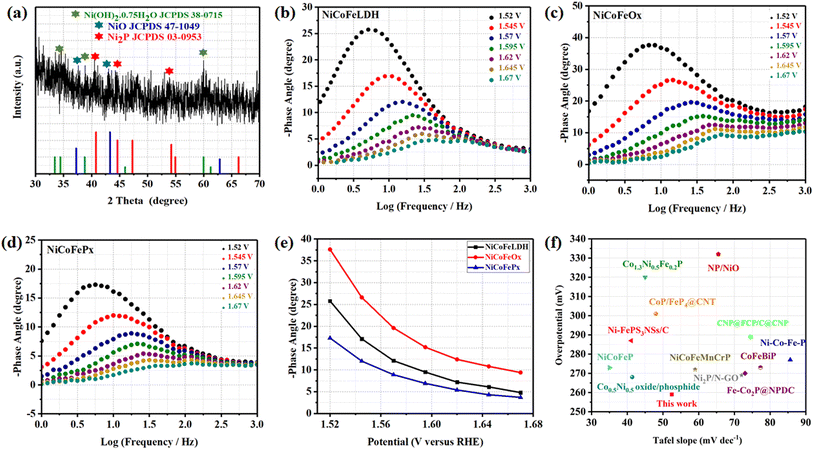 |
| | Fig. 6 (a) XRD of NiCoFePx after OER; Bode plots at range from 1.52 to 1.67 V (vs. RHE) of (b) NiCoFeLDH; (c) NiCoFeOx; (d) NiCoFePx; (e) potential dependence of the phase angle; (f) comparison of η10 overpotential and Tafel slope of NiCoFePx PNSs with some advanced NiCoFe-based OER electrocatalysts. | |
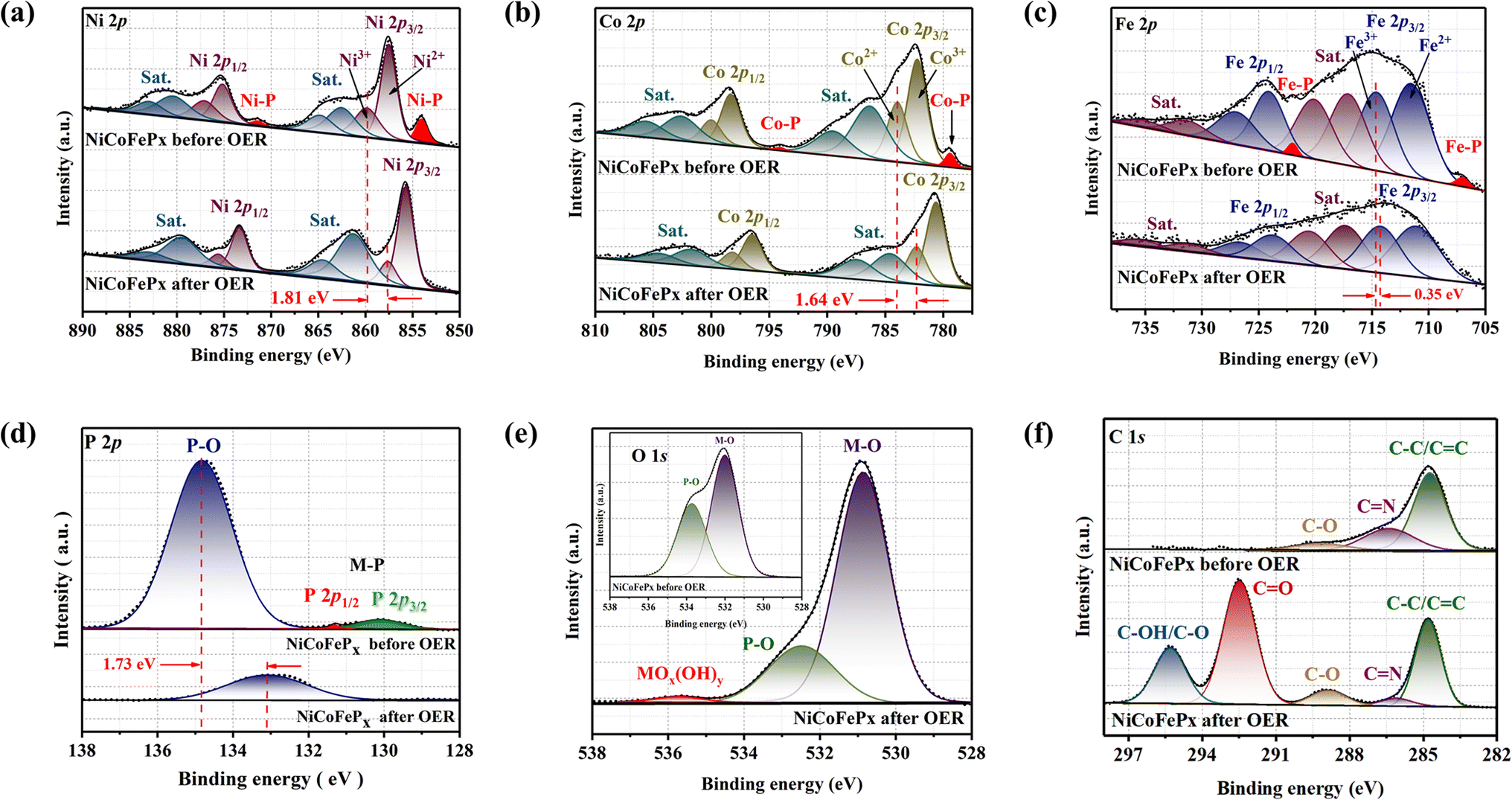
The XPS spectrum of the initial and post-OER of NiCoFePx PNSs catalysts after 1000 cycles was also studied to investigate the change in the electronic structure and the electrochemical surface reconstruction (Fig. S20, ESI†). The Ni 2p XPS profile (Fig. 7a) reveals a negative shift of Ni3+ binding energy, estimated to be 1.81 eV and the two peaks of Ni–P disappeared after OER. The Co 2p XPS profile (Fig. 7b) shows decreasing the intensity of the Co2+ peak and increasing the intensity of the Co3+ peak with a negative shift in binding energy estimated by 1.64 eV, which increases the high oxidation state Co3+ and decreases the lower oxidation state Co2+, and the two peaks of Co–P disappeared after OER. The Fe 2p XPS profile (Fig. 7c) shows a negative change in binding energy equal to 0.35 eV and the disappearance of the Fe–P peaks. The P 2p XPS profile (Fig. 7d) showed a negative shift by 1.73 eV of P–O with a decrease in peak intensity, and the M–P peaks disappeared.37 The O 1s (Fig. 7e) also showed a decrease in the intensity of P–O peak with a negative shift of binding energy, and a new peak of MOx(OH)y appears after OER, which proves the electrochemical surface reconstruction.38 C 1s XPS profile (Fig. 7f) is another piece of evidence of the surface reconstruction. The appearance of two new peaks at around 292.5 and 295 eV after the OER can be attributed to the chemical changes that occur on the catalyst surface during the reaction. The peak at around 292.5 eV typically corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, indicating the presence of carbon–oxygen species on the catalyst surface. This peak can be attributed to the adsorption of oxygen-containing species such as hydroxyls onto the catalyst surface during the OER. The peak at around 295 eV is often associated with C–OH or C–O bonds, indicating the presence of carbon–hydroxyl or carbon–oxygen species on the catalyst surface.39 By utilizing XPS data, we can investigate the electronic structure of the NiCoFePx PNSs catalyst before and after the formation of MOx(OH)y active species. Shedding light on the charge transfer pathways between the phosphide and surface species. The disappearance of M−P peaks during activation refers to the fact that metal phosphides are usually electrochemically oxidized into P-containing species (phosphates PO43−, PO3−, or P2O5), which is also the main mode of P–O binding, which dissolves in alkali and facilitates the electrochemical surface reconstruction, and generates MOx(OH)y active sites and facilitates the electron transfer during the OER.40 The charge transfers between phosphides and MOx(OH)y are very important for enhancing catalytic activity, electron transfer, and long-term stability. The exceptional electrocatalytic activity of the NiCoFePx PNSs can be attributed to their unique properties, such as mesoporous nanosheets with high ECSA, a high density of defects, and active sites resulting from electrochemical surface reconstruction, which lead to excellent stability and durability in alkaline conditions.
O bonds, indicating the presence of carbon–oxygen species on the catalyst surface. This peak can be attributed to the adsorption of oxygen-containing species such as hydroxyls onto the catalyst surface during the OER. The peak at around 295 eV is often associated with C–OH or C–O bonds, indicating the presence of carbon–hydroxyl or carbon–oxygen species on the catalyst surface.39 By utilizing XPS data, we can investigate the electronic structure of the NiCoFePx PNSs catalyst before and after the formation of MOx(OH)y active species. Shedding light on the charge transfer pathways between the phosphide and surface species. The disappearance of M−P peaks during activation refers to the fact that metal phosphides are usually electrochemically oxidized into P-containing species (phosphates PO43−, PO3−, or P2O5), which is also the main mode of P–O binding, which dissolves in alkali and facilitates the electrochemical surface reconstruction, and generates MOx(OH)y active sites and facilitates the electron transfer during the OER.40 The charge transfers between phosphides and MOx(OH)y are very important for enhancing catalytic activity, electron transfer, and long-term stability. The exceptional electrocatalytic activity of the NiCoFePx PNSs can be attributed to their unique properties, such as mesoporous nanosheets with high ECSA, a high density of defects, and active sites resulting from electrochemical surface reconstruction, which lead to excellent stability and durability in alkaline conditions.
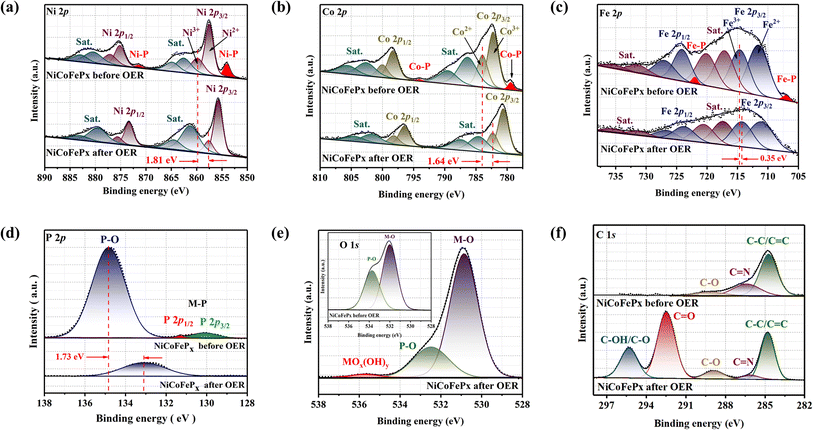 |
| | Fig. 7 XPS fine spectra of NiCoFePx PNSs before and after OER: (a) high resolution Ni 2p; (b) high resolution Co2p; (c) high resolution Fe 2p; and (d) high resolution P 2p; (e) high resolution O 2p; (f) high resolution C 1s. | |
4. Conclusion
In summary, the NiCoFe-based phosphides have been prepared by a simple one-step hydrothermal reaction using two different mixtures of solvents (oleylamine, ethanol and water) and (urea, ammonium fluoride and water) and chemical vapor deposition-based phosphorization. The NiCoFePx PNSs morphology revealed mesoporous ultrathin nanosheets with a low crystalline surface and abundant active sites, but the NiCoFePx-W PNSs revealed hexagonal nanosheets with a crystalline structure. The low crystalline NiCoFePx PNSs reveal the best electrocatalytic activity, which exhibits an overpotential of only 259 mV at 10 mA cm−2, a small Tafel slope of 50.47 mV dec−1 and excellent electrochemical durability in alkaline. The superior electrocatalytic activity of the NiCoFePx PNSs can be attributed to their morphology with mesoporous nanosheets with a high density of lattice defects and active sites, a high electrochemically active surface area (ECSA) caused by electrochemical surface reconstruction and the formation of MOx(OH)y active sites, and efficient charge transfer dynamics. These advantages ensure that the NiCoFePx PNSs electrocatalysts have good application prospects in practical electrochemical water splitting. This work provides a new path for optimizing metal phosphide electrocatalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Natural Science Foundation of Hubei Province (2021CFB144) and the Large-scale Instrument and Equipment Sharing Foundation of Wuhan University.
References
- N. Javed, T. Noor, N. Iqbal and S. R. Naqvi, RSC Adv., 2023, 13, 1137–1161 RSC
 .
. - Y. Zhang, L. Chen, B. Yan, F. Zhang, Y. Shi and X. Guo, Composites, Part B, 2023, 253, 110575 CrossRef CAS .
- X. Hu, X. Tian, Y. W. Lin and Z. Wang, RSC Adv., 2019, 9, 31563–31571 RSC .
- W. Liu, L. Wang and Y. Gong, Dalton Trans., 2023, 52, 8302–8310 RSC .
- A. Shekhawat, R. Samanta, S. Panigrahy and S. Barman, ACS Appl. Energy Mater., 2023, 6, 3135–3146 CrossRef CAS .
- D. Duan, D. Guo, J. Gao, S. Liu and Y. Wang, J. Colloid Interface Sci., 2022, 622, 250–260 CrossRef CAS PubMed .
- S. Sanati, A. Morsali and H. Garcia, Energy Environ. Sci., 2022, 15, 3119–3151 RSC .
- X. Yang, R. Guo, R. Cai, W. Shi, W. Liu, J. Guo and J. Xiao, Dalton Trans., 2022, 51, 4590–4607 RSC .
- W. Wu, X. Y. Wu, S. S. Wang and C. Z. Lu, RSC Adv., 2018, 8, 39291–39295 RSC .
- Z. Ahmad, A. Maqbool, M. A. Hussain, R. A. Pashameah, A. Shahzadi, N. Nazar, S. Iqbal, A. K. Alanazi, M. N. Ashiq and H. M. Abo-Dief, Fuel, 2023, 331, 125913 CrossRef .
- M. R. Kandel, U. N. Pan, P. P. Dhakal, R. B. Ghising, T. T. Nguyen, J. Zhao, N. H. Kim and J. H. Lee, Appl. Catal., B, 2023, 331, 122680 CrossRef CAS .
- J. Zhao, Y. He, J. Wang, J. Zhang, L. Qiu, Y. Chen, C. Zhong, X. Han, Y. Deng and W. Hu, Chem. Eng. J., 2022, 435, 134261 CrossRef CAS .
- S. Gupta, R. Fernandes, R. Patel, M. Spreitzer and N. Patel, Appl. Catal., A, 2023, 661, 119254 CrossRef CAS .
- S. Zhu, G. Duan, C. Chang, Y. Chen, Y. Sun, Y. Tang, P. Wan and J. Pan, ACS Sustain. Chem. Eng., 2020, 8, 9885–9895 CrossRef CAS .
- H. Zhao and Z. Y. Yuan, ChemCatChem, 2020, 12, 3797–3810 CrossRef CAS .
- B. Zhang, J. Shan, W. Wang, P. Tsiakaras and Y. Li, Small, 2022, 18, 2106012 CrossRef CAS PubMed .
- F. Tang, S. Guo, Y. Sun, X. Lin, J. Qiu and A. Cao, Small Struct., 2022, 3, 2100211 CrossRef CAS .
- H. Jia, N. Yao, J. Zhu and W. Luo, Chem.–Eur. J., 2023, 29, 202203073 CrossRef PubMed .
- N. Yao, G. Wang, H. Jia, J. Yin, H. Cong, S. Chen and W. Luo, Angew. Chem., 2022, 134, e202117178 CrossRef .
- X. Luo, X. Tan, P. Ji, L. Chen, J. Yu and S. Mu, EnergyChem, 2023, 5, 100091 CrossRef CAS .
- X. Lu, H. Xue, H. Gong, M. Bai, D. Tang, R. Ma and T. Sasaki, Nano-Micro Lett., 2020, 12, 1–32 CrossRef PubMed .
- K. Bera, A. Karmakar, S. Kumaravel, S. Sam Sankar, R. Madhu, H. N. Dhandapani, S. Nagappan and S. Kundu, Inorg. Chem., 2022, 61, 4502–4512 CrossRef CAS PubMed .
- M. Wang, Y. Long, H. Zhao, W. Zhang, L. Wang, R. Dong, H. Hou, H. Wang and X. Wang, ChemSusChem, 2022, 15, e202201518 CrossRef CAS PubMed .
- W. Zuo, Z. Xu, M. Hu, Y. Yu, J. Liu, G. Cheng and P. Zhao, Inorg. Chem. Front., 2023, 10, 2697–2707 RSC .
- N. Mushtaq, X. Ma, K. Aslam, W. Younas, Y. Zhu and C. Cao, Energy Technol., 2022, 10, 2200537 CrossRef CAS .
- S. Farokhi, M. Roushani and H. Hosseini, Electrochim. Acta, 2020, 362, 137218 CrossRef CAS .
- L. Bruno, M. Scuderi, F. Priolo and S. Mirabella, Sustain. Energy Fuels, 2022, 6, 4498–4505 RSC .
- X. Jin, J. Li, T. Xu, Y. Cui, N. Usoltseva, V. An, X. Zhang and B. Liu, Eur. J. Inorg. Chem., 2021, 34, 3481–3487 CrossRef .
- Y. Wang, S. Tao, H. Lin, S. Han, W. Zhong, Y. Xie, J. Hu and S. Yang, RSC Adv., 2020, 10, 33475–33482 RSC .
- J. A. Rajesh, Y. H. Lee, Y. H. Yun, V. H. V. Quy, S. H. Kang, H. Kim and K. S. Ahn, J. Ind. Eng. Chem., 2019, 79, 370–382 CrossRef CAS .
- J. Yin, J. Jin, H. Liu, B. Huang, M. Lu, J. Li, H. Liu, H. Zhang, Y. Peng, P. Xi and C. H. Yan, Adv. Mater., 2020, 32, 2001651 CrossRef CAS PubMed .
- R. Xiang, Y. Duan, C. Tong, L. Peng, J. Wang, S. S. A. Shah, T. Najam, X. Huang and Z. Wei, Electrochim. Acta, 2019, 302, 45–55 CrossRef CAS .
- J. Chang, Z. Hu, D. Wu, F. Xu, C. Chen, K. Jiang, Z. Gao, D. Duan, D. Guo, J. Gao, S. Liu and Y. Wang, J. Colloid Interface Sci., 2022, 622, 250–260 CrossRef PubMed .
- M. Kuang, J. Zhang, D. Liu, H. Tan, K. N. Dinh, L. Yang, H. Ren, W. Huang, W. Fang, J. Yao, X. Hao, J. Xu, C. Liu, L. Song, B. Liu and Q. Yan, Adv. Energy Mater., 2020, 10, 2002215 CrossRef CAS .
- M. Chen, D. Liu, J. Feng, P. Zhou, L. Qiao, W. Feng, Y. Chen, K. W. Ng, S. Wang, W. F. Ip and H. Pan, Chem. Eng. J., 2022, 443, 136432 CrossRef CAS .
- Z. Xu, W. Zuo, Y. Yu, J. Liu, G. Cheng and P. Zhao, Adv. Sci., 2024, 11, 2306758 CrossRef CAS PubMed .
- X. Zhou, Y. Zi, L. Xu, T. Li, J. Yang and J. Tang, Inorg. Chem., 2021, 60, 11661–11671 CrossRef CAS PubMed .
- G. Helal, Z. Xu, W. Zuo, Y. Yu, J. Liu, H. Su, J. Xu, H. Li, G. Cheng and P. Zhao, RSC Adv., 2024, 14(15), 10182–10190 RSC .
- C. Jin, H. Peng, X. Zeng, Z. Liu and D. Ding, ChemPhysMater, 2024, 3, 118–124 CrossRef .
- S. Zhao, Y. Zhao, J. Chen, R. Dai, W. Zhou, J. Yang, X. Zhao, Z. Chen, Y. Zhou, H. Zhang and A. Chen, Electrochim. Acta, 2022, 426, 140788 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Gongzhen Cheng
c,
Gongzhen Cheng