
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cellulose monolithic stir bar for sorptive extraction of glycerol from biodiesel†
Pablo H. S. Martinsa,
Maria A. Barrosa,
Caroline L. Silvaa,
Poliana Riccia,
Laís M. B. Castilhoa,
Allyson L. R. Santos a,
Hugo S. Rodriguesb,
Rosana M. N. Assunçãoab and
Anizio M. Faria*ab
a,
Hugo S. Rodriguesb,
Rosana M. N. Assunçãoab and
Anizio M. Faria*ab
aInstituto de Química, Universidade Federal de Uberlândia, 38304-402 Uberlândia, MG, Brazil. E-mail: anizio@ufu.br
bInstituto de Ciências Exatas e Naturais do Pontal, Universidade Federal de Uberlândia, 38304-402 Ituiutaba, MG, Brazil
First published on 29th May 2024
Abstract
This work presents an eco-friendly approach for determining free glycerol in biodiesel samples, using a cellulose monolith stir bar in the sorptive extraction method with analysis by high-performance liquid chromatography and a refractive index detector. The cellulose monolith was produced from cellulose acetate by non-solvent-induced phase separation and subsequent alkaline deacetylation. The cellulose monolith presented a hierarchically porous structure, with 68% porosity and almost total deacetylation, with morphological and polarity characteristics that favor an efficient extraction of free glycerol from biodiesel. The sorptive extraction method using a cellulose monolith stir bar was optimized, obtaining a total extraction time of 30 min at 70 °C, using ultrapure water as the desorption solvent, and extraction of free glycerol of 93.6 ± 2.3%. The proposed method showed selectivity in free glycerol extraction, with limits of detection and quantification of 6.60 × 10−5% w/w and 2.18 × 10−4% w/w, respectively. Compared with the official reference method, the proposed one presented similar precision and accuracy, with few manipulations and any reagent/solvents. Furthermore, it is compatible with the principles of green chemistry and can be considered an eco-friendly method for determining free glycerol in biodiesel.
Introduction
Biodiesel is a renewable fuel obtained by the transesterification reaction of vegetable oils and animal fat. Through this process, the triglycerides present in oils and animal fat react with primary alcohol, commonly methanol, under homogeneous alkaline catalysis, generating a mixture of fatty acid esters (biodiesel) and glycerol (1,2,3-propanetriol) as products in two immiscible phases with each other. The ester mixture can be marketed as biodiesel, intended for application in compression ignition engines only after the rigid purification processes.1–3 Purification is necessary to remove reagents and catalysts, especially glycerol. Removal of glycerol is desirable since it can cause occlusion of fuel filters, impairing the engine motors. Glycerol may also lead to the emission of hazardous substances during combustion, like acrolein, causing corrosion.4–7 Thus, the amount of glycerol and glycerides is critical in determining fuel quality. The maximum content of free glycerol allowed in biodiesel is 200 mg kg−1 or 0.02% (w/w), according to the regulations established in several countries.8–10 Consequently, quality control of this compound in biodiesel is essential.The official American Oil Chemists' Society (AOCS) method Ca is recommended to determine free glycerol in biodiesel. 4-56,11 and American Society for Testing and Materials (ASTM) D6584-21,12 and European Norm (EN) 14105:2020 (ref. 13) standard test methods. However, the AOCS 4-56 method is a volumetric one that uses several reagents, some with considerable toxicity, for example, sodium arsenite. In addition, this method is highly laborious and dependent on the analyst. ASTM D6584 and EN 14105 methods are gas chromatographic (GC) methods that require expensive derivatization agents and internal standards. In addition, the GC methods use high temperatures for the column oven and detector, close to the temperature limits for these components, and require thermally stable GC columns.14–17 Although the chromatographic methods are recognized for their accuracy in glycerol quantification, these conditions make their application difficult in most analytical laboratories since they make the process more expensive and less reproducible with the derivatization of glycerol before the chromatographic analysis.5,18
In this context, several methods for determining free glycerol in biodiesel have been proposed, aiming at a more viable, simple, and environmentally friendly.14–31 Among these methods, approaches employing electroanalytical,15,19–21 chromatographic,14,22–24 electrophoretic,17,25 spectrophotometric16,26–30 or volumetric31 techniques have been evaluated. However, most of these methods require the use of chemical reagents and organic solvents, such as derivatization reagents, dissolution solvents, and adsorbents, or specific devices (electrodes, apparatus, glassware, cartridges), which are not accessible and very expensive, maintaining necessary the search for a simple method for the determination of free glycerol in biodiesel.
A strategy that has been consolidated is the previous step of extracting glycerol from biodiesel since the joint determination can be limiting for samples of biodiesel produced from some types of raw materials (vegetable oils and animal fat).14,16,29 Solid phase extraction has been studied for the extraction of glycerol with subsequent quantification of the extract by high-performance liquid chromatography, in which the analysis time is shorter than GC one and sample derivatization is unnecessary.14 Stir-bar sorptive extraction (SBSE) is a promising alternative for free glycerol extraction from biodiesel.32,33 SBSE is an extraction method that uses a stir bar coated with an analyte-selective adsorbent in a complex matrix.34 As main advantages, this method is efficient, requires few steps and little analyst manipulation in the extraction process, and can concentrate the analyte during extraction, providing a gain in detectability to modern instrumentation.32–34 The stir bar coating can ensure analyte selectivity with the proper choice of adsorbent material. For glycerol, cellulose can be highly attractive for its extraction from biodiesel.35–39 Several studies show cellulose has a high adsorption capacity for glycerol due to its similar polarity and a strong tendency to form hydrogen bonds with the glycerol molecule. Cellulose has already been analyzed as a filtration membrane,35 pulp,36,37 and residual biomass4,38 as a biodiesel purification method. The use of cellulose as a stir bar coating can result in a process that is fully compatible with the principles of green chemistry, as in addition to using a biopolymer, which is widely available, it may require only water as a desorption solvent for glycerol extraction of biodiesel.
Due to the lack of a simple and eco-friendly method for the determination of glycerol in biodiesel, in this work, we propose a strategy based on the SBSE extraction of glycerol from biodiesel samples followed by HPLC analyses with a refractive index detector (RID) without the use of any chemical reagents, organic solvents, and sophisticated apparatus or devices. The monolithic cellulose-coated stirring bar will be prepared from cellulose acetate (CA) solutions using non-solvent-induced phase separation (NIPS),39 followed by deacetylation of the CA monolith. Magnets will be embedded in the cellulose monoliths during phase separation, and the bars will be employed for glycerol extraction.
Experimental
Chemicals
The following reagents were used to produce the monolith bars: cellulose acetate, degree of substitution 2.5, and average molecular weight of 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 was kindly provided by Rhodia Solvay Group (Santo André, Brazil). N,N-Dimethylformamide 99.9% (Synth, Diadema, Brazil), n-octyl alcohol P.A., ethanol P.A. 99.5%, n-heptane P.A., and sodium hydroxide in microbeads, all from Dinâmica (São Paulo, Brazil). The macroporosity of cellulosic stirring bars used n-butanol (Dinâmica) as solvent. Throughout the development of the sorptive extraction, P.A. glycerin, nitric acid, and methyl alcohol were used (Êxodo Científica, São Paulo, Brazil). 96% sulfuric acid (Synth) and HPLC-grade acetonitrile (Sigma-Aldrich, Darmstadt, Germany) were used for the mobile phase preparation. Potassium periodate, potassium iodide, sodium arsenite (Êxodo Científica), sodium bicarbonate, and soluble starch P.A. (Synth) were also used.
000 g mol−1 was kindly provided by Rhodia Solvay Group (Santo André, Brazil). N,N-Dimethylformamide 99.9% (Synth, Diadema, Brazil), n-octyl alcohol P.A., ethanol P.A. 99.5%, n-heptane P.A., and sodium hydroxide in microbeads, all from Dinâmica (São Paulo, Brazil). The macroporosity of cellulosic stirring bars used n-butanol (Dinâmica) as solvent. Throughout the development of the sorptive extraction, P.A. glycerin, nitric acid, and methyl alcohol were used (Êxodo Científica, São Paulo, Brazil). 96% sulfuric acid (Synth) and HPLC-grade acetonitrile (Sigma-Aldrich, Darmstadt, Germany) were used for the mobile phase preparation. Potassium periodate, potassium iodide, sodium arsenite (Êxodo Científica), sodium bicarbonate, and soluble starch P.A. (Synth) were also used.
Preparation of cellulose monolith stir bars
Commercial cellulose acetate (CA) with a degree of substitution of 2.5 was chosen as the starting material for cellulose monolith preparations due to its better solubility in common solvents. Therefore, the hierarchically porous CA monoliths were prepared from polymer solutions using non-solvent-induced phase separation (NIPS).39 Approximately 400 mg of cellulose acetate was dissolved in 2.0 mL of N,N-dimethylformamide (DMF) with slow magnetic stirring at room temperature (27 °C). Subsequently, 2.0 mL of n-octanol was added dropwise under constant stirring in a water bath at 70 °C. The solution was stirred until it turned transparent. Next, the transparent solution was poured into test tubes (molds) with an internal diameter of 5 mm, allowing it to reach a height of 10 mm for monolith formation. Neodymium magnets measuring 5 mm × 3 mm i.d. were inserted into the solution and suspended using a metallic wire with a thickness of 0.20 mm (Fig. S1, ESI†). The molds were left undisturbed at room temperature (27 °C) for 72 hours to allow for complete phase separation. After this period, the metallic wire was removed, leaving the magnet securely embedded inside the cellulose acetate monolith stir bar (CAMSB). Solvent exchanges, adding 2.0 mL of ethanol, were performed every eight hours for three days to replace n-octanol with ethanol. Then, CAMSB were removed from the molds and immersed in ethanol for 24 hours.Cellulose monolith stir bars (CMSB) were obtained through the alkaline hydrolysis of CAMSB. The CAMSB were immersed in 10 mL of a 2.0 mol L−1 solution of NaOH in ethanol and degassed for 5 min under a gentle inert flow. The mixtures were left at room temperature (∼27 °C) for 24 h to deacetylate the monoliths. Afterward, the CMSB was carefully removed from the solution and washed with ultrapure water until neutral (pH ∼7). Fig. S2 in ESI† presents a scheme for preparing CAMSB and CMSB.
Characterization of cellulosic monoliths
The CMSB porosity (ε) is defined as the pores volume divided by the total volume of the porous monolith. A small amount of dry cellulose monoliths was immersed in 20 mL of n-butanol, leaving them for 10 min at 27 °C. Every 10 min, the cellulose monoliths were removed from the bath, the excess solvent was dried with a paper towel, and the mass was measured. The measurements were performed for two hours until the wet CMSB masses were stable. The gravimetric method can determine the CMSB porosity (ε) using eqn (1).
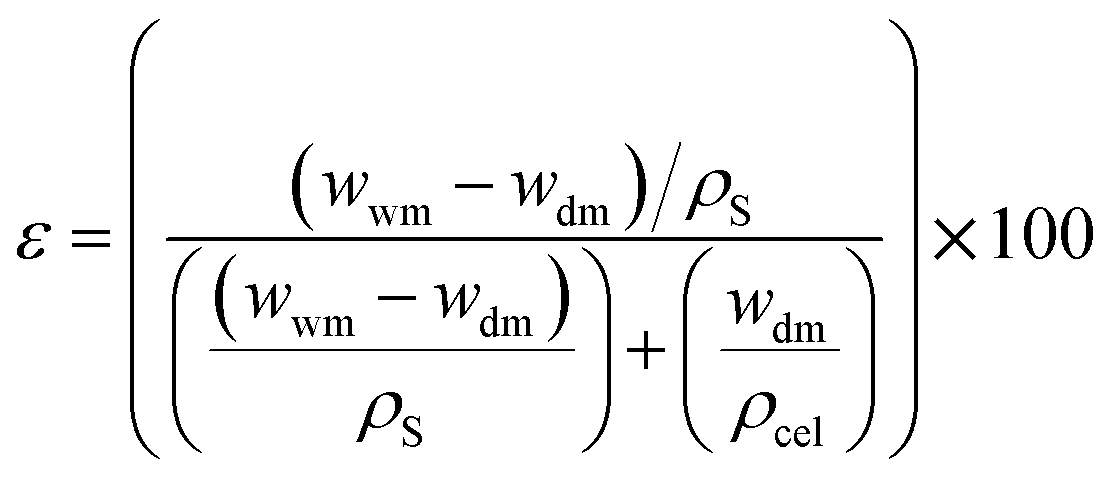 | (1) |
The infrared spectroscopic measurements were obtained using a Cary 630 FTIR spectrometer (Agilent, Santa Clara, CA, USA) with an attenuated total reflectance (ATR) accessory to evaluate spectral differences between CAMSB and CMSB. A small piece of the monoliths, ∼10 mg, was placed directly on the diamond crystal for the measurement. The FTIR spectrum background was corrected using a freshly prepared pure KBr pellet. The IR spectral range was evaluated from 650 to 4000 cm−1 using a resolution of 4 cm−1 and a scan rate of 32 scans min−1.
The thermal stability of CAMSB and CMSB was studied using samples of ∼5 mg, heating from 25 °C to 600 °C at 10 °C min−1 in a nitrogen gas atmosphere, using a TA instruments TGA-55 model (New Castle, DE, USA). The DSC experiments were performed using DSC-25 equipment (TA Instruments) at heating and cooling rates of 10 °C min−1 from 25 °C to 300 °C in a nitrogen gas atmosphere.
Images of the particles were used to evaluate CMSB morphologically. Samples were sputter-coated with gold and examined with a Tescan model Vega3 scanning electron microscope (Brno, Czech Republic) at 20 kV.
Optimization of the SBSE method
The effectiveness of CMSB for the sorptive extraction of glycerol from biodiesel samples was assessed. Methyl oleate was used as a matrix simulacrum for the biodiesel sample.40 The samples were spiked with 0.020% w/w of glycerol. Therefore, all the glycerol in the biodiesel sample originated solely from the fortification.The SBSE method, using the CMSB to extract glycerol from the biodiesel sample, was optimized to evaluate the adsorption and desorption time, desorption solvent, and temperatures of sorption and desorption. The extraction procedure initiates by contacting 5.0 mL of the spiked biodiesel sample with 0.02% w/w glycerol under magnetic stirring at 200 rpm with a CMSB for different times (15 min, 30 min, 60 min, and 2880 min) at various temperatures (30, 40, 60 and 70 °C). After that, excess methyl oleate from the CMSB was removed with paper towels. Then, the CMSB was placed in contact with 5.0 mL of desorption solvent (water, methanol, 0.02 mol L−1 NaOH sol, 0.02 mol L−1 HNO3 sol.), maintaining magnetic stirring at 200 rpm for different times (15 min, 30 min, 60 min, and 2880 min) under different temperatures (30, 40, 60 and 70 °C). Subsequently, HPLC-RID analyzed a 1.0 mL aliquot of the extract.
The reuse of CMSB was evaluated in the extraction of glycerol from biodiesel samples. A volume of biodiesel fortified with 0.020% w/w glycerol was prepared and divided into 5 mL portions. Triplicates of the portions were extracted using CMSB in the optimized SBSE method. After extraction, the CMSB was placed in 1 L of ultrapure water for 24 h, under gentle stirring. To evaluate total removal, the CMSB was placed in 5 mL of water at 70 °C for 15 min and analyzed by HPLC-RID. After that, the CMSB was again subjected to extraction of new portions of biodiesel fortified with glycerol, analyzing the extract by HPLC-RID. This procedure was repeated four times.
HPLC-RID analysis
Glycerol extracts were analyzed with a Waters HPLC system, model Alliance e2695 (Milford, MA, USA), equipped with an autosampler and refractive index detector (RID 2414). Chromatographic data were collected and recorded using the Empower3® software. The optimized separation conditions of glycerol were obtained in an Aminex HPX-87H column (300 mm × 7.6 mm ID, 9 μm particle size, 8% cross-linkage) at 35 °C using 5.0 mmol L−1 of sulfuric acid as isocratic mobile phase at a flow rate of 0.6 mL min−1. The refractive index detector was set at 35 °C with sensitivity = 16 and Hamming filter TC = 2.Standard glycerol solutions were prepared in ultrapure water at concentrations of 0.00022, 0.00044, 0.00087, 0.00435, 0.00870, 0.01740, and 0.03480% w/w for the analytical curve. The limits of detection and quantification of glycerol were obtained by sequentially injecting aqueous glycerol solutions in decreasing concentrations until a signal of 3× and 10× the noise level of a blank sample in the glycerol retention time, respectively.
Determination of glycerol by standard AOCS 14-56 method
The official AOCS (American Oil Chemist's Society) method Ca determined the free glycerol in biodiesel samples as a comparing method. 14-56,11 using an iodometric-periodic acid method. After the decantation step, the free glycerol was reacted with periodic acid. The resulting compound was titrated with a normalized sodium arsenite solution using an aqueous starch (1.0% w/v) indicator solution. The free glycerol was calculated based on eqn (2).
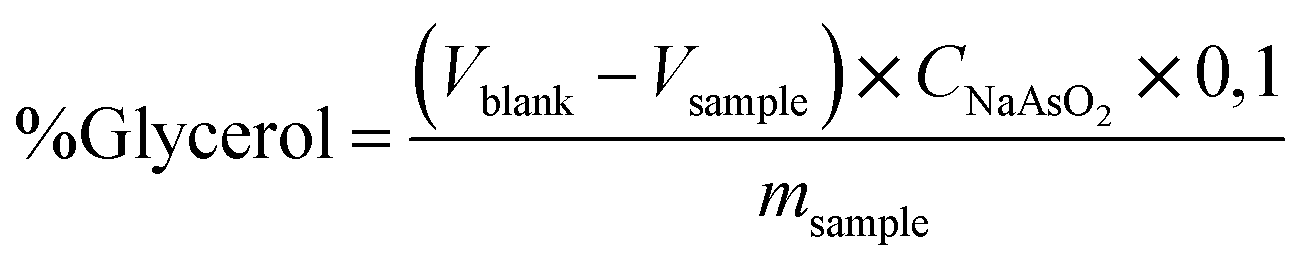 | (2) |
Application of the SBSE method for glycerol in biodiesel (B100) samples.
The proposed method for determining glycerol in biodiesel was applied to two biodiesel (b100) samples obtained from the transesterification of soybean oil with methanol and ethanol,41 resulting in methylic and ethylic soybean biodiesel samples. The total fatty acid esters and composition of biodiesel samples were obtained by gas chromatography-flame ionization detection using the EN: 14103 method.42 Samples without and with 0.02% w/w glycerol fortification were analyzed by optimized SBSE-HPLC-RID and standard AOCS 14-56 methods.
Results and discussion
Characterization of cellulosic monoliths
The monoliths were produced from the dissolution of cellulose acetate in N,N-dimethylformamide, using the non-solvent-induced phase separation (NIPS). NIPS is a method based on the non-solvent addition to the system, separating the phases. The non-solvent used was octanol since it results in monoliths with a hierarchically porous organic structure and does not compress when dry. After the phase separation, octanol was removed by solvent exchange with ethanol. As cellulose acetate has hydrophobic characteristics,43 it was necessary to convert to cellulose monolith by deacetylation reaction. This conversion was monitored by differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), Fourier transform infrared spectroscopy (FTIR), and scanning electron microscopy (SEM).Differential Scanning Calorimetry (DSC) was used to characterize the cellulosic monoliths through the heat flux in the monoliths, analyzing their phase transitions, changes in their properties, or their degradation. Fig. 1 shows the DSC curves for the cellulose acetate and cellulose monoliths.
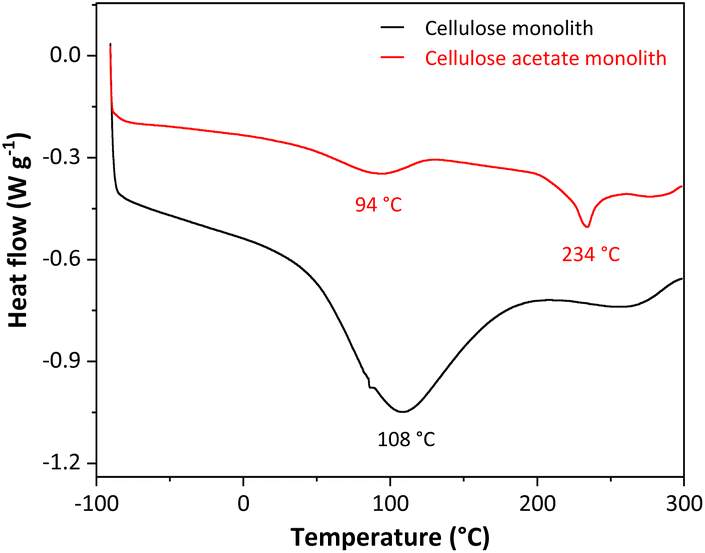 | ||
| Fig. 1 DSC curves for the cellulose acetate and cellulose monoliths. | ||
The two DSC curves show a primary thermal event related to the water outflow from the monoliths at 94 °C for cellulose acetate and 108 °C for cellulose as a high-intensity endothermic peak. A second endothermic peak on the DSC curve for the cellulose acetate monolith (CAM) was recorded at 234 °C and is associated with the melting of cellulose acetate.44 This same endothermic peak is not observed in the DSC curve for the cellulose monolith (CM), as expected, since the melting temperature of cellulose is above 300 °C.45
Thermogravimetric analysis of the monoliths was also performed, observing different TG curves for cellulose acetate and cellulose (Fig. 3S, ESI†). The TG curve for the CAM showed a thermal event in the range of 240–370 °C, corresponding to the decomposition of the cellulose acetate chains, representing a loss of 76% of the mass. For CM, the event occurs in the range of 230 to 320 °C and corresponds to the dehydration and decomposition of cellulose. The cellulose decomposition temperature was recorded in Tonset at 245 °C. This value is lower than the temperature at which cellulose begins to decompose and may be associated with the expanded structure of the monolith, which facilitates and accelerates the internal degradation process of cellulose.39,44
Fig. 2 presents the FTIR spectra of the cellulose acetate monoliths before and after the deacetylation reaction.
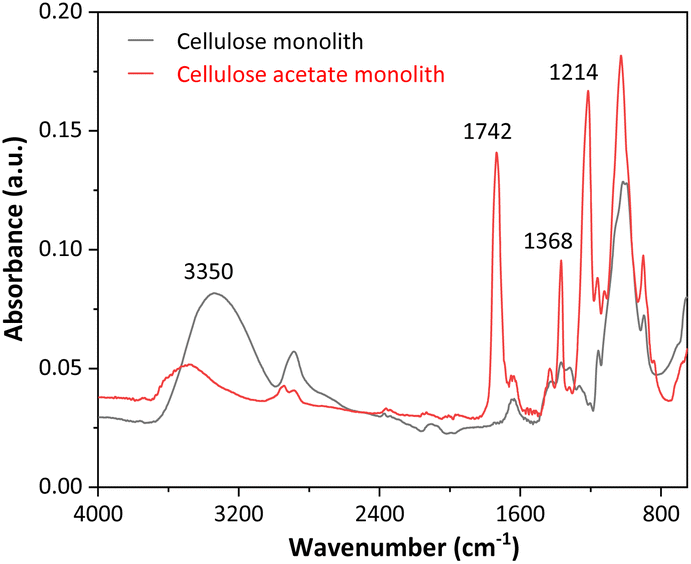 | ||
| Fig. 2 FTIR spectra of the cellulose acetate and cellulose monoliths. | ||
As seen in Fig. 2, the FTIR spectra for CAM and CM showed marked differences. At 3350 cm−1, high-intensity broadband is observed for the cellulose monolith, which is associated with the O–H stretching of adsorbed water. The first evidence of CAM deacetylation is that the signal for cellulose acetate is less intense. At 1742 cm−1, a peak is registered in the CAM spectrum, which is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the acetate groups, while in the CM spectrum, this signal is absent, indicating an effective removal of these groups with the alkaline deacetylation reaction. Peaks at 1368 and 1214 cm−1 in the CAM spectrum are attributed to the symmetric deformation of CH3 groups and C–O stretching of acetates, respectively. These signals are only present in the CAM FTIR spectra, confirming the efficiency of the deacetylation process and the production of cellulose monoliths.
O stretching of the acetate groups, while in the CM spectrum, this signal is absent, indicating an effective removal of these groups with the alkaline deacetylation reaction. Peaks at 1368 and 1214 cm−1 in the CAM spectrum are attributed to the symmetric deformation of CH3 groups and C–O stretching of acetates, respectively. These signals are only present in the CAM FTIR spectra, confirming the efficiency of the deacetylation process and the production of cellulose monoliths.
The degree of swelling (Sw) was measured for the cellulose monoliths by mass measures before and after water immersion. The Sw shows the polymer's ability to absorb solvent molecules and indicates the polymeric chain mobility and the conformational changes in the polymeric material.45 The results, shown in Fig. 3a, demonstrate that immersion of CM in water resulted in a mass increase of more than seven times on average compared to the initial CM dry mass, Sw of 619 ± 30%. These findings are significantly higher than those reported for particulate cellulosic materials (150–300%), suggesting that the expanded structure of CM allows for better water absorption.45,46 Cellulose monoliths quickly reach equilibrium when in contact with water and do not conform to the viscoelastic model.
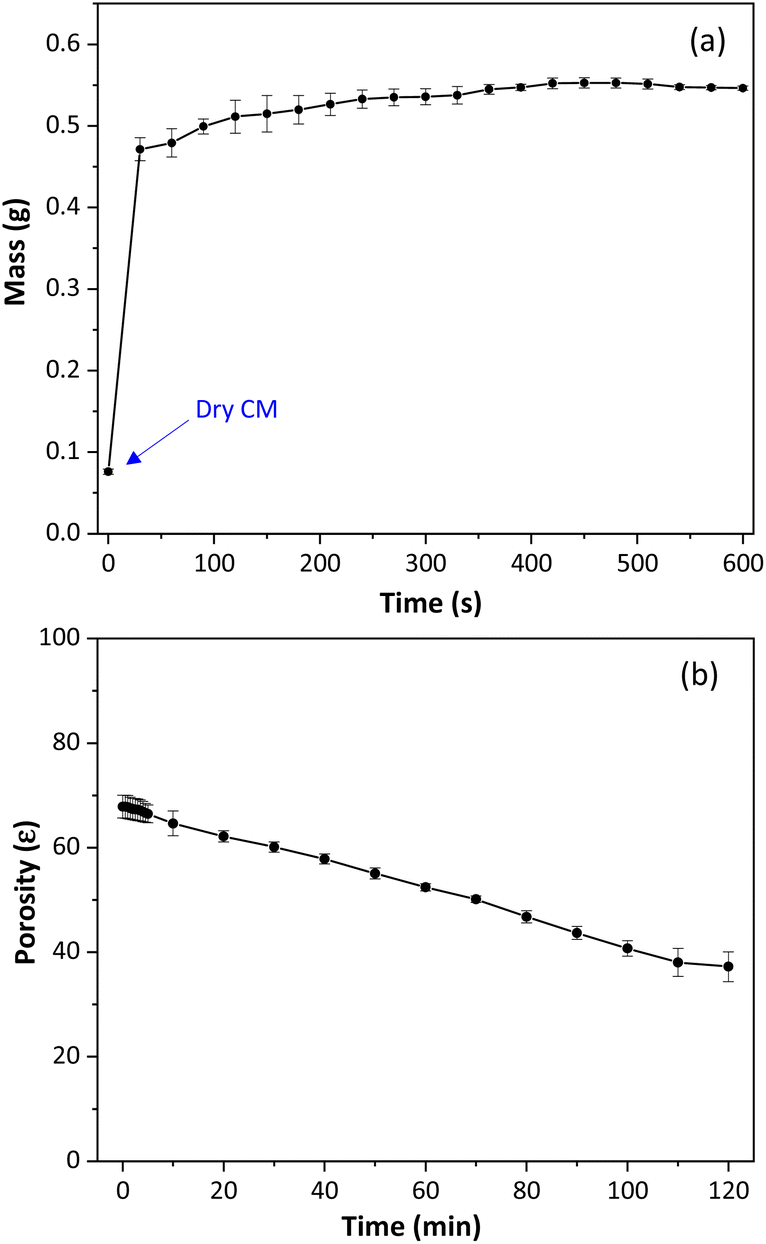 | ||
| Fig. 3 (a) Degree of swelling of cellulose monoliths. (b) Total porosity of cellulose monoliths used as sorptive stir bars. | ||
As seen, water causes an accentuated swelling of the monolithic structure of cellulose. For this reason, the total porosity of the CM was determined by the fluid saturation method, using n-butanol as solvent. Fig. 3b shows the variation of cellulose monolith porosity as a function of time. According to Fig. 3b, the cellulose monoliths' total porosity (ε) was 68 ± 2% and decreased over time. This result indicates that practically two-thirds of the monolith volume is empty.
The macroporous structure of the cellulose monoliths can be visualized in Fig. 4, which shows the SEM micrographs of the material. The CM is highly composed of cavities with 5–15 μm diameter along its entire surface and structure resembling a beehive. The solid CM structure presents a porous globular material that is sponge-like. Thus, the morphological and structural characteristics of the cellulose monolith are adequate for the extraction of glycerol in biodiesel samples.
 | ||
| Fig. 4 Electron micrographs of cellulose monolith used as a sorptive bar for glycerol extraction in biodiesel samples—cross-section at (a) 5k× and (b) 10k×. | ||
Optimization of the SBSE method using cellulose monolith stir bar
Cellulose monolith stir bars (CMSB) were manufactured with dimensions of 10 mm × 5 mm. A simple and environmentally friendly SBSE method was proposed for glycerol extraction. The CMSB's stirring speed was fixed at 200 rpm while the adsorption/desorption time, temperature, and extraction solvents varied.Initially, the best solvent for desorbing glycerol from CMSB was tested at room temperature (∼27 °C) for 15 minutes for glycerol adsorption and 15 minutes for desorption from CMSB. Ultrapure water, acidic and alkaline aqueous solutions, and methanol were evaluated as extraction solvents. Based on Fig. 5a, water was the most effective solvent for desorbing glycerol from the CMSB, resulting in a recovery rate of approximately 43%. Since glycerol does not dissociate within the pH range of 0–14, extraction with acidified or alkalized aqueous solutions was ineffective. Therefore, ultrapure water was chosen as the extraction solvent.
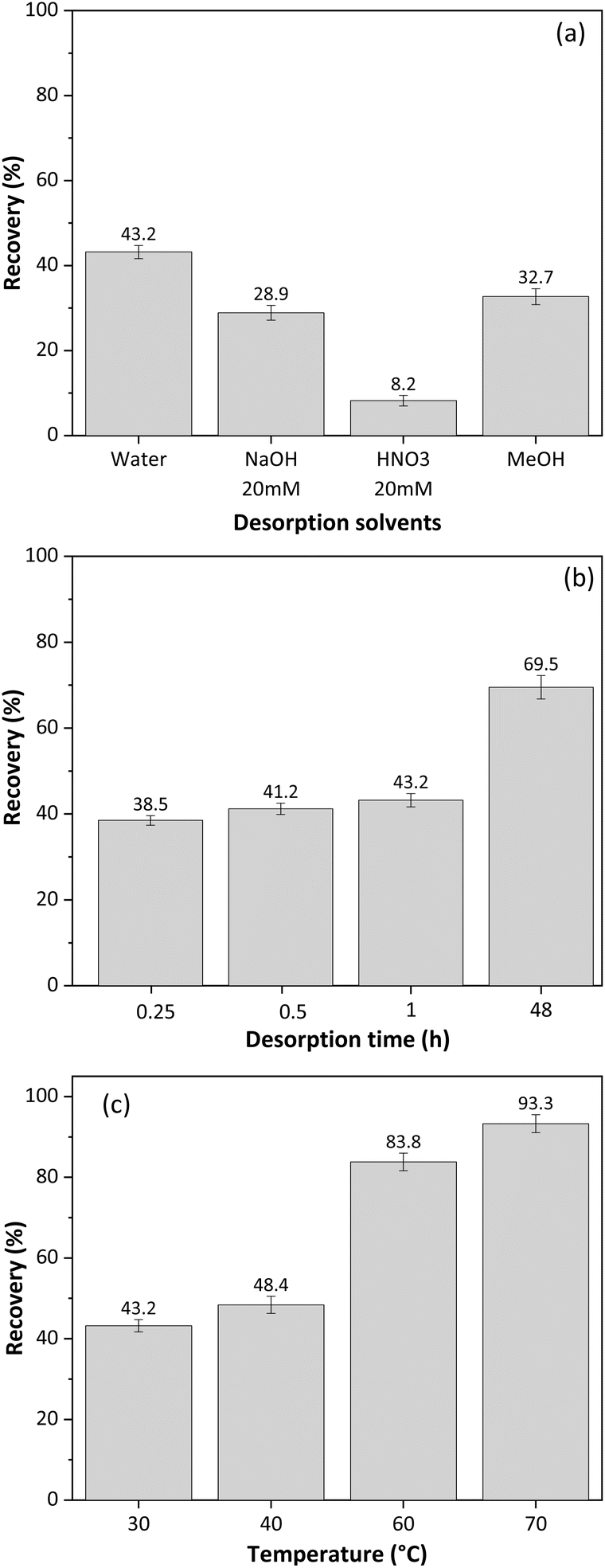 | ||
| Fig. 5 Effect of (a) desorption solvent, (b) desorption time, (c) adsorption and desorption temperature for SBSE method using cellulose monolith stir bar for free glycerol extraction from biodiesel. | ||
To further optimize the extraction process, the contact times of CMSB with the sample and the extraction solvent were varied simultaneously. For example, combinations of 30 min of adsorption and 30 min for desorption or 60 min of adsorption and 60 min for desorption were tested. Fig. 5b shows the effect of CMSB adsorption/desorption time on glycerol extraction from biodiesel samples using water as the extraction solvent.
According to Fig. 5b, increasing the contact time between the CMSB and the sample/extractor improves the glycerol extraction rate. However, the extraction increment was more significant after the adsorption and desorption times were extended up to 48 h, reaching a maximum extraction of 69.5%. Thus, we chose 15 min for adsorption and 15 min for glycerol desorption from CMSB. The high viscosity of biodiesel reduces the mass transfer rate and glycerol diffusion to the CMSB. Therefore, for a significant increase in the glycerol extraction rate, the decrease in biodiesel viscosity was performed with increases in the temperature of the solutions in the adsorption and desorption steps. Fig. 5c shows the glycerol extraction rates by the SBSE method with the CMSB as a function of temperature. The temperatures refer to the bottles containing the biodiesel sample and the ultrapure water. From 40 °C, a significant increase in glycerol extraction rate by CMSB was observed, reaching 93.6% at 70 °C. So, 70 °C was defined as the temperature at which glycerol adsorbs on CMSB and desorbs from it. The effect of sample and eluent volumes was not evaluated in the SBSE extraction process because volumes lower than 5 mL left the CMSB partially uncovered by the liquid. All measurements were performed, at least in triplicates, and the dispersion of the results in each experiment was always less than 3%, indicating an excellent precision of the SBSE method with the CMSB. This also shows the CMSB's good batch-to-batch reproducibility since at least five different preparations were carried out for these studies.
The reuse of CMSB in the extraction of glycerol from biodiesel samples is viable, as seen in Fig. S4 (ESI).† The analyses of the ultrapure water samples, interspersed between extractions of the fortified biodiesel samples, did not record the presence of glycerol, indicating the total desorption of glycerol from CMSB. So, the CMSB was prompted for new use after the regeneration procedure. Furthermore, the glycerol extraction efficiency remained at levels close to 100% extraction after three reuses.
Validation of the SBSE-HPLC-RID method
A validation of the SBSE method, using a CMSB, was performed to verify its quality for determining glycerol in biodiesel. Method selectivity was evaluated by comparing the chromatograms of the biodiesel sample and the SBSE extract of a fortified sample with 0.02% free glycerol, Fig. 6. As observed, the peak for glycerol presents a retention time of 12.9 min, while fatty acid esters at 17–19 min. Therefore, the method was selective for the glycerol quantitation, even if the biodiesel comes from raw material sources containing different compositions of fatty acids. The baseline instability in the chromatogram of Fig. 6b may be associated with poor miscibility of the sample in the mobile phase.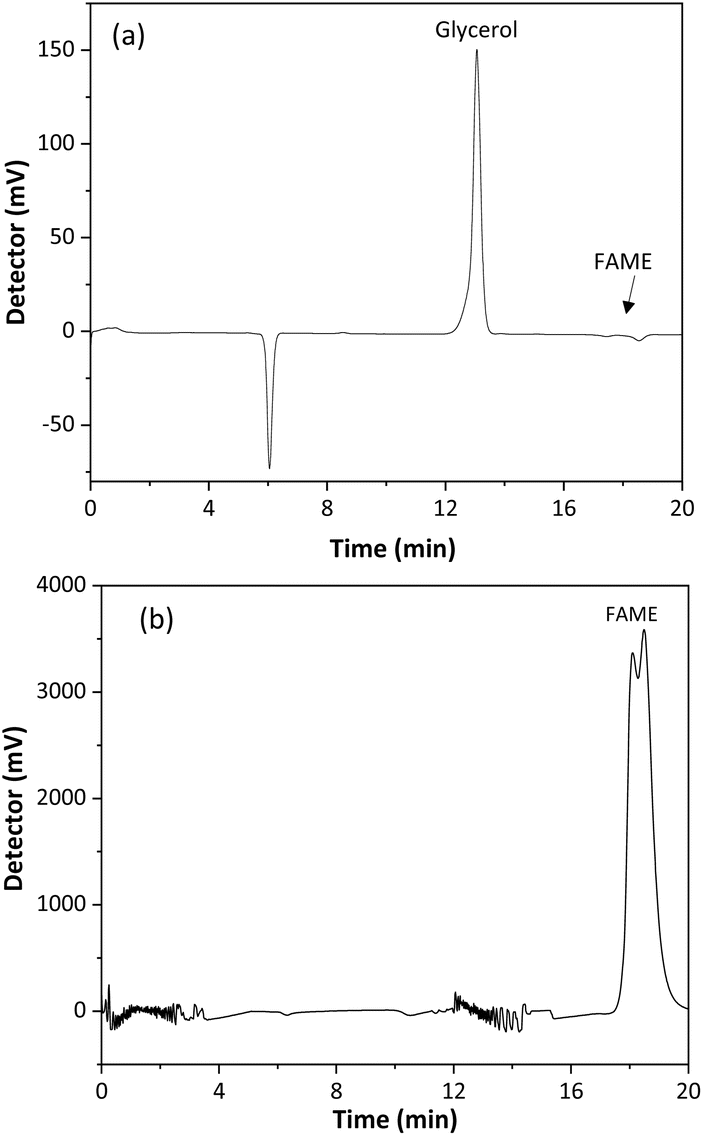 | ||
| Fig. 6 Chromatograms of the (a) extract of a sample spiked with 0.02% w/w free glycerol using the SBSE method and (b) biodiesel sample. Mobile phase: sulfuric acid at 0.005 mol L−1, flow rate at 0.6 mL min−1, injection volume 20 μL, oven temperature 35 °C, RI detection at 35 °C. FAME: fatty acid methyl ester. | ||
The method's limits of detection and quantification were obtained from the injection of decreasing concentrations of glycerol solutions in the HPLC-RID system. LOD and LOQ values were 6.6 × 10−5% w/w and 2.2 × 10−4% w/w, respectively. These values are below the reference concentrations for glycerol in biodiesel and the recommended methods. Furthermore, the detectability of the proposed method is lower than those other published methods using solvent extraction and capillary electrophoresis,25 gas chromatography-flame ionization detection,47 solid phase extraction (SPE) coupled with enzymatic-spectrophotometric technique,48 and HPLC with diode array detection.24 All used glycerol derivatization for detection. The proposed method shows lower detectability of other extraction methods with HPLC-RID analysis.14,49,50
Analytical parameters of linearity, range, and sensitivity were obtained from the construction of analytical curves for glycerol. Fig. S5 (ESI)† shows a typical analytical curve for glycerol using the SBSE-HPLC-RID method with a CMSB, in which excellent linearity is observed, with a linear regression coefficient (r) of 0.999, a range of 0.00022% to 0.03500% w/w and a sensitivity of 1.595 × 107% w/w.
The accuracy and precision of the method were determined by addition and recovery assays of free glycerol in a biodiesel sample and compared with the result obtained by the official AOCS Ca. 14-56 method. The results obtained are presented in Table 1. It is observed that the glycerol recovery rate was 94 ± 2% by the SBSE-HPLC-RID method using the CMSB, and the result did not show a statistical difference for the AOCS method at the 95% confidence level by the t-test.
The limit concentration of free glycerol in biodiesel is 0.0200% w/w according to several regulatory agencies for different countries,8–10 the AOCS Ca. 14-56 method is recognized as an official standard protocol since it is reliable for this determination. The method developed in this work presents similar results for determining free glycerol in biodiesel samples. However, the SBSE-HPLC-RID method with CMSB needs a few steps, low handling, low cost, and no chemical reagent. It is fully compatible with sustainability and can be considered an eco-friendly method.
Application of the SBSE method for glycerol in biodiesel B100 samples
CMSB was used to extract glycerol from two B100 biodiesel samples using the optimized SBSE method. Soybean oil methyl and ethyl biodiesel samples were characterized by GC-FID, showing 99.3 and 99.0% total fatty acid esters, respectively (Fig. S6 and Table S1†). The samples were subjected to the SBSE method before and after fortification with 0.0200% m/m of glycerol, and the results are in Table 2. HPLC-RID chromatograms of the biodiesel extracts are presented in Fig. S7.† The samples presented 0.0013 and 0.0021% w/w of glycerol for methyl and ethyl biodiesel, respectively, with recovery efficiency of ∼94%, comparable to other reported methods, in a range of 78–117%,18,19,25,26,51 but using a more straightforward procedure free of organic solvents.The method proposed for determining free glycerol in biodiesel, using SBSE with monolithic cellulose stir bars, is the simplest method available until now. Official methods either use various reagents, which can be toxic (e.g., AOCS Ca. 14-56 method)11 or require experimental conditions that are inaccessible to most analytical laboratories, such as the chromatographic conditions specified in EN: 14103 method.42 In contrast, our method requires only two steps to determine glycerol in B100 biodiesel, utilizing available experimental conditions such as a magnetic stirrer, water, and monolithic cellulose stir bars. Due to its simplicity, this method can be easily scaled and applied for industrial purposes. Thus, it presents a viable alternative for the efficient and safe determination of free glycerol in the biodiesel quality control process. Looking ahead, the automation of this determination by inline SBSE extraction with HPLC-RID analysis can facilitate the process even further.
Conclusions
The SBSE method with the cellulose monolith as a sorptive stir bar proved effective for extracting free glycerol from biodiesel samples using only hot water as the extracting solvent. The subsequent analysis of extracts by HPLC-RID makes it possible to quantify glycerol selectively, without any derivatization reagents, with superior detectability to gas chromatography methods. So, the SBSE-HPLC-RID method using CMSB does not require any reagent or extreme operational conditions to determine free glycerol. The extraction process takes about a half hour, with few steps and few manipulations, resulting in an environmentally friendly, simple, and economically feasible method like no other one to the best of our knowledge.Author contributions
Data curation: P. H. S. M., P. R., M. A. B., C. L. S.; formal analysis: P. H. S. M., P. R., C. L. S., A. L. R. S., M. A. B., L. M. B. C.; funding acquisition: R. M. N. A., A. M. F.; investigation: P. R., A. L. R. S., H. S. R.; methodology: P. H. S. M., C. L. S., P. R., L. M. B. C., H. S. R.; project administration: R. M. N. A., A. M. F.; resources: R. M. N. A., A. M. F.; supervision: A. M. F.; validation: H. S. R., R. M. N. A., A. M. F.; writing – original draft: P. H. S. M., A. M. F.; writing – review & editing: R. M. N. A., A. M. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fundação de Amparo à Pesquisa do Estado de Minas Gerais [grant numbers APQ-01901-22 and APQ-03286-21]; the Financiadora de Estudos e Projetos [grant numbers 01.13.0371.00 and 01.11.0135.00]; Coordenação de Aperfeiçoamento de Pessoal de Nível Superior [grant number 001], and the Conselho Nacional de Desenvolvimento Científico e Tecnológico.Notes and references
- G. M. Mathew, D. Raina, V. Narisetty, V. Kumar, S. Saran, A. Pugazhendi, R. Sindhu, A. Pandey and P. Binod, Sci. Total Environ., 2021, 794, 148751 CrossRef CAS PubMed.
- A. Mukhtar, S. Sagib, H. Lin, M. U. H. Shah, S. Ullah, M. Younas, M. Rezakazemi, M. Ibrahim, A. Mahmood, S. Asif and A. Bokhari, Renewable Sustainable Energy Rev., 2022, 157, 112012 CrossRef CAS.
- D. Singh, D. Sharma, S. L. Soni, S. Sharma, P. K. Sharma and A. Jhalani, Fuel, 2020, 262, 116553 CrossRef CAS.
- I. Atadashi, Alexandria Eng. J., 2015, 54, 1265–1272 CrossRef.
- M. R. Monteiro, A. R. P. Ambrozin, L. M. Lião and A. G. Ferreira, Talanta, 2008, 77, 593–605 CrossRef CAS.
- V. K. Mishra and R. Goswami, Biofuels, 2018, 9, 273–289 CrossRef CAS.
- I. M. Atadashi, M. K. Aroua and A. A. Aziz, Renewable Energy, 2011, 36, 437–443 CrossRef CAS.
- ASTM D6751-20a, Standard specification for biodiesel fuel blend stock (B100) for middle distillate fuels, ASTM International, available, https://www.astm.org.
- EN 14214:2012+A2:2019, Liquid petroleum products. Fatty acid methyl esters (FAME) for use in diesel engines and heating applications, Requirements and test methods, European Committee for Standardization, available, https://www.en-standard.eu/.
- Resolução ANP n° 920/2023, Estabelece a especificação do biodiesel e as obrigações quanto ao controle da qualidade a serem atendidas pelos agentes econômicos que comercializem o produto em território nacional, Agência Nacional do Petróleo, Gás Natural e Biocombustíveis, available, https://www.in.gov.br/.
- AOCS Standard Ca, 14-56, Total, free and combined glycerol iodometric-periodic acid method, American Oil Chemists' Society, Urbana, 2017, revised, available, https://www.aocs.org/.
- ASTM D6584-21, Standard test method for determination of total monoglycerides, total diglycerides, total triglycerides, and free and total glycerin in B-100 biodiesel methyl esters by gas chromatography, ASTM International, available, https://www.astm.org.
- EN 14105:2020, Fat and oil derivatives. Fatty acid methyl esters (FAME). Determination of free and total glycerol and mono-, di-, triglyceride contents, European Committee for Standardization, available, https://www.en-standard.eu/.
- D. A. Conzendey, R. O. Muniz, R. C. Santos, C. G. Souza, D. F. Andrade and L. A. d'Avila, Microchem. J., 2021, 168, 106347 CrossRef.
- G. G. Honório, J. N. Cunha, K. L. S. C. Assis, P. F. Aguiar, D. F. Andrade, C. G. Souza, L. A. d'Avila, B. S. Archanjo, C. A. Achete, R. N. C. Pradelle, F. Turkovics, R. Serralvo Neto and E. D'Elia, J. Solid State Electrochem., 2019, 23, 3057–3066 CrossRef.
- B. M. Moreira, J. N. Cunha, V. M. Paiva, G. G. Honório, R. T. Tarley and E. D'Elia, Anal. Lett., 2021, 54, 1654–1667 CrossRef CAS.
- A. V. F. Sako, D. A. Spudeit, M. Dupim, W. P. Oliveira Filho, T. D. Saint'Pierre, M. A. L. Oliveira and G. A. Micke, J. Chromatogr. A, 2018, 1570, 148–154 CrossRef CAS.
- M. D. Carabajal, A. Glorio, I. S. Marcipar and C. M. Lagier, Microchem. J., 2020, 158, 105148 CrossRef CAS.
- R. M. A. Tehrani and S. Ab Ghani, Electrochim. Acta, 2012, 70, 153–157 CrossRef CAS.
- T. G. G. Barbosa, E. M. Richter and R. A. A. Muñoz, Electroanalysis, 2012, 24, 1160–1163 CrossRef CAS.
- L. M. Lourenço and N. R. Stradiotto, Talanta, 2009, 79, 92–96 CrossRef.
- M. Hájek, F. Skopal and J. Machek, Eur. J. Lipid Sci. Technol., 2006, 108, 666–669 CrossRef.
- S. Hu, X. Luo, C. Wan and Y. Li, J. Agric. Food Chem., 2012, 60, 5915–5921 CrossRef CAS PubMed.
- M. A. Ahmed, I. Khan, J. Hashim and S. G. Musharraf, Anal. Methods, 2015, 7, 7805–7810 RSC.
- L. C. Gonçalves Filho and G. A. Micke, J. Chromatogr. A, 2007, 1154, 477–480 CrossRef.
- M. B. Lima, M. Insausti, C. E. Domini, M. F. Pistonesi, M. C. U. Araújo and B. S. F. Band, Talanta, 2012, 89, 21–26 CrossRef CAS.
- P. H. G. D. Diniz, M. F. Pistonesi, M. C. U. Araújo and B. S. F. Band, Talanta, 2013, 114, 38–42 CrossRef CAS.
- S. G. Silva and F. R. P. Rocha, Talanta, 2010, 83, 559–564 CrossRef CAS PubMed.
- E. J. Mercer and F. Halaweish, J. Am. Oil Chem. Soc., 2011, 88, 655–659 CrossRef CAS.
- P. Bondioli and L. Della Bella, Eur. J. Lipid Sci. Technol., 2005, 107, 153–157 CrossRef CAS.
- M. L. Pisarello, B. O. Dalla Costa, N. S. Veizaga and C. A. Querini, Ind. Eng. Chem. Res., 2010, 49, 8935–8941 CrossRef CAS.
- M. He, Y. Wang, Q. Zhang, L. Zang, B. Chen and B. Hu, J. Chromatogr. A, 2021, 1637, 461810 CrossRef CAS.
- F. David, N. Ochiai and P. Sandra, TrAC, Trends Anal. Chem., 2019, 112, 102–111 CrossRef CAS.
- C. K. Hasan, A. Ghiasvand, T. W. Lewis, P. N. Nesterenko and B. Paull, Anal. Chim. Acta, 2020, 1139, 222–240 CrossRef CAS PubMed.
- A. M. S. Reis, A. T. Vieira, A. L. R. Santos, M. V. Ferreira, A. C. F. Batista, R. M. N. Assunção, G. Rodrigues Filho, E. A. M. Ribeiro and A. M. Faria, J. Braz. Chem. Soc., 2020, 31, 1011–1020 CAS.
- M. G. Gomes, D. Q. Santos, L. C. Morais and D. Pasquini, Fuel, 2015, 155, 1–6 CrossRef CAS.
- A. L. Squissato, D. M. Fernandes, R. M. F. Sousa, R. R. Cunha, D. S. Serqueira, E. M. Richter, D. Pasquini and R. A. A. Muñoz, Cellulose, 2015, 22, 1263–1274 CrossRef CAS.
- M. J. Alves, I. V. Cavalcanti, M. M. Resende, V. L. Cardoso and M. H. Reis, Ind. Crops Prod., 2016, 89, 119–127 CrossRef CAS.
- Y. Xin, Q. Xiong, Q. Bai, M. Miyamoto, C. Li, Y. Shen and H. Uyama, Carbohydr. Polym., 2017, 157, 429–437 CrossRef CAS PubMed.
- C. C. Ferreira, L. M. Costa and P. J. S. Barbeira, Talanta, 2015, 138, 8–14 CrossRef.
- K. A. Borges, A. L. Squissato, D. Q. Santos, W. Borges Neto, A. C. F. Batista, T. A. Silva, A. T. Vieira, M. F. Oliveira and M. G. Hernández-Terrones, Energy, 2014, 67, 569–574 CrossRef CAS.
- F. Munari, D. Cavagnino, and A. Cadoppi, Thermo Fischer Scientific, 2007, Application Note 10212, 1–4, available, https://www.analiticaweb.com.br/downloads/literaturas/esteres_totais_biodiesel.pdf.
- X. Zhang, B. Wang, X. Qin, S. Ye, Y. Shi, Y. Feng, W. Han, C. Liu and C. Shen, Carbohydr. Polym., 2020, 241, 116361 CrossRef CAS PubMed.
- I. Chakraborty, S. Rongpipi, I. Govindaraju, S. S. Mal, E. W. Gomez, E. D. Gomez, R. D. Kalita, Y. Nath and N. Mazumder, Microsc. Res. Tech., 2022, 85, 1990–2015 CrossRef CAS PubMed.
- H. Geng, Carbohydr. Polym., 2018, 186, 208–216 CrossRef CAS PubMed.
- I. A. Udoetok, R. M. Dimmick, L. D. Wilson and J. V. Headley, Carbohydr. Polym., 2016, 136, 329–340 CrossRef CAS PubMed.
- L. Senila, M. Miclean, O. Cadar, M. Senila, M. Kovacs and M. A. Hoaghia, Studia UBB Chemia, 2016, 3, 345–353 Search PubMed.
- R. O. Muniz, S. B. Martins, G. G. Honório, J. N. Cunha, C. G. Souza, D. F. Andrade, R. N. C. Pradelle, F. Turkovics, R. Serralvo Neto, L. A. D'Avila and E. D'Elia, Anal. Methods, 2019, 11, 767–773 RSC.
- G. Santori, A. Arteconi, G. Di Nicola, M. Moglie and R. Stryjek, Energy Fuels, 2009, 23, 3783–3789 CrossRef CAS.
- C. J. Giertyas, D. S. Silva, C. L. F. Silva, M. R. Meneghetti, S. M. P. Meneghetti, R. M. Almeida and J. H. Bortoluzzi, Quim. Nova, 2019, 42, 729–735 CAS.
- S. G. Silva, A. Morales-Rubio, M. De La Guardia and F. R. P. Rocha, Anal. Bioanal. Chem., 2011, 401, 365–371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02985b |
| This journal is © The Royal Society of Chemistry 2024 |