
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and medicinal chemical characterisation of antiproliferative O,N-functionalised isopulegol derivatives†
Tam Minh Leab,
Isaac Kinyua Njangiruc,
Anna Vinczed,
István Zupkóc,
György T. Balogh*d and
Zsolt Szakonyi *a
*a
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös utca 6, H-6720 Szeged, Hungary. E-mail: szakonyi.zsolt@szte.hu; Fax: +36 62 545705; Tel: +36 62 546809
bHUN-REN–SZTE Stereochemistry, Research Group, University of Szeged, Eötvös u. 6, H-6720 Szeged, Hungary
cInstitute of Pharmacodynamics and Biopharmacy, University of Szeged, H-6720, Eötvös utca 6, Szeged, Hungary
dDepartment of Pharmaceutical Chemistry, Semmelweis University, Hőgyes Endre u. 9, H-1092 Budapest, Hungary
First published on 12th June 2024
Abstract
Benzylation of isopulegol furnished O-benzyl-protected isopulegol, which was transformed into aminodiols via epoxidation followed by ring opening of the corresponding epoxides and subsequent hydrogenolysis. On the other hand, (−)-isopulegol was oxidised to a diol, which was then converted into dibenzyl-protected diol derivatives. The products were then transformed into aminotriols by using a similar method. The antiproliferative activity of aminodiol and aminotriol derivatives was examined. In addition, structure–activity relationships were also explored from the aspects of substituent effects and stereochemistry on the aminodiol and aminotriol systems. The drug-likeness of the compounds was assessed by in silico and experimental physicochemical characterisations, completed by kinetic aqueous solubility and in vitro intestinal-specific parallel artificial membrane permeability assay (PAMPA-GI) measurements.
1 Introduction
Optically active aminodiols are of interest for the chiral pool and as partial structures of biologically active compounds.1–5 For example, their motif is found in the backbone of many bioactive molecules6–9 including pactamycin (antitumor),10 myriocin (antibiotic),11 the proteasome inhibitor TMC-95A12 and imino sugars,13 many of which are potent glycosidase inhibitors, just as miglustat and miglitol, which are in clinical development. In addition, the aminodiol subunit is present in several aza-sugars, polyhydroxylated indolizidine and pyrrolizidine alkaloids, many of which have interesting biological properties.14,15Furthermore, 2-amino-1,3-diol derivatives are common structural motifs present in nature. Relevant members of this family are sphingoid bases,16 sphinganines and clavaminol derivatives,17,18 which play important roles in physiological processes. In particular, sphingolipids,19,20 components of the cell membrane in living organisms, have been reported to be involved in cell recognition and signal transduction,21,22 and they show antitumor,23,24 immune-modulatory25 and immunosuppressive activities.26,27
Besides pharmacological interests, aromatic and aliphatic aminodiols bearing a 1,2- or a 1,3-aminoalcohol moiety have proven to be useful building blocks materials in the stereoselective synthesis of compounds with pharmacological interest.28–30
Besides their synthetic importance, aminodiols can also be applied as chiral ligands and auxiliaries in enantioselective transformation.31–33 In this regard, the asymmetric addition of organometallic reagents to aldehydes has become a highly investigated model reaction, applying chiral promoters such as 1,2- or 1,3-difunctionalised ligands.34–36
Several chemical methods exist to produce 3-amino-1,2-diols, starting from chiral pool materials such as serine37–39 and glucose.40 These include Sharpless asymmetric dihydroxylation with subsequent regioselective azide substitution,41,42 nucleophilic attack of α,β-epoxy carboxylic esters with azide followed by azide and ester reduction,43 nonenzymatic one-pot synthesis of α,α′-dihydroxyketones via a mimetic of the transketolase reaction44 and two-step stereoselective biocatalytic synthesis using a transketolase (TK) and a transaminase (TAm).45 However, the most popular approaches to these motifs often employ the chiral pools utilising the stereospecific (e.g. Sharpless) epoxidation46–52 followed by C-3 regioselective ring-opening of 2,3-epoxy alcohols with amines.53–68 In the last decade, a metal- and solvent-free protocol, 69 involving C-3 selective ring-opening of 2,3-epoxy alcohols with amines catalysed by W-salts,70 β-cyclodextrin,66 MgCl2,71 or the tungsten/bis(hydroxamic acid) system72 as well as a two-step combined epoxidation/ring-opening methodology73 were shown to generate virtually enantiopure functionalised 3-amino-1,2-diols. The ring-opening of chiral epoxy alcohols with nucleophiles74–82 provides direct access to versatile chiral building blocks for the synthesis of natural products and synthetic analogues with promising biological activities,83,84 such as cardiovascular,85 antibacterial86,87 and sedative effects.88
As a continuation of our research in structural modification of (−)-isopulegol for the development of anticancer agents,89 we herein report the preparation of a new library of isopulegol-based chiral aminodiols and, as an extension, aminotriols via asymmetric epoxidation and aminolysis, starting from commercially available natural (−)-isopulegol. We have also carried out a preliminary study investigating the effect of stereochemistry and substitution level of the amine and hydroxyl functions on the antiproliferative activity on multiple human cancer cell lines. In addition to exploring the pharmacological effect, the medicinal properties of novel (−)-isopulegol derivatives have also been examined using in silico medchem-related parameters and early-stage in vitro screening assays on a panel of human cancer cells.
2 Results and discussion
2.1. Chemistry
Benzylation of isopulegol 1 produced O-benzyl isopulegol 2a, which was subjected to epoxidation with m-CPBA to provide a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture of epoxides 3a and 3b in good yields. After chromatographic separation, ring opening of epoxide 3a with different primary amines in the presence of LiClO4 as catalyst delivered O-benzyl derivatives 6a–13a in moderate to good yields. LiClO4 shows enhanced reactivity for the ring-opening of epoxides through the coordination of Li+ with the epoxide oxygen, rendering the epoxide more susceptible to nucleophilic attack by amines and, therefore, dramatically reducing reaction times and improving yields.90 Although ClO4− can serve as an oxidative reagent, there is no oxidation under the applied condition. In addition to amines, the azole-mediated ring-opening of 3a with a variety of azoles was also performed. In a similar fashion, the oxirane ring could only be opened in the presence of K2CO3 owing to the lower reactivity of N-containing heterocycles. A possible reaction pathway through K2CO3-promoted azole nucleophilicity and subsequent nucleophilic addition to epoxide 3a afforded derivatives 14a and 15a.91 Debenzylation of 6a by hydrogenolysis over Pd/C in MeOH resulted in primary aminodiol 28a in moderate yields. The other epoxide 3b underwent similar reactions. Interestingly, the oxirane ring-opening with azoles was successfully achieved, while aminolysis with primary amines did not take place under the applied conditions. This is probably due to steric hindrance exerted by both the benzyl and the methyl group at the α position in epoxide 3b.92 The azole-based aminoalcohol adducts 14b and 15b were obtained with moderate yields (Scheme 1).
:1 diastereomeric mixture of epoxides 3a and 3b in good yields. After chromatographic separation, ring opening of epoxide 3a with different primary amines in the presence of LiClO4 as catalyst delivered O-benzyl derivatives 6a–13a in moderate to good yields. LiClO4 shows enhanced reactivity for the ring-opening of epoxides through the coordination of Li+ with the epoxide oxygen, rendering the epoxide more susceptible to nucleophilic attack by amines and, therefore, dramatically reducing reaction times and improving yields.90 Although ClO4− can serve as an oxidative reagent, there is no oxidation under the applied condition. In addition to amines, the azole-mediated ring-opening of 3a with a variety of azoles was also performed. In a similar fashion, the oxirane ring could only be opened in the presence of K2CO3 owing to the lower reactivity of N-containing heterocycles. A possible reaction pathway through K2CO3-promoted azole nucleophilicity and subsequent nucleophilic addition to epoxide 3a afforded derivatives 14a and 15a.91 Debenzylation of 6a by hydrogenolysis over Pd/C in MeOH resulted in primary aminodiol 28a in moderate yields. The other epoxide 3b underwent similar reactions. Interestingly, the oxirane ring-opening with azoles was successfully achieved, while aminolysis with primary amines did not take place under the applied conditions. This is probably due to steric hindrance exerted by both the benzyl and the methyl group at the α position in epoxide 3b.92 The azole-based aminoalcohol adducts 14b and 15b were obtained with moderate yields (Scheme 1).
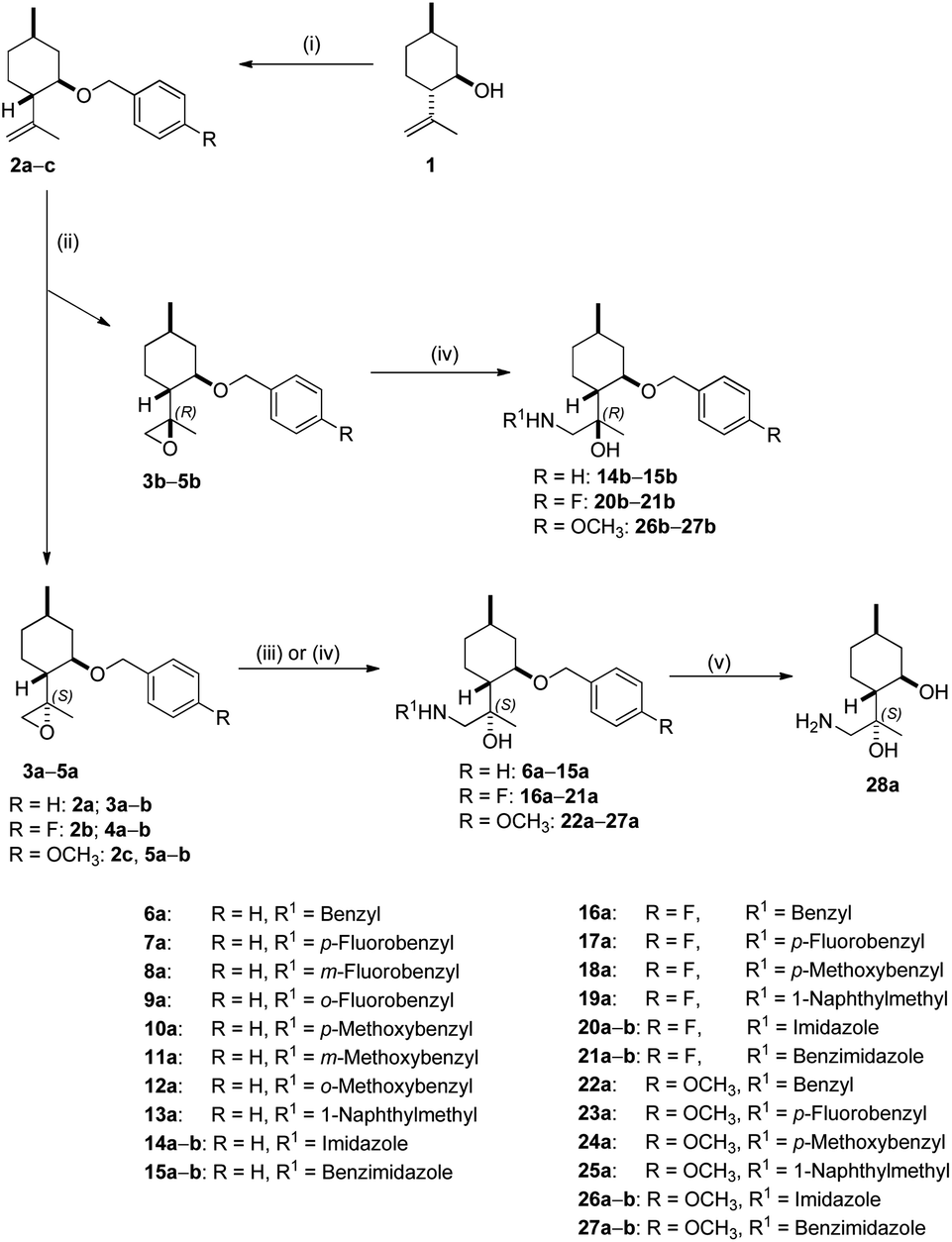 | ||
| Scheme 1 Isopulegol-based chiral aminodiol derivatives. Reagents and conditions: (i) NaH (1.0 eq.), substituted BnBr (1.5 eq.), KI (1.0 eq.), dry THF, 60 °C, 12 h, 70–88%; (ii) m-CPBA (2 eq.), Na2HPO4·12H2O (3 eq.), CH2Cl2, 25 °C, 2 h, 25–47%; (iii) R1NH2 (2 eq.), LiClO4 (1 eq.), CH3CN, 70–80 °C, 20 h, 55–92%; (iv) imidazole or benzimidazole (3 eq.), K2CO3 (5 eq.), dry DMF, 70–80 °C, 96 h, 25–71%; (v) 5% Pd/C, H2 (1 atm), CH3OH, 25 °C, 24 h, 67–87%. | ||
The literature revealed that various substituents of the benzyl group resulted in a significantly increased anticancer activity.93 Therefore, benzylations were performed by utilising p-fluoro- and p-methoxybenzyl bromides as reagents. Benzyl-protected isopulegol 2b and 2c were formed in good yields. In the same manner, 2b and 2c afforded aminodiol derivatives 16–21 and 22–27 by the method described in Scheme 1. Aminodiols 16a and 22a were then transformed into primary aminodiol 28a by hydrogenolysis over Pd/C. It has been reported that aminolysis and subsequent debenzylation gave the corresponding aminodiol with the same stereochemical configuration at the carbon atoms as that of the original moiety.92 The stereochemical structures of aminodiol 28a is well-known in the literature,92 therefore, the absolute configuration of epoxide 3a–5a could also be determined (Scheme 1).
Since the presence of the di-O-benzyl group could improve lipophilic properties leading to enhanced antiproliferative activity, derivative 30a was synthesised.94 However, the synthesis of 30a starting from 2a failed. Fortunately, we realised that it could be achieved starting from 29, prepared from 1 by a three-step sequence including acetylation with Ac2O/pyridine, followed by oxidation using SeO2/t-BuOOH (TBHP) as oxidant and subsequent reduction with LiAlH4.95 Diol 29 smoothly transformed into di-O-benzyl derivative 30a with an acceptable yield. Epoxidation of 30a with m-CPBA produced a 1:1 mixture of epoxides 31a and 31b. After purification, ring opening of oxirane 31a was accomplished with different primary amines as well as azoles resulting in a library of aminoalcohols 34a–41a and azole-based products 42a and 43a, respectively. Following this procedure, diastereoisomeric aminotriols 34b–41b and 42b–43b were prepared by ring opening of 31b with primary amines and azoles. Primary aminotriols 56a and 56b was obtained by debenzylation of the corresponding aminotriols 34a and 34b by hydrogenation in the presence of a Pd/C catalyst under standard conditions (Scheme 2).
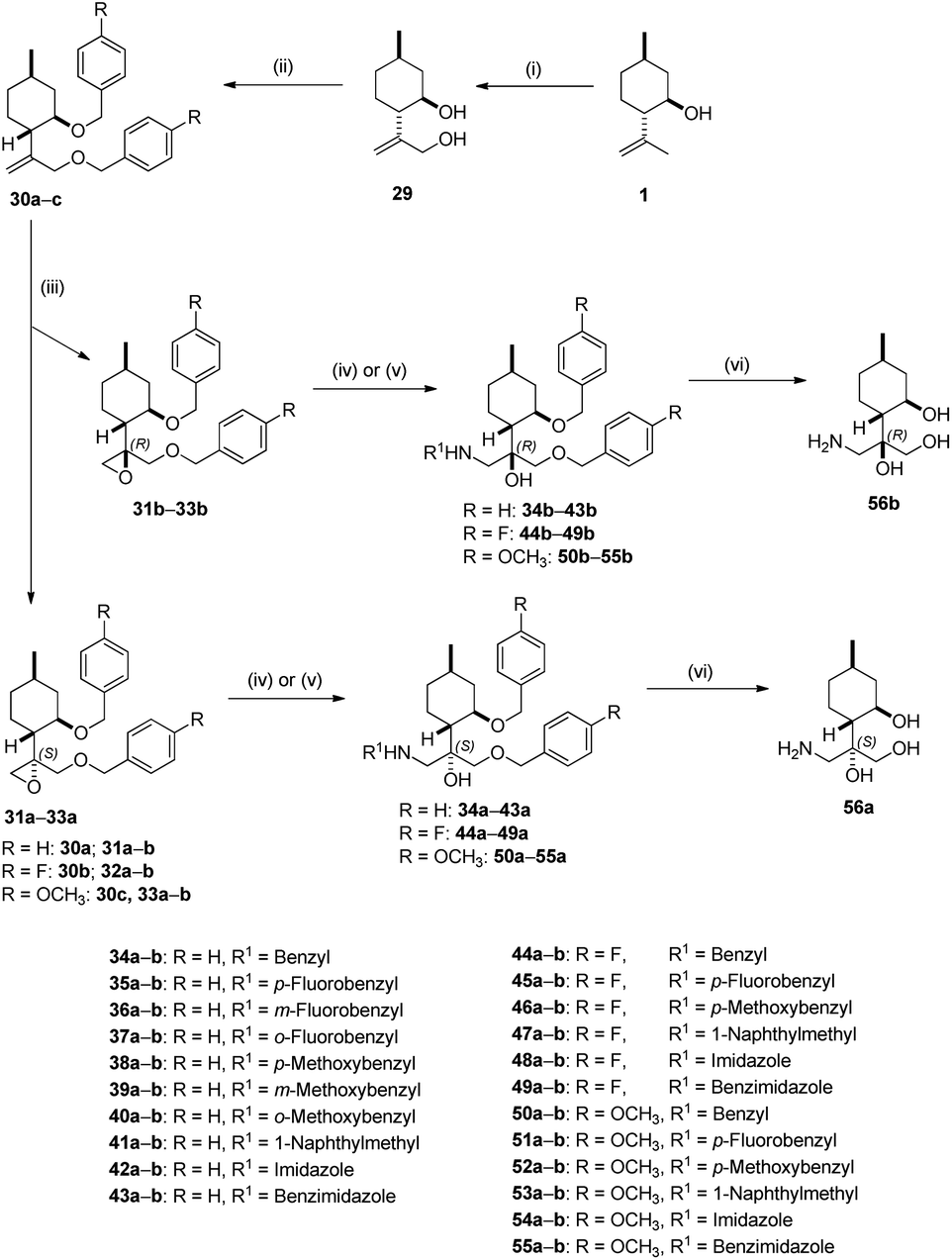 | ||
| Scheme 2 Isopulegol-based chiral aminotriol derivatives. Reagents and conditions: (i) (a) (CH3CO)2O (2 eq.), dry pyridine (1 eq.), 25 °C, 24 h, 95%, (b) SeO2 (0.24 eq.), 70% t-BuOOH (4 eq.), CHCl3, 60 °C, 20 h, (c) LiAlH4 (3 eq.), dry (CH3CH2)2O, 0 °C, 6 h, 60%; (ii) NaH (1.0 eq.), substituted BnBr (1.5 eq.), KI (1.0 eq.), dry THF, 60 °C, 24–72 h, 60–88%; (iii) m-CPBA (2 eq.), Na2HPO4·12H2O (3 eq.), CH2Cl2, 25 °C, 2 h, 28–48%; (iv) R1NH2 (2 eq.), LiClO4 (1 eq.), CH3CN, 70–80 °C, 8 h, 60–92%; (v) imidazole or benzimidazole (3 eq.), K2CO3 (5 eq.), dry DMF, 70–80 °C, 48 h, 45–84%; (vi) 5% Pd/C, H2 (1 atm), CH3OH, 25 °C, 24 h, 75–93%. | ||
In order to study the effects of substituents of the benzyl group on antiproliferative properties, substituted di-O-benzyl derivatives with either electron-withdrawing or electron-donating substituents were obtained. When diol 29 were treated with either p-fluorobenzyl bromide or p-methoxybenzyl bromide in the presence of a catalytic amount of KI under reflux condition, product 30b formed rapidly (24 h) in good yield, whereas longer reaction time (72 h) was required in the case of 30c. The probable reason is steric hindrance exerted by the p-methoxybenzyl group of 30c at the α position, which prevents the approach to the other hydroxyl group of the cyclohexyl ring. Di-O-benzyl derivatives 30b–c were transformed into aminoalcohols 44–49 and 50–55 in the same manner described in Scheme 2. Debenzylation via hydrogenolysis of compounds 44a, 50a as well as 44b, 50b over Pd/C resulted in primary aminotriol 56a and 56b in good yields, respectively. Since neither aminolysis of oxiranes 31a–b, 32a–b and 33a–b in alkaline condition nor the hydrogenolysis of N-benzyl analogues 33a–b, 44a–b and 50a–b had any effect on the absolute configuration, the relative configuration of the chiral centers of 56a–b is known to be the same as that of epoxides 31a–b, 32a–b and 33a–b.92 The stereochemical structure of aminotriols 56a–b is well-known in the literature,92 therefore, the absolute configuration of di-O-benzyl oxiranes 31a–b, 32a–b and 33a–b could also be determined (Scheme 2).
2.2. Antiproliferative activity
The in vitro antiproliferative activities of the synthesised aminodiols and aminotriols against a panel of different human cancer cell lines, including cervical (HeLa), breast (MCF7 and MDA-MB-231) and ovary (A2780) cancers, were assayed by the MTT method. Cisplatin, a clinically applied anticancer agent, was used as a positive control, and the results are summarised in Fig. 1 and Table S1 in the ESI.† | ||
| Fig. 1 Antiproliferative activities of the O,N-functionalised (−)-isopulegol analogues against cancer cells. *: data from reference.96 | ||
Our results indicated that compounds 10a and 11a were found to be inhibiting MCF-7 cell proliferation in a selective manner with IC50 values of 1.93 and 1.95 μM, respectively. In particular, N-benzyl-substituted di-O-benzyl derivatives exhibited remarkable antiproliferation of all tested cancer cell lines. Compounds 34a–b, 35a–b, 36a–b, 37a–b, 38a–b, 39a–b and 40a–b containing secondary amino function were the most active antiproliferative agents among the tested derivatives, while imidazole- or benzimidazole-derived congeners (42a–b, 43a–b) were ineffective. In the analysis of each individual cell line, HeLa cells were shown to be potently inhibited by compounds 34b, 37a, 38a–b and 40a with IC50 values in the range of 2.0–2.3 μM, in comparison with the positive control (cisplatin IC50 = 12.43 μM). Furthermore, compounds 34a, 37a, 38a–b and 40a–b could effectively inhibit MCF7 cells, with IC50 values around 2.0 μM, compared with cisplatin (IC50 = 5.78 μM). MDA-MB-231 cells were strongly inhibited by compounds 40a and 40b with IC50 values of 2.4 and 2.0 μM, respectively, in comparison with cisplatin (IC50 = 3.74 μM). Moreover, A2780 cells were highly susceptible to compounds 38a, 40a and 40b with IC50 values of 2.5, 2.2 and 2.0 μM, respectively, compared with cisplatin (IC50 = 1.3 μM).
2.3. In silico physicochemical characterisation and in vitro results
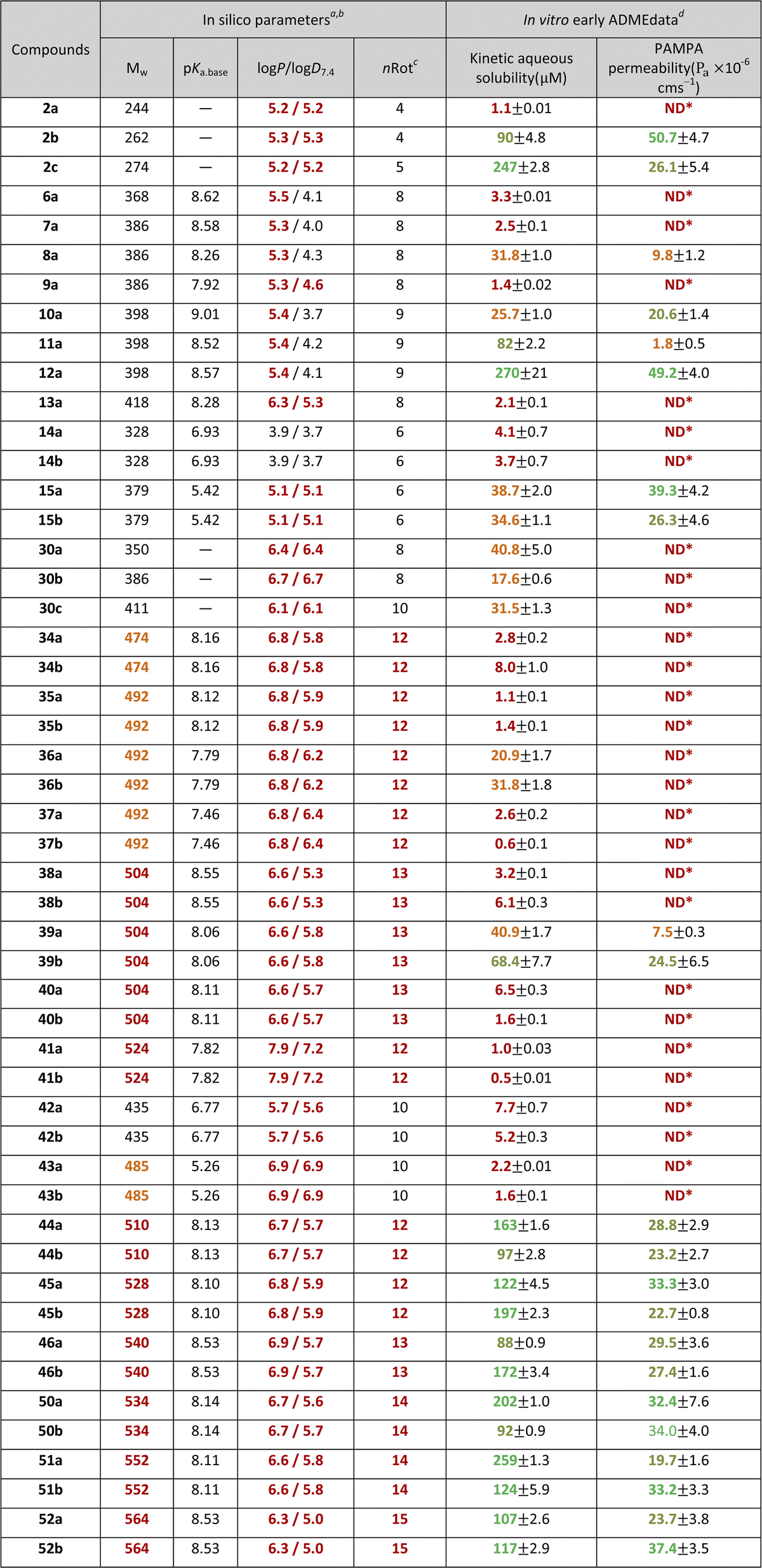
The characterisation for pre-screening of the novel isopulegol derivatives was primarily performed by mapping the in silico physicochemical properties and in vitro early ADME parameters that determine the drug-likeness (Table 1). Based on the overview of the in silico data, it can be seen that the starting structural element in O-benzyl isopulegol derivatives (2a–c) already carries an increased lipophilicity, which is one of the fundamental problems of medicinal chemists, that is, a unique approach to the lead optimisation process of the chemical space beyond the classical Lipinski rule of five (bRo5).97 However, it is worth noting that in natural product-based drug research programmes it is not unusual to exploit the bRo5 space.98 This, in our case, becomes particularly pronounced with N-benzyl O-benzyl aminodiol (6a–30c) and di-O-benzyl aminotriol (34a–52b) derivatives. The relationship between elevated lipophilicity and early ADME data can also be influenced by the basicity of the secondary amine moiety formed in the novel aminodiol–aminotriol derivatives, which can be identified on the basis of the pKa.base values and logP − logD7.4 differences. In our case, the unique behaviour of the bRo5-coupled physicochemical space is well reflected in the comparison of in silico parameters and in vitro characteristics. Regarding the lipophilicity values for neutral and ionic forms and the pKa values describing the basic nature, no correlation can be drawn based on the kinetic aqueous solubility (kin.sol.) measured in a buffered system (PBS, pH 7.4) or the apparent permeability values (Pa) obtained in the phosphatidylcholine:cholesterol-based artificial lipid system (PAMPA, pHdonor/acceptor 7.4), which could be clearly interpreted. Thus, in contrast to weak kin.sol. and Pa values, identified in the case of derivatives with favourable physicochemical parameters (e.g. 6a, 7a, 14a–b), several representatives of bRo5 also provided medium–high in vitro early ADME parameters (e.g. 8a, 15a–b, 39a–b). In the latter case, the N-benzyl di-O-benzyl derivative group 44a–52b should be mentioned as highlighted examples, where, in addition to the violation of the classic Ro5 (Mw and logP),99 the Veber's rule100 (number of rotatable bond: nRot <10 for good drug absorption) is also infringed, despite the fact that the kin.sol. and Pa values of these compounds are particularly high among all tested isopulegol derivatives.
| a All in silico parameters were calculated by ACD/Labs Percepta: release 2023.1.1 (Build 3666, accessed on 4 Jul 2023, https://www.acdlabs.com/products/percepta-platform/).b Classification system for in silico parameters: orange and red coloring indicates moderate and high violations of drug-likeness using Lipinski's99 and Veber's100 rules.c nRot – number of rotatable bonds.d Classification for experimental data: red – poor: kin.sol. ≤10 μM, Pa: *ND (non-detectable compound at the acceptor site) – practically non-permeable; orange – moderate: 10 μM < kin.sol. ≤ 50 μM or Pa (·10−6 cm s−1) ≤ 10; light green – high: 50 μM < kin.sol. ≤ 100 μM or 10 < Pa (·10−6 cm s−1) ≤ 30; green – increased: kin.sol. >100 μM or Pa (·10–6 cm s−1) >30. |
|---|
 |
Regarding the increased lipophilicity and general bRo5 nature of the tested isopulegol derivatives, it is also necessary to investigate separately their promiscuity risk and selectivity for individual cancer cell lines. In accordance, the compounds with an IC50 value less than 10 μM were evaluated and classified separately, according to their lipophilicity efficiency (LipE), which guides drug research programmes towards drug candidates that provide excellent in vivo efficacy and safety.101 As can be seen in Table 2, the 4-level classification system for both IC50 and LipE values proved to be sufficiently strict to identify selective and promiscuous isopulegol derivatives. Thus, it is particularly important to highlight compounds of our study that selectively provided a LipE value that was emphasised as a decreased (LipE >0.0) or moderate (−0.5 < LipE ≤ 0.0) promiscuity risk. These criteria are met by two compounds of high potency (10a, 11a) and one of low potency (15a) with high MCF-7 selectivity, which were identified as primary in vivo preclinical candidates. Compounds 12a and 15b showing HeLa and MCF-7 selectivity and compounds 36b and 39a–b, indicating MCF-7 and A2780 selectivity, were identified as secondary candidates with narrower selectivity and less favourable LipE values. Considering that several of the isopulegol derivatives showing selectivity also contain a methoxybenzyl moiety (10a, 11a, 39a), among the non-selective compounds with a high promiscuity risk, partial MCF-7 selectivity and sub-micromolar effect (IC50 < 1 μM), compounds 45a and 46a can be used for more detailed investigation as follow-up lead candidates. With a similar consideration, the partial selectivity of molecule 50b for the A2780 cell line can be highlighted as an alternative candidate for further preclinical investigations.
| a Levels of in vivo preclinical candidate: green – primary candidates, orange – secondary candidates, red – representants of non-selective compounds.b Classification for antiproliferative effectivity: grey – non-/poorly effective at 30 and 10 μM test concentration or IC50 > 10 μM; red – low: 6 μM ≤ IC50 ≤ 10 μM; orange – moderate: 3 μM ≤ IC50 < 6 μM; light green – high: 1 μM ≤ IC50 < 3 μM; green – very-high: IC50 < 1 μM.c Classification for LipE/promiscuity risk: purple – extreme risk: LipE ≥ −1.0, red – high risk: −1.0 < LipE ≤ −0.5, orange – moderate risk: −0.5 < LipE ≤ 0.0, light green – decreased risk: LipE > 0.0. |
|---|
 |
2.4. Structure–activity relationship
The SARs of these novel derivatives were analysed and summarised. Compounds 34a–b containing an N-benzyl-substituted aminoalcohol moiety showed enhanced antiproliferative activity compared with parent compound 30a. This observation further confirms that the secondary amino scaffold seems to be needed for the pronounced activity, since analogues without these elements are considerably less effective. Introduction of substituents with electron-donating effect (OCH3) into the N-benzyl ring could significantly improve the antiproliferative activity against most of the tested cancer cell lines (compared 34a–b with 38a–b), while compounds 35a–b with electron-withdrawing character (F) at the N-benzyl ring showed slightly diminished antiproliferative effect compared with 34a–b. This suggests that introduction of electron-donating substituents into the N-benzyl ring is beneficial and it improved in vitro antiproliferative activity. Compounds 39a–b, where the benzene ring has a methoxy group in the meta position, was slightly better than analogues 40a–b with the methoxy moiety at the ortho position. In contrast, switching the position of the methoxy substituent from the ortho to the para position (see compounds 38a–b) reduced the inhibitory effect compared with 40a–b, implying that the order of the activity was ranked as meta > ortho > para. Overall, electron-donating substituents at the meta position in the N-benzyl ring enhance activity. Replacement of the benzyl group (34a–b) by either a naphthylmethyl (41a–b) or an imidazole substituent (42a–b) was detrimental for inhibitory activity against all tested cancer cell lines. Modification of the imidazole moiety in 42a–b to benzimidazole (compounds 43a–b) gave more potent but not outstanding molecules. Furthermore, there is no substantial difference in activity between 34a and 34b with different stereochemistry of the hydroxyl group on the alkyl chain. This demonstrates that the stereochemistry of the hydroxyl group in the aminodiol and aminotriol function has no influence on the antiproliferative effect. Introduction of the O-benzyl moiety with fluoro or methoxy substituent in compounds 46a–b and 52a–b, respectively, contributes to improve the antiproliferative effect compared to compounds 38a–b, where R is hydrogen. However, compounds 46a–b with a fluoro substituent in the para position of the O-benzyl rings were more potent than compounds 52a–b, where the O-benzyl ring is p-methoxy-substituted. Taken together, these results demonstrate that the presence of electron-withdrawing R substituents of the O-benzyl moiety is beneficial and it improves in vitro antiproliferative activity. By contrast, replacing benzyl moieties in both 44a–b and 50a–b by either p-fluorobenzyl substituents (see 45a–b and 51a–b) or p-methoxybenzyl groups (see 45a–b and 51a–b) had no advantageous effects.3 Experimental section
3.1. Chemistry
Commercially available solvents were used as obtained from suppliers (Molar Chemicals Ltd, Halásztelek, Hungary; Merck Ltd, Budapest, Hungary and VWR International Ltd, Debrecen, Hungary), while applied solvents were dried according to standard procedures. Optical rotations were measured in MeOH at 20 °C with a PerkinElmer 341 polarimeter (PerkinElmer Inc., Shelton, CT, USA). Chromatographic separations and monitoring of reactions were carried out on Merck Kieselgel 60 (Merck Ltd, Budapest, Hungary). Elemental analyses for all prepared compounds were performed on a PerkinElmer 2400 Elemental Analyzer (PerkinElmer Inc., Waltham, MA, USA). GC measurements for direct separation of commercially available enantiomers of isopulegol to determine the enantiomeric purity of starting material 1 and separation of O-acetyl derivatives of enantiomers were performed on a Chirasil-DEX CB column (2500 × 0.25 mm I.D.) on a PerkinElmer Autosystem XL GC consisting of a Flame Ionization Detector (PerkinElmer Corporation, Norwalk, CT, USA) and a Turbochrom Workstation data system (PerkinElmer Corp., Norwalk, CT, USA). Melting points were determined on a Kofler apparatus (Nagema, Dresden, Germany) and are uncorrected. 1H- and 13C JMOD NMR were recorded on Brucker Avance DRX 500 spectrometer [500 MHz (1H) and 125 MHz (13C), δ = 0 (TMS)]. Chemical shifts are expressed in ppm (δ) relative to TMS as the internal reference. J values are given by Hz.(−)-Isopulegol 1 is available commercially from Merck Co with ee = 95%. Diol 27 were prepared according to literature procedures, and all spectroscopic data were similar to those described therein.95,102
The detailed experimental process, physical and chemical characterisation and all spectroscopic data (1H and 13C NMR together with HRMS) of new compounds can be found in the ESI.†
3.2. Physicochemical characterisation
L-α-Phosphatidylcholine (PC), cholesterol, phosphate buffered saline (PBS) powder and analytical grade solvents like DMSO, acetonitrile (AcN), dodecane, hexane and chloroform were purchased from Sigma Aldrich (Merck kGaA, Darmstadt, Germany). Phosphate buffered saline (PBS, pH 7.4) solutions were prepared by dissolving one bag of PBS powder in 1 L distilled water (provided by a Millipore Milli-Q® 140 Gradient Water Purification System). Donor buffer (pH 6.5) was made by mixing 0.01 M monosodium phosphate solution (0.01 M NaH2PO4, 0.0027 M KCl, 0.138 M NaCl) and 0.01 M disodium phosphate solution (0.01 M Na2HPO4, 0.0027 M KCl, 0.138 M NaCl) in a 3:1 ratio, and adjusting the pH with HCl.
For calculating the apparent permeability based on absorbance data, the following equation was used, described by Wohnsland and Faller.103
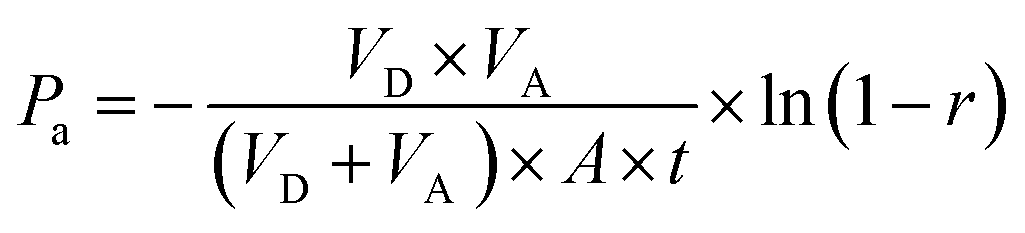 | (1) |
400 s) and 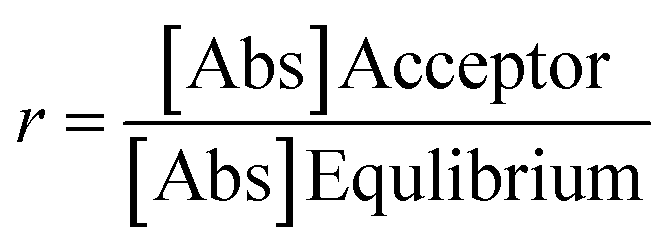 , where [Abs] is the absorbance (at λmax) of acceptor and equilibrium solutions. Equilibrium solutions were prepared by mixing 50 μL initial solution and 100 μL acceptor buffer together.
, where [Abs] is the absorbance (at λmax) of acceptor and equilibrium solutions. Equilibrium solutions were prepared by mixing 50 μL initial solution and 100 μL acceptor buffer together.
3.3. Determination of antiproliferative effect
The growth-inhibitory effects of the presented heterocyclic compounds were determined by a standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay on a panel of human adherent cancer cell lines of gynecological origin containing HeLa (cervical cancers), A2780 (ovarian cancer), MCF7 and MDA-MB-231 (breast cancer) cells.104 All cell lines were purchased from the European Collection of Cell Cultures (Salisbury, UK). The cells were cultivated in minimal essential medium (MEM) supplemented with fetal bovine serum (10%), non-essential amino acids, and penicillin–streptomycin (1% each) at 37 °C in a humidified atmosphere containing 5% CO2. All media and supplements were obtained from Lonza Group Ltd (Basel, Switzerland). Cancer cells were plated into 96-well plates at 5000 cells per well density. After overnight incubation, the test compound was added in two concentrations (10 μM and 30 μM) and incubated for 72 h under cell-culturing conditions. Then, MTT solution (5 mg mL−1, 20 μL) was added to each well and incubated for four hours. Finally, the medium was removed, and the precipitated formazan was dissolved in DMSO during 60 min of shaking at 37 °C. The absorbance was measured at 545 nm using a microplate reader (SpectoStarNano, BMG Labtech, Ortenberg, Germany). Two independent experiments were carried out with five wells for each condition. In the case of the agents eliciting more than 50% cancer cell growth inhibition at 10 μM the assays were repeated with a broad range of concentration (0.1–30 μM) to obtain their IC50 values. The clinically used anticancer agent cisplatin (Ebewe GmbH, Unterach, Austria) was included as a positive control. Calculations were performed using GraphPad Prism 5.01 software (GraphPad Software Inc., San Diego, CA, USA).4 Conclusions
A new library of isopulegol-based chiral aminodiols and aminotriols was developed from commercially available (−)-isopulegol. Di-O-benzyl aminotriols have shown significant antiproliferative effectiveness in all tested cancer cell lines. Among these derivatives, N-benzyl-substituted di-O-benzyl aminodiol derivatives exert outstanding antiproliferative properties with low, typically micromolar IC50 values.Furthermore, in vitro studies have clearly shown that substitution of the N-benzyl ring with electron-donating groups could enhance antiproliferative activity, whereas the stereochemistry of hydroxyl substituent in the aminodiol and aminotriol function has no influence on the antiproliferative effect. Through SAR study, we also found that electron-withdrawing substituents on the O-benzyl moiety increase the in vitro antiproliferative potency.
The in vitro values demonstrated that compound 11a has the potential to be developed into a clinically important therapeutic choice for the treatment of breast cancer.
Data availability
We declare that the data supporting this article have been included as part of the ESI† and available therein.Author contributions
Z. S., I. Z. and G. T. B. conceived and designed the experiments; T. M. L, N. I. K and A. V. performed the experiments, analysed the data and wrote the experimental part; T. M. L, Z. S., I. Z. and G. T. B. discussed the results and contributed to write the paper. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are grateful for financial support from the Hungarian Research Foundation (NKFI K138871). Project no. TKP2021-EGA-32 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme and also by University of Szeged Open Access Fund (grant No. 7057). The high-resolution mass spectrometric analysis was performed by Robert Berkecz. Project no. TKP2021-EGA-32 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.References
- D. Enders, A. Haertwig and J. Runsink, Eur. J. Org Chem., 1998, 1998, 1793–1802 CrossRef.
- M. E. González-Rosende, J. M. Jordá-Gregori, J. Sepúlveda-Arques and M. Orena, Tetrahedron: Asymmetry, 2004, 15, 419–422 CrossRef.
- W. Guarnieri, M. Sendzik, R. Fröhlich and D. Hoppe, Synthesis, 1998, 1274–1286 CrossRef CAS.
- A. Kamimura, K. Yoshihara, S. Marumo, A. Yamamoto, T. Nishiguchi, A. Kakehi and K. Hori, J. Org. Chem., 1992, 57, 5403–5413 CrossRef CAS.
- H. A. Walsh, P. L. Leslie, K. C. O'Shea and N. P. Botting, Bioorg. Med. Chem. Lett., 2002, 12, 361–363 CrossRef CAS PubMed.
- S. Hanessian, U. Soma, S. Dorich and B. Deschênes-Simard, Org. Lett., 2011, 13, 1048–1051 CrossRef CAS PubMed.
- A. Ohsaki, H. Ishiyama, K. Yoneda and J. Kobayashi, Tetrahedron Lett., 2003, 44, 3097–3099 CrossRef CAS.
- H. C. Kim and S. H. Kang, Angew. Chem., 2009, 121, 1859–1861 CrossRef.
- M. Azumi, K. Ogawa, T. Fujita, M. Takeshita, R. Yoshida, T. Furumai and Y. Igarashi, Tetrahedron, 2008, 64, 6420–6425 CrossRef CAS.
- J. T. Malinowski, R. J. Sharpe and J. S. Johnson, Science, 2013, 340, 180–182 CrossRef CAS PubMed.
- M. C. Jones and S. P. Marsden, Org. Lett., 2008, 10, 4125–4128 CrossRef CAS PubMed.
- B. K. Albrecht and R. M. Williams, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 11949–11954 CrossRef CAS PubMed.
- R. J. Nash, A. Kato, C.-Y. Yu and G. W. Fleet, Future Med. Chem., 2011, 3, 1513–1521 CrossRef CAS PubMed.
- K. Burgess and I. Herderson, Tetrahedron, 1992, 48, 4045–4066 CrossRef CAS.
- J. P. Michael, Nat. Prod. Rep., 2001, 18, 520–542 RSC.
- S. T. Pruett, A. Bushnev, K. Hagedorn, M. Adiga, C. A. Haynes, M. C. Sullards, D. C. Liotta and A. H. Merrill, J. Lipid Res., 2008, 49, 1621–1639 CrossRef CAS PubMed.
- A. Aiello, E. Fattorusso, A. Giordano, M. Menna, C. Navarrete and E. Muñoz, Tetrahedron, 2009, 65, 4384–4388 CrossRef CAS.
- T. Vijai Kumar Reddy, A. Jyotsna, B. L. A. Prabhavathi Devi, R. B. N. Prasad, Y. Poornachandra and C. Ganesh Kumar, Eur. J. Med. Chem., 2016, 120, 86–96 CrossRef CAS PubMed.
- T. Nakamura and M. Shiozaki, Tetrahedron, 2001, 57, 9087–9092 CrossRef CAS.
- Y. A. Hannun and L. M. Obeid, J. Biol. Chem., 2002, 277, 25847–25850 CrossRef CAS PubMed.
- A. J. Dyckman, J. Med. Chem., 2017, 60, 5267–5289 CrossRef CAS PubMed.
- M. P. Wymann and R. Schneiter, Nat. Rev. Mol. Cell Biol., 2008, 9, 162–176 CrossRef CAS PubMed.
- J. Newton, S. Lima, M. Maceyka and S. Spiegel, Exp. Cell Res., 2015, 333, 195–200 CrossRef CAS PubMed.
- C. Huang and C. Freter, Int. J. Mol. Sci., 2015, 16, 924–949 CrossRef CAS PubMed.
- L. M. Healy and J. P. Antel, Curr. Drug Targets, 2016, 17, 1841–1850 CrossRef CAS.
- L. Cavone, R. Felici, A. Lapucci, D. Buonvicino, S. Pratesi, M. Muzzi, B. Hakiki, L. Maggi, B. Peruzzi, R. Caporale, F. Annunziato, M. P. Amato and A. Chiarugi, Brain, Behav., Immun., 2015, 50, 78–86 CrossRef CAS PubMed.
- S. Ladisch, R. Li and E. Olson, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1974–1978 CrossRef CAS.
- V. A. Pavlov, Tetrahedron, 2008, 64, 1147–1179 CrossRef CAS.
- E. Gómez-Torres, D. A. Alonso, E. Gómez-Bengoa and C. Nájera, Org. Lett., 2011, 13, 6106–6109 CrossRef PubMed.
- X.-P. Hui, C. Yin, Z.-C. Chen, L.-N. Huang, P.-F. Xu and G.-F. Fan, Tetrahedron, 2008, 64, 2553–2558 CrossRef CAS.
- A. L. Braga, R. M. Rubim, H. S. Schrekker, L. A. Wessjohann, M. W. G. de Bolster, G. Zeni and J. A. Sehnem, Tetrahedron: Asymmetry, 2003, 14, 3291–3295 CrossRef CAS.
- A. Vidal-Ferran, A. Moyano, M. A. Pericàs and A. Riera, J. Org. Chem., 1997, 62, 4970–4982 CrossRef CAS.
- C. Yie-Jia, J.-M. Fang and Ta-J. Lu, Tetrahedron: Asymmetry, 1995, 6, 89–92 CrossRef.
- R. Roudeau, D. Gomez Pardo and J. Cossy, Tetrahedron, 2006, 62, 2388–2394 CrossRef CAS.
- I. Philipova, V. Dimitrov and S. Simova, Tetrahedron: Asymmetry, 1999, 10, 1381–1391 CrossRef CAS.
- A.-L. Zhang, L.-W. Yang, N.-F. Yang and D.-C. Liu, J. Organomet. Chem., 2014, 768, 50–55 CrossRef CAS.
- G. Veeresa and A. Datta, Tetrahedron Lett., 1998, 39, 8503–8504 CrossRef CAS.
- A. J. Ndakala, M. Hashemzadeh, R. C. So and A. R. Howell, Org. Lett., 2002, 4, 1719–1722 CrossRef CAS.
- H. Azuma, S. Tamagaki and K. Ogino, J. Org. Chem., 2000, 65, 3538–3541 CrossRef CAS PubMed.
- V. D. Chaudhari, K. S. Ajish Kumar and D. D. Dhavale, Org. Lett., 2005, 7, 5805–5807 CrossRef CAS PubMed.
- L. He, H.-S. Byun and R. Bittman, J. Org. Chem., 2000, 65, 7627–7633 CrossRef CAS PubMed.
- K. Smithies, M. E. B. Smith, U. Kaulmann, J. L. Galman, J. M. Ward and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 570–574 CrossRef CAS.
- T. Takanami, H. Tokoro, D. Kato, S. Nishiyama and T. Sugai, Tetrahedron Lett., 2005, 46, 3291–3295 CrossRef CAS.
- M. E. B. Smith, K. Smithies, T. Senussi, P. A. Dalby and H. C. Hailes, Eur. J. Org Chem., 2006, 2006, 1121–1123 CrossRef.
- M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99–107 CrossRef CAS.
- T. Katsuki and V. Martin, in Organic Reactions, ed. John Wiley & Sons, Inc., Hoboken, NJ, USA, 1996, pp. 1–299 Search PubMed.
- E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed.
- O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958–3987 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- W. Zhang, A. Basak, Y. Kosugi, Y. Hoshino and H. Yamamoto, Angew. Chem., Int. Ed., 2005, 44, 4389–4391 CrossRef CAS PubMed.
- H. Egami, T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 5886–5895 CrossRef CAS PubMed.
- J. L. Olivares-Romero, Z. Li and H. Yamamoto, J. Am. Chem. Soc., 2013, 135, 3411–3413 CrossRef CAS PubMed.
- R. M. Hanson, Chem. Rev., 1991, 91, 437–475 CrossRef CAS.
- P. Pena and S. Roberts, Curr. Org. Chem., 2003, 7, 555–571 CrossRef CAS.
- M. Caron and K. B. Sharpless, J. Org. Chem., 1985, 50, 1557–1560 CrossRef CAS.
- C. H. Behrens and K. B. Sharpless, J. Org. Chem., 1985, 50, 5696–5704 CrossRef CAS.
- S. Y. Ko and K. B. Sharpless, J. Org. Chem., 1986, 51, 5413–5415 CrossRef CAS.
- M. Caron, P. R. Carlier and K. B. Sharpless, J. Org. Chem., 1988, 53, 5185–5187 CrossRef CAS.
- M. Onaka, K. Sugita, H. Takeuchi and Y. Izumi, J. Chem. Soc., Chem. Commun., 1988, 1173 RSC.
- M. Canas, M. Poch, X. Verdaguer, A. Moyano, M. A. Pericàs and A. Riera, Tetrahedron Lett., 1991, 32, 6931–6934 CrossRef CAS.
- M. Chini, P. Crotti, L. A. Flippin, C. Gardelli, E. Giovani, F. Macchia and M. Pineschi, J. Org. Chem., 1993, 58, 1221–1227 CrossRef CAS.
- M. Sasaki, K. Tanino and M. Miyashita, Org. Lett., 2001, 3, 1765–1767 CrossRef CAS PubMed.
- M. Sasaki, K. Tanino, A. Hirai and M. Miyashita, Org. Lett., 2003, 5, 1789–1791 CrossRef CAS PubMed.
- M. Pastó, B. Rodríguez, A. Riera and M. A. Pericàs, Tetrahedron Lett., 2003, 44, 8369–8372 CrossRef.
- Y. Tomata, M. Sasaki, K. Tanino and M. Miyashita, Tetrahedron Lett., 2003, 44, 8975–8977 CrossRef CAS.
- K. Surendra, N. S. Krishnaveni and K. R. Rao, Synlett, 2005, 3, 506–510 Search PubMed.
- K. Samanta and G. Panda, Org. Biomol. Chem., 2011, 9, 7365 RSC.
- S. Uesugi, T. Watanabe, T. Imaizumi, M. Shibuya, N. Kanoh and Y. Iwabuchi, Org. Lett., 2014, 16, 4408–4411 CrossRef CAS PubMed.
- D. Li, J. Wang, S. Yu, S. Ye, W. Zou, H. Zhang and J. Chen, Chem. Commun., 2020, 56, 2256–2259 RSC.
- C. Wang and H. Yamamoto, J. Am. Chem. Soc., 2014, 136, 6888–6891 CrossRef CAS PubMed.
- A. Kumar and G. Panda, Tetrahedron Lett., 2021, 70, 153013 CrossRef CAS.
- C. Wang and H. Yamamoto, Angew. Chem., 2014, 126, 14140–14143 CrossRef.
- L. Luo and H. Yamamoto, Org. Biomol. Chem., 2015, 13, 10466–10470 RSC.
- B. Weber, Synthesis, 1999, 1593–1606 CrossRef CAS.
- H. Li, A. He, J. R. Falck and L. S. Liebeskind, Org. Lett., 2011, 13, 3682–3685 CrossRef CAS PubMed.
- B. F. Bonini, M. Comes-Franchini, M. Fochi, L. Lunazzi, A. Mazzanti, A. Ricci and G. Varchi, Synlett, 2001, 2001, 995–998 CrossRef.
- F. Benedetti, F. Berti, S. Budal, P. Campaner, F. Dinon, A. Tossi, R. Argirova, P. Genova, V. Atanassov and A. Hinkov, J. Med. Chem., 2012, 55, 3900–3910 CrossRef CAS PubMed.
- Z. Liu, H.-S. Byun and R. Bittman, J. Org. Chem., 2011, 76, 8588–8598 CrossRef CAS PubMed.
- K. S. Olivier and M. S. Van Nieuwenhze, Org. Lett., 2010, 12, 1680–1683 CrossRef CAS PubMed.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed.
- G. Sabitha, R. S. Babu, M. Rajkumar and J. S. Yadav, Org. Lett., 2002, 4, 343–345 CrossRef CAS PubMed.
- X. E. Hu, Tetrahedron, 2004, 60, 2701–2743 CrossRef CAS.
- S. Dinda, S. Das and G. Panda, Synthesis, 2009, 2009, 1886–1896 CrossRef.
- S. K. Das and G. Panda, Tetrahedron, 2008, 64, 4162–4173 CrossRef CAS.
- P. M. Manoury, J. L. Binet, J. Rousseau, F. M. Lefevre-Borg and I. G. Cavero, J. Med. Chem., 1987, 30, 1003–1011 CrossRef CAS PubMed.
- C.-L. J. Wang, W. A. Gregory and M. A. Wuonola, Tetrahedron, 1989, 45, 1323–1326 CrossRef CAS.
- W. A. Gregory, D. R. Brittelli, C. L. J. Wang, M. A. Wuonola, R. J. McRipley, D. C. Eustice, V. S. Eberly, A. M. Slee, M. Forbes and P. T. Bartholomew, J. Med. Chem., 1989, 32, 1673–1681 CrossRef CAS PubMed.
- R. Giani, E. Marinone, G. Melillo, M. Borsa and G. C. Tonon, Arzneimittelforschung, 1988, 38, 1139–1141 CAS.
- T. Minh Le and Z. Szakonyi, Chem. Rec., 2022, 22, e202100194 CrossRef CAS PubMed.
- N. Azizi, B. Mirmashhori and M. R. Saidi, Catal. Commun., 2007, 8, 2198–2203 CrossRef CAS.
- S. Wang, Z. Xie, M. Li and C. Wang, ChemistrySelect, 2020, 5, 6011–6015 CrossRef CAS.
- T. M. Le, T. Huynh, G. Endre, A. Szekeres, F. Fülöp and Z. Szakonyi, RSC Adv., 2020, 10, 38468–38477 RSC.
- L. Ciccone, G. Petrarolo, F. Barsuglia, C. Fruchart-Gaillard, E. Cassar Lajeunesse, A. T. Adewumi, M. E. S. Soliman, C. La Motta, E. Orlandini and S. Nencetti, Biomolecules, 2022, 12, 448 CrossRef CAS PubMed.
- T. M. Le, T. Huynh, F. Z. Bamou, A. Szekeres, F. Fülöp and Z. Szakonyi, Int. J. Mol. Sci., 2021, 22, 5626 CrossRef CAS PubMed.
- D. Friedrich and F. Bohlmann, Tetrahedron, 1988, 44, 1369–1392 CrossRef CAS.
- D. Bai, Z. Schelz, D. Erdős, A. K. Kis, V. Nagy, I. Zupkó, G. T. Balogh and Z. Szakonyi, Int. J. Mol. Sci., 2023, 24, 1121 CrossRef CAS PubMed.
- D. A. DeGoey, H.-J. Chen, P. B. Cox and M. D. Wendt, J. Med. Chem., 2018, 61, 2636–2651 CrossRef CAS PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- T. W. Johnson, R. A. Gallego and M. P. Edwards, J. Med. Chem., 2018, 61, 6401–6420 CrossRef CAS PubMed.
- B. R. Travis, R. S. Narayan and B. Borhan, J. Am. Chem. Soc., 2002, 124, 3824–3825 CrossRef CAS PubMed.
- F. Wohnsland and B. Faller, J. Med. Chem., 2001, 44, 923–930 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03467h |
| This journal is © The Royal Society of Chemistry 2024 |