
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChlorine-containing polyacetoxybriarane diterpenoids from the octocoral Junceella fragilis†
Hai Nhat Do‡
ab,
Yu-Ta Chen‡c,
Su-Ying Chiend,
You-Ying Chenb,
Mingzi M. Zhang e,
Lun Kelvin Tsouf,
Jih-Jung Cheng,
Zhi-Hong Wena,
Yi-Hao Lochi,
Li-Guo Zheng*j and
Ping-Jyun Sung*abklm
e,
Lun Kelvin Tsouf,
Jih-Jung Cheng,
Zhi-Hong Wena,
Yi-Hao Lochi,
Li-Guo Zheng*j and
Ping-Jyun Sung*abklm
aDepartment of Marine Biotechnology and Resources, National Sun Yat-sen University, Kaohsiung 804201, Taiwan
bNational Museum of Marine Biology & Aquarium, Pingtung 944401, Taiwan. E-mail: pjsung@nmmba.gov.tw
cDepartment of Family Medicine, Zuoying Armed Forces General Hospital, Kaohsiung 813204, Taiwan
dInstrumentation Center, National Taiwan University, Taipei 106319, Taiwan
eInstitute of Molecular and Genomic Medicine, National Health Research Institutes, Miaoli 350401, Taiwan
fInstitute of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Miaoli 350401, Taiwan
gDepartment of Pharmacy, School of Pharmaceutical Sciences, National Yang Ming Chia Tung University, Taipei 112304, Taiwan
hShu-Zen Junior College of Medicine and Management, Kaohsiung 821004, Taiwan
iInstitute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung 804201, Taiwan
jDoctoral Degree Program in Marine Biotechnology, National Sun Yat-sen University, Kaohsiung 804201, Taiwan. E-mail: t0919928409@gmail.com
kChinese Medicine Research and Development Center, China Medical University Hospital, Taichung 404394, Taiwan
lGraduate Institute of Natural Products, Kaohsiung Medical University, Kaohsiung 807378, Taiwan
mPhD Program in Pharmaceutical Biotechnology, Fu Jen Catholic University, New Taipei City 242062, Taiwan
First published on 28th May 2024
Abstract
The chemical screening of an octocoral identifed as Junceella fragilis has led to the isolation of five chlorinated briarane-type diterpenoids, including three known metabolites, gemmacolide X (1), frajunolide I (2), and fragilide F (3), along with two new analogs, 12α-acetoxyfragilide F (4) and 12α-acetoxyjunceellin (5). Single-crystal X-ray diffraction analysis was carried out to determine the absolute configurations of 1 and 2, while the structures of new compounds 4 and 5 were ascertained with 2D NMR experiments. Briaranes 1 and 3–5 were active in enhancing alkaline phosphatase (ALP) activity.
1 Introduction
The octocorals belonging to genus Junceella (phylum Cnidaria, sub-phylum Anthozoa, class Octocorallia, order Scleralcyonacea, family Ellisellidae),1 distributed in the shallow waters of the tropical Indo-Pacific Ocean, have been proven to be a rich source of briarane-type diterpenoids with uncommon structures.2 This study explored further new substances from Junceella fragilis (Ridley 1884), collected from waters off the coast of Taiwan, an area with high biodiversity at the intersection of Kuroshio and South China Sea surface currents. The study successfully isolated five chlorinated briaranes, including three known metabolites, gemmacolide X (1),3 frajunolide I (2),4 and fragilide F (3),5 as well as two new analogs, 12α-acetoxyfragilide F (4) and 12α-acetoxyjunceellin (5) (Fig. 1) and ascertained their structures and ALP activity. The absolute configurations of 1 and 2 were further determined via single-crystal X-ray diffraction analysis with a diffractometer equipped with a copper (Cu Kα) source.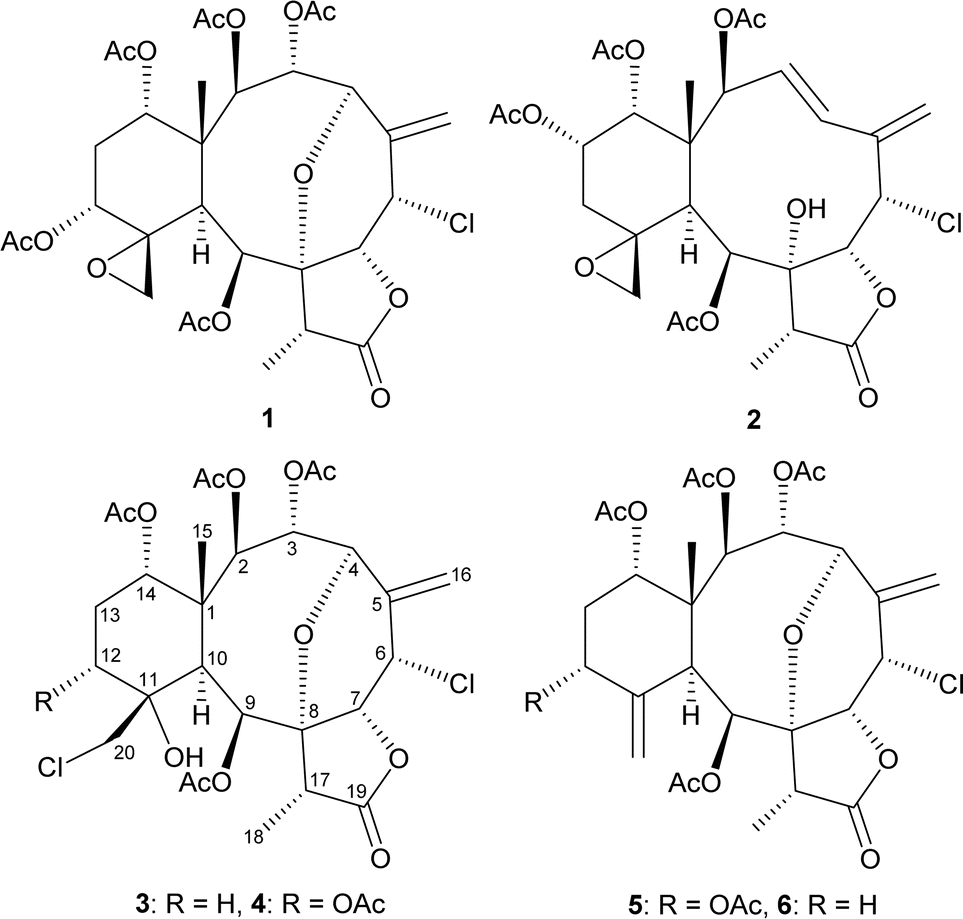 | ||
| Fig. 1 Structures of gemmacolide X (1), frajunolide I (2), fragilide F (3), 12α-acetoxyfragilide F (4), 12α-acetoxyjunceellin (5), and junceellin (6). | ||
2 Results and discussion
Gemmacolide X (1) and frajunolide I (2) were originally isolated from the octocorals Dichotella gemmacea and Junceella fragilis, respectively; and the structures of these two briaranes, including the relative configuration, were elucidated by spectroscopic analysis.3,4 The absolute configuration of these two compounds was supported in this study by single-crystal X-ray diffraction analysis (Flack parameter x = 0.000(5) for 1 and −0.002(13) for 2).6 The ORTEP diagram (Fig. 2) showed that the absolute configuration of stereogenic carbons of 1 and 2 are assigned as 1R, 2R, 3R, 4R, 6S, 7R, 8R, 9S, 10S, 11R, 12R, 14S, 17R and 1S, 2S, 6S, 7R, 8R, 9S, 10S, 11R, 13S, 14R, 17R, respectively. | ||
| Fig. 2 ORTEP demonstrates the structures of gemmacolide X (1) and frajunolide I (2). | ||
The (+)-ESIMS of 3 showed sodiated peaks at m/z 657/659/661 ([M + Na]+/[M + 2 + Na]+/[M + 4 + Na]+) (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:1) with a relative intensity suggestive of two chlorine atoms. Strong bands at 3478, 1790, and 1742 cm−1 observed in the IR spectrum confirmed the presence of hydroxy, γ-lactone, and ester groups in 3. It was found that the spectroscopic data of 3 were identical to those of a known briarane, fragilide F, and these two compounds possessed negative optical values ([α] −15 for 3 and [α] −19 for fragilide F);5 thus, compound 3 was identified as fragilide F.
:6:1) with a relative intensity suggestive of two chlorine atoms. Strong bands at 3478, 1790, and 1742 cm−1 observed in the IR spectrum confirmed the presence of hydroxy, γ-lactone, and ester groups in 3. It was found that the spectroscopic data of 3 were identical to those of a known briarane, fragilide F, and these two compounds possessed negative optical values ([α] −15 for 3 and [α] −19 for fragilide F);5 thus, compound 3 was identified as fragilide F.
12α-Acetoxyfragilide F (4) was isolated as an amorphous powder and its molecular formula was determined to be C30H38Cl2O14 (Ω = 11) by (+)-HRESIMS at m/z 715.15302 (calcd for C30H38Cl2O14 + Na, 715.15308). Comparison of the 1H NMR, HSQC, and HMBC data with the molecular formula indicated that there must be an exchangeable proton, requiring the presence of a hydroxy group, and this deduction was supported by a broad absorption in the IR spectrum at 3466 cm−1. The IR spectrum of 4 also showed strong bands at 1791 and 1740 cm−1, consistent with the presence of γ-lactone and ester groups. The presence of an exocyclic olefin was deduced from the signals of an sp2 methylene carbon at δC 119.6 (CH2-16). Six carbonyl resonances at δC 175.2 (C-19), 171.2, 170.2, 170.2, 169.8, and 169.6, confirmed the presence of a γ-lactone and five ester groups; five acetate methyls (δH 2.36, 2.07, 2.04, 2.03, and 2.01, each 3H × s) were also observed. From the above NMR data (Table 1), seven degrees of unsaturation were accounted for, and 4 must be tetracyclic.
| Position | δHa (J in Hz) | δCb, Mult.c |
|---|---|---|
| a Spectra recorded at 600 MHz in CDCl3 at 25 °C.b Spectra recorded at 150 MHz in CDCl3 at 25 °C.c Data assigned with the assistance of HSQC and HMBC spectra.d N. o. = not observed. | ||
| 1 | 45.1, C | |
| 2 | 5.48 d (6.6) | 73.1, CH |
| 3 | 6.19 dd (10.8, 6.6) | 63.8, CH |
| 4 | 4.47 d (10.8) | 78.5, CH |
| 5 | N. o.d | |
| 6 | 5.04 ddd (2.4, 2.4, 2.4) | 53.9, CH |
| 7 | 4.37 d (2.4) | 78.9, CH |
| 8 | 83.9, C | |
| 9 | 6.40 s | 73.2, CH |
| 10 | 2.99 s | 41.6, CH |
| 11 | 74.6, C | |
| 12 | 5.52 dd (3.0, 3.0) | 68.6, CH |
| 13α/β | 2.20 ddd (16.8, 3.0, 3.0); 1.97 ddd (16.8, 3.0, 3.0) | 26.2, CH2 |
| 14 | 4.83 dd (3.0, 3.0) | 72.6, CH |
| 15 | 1.30 s | 16.4, CH3 |
| 16a/b | 5.36 d (2.4); 5.57 d (2.4) | 119.6, CH2 |
| 17 | 2.82 q (7.2) | 49.5, CH |
| 18 | 1.39 d (7.2) | 7.2, CH3 |
| 19 | 175.2, C | |
| 20a/b | 3.52 d (12.0); 3.84 d (12.0) | 48.3, CH2 |
| OH-11 | 3.11 s | |
| Acetoxy groups | 2.36 s | 21.0, CH3 |
| 169.6, C | ||
| 2.07 s | 21.2, CH3 | |
| 171.2, C | ||
| 2.04 s | 21.0, CH3 | |
| 170.2, C | ||
| 2.03 s | 20.4, CH3 | |
| 170.2, C | ||
| 2.01 s | 21.0, CH3 | |
| 169.8, C | ||
In addition, a tertiary methyl singlet, a methyl doublet, a pair of aliphatic methylene protons, two aliphatic methine protons, seven oxymethine protons, a downfield methine proton (δH 5.04, 1H, ddd, J = 2.4, 2.4, 2.4 Hz, H-6), a pair of low field methylene protons (δH 3.52, 1H, d, J = 12.0 Hz; 3.84, 1H, d, J = 12.0 Hz, H-20a/b), and a hydroxy proton (δH 3.11, 1H, s, OH-11) were observed in the 1H NMR spectrum of 4 (Table 1).
The gross structure of 4 was verified by 2D NMR studies. 1H NMR coupling information in the 1H–1H COSY spectrum of 4 enabled identification of C2–C3–C4, C6–C7, C12–C13–C14, and C17–C18 units, which were assembled with the assistance of an HMBC experiment (Fig. 3). The HMBC between protons and non-protonated carbons of 4, such as H-2, H-9, H-10, H3-15/C-1; H-4, H-10, H3-18/C-8; H-9, H-10, H-20a/C-11; and H-17, H3-18/C-19, permitted elucidation of the carbon skeleton. An exocyclic double bond attached at C-5 was confirmed by the allylic coupling between H2-16 and H-6 in the 1H–1H COSY experiment and by the HMBC between H-16a/C-4, C-6; and H-16b/C-6. The ring junction C-15 methyl group was positioned at C-1 from the HMBC between H3-15/C-1, C-2, C-10, C-14. The presence of a hydroxy group at C-11 was deduced from the HMBC between a hydroxy proton (δH 3.11) with C-10 methine (δC 41.6). The acetate ester at C-9 was established by a correlation between H-9 (δH 6.40) and the acetate carbonyl (δC 169.6) observed in the HMBC spectrum. Thus, the remaining four acetoxy groups should be positioned at C-2, C-3, C-12, and C-14, as indicated by the characteristic NMR signal analysis of the oxymethines CH-2 (δH 5.48/δC 73.1), CH-3 (δH 6.19/δC 63.8), CH-12 (δH 5.52/δC 68.6), and CH-14 (δH 4.83/δC 72.6), respectively, although no HMBC was observed between the oxymethine protons H-2, H-3, H-12, and H-14 and those acetate carbonyls.
 | ||
| Fig. 3 Key HMBC and COSY correlations of 4. | ||
The intensity of sodiated molecules (M + 2 + Na)+ and (M + 4 + Na)+ isotope peaks observed in (+)-ESIMS spectrum [(M + Na)+:(M + 2 + Na)+:(M + 4 + Na)+ = 9:6:1] were strong evidence of the presence of two chlorine atoms in 4. The methine unit at δC 53.9 was more shielded than that expected for an oxygenated C-atom and was correlated to the methine proton at δH 5.04 in the HSQC spectrum and this proton signal was 3J-correlated with H-7 (δH 4.37) (J = 2.4 Hz), proving the attachment of a chlorine atom at C-6. In addition, the methylene unit at δC 48.3 was also more shielded than that expected for an oxygenated C-atom and was correlated to the methylene protons at δH 3.52 and 3.84 in the HSQC spectrum and one of the methylene proton signals (δH 3.52, H-20a) exhibited HMBC with C-11 and C-12, proving the attachment of a chloromethyl group at C-11. Furthermore, an HMBC between H-4 (δH 4.47) and an oxygenated quaternary carbon at δC 83.9 (C-8) suggested the presence of a C-4/8 ether linkage.
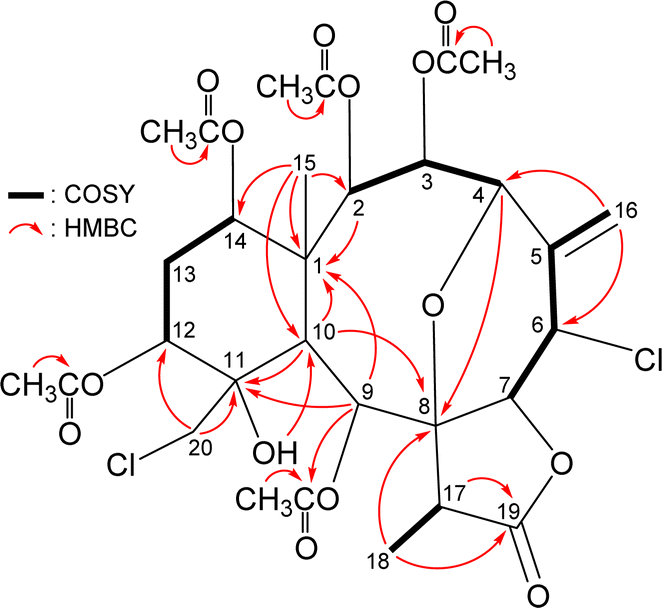
The relative stereochemistry of 4 was elucidated by analysis of NOESY correlations and by vicinal 1H–1H proton coupling constants analysis. In the NOESY experiment (Fig. 4), H-10 correlated with H-2, H-9, and H3-18 indicated that these protons were situated on the same face; they were assigned as α-protons, as C-15 methyl was β-oriented at C-1 and H3-15 did not show correlation with H-10. Also, no coupling was found between H-9 and H-10, indicating that the dihedral angle between these two protons was approximately 90°, further confirmed that H-9 had an α-orientation. Due to H-14 proton being correlated with H3-15, this proton was of a β-orientation at C-14. The C-13 methylene protons displayed identical coupling constants with H-14 (J = 3.0, 3.0 Hz) and H-12 (J = 3.0, 3.0 Hz), respectively, indicating that both H-14 and H-12 should be positioned on the β-equatorial direction in the six-membered ring of 4.
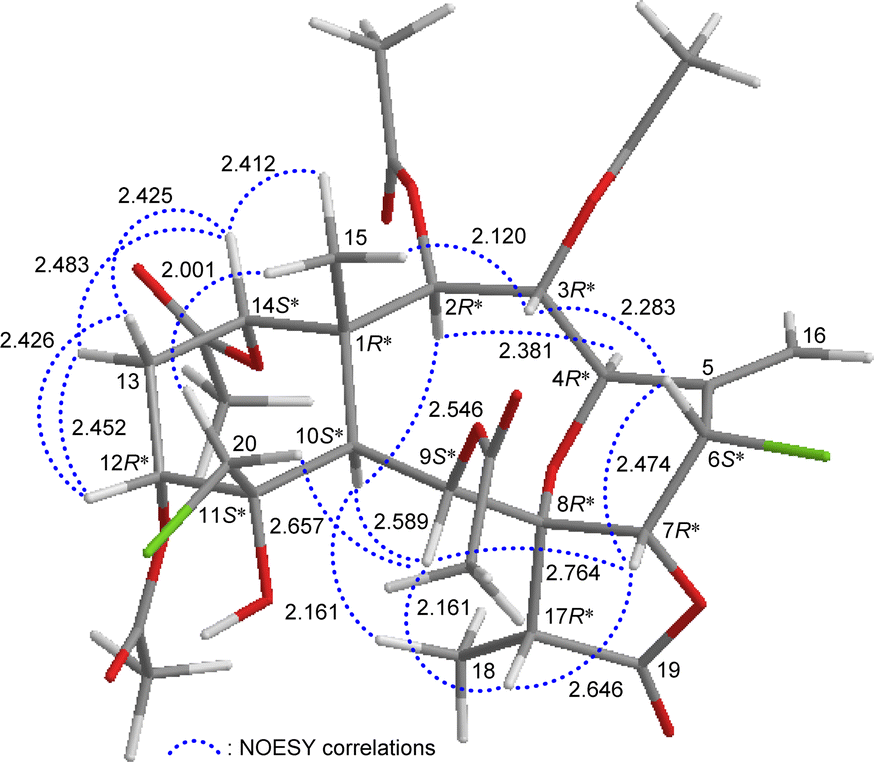 | ||
| Fig. 4 Stereo-view of 4 (generated by computer modeling) and calculated distances (Å) between selected protons with key NOESY correlations. | ||
The oxymethine proton H-3 and one of the chlorinated C-20 methylene protons (δH 3.84, H-20b) were found to exhibit responses with H3-15 but not with H-10, revealing H-3 and C-20 methylene were β-oriented at C-3 and C-11, respectively. H-9 was found to show correlations with H-7, H-17, and one proton of C-20 methylene protons (δH 3.52, H-20a). From modeling analysis, H-9 was found to be reasonably close with H-7, H-17, and H-20a and can therefore be placed on the α face in the 10-membered ring and both H-7 and H-17 are β-oriented in the γ-lactone moiety. H-7 exhibited interactions with H-6 and H-17; and H-6 correlated with H-3, indicating that H-7 and H-6 are on the β face. Furthermore, H-4 showed a correlation with H-2; and a large coupling constant was found between H-4 and H-3 (J = 10.8 Hz), indicating the dihedral angle between H-4 and H-3 is approximately 180° and H-4 has an α-orientation at C-4. The above interpretation enables the identification of the relative configuration of all stereogenic centers of 4 as 1R*, 2R*, 3R*, 4R*, 6S*, 7R*, 8R*, 9S*, 10S*, 11S*, 12R*, 14S*, 17R*. According to the above and comparing the NMR data of 4 with those of the literature, the structure of 4 was similar to that of fragilide F (3) (Fig. 1),5 except for the 12α-proton in 3 was instead of an acetoxy group in 4. Hence, 4 was found to be the 12α-acetoxy derivative of 3 and named 12α-acetoxyfragilide F.
12α-Acetoxyjunceellin (5) was isolated as an amorphous powder. Its (+)-HRESIMS peak was at m/z 663.18121, consistent with the molecular formula C30H37ClO13 (calcd for C30H37ClO13 + Na, 663.18149) with 12 degrees of unsaturation. The IR spectrum of 5 contained signals of γ-lactone (νmax 1791 cm−1) and ester (νmax 1740 cm−1) functionalities. Analyzing the 1H NMR (Table 2), HSQC, and HMBC spectra of 5 led to the assignment of five acetoxy groups; as well as two exocyclic carbon–carbon double bonds, a γ-lactone moiety, and other 15 carbon signals (Table 2).
| Position | δHa (J in Hz) | δCb, Mult.c |
|---|---|---|
| a Spectra recorded at 600 MHz in CDCl3 at 25 °C.b Spectra recorded at 150 MHz in CDCl3 at 25 °C.c Data assigned with the assistance of HSQC and HMBC spectra.d Signals overlapped.e The coupling pattern and coupling constant for H-12 were assigned by its vicinal couplings with H-13α/β.f The coupling pattern and coupling constant for H-16a/b were assigned by their allylic long-range 4J-coupling with H-6.g Signals overlapped. | ||
| 1 | 47.1, C | |
| 2 | 5.55 d (6.6) | 72.6, CH |
| 3 | 6.16 dd (10.8, 6.6) | 63.6, CH |
| 4 | 4.51 d (10.8) | 79.0, CHg |
| 5 | 134.1, C | |
| 6 | 5.01 ddd (3.0, 1.8, 1.8) | 53.8, CH |
| 7 | 4.52 d (3.0) | 79.0, CHg |
| 8 | 82.8, C | |
| 9 | 5.89 s | 77.5, CH |
| 10 | 3.56 s | 39.7, CH |
| 11 | 144.5, C | |
| 12 | 5.38 dd (3.6, 3.6)de | 74.8, CH |
| 13α/β | 2.19 ddd (16.2, 3.6, 3.0); 1.90 ddd (16.2, 3.6, 2.4) | 30.6, CH2 |
| 14 | 4.93 dd (3.0, 2.4) | 73.6, CH |
| 15 | 1.14 s | 14.5, CH3 |
| 16a/b | 5.38 d (1.8)df; 5.59 d (1.8)f | 119.7, CH2 |
| 17 | 2.78 q (7.2) | 49.9, CH |
| 18 | 1.35 d (7.2) | 6.9, CH3 |
| 19 | 173.9, C | |
| 20a/b | 5.44 s; 5.03 br s | 117.5, CH2 |
| Acetoxy groups | 2.34 s | 21.2, CH3 |
| 169.8, C | ||
| 2.05 s | 21.0, CH3 | |
| 170.7, C | ||
| 2.05 s | 21.0, CH3 | |
| 169.4, C | ||
| 2.01 s | 21.0, CH3 | |
| 170.4, C | ||
| 2.01 s | 21.0, CH3 | |
| 169.6, C | ||
The carbon skeleton of 5 was fully established by following correlations observed in the 1H–1H COSY and HMBC spectra (Fig. 5). The oxymethine protons H-3 (δH 6.16), H-9 (δH 5.89), and H-2 (δH 5.55) showed HMBC to the acetate carbonyls at δC 169.6, 169.8, and 170.4, confirmed the position of acetoxy groups at C-3, C-9, and C-2, respectively. Evaluated on the NMR chemical shifts of oxymethines CH-12 (δH 5.38/δC 74.8) and CH-14 (δH 4.93/δC 73.6), the remaining acetoxy groups should be positioned at C-12 and C-14, respectively.
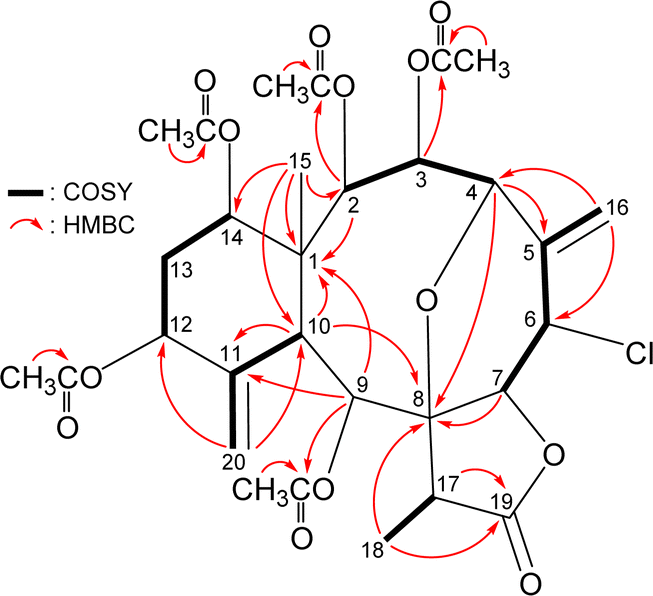 | ||
| Fig. 5 Key HMBC and COSY correlations of 5. | ||
The relative stereochemistry of 5 was established by analyzing the NOESY information in combination with the computer-generated model structure. We have noticed that all naturally-occurring briaranes possess a β-Me-15 placed at C-1 and have an α-orientation of H-10. In the NOESY spectrum (Fig. 6), H-10 showed correlations with H-2, H-9, and H3-18; H3-15 was correlated with H-3, H-14, and one of the C-13 methylene protons (δH 1.90, H-13β); and H-13β was correlated with H-12, proving the α-orientation of OAc-3, OAc-12, and OAc-14; and β-orientation of OAc-2 and OAc-9. H-3 exhibited an interaction with H-6; and H-6 correlated with H-7, indicating that H-6 and H-7 are on the β face. Furthermore, H-4 showed a correlation with H-2; and a large coupling constant was found between H-4 and H-3 (J = 10.8 Hz), indicating the dihedral angle between H-4 and H-3 is approximately 180° and H-4 has an α-orientation at C-4. Additionally, there was a correlation between H-7 and H-17, suggesting that H-17 is situated on the β face in the γ-lactone moiety. The above interpretation enables the identification of the relative configuration of all stereogenic centers of 5 as 1R*, 2R*, 3R*, 4R*, 6S*, 7R*, 8R*, 9S*, 10S*, 12R*, 14S*, 17R*. It was found that the NMR signals of 5 were similar to those of a known briarane, junceellin (6),7,8 except that the signals corresponding to the α-proton at C-12 in 6 were replaced by signals for an acetoxy group in 5. Thus, 5 was found to be the 12α-acetoxy derivative of 6 and named 12α-acetoxyjunceellin.
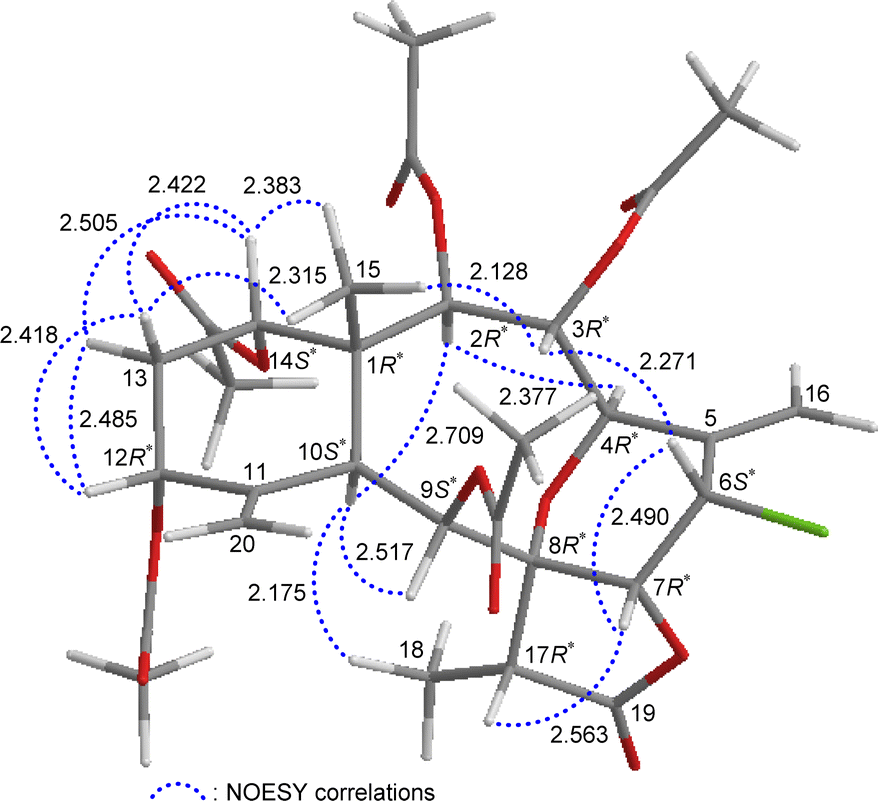 | ||
| Fig. 6 Stereo-view of 5 (generated by computer modeling) and calculated distances (Å) between selected protons with key NOESY correlations. | ||
As briaranes 4 and 5, in addition to 1 and 2, were isolated from the same target organism, J. fragilis, it is reasonable to assume on biogenetic grounds that briaranes 4 and 5 have the same absolute configuration as 1 and 2. Therefore, the absolute configurations of 4 and 5 were suggested to be (1R, 2R, 3R, 4R, 6S, 7R, 8R, 9S, 10S, 11S, 12R, 14S, 17R) and (1R, 2R, 3R, 4R, 6S, 7R, 8R, 9S, 10S, 12R, 14S, 17R), respectively.
Previous studies have found briarane-type natural products to be a natural remedy for osteoclastogenic disease.9,10 Via an ALP ELISA assay with MG63 human mesenchymal stem cells (Table 3), the study found that briaranes 1 and 3–5 were active in enhancing ALP activity at a concentration of 10 μM.
| Compounds | ALP activity (king unit per mg prot.) |
|---|---|
| a Data are expressed with the mean standard error of the mean (SEM) (n = 3). The significance was determined with Student's t-test. **p < 0.01, ***p < 0.001 and comparison with untreated cells. | |
| 1 | 4.2 ± 1.0** |
| 2 | 2.9 ± 1.1** |
| 3 | 6.6 ± 0.2*** |
| 4 | 5.8 ± 0.5*** |
| 5 | 4.8 ± 0.7*** |
| Rutin | 3.1 ± 0.2 |
| Control | −5.0 ± 0.5 |
3 Conclusions
The octocorals belonging to the genus Junceella have demonstrated a wide structural diversity of briarane diterpenoids with various pharmacological properties.2 In our ongoing research on J. fragilis, we isolated five chlorinated briaranes, including two previously undiscovered briaranes: 12α-acetoxyfragilide F (4) and 12α-acetoxyjunceellin (5). Three known analogs were also identified: gemmacolidex (1),3 frajunolide I (2),4 and fragilide F (3).5 The structures, including the absolute configurations, of 1 and 2 were further established through single-crystal X-ray diffraction analysis. For compounds 4 and 5, their structures were confirmed using various spectroscopic techniques, particularly 2D NMR experiments and comparison with existing literature data. Briaranes 1 and 3–5 were active in enhancing ALP activity.4 Experimental
4.1 General experimental procedures
Optical rotation values were measured using a JASCO P-1010 digital polarimeter. IR spectra were obtained with a Thermo Scientific Nicolet iS5 FT-IR spectrophotometer. NMR spectra were recorded on a 600 MHz Jeol ECZ NMR spectrometer using the residual CHCl3 (δH 7.26 ppm) and CDCl3 (δC 77.0 ppm) as internal standards for 1H and 13C NMR, respectively; coupling constants (J) are presented in Hertz (Hz). The ESIMS and HRESIMS spectra were ascertained with Thermo Fisher orbitrap Exploris 120 mass spectrometer equipped with an ESI ion source in positive ionization mode. The extracted samples were separated via column chromatography with silica gel (particle size, 230–400 mesh; Merck). TLC was performed on plates precoated with silica gel 60 (DC-Fertigfolien Alugram Xtra SIL G/UV254, layer thickness 0.20 mm, Macherey-Nagel) and RP-18 F254s (layer thickness 0.16–0.20 mm, Merck), and visualization of the TLC plates was conducted using an aqueous solution of 10% H2SO4, subsequently to be heated to show the spots of signals. Reverse-phase HPLC (RP-HPLC) separation was carried out with a system containing a pump (Hitachi, model L-7110) with a photo-diode array detector (Hitachi, model L-2400), equipped with a reverse-phase column (Luna, 5 μm, C18(2) 100 Å, 250 × 21.2 mm). Normal-phase HPLC (NP-HPLC) separation was carried out with a system containing a pump (Hitachi, model L-5110), equipped with a normal-phase column (Galaksil, EF-SiO2, 5 μm 120 Å, 250 × 10 mm).4.2 Animal material
Specimen of J. fragilis was collected manually via SCUBA diving off the coast of Southern Taiwan in 2012. A voucher specimen was deposited at the National Museum of Marine Biology & Aquarium, Taiwan. To identify the species, we compared its physical characteristics and microscopic images of the coral sclerites with those mentioned in previous studies.1,11–134.3 Extraction and isolation
The freeze-dried specimen (wet/dry weight = 6.01/2.39 kg) was sliced and treated with a 1:1 mixture solvent of MeOH and CH2Cl2 at room temperature to produce crude extract weighing 140.1 g, which was then subjected to liquid–liquid partition between EtOAc and H2O. The EtOAc phase (19.2 g) was applied to a silica gel column chromatography (Si. C. C.). Elution was carried out with a gradient solvent system containing n-hexane, followed by increasing polarity mixtures of n-hexane and EtOAc, pure acetone, and pure methanol for use as eluting solvents. The process yielded 13 fractions A–M. Fraction D was chromatographed with NP-HPLC via an isocratic solvent system, n-hexane/acetone mixture (6:1). The process yielded 7 fractions D1–D7. Fraction D6 was separated with NP-HPLC via an isocratic solvent system, DCM/acetone mixture (18:1). The process yielded 3 fractions D6A–D6C. Fraction D6A was purified with RP-HPLC via an isocratic solvent system, ACN/H2O mixture (60:40; flow rate = 3 mL min−1), to afford 3 (0.4 mg). Fraction E was chromatographed with Si. C.C. and eluted with an isocratic solvent system, DCM/acetone mixture (10:1). The process yielded 8 fractions E1–E8. Fraction E2 was separated by NP-HPLC via an isocratic solvent system, n-hexane/EtOAC mixture (2:1) to obtain 5 fractions E2A–E2E. Fraction E2E was purified by RP-HPLC with an isocratic solvent system, ACN/H2O mixture (50:50; flow rate = 3 mL min−1), to obtain 2 (0.3 mg). Fraction E2D was purified by RP-HPLC with an isocratic solvent system, ACN/H2O mixture (60:40; flow rate = 3 mL min−1), to obtain 5 (0.4 mg). Fraction F was separated on Si. C. C. with an isocratic solvent system, DCM/acetone mixture (15:1). The process yielded 8 fractions F1–F8. Fraction F4 was separated Si C. C. and eluted with DCM/EtOAc mixture (20:1) to yield 6 fraction F4A–F4F. Fraction F4E was purified by RP-HPLC with an isocratic solvent system, MeOH/H2O mixture (70:30; flow rate = 3 mL min−1) to obtain 1 (0.8 mg) and 4 (0.4 mg), respectively.
4.4 Structural characterization of undescribed compounds
4.5 Single-crystal X-ray crystallography of gemmacolide X (1)
Suitable colorless prisms of 1 were obtained from a solution of MeOH. The crystal (0.363 × 0.235 × 0.198 mm3) was identified as being of the orthorhombic system, space group P212121 (#19),14 with a = 12.2945(2) Å, b = 12.4454(2) Å, c = 22.2997(4) Å, V = 3412.08(10) Å3, Z = 4, Dcalcd = 1.297 Mg m−3 and λ (Cu Kα) = 1.54178 Å. Intensity data were obtained on a crystal diffractometer (Bruker, model: D8 Venture) up to a θmax of 68.349°. All measurement data of 34394 reflections were collected, of which 6232 were independent. The structure was solved by direct methods and refined by a full-matrix least-squares on F2 procedure.15,16 The refined structural model converged to a final R1 = 0.0292; wR2 = 0.0758 for 5969 observed reflections [I > 2σ(I)] and 425 variable parameters; and the absolute configuration was established from the Flack parameter x = 0.000(5).6,17,18 Crystallographic data for the structure of gemmacolide X (1) were submitted to the Cambridge Crystallographic Data Center (CCDC) with ESI publication number CCDC 2323829 (data can be obtained from the CCDC website at https://www.ccdc.cam.ac.uk/conts/retrieving.html).
4.6 Single-crystal X-ray crystallography of frajunolide I (2)
Suitable colorless prisms of 2 were obtained from a solution of MeOH. The crystal (0.191 × 0.102 × 0.049 mm3) was identified as being of the hexagonal system, space group P61 (#169),14 with a = b = 22.8317(3) Å, c = 10.2937(2) Å, V = 4647.06(15) Å3, Z = 6, Dcalcd = 1.284 Mg m−3 and λ (Cu Kα) = 1.54178 Å. Intensity data were obtained on a crystal diffractometer (Bruker, model: D8 Venture) up to a θmax of 74.402°. All measurement data of 47636 reflections were collected, of which 6221 were independent. The structure was solved by direct methods and refined by a full-matrix least-squares on F2 procedure.15,16 The refined structural model converged to a final R1 = 0.0358; wR2 = 0.0960 for 5789 observed reflections [I > 2σ(I)] and 377 variable parameters; and the absolute configuration was established from the Flack parameter x = −0.002(13).6,17,18 Crystallographic data for the structure of frajunolide I (2) were submitted to CCDC with ESI publication number CCDC 2326820 (data can be obtained from the CCDC website at https://www.ccdc.cam.ac.uk/conts/retrieving.html).
4.7 Alkaline phosphatase (ALP) activity assay
The ALP assay was released to assess the activity of compounds 1–5 from MG63 human mesenchymal stem cells, in line with suggestion of previous studies.19Author contributions
Hai Nhat Do and Yu-Ta Chen: methodology, software, analysis, investigation, data curation, original draft writing. Su-Ying Chien: analysis, investigation. You-Ying Chen: investigation. Mingzi M. Zhang, Lun Kelvin Tsou, Jih-Jung Chen, Zhi-Hong Wen, and Yi-Hao Lo: methodology, software, analysis. Li-Guo Zheng and Ping-Jyun Sung: conceptualization, resources, draft review & editing, visualization, supervision, project administration, fundraising.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Ms. Hsiao-Ching Yu and Chao-Lien Ho, of the High Valued Instrument Center, National Sun Yat-sen University, for the mass (MS 006500) and NMR (NMR 001100) spectra (NSTC 112-2740-M-110-002), and to the Instrumentation Center, National Taiwan University, for providing X-ray facilities (NSTC 112-2740-M-002-006, XRD 000200). This research has been principally supported by grants from the National Museum of Marine Biology & Aquarium, the National Science and Technology Council (NSTC 111-2320-B-291-001, 112-2320-B-291-001, and 112-2811-B-291-002) and the Zuoying Armed Forces General Hospital (KAFGH-ZY-A-112020), Taiwan, awarded to Yu-Ta Chen and Ping-Jyun Sung. All funding is gratefully acknowledged.Notes and references
- C. S. McFadden, L. P. van Ofwegen and A. M. Quattrini, Revisionary systematics of octocorallia (Cnidaria: Anthozoa) guided by phylogenomics, Bull. Soc. Syst. Biol., 2022, 1, 8735 Search PubMed.
- H.-M. Chung, Y.-C. Wang, C.-C. Tseng, N.-F. Chen, Z.-H. Wen, L.-S. Fang, T.-L. Hwang, Y.-C. Wu and P.-J. Sung, Natural product chemistry of gorgonian corals of genus Junceella–part III, Mar. Drugs, 2018, 16, 339 CrossRef PubMed.
- C. Li, M.-P. La, H. Tang, W.-H. Pan, P. Sun, K. Krohn, Y.-H. Yi, L. Li and W. Zhang, Bioactive briarane diterpenoids from the South China Sea gorgonian Dichotella gemmacea, Bioorg. Med. Chem. Lett., 2012, 22, 4368–4372 CrossRef CAS PubMed.
- C.-C. Liaw, Y.-C. Shen, Y.-S. Lin, T.-L. Hwang, Y.-H. Kuo and A. T. Khalil, Frajunolides E–K, briarane diterpenes from Junceella fragilis, J. Nat. Prod., 2008, 71, 1551–1556 CrossRef CAS PubMed.
- P.-J. Sung, S.-H. Wang, M. Y. Chiang, Y.-D. Su, Y.-C. Chang, W.-P. Hu, C.-Y. Tai and C.-Y. Liu, Discovery of new chlorinated briaranes from Junceella fragilis, Bull. Chem. Soc. Jpn., 2009, 82, 1426–1432 CrossRef CAS.
- S. Parsons, H. D. Flack and T. Wagner, Use of intensity quotients and differences in absolute structure refinement, Acta Crystallogr., 2013, B69, 249–259 Search PubMed.
- Y. Lin and K. Long, Studies of the chemical constituents of the Chinese gorgonian (IV)—Junceellin, a new chlorine-containing diterpenoid from Junceella squamata, Zhongshan Daxue Xuebao, Ziran Kexueban Acta Sci. Nat. Univ. Sunyatseni, 1983, 22(2), 46–51 Search PubMed.
- J. Shin, M. Park and W. Fenical, The junceellolides, new anti-inflammatory diterpenoids of the briarane class from the Chinese gorgonian Junceella fragilis, Tetrahedron, 1989, 45, 1633–1638 CrossRef CAS.
- Y.-Y. Lin, Y.-H. Jean, H.-P. Lee, S.-C. Lin, C.-Y. Pan, W.-F. Chen, S.-F. Wu, J.-H. Su, K.-H. Tsui, J.-H. Sheu, P.-J. Sung and Z.-H. Wen, Excavatolide B attenuates rheumatoid arthritis through the Inhibition of osteoclastogenesis, Mar. Drugs, 2017, 15, 9 CrossRef PubMed.
- X. Qi, X. Zhang, J. Meng, J. Wu, W. Cheng, J. Huang and W. Lin, Briarane-type diterpenoids, the inhibitors of osteoclast formation by interrupting Keap1-Nrf2 interaction and activating Nrf2 pathway, Eur. J. Med. Chem., 2023, 246, 114948 CrossRef CAS PubMed.
- C.-F. Dai, Octocoral Fauna of Taiwan, Ocean Center, National Taiwan University, Taipei, Taiwan, 2019, pp. 636–637 Search PubMed.
- C.-F. Dai and C.-H. Chin, Octocoral Fauna of Kenting National Park, Kenting National Park Headquarters, Kenting, Pingtung, 2019, pp. 492–493 Search PubMed.
- C.-F. Dai, Corals of Taiwan, Octocorallia, Owl Publishing House Co., Ltd, Taipei, 2022, vol. 2, p. 381 Search PubMed.
- D. Hestenes and J. W. Holt, Crystallographic space groups in geometric algebra, J. Math. Phys., 2007, 48, 023514 CrossRef.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, SHELXT-Integrated space-group and crystal-structure determination, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- H. D. Flack, On enantiomorph-polarity estimation, Acta Crystallogr., 1983, A39, 876–881 CrossRef CAS.
- H. D. Flack and G. Bernardinelli, Absolute structure and absolute configuration, Acta Crystallogr., 1999, A55, 908–915 CrossRef CAS PubMed.
- Y. Wang, B. Kong, X. Chen, R. Liu, Y. Zhao, Z. Gu and Q. Jiang, BMSC exosome-enriched acellular fish scale scaffolds promote bone regeneration, J. Nanobiotechnol., 2022, 20, 444 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESIMS, 1D and 2D NMR spectra of 4 and 5; X-ray crystallographic data of 1 and 2. CCDC 2323829 and 2326820, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra03062a |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |