DOI:
10.1039/D4RA04277H
(Paper)
RSC Adv., 2024,
14, 23583-23591
Towards combining backbone and sugar constraint in 3′-3′ bis-phosphonate tethered 2′-4′ bridged LNA oligonucleotide trimers†
Received
12th June 2024
, Accepted 22nd July 2024
First published on 26th July 2024
Abstract
Therapeutic oligonucleotides are chemically modified to enhance their drug-like properties – including binding affinity for target RNA. Many nucleic acid analogs that enhance RNA binding affinity constrain the furanose sugar in an RNA-like sugar pucker. The improvements in binding affinity result primarily from increased off-rates with minimal effects on on-rates for hybridization. To identify alternate chemical modification strategies that can modulate on- and off-rates for oligonucleotide hybridization, we hypothesized that extending conformational restraint across multiple nucleotides could modulate hybridization kinetics by restricting rotational freedom of the sugar-phosphate backbone. As part of that effort, we recently reported that using hydrocarbon tethers to bridge adjacent phosphodiester linkages as phosphonate tethered bridges can pre-organize nucleic acids in conformations conducive for Watson–Crick base-pairing and modulate hybridization kinetics. In this report, we describe the synthesis of locked nucleic acid (LNA) trimers linked through alkylphosphonate tethers which restrict conformation of the furanose sugar in addition to restricting conformational mobility of the sugar-phosphate backbone across three nucleotide units.
Introduction
RNA-targeted oligonucleotide therapies are making rapid progress in the clinic with several approved medicines and >100 candidates in clinical development.1,2 RNA-targeted therapeutic oligonucleotides can be classified into at least two broad classes. The first class typically comprises single stranded antisense oligonucleotides (ASOs) that first bind their target RNA in cells through Watson–Crick base-pairing and recruit an effector protein to modulate RNA processing. Examples of this class include gapmer ASOs that degrade RNA via RNAse-H1 mediated hydrolysis,3 ASOs that modulate splicing patterns to promote exon skipping or inclusion,4 ASOs that promote RNA-editing via recruiting endogenous ADARs5 (adenosine deaminase acting on RNA) and ASOs that antagonize natural miRNAs.6 In contrast, a second class of RNA-targeted therapeutic oligonucleotides are first loaded into a protein effector complex prior to interacting with their cognate RNA. Examples of this class includes double stranded siRNA, where the guide strand of the duplex is first loaded into argonaute 2, which facilitates RNA binding and subsequent RNA cleavage.7 Interestingly, oligonucleotide therapeutics that promote genome editing are also loaded into effector proteins such as CAS9, which in turn facilitate DNA binding and subsequent single or double stranded cleavage or base-editing.8
For oligonucleotide therapeutics that belong to the first class described above, ASOs must bind to their cognate RNA while avoiding interactions with mismatched RNA, to elicit sequence-specific antisense effects.9 The RNA-binding affinity of ASOs is in turn governed by rates of association and dissociation for their RNA-targets. A recent study demonstrated that short oligonucleotides can form partially hybridized meta-stable intermediates with mismatched RNA.10 These intermediate partially complementary duplexes were sufficiently long-lived such that they could be intercepted by enzymatic processes. In the context of oligonucleotide therapeutics, this could result in off-target effects. One strategy to avoid these issues would be to modulate the kinetics of oligonucleotide hybridization such that the ASO could sample the RNA sequence space relatively quickly to identify its fully complementary binding site without forming meta-stable partial duplexes of sufficient stability with mis-matched RNA.
Therapeutic ASOs are typically chemically modified to enhance their drug-like properties – including binding affinity for target RNA.11 Many of these modifications enhance binding affinity by enforcing conformational constraint within the nucleoside structure. Some notable examples include bi- and tri-cyclo DNA, one of the first examples of conformationally restricted nucleic acid analogs, described by Leumann et al. where torsion angle γ was restricted in a non-canonical orientation to enhance duplex stability;12–14 conformational restriction of the furanose sugar moiety in the RNA-like C3′-endo sugar pucker to provide the family of locked nucleic acids (LNA);15,16 restriction of backbone α and β torsion angles to provide constrained nucleic acids (α,β-CNA)17,18 and dual constrained nucleic acids where both the sugar ring and the backbone torsion angles were restricted to enhance duplex stability.19–21 Interestingly, as demonstrated for LNA-modified oligonucleotides, the improvement in duplex stability was primarily a result of increased off-rates with negligible effects on on-rates.22
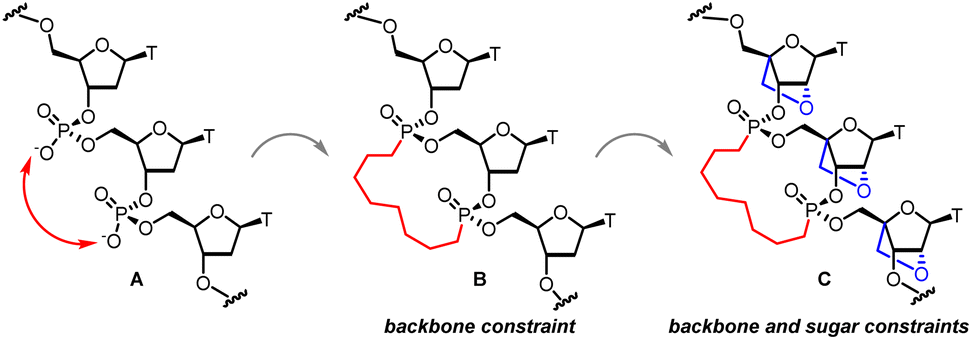
All the examples of conformationally constrained nucleic acid analogs described above restrict conformation across a single nucleotide unit within an oligonucleotide. We postulated that extending conformational restraint across multiple nucleotides could modulate hybridization kinetics by restricting rotational freedom of the sugar-phosphate backbone. As part of that effort, we recently reported that using hydrocarbon tethers to bridge adjacent phosphodiester linkages as phosphonate tethered bridges can pre-organize nucleic acids in conformations conducive for Watson–Crick base-pairing and modulate hybridization kinetics (Fig. 1A).22 It was concluded that 15-membered octyl 3′,3′-phosphonate linked macrocycles with stereochemically defined phosphorus atoms were preferred to shorter 11-membered counterparts. These results were corroborated with detailed molecular dynamic simulation measurements. In a subsequent disclosure involving a systematic study of the length of the alkyl tether and the configuration of the phosphorus atom of 12, 13, 14, and 16-membered macrocycles it was concluded that the 15-membered macrocycle harboring S,R-stereochemistry at the two phosphorus atoms was preferred compared to smaller macrocycles (Fig. 1B).23 However, these macrocyclic analogues showed reduced RNA-binding affinity and duplex stability, because of increased off-rates relative to unmodified DNA.
 |
| | Fig. 1 (A) Trinucleotide DNA unit; (B) S/R phosphonate bridged 15-membered macrocycle DNA unit; (C) S/R phosphonate tethered 15-membered macrocycle LNA unit. | |
To address this, we further hypothesized that combining backbone constraint with sugar constraint could provide analogues that could effectively modulate on- and off-rates of hybridization without reducing overall duplex stability. We chose the arduously accessible LNA 15-membered octyl bis-phosphonate macrocyclic trimer as a synthetic prototype to restrict the conformation of the furanose sugar in addition to restricting the conformational mobility of the sugar-phosphate backbone across three nucleotide units (Fig. 1C). In this report, we describe the synthesis of LNA trimers linked through octyl 3′3′-bis-alkylphosphonate tethers which restrict conformation of the furanose sugar in addition to restricting conformational mobility of the sugar-phosphate backbone across three nucleotide units. In view of the steric environment presented by the oxacyclic linking C2′ and C4′, the challenge was to explore the feasibility of synthesizing two pairs of stereochemically pure 15-membered macrocyclic octyl 3′-3′-bis-phosphonates.
Results and discussion
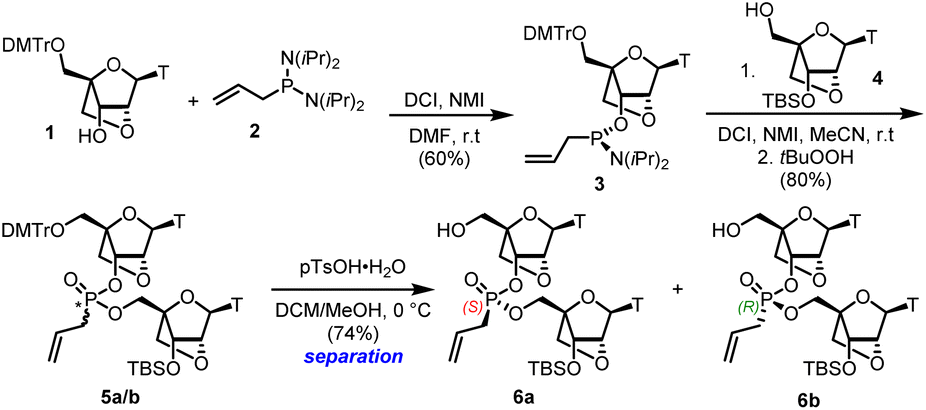
The general synthetic route consisted of previously adopted phosphoramidite coupling protocols.24 Thus, the known 2′,5′-anhydro nucleoside 1 was converted to the phosphoramidite 3, and the product was coupled with the known 3′-OTBS-2′,5′-anhydro nucleoside 4 (ref. 25) to give the corresponding mixture of S- and R-epimeric allyl phosphonates 5a/5b (Scheme 1).
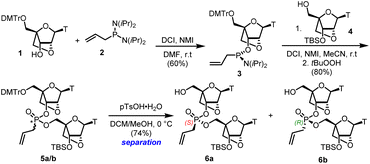 |
| | Scheme 1 Synthesis of LNA dimers from an LNA monomer unit. | |
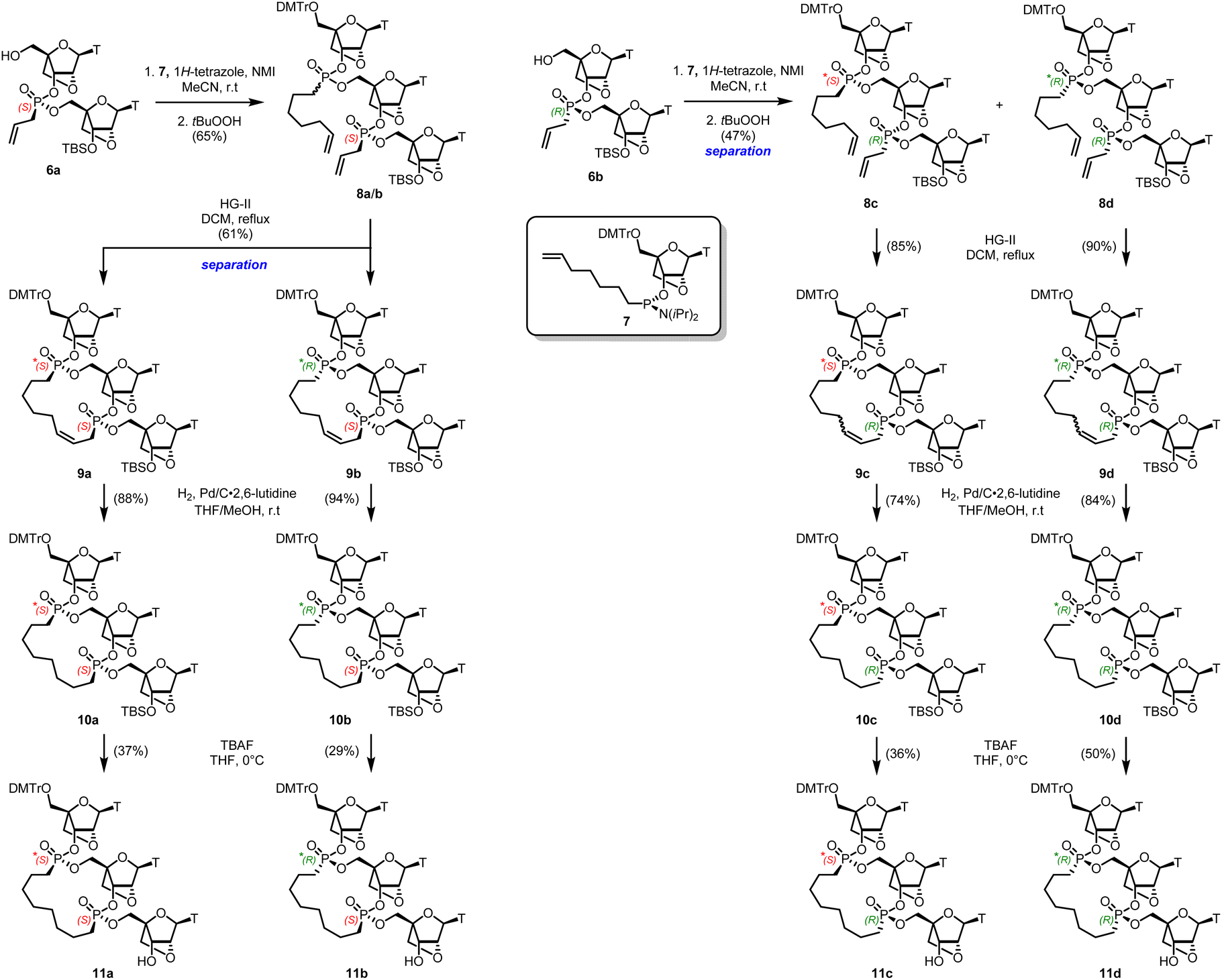
Cleavage of the DMT with p-toluenesulfonic acid hydrate gave both dimers 6a/6b in high yield after separation by silica gel chromatography without deprotection of the silyl ether group. The assignment of stereochemistry at phosphorus in each isomer followed conventional methods described in our previous work,22,23 and correlating with data reported by Baran.26 A coupling reaction of the S-6a isomer with an excess of 1-heptenyl phosphoramidite 7 and tetrazole as the activator was performed on a small scale in batches (Scheme 2).27 The resulting mixture S*,S and R*,S diastereoisomers 8a and 8b could not be separated by chromatography. However, treatment of this mixture with the Hoveyda–Grubbs II catalyst28 in refluxing DCM under high dilution led to two compounds which were arbitrarily designated as bis-phosphonate macrocycles S*,S-9a and R*,S-9b in 30% and 31% yield respectively, following the trend observed in the DNA macrocycles.22
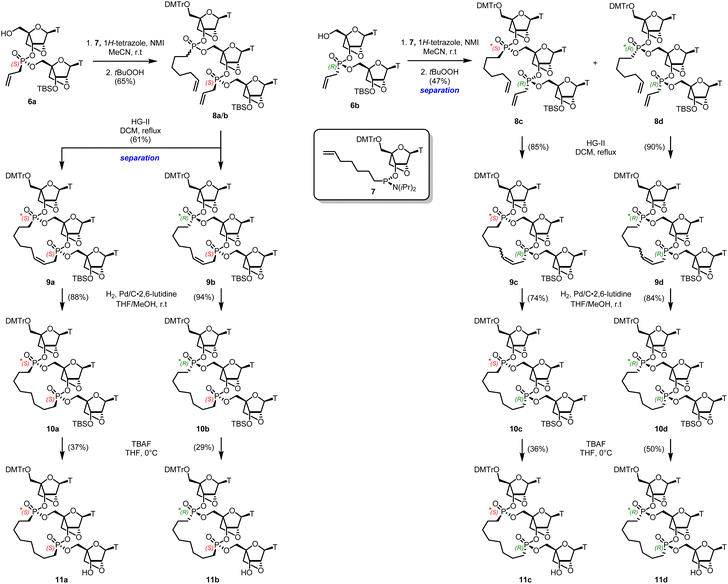 |
| | Scheme 2 Synthesis of bis-phosphonate LNA trimer macrocycles. The asterisk denotes uncertain R- or S- stereochemistry. | |
Similar phosphoramidite 7 coupling of the R-6b isomer, led to the S*,R-8c and the R*,S-8d diastereomers respectively, which could be separated by flash column chromatography. Ring-closing metathesis using the Hoveyda–Grubbs II catalyst at high dilution led to a mixture of cis and trans unsaturated macrocycles 9c and 9d.28 To avoid deprotection of the DMT group, reduction of the macrocyclic olefins was done by hydrogenolysis with Pd/C deactivated by 2,6-lutidine in a mixture of THF/MeOH.29 This was followed by removal of the TBS protecting group, leading to the four diastereomers 11a–d, which were isolated by reverse phase flash chromatography and individually characterized spectroscopically. The four isomers were chromatographically different by TLC.
We can only designate the absolute stereochemistry of the phosphorus atom of the “lower” nucleotide unit within each macrocycle 11a–d relying on the known stereochemistry of the monomers 6a and 6b from which they are synthesized (Scheme 2). The stereochemistry of the phosphorus atom in the “upper” nucleoside unit in the trimers 8a–d as well as the macrocycles 11a–d is currently unknown. It would be misleading to compare the 31P values of the current LNA macrocyclic compounds 11a–d with their counterparts in the DNA P-macrocycles series.22 Furthermore, the differences in 31P chemical shifts within each series are too close to establish a reliable correlation or trend. Nevertheless, each of the bis-phosphonate LNA macrocyles 11a–d are individually characterized as chromatographically homogeneous and distinguishable compounds. The difficulty of ascertaining the absolute configuration at phosphorus in the “upper” nucleoside in each trimeric unit warrants an independent synthesis possibly relying on X-ray crystallography analogues such as 3′-esters.
Conclusions
We have developed methods to synthesize 15-membered bis-phosphonate macrocyclic nucleosides tethered between 3′ and 3′ of a trimeric LNA unit as constrained analogues of the corresponding LNA trimers. Incorporation into oligonucleotides and measurement of Tm values along with kinetic parameters of binding for one of the analogues will determine which of the macrocyclic analogues may be pursued and to establish the absolute stereochemistry at phosphorus in the “upper” nucleoside unit by independent synthesis.
Experimental section
General information
Anhydrous tetrahydrofuran, diethyl ether, toluene and dichloromethane were obtained through activated columns of alumina, while all other solvents were used as received from chemical suppliers. Reagents were purchased and used without further purification unless otherwise specified. Yields refer to chromatographically and spectroscopically homogeneous material, unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on Silicycle SiliaPlate 60 Å, 250 μm (indicator F-254) that were visualized using a UVP UVG-11 compact UV lamp (254 nm) and developed with usual revealing agents (cerium ammonium molybdate, potassium permanganate or ninhydrin). Flash chromatography was performed using Silicycle SiliaFlash F60 (230–400 mesh) silica gel or using TELEDYNE ISCO CombiFlash Rf+ when a gradient was used. NMR spectra were recorded on Bruker AV-300, AV-400 or AV-500, calibrated using residual undeuterated solvent as internal reference (CHCl3, δ = 7.26 ppm for 1H NMR; δ = 77.16 ppm for 13C NMR; DMSO, δ = 2.50 ppm for 1H NMR; δ = 39.52 ppm for 13C NMR; acetone, δ = 2.05 ppm for 1H NMR; δ = 29.84 ppm for 13C NMR) and reported in parts per million from tetramethylsilane as follows: chemical shift (multiplicity, coupling constant in Hz, integration). The following abbreviations were used to describe multiplicities: s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet, br = broad. High-resolution mass spectra (HRMS) were recorded at the Centre Régional de Spectrométrie de Masse de l’Université de Montréal on an Agilent LC-MSD TOF mass spectrometer by electrospray ionization time of flight reflectron experiments. Specific rotations were measured with an Anton Paar MCP100 polarimeter.
Characterization data and procedures
Synthesis of phosphoramidite (3). 5′-ODMT LNA thymidine 1 (0.16 g, 0.28 mmol) was co-evaporated with toluene twice. Compound 1 was dissolved in anhydrous DMF (0.2 mL) under Ar. In a separate flask under Ar, a solution of 4,5-dicyanoimidazole (0.02 g, 0.17 mmol) and N-methylimidazole (10 μL, 0.12 mmol) in anhydrous DMF (0.3 mL) was prepared. In a third flask under Ar, a solution of allyl phosphordiamine 2 (0.12 g, 0.43 mmol) in anhydrous DMF (0.2 mL), was prepared. The solution containing 4,5-dicyanoimidazole was added dropwise to the reaction solution, followed by a dropwise addition of the phosphordiamine solution. The reaction was stirred at r.t overnight until consumption of 5′-ODMT LNA thymidine 1 (TLC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane/ethyl acetate 7:3). A few drops of Et3N were added, and the solvent was evaporated in vacuo. The resulting oil was dissolved in a minimum amount of CH2Cl2 with Et3N, and purified by flash chromatography (gradient, from 100% hexanes + 1% Et3N to hexanes/EtOAc: 7/3 + 1% Et3N). White powder (120 mg; 60%). 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.73 (dd, J = 14.9, 1.4 Hz, 1H), 7.55–7.31 (m, 7H), 6.87 (td, J = 9.0, 2.9 Hz, 4H), 5.74–5.61 (m, 1H), 5.57–5.35 (m, 1H), 5.13–5.02 (m, 1H), 4.99–4.78 (m, 1H), 4.44–4.33 (m, 1H), 4.17 (dd, J = 15.4, 6.8 Hz, 1H), 3.83 (s, 6H), 3.69–3.34 (m, 4H), 2.99 (s, 1H), 2.91 (s, 1H), 2.56–2.19 (m, 2H), 1.75–1.72 (m, 1H), 1.36–1.19 (m, 3H), 1.19–0.95 (m, 12H). 31P NMR (160 MHz, CDCl3) δ 130.9, 125.2.
:hexane/ethyl acetate 7:3). A few drops of Et3N were added, and the solvent was evaporated in vacuo. The resulting oil was dissolved in a minimum amount of CH2Cl2 with Et3N, and purified by flash chromatography (gradient, from 100% hexanes + 1% Et3N to hexanes/EtOAc: 7/3 + 1% Et3N). White powder (120 mg; 60%). 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.73 (dd, J = 14.9, 1.4 Hz, 1H), 7.55–7.31 (m, 7H), 6.87 (td, J = 9.0, 2.9 Hz, 4H), 5.74–5.61 (m, 1H), 5.57–5.35 (m, 1H), 5.13–5.02 (m, 1H), 4.99–4.78 (m, 1H), 4.44–4.33 (m, 1H), 4.17 (dd, J = 15.4, 6.8 Hz, 1H), 3.83 (s, 6H), 3.69–3.34 (m, 4H), 2.99 (s, 1H), 2.91 (s, 1H), 2.56–2.19 (m, 2H), 1.75–1.72 (m, 1H), 1.36–1.19 (m, 3H), 1.19–0.95 (m, 12H). 31P NMR (160 MHz, CDCl3) δ 130.9, 125.2.
Synthesis of dimers (5). Before the reaction, nucleoside 4 was co-evaporated with toluene twice. To a solution of nucleoside 4 (60 mg, 0.15 mmol) in anhydrous acetonitrile (0.5 mL), a solution of phosphoramidite 3 (200 mg, 0.27 mmol) in anhydrous MeCN (0.5 mL) was added dropwise under Ar, followed by a dropwise addition of a solution containing 4,5-dicyanoimidazole (30 mg, 0.25 mmol) and N-methylimidazole (2 μL, 0.024 mmol) in anhydrous MeCN (0.3 mL). The progress of the reaction was monitored by TLC analysis and mass spectrometry. Upon completion, a 5 M solution of t-BuOOH in decanes (30 μL, 0.015 mmol) was added dropwise to the reaction mixture and stirred for 45 min. The reaction mixture was diluted with EtOAc, cooled to 0 °C, and aq. 10% NaHSO3 solution (1 mL) was added. After stirring for 5 min, the reaction mixture was washed with saturated aq. NaHCO3 solution (5 mL). The aqueous layer was extracted with EtOAc, the combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was dissolved in a minimum amount of CH2Cl2 and purified by flash column chromatography (gradient from 100% hexanes to hexanes/CH2Cl2/MeOH: 7/2/0.5) to give a mixture of dimers. White powder (120 mg; 80%). 1H NMR (500 MHz, CDCl3) δ 9.47 (s, 2H), 7.62 (t, J = 1.8 Hz, 1H), 7.46 (ddd, J = 7.5, 5.6, 1.7 Hz, 2H), 7.40–7.21 (m, 9H), 6.97–6.81 (m, 5H), 5.80–5.45 (m, 3H), 5.31–5.19 (m, 1H), 5.10–4.79 (m, 3H), 4.49–4.28 (m, 3H), 4.09–3.84 (m, 5H), 3.84–3.78 (m, 8H), 3.71 (dd, J = 11.0, 5.4 Hz, 1H), 3.54–3.35 (m, 2H), 2.79 (dd, J = 22.2, 7.3 Hz, 1H), 2.52 (dqd, J = 22.5, 15.2, 7.4 Hz, 1H), 1.99–1.88 (m, 3H), 1.58 (dd, J = 2.5, 1.2 Hz, 3H), 1.35–1.11 (m, 2H), 0.90 (d, J = 5.6 Hz, 2H), 0.87 (d, J = 2.2 Hz, 9H), 0.07 (dd, J = 8.3, 3.2 Hz, 6H). 31P NMR (200 MHz, CDCl3) δ 29.1, 28.0. HRMS (ESI): calcd for C52H63N4O15PSiNa [M + Na]+: 1065.3689; found 1065.3721.
Synthesis of dimers (6a) and (6b). A solution of compound 5 (120 mg, 0.11 mmol) in dichloromethane (2.5 mL) was cooled down to 0 °C, then a solution of p-toluenesulfonic acid monohydrate (24 mg, 0.12 mmol) in MeOH (1.2 mL) was added slowly. The reaction was kept at 0 °C for 30 min, until consumption of the starting material (monitored by TLC analysis). Solid Na2CO3 was added to the cold reaction mixture and the resulting suspension was stirred until the orange color disappeared (5–15 min). The volatiles were removed by rotary evaporation, the solid residue was dissolved in a minimum amount of DCM, and then purified by flash column chromatography (hexanes/CH2Cl2/MeOH: 7/3/0.5) to give mixed dimers 6a and 6b (60 mg, 74% yield). The mixture of dimers was suspended in (20 mL) EtOAc/MeCN: 95/5. The upper isomer (6b) (20 mg) slowly precipitated out from the solution while the bottom isomer remained in solution. To further purify the bottom isomer (6a), the collected filtrate was evaporated under reduced pressure. The residue was dissolved in EtOAc/acetone: 9/1 and loaded on silica gel. The column was eluted with EtOAc/acetone: 9/1 run by gravity to obtain pure 6a (30 mg) (6a): white powder (30 mg; 37%). 1H NMR (500 MHz, acetone-d6) δ 10.17 (d, J = 55.3 Hz, 2H), 7.67 (q, J = 1.2 Hz, 1H), 7.64 (q, J = 1.2 Hz, 1H), 5.88 (ddq, J = 17.3, 10.2, 7.2 Hz, 1H), 5.57–5.48 (m, 2H), 5.37 (ddq, J = 17.1, 5.5, 1.5 Hz, 1H), 5.28 (ddq, J = 10.1, 3.9, 1.2 Hz, 1H), 4.85 (d, J = 7.0 Hz, 1H), 4.76 (s, 1H), 4.61–4.45 (m, 3H), 4.39 (d, J = 0.8 Hz, 1H), 4.29 (s, 1H), 4.06–3.95 (m, 4H), 3.84 (dd, J = 17.9, 8.0 Hz, 2H), 2.96 (dddt, J = 22.4, 7.4, 2.2, 1.3 Hz, 2H), 1.86 (s, 3H), 1.78 (d, J = 1.3 Hz, 3H), 0.90 (s, 10H), 0.12 (d, J = 5.3 Hz, 6H). 13C NMR (125 MHz, acetone-d6) δ 205.3, 133.99, 133.96, 120.4, 120.3, 109.6, 87.6, 87.0, 86.9, 79.1, 78.4, 72.1, 71.2, 71.1, 70.8, 56.2, 31.3, 30.2, 25.1, 17.6, 11.9, 11.7, −5.6, −5.9. 31P NMR (200 MHz, acetone-d6) δ 28.8 (6b): white powder (20 mg; 25%). 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H), 7.61 (d, J = 1.5 Hz, 1H), 7.39 (d, J = 1.4 Hz, 1H), 5.72 (ddq, J = 17.3, 10.1, 7.2 Hz, 1H), 5.47 (d, J = 17.9 Hz, 2H), 5.39 (t, J = 5.8 Hz, 1H), 5.30–5.13 (m, 2H), 4.57 (d, J = 6.3 Hz, 2H), 4.45 (dd, J = 12.2, 5.2 Hz, 1H), 4.33 (dd, J = 12.2, 6.4 Hz, 1H), 4.25 (s, 1H), 4.07 (s, 1H), 3.88 (d, J = 7.9 Hz, 1H), 3.86–3.75 (m, 4H), 3.74 (d, J = 8.0 Hz, 1H), 2.92 (d, J = 7.3 Hz, 1H), 2.87 (d, J = 7.3 Hz, 1H), 1.78 (dd, J = 16.1, 1.1 Hz, 6H), 0.85 (s, 9H), 0.08 (d, J = 4.1 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 163.8, 157.1, 150.6, 134.8, 127.7, 127.6, 120.9, 120.8, 109.22, 109.20, 88.9, 87.2, 86.8, 79.2, 78.3, 72.8, 71.5, 71.0, 25.9, 18.1, 12.8, 12.8, −4.5, −4.8. 31P NMR (160 MHz, DMSO-d6) δ 28.3.
Synthesis of phosphoramidite (7). 5′-ODMT thymidine 1 (1.2 g, 2.09 mmol) was co-evaporated with toluene twice. 5′-ODMT thymidine was dissolved in anhydrous DMF (0.5 mL) under Ar. In a separate flask under Ar, a solution of 4,5-dicyanoimidazole (0.24 g, 2.03 mmol) and 1-methyl imidazole (80 μL, 0.97 mmol) in anhydrous DMF (1 mL) was prepared. In a third flask under Ar, a solution of phosphordiamine S1 (1.12 g, 3.41 mmol) in anhydrous DMF (1 mL) was prepared. The solution containing 4,5-dicyanoimidazole and 1-methyl imidazole was added dropwise to the 5′-ODMT thymidine solution, followed by a dropwise addition of the phosphordiamine S1 solution. The reaction was allowed to stir at r.t for approximately 12 h until full consumption of the 5′-ODMT thymidine (monitored by TLC analysis). The volatiles were removed under reduced pressure (keeping the temperature of the rotavapor water bath under 35 °C). The resulting oil was dissolved in a minimum amount of CH2Cl2 and purified by flash silica gel column chromatography (from hexanes + 1% Et3N to hexanes/EtOAc: 7/3 + 1% Et3N). Mixture of P-diastereoisomers. White powder (1.4 g; 87%). 1H NMR (500 MHz, CDCl3) δ 8.55 (s, 1H), 7.56–7.45 (m, 2H), 7.42–7.38 (m, 3H), 7.37–7.29 (m, 4H), 6.90–6.83 (m, 4H), 5.81 (ddtd, J = 16.9, 10.2, 6.7, 3.6 Hz, 1H), 5.66 (s, 1H), 5.04–4.91 (m, 2H), 4.39 (t, J = 5.0 Hz, 2H), 3.87–3.76 (m, 9H), 3.61 (d, J = 10.8 Hz, 1H), 3.37 (dd, J = 18.1, 10.9 Hz, 1H), 2.09–1.98 (m, 2H), 1.74 (d, J = 1.2 Hz, 1H), 1.55 (d, J = 1.2 Hz, 3H), 1.50–1.23 (m, 8H), 1.19–1.04 (m, 14H), 1.00 (d, J = 6.7 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 163.7, 158.7, 149.6, 144.2, 138.9, 135.3, 134.8, 130.4, 130.3, 130.2, 130.1, 128.5, 128.2, 127.9, 127.2, 114.4, 113.21, 113.15, 110.3, 87.9, 87.2, 86.8, 74.6, 74.4, 72.0, 58.1, 55.2, 33.73, 33.65, 31.32, 31.32, 31.25, 30.8, 30.7, 28.7, 28.6, 24.0, 12.3. 31P NMR (200 MHz, CDCl3) δ 135.3, 128.2. HRMS (ESI): calcd for C45H59N3O8P [M + H]+: 800.4034; found 800.4062.
Synthesis of trimers (8a) and (8b). In a flame-dried 50 mL round-bottom flask under Ar, S-dimer 6a (100 mg, 0.135 mmol) and phosphoramidite 7 (340 mg, 0.425 mmol, 3.2 eq.) were combined with MeCN (7 mL, c = 19 mM) to give a colorless solution. Tetrazole (0.48 mL, 0.45 M in MeCN, 0.216 mmol, 1.6 eq.) was added dropwise followed by NMI (10 mg, 10 μL, 0.124 mmol, 0.9 eq.). The mixture was stirred at r.t overnight. The mixture was quenched with t-BuOOH (0.2 mL, 5.5 M in decanes, 1.08 mmol, 8 eq.) and stirred for 30 min. The solution was diluted with EtOAc and an aq. solution of 10% NaHSO4/sat. NaHCO3: 1/1 was added. After 5 min, the aq. phase was extracted with EtOAc (x3). The comb. org. layers were dried over MgSO4, filtered and evaporated in vacuo. The crude white solid (468 mg) was purified on silica (10 × 2.5 cm; hexanes/EtOAc: 1/2 then EtOAc then DCM/MeOH: 95/5). Mixture of diastereoisomers 8a/8b. White powder (128 mg; 65%). 31P NMR (200 MHz, CDCl3) δ 34.6 (minor), 34.1 (major), 28.0 (minor), 27.7 (major).
Synthesis of trimers (8c) and (8d). In a flame-dried 100 mL round-bottom flask under Ar, R-dimer 6b (300 mg, 0.405 mmol) and phosphoramidite 7 (1.0 g, 1.26 mmol, 3.1 eq.) were combined with MeCN (24 mL, c = 17 mM) to give a white suspension. Tetrazole (1.44 mL, 0.45 M in MeCN, 0.648 mmol, 1.6 eq.) was added dropwise followed by NMI (31 mg, 30 μL, 0.373 mmol, 0.9 eq.). The mixture was stirred at r.t overnight. The mixture was quenched with t-BuOOH (0.52 mL, 5.5 M in decanes, 2.83 mmol, 7 eq.) and stirred for 30 min. The solution was diluted with EtOAc and an aq. solution of 10% NaHSO4/sat. NaHCO3: 1/1 was added. After 5 min, the aq. phase was extracted with EtOAc (x3). The comb. org. layers were dried over MgSO4, filtered and evaporated in vacuo. The crude white solid (1.3 g) was purified on silica (9 × 3 cm; hexanes/EtOAc: 1/2 then EtOAc). The recovered mixture of trimers 8c/8d, a white solid (360 mg), was purified again using 40 g Premium silica column (from 0 to 6% EtOH over 10 min then gradient to 10% EtOH over 20 min after 50 min). First fraction (8c): white powder (157 mg; 27%). 1H NMR (400 MHz, CDCl3) δ 10.04 (s, 1H), 9.58 (s, 1H), 8.99 (s, 1H), 7.64 (s, 1H), 7.43 (d, J = 7.4 Hz, 2H), 7.37 (s, 1H), 7.35–7.28 (m, 6H), 7.21 (s, 1H), 6.90–6.82 (m, 4H), 5.80–5.68 (m, 2H), 5.64 (s, 1H), 5.56 (s, 1H), 5.51 (s, 1H), 5.30–5.21 (m, 2H), 4.99–4.86 (m, 4H), 4.80–4.75 (m, 2H), 4.63 (dd, J = 12.2, 7.8 Hz, 1H), 4.48 (dd, J = 12.0, 6.3 Hz, 1H), 4.34 (s, 1H), 4.24 (dd, J = 12.1, 5.2 Hz, 1H), 4.18–4.11 (m, 1H), 3.97 (s, 1H), 3.93–3.84 (m, 3H), 3.78 (s, 6H), 3.74–3.69 (m, 2H), 3.66 (s, 1H), 3.64–3.60 (m, 1H), 3.39 (d, J = 11.1 Hz, 1H), 2.84–2.80 (m, 1H), 2.79–2.75 (m, 1H), 2.05–1.98 (m, 3H), 1.92 (s, 3H), 1.80 (s, 3H), 1.61 (s, 8H), 1.42–1.31 (m, 4H), 0.86 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H). 31P NMR (160 MHz, CDCl3) δ 34.4, 30.0. HRMS (ESI): calcd for C70H88N6O22P2SiNa [M + Na]+: 1477.5088; found 1477.5107. Second fraction (8d): white powder (118 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 10.02 (s, 1H), 9.55 (s, 1H), 9.30 (s, 1H), 7.58 (s, 1H), 7.43 (d, J = 7.4 Hz, 2H), 7.37–7.27 (m, 8H), 6.90–6.82 (m, 4H), 5.79–5.67 (m, 2H), 5.63 (s, 1H), 5.60 (s, 1H), 5.59 (s, 1H), 5.31–5.20 (m, 2H), 5.03 (d, J = 5.2 Hz, 1H), 4.99–4.90 (m, 3H), 4.87 (s, 1H), 4.67 (d, J = 4.5 Hz, 1H), 4.55–4.39 (m, 3H), 4.38–4.32 (m, 2H), 4.02 (s, 1H), 4.00–3.92 (m, 3H), 3.89–3.82 (m, 3H), 3.79 (s, 6H), 3.64 (d, J = 11.0 Hz, 1H), 3.35 (d, J = 11.0 Hz, 1H), 2.82 (d, J = 7.3 Hz, 1H), 2.77 (d, J = 7.3 Hz, 1H), 2.06–1.93 (m, 3H), 1.92 (s, 3H), 1.89 (s, 3H), 1.72–1.63 (m, 5H), 1.58 (s, 3H), 1.29–1.21 (m, 6H), 0.87 (s, 9H), 0.08 (s, 6H). 31P NMR (160 MHz, CDCl3) δ 34.5, 29.0. HRMS (ESI): calcd for C70H88N6O22P2SiNa [M + Na]+: 1477.5088; found 1477.5095.
Synthesis of S*,S-macrocycle (9a) and R*,S-macrocycle (9b). In a flame-dried 1 L round-bottom flask under Ar, trimers 8a/8b (380 mg, 0.261 mmol) were dissolved in CH2Cl2 (390 mL, c = 671 μM) to give a colorless solution. The mixture was degassed for 15 min and Hoveyda–Grubbs-II (50 mg, 0.078 mmol, 0.3 eq.) was added. The mixture was stirred under reflux overnight. The green solution was evaporated in vacuo and the crude green solid (407 mg) was partitioned in two portions and each was purified on silica (40 g; from 0 to 5% EtOH in CH2Cl2 over 10 min, then 6% EtOH after 70 min). First fraction (9a): light brown solid (112 mg; 30%). 1H NMR (700 MHz, acetone-d6) δ 10.27 (br s, 1H), 10.19 (br s, 1H), 10.07 (br s, 1H), 7.68 (s, 1H), 7.64 (s, 1H), 7.56–7.52 (m, 2H), 7.42–7.38 (m, 4H), 7.38–7.33 (m, 3H), 7.30–7.26 (m, 1H), 6.93–6.89 (m, 4H), 5.69–5.66 (m, 1H), 5.65 (s, 1H), 5.56 (s, 1H), 5.48 (s, 1H), 5.42 (td, J = 14.7, 7.1 Hz, 2H), 5.11 (d, J = 6.4 Hz, 1H), 4.94 (s, 1H), 4.76 (s, 1H), 4.58 (dd, J = 12.0, 7.2 Hz, 2H), 4.56–4.54 (m, 1H), 4.50 (dd, J = 11.9, 6.2 Hz, 1H), 4.40 (s, 1H), 4.30 (s, 1H), 4.16–4.13 (m, 1H), 4.05 (d, J = 7.8 Hz, 1H), 3.95 (d, J = 8.1 Hz, 1H), 3.89–3.87 (m, 1H), 3.85 (d, J = 7.7 Hz, 1H), 3.80 (s, 6H), 3.77 (d, J = 11.0 Hz, 1H), 3.60 (d, J = 11.0 Hz, 1H), 3.57 (d, J = 8.2 Hz, 1H), 2.74–2.65 (m, 2H), 2.21–2.15 (m, 1H), 2.13–2.09 (m, 1H), 1.94–1.86 (m, 2H), 1.83 (s, 3H), 1.80 (s, 3H), 1.74–1.46 (m, 7H), 1.44 (s, 3H), 1.43–1.35 (m, 3H), 0.90 (s, 9H), 0.12 (s, 3H), 0.11 (s, 3H). 13C NMR (175 MHz, acetone-d6) δ 164.4, 164.3, 164.1, 159.8, 150.9, 150.8, 145.6, 137.3, 136.3, 136.2, 135.3, 134.9, 134.1, 131.4, 129.4, 128.7, 128.0, 119.2, 119.1, 114.0, 110.8, 110.4, 110.3, 88.4, 88.0, 87.9, 87.8, 87.6, 87.5, 87.4, 80.2, 79.5, 79.2, 74.1, 73.7, 73.6, 72.7, 72.2, 72.1, 71.8, 62.1, 59.3, 58.7, 55.6, 34.2, 32.6, 32.0, 30.8, 30.6, 30.3, 28.6, 28.4, 26.4, 26.0, 25.7, 25.6, 23.3, 23.1, 23.0, 18.5, 14.4, 13.1, 12.7, −4.7, −4.9. 31P NMR (160 MHz, acetone-d6) δ 35.6, 29.0. HRMS (ESI): calcd for C68H84N6O22P2SiNa [M + Na]+: 1449.4775; found 1449.4787. Second fraction (9b): light brown solid (115 mg; 31%). 1H NMR (700 MHz, acetone-d6) δ 7.76 (s, 1H), 7.72 (s, 1H), 7.59 (s, 1H), 7.55 (d, J = 7.5 Hz, 2H), 7.43–7.40 (m, 4H), 7.39–7.35 (m, 2H), 7.32–7.29 (m, 2H), 6.95–6.92 (m, 4H), 5.70–5.64 (m, 2H), 5.63 (s, 1H), 5.58 (s, 1H), 5.56 (s, 1H), 5.46–5.39 (m, 3H), 5.21 (d, J = 6.6 Hz, 1H), 5.04 (s, 1H), 5.02 (s, 1H), 4.71 (d, J = 4.4 Hz, 1H), 4.62 (dd, J = 12.2, 7.7 Hz, 2H), 4.55 (dd, J = 11.7, 6.8 Hz, 2H), 4.44–4.35 (m, 4H), 4.26 (s, 1H), 4.09–4.00 (m, 4H), 3.94–3.87 (m, 4H), 3.85 (d, J = 7.7 Hz, 1H), 3.81 (dd, J = 6.0, 1.8 Hz, 4H), 3.70–3.66 (m, 1H), 3.59–3.53 (m, 4H), 2.72–2.64 (m, 3H), 2.14–2.12 (m, 1H), 2.02–1.97 (m, 2H), 1.88 (s, 3H), 1.79 (s, 3H), 1.78–1.55 (m, 8H), 1.53 (s, 3H), 0.88 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H). 13C NMR (175 MHz, acetone-d6) δ 174.6, 172.0, 164.1, 159.88, 159.86, 150.9, 145.4, 137.0, 136.1, 136.0, 135.1, 134.6, 131.2, 131.1, 129.22, 129.16, 128.9, 128.0, 119.53, 119.46, 114.08, 114.07, 111.4, 111.0, 110.6, 88.49, 88.45, 88.4, 88.3, 88.1, 88.0, 87.6, 79.9, 79.0, 72.5, 72.0, 71.9, 71.8, 62.2, 58.6, 57.7, 55.6, 55.5, 45.6, 34.2, 33.1, 32.6, 32.0, 30.6, 30.3, 30.2, 26.0, 23.9, 23.43, 23.41, 23.3, 20.5, 18.9, 18.5, 14.4, 12.9, 12.8, 12.6, −4.7, −4.9. 31P NMR (160 MHz, acetone-d6) δ 32.9, 28.2. HRMS (ESI): calcd for C68H84N6O22P2SiNa [M + Na]+: 1449.4775; found 1449.4787.
Synthesis of S*,R-macrocycle (9c). In a flame-dried 500 mL round-bottom flask under Ar, trimer 8c (308 mg, 0.212 mmol) was dissolved in CH2Cl2 (300 mL, c = 705 μM) to give a colorless solution. The mixture was degassed for 15 min and Hoveyda–Grubbs-II (41 mg, 0.064 mmol, 0.3 eq.) was added. The mixture was stirred under reflux overnight. The green solution was evaporated in vacuo and the crude green solid (421 mg) was purified on silica (6 × 3 cm; CH2Cl2 then CH2Cl2/MeOH: 96/4). Brown solid (258 mg; 85%). 31P NMR (101 MHz, acetone-d6) δ 35.7 (major), 35.1 (minor), 28.6 (minor), 27.6 (major). HRMS (ESI): calcd for C68H84N6O22P2SiNa [M + Na]+: 1449.4775; found 1449.4755.
Synthesis of R*,R-macrocycle (9d). In a flame-dried 500 mL round-bottom flask under Ar, trimer 8d (391 mg, 0.269 mmol) was dissolved in CH2Cl2 (400 mL, c = 671 μM) to give a colorless solution. The mixture was degassed for 15 min and Hoveyda–Grubbs-II (52 mg, 0.081 mmol, 0.3 eq.) was added. The mixture was stirred under reflux overnight. The green solution was evaporated in vacuo and the crude green solid (421 mg) was purified on silica (8 × 3 cm; CH2Cl2 then CH2Cl2/MeOH: 96/4). Light brown solid (346 mg; 90%). 31P NMR (101 MHz, acetone-d6) δ 33.4 (major), 33.2 (minor), 27.5 (major), 26.8 (minor). HRMS (ESI): calcd for C68H84N6O22P2SiNa [M + Na]+: 1449.4775; found 1449.4790.
Synthesis of S*,S-macrocycle (10a). In a 50 mL round-bottom flask under Ar, macrocycle 9a (66 mg, 0.046 mmol) was combined with THF/MeOH: 1/1 (8 mL) to give a light brown solution. Pd/C 5% deactivated with 2,6-lutidine (295 mg, 0.139 mmol, 3 eq.) was charged and the flask was purged with Ar (x3). The flask was then purged with H2 (x3) and the mixture was stirred at r.t for 3 h. The mixture was filtered on Celite and the filtrate was evaporated in vacuo. The crude white solid (58 mg) was used in the next step without any further purification. White solid (58 mg; 88%). 1H NMR (700 MHz, acetone-d6) δ 10.45 (s, 1H), 10.27 (s, 1H), 10.08 (s, 1H), 7.69 (s, 1H), 7.67 (s, 1H), 7.55–7.51 (m, 2H), 7.42–7.37 (m, 5H), 7.38–7.33 (m, 2H), 7.30–7.25 (m, 1H), 6.94–6.89 (m, 4H), 5.75 (s, 1H), 5.65 (s, 1H), 5.56 (s, 1H), 5.44 (s, 1H), 5.15 (d, J = 6.5 Hz, 1H), 4.89 (s, 1H), 4.85 (s, 1H), 4.78 (s, 1H), 4.57 (dd, J = 12.1, 7.1 Hz, 1H), 4.48 (dd, J = 12.1, 6.5 Hz, 1H), 4.40 (s, 1H), 4.31 (s, 1H), 4.22–4.15 (m, 2H), 4.06 (d, J = 7.8 Hz, 1H), 3.93 (d, J = 8.1 Hz, 1H), 3.90–3.87 (m, 2H), 3.86 (d, J = 7.8 Hz, 1H), 3.80 (s, 6H), 3.78 (d, J = 11.0 Hz, 1H), 3.60 (d, J = 11.0 Hz, 1H), 3.53 (d, J = 8.1 Hz, 1H), 2.19–2.10 (m, 3H), 1.99–1.87 (m, 2H), 1.85 (s, 3H), 1.79 (s, 3H), 1.73–1.49 (m, 7H), 1.44 (s, 3H), 1.43–1.36 (m, 3H), 0.91 (s, 9H), 0.13 (s, 3H), 0.12 (s, 3H). 13C NMR (175 MHz, acetone-d6) δ 164.5, 164.4, 164.3, 159.8, 159.8, 150.9, 150.8, 150.7, 145.5, 136.3, 136.2, 135.4, 134.9, 134.7, 131.3, 129.5, 128.7, 128.0, 114.0, 110.3, 110.2, 110.2, 88.4, 88.4, 88.3, 88.1, 88.0, 87.9, 87.9, 87.6, 80.2, 79.7, 79.2, 74.2, 74.1, 73.5, 73.5, 72.7, 72.3, 72.1, 71.9, 61.6, 61.5, 59.4, 58.9, 58.9, 55.6, 53.6, 28.5, 28.4, 27.9, 27.9, 27.2, 27.0, 26.0, 25.5, 25.2, 24.7, 22.8, 22.7, 21.9, 21.9, 18.5, 13.3, 12.7, 12.7, −4.7, −4.9. 31P NMR (101 MHz, acetone-d6) δ 36.2, 34.8. HRMS (ESI): calcd for C68H86N6O22P2SiNa [M + Na]+: 1451.4932; found 1451.4926.
Synthesis of R*,S-macrocycle (10b). In a 50 mL round-bottom flask under Ar, macrocycle 9b (118 mg, 0.083 mmol) was combined with THF/MeOH: 1/1 (10 mL) to give a light brown solution. Pd/C 5% deactivated with 2,6-lutidine (528 mg, 0.248 mmol, 3 eq.) was charged and the flask was purged with Ar (x3). The flask was then purged with H2 (x3) and the mixture was stirred at r.t for 3 h. The mixture was filtered on Celite and the filtrate was evaporated in vacuo. The crude white solid (111 mg) was used in the next step without any further purification. White solid (111 mg; 94%). 1H NMR (700 MHz, acetone-d6) δ 7.88 (s, 1H), 7.73 (s, 1H), 7.63 (s, 1H), 7.56–7.53 (m, 2H), 7.44–7.40 (m, 4H), 7.39–7.35 (m, 3H), 7.32–7.28 (m, 2H), 6.96–6.92 (m, 4H), 5.61 (s, 1H), 5.59 (s, 1H), 5.55 (s, 1H), 5.27 (d, J = 6.7 Hz, 1H), 5.04 (s, 1H), 5.02 (s, 1H), 4.87 (d, J = 3.6 Hz, 1H), 4.75–4.70 (m, 2H), 4.55 (dd, J = 11.7, 6.7 Hz, 1H), 4.43–4.39 (m, 3H), 4.26 (s, 1H), 4.08 (d, J = 8.1 Hz, 1H), 4.01 (d, J = 7.7 Hz, 1H), 3.92–3.88 (m, 3H), 3.85 (d, J = 7.6 Hz, 1H), 3.81 (s, 6H), 3.72 (d, J = 11.0 Hz, 1H), 3.54 (d, J = 11.1 Hz, 1H), 2.27 (t, J = 7.4 Hz, 1H), 2.01–1.93 (m, 3H), 1.89 (s, 3H), 1.87–1.82 (m, 2H), 1.80 (s, 3H), 1.73–1.52 (m, 7H), 1.50 (s, 3H), 0.88 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H). 13C NMR (175 MHz, acetone-d6) δ 165.1, 164.2, 164.1, 159.9, 159.9, 151.7, 151.3, 150.9, 145.3, 136.1, 136.0, 135.3, 134.9, 134.6, 131.2, 129.2, 128.9, 128.0, 114.1, 114.1, 111.2, 111.0, 110.5, 88.5, 88.4, 88.4, 88.3, 88.1, 88.1, 87.6, 79.9, 79.2, 79.2, 79.0, 72.5, 72.0, 72.0, 71.8, 61.9, 61.8, 61.2, 58.6, 55.6, 55.5, 34.2, 32.6, 32.0, 28.8, 28.7, 28.4, 28.4, 27.8, 27.2, 25.7, 25.6, 25.3, 24.8, 24.5, 22.6, 22.3, 22.3, 18.5, 13.0, 12.8, 12.6, −4.7, −5.0. 31P NMR (101 MHz, acetone-d6) δ 33.3, 33.1. HRMS (ESI): calcd for C68H86N6O22P2SiNa [M + Na]+: 1451.4932; found 1451.4910.
Synthesis of S*,R-macrocycle (10c). In a 50 mL round-bottom flask under Ar, macrocycle 9c (247 mg, 0.173 mmol) was combined with THF/MeOH: 1/1 (20 mL) to give a light brown solution. Pd/C 5% deactivated with 2,6-lutidine (1.10 g, 0.519 mmol, 3 eq.) was charged and the flask was purged with Ar (x3). The flask was then purged with H2 (x3) and the mixture was stirred at r.t for 3 h. The mixture was filtered on Celite and the filtrate was evaporated in vacuo. The crude white solid (184 mg) was used in the next step without any further purification. White solid (184 mg; 74%). 1H NMR (700 MHz, acetone-d6) δ 7.68 (s, 1H), 7.55–7.54 (m, 2H), 7.48 (s, 1H), 7.42–7.39 (m, 4H), 7.39–7.35 (m, 3H), 7.30–7.28 (m, 1H), 6.93–6.92 (m, 4H), 5.66 (s, 1H), 5.58 (s, 1H), 5.47 (s, 1H), 5.11 (d, J = 6.4 Hz, 1H), 4.84 (s, 1H), 4.78 (s, 1H), 4.57 (dd, J = 11.9, 5.9 Hz, 1H), 4.51 (d, J = 3.9 Hz, 1H), 4.43 (dd, J = 11.9, 5.1 Hz, 1H), 4.39 (d, J = 4.1 Hz, 1H), 4.37 (s, 1H), 4.27 (s, 1H), 4.16 (dd, J = 12.2, 7.9 Hz, 1H), 4.05 (d, J = 7.8 Hz, 1H), 3.98 (d, J = 8.2 Hz, 1H), 3.92 (s, 2H), 3.84 (d, J = 7.8 Hz, 1H), 3.81 (s, 6H), 3.76 (d, J = 11.1 Hz, 1H), 3.65–3.61 (m, 2H), 2.60 (s, 1H), 2.42 (s, 1H), 2.19–2.10 (m, 3H), 1.97–1.86 (m, 3H), 1.80 (s, 3H), 1.78 (s, 3H), 1.76–1.48 (m, 10H), 1.45 (s, 3H), 0.89 (s, 9H), 0.12 (s, 3H), 0.10 (s, 3H). 13C NMR (175 MHz, acetone-d6) δ 164.3, 164.3, 164.2, 159.8, 159.8, 150.9, 150.8, 150.8, 145.6, 136.3, 136.2, 135.0, 134.9, 134.3, 131.3, 131.3, 129.4, 128.8, 128.0, 126.1, 120.6, 114.0, 110.8, 110.4, 110.0, 88.4, 88.4, 88.3, 88.1, 88.0, 87.7, 87.7, 87.5, 80.2, 79.2, 79.1, 74.1, 74.0, 73.6, 73.6, 72.7, 72.3, 71.9, 71.7, 61.2, 61.2, 59.5, 59.3, 59.3, 55.9, 28.0, 28.0, 27.8, 27.8, 26.5, 26.0, 25.3, 23.8, 23.3, 23.0, 21.9, 21.9, 21.5, 21.5, 18.5, 13.2, 13.0, 12.7, −4.7, −5.0. 31P NMR (101 MHz, acetone-d6) δ 35.2, 31.9. HRMS (ESI): calcd for C68H90N7O22P2Si [M + NH4]+: 1446.5378; found 1446.5367.
Synthesis of R*,R-macrocycle (10d). In a 50 mL round-bottom flask under Ar, macrocycle 9d (340 mg, 0.238 mmol) was combined with THF/MeOH: 1/1 (30 mL) to give a light brown solution. Pd/C 5% deactivated with 2,6-lutidine (1.52 g, 0.715 mmol, 3 eq.) was charged and the flask was purged with Ar (x3). The flask was then purged with H2 (x3) and the mixture was stirred at r.t for 3 h. The mixture was filtered on Celite and the filtrate was evaporated in vacuo. The crude white solid (150 mg) was used in the next step without any further purification. White solid (285 mg; 84%). 1H NMR (700 MHz, acetone-d6) δ 7.83 (s, 1H), 7.74 (s, 1H), 7.56–7.53 (m, 2H), 7.42–7.37 (m, 7H), 7.32–7.29 (m, 1H), 6.96–6.94 (m, 4H), 5.61 (s, 1H), 5.60 (s, 1H), 5.59 (s, 1H), 5.26 (d, J = 6.7 Hz, 1H), 4.98 (d, J = 9.7 Hz, 2H), 4.75 (dd, J = 12.0, 8.5 Hz, 1H), 4.57–4.53 (m, 2H), 4.51 (dd, J = 12.4, 6.6 Hz, 1H), 4.42 (dd, J = 11.8, 4.4 Hz, 1H), 4.36 (s, 1H), 4.23 (s, 1H), 4.09 (d, J = 8.3 Hz, 1H), 4.03 (d, J = 7.8 Hz, 1H), 3.95 (d, J = 8.2 Hz, 1H), 3.92 (d, J = 8.2 Hz, 1H), 3.90 (d, J = 8.2 Hz, 1H), 3.87 (d, J = 7.7 Hz, 1H), 3.81 (s, 6H), 3.75 (d, J = 11.1 Hz, 1H), 3.54 (d, J = 11.1 Hz, 1H), 2.22 (s, 2H), 2.03–1.89 (m, 3H), 1.87 (s, 3H), 1.86–1.80 (m, 3H), 1.77 (s, 3H), 1.74–1.50 (m, 8H), 1.48 (s, 3H), 0.87 (s, 9H), 0.10 (s, 3H), 0.07 (s, 3H). 13C NMR (175 MHz, acetone-d6) δ 164.7, 164.5, 164.3, 159.9, 159.9, 151.5, 151.3, 150.8, 145.3, 136.1, 136.0, 134.9, 134.6, 131.2, 131.2, 129.2, 128.9, 128.0, 126.1, 114.1, 111.4, 111.0, 110.1, 88.5, 88.4, 88.3, 88.2, 88.0, 87.7, 87.7, 87.6, 80.1, 79.1, 78.9, 73.9, 73.8, 73.2, 73.2, 72.6, 72.2, 71.9, 71.7, 61.2, 61.2, 61.0, 61.0, 58.7, 55.6, 28.4, 28.3, 28.1, 28.1, 27.2, 27.1, 26.0, 25.0, 24.5, 23.7, 22.4, 22.3, 22.2, 22.2, 18.5, 13.2, 12.9, 12.7, −4.7, −5.0. 31P NMR (101 MHz, acetone-d6) δ 33.4, 31.4. HRMS (ESI): calcd for C68H86N6O22P2SiNa [M + Na]+: 1451.4932; found 1451.4913.
Synthesis of S*,S-LNA macrocyle (11a). In a 50 mL round-bottom flask under Ar, macrocycle 10a (80 mg, 56.0 μmol) was combined with THF (5 mL) to give a colorless solution. The mixture was cooled down to 0 °C and TBAF (1 M in THF, 70 μL, 70.0 μmol, 1.25 eq.) was added. The mixture was stirred for 1 h and 2 drops of sat. aq. NH4Cl soln. were added. The mixture was evaporated in vacuo and the crude white solid residue (137 mg) was purified on silica (12 × 1 cm; CH2Cl2 then CH2Cl2/MeOH: 94/6). The recovered 57 mg were purified by reverse phase chromatography (SiO2 C18; 4 g; from 10 to 80% MeCN in water over 15 min). White solid (27 mg; 37%). 1H NMR (500 MHz, acetone-d6) δ 10.40 (br s, 1H), 10.21 (br s, 1H), 10.04 (br s, 1H), 7.69 (s, 1H), 7.60 (s, 1H), 7.56–7.51 (m, 2H), 7.43–7.32 (m, 8H), 7.28 (t, J = 7.4 Hz, 1H), 6.94–6.88 (m, 4H), 5.65 (s, 1H), 5.55 (s, 1H), 5.45 (s, 1H), 5.14 (d, J = 6.6 Hz, 1H), 4.91 (s, 1H), 4.83 (s, 1H), 4.78 (s, 1H), 4.59–4.48 (m, 2H), 4.34 (s, 1H), 4.26–4.19 (m, 2H), 4.19–4.11 (m, 1H), 4.06 (d, J = 7.8 Hz, 1H), 3.99 (d, J = 8.0 Hz, 1H), 3.89 (s, 2H), 3.84 (d, J = 8.0 Hz, 1H), 3.82–3.75 (m, 7H), 3.60 (d, J = 11.2 Hz, 1H), 3.52 (d, J = 7.9 Hz, 1H), 2.19–2.08 (m, 3H), 2.01–1.88 (m, 2H), 1.86 (s, 3H), 1.79 (s, 3H), 1.76–1.46 (m, 7H), 1.45 (s, 3H), 1.44–1.28 (m, 4H). A good quality 13C NMR spectrum could not be obtained due to poor solubility of the compound in usual NMR solvents. 31P NMR (202 MHz, acetone-d6) δ 36.1, 35.1. HRMS (ESI): calcd for C62H72N6O22P2Na [M + Na]+: 1337.4067; found 1337.4130. [α]D20 = +40.000 (c 0.50, acetone).
Synthesis of R*,S-LNA macrocycle (11b). In a 50 mL round-bottom flask under Ar, macrocycle 10b (75 mg, 52.5 μmol) was combined with THF (4 mL) to give a colorless solution. The mixture was cooled down to 0 °C and TBAF (1 M in THF, 66 μL, 65.6 μmol, 1.25 eq.) was added. The mixture was stirred for 1 h and 2 drops of sat. aq. NH4Cl soln were added. The mixture was evaporated in vacuo and the crude white solid residue (140 mg) was purified on silica (12 × 1 cm; CH2Cl2 then CH2Cl2/MeOH: 94/6). The recovered 50 mg were purified by reverse phase chromatography (SiO2 C18; 4 g; from 10 to 80% MeCN in water over 15 min). White solid (20 mg; 29%). 1H NMR (500 MHz, acetone-d6) δ 10.76–10.29 (m, 3H), 7.83 (s, 1H), 7.72 (s, 1H), 7.59 (s, 1H), 7.56–7.53 (m, J = 7.3 Hz, 2H), 7.44–7.35 (m, 6H), 7.33–7.28 (m, 1H), 6.94 (dd, J = 9.0, 2.0 Hz, 4H), 5.59 (s, 1H), 5.59 (s, 1H), 5.54 (s, 1H), 5.26 (d, J = 7.0 Hz, 1H), 5.00 (s, 1H), 4.97 (s, 1H), 4.81 (d, J = 4.0 Hz, 1H), 4.68 (dd, J = 12.4, 7.9 Hz, 1H), 4.49 (d, J = 6.2 Hz, 2H), 4.40 (dd, J = 12.6, 6.7 Hz, 1H), 4.37 (s, 1H), 4.19 (s, 1H), 4.13 (d, J = 8.3 Hz, 1H), 4.02 (d, J = 7.9 Hz, 1H), 3.90 (t, J = 5.7 Hz, 3H), 3.82 (s, 1H), 3.81 (s, 6H), 3.73 (d, J = 11.4 Hz, 1H), 3.54 (d, J = 11.2 Hz, 1H), 2.03–1.96 (m, 2H), 1.87 (s, 3H), 1.82 (s, 3H), 1.79–1.53 (m, 7H), 1.49 (s, 3H), 1.43–1.31 (m, 8H). 13C NMR (100 MHz, acetone-d6) δ 164.0, 163.6, 163.3, 159.0, 150.6, 150.3, 150.0, 144.4, 135.2, 135.1, 134.4, 134.0, 133.7, 130.4, 128.4, 128.0, 127.1, 113.2, 110.2, 110.0, 109.4, 87.5, 87.3, 87.25, 87.16, 87.1, 86.8, 79.3, 78.4, 78.3, 72.2, 72.0, 71.7, 71.2, 70.9, 69.8, 60.9, 60.2, 58.5, 57.8, 54.7, 27.9, 27.8, 27.4, 27.3, 26.7, 26.4, 25.0, 24.9, 23.7, 23.5, 21.7, 21.5, 12.0, 11.9. 31P NMR (202 MHz, acetone-d6) δ 34.0, 33.4. HRMS (ESI): calcd for C62H72N6O22P2Na [M + Na]+: 1337.4067; found 1337.4020. [α]D20 = +57.308 (c 0.52, acetone).
Synthesis of S*,R-LNA macrocycle (11c). In a 50 mL round-bottom flask under Ar, macrocycle 10c (106 mg, 74.2 μmol) was combined with THF (4 mL) to give a colorless solution. The mixture was cooled down to 0 °C and TBAF (1 M in THF, 93 μL, 92.7 μmol, 1.25 eq.) was added. The mixture was stirred for 1 h and 2 drops of sat. aq. NH4Cl soln. were added. The mixture was evaporated in vacuo and the crude white solid residue (228 mg) was purified on silica (5 × 3 cm; CH2Cl2 then CH2Cl2/MeOH: 94/6). The recovered 92 mg were purified by reverse phase chromatography (SiO2 C18; 4 g; from 10 to 80% MeCN in water over 20 min). White solid (35 mg; 36%). 1H NMR (700 MHz, acetone-d6) δ 7.69–7.67 (m, 1H), 7.56–7.53 (m, 2H), 7.47–7.46 (m, 1H), 7.43–7.39 (m, 4H), 7.39–7.35 (m, 3H), 7.31–7.28 (m, 1H), 6.94–6.91 (m, 4H), 5.65 (s, 1H), 5.53 (s, 1H), 5.49 (s, 1H), 5.11 (t, J = 3.1 Hz, 2H), 4.83 (s, 1H), 4.77 (s, 1H), 4.63–4.60 (m, 1H), 4.60–4.59 (m, 1H), 4.41 (dd, J = 12.5, 9.1 Hz, 1H), 4.38 (dd, J = 12.4, 4.5 Hz, 1H), 4.33 (s, 1H), 4.18–4.15 (m, 1H), 4.14 (s, 1H), 4.02 (d, J = 7.9 Hz, 1H), 3.99 (d, J = 8.3 Hz, 1H), 3.90 (s, 2H), 3.81 (s, 6H), 3.78 (d, J = 7.9 Hz, 1H), 3.76 (d, J = 11.1 Hz, 1H), 3.62 (d, J = 5.0 Hz, 1H), 3.60 (d, J = 2.0 Hz, 1H), 2.21–2.16 (m, 1H), 2.14–2.10 (m, 1H), 1.99–1.91 (m, 3H), 1.81 (s, 3H), 1.79 (s, 3H), 1.77–1.50 (m, 10H), 1.46 (s, 3H). 13C NMR (175 MHz, acetone-d6) δ 164.3, 164.3, 164.2, 159.8, 159.8, 150.9, 150.8, 150.8, 145.6, 136.3, 136.2, 134.9, 134.8, 134.4, 131.4, 131.3, 129.4, 128.8, 128.0, 114.0, 114.0, 110.8, 110.4, 109.9, 88.4, 88.4, 88.3, 88.3, 88.2, 88.0, 88.0, 87.5, 87.3, 80.1, 79.2, 79.1, 77.2, 74.1, 74.1, 73.9, 73.8, 72.7, 71.8, 70.9, 61.4, 61.4, 59.4, 59.2, 59.2, 55.9, 55.6, 28.0, 28.0, 26.6, 26.3, 25.9, 25.1, 23.7, 23.3, 22.9, 22.1, 22.0, 21.6, 21.5, 13.1, 13.0, 12.7. 31P NMR (101 MHz, acetone-d6) δ 35.1, 33.5. HRMS (ESI): calcd for C62H72N6O22P2Na [M + Na]+: 1337.4067; found 1337.4040. [α]D20 = +30.000 (c 0.46, acetone).
Synthesis of R*,R-LNA macrocycle (11d). In a 50 mL round-bottom flask under Ar, macrocycle 10d (140 mg, 97.9 μmol) was combined with THF (6 mL) to give a colorless solution. The mixture was cooled down to 0 °C and TBAF (1 M in THF, 0.12 mL, 0.122 mmol, 1.25 eq.) was added. The mixture was stirred for 1 h and 2 drops of sat. aq. NH4Cl soln. were added. The mixture was evaporated in vacuo and the crude white solid residue (231 mg) was purified on silica (12 × 1 cm; CH2Cl2 then CH2Cl2/MeOH: 94/6). The recovered 144 mg were purified by reverse phase chromatography (SiO2 C18; 4 g; from 10 to 80% MeCN in water over 20 min). White solid (65 mg; 50%). 1H NMR (700 MHz, acetone-d6) δ 10.57 (br s, 1H), 10.38 (br s, 1H), 10.21 (br s, 1H), 7.77 (s, 1H), 7.72 (s, 1H), 7.56–7.52 (m, 2H), 7.47 (s, 1H), 7.42–7.36 (m, 6H), 7.32–7.28 (m, 1H), 6.98–6.92 (m, 4H), 5.63 (s, 1H), 5.58 (s, 1H), 5.54 (s, 1H), 5.22 (d, J = 6.5 Hz, 1H), 4.91 (s, 1H), 4.89 (s, 1H), 4.79–4.73 (m, 1H), 4.67–4.64 (m, 1H), 4.64–4.59 (m, 1H), 4.51–4.46 (m, 1H), 4.46–4.41 (m, 1H), 4.33 (s, 1H), 4.12 (s, 1H), 4.11 (d, J = 8.4 Hz, 1H), 4.04 (d, J = 7.9 Hz, 1H), 3.96–3.89 (m, 3H), 3.81 (s, 6H), 3.76 (d, J = 11.2 Hz, 1H), 3.53 (d, J = 11.2 Hz, 1H), 2.85 (s, 6H), 2.81 (s, 3H), 2.14–2.08 (m, 2H), 2.02–1.93 (m, 2H), 1.86 (s, 3H), 1.80 (s, 3H), 1.78–1.50 (m, 8H), 1.48 (s, 3H), 1.45–1.29 (m, 8H). 13C NMR (175 MHz, acetone-d6) δ 164.4, 164.3, 159.90, 159.88, 151.2, 151.1, 150.8, 145.3, 136.1, 136.0, 135.1, 135.0, 134.7, 131.24, 131.2, 129.3, 128.9, 128.0, 114.1, 111.1, 110.8, 110.1, 88.4, 88.36, 88.32, 88.19, 88.16, 88.05, 87.7, 87.6, 80.2, 79.3, 79.1, 74.21, 74.17, 73.5, 73.4, 72.6, 72.0, 71.9, 71.0, 61.8, 61.1, 58.8, 55.6, 28.3, 28.28, 28.23, 28.18, 27.12, 27.10, 25.9, 25.1, 24.6, 23.8, 22.4, 22.2, 13.0, 12.9, 12.7. 31P NMR (101 MHz, acetone-d6) δ 33.7, 33.0. HRMS (ESI): calcd for C62H72N6O22P2Na [M + Na]+: 1337.4067; found 1337.4040. [α]D20 = +37.197 (c 0.48, acetone).
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Notes and references
- S. T. Crooke, B. F. Baker, R. M. Crooke and X. H. Liang, Nat. Rev. Drug Discovery, 2021, 20, 427–453 CrossRef CAS PubMed.
- R. L. Setten, J. J. Rossi and S.-P. Han, Nat. Rev. Drug Discovery, 2019, 18, 421–446 CrossRef CAS PubMed.
- S. T. Crooke and C. F. Bennett, Annu. Rev. Pharmacol. Toxicol., 1996, 36, 107–129 CrossRef CAS PubMed.
- M. A. Havens and M. L. Hastings, Nucleic Acids Res., 2016, 44, 6549–6563 CrossRef PubMed.
- B. J. Booth, S. Nourreddine, D. Katrekar, Y. Savva, D. Bose, T. J. Long, D. J. Huss and P. Mali, Mol. Ther., 2023, 31, 1533–1549 CrossRef CAS PubMed.
- E. C. Lee, T. Valencia, C. Allerson, A. Schairer, A. Flaten, M. Yheskel, K. Kersjes, J. Li, S. Gatto, M. Takhar, S. Lockton, A. Pavlicek, M. Kim, T. Chu, R. Soriano, S. Davis, J. R. Androsavich, S. Sarwary, T. Owen, J. Kaplan, K. Liu, G. Jang, S. Neben, P. Bentley, T. Wright and V. Patel, Nat. Commun., 2019, 10, 4148 CrossRef CAS PubMed.
- H. Dana, G. M. Chalbatani, H. Mahmoodzadeh, R. Karimloo, O. Rezaiean, A. Moradzadeh, N. Mehmandoost, F. Moazzen, A. Mazraeh, V. Marmari, M. Ebrahimi, M. M. Rashno, S. J. Abadi and E. Gharagouzlo, Int. J. Biomed. Sci., 2017, 13, 48–57 CrossRef PubMed.
- M. Asmamaw and B. Zawdie, Biologics, 2021, 15, 353–361 Search PubMed.
- S. T. Crooke, J. L. Witztum, C. F. Bennett and B. F. Baker, Cell Metab., 2018, 27, 714–739 CrossRef CAS PubMed.
- M. Todisco, D. Ding and J. W. Szostak, Nucleic Acids Res., 2024, 52, 2174–2187 CrossRef PubMed.
- W. B. Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef CAS PubMed.
- J. C. Litten and C. Leumann, Helv. Chim. Acta, 1996, 79, 1129–1146 CrossRef.
- D. Renneberg, E. Bouliong, U. Reber, D. Schümperli and C. J. Leumann, Nucleic Acids Res., 2002, 30, 2751–2757 CrossRef CAS PubMed.
- P. S. Pallan, D. Ittig, A. Héroux, Z. Wawrzak, C. J. Leumann and M. Egli, Chem. Commun., 2008, 883–885 RSC.
- A. A. Koshkin, P. Nielsen, M. Meldgaard, V. K. Rajwanshi, S. K. Singh and J. Wengel, J. Am. Chem. Soc., 1998, 120, 13252–13253 CrossRef CAS.
- S. Obika, T. Uneda, T. Sugimoto, D. Nanbu, T. Minami, T. Doi and T. Imanishi, Bioorg. Med. Chem., 2001, 9, 1001–1011 CrossRef CAS PubMed.
- C. Dupouy, N. Iché-Tarrat, M.-P. Durrieu, F. Rodriguez, J.-M. Escudier and A. Vigroux, Angew. Chem., Int. Ed., 2006, 45, 3623–3627 CrossRef CAS PubMed.
- D. A. Catana, B. L. Renard, M. Maturano, C. Payrastre, N. Tarrat and J. M. Escudier, J. Nucleic Acids, 2012, 2012, 215876 Search PubMed.
- S. Hanessian, B. R. Schroeder, R. D. Giacometti, B. L. Merner, M. Ostergaard, E. E. Swayze and P. P. Seth, Angew. Chem., Int. Ed., 2012, 51, 11242–11245 CrossRef CAS PubMed.
- R. D. Giacometti, J. C. Salinas, M. E. Østergaard, E. E. Swayze, P. P. Seth and S. Hanessian, Org. Biomol. Chem., 2016, 14, 2034–2040 RSC.
- J. C. Salinas, J. Yu, M. Østergaard, P. P. Seth and S. Hanessian, Org. Lett., 2018, 20, 5296–5299 CrossRef CAS PubMed.
- T. Rajasekaran, G. C. Freestone, R. Galindo-Murillo, B. Lugato, L. Rico, J. C. Salinas, H. Gaus, M. T. Migawa, E. E. Swayze, T. E. Cheatham III, S. Hanessian and P. P. Seth, J. Am. Chem. Soc., 2022, 144, 1941–1950 CrossRef CAS PubMed.
- T. Rajasekaran, G. C. Freestone, R. Galindo-Murillo, B. Lugato, H. Gaus, M. T. Migawa, E. E. Swayze, T. E. Cheatham III, P. P. Seth and S. Hanessian, J. Org. Chem., 2023, 88, 3599–3614 CrossRef CAS PubMed.
-
(a) S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS;
(b) S. P. Adams, K. S. Kavka, E. J. Wykes and S. B. Holder, J. Am. Chem. Soc., 1983, 105, 661–663 CrossRef CAS;
(c) For a review on the coupling of phosphoramidites, see: X. Wei, Tetrahedron, 2013, 69, 3615–3637 CrossRef CAS.
- C. Desnous, B. R. Ravindra, C. Moriou, J. U. O. Mayo, A. Favre, J. Wengel and P. Clivio, J. Am. Chem. Soc., 2008, 130, 30–31 CrossRef CAS PubMed.
- D. Xu, N. Rivas-Bascón, N. M. Padial, K. W. Knouse, B. Zheng, J. C. Vantourout, M. A. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 5785–5792 CrossRef CAS PubMed.
-
(a) B. H. Dahl, J. Nielsen and O. Dahl, Nucleic Acids Res., 1987, 15, 1729–1743 CrossRef CAS PubMed;
(b) S. Berner, K. Mühlegger and H. Seliger, Nucleic Acids Res., 1989, 17, 853–864 CrossRef CAS PubMed.
- R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446–452 CrossRef CAS.
- M. K. Lakshman and B. Zajc, Nucleosides Nucleotides, 1996, 15, 1029–1039 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ad
*ad