
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRationale, in silico docking, ADMET profile, design, synthesis and cytotoxicity evaluations of phthalazine derivatives as VEGFR-2 inhibitors and apoptosis inducers†
Hatem Hussein Bayoumi a,
Mohamed-Kamal Ibrahima,
Mohammed A. Dahaba,
Fathalla Khedra and
Khaled El-Adl*ba
a,
Mohamed-Kamal Ibrahima,
Mohammed A. Dahaba,
Fathalla Khedra and
Khaled El-Adl*ba
aPharmaceutical Medicinal Chemistry and Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Nasr City 11884, Cairo, Egypt. E-mail: eladlkhaled74@yahoo.com; eladlkhaled74@azhar.edu.eg; khaled.eladl@hu.edu.eg
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt
First published on 27th August 2024
Abstract
New phthalazine derivatives as vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors were synthesized joined to different spacers including pyrazole, α,β-unsaturated ketonic fragment, pyrimidinone and/or pyrimidinthione. A docking study was carried out to explore the suggested binding orientations of the novel derivatives inside the active site of VEGFR-2. The obtained biological data were extremely interrelated to that of the docking study. In particular, compounds 4b and 3e showed the highest activities against Michigan Cancer Foundation-7 (MCF-7) and Hepatocellular carcinoma G2 (HepG2) with half maximal inhibitory concentration (IC50) = 0.06, 0.06 μM and 0.08, 0.19 μM respectively. Our derivatives 3a–e, 4a,b and 5a,b were evaluated for their cytotoxicity against normal VERO cells. Our compounds exhibited low toxicity concerning normal VERO cells with IC50 = 3.00–4.75 μM. In addition, our final derivatives 3a–e, 4a, 4b, 5a and 5b were investigated for their VEGFR-2 inhibitory activities. Derivative 4b exhibited the highest VEGFR-2 inhibitory activities at an IC50 value of 0.09 ± 0.02 μM. Derivatives 3e, 4a and 5b demonstrated good activities with IC50 values = 0.12 ± 0.02, 0.15 ± 0.03 and 0.13 ± 0.03 μM respectively. Furthermore, the activities of 4b were assessed against MCF-7 cancer cells for apoptosis induction, cell cycle distribution and growth inhibition. Compound 4b caused cell growth arrest in growth 2-mitosis (G2-M) phase; accumulation of cells at that phase became 6.92% after being 13.2 in control cells. Moreover, our derivatives 3e, 4b and 5b revealed a good in silico considered absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile in comparison to sorafenib.
1. Introduction
Vascular endothelial growth factors (VEGFs) are transmembrane proteins that trigger angiogenesis through VEGF receptor signaling.1 To date, three types of VEGF tyrosine kinase receptors are known, VEGFR-1, VEGFR-2, and VEGFR-3. All of them display a high affinity towards VEGF. However, VEGFR-2 solely remains the only one that transmits the angiogenic signals.2 The VEGFR-2 receptor has been documented to play major roles in both physiological and many pathological angiogenesis, such as cancer.1 Overexpression of this receptor has been ascertained in a number of solid cancers3 and hematological malignancies.4 Consequently, VEGFR-2 has emerged as an ideal target for the development of novel chemotherapeutic agents.3 Tyrosine kinase inhibitors are grouped into three types, type I, II and type III.5 Type II inhibitors interact with the adenosine triphosphate (ATP) binding site as well as the allosteric hydrophobic site and display high selectivity.6,7 The architecture design of VEGFR-2 active site comprises of two pockets, the front and the back. The front pocket has two associated key residues, cysteine919 (Cys919) while the back hydrophobic pocket has another two key residues, glutamate885 (Glu885) and aspartate1046 (Asp1046).8 The pharmacophoric features shared by all of the reported VEGFR-2 inhibitors showed the presence of four main features as shown in Fig. 1;9,10 (i) the core structure occupies the catalytic ATP-binding domain and consists of a flat aromatic ring; (ii) the central hydrophobic spacer occupies the linker region between the ATP-binding domain and the DFG domain; (iii) a hydrophilic linker e.g., amino or urea to act as both H-bond donor and acceptor with the above-mentioned two key amino acid residues, Glu885 and Asp1046; (iv) a terminal hydrophobic moiety to occupy the allosteric hydrophobic pocket.11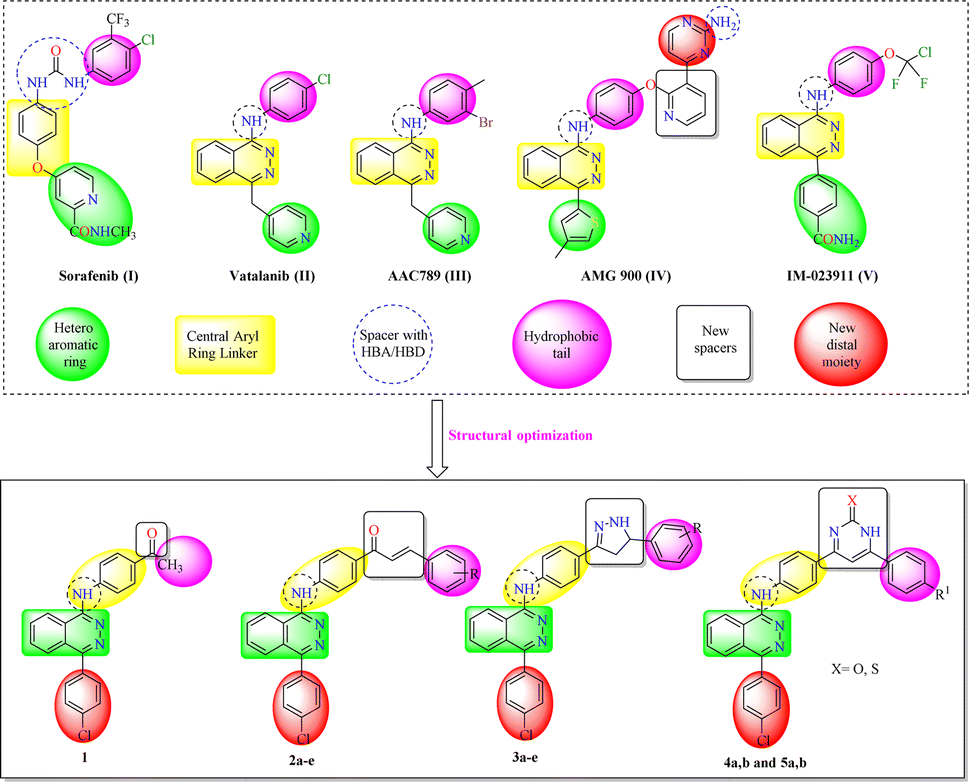 | ||
| Fig. 1 Rationale design structures of our compounds and pharmacophoric features of VEGFR-2 inhibitors. | ||
Over the last two decades, a number of VEGFR-2 inhibitors have demonstrated success as targeted anticancer therapeutics. Among which, sorafenib (I), the 1,4-disubstituted phthalazine derivatives, vatalanib (II), AAC789 (III), AMG900 (IV), and IM-023911 (V) revealed potent inhibitory activity against VEGFR-2 in nanomolar levels of IC50.12,13 Sorafenib is a food and drug administration (FDA) approved antiangiogenic drug that shares the above pharmacophoric features.14
Phthalazine ring is a vital pharmacophoric scaffold present in the core structures of many anticancer molecules15–19 with potent activity against hepatocellular carcinoma,18 colon cancer,11 breast cancer,19 and as degraders of VEGFR-2.15,20 Many phthalazine derivatives were reported to have VEGFR-2 and EGFR kinase inhibitory activity with IC50 values in the nanomolar range, however, compounds were more selective and better inhibitors of VEGFR-2 compared to epidermal growth factor receptor (EGFR).21,22 Other pharmacophoric heterocycles are implanted in the structures of recently reported anticancer with VEGFR-2 inhibitory effects as pyrazole23,24 and pyrimidine.25,26 These latter were reported to have the spatial configurations that enable both to interact with the VEGFR-2 binding site.26,27 Moreover, the α,β-unsaturated ketonic fragment is also presented in a number of synthetic derivatives with potent VEGFR-2 inhibitory activity.28,29
Over the last few years, our research group members were interested in the construction and evaluation of heterocyclic molecules of expected biological activity particularly as anticancer.30–50
The aim of this study is to develop novel inhibitors of VEGFR-2 protein. Accordingly, the abovementioned facts have encouraged us to design novel phthalazine derivatives linked with fragments of verified VEGFR-2 inhibitory potentials, including α,β-unsaturated ketonic fragment, pyrazole, pyrimidinone and/or pyrimidinthione to investigate the anticancer and VEGFR-2 inhibitory potentials of the designed compounds. All the designed compounds retained the essential pharmacophoric features of the reported and clinically used VEGFR-2 inhibitors (Fig. 1) with two additional bioisosteric modifications: (i) insertion of a new bioisosteric spacer with that reported in the most recent molecular docking and molecular dynamic simulation studies.13 The new spacer has inserted in the form of pharmacophoric fragments of reported anticancer potentials including α,β-unsaturated ketonic fragment, dihydropyrazole, pyrimidinone and/or pyrimidinthione; (ii) attachment of a new substituted tail bioisosteric hydrophobic moieties with that of AMG 900 (IV) which are expected to increase the hydrophobic interaction with VEGFR-2 and consequently the affinity. Substitution pattern of the new tail bioisosteric hydrophobic moieties was selected to ensure different electronic and lipophilic environments that could influence the activity of the target compounds. These modifications were performed in order to carry out further elaboration of the phthalazine scaffolds and to explore a valuable SAR.
The designed target derivatives were synthesized and evaluated as potential VEGFR-2 inhibitors and anti-tumors against Hepatocellular carcinoma G2 (HepG2) and Michigan Cancer Foundation-7 (MCF-7).
2. Results and discussion
2.1. Docking studies
All modeling experiments in the present work were performed using Molsoft software. VEGFR-2 experimental structure was downloaded from the Protein Databank (PDB ID 4ASD).51 All studied ligands have similar position and orientation inside the recognized binding site of VEGFR-2.Sorafenib exhibited −99.50 kcal mol−1 (Table 1) and 5 H-bonds with Asp1046 (1.50 Å), Glu885 (1.77 Å and 2.75 Å), and Cys919 (2.51 Å and 2.10 Å). The N-methylpicolinamide group placed in the pocket produced by Glu917, Val848, Lys920, Leu1035, Cys919, Phe918 and Leu840. As well, the hydrophobic hollow constructed by Val848, Lys868, Thr916, Leu1035, and Cys1045 filled by the central phenyl spacer. As well, Ile892, Ile888, Hie1026, Glu885, Cys1045 and Asp1046 constructed a hydrophobic canal which filled with the terminal 3-trifluoromethyl-4-chlorophenyl moiety (Fig. 2).
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 1 | −70.11 | 3c | −89.68 |
| 2a | −72.77 | 3d | −93.44 |
| 2b | −72.80 | 3e | −97.75 |
| 2c | −71.90 | 4a | −89.69 |
| 2d | −74.04 | 4b | −107.95 |
| 2e | −77.85 | 5a | −83.61 |
| 3a | −92.01 | 5b | −94.27 |
| 3b | −90.91 | Sorafenib | −99.55 |
 | ||
| Fig. 2 Binding of sorafenib with VEGFR-2. H-bonds are indicated by dotted lines. | ||
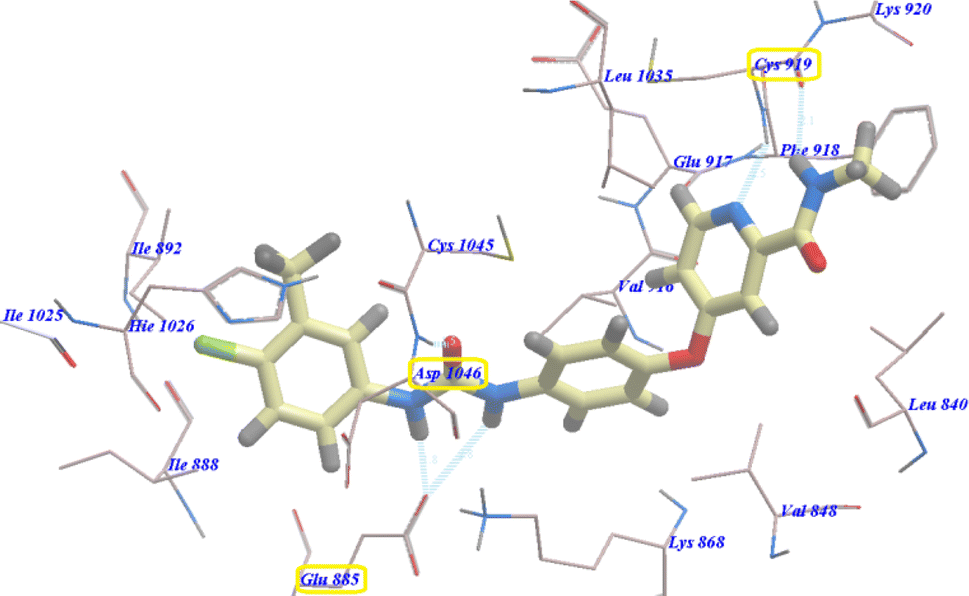
As planned, compound 4b showed virtually binding mode as that of sorafenib. It revealed −107.95 kcal mol−1 and formed 10 H-bonds with the key amino acid Glu885 (2.10 Å), Asp1046 (2.54 Å), Cys919 (2.24 Å and 2.31 Å), Glu917 (2.41 Å) and Lys868 (0.96 Å, 1.07 Å, 1.19 Å, 2.12 Å and 2.63 Å). The 4-chlorophenylphthalazine scaffold occupied the hydrophobic ATP binding groove formed by Leu1035, Cys919, Phe918, Glu917, Val848, Lys920 and Leu840. Moreover, the central phenyl group occupied the linker hydrophobic pocket formed by Asp1046, Cys1045, Thr916, Glu917, Lys868 and Val848. Furthermore, the 4-chlorophenyl tail occupied the new hydrophobic groove formed by Asp1046, Cys1045, His1026, Ile1025, Glu885 and Ile888 (Fig. 3). These interactions may explain the highest anticancer activity of compound 4b.
 | ||
| Fig. 3 Predicted binding mode for 4b with 4ASD. | ||
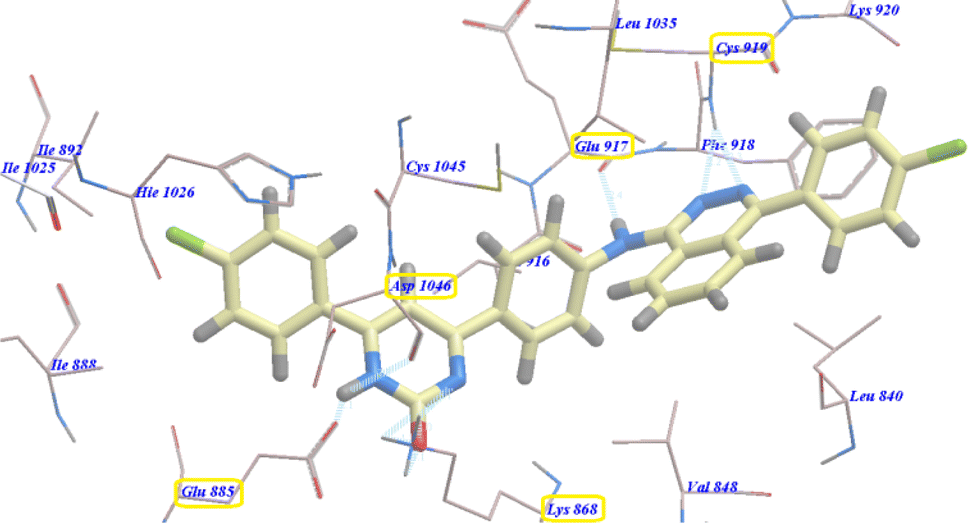
Also compound 3e showed virtually binding mode as that of 4b. It revealed −97.75 kcal mol−1 and formed 7 H-bonds with Glu885 (2.96 Å), Asp1046 (1.01 Å, 1.72 Å and 2.43 Å), Cys919 (1.87 Å and 2.45 Å), and Glu917 (2.61 Å) (Fig. 4).
 | ||
| Fig. 4 Predicted binding mode for 3e with 4ASD. | ||
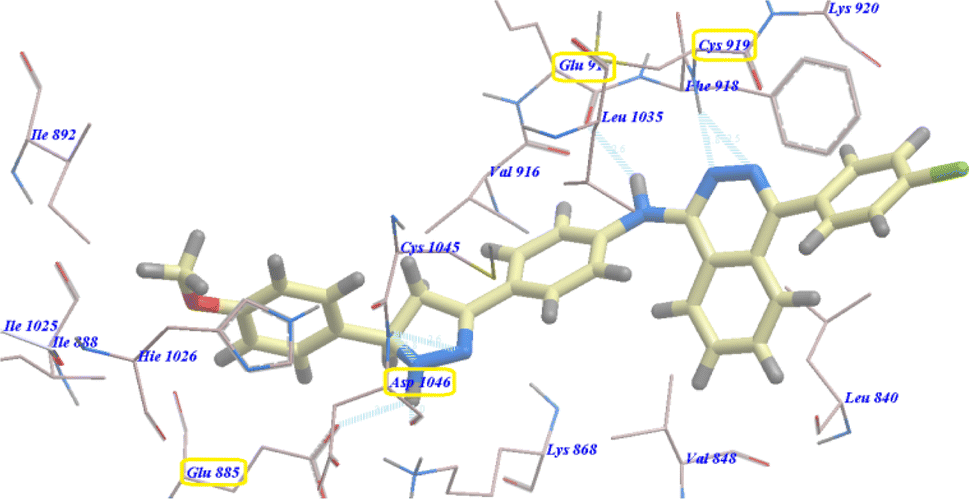
Moreover, compound 5b revealed −94.27 kcal mol−1 and formed 7 with Glu885 (2.98 Å), Asp1046 (2.97 Å), Cys919 (2.57 Å and 2.95 Å), Glu917 (2.78 Å) and Lys868 (1.93 Å and 2.02 Å) (Fig. 5).
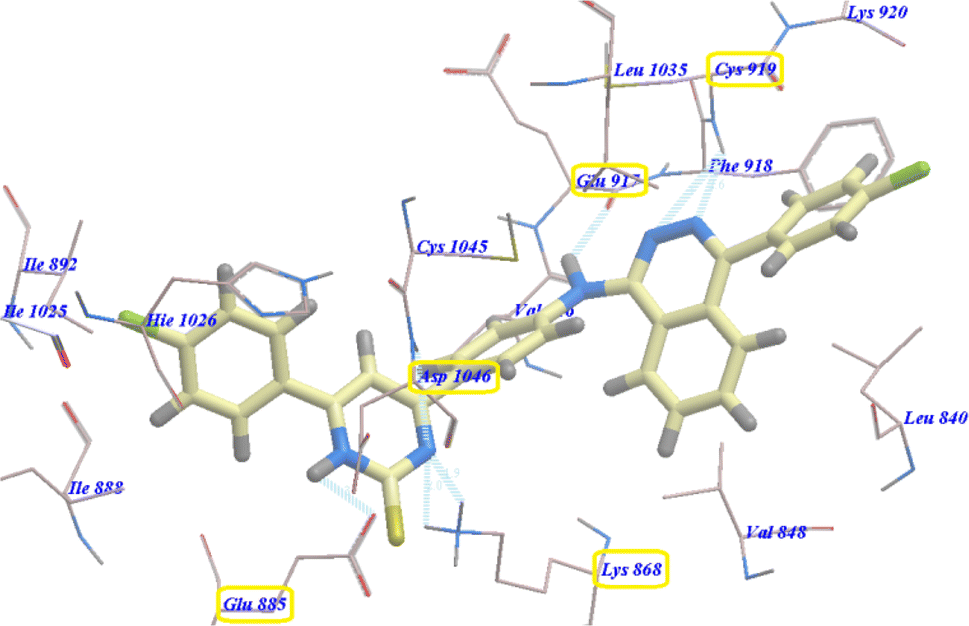 | ||
| Fig. 5 Predicted binding mode for 5b with 4ASD. | ||
The obtained results showed that our derivatives inhibited the ATP binding domain and forming H-bond with Cys919 and extended over the gate area into the adjacent allosteric hydrophobic which approved that our compound were considered as type II inhibitors of VEGFR-2.
2.2. Validation of the accuracy of docking
As cited in literature52 if the RMSD (root mean square deviation) of the best docked conformation is ≤2.0 Å from the bound ligand in the experimental crystal, the used scoring function is successful. Therefore, the docked results were compared to the crystal structure of the bound ligand–protein complex. The obtained success rates were highly excellent as cited in Table 1. The sorafenib ligand was docked in VEGFR-2 receptor (pdb code: 4ASD). The RMSD of the docked sorafenib was 0.64 Å as it seems exactly superimposed on the co-crystallized native bound one (Fig. 6). These results indicated the high accuracy of the docking simulation in comparison with the biological methods.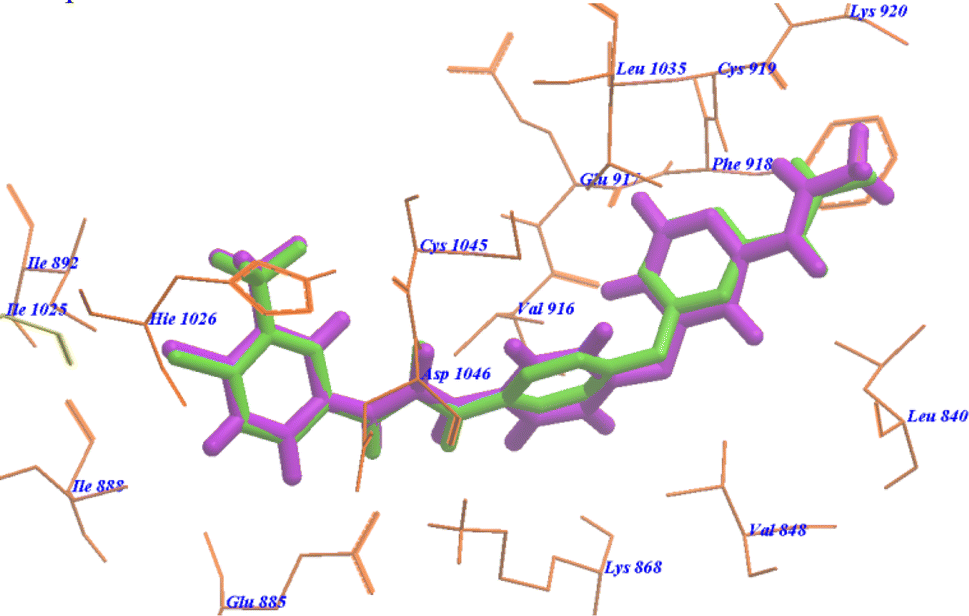 | ||
| Fig. 6 Superimposition of sorafenib on the co-crystallized native bound one with 4ASD. | ||
2.3. Chemistry
The adopted synthetic strategies for the preparation of target compounds (1–5) are depicted in Schemes 1 and 2. The synthesis was initiated by cyclocondensation of 2-(4-chlorobenzoyl)benzoic acid with hydrazine hydrate to afford the corresponding 4-(4-chlorophenyl)phthalazin-1(2H)-one11,53,54 which underwent chlorination by reaction with phosphorous oxychloride11,55 to afford 1-chloro-4-(4-chlorophenyl)phthalazine. The chloro derivative was heated under reflux with the 4-aminoacetophenone to afford the corresponding acetyl derivative 1. On the other hand, the acetyl derivative 1 was condensed with the appropriate benzaldehyde to yield the corresponding chalcone derivatives 2a–e following the reported procedure53,54,56 (Scheme 1). The produced chalcone derivatives 2a–e underwent binucleophilic cyclocondensation reactions with hydrazine hydrate, urea and/or thiourea57,58 to give the corresponding pyrazoline 3a–e, pyrimidin-2(1H)-one 4a,b and/or pyrimidine-2(1H)-thione 5a,b respectively (Scheme 2).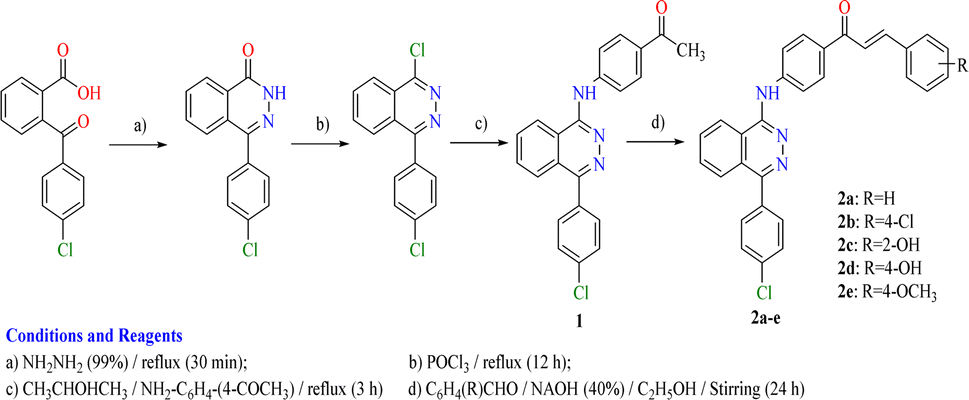 | ||
| Scheme 1 Synthetic route for preparation of the target compounds 1 and 2a–e. | ||
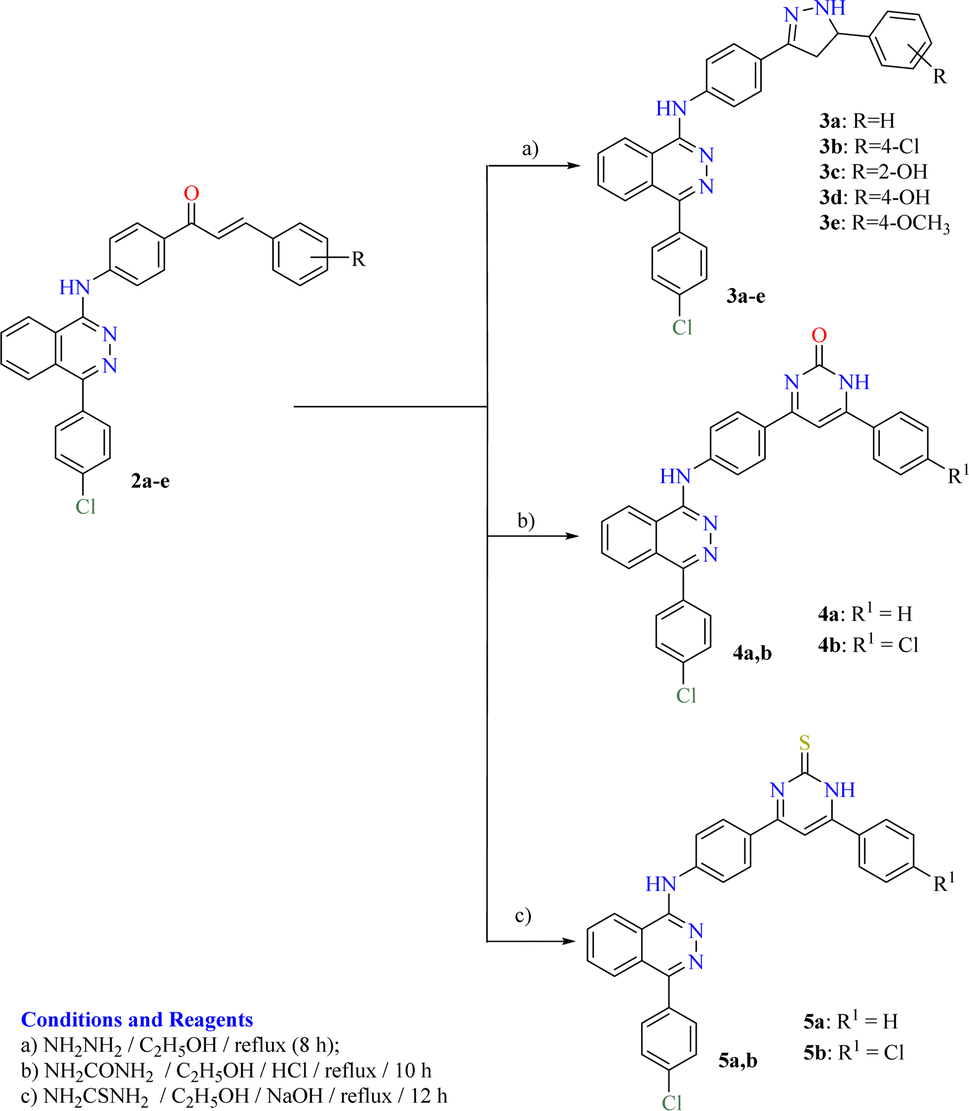 | ||
| Scheme 2 Synthetic route for preparation of the target compounds 3a–e, 4a,b and 5a,b. | ||
2.4. In vitro cytotoxic activity
The anti-proliferative activity of the newly synthesized phthalazine derivatives was examined against MCF-7 and HepG2 cell lines using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay.59 Sorafenib was used as a reference cytotoxic drug. The results were expressed as half maximal inhibitory concentration (IC50) values which represent the compound concentrations required to produce a 50% inhibition of cell growth after 72 h of incubation calculated from the concentration-inhibition response curve and summarized in Table 2. From the obtained results, it was explicated that most of the prepared compounds displayed excellent to moderate growth inhibitory activity against the tested cancer cell lines. In particular, compounds 4b and 3e were found to be the most potent derivatives over all the tested compounds against the two MCF-7 and HepG2 cancer cell lines with IC50 = 0.06, 0.06 μM and 0.08, 0.19 μM respectively.| Comp. | IC50a (μM) | |||
|---|---|---|---|---|
| MCF-7 | HepG2 | VERO | VEGFR-2 | |
| a IC50 values are the mean ± SD of three separate experiments.b NT = not tested. | ||||
| 3a | 1.16 ± 0.01 | 1.27 ± 0.03 | 4.75 ± 0.31 | 1.25 ± 0.05 |
| 3b | 0.83 ± 0.02 | 0.80 ± 01 | 4.25 ± 0.31 | 0.80 ± 0.05 |
| 3c | 0.88 ± 0.03 | 1.25 ± 0.02 | 4.11 ± 0.43 | 0.84 ± 0.05 |
| 3d | 0.74 ± 0.01 | 0.15 ± 01 | 4.35 ± 0.31 | 0.70 ± 0.05 |
| 3e | 0.08 ± 0.01 | 0.19 ± 01 | 3.22 ± 0.31 | 0.12 ± 0.02 |
| 4a | 0.16 ± 01 | 0.29 ± 0.01 | 4.00 ± 0.32 | 0.15 ± 0.03 |
| 4b | 0.06 ± 0.01 | 0.06 ± 0.01 | 3.00 ± 0.31 | 0.09 ± 0.02 |
| 5a | 0.86 ± 0.01 | 0.80 ± 0.02 | 4.12 ± 0.42 | 0.75 ± 0.05 |
| 5b | 0.11 ± 0.01 | 0.15 ± 0.01 | 3.15 ± 0.31 | 0.13 ± 0.03 |
| Sorafenib | 0.05 ± 0.01 | 0.03 ± 0.01 | NTb | 0.022 ± 0.06 |
With respect to the MCF-7 cell line, compounds 4a and 5b displayed very good anticancer activities with IC50 = 0.16 and 0.11 μM respectively. Compounds 3b, 3c, 3d and 5a, with IC50 ranging from 0.74 to 0.88 μM exhibited good cytotoxicity. Compound 3a with IC50 = 1.16 μM displayed moderate cytotoxicity.
With respect to the HepG2 hepatocellular carcinoma cell line, compounds 3d, 4a and 5b displayed very good anticancer activities with IC50 = 0.15, 0.29 and 0.15 μM respectively. Compounds 3b and 5a with the same IC50 = 0.80 μM for each, exhibited good cytotoxicity. Compounds 3a and 3c, with IC50 = 1.27 and 1.25 μM respectively displayed moderate cytotoxicity.
2.5. In vitro VEGFR-2 kinase assay
The nine derivatives 3a–e, 4a, 4b, 5a and 5b were evaluated for their inhibitory activities against VEGFR-2 by using an anti-phosphotyrosine antibody with the Alpha Screen system (PerkinElmer, USA).60 The results were reported as a 50% inhibition concentration value (IC50) calculated from the concentration-inhibition response curve. Results of VEGFR-2 enzyme assay are summarized in Table 2. Sorafenib was used as positive control in this assay. The tested compounds displayed high and low inhibitory activities with IC50 values ranging from 0.09 ± 0.02 to 1.25 ± 0.05 μM. Among them, compound 4b was found to be the most potent derivative that inhibited VEGFR-2 at IC50 value of 0.09 ± 0.02 μM. Compounds 3e, 4a and 5b exhibited good activity with IC50 values = 0.12 ± 0.02, 0.15 ± 0.03 and 0.13 ± 0.03 μM respectively. Finally, the other compounds 3a, 3b, 3c, 3d and 5a exhibited low activities with IC50 values ranging from 0.70 ± 0.05 to 1.25 ± 0.05 μM.2.6. Structure activity relationship (SAR)
The preliminary SAR study has focused on the effect of position, hydrophobic and/or electronic nature of the substituents used in this study. Also, it focused on the effect of the type, length and number of spacers used. Generally, the 4-(4-chlorophenyl)phthalazine scaffold, bearing different 4-substituted anilines joined to the hydrophobic tails moieties through the new spacers; pyrazoline, pyrimidin-2(1H)-one and/or pyrimidin-2(1H)-thione.In molecular docking studies, generally compound 4b with the new pyrimidin-2(1H)-one spacer impart higher VEGFR-2 binding affinity and consequently higher anticancer activity (Fig. 7) than compounds 3e with pyrazoline and compounds 5b with pyrimidin-2(1H)-thione respectively.
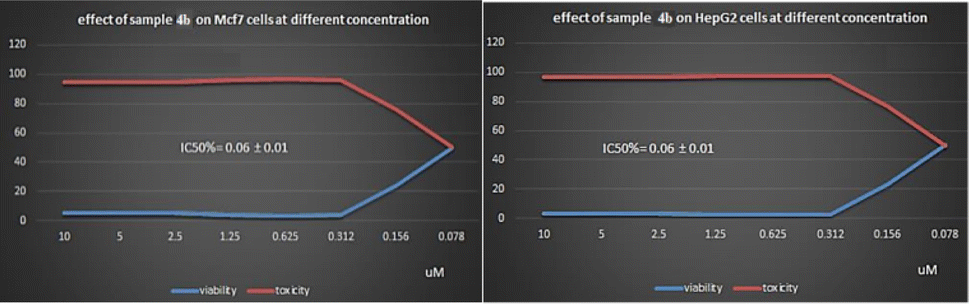 | ||
| Fig. 7 Effect of 4b on MCF-7 and HepG-2 cells at different concentrations. | ||
The data obtained from biological testing highly matched with that obtained from molecular modeling studies. Compound 4b with pyrimidin-2(1H)-one spacer joined to 4-chlorophenyl tail moiety exhibited higher anticancer activities against both HepG2 and MCF-7 cell lines than derivative 3e with pyrazoline spacer and 4-methoxyphenyl tail moiety and derivative 5b with pyrimidin-2(1H)-thione spacer and 4-chlorophenyl tail moiety.
From the structure of the synthesized derivatives and the data shown in Table 2 we can divide these tested compounds into three groups. The first group contains the new pyrazoline spacers as in compounds 3a–e. Generally, in this group the substituents at position-4 of the tail moieties exhibited higher activities than that at position-2 against the two cancer cell lines. Compound 3e with the hydrophobic electron donating +mesomeric (+M) and +inductive (+I) 4-methoxy group showed higher anticancer activities than compound 3b with hydrophobic electron withdrawing (+M) and (−I) 4-chloro one and compound 3c with hydrophilic electron donating 2-OH group against the two cancer cell lines. Also, compound 3e (+M and +I) showed higher anticancer activities than compound 3d with hydrophilic electron donating (+M and −I) 4-OH group against MCF-7 while against HepG2 3d showed higher activities than 3e. Compound 3d with 4-OH substitution exhibited higher anticancer activities than compound 3c with 2-OH substituent which enables us to conclude that position-4 play an important role in anticancer activities. The unsubstituted compound 3a displayed the lowest anticancer activities among this group against both tested cell lines.
The second group 4a,b contains the new pyrimidin-2(1H)-one spacers. Compound 4b with hydrophobic electron withdrawing (+M and −I) 4-chloro group at tail moiety showed higher anticancer activities than the unsubstituted one 4a against the two cancer cell lines.
In the same manner, in the third group compound 5b with pyrimidin-2(1H)-thione spacer and hydrophobic electron withdrawing (+M and −I) 4-chloro group at tail moiety showed higher anticancer activities than the unsubstituted one 5a against the two cancer cell lines.
2.7. Effect on cell cycle progression
To get a better insight regarding the effect of 4b on growth inhibition of cancer cells, its effect on the cell cycle distribution and apoptosis induction were evaluated in MCF-7 cells according to the procedure described by Wang et al.61 MCF-7 cells were treated with 2.3 μM of compound 4b for 24 h. Then, the cells were harvested, stained with propidium iodide, and analyzed for cell distribution during the various phases of the cell cycle. The results (Table 3 and Fig. 8A) showed that compound 4b arrested cell growth in growth 2-mitosis (G2-M) phase; accumulation of cells at that phase became 6.92% after being 13.2 in control cells.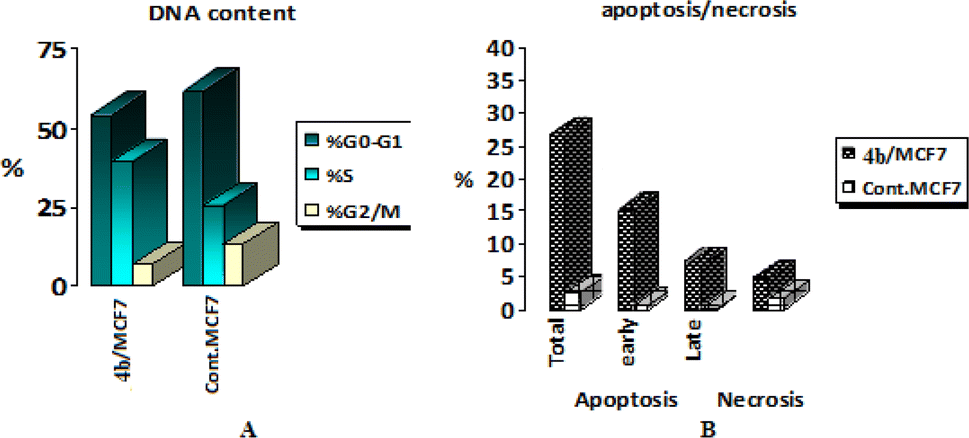 | ||
| Fig. 8 Analysis of (A) cell cycle and (B) apoptosis in MCF-7 cells exposed to compound 4b. | ||
2.8. Induction of apoptosis and necrosis
To explore the mode of induced cell death, induced apoptosis in MCF-7 cells by compound 4b was evaluated using Annexin V and propidium iodide (PI) double staining assay.62 MCF-7 cells were treated with the IC50 of compound 4b for 24 h, harvested, stained with Annexin-V/PI and analyzed for apoptosis using Flowing Software. The obtained results are represented in Table 4 and Fig. 8B. The obtained results showed that compound 4b induced early apoptosis (15.06%) by more than 25 folds over the control (0.59%). On the other hand, the obtained results showed that compound 4b induced necrotic effect (4.66%) twice the effect of the control (1.75%).2.9. Effects on mitochondrial apoptosis pathway (Bcl-2 family) proteins
Mitochondrial apoptotic pathway is chiefly regulated by the members of the B-cell lymphoma-2 (BCL-2) family.63 Among these, Bcl2 and Bcl2 associated X, apoptosis regulator (BAX) finely tune this programmed process. The Bcl2 protein inhibits apoptosis (anti-apoptotic) while Bax stimulates it (pro-apoptotic). Thus, the balance between these two different opposing proteins regulates the cell fate.64,65 Increments in the Bax/Bcl2 ratio trigger the release of mitochondrial cytochrome C into the cytosol which in turn potentiates a cascade of caspases that ultimately leads to activation of caspase 3; the apoptosis executioner.66,67 Accordingly, in the current study, MCF-7 cells were treated with the IC50 of compound 4b and their effect on the expression levels of Bcl-2 and Bax were determined as illustrated in Table 5 and Fig. 9. As shown by the results, compound 4b boosted the level of the pro-apoptotic protein; Bax by approximately 3 folds. Moreover, compound 4b markedly reduced the levels of the anti-apoptotic proteins Bcl-2 by approximately 3 folds compared to the control. A rather more precise value for apoptosis induction is the Bax/Bcl2 ratio as it gives a more accurate estimation of the overall proapoptotic activity of the molecule. Analyzing the results reveals that compound 4b interestingly boosted the Bax/Bcl2 ratio by approximately 8 folds, as compared to the control. Moreover, compound 4b markedly reduced the levels of the anti-apoptotic proteins Bcl-2 by approximately 4 folds compared to the control. A relatively more indicative and precise value for apoptosis induction is the Bax/Bcl2 ratio as it gives a more accurate estimation of the overall proapoptotic activity of the molecule. Collectively, these findings that compound 4b markedly increased Bax level and downregulated Bcl2 level, concomitantly with tremendously augmenting the Bax/Bcl2 ratio proved undoubtedly their pro-apoptotic effect.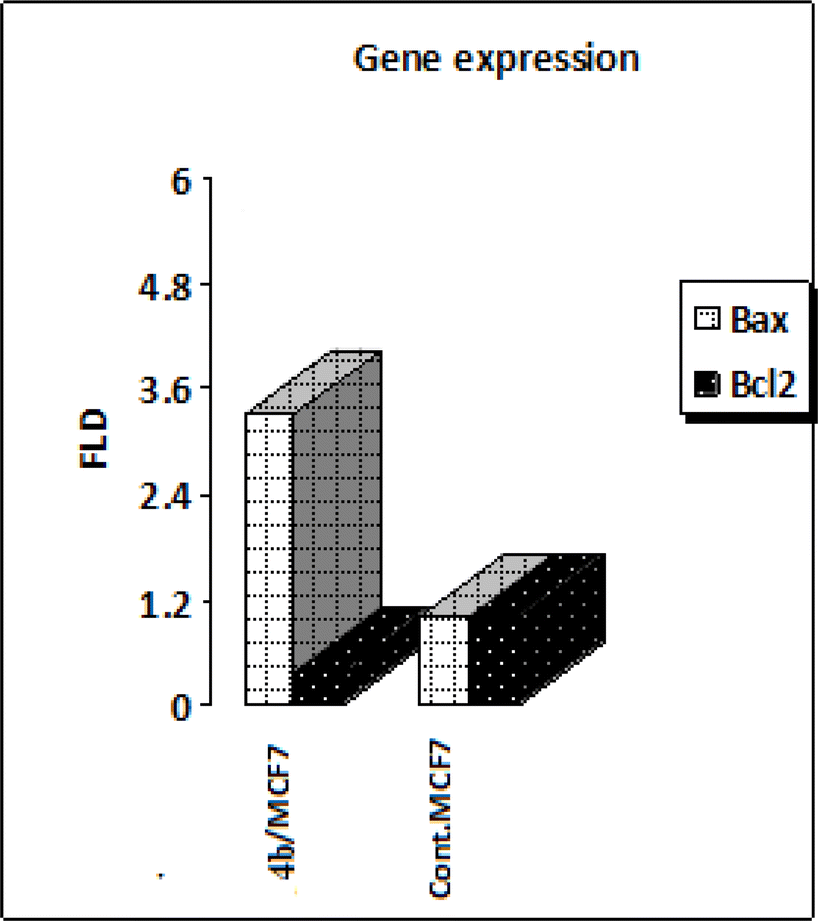 | ||
| Fig. 9 Analysis of the expression levels of Bax and Bacl2 in MCF-7 cells exposed to compound 4b. | ||
It should be noted that all experiments were performed in compliance with relevant laws or guidelines; all experiments followed institutional guidelines; Research Ethics Committee of Al-Azhar University approved the experiments as there is no human subjects were used.
2.10. ADMET profiling study
In silico report of the highly active derivatives 3e, 4b and 5b was conducted for their physicochemical character evaluation and the proposed absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile. It was predicted using pkCSM descriptor algorithm procedures68 and matched to the rule of five described by Lipinski.69 Good absorption properties were expected for the molecules that accomplish at least three rules: (i) hydrogen bond donors ≤5, (ii) hydrogen bond acceptors ≤10, (iii) molecular weight <500, (iv) log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P ≤ 5. In the current work, the standard anticancer agent sorafenib violated logP rule and our compounds violated molecular weight and logP rules.
P ≤ 5. In the current work, the standard anticancer agent sorafenib violated logP rule and our compounds violated molecular weight and logP rules.
As a result of obtaining data from Table 6, we can assume that compounds 3e, 4b and 5b have excellent gastrointestinal tract (GIT) absorption in human (90.574–87.184) which indicates easier to cross different biological membranes.70 So, they may show a significantly high bioavailability through GIT. Concerning central nervous system (CNS) penetrability, our prepared compounds can reach CNS (CNS permeability values −1.384 to −0.863).
| Parameter | 3e | 4b | 5b | Soraf. |
|---|---|---|---|---|
| Physicochemical properties | ||||
| Molecular weight | 506.009 | 536.422 | 552.49 | 464.831 |
| LogP | 7.1411 | 7.7645 | 9.13379 | 5.5497 |
| Rotatable bonds | 6 | 5 | 5 | 5 |
| Acceptors | 6 | 5 | 5 | 4 |
| Donors | 2 | 2 | 2 | 3 |
| Surface area | 219.528 | 227.506 | 233.704 | 185.111 |
|
||||
| Absorption | ||||
| Water solubility | −3.586 | −2.97 | −2.969 | −4.822 |
| Human intest. Absorption | 90.574 | 89.92 | 87.184 | 89.043 |
|
||||
| Distribution | ||||
| Permeability throughout BBB | 0.015 | −1.61 | 1.027 | −1.684 |
| Permeability to CNS | −1.384 | −1.211 | −0.863 | −2.007 |
|
||||
| Metabolism | ||||
| CYP2D6 substrate | No | No | No | No |
| CYP3A4 substrate | Yes | Yes | Yes | Yes |
| Inhibition of CYP1A2 | Yes | No | No | Yes |
| Inhibition of CYP2C19 | Yes | Yes | Yes | Yes |
| Inhibition of CYP2C9 | Yes | Yes | Yes | Yes |
| Inhibition of CYP2D6 | No | No | No | No |
| Inhibition of CYP3A4 | No | No | No | Yes |
|
||||
| Excretion | ||||
| Clearance | 0.107 | −0.026 | −0.067 | −0.219 |
|
||||
| Toxicity | ||||
| Human max. Tolerated dose | 0.523 | 0.431 | 0.431 | 0.549 |
| Acute toxic activity (LD50) | 3.443 | 2.751 | 2.747 | 2.538 |
| Chronic toxic activity (LOAEL) | 1.643 | 1.62 | 1.418 | 1.198 |
| Hepatotoxic effect | Yes | Yes | No | Yes |
It is well known that cytochrome P450 3A4 (CYP3A4), the major drug-metabolizing enzyme, could be inhibited by sorafenib, but our derivatives couldn't. Elimination was expected depending on the total clearance which is a considerable factor in deciding dose intervals. Unlike, sorafenib our compounds exhibited slowly clearance rate, which signifies a long duration of action and extended dosing intervals. Toxicity is the final ADMET profile studied factor. Sorafenib and the novel compounds 3e and 4b shared the drawback of unwanted hepatotoxic actions while our derivative 5b showed no hepatotoxicity. Sorafenib and our compounds demonstrated high maximum tolerated dose. These involve the advantage of the broad therapeutic index of, sorafenib, and our derivatives. The oral acute and chronic toxic doses of the novel compounds 3e, 4b and 5b are higher than that of sorafenib.
3. Conclusion
In this work, we describe the design and synthesis of fifteen new phthalazine derivatives. In the designed compounds, new spacers in the form of fragments with verified VEGFR-2 inhibitory potentials, including α,β-unsaturated ketonic fragment, pyrazole, pyrimidinone and/or pyrimidinthione were introduced at position-4 of the phenyl tail. Also, new tail hydrophobic moieties were attached to these spacers that are expected to increase the hydrophobic interaction with VEGFR-2 enzyme. These structural optimizations have led to the identification of the novel derivatives 4b and 3e as the most promising hit molecules. The designed compounds were evaluated for their anticancer activities against two human tumor cell lines: HepG2 and MCF-7. All the tested compounds showed low to high anticancer activities against the selected cancer cells. Also, molecular docking study was performed to investigate the proposed binding mode of the new compounds with VEGFR-2 active site. The data obtained from biological testing were highly correlated with that obtained from docking study. In particular, compounds 4b and 3e were found to be the most potent derivatives over all the tested compounds against the two MCF-7 and HepG2 cancer cell lines with IC50 = 0.06, 0.06 μM and 0.08, 0.19 μM respectively. Our final compounds 3a–e, 4a,b and 5a,b were evaluated for their cytotoxicity against normal VERO cells. Our compounds exhibited low toxicity concerning normal VERO cells with IC50 = 3.00–4.75 μM. All compounds displayed high selectivity except compounds 3a and 3c against both tested cancer cell lines and compound 5a against MCF-7 which exhibited moderate selectivity. The nine derivatives 3a–e, 4a, 4b, 5a and 5b were evaluated for their inhibitory activities against VEGFR-2. Compound 4b was found to be the most potent derivative that inhibited VEGFR-2 at IC50 value of 0.09 ± 0.02 μM. Compounds 3e, 4a and 5b exhibited good activity with IC50 values = 0.12 ± 0.02, 0.15 ± 0.03 and 0.13 ± 0.03 μM respectively. To get a better insight regarding the effect of 4b on growth inhibition of cancer cells, its effect on the cell cycle distribution and apoptosis induction were evaluated in MCF-7 cells. Compound 4b arrested cell growth in G2-M phase; accumulation of cells at that phase became 6.92% after being 13.2 in control cells. In addition, our derivatives 3e, 4b and 5b showed good in silico calculated ADMET profile in comparing to sorafenib.4. Experimental
All melting points were carried out by open capillary method on a Gallen kamp Melting point apparatus at faculty of pharmacy Al-Azhar University and were uncorrected. The infrared spectra were recorded on pye Unicam SP 1000 IR spectrophotometer at Pharmaceutical analytical Unit, Faculty of Pharmacy, Al-Azhar University using potassium bromide disc technique. Proton magnetic resonance 1H NMR spectra were recorded on a Bruker 400 Megahertz-nuclear magnetic resonance (400 MHZ-NMR) spectrophotometer at Faculty of pharmacy, Mansoura University. Carbon-13 (C13) nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker 100 Megahertz-nuclear magnetic resonance (100 MHZ-NMR) spectrophotometer at Faculty of pharmacy, Mansoura University. The mass spectra were carried out on Direct Probe Controller Inlet part to Single Quadropole mass analyzer in Thermo Scientific Gas chromatography-mass spectrometry (GCMS) model ISQ LT using Thermo X-Calibur software at the Mycology and Biotechnology Regional Center, Al-Azhar University. Elemental analyses (C, H, N) were performed on a carbon hydrogen and nitrogen (CHN) analyzer at Mycology and Biotechnology Regional Center, Al-Azhar University.4.1. Chemistry
4-(4-Chlorophenyl)phthalazin-1(2H)-one, and 1-chloro-4-(4-chlorophenyl)phthalazine were obtained according to the reported procedures.11Yield, 80%; m.p. 176–178 °C; IR νmax (cm−1): 3426 (NH), 3049 (C–H aromatic), 2915 (C–H aliphatic), 1679 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1592 (CN); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 9.06 (d, J = 8.3 Hz, 1H), 8.19 (t, J = 7.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 3H), 8.02 (d, J = 8.5 Hz, 2H), 7.96 (d, J = 8.2 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.70 (d, J = 8.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 197.26, 153.14, 152.61, 142.96, 136.17, 135.41, 133.62, 132.46, 130.51, 129.99, 129.50, 128.55, 127.57, 125.46, 122.74, 121.61, 27.13; MS (m/z): 373.86 (M+, 8.13%), 79.04 (100%, base peak); anal. calcd for C22H16ClN3O (373.84): C, 70.68; H, 4.31; N, 11.24. Found: C, 70.51; H, 4.43; N, 11.45%.
O), 1592 (CN); 1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 1H), 9.06 (d, J = 8.3 Hz, 1H), 8.19 (t, J = 7.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 3H), 8.02 (d, J = 8.5 Hz, 2H), 7.96 (d, J = 8.2 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.70 (d, J = 8.2 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 197.26, 153.14, 152.61, 142.96, 136.17, 135.41, 133.62, 132.46, 130.51, 129.99, 129.50, 128.55, 127.57, 125.46, 122.74, 121.61, 27.13; MS (m/z): 373.86 (M+, 8.13%), 79.04 (100%, base peak); anal. calcd for C22H16ClN3O (373.84): C, 70.68; H, 4.31; N, 11.24. Found: C, 70.51; H, 4.43; N, 11.45%.
4.1.2.1. 1-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}amino]phenyl)-3-phenyl-prop-2-en-1-one (2a). Light yellow powder; yield, 75%; m.p. 245–247 °C; IR νmax (cm−1): 3440 (NH), 2917 (CH aromatic), 2851 (CH aliphatic), 1654 (C
O), 1600 (CN); 1H NMR (300 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.72 (t, J = 7.3 Hz, 1H), 8.22 (s, 2H), 8.11 (d, J = 8.8 Hz, 2H), 7.99 (dq, J = 9.0, 3.3 Hz, 4H), 7.90 (dd, J = 6.8, 3.8 Hz, 2H), 7.78–7.69 (m, 3H), 7.70–7.56 (m, 4H), 7.47 (dd, J = 5.1, 1.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 196.84, 187.72, 152.49, 143.48, 135.84, 134.16, 133.15, 132.52, 132.05, 130.29, 129.81, 129.41, 129.27, 129.05, 128.78, 126.24, 122.59, 119.80, 119.67; anal. calcd for C29H20ClN3O (461.95): C, 75.40; H, 4.36; N, 9.10. Found: C, 75.62; H, 4.51; N, 9.27%.
4.1.2.2. 3-(4-Chlorophenyl)-1-(4-[{4-(4-chlorophenyl)phthalazin-1-yl}amino]phenyl)prop-2-en-1-one (2b). Light yellow powder, yield, 82%; m.p. 244–246 °C; IR νmax (cm−1): 3443 (NH), 2918 (CH aromatic), 2851 (CH aliphatic), 1604 (C
O), 1447 (CN); 1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 1H), 8.73 (d, J = 8.1 Hz, 1H), 8.23 (s, 2H), 8.09 (t, J = 7.4 Hz, 2H), 8.03 (d, J = 10.6 Hz, 2H), 8.00–7.96 (m, 2H), 7.93 (d, J = 10.1 Hz, 2H), 7.77–7.74 (m, 2H), 7.70 (d, J = 10.9 Hz, 1H), 7.68–7.63 (m, 2H), 7.55 (d, J = 8.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 196.84, 187.50, 141.99, 135.83, 135.33, 134.36, 134.15, 133.14, 132.52, 132.50, 132.06, 132.04, 130.98, 130.84, 130.32, 129.79, 129.43, 129.04, 126.21, 123.33, 119.67; anal. calcd for C29H19Cl2N3O (496.39): C, 70.17; H, 3.86; N, 8.47. Found: C, 70.39; H, 4.03; N, 8.70%.
4.1.2.3. 1-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}amino]phenyl)-3-(2-hydroxyphenyl)prop-2-en-1-one (2c). Reddish yellow powder; yield, 77%; m.p. 246–248 °C; IR νmax (cm−1): 3419 (OH), 3261 (NH), 3057 (CH aromatic), 2959, 2918 (CH aliphatic), 1674 (C
O), 1601 (CN); 1H NMR (400 MHz, DMSO-d6) δ 9.75 (s, 1H), 9.39 (s, 1H), 8.73 (d, 1H), 8.16–7.65 (m, 17H); 13C NMR (101 MHz, DMSO-d6) δ 196.84, 157.58, 153.78, 152.19, 148.83, 145.93, 135.84, 134.15, 133.13, 132.50, 132.04, 130.85, 129.81, 129.04, 126.23, 126.17, 123.40, 119.67, 119.32, 116.07; anal. calcd for C29H20ClN3O2 (477.95): C, 72.88; H, 4.22; N, 8.79. Found: C, 73.04; H, 4.29; N, 9.02%.
4.1.2.4. 1-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}amino]phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (2d). Yellow powder; yield, 78%; m.p. 247–249 °C; IR νmax (cm−1): 3417 (OH), 3263 (NH), 2918 (CH aromatic), 2851 (CH aliphatic), 1674 (C
O), 1601 (CN); 1H NMR (400 MHz, DMSO-d6) δ 9.78 (s, 1H), 9.24 (s, 1H), 8.73 (d, 1H), 8.16–7.64 (m, 17H); 13C NMR (101 MHz, DMSO-d6) δ 196.83, 186.67, 153.78, 152.14, 148.89, 146.09, 135.83, 134.14, 133.10, 132.48, 132.03, 130.83, 129.79, 129.03, 126.20, 126.16, 123.40, 119.67, 119.36, 115.87. Anal. calcd for C29H20ClN3O2 (477.95): C, 72.88; H, 4.22; N, 8.79. Found: C, 73.07; H, 4.31; N, 9.05%.
4.1.2.5. 1-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}amino]phenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (2e). Orange powder; yield, 75%; m.p. 250–252 °C; IR νmax (cm−1): 3421 (NH), 2919 (CH aromatic), 2851 (CH aliphatic), 1602 (C
O), 1447 (CN); 1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 1H), 8.75 (d, 1H), 8.22–8.07 (m, 4H), 8.01–7.86 (m, 4H), 7.76–7.65 (m, 7H), 7.03 (d, 2H); 13C NMR (101 MHz, DMSO-d6) δ 196.83, 187.64, 161.67, 145.88, 143.47, 135.84, 134.16, 133.15, 132.52, 132.05, 131.14, 130.85, 130.12, 129.81, 129.05, 128.00, 126.24, 126.18, 123.39, 120.05, 119.78, 119.65, 114.88, 55.86; anal. calcd for C30H22ClN3O2 (491.98): C, 73.24; H, 4.51; N, 8.54. Found: C, 73.45; H, 4.63; N, 8.81%.
4.1.3.1. 4-(4-Chlorophenyl)-N-(4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenyl)phthalazin-1-amine (3a). Yellow powder; yield, 70%; m.p. 271–273 °C; IR νmax (cm−1): 3424 (NH), 2919 (CH aromatic), 2851 (CH aliphatic), 1600 (C
N); 1H NMR (300 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.75 (s, 1H), 8.07–7.85 (m, 7H), 7.74–7.58 (m, 7H), 7.49–7.13 (m, 3H), 6.21 (m, 2H), 4.84 (m, 1H); MS (m/z): 475.29 (M+, 12.81%), 417.60 (43.00%), 395.22 (57.82%), 368.38 (100%, base peak); anal. calcd for C29H22ClN5 (475.16): C, 73.18; H, 4.66; N, 14.71. Found: C, 73.40; H, 4.83; N, 14.85%.
4.1.3.2. 4-(4-Chlorophenyl)-N-(4-(5-(4-chlorophenyl)-4,5-dihydro-1H-pyrazol-3-yl)phenyl)phthalazin-1-amine (3b). Yellow powder; yield, 70%; m.p. 270–272 °C; IR νmax (cm−1): 3424 (NH), 2919 (CH aromatic), 2852 (CH aliphatic), 1597, 1548 (C
N); 1H NMR (300 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.74 (s, 1H), 8.05–7.90 (m, 8H), 7.73–7.71 (m, 2H), 7.68–7.65 (m, 4H), 7.44–7.43 (m, 2H), 6.26 (m, 2H), 4.67 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 158.67, 152.36, 145.86, 142.92, 136.10, 134.40, 132.53, 132.24, 132.01, 129.83, 129.05, 129.03, 127.89, 125.36, 123.57, 123.26, 120.87, 118.93, 113.91, 109.82, 99.73, 43.54, 36.38; MS (m/z): 509.47 (M+, 4.00%), 132.12 (46.22%), 77.09 (100%, base peak), 43.09 (97.05%); anal. calcd for C29H21Cl2N5 (509.12): C, 68.24; H, 4.15; N, 13.72. Found: C, 68.43; H, 4.37; N, 13.99%.
4.1.3.3. 2-(3-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]-4,5-dihydro-1H-pyrazol-5-yl)phenol (3c). Reddish yellow powder; yield, 69%; m.p. 266–268 °C; IR νmax (cm−1): 3753 (OH), 3422 (NH), 2918 (CH aromatic), 2852 (CH aliphatic), 1643, 1620 (C
N); 1H NMR (300 MHz, DMSO-d6) δ 9.76 (s, 1H), 9.38 (s, 1H), 8.72 (s, 1H), 8.18–7.64 (m, 16H), 6.14 (m, 2H), 4.75 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 196.85, 153.84, 152.77, 152.35, 142.96, 140.13, 136.09, 134.39, 134.15, 133.14, 132.80, 132.23, 131.98, 130.86, 129.81, 129.04, 128.99, 125.36, 120.87, 119.65, 118.92, 43.18, 31.19; MS (m/z): 491.94 (M+, 12.81%), 165.80 (49.57%), 129.96 (53.75%), 103.84 (100%, base peak), 76.17 (69.04%); anal. calcd for C29H22ClN5O (491.15): C, 70.80; H, 4.51; N, 14.24. Found: C, 71.02; H, 4.67; N, 14.51%.
4.1.3.4. 4-(3-[4-{(4-[4-Chlorophenyl]phthalazin-1-yl)amino}phenyl]-4,5-dihydro-1H-pyrazol-5-yl)phenol (3d). Orange powder; yield, 61%; m.p. 265–267 °C; IR νmax (cm−1): 3439 (OH), 3303 (NH), 3081 (CH aromatic), 2962 (CH aliphatic), 1599, 1574 (C
N); 1H NMR (300 MHz, DMSO-d6) δ 9.76 (s, 1H), 9.38 (s, 1H), 8.72 (s, 1H), 8.18–7.65 (m, 16H), 6.26 (m, 2H), 4.74 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 196.85, 134.40, 134.16, 133.94, 133.16, 132.53, 132.25, 132.05, 131.99, 130.87, 129.82, 129.05, 129.00, 126.25, 126.18, 126.07, 125.36, 123.38, 120.87, 119.64, 45.00, 31.19; MS (m/z): 491.96 (M+, 0.58%), 127.06 (60.97%), 115.07 (98.22%), 91.10 (97.78%), 43.08 (100%, base peak); anal. calcd for C29H22ClN5O (491.15): C, 70.80; H, 4.51; N, 14.24. Found: C, 68.69; H, 4.68; N, 14.45%.
4.1.3.5. 4-(4-Chlorophenyl)-N-(4-[5-{4-methoxyphenyl}-4,5-dihydro-1H-pyrazol-3-yl]phenyl)phthalazin-1-amine (3e). Yellow powder; yield, 67%; m.p. 353–355 °C; IR νmax (cm−1): 3436 (NH), 2918 (CH aromatic), 2851 (CH aliphatic), 1601, 1547 (C
N); 13C NMR (101 MHz, DMSO-d6) δ 197.03, 153.41, 134.92, 134.22, 134.21, 133.99, 133.65, 132.27, 132.19, 131.89, 131.71, 129.92, 129.30, 129.24, 129.13, 128.33, 127.15, 126.90, 126.67, 123.96, 120.75, 43.89, 31.19, 26.99; MS (m/z): 505.71 (M+, 13.43%), 476.19 (65.66%), 413.94 (100%, base peak), 312.93 (73.97%), 295.66 (67.05%); anal. calcd for C30H24ClN5O (505.17): C, 71.21; H, 4.78; N, 13.84. Found: C, 71.38; H, 5.01; N, 14.02%.
4.1.4.1. 4-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}amino]phenyl)-6-phenylpyrimidin-2(1H)-one (4a). Yellow powder; yield, 65%; m.p. 265–267 °C; IR νmax (cm−1): 3437 (NH), 3070 (CH aromatic), 2917 (CH aliphatic), 1652 (C
O), 1602 (CN); 1H NMR (300 MHz, DMSO-d6) δ 9.75 (s, 1H), 8.72 (s, 1H), 8.26–7.48 (m, 18H); 13C NMR (101 MHz, DMSO-d6) δ 196.85, 187.74, 143.52, 135.67, 135.37, 134.23, 133.24, 132.63, 132.07, 131.71, 130.97, 130.30, 129.82, 129.55, 129.41, 129.28, 129.13, 129.07, 126.33, 126.22, 123.41, 122.57, 119.75; MS (m/z): 501.84 (M+, 48.68%), 493.76 (52.76%), 187.40 (100%, base peak), 149.91 (74.31%), 130.84 (97.80%); anal. calcd for C30H20ClN5O (501.97): C, 71.78; H, 4.02; N, 13.95. Found: C, 72.04; H, 4.13; N, 14.17%.
4.1.4.2. 6-(4-Chlorophenyl)-4-(4-[{4-(4-chlorophenyl)phthalazin-1-yl}amino]phenyl)pyrimidin-2(1H)-one (4b). Yellow powder; yield, 56%; m.p. 270–272 °C; IR νmax (cm−1): 3429 (NH), 3067 (CH aromatic), 2917 (CH aliphatic), 1657 (C
O), 1598 (CN); 1H NMR (300 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.95 (s, 1H), 8.30–7.55 (m, 17H); 13C NMR (101 MHz, DMSO-d6) δ 187.81, 153.29, 152.62, 142.39, 135.45, 135.20, 134.29, 134.22, 134.06, 132.27, 131.71, 131.05, 130.71, 130.44, 129.94, 129.45, 129.30, 129.14, 127.45, 126.88, 124.26, 123.23, 121.22, 113.29; MS (m/z): 536.28 (M+, 26.02%), 533.62 (55.08%), 367.24 (100%, base peak), 296.64 (79.61%), 232.05 (93.84%); anal. calcd for C30H19Cl2N5O (536.42): C, 67.17; H, 3.57; N, 13.06. Found: C, 67.41; H, 3.62; N, 13.29%.
4.1.5.1. 4-(4-[{4-(4-Chlorophenyl)phthalazin-1-yl}amino]phenyl)-6-phenylpyrimidin-2(1H)-thione (5a). Yellow powder; yield, 60%; m.p. 251–253 °C; IR νmax (cm−1): 3423 (NH), 2917 (CH aromatic), 2850 (CH aliphatic), 1597, 1543 (C
N), 1402 (CS); 1H NMR (300 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.83 (s, 1H), 8.19–7.69 (m, 18H); 13C NMR (101 MHz, DMSO-d6) δ 196.98, 135.06, 134.82, 134.20, 133.88, 133.51, 132.95, 132.17, 131.80, 131.76, 130.19, 129.91, 129.31, 129.21, 127.04, 125.65, 126.62, 123.89, 123.87, 120.62, 120.05; MS (m/z): 518.06 (M+, 22.25%), 471.49 (88.13%), 451.77 (73.13%), 448.63 (100%, base peak), 152.60 (98.44%); anal. calcd for C30H20ClN5S (518.04): C, 69.56; H, 3.89; N, 13.52. Found: C, 69.78; H, 4.06; N, 13.74%.
4.1.5.2. 6-(4-Chlorophenyl)-4-(4-[{4-(4-chlorophenyl)phthalazin-1-yl}amino]phenyl)pyrimidin-2(1H)-thione (5b). Light yellow powder; yield, 71%; m.p. 260–262 °C; IR νmax (cm−1): 3424 (NH), 3065 (CH aromatic), 2918 (CH aliphatic), 1596, 1547 (C
N), 1402 (CS); 1H NMR (300 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.82 (s, 1H), 8.36–8.11 (m, 4H), 8.06–8.02 (m, 4H), 7.96–7.90 (m, 2H), 7.76–7.68 (m, 6H), 7.63 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 196.94, 159.72, 152.39, 145.84, 145.15, 134.69, 134.41, 134.29, 134.20, 133.73, 133.30, 132.15, 131.70, 131.58, 129.87, 129.28, 129.17, 129.13, 128.33, 126.87, 126.62, 126.52, 123.82, 120.43; MS (m/z): 552.91 (M+, 6.08%), 531.17 (58.53%), 348.27 (100%, base peak), 121.10 (99.95%); anal. calcd for C30H19Cl2N5S (552.48): C, 65.22; H, 3.47; N, 12.68. Found: C, 65.43; H, 3.65; N, 12.90%.
4.2. Docking studies
VEGFR-2 (PDB ID 4ASD)51 was used by Molsoft program to carry out docking studies.4.3. Biological testing
4.4. ADMET profile
In silico ADMET profile of the highly active derivatives 3e, 4b and 5b was predicted using pkCSM descriptor algorithm procedures.68Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no interest to declare.Acknowledgements
The authors express their gratitude and admiration Dr Mohamed Morsy Faculty of Science, Al-Azhar University for supporting the pharmacological part.References
- F. S. G. A. Cuthbert, S. Homer-Vanniasinkam, S. P. Muench, S. Ponnambalam and M. A. Harrison, Biomolecules, 2020, 10(12), 1673, DOI:10.3390/biom10121673.
- X. Ye, J. F. Gaucher, M. Vidal and S. Broussy, Molecules, 2021, 26(22), 6759, DOI:10.3390/molecules26226759.
- B. Wang, S. Hu, Y. Teng, J. Chen, H. Wang, Y. Xu, K. Wang, J. Xu, Y. Cheng and X. Gao, Signal Transduction Targeted Ther., 2024, 9, 200, DOI:10.1038/s41392-024-01889-y.
- L. Peng, G. Sferruzza, L. Yang, L. Zhou and S. Chen, Cell. Mol. Immunol., 2024 DOI:10.1038/s41423-024-01207-0.
- C. Pottier, M. Fresnais, M. Gilon, G. Jérusalem, R. Longuespée and N. E. Sounni, Cancers, 2020, 12(3), 731, DOI:10.3390/cancers12030731.
- M. Li, A. Ur Rehman, Y. Liu, K. Chen and S. Lu, Adv. Protein Chem. Struct. Biol., 2021, 124, 87–119, DOI:10.1016/bs.apcsb.2020.09.005.
- I. Abdelbaky, H. Tayara and K. T. Chong, Sci. Rep., 2021, 11, 706, DOI:10.1038/s41598-020-80758-4.
- M. A. Zeidan, A. S. Mostafa, R. M. Gomaa, L. A. Abou-zeid, M. El-Mesery, M. A.-A. El-Sayed and K. B. Selim, Eur. J. Med. Chem., 2019, 168, 315, DOI:10.1016/j.ejmech.2019.02.050.
- A.-G. A. El-Helby, H. Sakr, I. H. Eissa, H. Abulkhair, A. A. Al-Karmalawy and K. El-Adl, Arch. Pharm., 2019, 352, 1900113, DOI:10.1002/ardp.201900113.
- A. E. Abdallah, R. R. Mabrouk, M. R. Elnagar, A. M. Farrag, M. H. Kalaba, M. H. Sharaf, E. M. El-Fakharany, D. A. Bakhotmah, E. B. Elkaeed and M. M. S. Al Ward, Drug Des., Dev. Ther., 2022, 16, 587–606, DOI:10.2147/DDDT.S344750.
- K. El-Adl, M. K. Ibrahim, F. Khedr, H. S. Abulkhair and I. H. Eissa, Arch. Pharm., 2021, 354(3), e202000219, DOI:10.1002/ardp.202000219.
- M. Payton, H.-K. Cheung, M. S. S. Ninniri, C. Marinaccio, W. C. Wayne, K. Hanestad, J. D. Crispino, G. Juan and A. Coxon, Mol. Cancer Ther., 2018, 17, 2575, DOI:10.1158/1535-7163.MCT-18-0186.
- S. Rampogu, A. Baek, A. Zeb and K. W. Lee, BMC Cancer, 2018, 18, 264, DOI:10.1186/s12885-018-4050-1.
- S. M. Mennen, M. L. Mak-Jurkauskas, M. M. Bio, L. S. Hollis, K. A. Nadeau, A. M. Clausen and K. B. Hansen, Org. Process Res. Dev., 2015, 19, 884, DOI:10.1021/acs.oprd.5b00135.
- A.-G. A. El-Helby, R. R. A. Ayyad, H. Sakr, K. El-Adl, M. M. Ali and F. Khedr, Arch. Pharm., 2017, 350(12), 1–16, DOI:10.1002/ardp.201700240.
- K. El-Adl, A.-G. A. El-Helby, R. R. A. Ayyad, H. Sakr, M. M. Ali and F. Khedr, Anticancer Agents Med. Chem., 2018, 18, 1184, DOI:10.2174/1871520618666180412123833.
- H. S. Abulkhair, A. Turky, A. Ghiaty, H. E. A. Ahmed and A. H. Bayoumi, Bioorg. Chem., 2020, 100, 103899, DOI:10.1016/j.bioorg.2020.103899.
- A. Turky, A. H. Bayoumi, A. Ghiaty, A. S. El-Azab, A. A.-M. Abdel-Aziz and H. S. Abulkhair, Bioorg. Chem., 2020, 101, 104019, DOI:10.1016/j.bioorg.2020.104019.
- H. G. Ezzat, A. H. Bayoumi, F. F. Sherbiny, A. M. El-Morsy, A. Ghiaty, M. Alswah and H. S. Abulkhair, Mol. Diversity, 2021, 25, 291, DOI:10.1007/s11030-020-10070-w.
- S. Elmeligie, A. M. Aboul-Magd, D. S. Lasheen, T. M. Ibrahim, T. M. Abdelghany, S. M. Khojah and K. A. M. Abouzid, J. Enzyme Inhib. Med. Chem., 2019, 34, 1347, DOI:10.1080/14756366.2019.1642883.
- S. Zaib and I. Khan, Bioorg. Chem., 2020, 105, 104425, DOI:10.1016/j.bioorg.2020.104425.
- S. Elmeligie, A. M. Aboul-Magd, D. S. Lasheen, T. M. Ibrahim, T. M. Abdelghany, S. M. Khojah and K. A. M. Abouzid, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 1347–1367, DOI:10.1080/14756366.2019.1642883.y.
- P. Ravula, H. B. Vamaraju, M. Paturi and J. N. G. N. Sharath Chandra, Arch. Pharm., 2018, 351, 1700234, DOI:10.1002/ardp.201700234.
- N. M. Saleh, M. G. El-Gazzar, H. M. Aly and R. A. Othman, Front. Chem., 2020, 7, 917, DOI:10.3389/fchem.2019.00917.
- W. Sun, S. Hu, S. Fang and H. Yan, Bioorg. Chem., 2018, 78, 393, DOI:10.1016/j.bioorg.2018.04.005.
- J. Li, W. Gu, X. Bi, H. Li, C. Liao, C. Liu, W. Huang and H. Qian, Bioorg. Med. Chem., 2017, 25, 6674, DOI:10.1016/j.bmc.2017.11.010.
- V. Asati, A. Anant, P. Patel, K. Kaur and G. D. Gupta, Eur. J. Med. Chem., 2021, 225, 113781, DOI:10.1016/j.ejmech.2021.113781.
- E. B. Elkaeed, R. G. Yousef, M. M. Khalifa, A. Ibrahim, A. B. M. Mehany, I. M. M. Gobaara, B. A. Alsfouk, W. M. Eldehna, A. M. Metwaly, I. H. Eissa and M. A. El-Zahabi, Molecules, 2022, 27(19), 6203, DOI:10.3390/molecules27196203.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762, DOI:10.1021/acs.chemrev.7b00020.
- M. A. Abdelgawad, K. El-Adl, S. S. El-Hddad, M. M. Elhady, N. M. Saleh, M. M. Khalifa, F. Khedr, M. Alswah, A. A. Nayl and M. M. Ghoneim, Pharmaceuticals, 2022, 15, 226, DOI:10.3390/ph15020226.
- F. Khedr, M.-K. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Arch. Pharm., 2021, 354(11), e2100201, DOI:10.1002/ardp.202100201.
- A. K. B. Aljohani, K. El-Adl, B. Almohaywi, O. M. Alatawi, M. Alsulaimany, A. El-morsy, S. A. Almadani, H. Y. Alharbi, M. S. Aljohani, F. A. M. Abdulhaleem, H. E. M. Osman and S. Mohamady, RSC Adv., 2024, 14, 7964–7980, 10.1039/D4RA00502C.
- M. M. Ghorab, A. M. Soliman, K. El-Adl and N. S. Hanafy, Bioorg. Chem., 2023, 140, 106791, DOI:10.1016/j.bioorg.2023.106791.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem, S. I. Eissa, H. Elkady and I. H. Eissa, Bioorg. Chem., 2020, 104, 104218, DOI:10.1016/j.bioorg.2020.104218.
- N. M. Saleh, M. S. A. El-Gaby, K. El-Adl and N. E. A. Abd El-Sattar, Bioorg. Chem., 2020, 104, 104350, DOI:10.1016/j.bioorg.2020.104350.
- A. E. Abdallah, I. H. Eissa, A. B. M. Mehany, H. Sakr, A. Atwa, K. El-Adl and M. A. El-Zahabi, J. Mol. Struct., 2023, 1281, 135164, DOI:10.1016/j.molstruc.2023.135164.
- N. S. Hanafy, N. A. A. M. Aziz, S. S. A. El-Hddad, M. A. Abdelgawad, M. M. Ghoneim, A. F. Dawood, S. Mohamady, K. El-Adl and S. Ahmed, Arch. Pharm., 2023, 356, e2300137, DOI:10.1002/ardp.202300137.
- D. Adel, K. El-Adl, T. Nasr, T. M. Sakr and W. Zaghary, J. Mol. Struct., 2023, 1291, 136047, DOI:10.1016/j.molstruc.2023.136047.
- H. Elkady, K. El-Adl, H. Sakr, A. S. Abdelraheem, S. I. Eissa and M. A. El-Zahabi, Arch. Pharm., 2023, 356(9), e2300097, DOI:10.1002/ardp.202300097.
- M. A. El-Zahabi, H. Elkady, H. Sakr, A. S. Abdelraheem, S. I. Eissa and K. El-Adl, J. Biomol. Struct. Dyn., 2023, 41(24), 15106–15123, DOI:10.1080/07391102.2023.2187217.
- M. Alsulaimany, K. El-Adl, A. K. B. Aljohani, H. Y. Alharbi, O. M. Alatawi, M. S. Aljohani, A. El-Morsy, S. A. Almadani, A. A. Alsimaree, S. A. Salama, D. E. Keshek and A. A. Mohamed, RSC Adv., 2023, 13(51), 36301–36321, 10.1039/d3ra07700d.
- K. E. Anwer, S. S. A. El-Hddad, N. E. A. Abd El-Sattar, A. M. El-Morsy, F. Khedr, S. Mohamady, D. E. G. Keshek, S. A. Salama, K. El-Adl and N. S. Hanafy, RSC Adv., 2023, 13, 35321–35338 RSC , https://api.semanticscholar.org/CorpusID:265657522.
- M. S. A. El-Gaby, M. A. M. Abdel Reheim, Z. S. M. Akrim, B. H. Naguib, N. M. Saleh, A. A. A. M. El-Adasy, K. El-Adl and S. Mohamady, Drug Dev. Res., 2024, 85(1), e22143, DOI:10.1002/ddr.22143.
- A. A. Mohamed, S. S. A. El-Hddad, A. K. B. Aljohani, F. Khedr, O. M. Alatawi, D. E. Keshek, S. Ahmed, M. Alsulaimany, S. A. Almadani, K. El-Adl and N. S. Hanafy, Bioorg. Chem., 2024, 143, 107062, DOI:10.1016/j.bioorg.2023.107062.
- K. El-Adl, H. Sakr, S. S. A. El-Hddad, A. A. El-Helby, M. Nasser and H. S. Abulkhair, Arch. Pharm., 2021, 354(7), e2000491, DOI:10.1002/ardp.202000491.
- K. El-Adl, H. Sakr, M. Nasser, M. Alswah and F. M. A. Shoman, Arch. Pharm., 2020, 353, e2000079, DOI:10.1002/ardp.202000079.
- K. El-Adl, A.-G. A. El-Helby, H. Sakr and S. S. A. El-Hddad, Arch. Pharm., 2020, 353, e2000068, DOI:10.1002/ardp.202000068.
- K. El-Adl, A.-G. A. El-Helby, H. Sakr, R. R. Ayyad, H. A. Mahdy, M. Nasser, H. S. Abulkhair and S. S. A. El-Hddad, Arch. Pharm., 2021, 354(2), e2000279, DOI:10.1002/ardp.202000279.
- K. El-Adl, A. A. El-Helby, R. R. Ayyad, H. A. Mahdy, M. M. Khalifa, H. A. Elnagar, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly and I. H. Eissa, Bioorg. Med. Chem., 2020, 29, 115872, DOI:10.1016/j.bmc.2020.115872.
- N. E. A. Abd El-Sattar, K. El-Adl, M. A. El-Hashash, S. A. Salama and M. M. Elhady, Bioorg. Chem., 2021, 115, 105186, DOI:10.1016/j.bioorg.2021.105186.
- V. A. Machado, D. Peixoto, R. Costa, H. J. C. Froufe, R. C. Calhelha, R. M. V. Abreu, I. C. F. R. Ferreira, R. Soares and M.-J. R. P. Queiroz, Bioorg. Med. Chem., 2015, 23, 6497, DOI:10.1016/j.bmc.2015.08.010.
- K. El-Adl, A.-G. A. El-Helby, H. Sakr and A. Elwan, Bioorg. Chem., 2020, 105, 104399, DOI:10.1016/j.bioorg.2020.104399.
- M. McTigue, B. W. Murray, J. H. Chen, Y. Deng, J. Solowiej and R. S. Kania, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(45), 18281–18289, DOI:10.1073/pnas.1207759109.
- A.-G. A. El-Helby, R. R. A. Ayyad, M. F. Zayed, H. S. Abulkhair, H. Elkady and K. El-Adl, Arch. Pharm., 2019, 352, 1800387, DOI:10.1002/ardp.201800387.
- M. H. El-Shershaby, K. M. El-Gamal, A. H. Bayoumi, K. El-Adl, M. Alswah, H. E. A. Ahmed, A. A. Al-Karmalamy and H. S. Abulkhair, New J. Chem., 2021, 45, 13986–14004, 10.1039/D1NJ02838C.
- A. A. El-Helby, H. Sakr, R. R. A. Ayyad, K. El-Adl, M. M. Ali and F. Khedr, Anticancer Agents Med. Chem., 2018, 18(8), 1184–1196, DOI:10.2174/1871520618666180412123833.
- M. K. Ibrahim, K. El-Adl and A. A. Al-Karmalawy, Bull. Fac. Pharm. (Cairo Univ.), 2015, 53, 101, DOI:10.1016/j.bfopcu.2015.05.001.
- K. M. El-Gamal, M. S. Hagrs and H. S. Abulkhair, Bull. Fac. Pharm. (Cairo Univ.), 2016, 54, 263, DOI:10.1016/j.bfopcu.2016.08.002.
- M. Ghasemi, T. Turnbull, S. Sebastian and I. Kempson, Int. J. Mol. Sci., 2021, 22(23), 12827, DOI:10.3390/ijms222312827.
- H. H. Bayoumi, M.-K. Ibrahim, M. A. Dahab, F. Khedra and K. El-Adl, RSC Adv., 2024, 14, 21668, 10.1039/d4ra03459g.
- J. Wang and M. J. Lenardo, J. Cell Sci., 2000, 113(5), 753–757, DOI:10.1242/jcs.113.5.753.
- A. M. Rieger, K. L. Nelson, J. D. Konowalchuk and D. R. Barreda, J. Visualized Exp., 2011, 50, 2597, DOI:10.3791/2597.
- P. E. Czabotar and A. J. Garcia-Saez, Nat. Rev. Mol. Cell Biol., 2023, 24, 732–748, DOI:10.1038/s41580-023-00629-4.
- M. A. Lemmon and J. Schlessinger, Cell, 2010, 141(7), 1117–1134, DOI:10.1016/j.cell.2010.06.011.
- R. Singh, A. Letai and K. Sarosiek, Nat. Rev. Mol. Cell Biol., 2019, 20(3), 175–193, DOI:10.1038/s41580-018-0089-8.
- R. A. Sikes, Br. J. Cancer, 2007, 97(12), 1713, DOI:10.1038/sj.bjc.6604075.
- Z. K. Otrock, J. A. Makarem and A. I. Shamseddine, Blood Cells, Mol., Dis., 2007, 38(3), 258–268, DOI:10.1016/j.bcmd.2006.12.003.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066, DOI:10.1021/acs.jmedchem.5b00104.
- T. K. Karami, S. Hailu, S. Feng, R. Graham and H. J. Gukasyan, J. Ocul. Pharmacol. Ther., 2022, 38(1), 43–55, DOI:10.1089/jop.2021.0069.
- A. Beig, R. Agbaria and A. Dahan, PLoS One, 2013, 8, e68237, DOI:10.1371/journal.pone.0068237.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04956j |
| This journal is © The Royal Society of Chemistry 2024 |