
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntact quantitation and evaluation of a PEG-glycosulfopeptide as a therapeutic P-selectin antagonist†
Diane D. Park ab,
Simon S. Parkab,
Erbin Daiab,
Carolyn A. Hallerab,
Daniel J. Wongab,
Walter J. Weverab,
Richard D. Cummingsac and
Elliot L. Chaikof*abcd
ab,
Simon S. Parkab,
Erbin Daiab,
Carolyn A. Hallerab,
Daniel J. Wongab,
Walter J. Weverab,
Richard D. Cummingsac and
Elliot L. Chaikof*abcd
aDepartment of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, 110 Francis Street, Suite 9F, 330 Brookline Ave, Boston, MA 02215, USA. E-mail: echaikof@bidmc.harvard.edu; Tel: +1-617-632-9581; Fax: +1-617-632-9701
bWyss Institute for Biologically Inspired Engineering, Harvard University, 3 Blackfan Circle, Boston, MA 02115, USA
cHarvard Medical School Center for Glycoscience, Harvard Medical School, Boston, MA 02215, USA
dDivision of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 28th October 2024
Abstract
Peptide-based therapeutics are recognized as potent and selective molecules but are often limited by short circulating half-lives, instability towards enzymatic degradation, and immunogenicity. To address these limitations and improve their pharmacological properties, peptides are commonly modified by the covalent attachment of polyethylene glycol (PEG). However, the large molecular weight and polydispersity of PEG chains complicate the interpretation of the full structure of PEGylated peptide therapeutics using standard analytical techniques. Here, we developed a mass spectrometric-based workflow in negative ion mode to identify and quantify GSnP-6, a P-selectin antagonist, with a linear 10 kDa PEG (PEG10) attached at the N-terminus of the glycopeptide. Intact mass analysis with multiple microscans allowed accurate measurements of precursor ions in complex biological mixtures with baseline resolution. Utilizing stepped collision energies improved sequence coverage and enabled identification of key amino acid modifications. We show the utility of this approach in evaluating the properties of PEG10-GSnP-6 in vitro and in vivo. Inhibitory capacity was preserved while extending the half-life of this glycopeptide, as shown by the reduction of P-selectin/PSGL-1 binding. By sustaining effective circulating concentrations, PEG conjugation of a P-selectin glycopeptide antagonist represents a promising therapeutic strategy to target diseases linked to inflammatory processes.
Introduction
Peptide-based therapeutics are recognized as potent and selective molecules but are often limited by short circulating half-lives. Consequently, high doses are needed with high frequency of administration, which pose burdens on pharmaceutical manufacturing and increased risks of a negative therapeutic response. To improve pharmacological properties, peptides are commonly modified by the covalent attachment of polyethylene glycol (PEG), an uncharged, water soluble, and nontoxic polymer. Compared to other polymers, PEG is commercially available in large scale and can be prepared in a wide range of molecular weights with low polydispersity, and as polymers with either linear or branch architectures. Additionally, a long history of safety assessments has shown that PEG is safe for use in humans as an additive in foods, medications, cosmetics, and other consumer products.1–3 PEGylation in turn increases the hydrodynamic radius, slowing renal clearance, while retaining biological activity.4,5 A number of factors can be customized to alter the properties of PEGylated peptides, including molecular weight, branching, number of chains, site of attachment, and conjugation chemistry. Compared to non-PEGylated therapeutic molecules, conjugation of PEGs varying from 10 to 1000 kDa has been shown to protect peptide therapeutics from metabolic degradation, shield antigenic sites of proteins, and promote accumulation in tissues.6–8Although the PEGylation of biopharmaceuticals in most cases is simple and fast, structural analysis and quantification of the conjugated products is challenging due to the effects of its high molecular weight and inherent polydispersity. Conventional techniques based on ELISA provide highly accurate pre-clinical measurements that are superior to alternative fluorometric quantification methods9–11 but are not universally applicable for all PEGylated biopharmaceuticals due to interference from unconjugated PEGs, epitope-masking effects, or the lack of available antibodies. Anti-PEG ELISA assays allow highly sensitive quantification of a range of PEG-therapeutics in biological samples.12–15 However, as an indirect measurement, the assay does not provide essential structural information about the therapeutic, which includes site-specificity or transformations of drug molecules in vivo.
Advanced tools are thus needed to be able to assess critical pre-clinical parameters of PEGylated biotherapeutics in vivo. Nanoflow liquid chromatography coupled with high resolution mass spectrometry is a highly sensitive and precise analytical tool for profiling various biomolecules within complex biological matrices. Prior approaches have utilized fragmentation techniques with high in-source collision energy16–19 or enzymatic digestion20–24 for quantitative analyses of partially PEGylated biomolecules or de-PEGylated peptides, respectively. In the process, the original connectivity and orientation is lost and as a result, the evaluation of PEGylation site localization, efficiency, and stability may be impaired. While alternative techniques that incorporate fragmentation25–27 and derivatization28 have been able to successfully determine the location of PEG attachment sites, using the methodology to determine potential in vivo PEG cleavage is challenging and enzymatic approaches are limited in application only to a specific range of substrates. In addition to site mapping, analytical techniques are necessary for confirming the correct molecular weight and structural integrity of therapeutics. Here we utilized intact MS analysis of PEG constructs to detect the full-length molecule and preselected charge-defined parent ions for MS/MS to determine structural motifs. The unique features of the analysis included performing MS and MS/MS experiments in negative mode with PEGylated peptides remaining fully intact. This type of analysis facilitates confirmation of molecular composition and linkages, enabling accurate characterization of biotherapeutics and their modifications throughout therapeutic development and manufacturing.
GSnP-6 is a therapeutic glycosulfopeptide modeled after the N-terminus of P-selectin glycoprotein ligand-1 (PSGL-1), a leukocyte membrane protein that facilitates recruitment to sites of inflammation.29 In this study, we characterized constructs of GSnP-6 bearing a linear 10 kDa PEG chain at the N-terminus (PEG10-GSnP-6) and quantified its circulating amounts in vivo following intravenous bolus administration. We demonstrated the applicability of the method in intact analysis of other similarly sized peptides and proteins that have bioactivity. Samples required minimal processing, rendering it amenable to large pre-clinical sample sets. Significantly, the inhibitory capacity of PEG10-GSnP-6 was demonstrated using a mouse model of inflammation. In addition to favorable pharmacological properties, PEGylated GSnP-6 has potent antagonist activity, with potential to be used as a disease modifying therapeutic for a variety of disorders of thromboinflammation, including for the prevention of venous thromboembolism.
Experimental
General
All commercially available reagents and solvents were used without further purification. N,N-Dimethylformamide, N,N′-diisopropylethylamine (DIPEA), acetic acid, Super-DHB (2,3-dihydroxylbenzoic acid), aprotinin from bovine lung, and ethylenediaminetetraacetic acid (EDTA) were purchased from Sigma-Aldrich (St. Louis, MO). Ultra-high performance liquid chromatography (UHPLC) grade acetonitrile, methanol, and water were purchased from Fisher Scientific (Pittsburgh, PA). High performance liquid chromatography (HPLC) grade trifluoroacetic acid (TFA) and ethanol were purchased from Sigma-Aldrich. HPLC grade ammonium bicarbonate, ammonium acetate, ammonium formate, and ammonia were purchased from Honeywell Fluka. HPLC grade acetone, triethylamine, and 1,2-butylene carbonate were purchased from Fisher Scientific. The PEGylation reagent, 10 kDa methoxy poly(ethylene glycol)-succinimidyl valerate (mPEG-SVA), was purchased from Advanced BioChemicals (PDI 1.02; Lawrenceville, GA). All compounds were >95% pure by HPLC analysis. Sterile saline was purchased from Teleflex (Wayne, PA). Recombinant human ubiquitin L73P protein was purchased from R&D Systems (Minneapolis, MN). Recombinant human thioredoxin-1 protein and enfuvirtide were purchased from Thermo Fisher Scientific. Recombinant human alpha-synuclein was purchased from rPeptide (Watkinsville, GA). Sulfated big gastrin was purchased from Echelon Biosciences (Salt Lake City, UT). All murine protocols were approved by the Institutional Animal Care and Use Committee and Institutional Review Board of Beth Israel Deaconess Medical Center.Synthesis of PEG10-GSnP-6
GSnP-6 was synthesized as previously described.29 PEGylation of GSnP-6 was performed by adding mPEG-SVA (1 eq.) to GSnP-6 (2 eq.) in N,N-dimethylformamide (1.25 mM) at 0 °C. Subsequently, DIPEA was slowly added and shaken for 22 h at 25 °C to a final concentration of GSnP-6 of 20.1 μL mg−1. The reaction was quenched using acetic acid and the crude material was purified to afford the final product, PEG10-GSnP-6, using reverse phase (RP)-HPLC (preparative). For preparative purification, a binary gradient was applied to a C18 100 Å (250 × 30 mm) column (Phenomenex, Torrance, CA) using (A) water and 0.1% (v/v) TFA and (B) acetonitrile as follows: 0–10 min, 25% (B); 10–40 min, 25–80% (B); 40–41 min, 80–98% (B); 41–50 min, 98% (B); 50–51 min, 98–25% (B); 51–60 min, 25% (B). The Waters 2767 Gradient Purification System, Waters 2489 UV/Vis detection module, and a Waters 2545 Binary Gradient Module was used for loading, separation, and fraction collection (Waters, Milford, MA). A Bruker Ultraflex II matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer (Bruker, Billerica, MA) was used to analyze samples co-crystallized with Super-DHB matrix. Purity was assessed using RP-HPLC (analytical). For analytical analysis, a binary gradient was applied to a C18 100 Å (50 × 4.6 mm) column (Phenomenex) using (A) water and 0.1% (v/v) TFA and (B) acetonitrile as follows: 0–2 min, 5% (B); 2–6 min, 5–98% (B); 6–20 min, 98% (B); 20–21 min, 98–5% (B); 21–22 min, 5% (B).Flow cytometry
Flow cytometry was used to quantify binding inhibition of P-selectin Fc chimeras to human U-937 monocytes (ATCC, Manassas, VA). A total of 3 × 105 cells were incubated with either serial dilutions of GSnP-6 or PEG10-GSnP-6 (0–150 μM), EDTA (20 mM; Thermo Scientific), anti-P-selectin antibody (2 μg; R&D Systems), or anti-PSGL-1 antibody (anti-human CD162) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5; BD Biosciences, Rahway, NJ) along with Fc chimeras of human P-selectin (2 μg mL−1; R&D Systems) followed by PE-conjugated anti-Fc (1:100; BD Biosciences, Rahway, NJ) in HBSS/calcium/magnesium (Life Technologies). U-937 interactions with P-selectin were analyzed using a CytoFLEX LX flow cytometer (Beckman Coulter, Brea, CA) and quantified as percent inhibition using FlowJo v.10.7.1 (BD, Franklin Lakes, NJ). Relative IC50 values were calculated using Prism v.9.0.2 (GraphPad Software, San Diego, CA). Inhibition experiments were conducted in triplicate and representative curves presented.
:5; BD Biosciences, Rahway, NJ) along with Fc chimeras of human P-selectin (2 μg mL−1; R&D Systems) followed by PE-conjugated anti-Fc (1:100; BD Biosciences, Rahway, NJ) in HBSS/calcium/magnesium (Life Technologies). U-937 interactions with P-selectin were analyzed using a CytoFLEX LX flow cytometer (Beckman Coulter, Brea, CA) and quantified as percent inhibition using FlowJo v.10.7.1 (BD, Franklin Lakes, NJ). Relative IC50 values were calculated using Prism v.9.0.2 (GraphPad Software, San Diego, CA). Inhibition experiments were conducted in triplicate and representative curves presented.
Intravital microscopy
Surgical preparation of the mouse cremaster was performed as previously described.30 Mice were anesthetized with an intraperitoneal injection of ketamine HCl (125 mg kg−1) and xylazine (12.5 mg kg−1) and placed on a 37 °C surgical blanket. The jugular vein was cannulated with PE10 tubing to allow the introduction of PEG10-GSnP-6 (4 μmol kg−1 in saline), saline vehicle control, or 0.5 μm Dragon Green microspheres (Bangs Laboratories, Inc.). The trachea was intubated with PE90 to facilitate breathing. The cremaster muscle was exteriorized, immobilized, and superfused with thermocontrolled bicarbonate-buffered saline equilibrated with 5% CO2 in N2. Images were obtained using an Olympus AX microscope (Olympus, Tokyo, Japan) with 60× water immersion objectives recorded with a Hamamatsu C9300-201/GenIII videoscope (Hamamatsu Photonics, Hamamatsu City, Japan) with coordinated image acquisition and analysis using SlideBook software (Intelligent Imaging Innovations, Denver, CO). At the termination of the experiment, blood was drawn via cardiac puncture and analyzed by LC-MS to determine the plasma concentration of PEG10-GSnP-6. Venules (30–40 μm in diameter) were observed 15–25 min after surgical stimulation of tissue to study P-selectin-dependent rolling. Vessel diameter was measured using SlideBook. Centerline blood flow velocity (Vcl) was determined by measuring frame-to-frame displacement of single fluorescent microspheres in the center of the vessel wall. Volumetric blood flow rate (Q) was calculated using the equation| Q = 0.625 × Vcl × Acs, |
| γw = 2.12 × [(8 × 0.625 × Vcl)/Dv], |
Reduction of proteins
Alpha-synuclein and thioredoxin were incubated in 550 mM dithiothreitol (DTT) in 50 mM ammonium bicarbonate at 56 °C for 25 min. Subsequently, 450 mM iodoacetamide was added and samples were incubated in the dark at room temperature for 20 min. Reactions were quenched with the addition of 550 mM DTT and incubated in the dark for 15 minutes. All samples were filtered through an Amicon 3k molecular weight cutoff centrifugal filters (MilliporeSigma, Burlington, MA) following the manufacturer's protocol.Plasma collection
Blood was collected from male C57BL/6 mice aged 6–10 weeks into heparin-coated tubes (BD Biosciences) and centrifuged at 1500g for 10 min at 25 °C. The plasma layer was isolated, and PEG10-GSnP-6 was spiked into 20 μL aliquots at specified concentrations to generate a standard curve.Plasma stability
Pooled C57BL/6 mouse plasma was purchased from Innovative Research (Novi, MI). Plasma was spiked with 200 μg mL−1 PEG10-GSnP-6 in saline (1:10) and incubated in 37 °C with gentle rocking for 0, 15, 30, 60, 120, and 180 min prior to compound extraction. Each condition was prepared in triplicates.
In vivo detection
A single dose (0.6 mg/44.4 nmol) of PEG10-GSnP-6 was administered intravenously via the penile vein to anesthetized male C57BL/6 mice aged 6–10 weeks. Saline was administered to age matched male mice via the same route. Blood was collected serially three times per mouse into heparin-coated tubes at defined time points via submandibular vein (alternating sides) followed by a terminal blood draw via cardiac puncture under deep anesthesia. Tubes were centrifuged at 1500g for 10 min at 25 °C. Per collection, 20 μL of the plasma layer was isolated for compound extraction.Compound extraction from plasma
To remove abundant plasma proteins, cold methanol, ethanol, or acetone was added to plasma at a ratio of 1:4 (plasma:organic solvent). Samples were incubated at −80 °C overnight and centrifuged at 17000g for 20 min at °C. The supernatant containing PEG10-GSnP-6 was isolated, dried in vacuo, and solubilized in water prior to mass spectrometric analysis.
Compound extraction from urine
Urine was collected from male C57BL/6 mice following IV administration of PEG10-GSnP-6 and centrifuged at 3000g for 15 minutes. Samples were filtered through an Amicon 3k molecular weight cutoff centrifugal filters (MilliporeSigma) following the manufacturer's protocol. Proteins were precipitated with the addition of cold methanol (1:4), incubated at −80 °C for 1.5 hours, and centrifuged at 17000g for 20 min at 4 °C. Supernatants were filtered through a 0.45 μm centrifugal filter.
Nano-LC-MS/MS
Samples were loaded onto a C18 trap column (Thermo Fisher Scientific, Waltham, MA) at 5 μL min−1 using 3% (v/v) acetonitrile and 0.05% (v/v) TFA in water connected to an UltiMate 3000 RSLCnano UHPLC system (Dionex, Sunnyvale, CA). A binary gradient was applied to an Accucore 2.6 μm amide column (75 μm × 50 mm; Thermo Fisher Scientific) as follows: 0–3 min, 99% (B); 3–25 min, 99–49% (B); 25–34 min, 49% (B); 34–44 min, 49–99% (B). The composition of solvent (A) and solvent (B) varied as follows: (A) 5 mM ammonium formate/ammonia in water (pH 8.3) and (B) 5 mM ammonium formate/ammonia in 20:80 water:acetonitrile (pH 8.3), (A) 0.05% (v/v) triethylamine in 5 mM ammonium formate, and (B) 0.05% (v/v) triethylamine in 5 mM ammonium formate in 20:80 water:acetonitrile, 5 mM ammonium acetate/acetic acid in water (pH 3.8) and (B) 5 mM ammonium acetic/acetic acid in 20:80 water:acetonitrile (pH 3.8), or (A) 10 mM ammonium bicarbonate in water and (B) 10 mM ammonium bicarbonate in 20:80 water:acetonitrile. Spectra were acquired in negative ionization mode using a Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) with the following acquisition settings: ion transfer tube, 300 °C; scan range, 900–6000 m/z or 900–3000 m/z; RF lens, 24%; MS AGC target, 4e5; MS max injection time, 150 ms; MS microscans, 3; MS resolution, 120000; isolation window, 2 m/z; HCD collision energy, 30, 40, 50; MS/MS resolution, 30000; MS/MS AGC target, 5e4; MS/MS max injection time, 200 ms; MS/MS microscans, 2. The column temperature was maintained at 40 °C. The autosampler temperature was maintained at 4 °C.
Data analysis
MALDI-TOF MS spectra were analyzed using FlexControl v.3.4 (Bruker). Orbitrap Fusion Lumos MS spectra were deconvoluted using Protein Deconvolution 4.0 (Thermo Fisher Scientific). Chromatographic peaks were quantified using Processing Setup (Thermo Fisher Scientific). A linear regression curve was fitted to the standard curve and used to determine the concentrations of PEG10-GSnP-6 in collected plasma samples. Compound half-life was determined using a one compartment model. Statistical analysis was performed using Prism v.9.0.2 (GraphPad Software).Results & discussion
Intact MS analysis of PEG10-GSnP-6
A linear 10 kDa PEG was conjugated to the ε-amine of the N-terminal lysine in GSnP-6 (Fig. 1A–C) and analyzed by nanoflow hydrophilic interaction chromatography (HILIC)-electrospray ionization (ESI)-mass spectrometry. Notably, the size and chemical properties of PEG10-GSnP-6 enabled coupling of a C18 bidirectional trap column and a HILIC analytical column (ESI Fig. S1†). The main advantages of this coupling were the ability to inject aqueous solutions, eliminating concerns of evaporation or low solubility with organic solvents, and the ability to add an online desalting step. Loading and elution conditions were optimized to allow complete sample transfer using low and high organic strength, respectively. Backflush pre-column elution was used to improve retention time reproducibility and to reduce sample matrix interferences.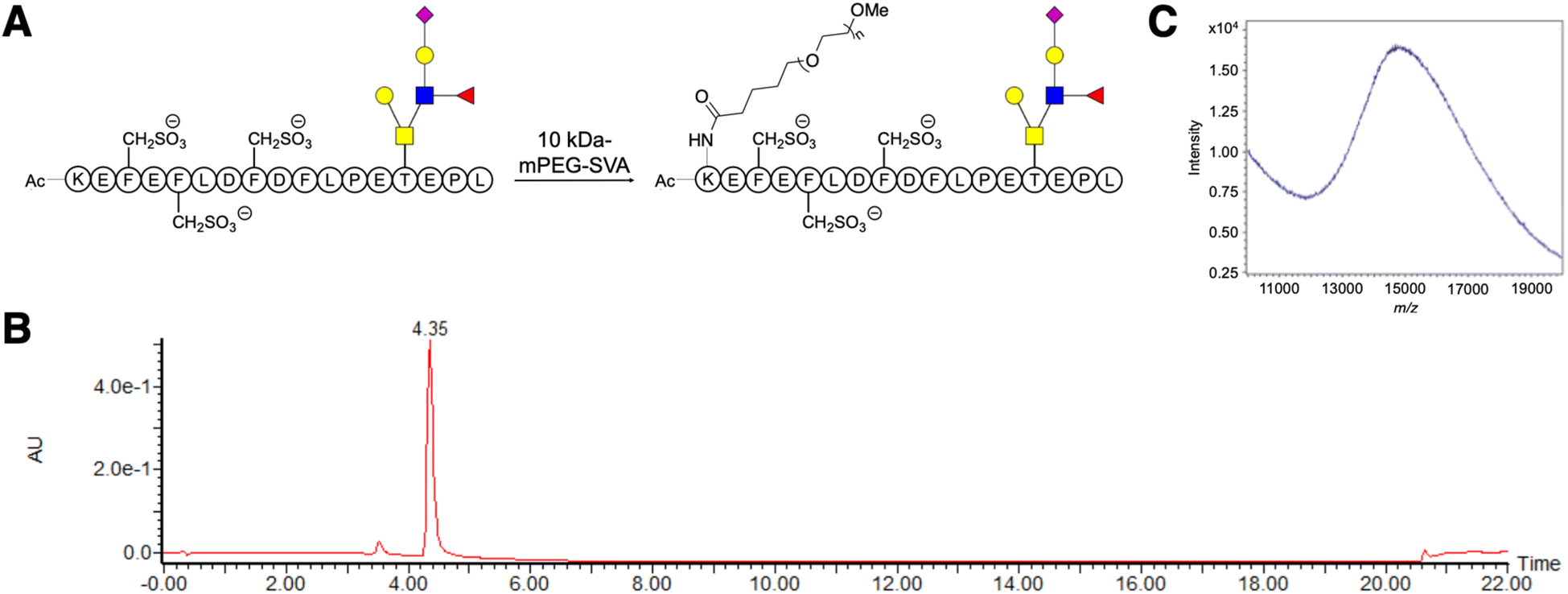 | ||
| Fig. 1 Synthesis of PEG10-GSnP-6. (A) Conjugation of a linear 10 kDa monomethoxy-PEG-succinimidyl valerate (mPEG-SVA) to the N-terminus of GSnP-6. Glycans are represented using symbol notation. (B) Chromatogram of PEG10-GSnP-6 purified by HPLC. (C) MALDI-MS spectrum of PEG10-GSnP-6. | ||
Pure solutions were used to evaluate the chromatographic behavior of PEG10-GSnP-6 and obtain distinguishing structural information. Using various buffered solvent preparations to control pH and ionic strength, we observed charge states of four to ten at high abundance within the detectable mass range in negative mode (Fig. 2A and B). The addition of triethylamine (TEA) resulted in the reduction of charge states with distinct charge state group separation across a wider m/z range. The charge reduction has been associated with the formation of triethylammoniated species over protonated species.31 Likewise, in negative mode, the addition of TEA likely competes with deprotonation. Other modifications including additives (1,2-butylene carbonate) were also suitable for the detection of PEG10-GSnP-6 (ESI Fig. S2†).
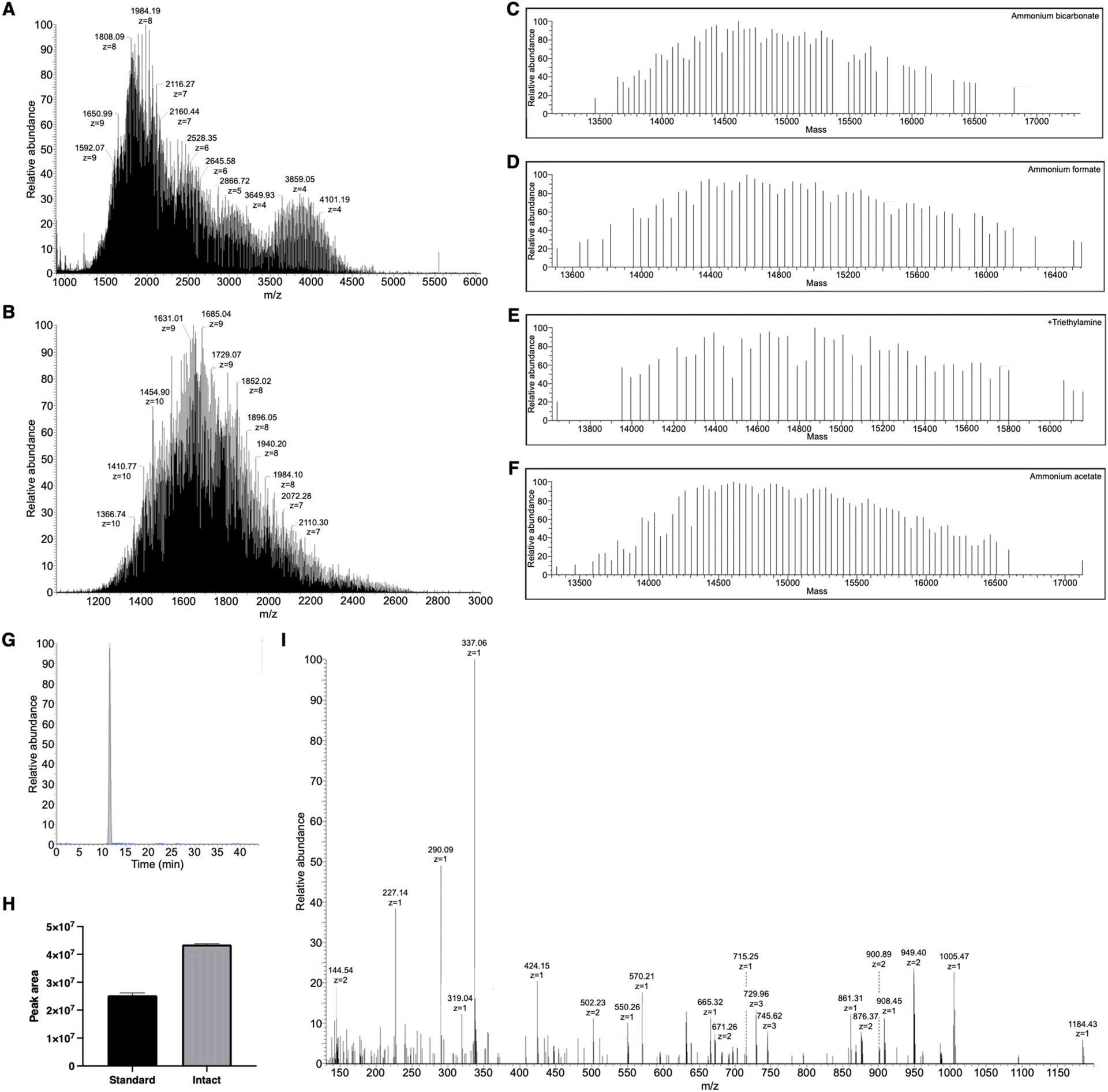 | ||
| Fig. 2 Mass spectrometric analysis of PEG10-GSnP-6. (A) Full mass spectrum of PEG10-GSnP-6 analyzed using triethylamine as an additive. (B) Full mass spectrum of PEG10-GSnP-6 analyzed using ammonium bicarbonate. (C–F) Deconvoluted mass spectra of PEG10-GSnP-6 analyzed using various LC-MS solvents. (G) Extracted chromatogram of PEG10-GSnP-6 in solution. (H) Comparison of PEG10-GSnP-6 ion counts with analysis by standard mode and by intact protein low pressure mode. (I) MS/MS spectrum of PEG10-GSnP-6 employing higher-energy collisional dissociation (HCD) with stepped collision energies in negative mode. | ||
MS spectral acquisition settings were optimized to achieve isotopic resolution and cover entire charge envelopes without fragmenting the peptide, compromising signal, or dragging scan time. Full MS data were acquired using a high-resolution scan of at least 120000. As the peptide was split between multiple charge states, spectral signal was improved by performing three microscans. Deconvolutions revealed sufficiently resolved signals with mass difference values of 44 Da, corresponding to the ethylene glycol units (Fig. 2C–F). Both standard pressure and low mTorr pressure control in the ion-routing multipole could be employed for efficient trapping of the PEGylated conjugate (Fig. 2G and H). In all subsequent analysis, PEG10-GSnP-6 was detected in its full-length form. In this way, the native state was preserved without chemical modifications or derivatization. In addition, our analysis prevented potential misrepresentation of the parent ion as in fragmentation approaches.
Fast scan speeds allowed more opportunities for MS/MS experiments to obtain key structural information. Stepped higher-energy collisional dissociation (HCD) was employed to generate product ions of the constituent parts of PEG10-GSnP-6 in the MS/MS spectra, which was used along with column retention time as a signature profile for facile matching in subsequent samples (Fig. 2I). High fragment ion coverage and accurate mass measurements of product ions facilitated the localization of sites of amino acid modifications, including the sulfonation of phenylalanine and O-glycosylation of threonine.
PEG10-GSnP-6 is a multiply charged, polydisperse bioconjugate with high molecular mass. Nanoflow HILIC, which offered high sensitivity and high retention of polar compounds, may be widely suitable for analyzing peptides conjugated to linear PEG polymers of varying lengths. Moreover, negative mode detection, which is advantageous for lowering background noise and for the detection of acidic compounds, will be suitable for similarly acidic compounds as GSnP-6, a tri-sulfonated and sialylated peptide.
In addition to PEG10-GSnP-6, we analyzed negatively charged/neutral protein standards with a molecular weight ranging from 8 to 14 kDa to assess the applicability of the method. Alpha synuclein (14 kDa), thioredoxin (12 kDa), and ubiquitin (8.5 kDa) were detected intact following reduction and carboxyamidation of disulfide bonds (ESI Fig. S3–S5†). The method was also amenable to analysis of large polypeptide standards (<7 kDa), aprotinin (6.5 kDa), enfuvirtide (4 kDa), and sulfated big gastrin (3.9 kDa), in their full-length form (ESI Fig. S6–S8†).
Quantification of PEG10-GSnP-6 in plasma
Structural identification was followed by quantification of PEG10-GSnP-6 in murine plasma. Compound recovery according to mean peak areas was consistently high (>86%) using methanol or ethanol precipitation as a method to extract PEG10-GSnP-6 from plasma (Fig. 3A–C). Gradient elution of repeat injections of extracted PEG10-GSnP-6 yielded highly reproducible retention times (ESI Fig. S9†). External standard curves were constructed from pooled plasma spiked with serial dilutions of PEG10-GSnP-6. Linearity was demonstrated over a dynamic range that spanned three orders of magnitude (Fig. 3D). The validity of the linear regression model was supported by a residual plot (Fig. 3E).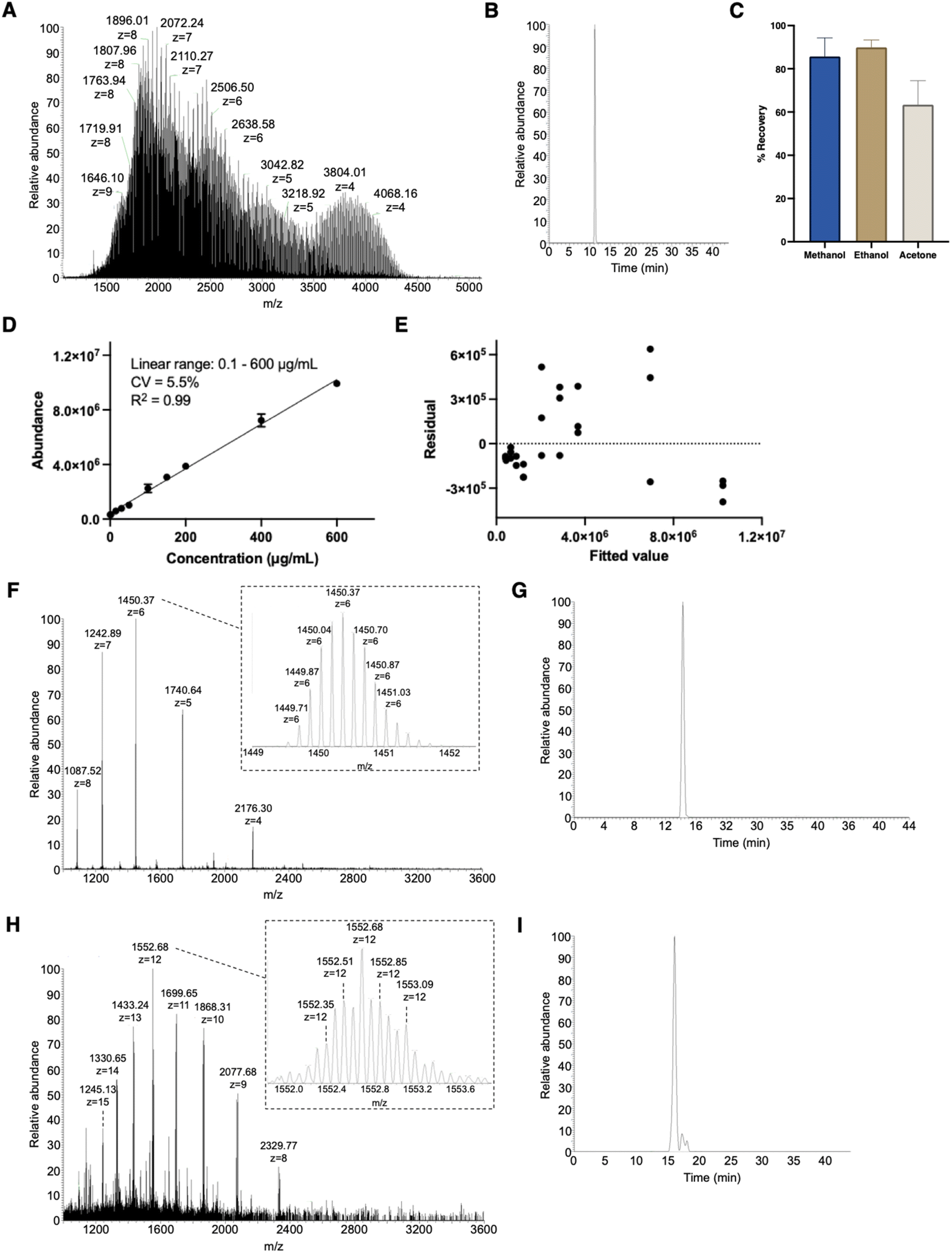 | ||
| Fig. 3 Quantification PEG10-GSnP-6 in plasma. (A) Full mass spectrum and (B) extracted chromatogram of PEG10-GSnP-6 spiked in plasma. (C) Percent compound recovery following protein precipitation. Data are represented as mean ± SD (n = 3). (D) Standard curve of PEG10-GSnP-6 extracted from plasma. CV, coefficient of variation. (E) A residual plot from the linear regression is confirmatory of the model. (F and G) Murine plasma proteins and (H and I) urine proteins extracted using protein precipitation and analyzed by intact protein low pressure mode. | ||
During the extraction of PEG10-GSnP-6 from biological fluids, we assessed the ability to isolate endogenous proteins in addition to the target analyte. Mid molecular weight proteins were detected in all extractions from plasma (Fig. 3F and G) and from urine (Fig. 3H and I). Sufficient chromatographic separation enables subsequent quantification in low pressure intact mode or standard mode. Additional structural information was obtained using tandem mass spectrometry (ESI Fig. S10 and S11†).
PEG10 extends the terminal half-life of GSnP-6 in mice
A single dose of PEG10-GSnP-6 was administered intravenously to adult mice and in vivo plasma concentrations were measured serially over time. Mass spectrometric profiles of PEG10-GSnP-6 in plasma collected from mice post-injection were consistent with those observed from spiked plasma (Fig. 4A). Time-dependent changes in the plasma concentration of PEG10-GSnP-6 were consistent with a terminal half-life of 83.9 ± 1.2 minutes (Fig. 4B). In contrast, structurally similar non-PEGylated glycosulfopeptide mimics of PSGL-1 modeled after the N-terminus region exhibited a half-life of less than 15 minutes.32 Loss resulting from degradation was unlikely as PEG10-GSnP-6 remained intact in plasma ex vivo at 37 °C over time (Fig. 4C).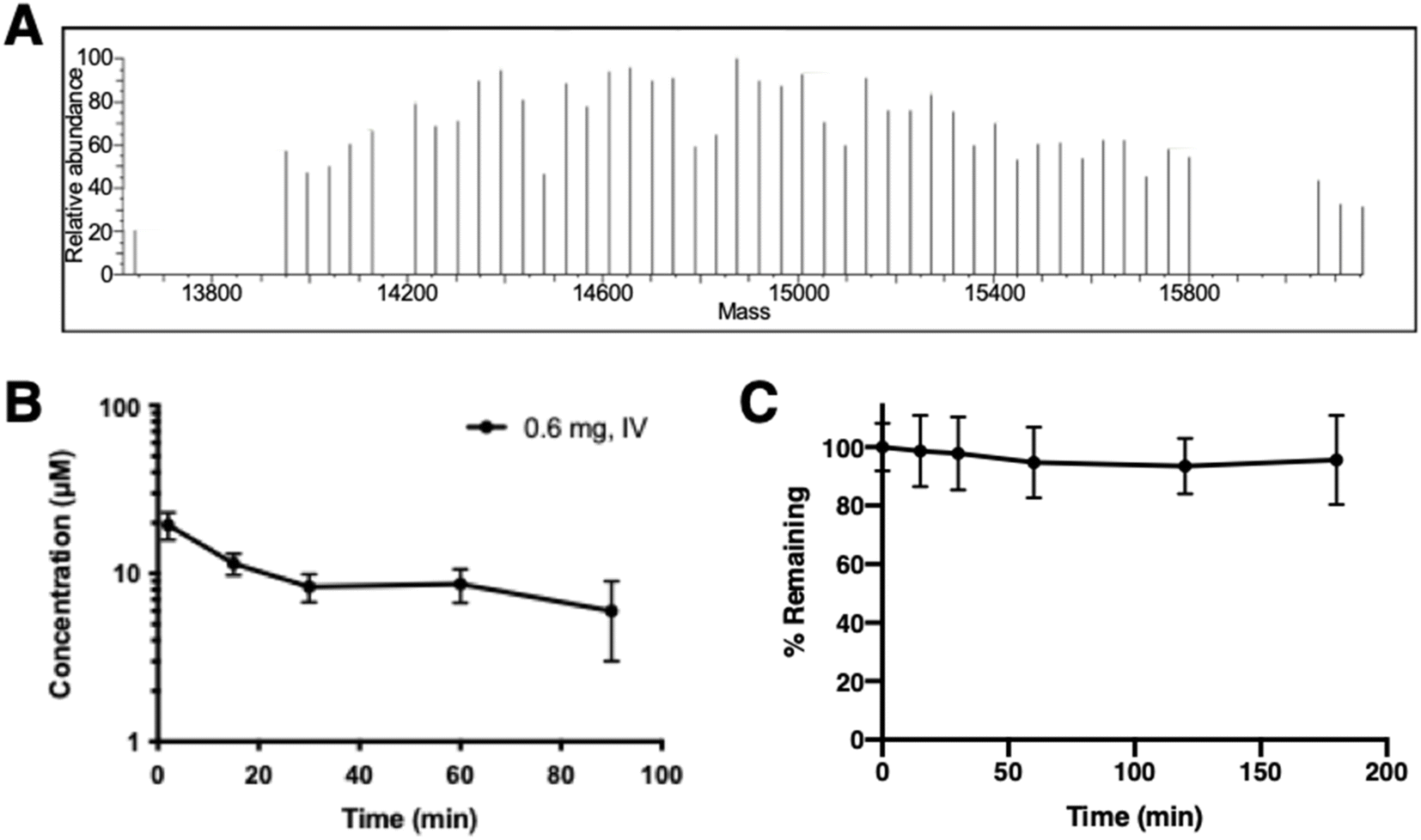 | ||
| Fig. 4 Pharmacokinetic profile of PEG10-GSnP-6. (A) Full-length detection of PEG10-GSnP-6 in plasma collected post-IV administration of 0.6 mg PEG10-GSnP-6 using HILIC-MS. (B) Concentration of PEG10-GSnP-6 in plasma over time in vivo. Data are represented as mean ± SD (n = 3). (C) Stability of PEG10-GSnP-6 in plasma over time ex vivo. Data are represented as mean ± SD (n = 3). | ||
The generality of this LC-MS-based method allows for broad use in in vivo screening studies and evaluation of terminal half-lives. We observed a wide dynamic range with minimal sample processing, allowing serial collections to be analyzed concurrently without further dilution or large starting amounts. Short acquisition times and small injection volumes enable analysis of a variety of biological samples. Significantly, high sensitivity and specificity of the method makes it amenable to samples sets that require limited sampling strategies to accurately determine in vivo concentrations. In addition to measuring concentrations, with full scan mass spectrometric analysis, the composition of the sample, relative purity, and the integrity of the therapeutic can be inspected under different biological conditions. For example, we determined that de-PEGylation events did not occur and the biotherapeutic was stable over time. This information will be useful for detecting PEG10-GSnP-6 or similar therapeutics in other tissues or biological fluids that will inform decisions related to drug distribution or clearance.
PEG10-GSnP-6 inhibits P-selectin binding in vitro
Competitive inhibition of human leukocyte binding to P-selectin by GSnP-6 and PEG10-GSnP-6 was assessed using flow cytometry. U-937 monocytes that bound to recombinant human P-selectin-Fc chimeras (2 μg mL−1) in the presence of GSnP-6 or PEG10-GSnP-6 (0–100 μM) were measured using a fluorescently labeled anti-Fc antibody, quantified as mean fluorescent intensity, and presented as percent inhibition of binding. P-selectin binding in the absence of GSnP-6 or PEG10-GSnP-6 was defined as 0% inhibition. Dose-dependent decreases in mean fluorescence intensity were observed with GSnP-6 or PEG10-GSnP-6, demonstrating inhibitory activity against cell surface PSGL-1/P-selectin interactions. GSnP-6 inhibited P-selectin binding to U-937 monocytes with an IC50 of 11.07 μM (Fig. 5A and B). PEG10-GSnP-6 exhibited similar inhibition kinetics as compared to GSnP-6, with an IC50 of 12.37 μM. Slight differences in activity at near-saturating concentrations may be related to steric effects in vitro. Positive controls included EDTA (20 mM), as well as anti-P-sel (2 μg) and anti-PSGL-1 (1:5) monoclonal antibodies (Fig. 5C and ESI Fig. S12†).
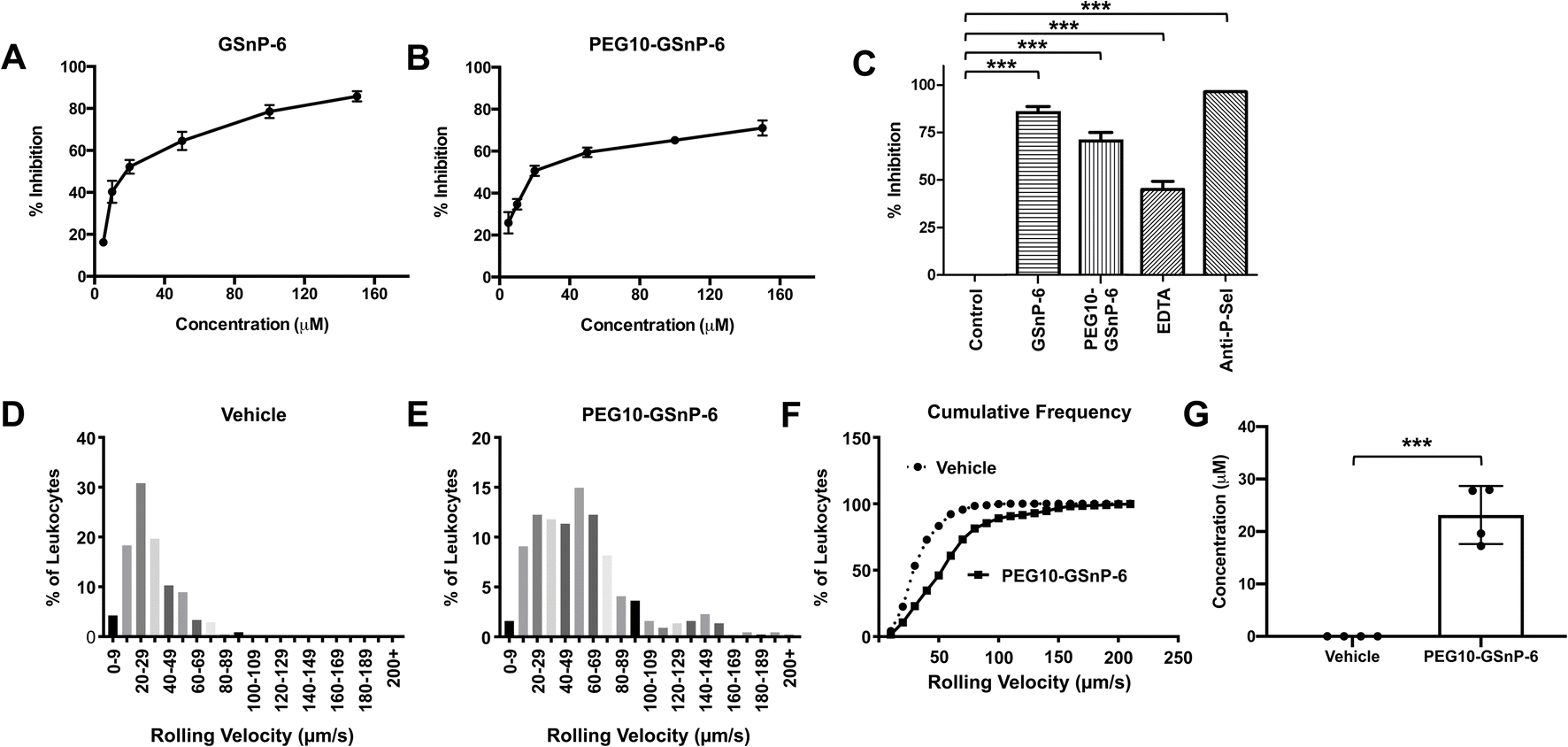 | ||
| Fig. 5 Inhibitory capacity of PEG10-GSnP-6. (A and B) Percent inhibition of P-selectin binding to U-937 monocytes by GSnP-6 or PEG10-GSnP-6 in vitro. Data are represented as mean ± SD (n = 3). (C) Percent inhibition of P-selectin binding to U-937 monocytes in vitro by GSnP-6, PEG10-GSnP-6 (150 μM), EDTA (20 mM), anti-P-selectin (2 μg), or with the absence of inhibitor (control). Data are represented as mean ± SD (n = 3). ***p < 0.001, Student's t-test. (D–F) Reduction of leukocyte rolling velocities by PEG10-GSnP-6 in vivo. (G) Measurement of circulating PEG10-GSnP-6 in control and stimulated mice at the termination of recording (t = 37.3 ± 4.6 min). Data are represented as mean ± SD (n = 4). ***p < 0.001, Student's t-test. | ||
PEG10-GSnP-6 inhibits leukocyte rolling and arrest in vivo
To determine the activity of GSnP-6 against leukocyte binding to the microvascular endothelium in vivo, intravital microscopy was performed post-surgical stimulation of the murine cremaster. PEG10-GSnP-6 (4 μmol kg−1) or saline was administered intravenously prior to induction of inflammation and leukocyte rolling velocity was recorded by individually tracking leukocyte distance/time (μm/s; n = 3–4 mice/group, 7–9 vessels/mouse). PEG10-GSnP-6 significantly increased mean rolling velocities compared to saline vehicle control (p < 0.01) (Fig. 5D and E). Based on a cumulative frequency histogram analysis, a two-fold increase in median rolling velocities was observed with administration of PEG10-GSnP-6 (Fig. 5F). PEG10-GSnP-6 plasma concentrations were measured at the termination of recording using nanoflow HILIC-MS/MS, with an average of 23 μM (Fig. 5G).Blocking PSGL-1/P-selectin interactions is a potential therapeutic strategy for the prevention and treatment of adverse thrombo-inflammatory events. Fundamentally, this interaction requires key structural motifs, including clustered tyrosine sulfates and sialyl Lewis X, that must be displayed in a specific orientation. GSnP-6 preserves these critical interactions and holds therapeutic promise as a high affinity P-selectin antagonist. PEGylation provides the advantage of less frequent administrations as compared to unmodified peptides. Conjugation does not interfere with peptide function and establishes a viable route to further develop peptide-based therapeutic products with tailored drug exposure levels. In this regard, PEG-10-GSnP-6 represents a potential candidate to selectively block PSGL-1/P-selectin interactions. Significantly, native mass spectrometry of PEGylated peptides is a useful tool to understand drug behavior in various tissues and over various lengths of time.
Conclusions
PEGylation is frequently used to improve the pharmacological properties of therapeutics. However, the analysis of high molecular weight proteins by mass spectrometry is often hampered by poor resolution, poor sensitivity, and complicated analysis. We developed a mass spectrometric method to detect and quantify PEGylated acidic peptides in their native form. Concentrations of PEG10-GSnP-6 were measured in plasma as a function of time to determine its circulating half-life in mice. Peptide stability was maintained with PEG conjugation. In addition, its inhibitory capacity against P-selectin was unaffected by the conjugation of a linear 10 kDa PEG. PEG10-GSnP-6 can potentially be used in vivo to directly inhibit PSGL-1/P-selectin interactions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. D. P., S. S. P., C. A. H., R. D. C., and E. L. C. conceived the research. D. D. P., S. S. P., E. D., W. J. W., and D. J. W. performed the research. D. D. P. analyzed the data. R. D. C. and E. L. C. supervised the research. D. D. P. and E. L. C. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge support from the U.S. National Institutes of Health under award number R01HL128237 (E. L. C.), R01DK107405 (E. L. C.), U01GM116196 (R. D. C. and E. L. C.), and T32HL007734 (D. J. W.) and the Blavatnik Therapeutic Challenge Award from Harvard Medical School (E. L. C.). D. D. P. was supported by the Charles A. King Postdoctoral Fellowship. We thank Sunia Trauger at the Small Molecule Mass Spectrometry Facility at Harvard University for Proteome Discoverer.References
- C. Fruijtier-Pölloth, Toxicology, 2005, 214, 1–38 CrossRef.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef CAS.
- J. R. Hecht, S. Lonardi, J. Bendell, H. W. Sim, T. Macarulla, C. D. Lopez, E. van Cutsem, A. J. Muñoz Martin, J. Oh Park, R. Greil, H. Wang, R. R. Hozak, I. Gueorguieva, Y. Lin, S. Rao and B. Y. Ryoo, J. Clin. Oncol., 2021, 39, 1108–1118 CrossRef CAS.
- J. Milton Harris, N. E. Martin and M. Modi, Clin. Pharmacokinet., 2001, 40, 539–551 CrossRef PubMed.
- J. Milton Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef.
- F. J. C. Bayard, W. Thielemans, D. I. Pritchard, S. W. Paine, S. S. Young, P. Bäckman, P. Ewing and C. Bosquillon, J. Controlled Release, 2013, 171, 234–240 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177–1188 CrossRef CAS.
- P. Calceti, S. Salmaso, G. Walker and A. Bernkop-Schnürch, Eur. J. Pharm. Sci., 2004, 22, 315–323 CrossRef CAS PubMed.
- M. C. Gauduin, P. W. H. I. Parren, R. Weir, C. F. Barbas, D. R. Burton and R. A. Koup, Nat. Med., 1997, 3, 1389–1393 CrossRef CAS PubMed.
- L. D. Curtis, A. Brown, M. B. Comer, J. M. Senior, S. Warrington and K. M. Dawson, J. Thromb. Haemostasis, 2005, 3, 1180–1186 CrossRef CAS.
- N. Siebert, C. Eger, D. Seidel, M. Jüttner, M. Zumpe, D. Wegner, S. Kietz, K. Ehlert, G. J. Veal, W. Siegmund, M. Weiss, H. Loibner, R. Ladenstein and H. N. Lode, MAbs, 2016, 8, 604–616 CrossRef CAS PubMed.
- C. Danika, M. A. El Mubarak, I. Leontari and G. B. Sivolapenko, Anal. Biochem., 2019, 564–565, 72–79 CrossRef CAS PubMed.
- T. C. Cheng, K. H. Chuang, M. Chen, H. E. Wang, S. C. Tzou, Y. C. Su, C. H. Chuang, C. H. Kao, B. M. Chen, L. Sen Chang, S. R. Roffler and T. L. Cheng, Bioconjugate Chem., 2013, 24, 1408–1413 CrossRef CAS.
- W. W. Lin, Y. C. Hsieh, Y. A. Cheng, K. H. Chuang, C. C. Huang, C. H. Chuang, I. J. Chen, K. W. Cheng, Y. C. Lu, T. C. Cheng, Y. T. Wang, S. R. Roffler and T. L. Cheng, Anal. Chem., 2016, 88, 12371–12379 CrossRef CAS.
- X. H. Song, G. Huhle, L. C. Wang and J. Harenberg, Thromb. Res., 2000, 99, 195–202 CrossRef CAS.
- M. Shi, H. Jiang, L. Yin, Y. Liu and M. Xu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1125, 121716 CrossRef CAS PubMed.
- J. Gong, X. Gu, W. E. Achanzar, K. D. Chadwick, J. Gan, B. J. Brock, N. S. Kishnani, W. G. Humphreys and R. A. Iyer, Anal. Chem., 2014, 86, 7642–7649 CrossRef CAS PubMed.
- B. M. Warrack, B. P. Redding, G. Chen and M. S. Bolgar, Anal. Bioanal. Chem., 2013, 405, 4283–4287 CrossRef CAS.
- H. Li, M. J. Rose, J. R. Holder, M. Wright, L. P. Miranda and C. A. James, J. Am. Soc. Mass Spectrom., 2011, 22, 1660–1667 CrossRef CAS PubMed.
- Z. Yang, J. Ke, M. Hayes, M. Bryant and F. L. S. Tse, J. Chromatogr. B, 2009, 877, 1737–1742 CrossRef CAS PubMed.
- Y. Xu, J. T. Mehl, R. Bakhtiar and E. J. Woolf, Anal. Chem., 2010, 82, 6877–6886 CrossRef CAS PubMed.
- S. T. Wu, Z. Ouyang, T. V. Olah and M. Jemal, Rapid Commun. Mass Spectrom., 2011, 25, 281–290 CrossRef CAS.
- Y. Chang, G. M. Maylin, G. Matsumoto, S. M. Neades and D. H. Catlin, Drug Test. Anal., 2011, 3, 68–73 CrossRef CAS PubMed.
- N. H. Yu, E. N. M. Ho, T. S. M. Wan and A. S. Y. Wong, Anal. Bioanal. Chem., 2010, 396, 2513–2521 CrossRef CAS PubMed.
- X. Zhou, X. Meng, L. Cheng, C. Su, Y. Sun, L. Sun, Z. Tang, J. P. Fawcett, Y. Yang and J. Gu, Anal. Chem., 2017, 89, 5193–5200 CrossRef CAS PubMed.
- V. Vijaya Bhaskar, A. Middha, S. Tiwari and S. Shivakumar, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 926, 68–76 CrossRef CAS PubMed.
- X. Lu, P. C. Gough, M. R. DeFelippis and L. Huang, J. Am. Soc. Mass Spectrom., 2010, 21, 810–818 CrossRef CAS PubMed.
- M. Cindrić, T. Čepo, N. Galić, M. Bukvić-Krajačić, N. Tomczyk, J. P. C. Vissers, L. Bindila and J. Peter-Katalinić, J. Pharm. Biomed. Anal., 2007, 44, 388–395 CrossRef.
- V. R. Krishnamurthy, M. Y. R. Sardar, Y. Ying, X. Song, C. Haller, E. Dai, X. Wang, D. Hanjaya-Putra, L. Sun, V. Morikis, S. I. Simon, R. J. Woods, R. D. Cummings and E. L. Chaikof, Nat. Commun., 2015, 6, 6387 CrossRef CAS PubMed.
- V. C. Ridger, P. G. Hellewell and K. E. Norman, Am. J. Pathol., 2005, 166, 945–952 CrossRef CAS PubMed.
- L. Huang, P. C. Gough and M. R. DeFelippis, Anal. Chem., 2009, 81, 567–577 CrossRef CAS PubMed.
- A. E. R. Hicks, A. Leppänen, R. D. Cummings, R. P. McEver, P. G. Hellewell and K. E. Norman, FASEB J., 2002, 16, 1461–1462 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05000b |
| This journal is © The Royal Society of Chemistry 2024 |