
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, anticancer and antibacterial evaluation of novel spiramycin-acylated derivatives†
Zhiwei Wang‡
 ab,
Junxiang Cheng‡b,
Hui Wenb,
Tao Houab,
Fengbin Luob,
Yaodong Wangb,
Xingjun Xuab,
Yanfang Liuab,
Yaopeng Zhao*ab and
Xinmiao Liang*ab
ab,
Junxiang Cheng‡b,
Hui Wenb,
Tao Houab,
Fengbin Luob,
Yaodong Wangb,
Xingjun Xuab,
Yanfang Liuab,
Yaopeng Zhao*ab and
Xinmiao Liang*ab
aKey Lab of Phytochemistry and Natural Medicines, Dalian Institute of Chemical Physics, CAS, Dalian 116023, China. E-mail: ypzhao@dicp.ac.cn; liangxm@dicp.ac.cn; Fax: +86 411 84379539; Tel: +86 411 84379519
bGanjiang Chinese Medicine Innovation Center, Nanchang, 330000, China
First published on 9th December 2024
Abstract
Spiramycin and its derivatives are commonly used antimicrobials, and its derivative, carrimycin, has recently been found to have good anticancer potential. Here, we found that the 4′′-OH of spiramycin can be selectively acylated, resulting in a series of novel spiramycin derivatives with a structure similar to carrimycin. Anticancer studies showed that most of the derivatives exhibited moderate to good anti-proliferative activity against four cancer cell lines, including HGC-27, HT-29, HCT-116 and HeLa, especially compound 14, which has the strongest activity against HGC-27 cells with an IC50 value of 0.19 ± 0.02 μM. Pharmacological studies on HGC-27 cells revealed that compound 14 could arrest the cell cycle in the S phase, raise ROS levels, and induce cell apoptosis via activation of Erk/p38 MAPK signaling pathways. In addition, antibacterial studies showed that most of the spiramycin I derivatives modified at the 4′′-OH group enhanced antibacterial activity on the four tested strains, including S. aureus, S. aureus MRSA, S. epidermidis, and B. subtilis. In particular, compound 16 was the most effective one and comparable to linezolid, a commonly used first-line antimicrobial. These results suggest that spiramycin I derivatives may provide an opportunity to design new anticancer or antibacterial agents, even dual-function agents.
Introduction
Spiramycin is a 16-membered macrolide antibiotic, produced by Streptomyces ambofaciens in the form of a mixture of three main compounds: spiramycins I, II and III,1,2 of which spiramycin I is dominant (∼80%) (Scheme 1).3 As a widely used drug, spiramycin has potent antibacterial activity against most of Gram-(+) cocci and rods, mycoplasmas, and Toxoplasma gondii,4,5 and exhibit good gastrointestinal tolerance, high affinity for tissues, and few adverse effects.6,7 Several chemical modifications of spiramycin have been developed to improve its antibacterial properties, such as etherification, esterification or sulphonylation of the mycaminosyl-mycarose moiety and lactone ring,8–11 N-demethylation or N-substitution of the forosamine and/or mycaminose moieties,12 amination or alkenylation of the aldehyde,13,14 and incorporation of nitrogen into the lactone macrocyclic ring.15 Therein, Sano et al. found that 3,3′′,4′′-triacyl and 4′′-substituted derivatives with relatively small acyl, sulfonyl and alkyl groups showed superior or comparable antibacterial effects to spiramycin I.8,9,16 It is well known that many macrolide antibiotics have anticancer or synergistic anticancer effects, in addition to being antibacterial. For example, clarithromycin can suppress hepatocellular carcinoma, and azithromycin can enhance the anticancer activity of TRAIL (tumor necrosis factor-related apoptosis-inducing ligand).17–22 Unlike those macrolide antibiotics, which have dual anti-infection and anticancer functions, spiramycin has only a weak anticancer effect in several human cancer cell lines with IC50 > 30 μM.23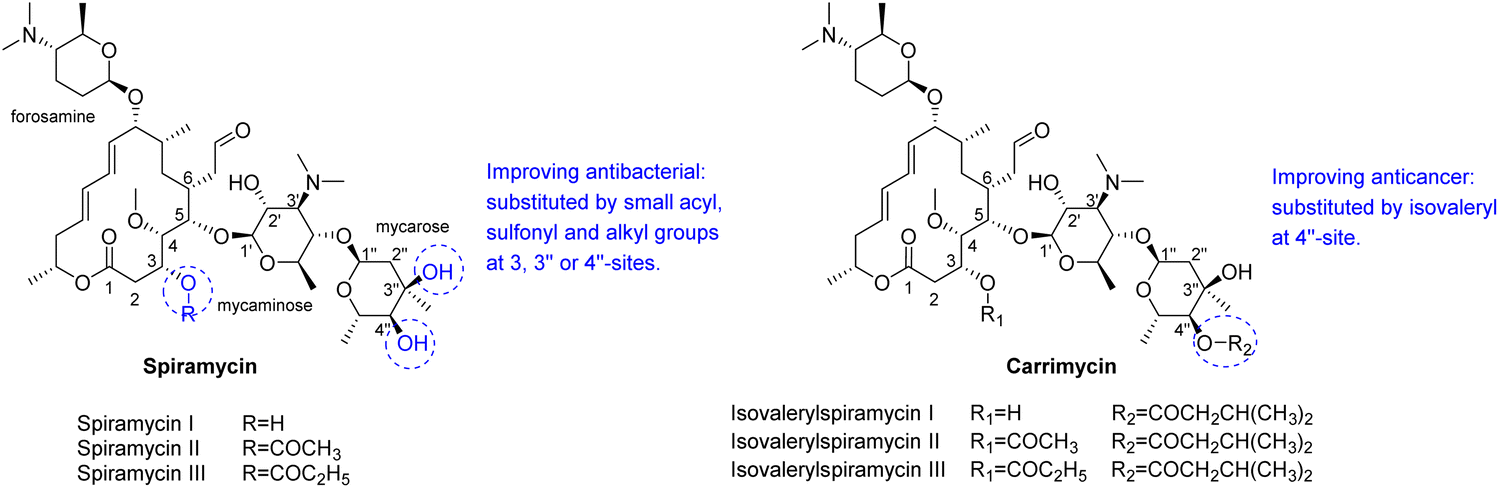 | ||
| Scheme 1 Structures of main components of spiramycin and carrimycin, and some known structure–activity relationships. | ||
Recently, carrimycin and its active ingredient isovalerylspiramycin I have received attention for their relatively potent anticancer effects in addition to being antibacterial.24–27 Carrimycin is a genetically engineered macrolide antibiotic that can be regarded as a bio-derivative of spiramycin, mainly consisting of 4′′-isovalerylspiramycin I, II, III (Scheme 1). Liang et al. reported that carrimycin can regulate the PI3K/AKT/mTOR and MAPK pathways, thus causing apoptosis of OSCC (oral squamous cell carcinoma) cells in vitro and in vivo.24 Jin et al. further confirmed the anticancer capacity of carrimycin and its monomer (isovalerylspiramycin I) by inhibiting the proliferation, migration, and invasion of hepatocellular carcinoma cells.25 Liu et al. reported that isovalerylspiramycin I could suppress non-small cell lung carcinoma (NSCLC) growth through ROS-mediated inhibition of PI3K/AKT signaling pathway.26 Cui et al. showed that isovalerylspiramycin I could link the cancer cell vulnerability to oxidative stress and RNA biogenesis by targeting the nucleolar protein selenoprotein H.27 Based on these results, further exploration of the anticancer potential of spiramycin derivatives is encouraged.
However, the anticancer potency of most known spiramycin derivatives to date remains moderate or weak, and their structural diversity is also very limited, which limits their drug development. Here, we are very interested in developing more spiramycin derivatives by means of chemical modification, and investigating their potential as both anticancer and antimicrobial agents.
Results and discussion
Chemistry
Spiramycin I has four hydroxyl groups, including 2′-, 3-, 3′′- and 4′′-OH (Scheme 1). Sano et al. reported that spiramycin I is esterified in a reactive order of 2′ > 4′′ > 3 > 3′′; therefore, the selective esterification of the 4′′-OH group usually involves additional prior protection and subsequent deprotection of the 2′- and 3-OH groups.9–11 To rapidly explore the modifiable scope of spiramycin I, we tried the direct esterification of spiramycin I. We first attempted the chemical synthesis of isovalerylspiramycin I using spiramycin I and isovaleryl chloride as starting materials. Results showed that by controlling the equivalent of isovaleryl chloride, a mono-isovalerylated spiramycin I (1) was obtained as the main product with a 30% yield. This was confirmed to be 4′′-isovalerylspiramycin I by HPLC, HR-ESI-MS and NMR analysis (Fig. S2†). In another words, the main active component of carrimycin, 4′′-isovalerylspiramycin I was chemically synthesized. This result is inconsistent with the order of hydroxyl activity in previous reports,9 probably due to the use of a larger isovalery group, instead of the smaller substituents used by Sano et al. The steric hindrance of the 2′-OH was possibly larger than that of the 4′′-OH group, leading to the isovaleryl group to preferentially bind to 4′′-OH. Based on this finding, various acylated reagents with the number of carbon and heteroatoms ≥5 were selected to synthesize 4′′-acylated sipramycin I derivatives 2–20 (Table 1).
|
|||||
|---|---|---|---|---|---|
| Acylation conditions: (1) acyl chloride, DMAP, Et3N, DCM; (2) acid, DCC, 4-PPY, DMF; (3) isocyanates, Et3N, DMF | |||||
| Cmpd | R1 | R2 | Cmpd | R1 | R2 |
| 1 | 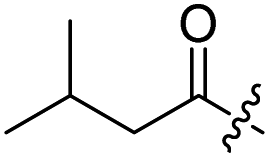 |
H | 2 | 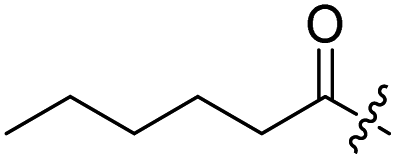 |
H |
| 3 | 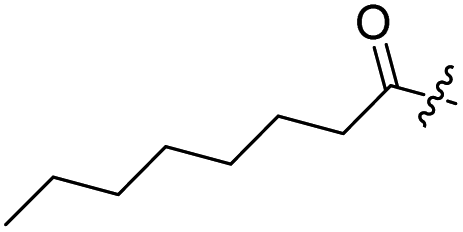 |
H | 4 | 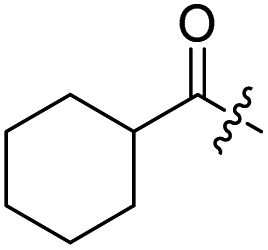 |
H |
| 5 | 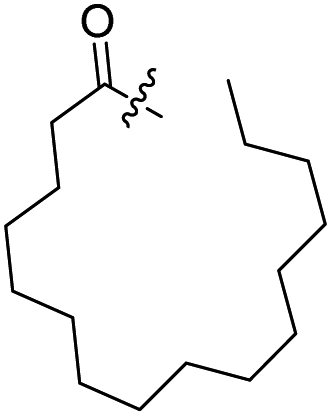 |
H | 6 | 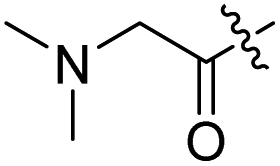 |
H |
| 7 | 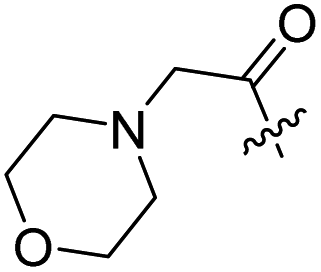 |
H | 8 | 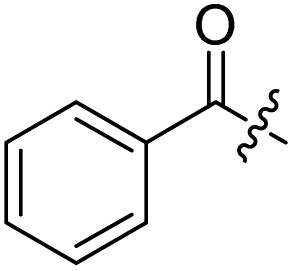 |
H |
| 9a | 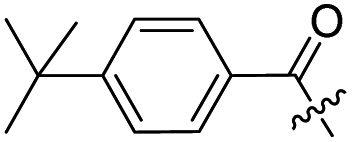 |
H | 9b | H |  |
| 10a | 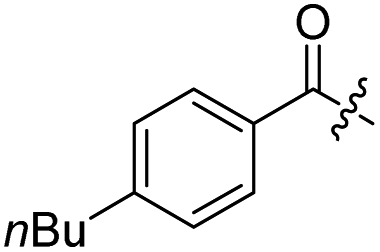 |
H | 10b | H | 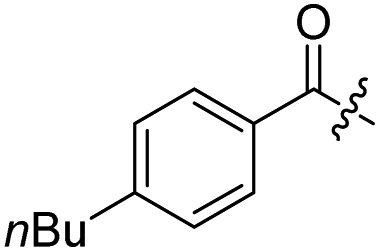 |
| 11 | 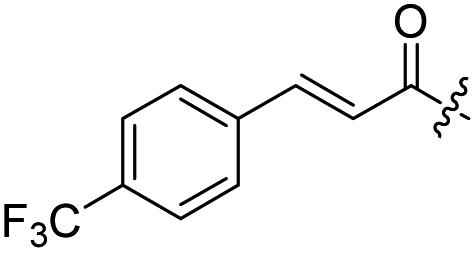 |
H | 12 | 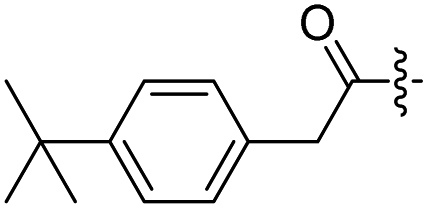 |
H |
| 13 | 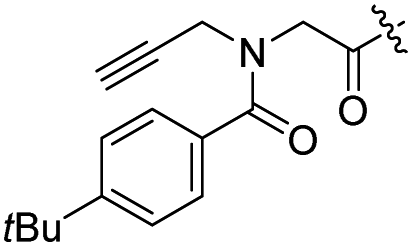 |
H | 14 | 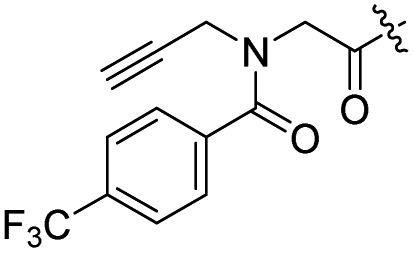 |
H |
| 15 | 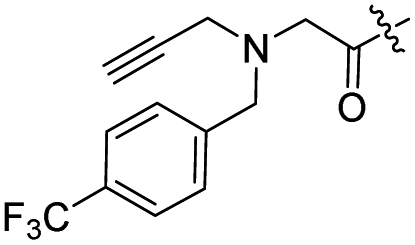 |
H | 16 |  |
H |
| 17 | 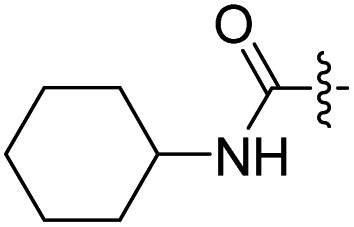 |
H | 18 | 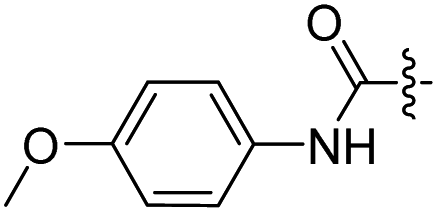 |
H |
| 19 | 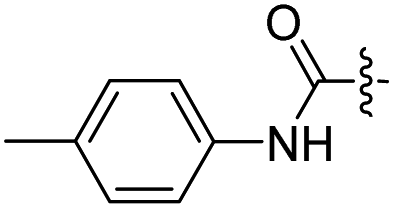 |
H | 20 | 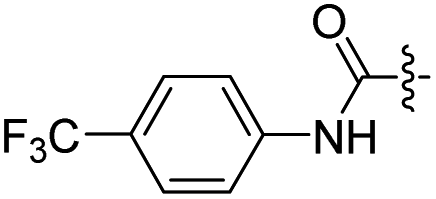 |
H |

Although only one mono-substituted product was obtained in most modification reactions, two mono-substituted products were obtained with comparable yields in the preparation of 9 (9a, 20% yield; 9b, 20% yield) and 10 (10a, 25% yield; 10b, 13% yield). 1D and 2D NMR studies of the two isomers 9a and 9b indicated that they were 3- and 4′′-p-(tert-butyl)benzoyl spiramycin I, respectively (Fig. S3†). HPLC profile showed that 9a had a shorter retention time than 9b (Fig. S6†). Based on a similar HPLC analysis, 10a and 10b were attributed as 3- and 4′′-p-butylbenzoyl spiramycin I, respectively. These results further contradicted the order of hydroxyl reactivity previously reported,9 indicating that the steric hindrance of 3-OH is also smaller than that of 2′-OH. In addition to acyl chlorides and acids, several isocyanates can be used to react with spiramycin I to form 4′′-carbamate derivatives 16–20.
Pharmacology
| Cmpd | IC50 (μM) | SI (GES-1/HGC-27) | ||||
|---|---|---|---|---|---|---|
| HGC-27 | HT-29 | HeLa | HCT-116 | GES-1 | ||
| 1 | 8.08 ± 0.30 | 13.15 ± 1.29 | 6.23 ± 0.36 | 12.66 ± 1.34 | 18.54 ± 2.56 | 2.29 |
| 2 | 5.23 ± 0.77 | 6.44 ± 0.36 | 23.67 ± 0.58 | 38.46 ± 6.36 | 38.82 ± 26.69 | 7.42 |
| 3 | 3.96 ± 0.17 | 5.42 ± 0.31 | 7.13 ± 0.24 | 10.62 ± 0.28 | 12.09 ± 0.60 | 3.05 |
| 4 | 4.27 ± 0.15 | 5.01 ± 0.28 | 8.50 ± 0.27 | 12.98 ± 0.30 | 14.51 ± 0.72 | 3.40 |
| 5 | 4.20 ± 0.08 | 3.94 ± 0.20 | 8.81 ± 0.17 | 14.56 ± 0.46 | 10.82 ± 0.35 | 2.58 |
| 6 | >50 | >50 | >50 | >50 | >50 | — |
| 7 | 5.78 ± 0.15 | 4.19 ± 0.21 | 9.71 ± 0.31 | 8.0 ± 0.12 | 9.56 ± 0.323 | 1.65 |
| 8 | 5.31 ± 0.16 | 3.49 ± 0.16 | 7.18 ± 0.22 | 9.21 ± 0.14 | 13.08 ± 0.73 | 2.40 |
| 9a | 2.31 ± 0.25 | 0.91 ± 0.02 | 1.30 ± 0.07 | 4.78 ± 0.52 | 3.34 ± 0.16 | 1.44 |
| 9b | 7.97 ± 0.61 | 5.95 ± 0.61 | 26.37 ± 0.72 | 10.94 ± 0.23 | 31.25 ± 10.65 | 3.92 |
| 10a | 1.66 ± 0.29 | 4.67 ± 0.40 | 4.22 ± 0.26 | 3.51 ± 0.49 | 2.75 ± 0.31 | 1.66 |
| 10b | 0.92 ± 0.06 | 1.45 ± 0.12 | 1.00 ± 0.06 | 12.43 ± 0.68 | 4.68 ± 1.26 | 5.09 |
| 11 | 0.96 ± 0.20 | 2.32 ± 0.16 | 1.30 ± 0.08 | 2.36 ± 0.46 | 2.34 ± 0.36 | 2.44 |
| 12 | 0.49 ± 0.04 | 1.64 ± 0.13 | 2.05 ± 0.22 | 2.24 ± 0.45 | 2.29 ± 0.48 | 4.67 |
| 13 | 0.53 ± 0.15 | 1.84 ± 0.10 | 1.76 ± 0.70 | 10.98 ± 0.64 | 2.28 ± 0.47 | 4.30 |
| 14 | 0.19 ± 0.02 | 0.71 ± 0.06 | 1.24 ± 0.10 | 1.03 ± 0.05 | 1.96 ± 0.11 | 10.32 |
| 15 | 1.20 ± 0.17 | 2.15 ± 0.54 | 2.16 ± 0.44 | 4.39 ± 0.69 | 4.65 ± 0.18 | 3.88 |
| 16 | >50 | >50 | >50 | >50 | >50 | — |
| 17 | >50 | >50 | >50 | >50 | >50 | — |
| 18 | >50 | >50 | >50 | >50 | >50 | — |
| 19 | 1.26 ± 0.19 | 3.27 ± 0.95 | 9.91 ± 0.66 | 22.01 ± 4.58 | 41.66 ± 13.93 | 33.06 |
| 20 | 1.76 ± 0.34 | 3.20 ± 0.24 | 2.29 ± 0.14 | 5.79 ± 0.38 | 6.64 ± 0.56 | 3.77 |
| 5-FU | 4.75 ± 0.18 | 4.60 ± 0.55 | 3.43 ± 0.33 | 3.32 ± 0.30 | 4.03 ± 0.19 | 0.85 |
Compounds 1–20 were further evaluated on three other human cancer cell lines (colorectal HT-29 and HCT-116, and cervical HeLa) and one normal gastric epithelial cell line (GES-1). Results showed that these compounds also exhibited good to moderate anti-proliferative properties, but in general, most compounds were more sensitive to HGC-27 (Table 2). The selectivity index (SI) was calculated as the IC50 ratio of normal cells to cancer cells, SI = IC50 (GES-1)/IC50 (HGC-27). Results showed that compounds 14 and 19 had the highest selectivity against cancer cells, which suppressed the growth of HGC-27 cell lines with SI = 10.32 and 33.06, respectively. Considering the highest activity and selectivity, compound 14 was selected for further pharmacological studies on HGC-27.
 | ||
| Fig. 1 Compound 14 induced apoptosis of HGC-27 cells. (A) Flow cytometry and (B) bar graph analysis of cell apoptosis in HGC-27 after treatment with 14 (0, 0.22 0.45, 0.9 μM) for 72 h. (C) Western blot images and (D) bar graph analysis of PARP, cleaved PARP, caspase-3 and cleaved caspase-3 protein expressions in HGC-27 after treatment with 14 (0, 0.10, 0.45, 0.90 μM) for 24 h, *p < 0.05; **p < 0.01; ***p < 0.001. | ||
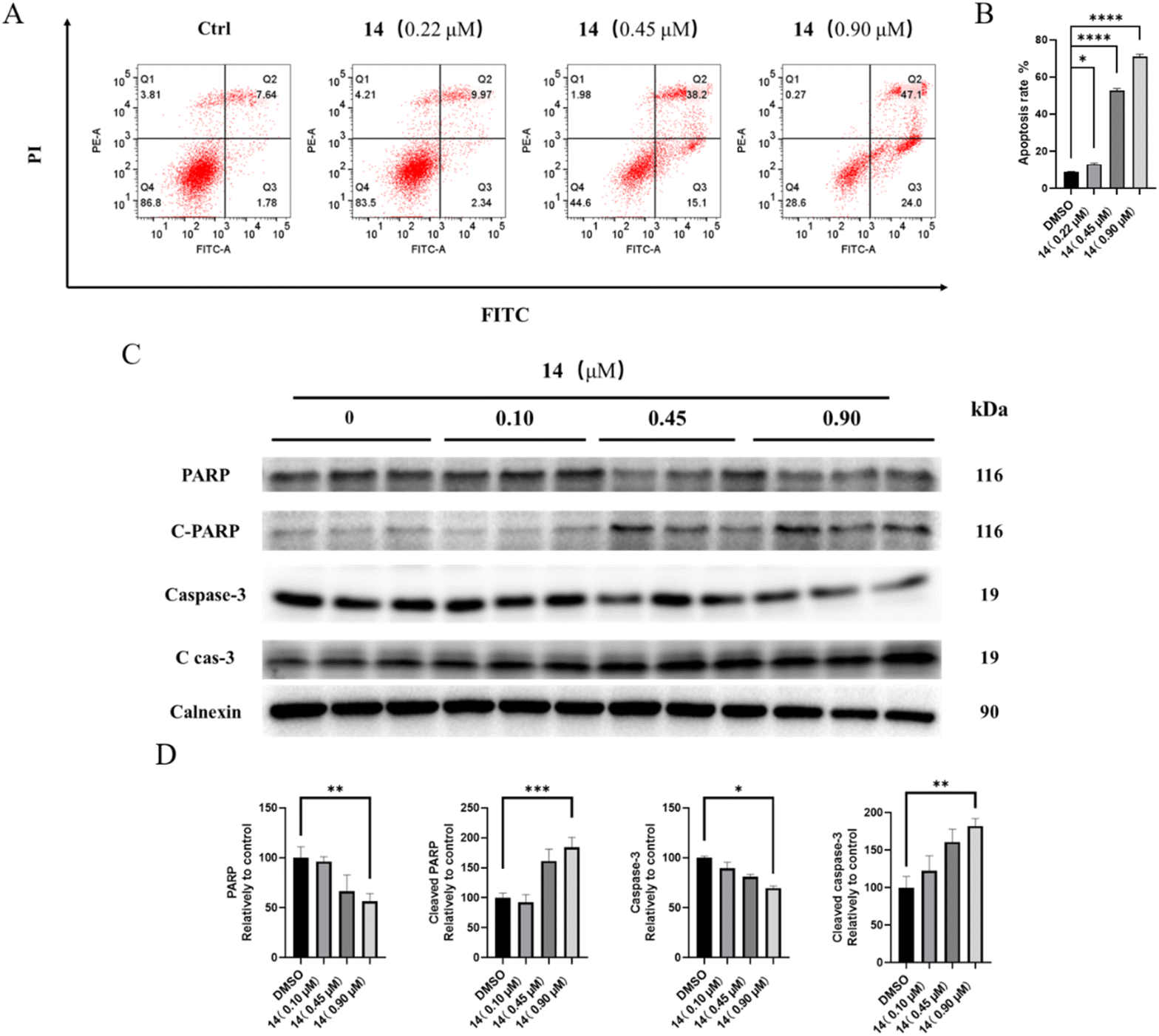
 | ||
| Fig. 2 The effect of compound 14 on HGC-27 cell cycle. (A) Flow cytometry and (B) bar graph analysis of cell cycle distribution in HGC-27 after treatment with compound 14 (0, 0.10, 0.45, 0.90 μM) for 48 h. (C) Western blot images and (D) bar graph analysis of p53, CDK2 and Cyclin A protein expressions in HGC-27 after treatment with compound 14 (0, 0.10, 0.45, 0.90 μM) for 24 h. *p < 0.05; **p < 0.01; ****p < 0.0001. | ||
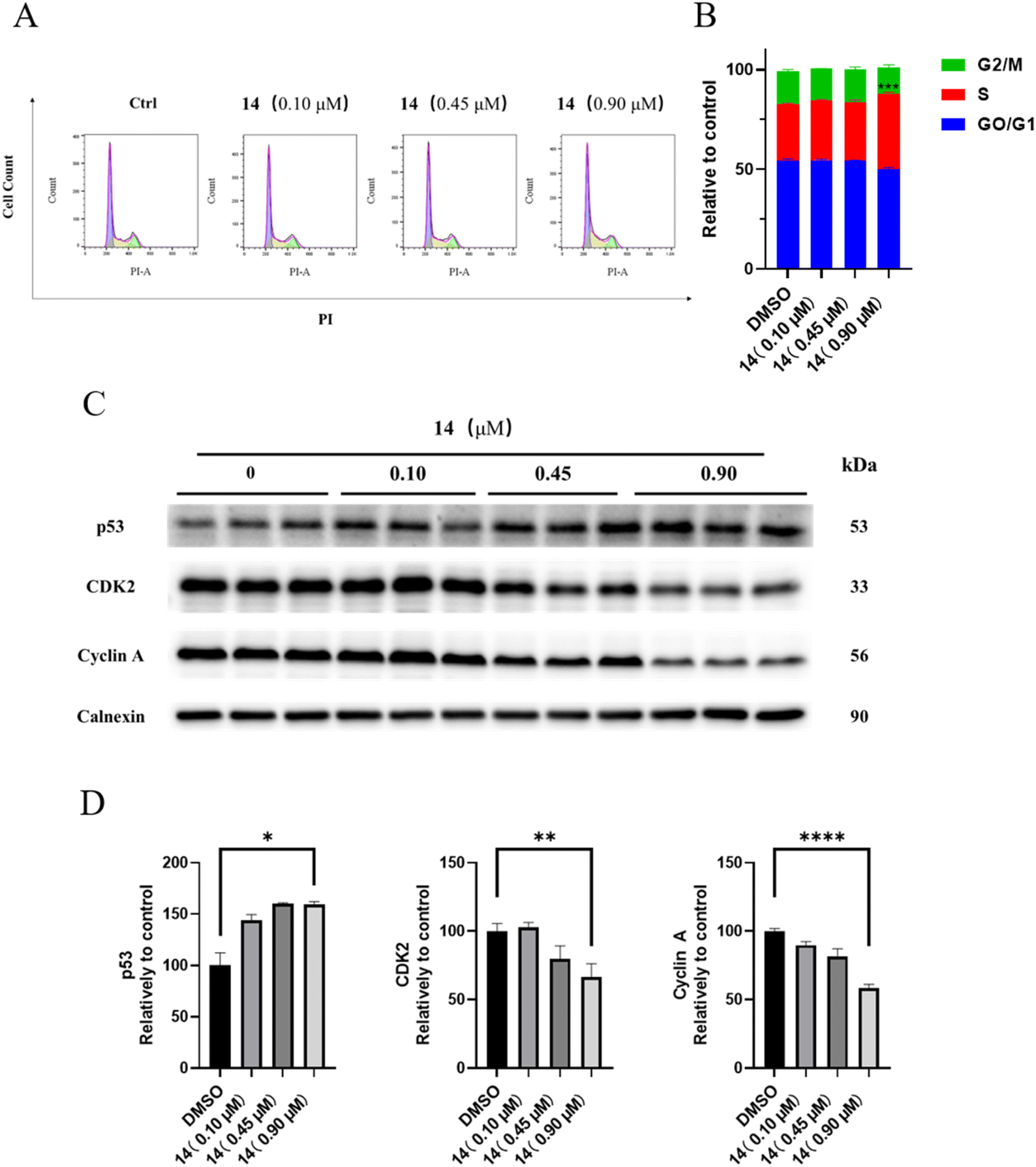
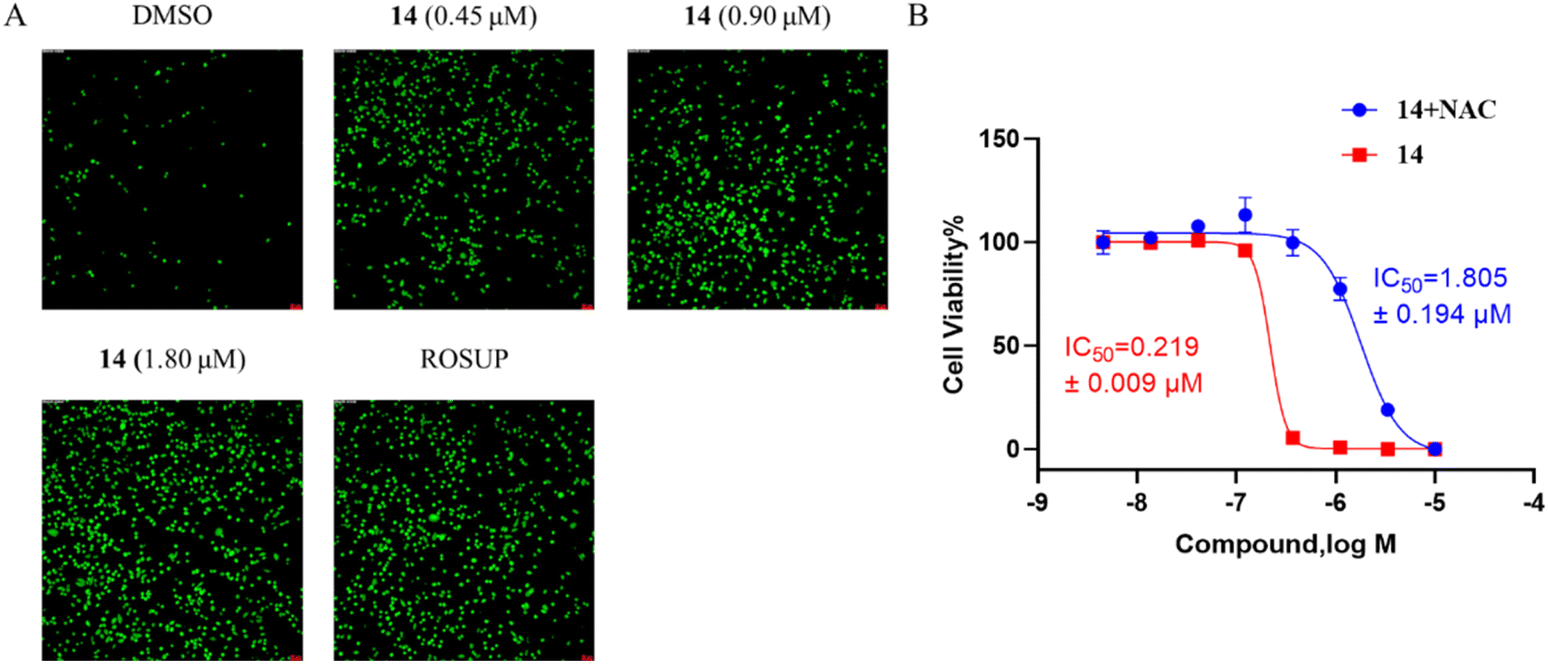 | ||
| Fig. 3 The effect of compound 14 on HGC-27 cell ROS generation. (A) Detection of ROS generation by DCFH-DA in HGC-27 cells after incubation with compound 14 (0, 0.45, 0.90, 1.80 μM) for 48 h, (B) ROS scavenger NAC increase the IC50 value of compound 14. | ||
 | ||
| Fig. 4 The effect of compound 14 on Erk/p38 MAPK signaling pathways. (A) Western blot images of Erk1/2, p-Erk1/2, p38 and p-p38 protein expressions in HGC-27 after treatment with (A) compound 14 (0, 0.10, 0.45, 0.90 μM) for 24 h. (B) Corresponding bar graph analysis of the signaling pathways. **p < 0.01; ****p < 0.0001. | ||
| S. aureus ATCC25923 | S. aureus MRSA ATCC515992 | S. epidermidis ATCC12228 | B. subtilis ATCC6633 | |
|---|---|---|---|---|
| Spiramycin I | 53.96 [64] | 53.96 [64] | 13.49 [16] | 13.49 [16] |
| 1 | 7.40 [8] | 14.80 [16] | 3.70 [4] | 3.70 [4] |
| 2 | 3.77 [4] | 7.54 [8] | 3.77 [4] | 1.89 [2] |
| 3 | 7.75 [8] | 15.51 [16] | 7.75 [8] | 1.94 [2] |
| 4 | 7.63 [8] | 15.25 [16] | 3.81 [4] | 3.81 [4] |
| 5 | 14.85 [16] | 29.70 [32] | 7.43 [8] | 3.71 [4] |
| 6 | 7.76 [8] | 15.52 [16] | 7.76 [8] | 1.94 [2] |
| 9a | >128.42 [>128] | 16.05 [16] | 4.01 [4] | 4.01[4] |
| 10a | 8.02 [8] | 32.08 [32] | 4.01 [4] | 4.01 [4] |
| 10b | 16.04 [16] | 16.04 [16] | 8.02 [8] | 2.00 [2] |
| 11 | >133.12 [>128] | 33.30 [32] | 4.16 [4] | 8.32 [8] |
| 12 | 8.13 [8] | 16.27 [16] | 4.07 [4] | 4.07 [4] |
| 13 | 17.56 [16] | 35.12 [32] | 8.78 [8] | 4.39 [4] |
| 14 | 17.75 [16] | 17.75 [16] | 8.88 [8] | 4.44 [4] |
| 16 | 3.87 [4] | 7.75 [8] | 0.97 [1] | 0.97 [1] |
| 17 | 15.61 [16] | 62.44 [64] | 7.80 [8] | 15.61 [16] |
| 20 | 8.24 [8] | 32.97 [32] | 8.24 [8] | 2.06 [2] |
| Linezolid | 1.35 [4] | 2.67 [8] | 0.34 [1] | 0.17 [0.5] |
Conclusion
In conclusion, we found that the 4′′-OH of spiramycin I can be preferably acylated under controlled conditions, leading to the preparation of 21 novel spiramycin I derivatives, which greatly expand its chemical space. More importantly, the 4′′-OH acylation greatly improves its anti-proliferative activity across four different cancer cell lines. The structure–activity relationship study indicated that the aryl-acylated derivatives of spiramycin I appear to have more potent anti-proliferative activity against the HGC-27 cell line compared to the alkyl-acylated derivatives. The most potent anti-proliferative compound 14 was found to arrest the HGC-27 cell cycle in the S phase, increase ROS levels, inhibit the phosphorylation of Erk and p38, thus lead to the apoptosis of HGC-27 cells. In addition to its anti-proliferative activity, most of these derivatives gave rise to enhanced antibacterial activity against four common strains compared to spiramycin I. Therein, compound 16 showed very potent antimicrobial activity comparable to that of linezolid. Considering that patients with malignant tumors are more likely to develop infection during surgery, drugs that have the dual functions of anticancer and antibacterial appear to be very advantageous in cancer treatment.Experimental
Chemistry
Method A: general synthesis procedure by using acyl chloride reagents. Spiramycin I (0.5 mmol, 1.0 equiv.) and the appropriate acyl chloride reagent (1.0 mmol, 2.0 equiv.) were dissolved in 10 ml of DCM. To this solution, N,N-dimethylaminopyridine (DMAP, 1.0 mmol, 2.0 equiv.) and triethylamine (TEA, 1.0 mmol, 2.0 equiv.) were added, and the mixture was stirred at 30 °C for 24 h. Once the acyl chloride reagent was completely consumed, the reaction mixture was concentrated under reduced pressure. The oil residue was purified by flash silica chromatography using a 5% ACN/CH2Cl2 eluent. The resulting crude product was further purified by a C18 pre-HPLC to give the target products.
Method B: general synthesis procedure using acid reagents. Spiramycin I (0.5 mmol, 1.0 equiv.) and the appropriate carboxylic acid (1.0 mmol, 2.0 equiv.) were dissolved in 10 ml DMF. To this solution, dicyclohexylcarbodiimide (DDC, 0.5 mmol, 1.0 equiv.) and 4-pyrrolidinopyridine (4-PPY, 0.25 mmol, 0.5 equiv.) were added, and the resulting mixture was stirred at 30 °C for 24 h. When the reaction was finished, the mixture was concentrated under reduced pressure. The oil residue was purified by flash silica chromatography using a 5% ACN/CH2Cl2 eluent. The resulting crude product was further purified by C18 pre-HPLC to give the target products.
Method C: general synthesis procedure by using isocyanate reagents. Spiramycin I (0.5 mmol, 1.0 equiv.) and the appropriate isocyanate (1.0 mmol, 2.0 equiv.) were dissolved in 10 ml of DMF. To this solution, triethylamine (TEA, 1.0 mmol, 2.0 equiv.) was added, the resulting mixture was stirred at 30 °C for 24 h. When the reaction was finished, the mixture was concentrated under reduced pressure. The oil residue was purified by flash silica chromatography using a 5% ACN/CH2Cl2, eluent. The resulting crude product was further purified by C18 pre-HPLC to give the target products.
Reagents, cell lines and bacteria strains. Cancer cell lines HGC-27, HT-29, HeLa, HCT-116 and the normal cell line GES-1 were purchased from the National Collection of Authenticated Cell Cultures (Shanghai, China). Fetal bovine serum (FBS), penicillin–streptomycin, and trypsin were purchased from Thermo Fisher Scientific Inc (Waltham, USA). Bacteria strains S. aureus ATCC25923, S. aureus MRSA ATCC515992, S. epidermidis ATCC12228 and B. subtilis ATCC6633 were purchased from BeNa Culture Collection (Beijing, China). MH broth and NB broth were also purchased from BeNa Culture Collection (Beijing, China). WST-8 and 5-FU were purchased from Yuanye Biotechnology (Shanghai, China). Protease inhibitors, phosphatase inhibitors, western blocking buffer, enhanced BCA protein assay kit, SDS-PAGE gel kit, BeyoECL Plus kit, Annexin V-FITC cell apoptosis detection kit and PI cell cycle detection kit were purchased from Beyotime Biotechnology (Shanghai, China). TBST buffer, glycine, Tris and SDS were purchased from Solarbio (Beijing, China). Antibodies against Erk1/2, p-Erk1/2, p38, p-p38, caspase-3, cleaved caspase-3, PARP, cleaved PARP, GAPDH, tubulin, anti-mouse IgG-HRP-linked antibody, and anti-rabbit IgG-HRP-linked antibody were purchased from Cell Signaling Technology (Boston, USA). Antibodies against p53, CDK2, Cyclin A and Calnexin were purchased from Proteintech (Wuhan, China).
Cell culture and anti-proliferation assay. HGC-27 and GES-1 were cultured in the RPMI 1640 medium. HT-29 and HCT116 were cultured in the McCoy's 5A medium. HeLa cells were cultured in the DMEM medium. All cell culture media contained 10% FBS and 1% penicillin–streptomycin. All cell lines were cultured in 5% CO2 incubator at 37 °C. When the cell confluency reached more than 80%, cells were digested and plated on 96 well plates (4000 cells per well), then incubated overnight for adherence. Different concentrations of 5-FU (0–50 μM, 2-fold dilutions) and derivatives (0–500 μM, 3-fold dilutions) were added to each well to measure the anti-proliferation ability. After 72 h treatment, old media was exchanged with the fresh media containing cell counting kit (CCK8, consist 0.5 mM wst-8 and 10 μM pms). After 0.5–2 h incubation, absorbance at wavelength 450 nm was recorded by the microplate reader. The cell viability and anti-proliferative activity were calculated.
Cell cycle distribution assay. The effects of compound 14 on cell cycle distribution were determined by flow cytometry. HGC-27 was seeded in 6-well plates (2 × 105 cells per well) and incubated overnight for adherence. Then, cells were treated with compound 14 (0, 0.10, 0.45, 0.90 μM) for 48 h. After treatment, cells were harvested, fixed in 70% ethanol for 1 h at 4 °C, and stained with a solution containing PI and RNase for 0.5 h. The stained cells were then examined on a FACS Calibur flow cytometer using CellQuest Pro software (BD Biosciences, California, USA). The percentage of cells in different phases of the cell cycle was determined using FlowJo software (BD Biosciences).
Cell apoptosis assay. The effects of compound 14 on cell apoptosis were determined by flow cytometry. HGC-27 cells were seeded in 6-well plates (2 × 105 cells per well) and incubated overnight for adherence. Then, cells were treated with compound 14 (0, 0.22, 0.45, 0.9 μM) for 48 h. After treatment, cells were harvested and stained with a solution containing Annexin V-FITC and PI for 0.5 h. The stained cells were then examined using a FACS Calibur flow cytometer with CellQuest Pro software (BD Biosciences). The percentage of apoptosis population was determined using FlowJo software (BD Biosciences).
Western blot assay. HGC-27 were seeded in 6 well plates (1 × 106 cells per well) and incubated overnight for adherence. Then, cells were treated with compound 14 (0, 0.10, 0.45, 0.9 μM) for 24 h. Cells were harvested and lysed with RIPA lysis buffer containing protease and phosphatase inhibitors. Lysates were centrifuged for 10 min at 15
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, and the supernatants were collected. Total protein concentration were measured using the enhanced BCA protein assay kit. Equal amounts of protein (20–50 μg) were loaded onto an SDS-PAGE gel (8%, 10%, 12%, 15%). After electrophoresis, the gels were transferred to a polyvinylidene difluoride (PVDF) membrane, which was then blocked by western blocking buffer for 1 h. Then PVDF membranes were interacted with primary antibodies overnight at 4 °C, and washed three times for 10 min in 1× TBST solution. After that, PVDF membranes were mixed with peroxidase-labeled secondary antibodies for 1 h at room temperature, then washed three times for 10 min in 1× TBST solution. Protein bands were acquired using BeyoECL Plus kit by ChemiDocTM XRS+ (Bio-Rad, California, USA). Band intensities were quantified using the Image J (National Institute of Health, Maryland, USA).
000 rpm, and the supernatants were collected. Total protein concentration were measured using the enhanced BCA protein assay kit. Equal amounts of protein (20–50 μg) were loaded onto an SDS-PAGE gel (8%, 10%, 12%, 15%). After electrophoresis, the gels were transferred to a polyvinylidene difluoride (PVDF) membrane, which was then blocked by western blocking buffer for 1 h. Then PVDF membranes were interacted with primary antibodies overnight at 4 °C, and washed three times for 10 min in 1× TBST solution. After that, PVDF membranes were mixed with peroxidase-labeled secondary antibodies for 1 h at room temperature, then washed three times for 10 min in 1× TBST solution. Protein bands were acquired using BeyoECL Plus kit by ChemiDocTM XRS+ (Bio-Rad, California, USA). Band intensities were quantified using the Image J (National Institute of Health, Maryland, USA).
ROS detection assay and NAC inhibition assay. HGC-27 were seeded in 96 well plates (1 × 104 cells per well) and incubated overnight for adherence. Then, cells were treated with compound 14 (0, 0.45, 0.90, 1.80 μM) for 24 h. As control, cells were treated with ROSUP (50 μg mL−1) for 0.5 h. After treatment, DCFH-DA was added for ROS detection, the green fluorescence was observed using IFM (Leica, Wetzlar, German). HGC-27 were seeded in 96-well plates (4 × 103 cells per well) and incubated overnight for adherence. Then, cells were treated with different compounds (group compound 14 cell were only treated with compound 14, group Cotreat cells were treated with compound 14 and 3 mM NAC). Cell viability was measured using the CCK8 assay and the IC50 was calculated after 72 h.
Bacterial culture and MIC assay. S. aureus ATCC25923, S. aureus MRSA ATCC515992, and B. subtilis ATCC6633 were cultured in MH broth, while S. epidermidis ATCC12228 was cultured in NB broth. All bacterial strains were cultured at 37 °C under aerobic conditions. Concentrated solutions of the tested compounds (positive control linezolid) were dissolved in DMSO and diluted in MH/NB broth to obtain the required concentrations (0.5–128 μM). Four bacterial strains were diluted (final concentration OD600 = 0.05) and plated in 96-well plate. Different concentrations of the tested compounds were added to each well. The minimal inhibitory concentration (MIC) was determined after 18 h of incubation at 37 °C.
Data availability
All relevant data are within the manuscript and its additional files.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend their sincere appreciation to the Jiangxi Provincial Natural Science Foundation (20224BAB216003).References
- A. Thibessard, D. Haas, C. Gerbaud, B. Aigle, S. Lautru, J. L. Pernodet and P. Leblond, J. Biotechnol., 2015, 214, 117–118 CrossRef CAS.
- T. Mazzei, E. Mini, A. Novelli and P. Periti, J. Antimicrob. Chemother., 1993, 31(Suppl C), 1–9 CrossRef CAS.
- L. Liu, E. Roets and J. Hoogmartens, J. Chromatogr. A, 1997, 764, 43–53 CrossRef CAS.
- K. Ramu, S. Shringarpure and J. S. Williamson, Pharm. Res., 1995, 12, 621–629 CrossRef CAS.
- T. Miura, K. Kanemoto, S. Natsume, K. Atsumi, H. Fushimi, T. Yoshida and K. Ajito, Bioorg. Med. Chem., 2008, 16, 10129–10156 CrossRef CAS PubMed.
- E. Rubinstein and N. Keller, J. Antimicrob. Chemother., 1998, 42, 572–576 CrossRef CAS PubMed.
- M. A. Pilot and X. Y. Qin, J. Antimicrob. Chemother., 1988, 22(Suppl B), 201–206 CrossRef PubMed.
- H. Sano, T. Sunazuka, H. Tanaka, K. Yamashita, R. Okachi and S. Omura, J. Antibiot., 1984, 37, 750–759 CrossRef CAS PubMed.
- H. Sano, T. Sunazuka, H. Tanaka, K. Yamashita, R. Okachi and S. Omura, J. Antibiot., 1984, 37, 760–772 CrossRef CAS.
- M. Sano, T. Sunazuka, H. Tanaka, K. Yamashita, R. Okachi and S. Omura, J. Antibiot., 1985, 38, 1350–1358 CrossRef CAS PubMed.
- T. Hirose, T. Sunazuka, Y. Noguchi, Y. Yamaguchi, H. Hanaki, K. B. Sharpless and S. Omura, Heterocycles, 2006, 69, 55–61 CrossRef CAS PubMed.
- H. Sano, H. Tanaka, K. Yamashita, R. Okachi and S. Omura, J. Antibiot., 1985, 38, 186–196 CAS.
- K. I. Rivera-Márquez, C. Godoy-Alcántar, M. Á. Claudio-Catalán and F. Medrano, J. Inclusion Phenom. Macrocyclic Chem., 2016, 86, 211–219 CrossRef.
- S. K. Murphy, J. W. Park, F. A. Cruz and V. M. Dong, Science, 2015, 347, 56–60 CrossRef CAS PubMed.
- G. Castro-Falcón, N. Millán-Aguiñaga, C. Roullier, P. R. Jensen and C. C. Hughes, ACS Chem. Biol., 2018, 13, 3097–3106 CrossRef PubMed.
- H. Takahira, Jpn. J. Antibiot., 1970, 23, 424–428 CAS.
- U. Galm, M. H. Hager, S. G. Van Lanen, J. Ju, J. S. Thorson and B. Shen, Chem. Rev., 2005, 105, 739–758 CrossRef CAS PubMed.
- A. Hussain, M. S. Dar, N. Bano, M. M. Hossain, R. Basit, A. Q. Bhat, M. A. Aga, S. Ali, Q. P. Hassan and M. J. Dar, Cancer Chemother. Pharmacol., 2019, 84, 551–559 CrossRef CAS.
- X. Qiao, X. Wang, Y. Shang, Y. Li and S. Z. Chen, Cancer Commun., 2018, 38, 43 Search PubMed.
- X. Zhou, Y. Zhang, Y. Li, X. Hao, X. Liu and Y. Wang, Cancers, 2012, 4, 1318–1332 CrossRef CAS PubMed.
- N. I. Abdel-Hamid, M. F. El-Azab and Y. M. Moustafa, Naunyn-Schmiedeberg's Arch. Pharmacol., 2017, 390, 379–395 CrossRef CAS PubMed.
- R. Lamb, B. Ozsvari, C. L. Lisanti, H. B. Tanowitz, A. Howell, U. E. Martinez-Outschoorn, F. Sotgia and M. P. Lisanti, Oncotarget, 2015, 6, 4569–4584 Search PubMed.
- K. Klich, K. Pyta, M. M. Kubicka, P. Ruszkowski, L. Celewicz, M. Gajecka and P. Przybylski, J. Med. Chem., 2016, 59, 7963–7973 CAS.
- S. Y. Liang, T. C. Zhao, Z. H. Zhou, W. T. Ju, Y. Liu, Y. R. Tan, D. W. Zhu, Z. Y. Zhang and L. P. Zhong, Transl. Oncol., 2021, 14, 101074 CAS.
- Y. Jin, H. X. Zuo, M. Y. Li, Z. H. Zhang, Y. Xing, J. Y. Wang, J. Ma, G. Li, H. Piao, P. Gu and X. Jin, Front. Pharmacol, 2021, 12, 774231 CAS.
- Z. Liu, M. Huang, Y. Hong, S. Wang, Y. Xu, C. Zhong, J. Zhang, Z. Zhuang, S. Shan and T. Ren, Int. J. Biol. Sci., 2022, 18, 3714–3730 CAS.
- J. Cui, J. Zhou, W. He, J. Ye, T. Westlake, R. Medina, H. Wang, B. L. Thakur, J. Liu, M. Xia, Z. He, F. E. Indig, A. Li, Y. Li, R. J. Weil, M. I. Aladjem, L. Zhong, M. R. Gilbert and Z. Zhuang, J. Exp. Clin. Cancer Res., 2022, 41, 126 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03126a |
| ‡ Zhiwei Wang and Junxiang Cheng were contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |