DOI:
10.1039/D4RA05017G
(Paper)
RSC Adv., 2024,
14, 31422-31428
Inductively coupled plasma optical emission spectroscopic determination of trace rare earth elements in highly refractory gadolinium zirconate (Gd2Zr2O7)†
Received
11th July 2024
, Accepted 27th September 2024
First published on 3rd October 2024
Abstract
Gadolinium zirconate (Gd2Zr2O7) belongs to the category of burnable absorber (BA) material in nuclear reactors. The high neutron-absorption cross sections of Gd isotopes (155Gd and 157Gd) implement negative reactivity in the reactor core to control the excess reactivity of the fuel at the beginning of the fission cycle. The presence of other rare earth elements, which can come from the starting material and/or may be taken up during the synthesis steps, will affect the negative reactivity calculation. Thus the chemical quality assurance of Gd2Zr2O7 concerning the other trace rare earth impurities is indispensable. Here in this work, we decided to quantify thirteen trace rare earth elements in Gd2Zr2O7 by inductively coupled plasma optical emission spectroscopy (ICP-OES). One of the matrix elements viz., zirconium was separated via precipitation using D,L-mandelic acid. The trace rare earths were determined in the presence of gadolinium matrix in solution. It was found that all thirteen rare earth elements can be quantified in the range of 0.25 to 2.5 mg L−1 in the presence of 516 mg L−1 of Gd with a relative standard deviation of 1–3%. This corresponds to the determination of a minimum of 0.25 mg analyte per g of Gd2Zr2O7. The method detection limits of all thirteen rare earth elements range between 0.01 and 0.075 mg g−1. The proposed methodology was validated by analyzing synthetic standards and real samples with spike addition.
1. Introduction
In the scenario of depleting fossil fuel resources, nuclear reactors are one of the potential candidates for fulfilling the huge worldwide energy demand. The nuclear fuel meat which is the target component in fission reaction offers excess reactivity at the beginning of the reactor cycle. This excess reactivity is required to increase the fuel burn-up and to recompense for the loss of reactivity due to fuel depletion and fission product buildup.1–3 In nuclear reactors, this excess reactivity is being controlled by burnable absorbers (BAs) which are materials containing non-fissile nuclei with large neutron-absorption cross-sections (σa). Typically, a BA should burn out at the same rate as fuel to keep the net reactivity constant with time.1 When the BA burns out too quickly, a positive reactivity swing in the fuel cycle will hamper the reactor operation in the later stages. Similarly, if the BA burns out too slowly, then a negative reactivity of the remaining BA will hinder the end of the fuel's operational cycle. Other than their control over excess reactivity, varieties of BAs are reported to offer benefits like tritium production and burning of long-lived radionuclides from spent nuclear fuel.4–6 There are two modes of using BA material in the reactor namely, homogeneous and heterogeneous modes.7–11 In homogeneous mode, the BA material is either homogeneously dispersed in the fuel or used as a coating. On the other hand, in heterogeneous mode, the BA material is dispersed in an inert matrix and the discrete rods are placed in the fuel assembly. The homogeneous mode has the advantage of even power distribution over the heterogeneous one.2 But the former mode has direct affect on the sinterability and thermal conductivity of the fuel.2 Hence the heterogeneous mode is more widely explored in the nuclear industry as it does not affect the fuel properties. Also in this mode, the BA material property can be tuned according to the reactor's requirement.1–3
BA materials consist of burnable poison (BP) nuclides like 10B, 113Cd, 151Eu and 153Eu, 155Gd and 157Gd, and 167Er etc.1 Among the available BP nuclides, the most widely used one is gadolinium (Gd) due to the significant natural abundance and high σa values of two of its isotopes.1,2 Gd2O3 is being used with some inert matrix to prepare the BA pellet and loaded to the BA rods. The inert matrices are chosen to form pyrochlore structures of the type GdxMyOz (where M = Ti, Zr or Al).2,12–15 Gd2Zr2O7 is one such BA material that was prepared by various scientists and tested for its performance.16–18 This pyrochlore is highly refractory and has low swelling properties, excellent irradiation, and chemical stability.12–15 In India, this material has been proposed to be the BA rod material for compact high-temperature reactor (CHTR, a light water reactor). The chemical quality assurance (CQA) of any BA material includes the checking of burnable poison concentration and material properties before being used in the reactor. Thus the characterization of trace critical impurities (elements other than Gd which have high σa values) which can affect the negative reactivity calculation of the BA material is one of the important aspects of CQA. These impurities can come directly from the precursor viz., Gd2O3 and ZrO2, or from the production steps. The precursor materials can ingress impurities with high σa values like rare earth elements and hafnium. Hence we focused our research on quantifying thirteen trace rare earth impurities viz., La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, and Lu in Gd2Zr2O7.
The determination of thirteen trace rare earth impurities in Gd2Zr2O7 presented challenges in the selection of suitable digestion procedure, difficulties in the separation of trace rare earth impurities from matrix elements, and matrix interference on analyte signals. The method commenced with the microwave-assisted dissolution of this highly refractory material. The dissolution procedure was optimized concerning the various acid combinations and proportions. Once the material was dissolved, zirconium(IV) was selectively precipitated using D,L-mandelic acid. This step also eliminated hafnium from the solution and hence we did not carry out hafnium estimation. The separation of thirteen trace rare earth elements from the Gd matrix is a highly difficult task due to their similar chemical behavior. Hence, we decide to analyze all thirteen trace rare earth elements in the presence of gadolinium. The estimation thus needed an analytical technique with high sample throughput, high sensitivity, and low detection limit. A technique like inductively coupled plasma mass spectrometry (ICP-MS)19–22 qualifies for all the above-mentioned requirements. However, the high matrix concentration can clog its cones and affect the ion beam in the ion optics region which may lead to unreliable results. In the case of X-ray fluorescence (XRF) spectroscopy,23,24 the low fluorescence yield of lanthanide L lines will be further reduced in presence of a high Gd matrix. On the other hand, ICP optical emission spectroscopy (ICP-OES)25–29 can handle higher matrix concentration compared to both ICP-MS and XRF techniques. Hence the digested sample solution after zirconium precipitation was directly analyzed by ICP-OES. The ICP-OES procedure was optimized for the determination of minimum analyte concentration in the presence of maximum matrix concentration. The matrix interferences on all thirteen analyte spectra were examined and corrected for the required ones. The method was established by analyzing synthetic standards of Gd and Zr matrix containing thirteen trace rare earth elements. The proposed analytical methodology was validated by analyzing real samples via spike addition and recovery studies.
2. Experimental
2.1. Instrumentation
A high resolution ICP-OES of model no: Ultima 2 from HORIBA Scientific (Jobin Yvon Technology), France was employed for the quantification of analytes. The spectrometer has a spectral range of 160–800 nm. The complete instrumental details are tabulated in Table 1. An Anton Par Multiwave 5000 microwave digestion system with 8× rotor was used for dissolution of Gd2Zr2O7. A Mettler Toledo microbalance, model: XP26, was used for weighing the solids.
Table 1 ICP-OES specifications and operating parameters
| Spectrometer |
| Optical design |
1 m Czerny Turner |
| Grating |
Holographic, dual back-to-back gratings |
| Groove density |
4320 g mm−1 and 2400 g mm−1 |
| Resolution |
1st and 2nd order with optic resolution <0.005 nm for 160–320 nm and <0.01 nm for 320–800 nm |
| Thermal regulation |
Controlled to 32.5 °C |
| Plasma parameters |
| Radio frequency |
40.68 MHz |
| Applied power |
1000 W |
| Reflected power |
<1 W |
| Ar gas flow rates |
| Plasma gas |
13 L min−1 |
| Nebulizer gas |
1.5 L min−1 |
| Sheath gas |
0.2 L min−1 |
| Acquisition parameters |
| Nebulizer |
Concentric |
| Spray chamber |
Cyclonic |
| Sample rate |
0.9 L min−1 |
| Detection |
Dual PMT detection with high dynamic detection system (HDD) |
| Gain |
3 |
| Entrance slit |
20 μm |
| Exit slit |
15 μm |
| Measurement |
Peak top, background |
| Integration time |
3 s |
| No. of replicates |
5 |
2.2. Reagents and solutions
Suprapur® sulfuric acid (96% assay), hydrochloric acid (30% assay), nitric acid (65% assay) and hydrofluoric acid (40% assay) were purchased from Merck. Ultrapure water (18.2 MΩ cm) from Milli-Q system of Millipore Corporation was used throughout the experiments. 1000 mg L−1 ICP standard solutions (Alfa Aesar) of thirteen rare earth elements was used to prepare the synthetic standard solutions and for spike addition. High purity Gd2O3 (99.999%, Sigma-Aldrich) and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg L−1 ICP standard solution of Zr (Aldrich Chemical Company, Inc.) were utilized for preparing synthetic standards. D,L-Mandelic acid (99%) was procured from Sigma-Aldrich. All the solutions were prepared in Nalgene® PP volumetric flasks.
000 mg L−1 ICP standard solution of Zr (Aldrich Chemical Company, Inc.) were utilized for preparing synthetic standards. D,L-Mandelic acid (99%) was procured from Sigma-Aldrich. All the solutions were prepared in Nalgene® PP volumetric flasks.
2.3. Preparation of synthetic standard and sample solutions
About 29.5, 59, and 119 mg of Gd2O3 were weighed in the microbalance and taken in different beakers. In these beakers, calculated volumes of 1000 mg L−1 rare earth standards and 10000 mg L−1 Zr standard were added to prepare synthetic standards according to Table T1 (see ESI†). The mixtures were dissolved by adding 2 mL of conc. HNO3 under heating at 80 °C for 5 min. The clean solution was then evaporated near to dryness and re-dissolved by conc. HCl. This step was repeated for 2–3 times to eliminate the nitric acid. The solution was then made up to 20 mL by 6.0 mol L−1 HCl solution. To this solution, 50 mL of 16% (w/v) mandelic acid was added at room temperature and the total volume was made 100 mL by Milli-Q water. The temperature was then slowly raised to 85 °C, and held there for 30 min. The white precipitate was then filtered and washed by using 2% (v/v) HCl and 5% (w/v) mandelic acid solution. The raffinate was then evaporated to dryness and then heated in a muffle furnace at 850 °C for 4 h. The residue was then completely dissolved by 5 mL of conc. HNO3 under heating at 80 °C for 5 min. The solution was then made up to 100 mL in a volumetric flask with Milli-Q water for ICP-OES analysis.
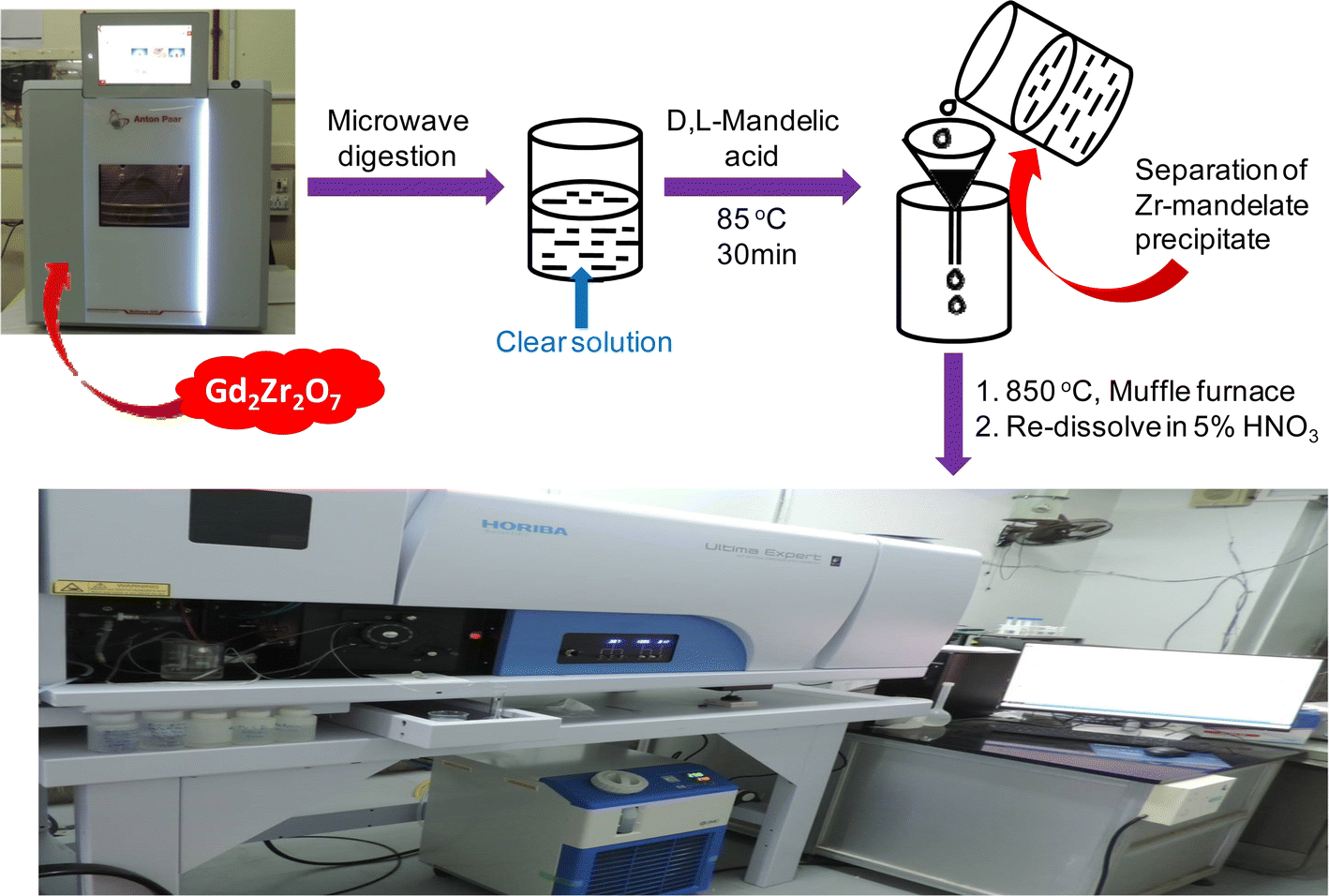
About 100 mg of Gd2Zr2O7 was taken in NXF100 Teflon vessel along with 3 mL of conc. H2SO4, 5 mL of conc. HCl, 1 mL of conc. HF and 1 mL of Milli-Q water. In case of spike addition appropriate volumes of 1000 mg L−1 individual rare earth ICP-standard solutions were added along with the sample and reagents into the NXF100 Teflon vessel. All eight vessels along with their attachments were kept in the microwave at 700 W for 30 min and then at 1400 W for 60 min. The complete dissolution parameters have been discussed in Table 2 in the results and discussion section. The digested sample was then taken out in a beaker and heated at 275 °C in a fume hood for 1 h. The fumes came out during this heating procedure were trapped in a NaOH solution. This method removes all the HF and HCl from the solution leaving all the elements in the H2SO4 medium. To this 20 mL of 6.0 mol L−1 HCl solution was added and then zirconium was precipitated using mandelic acid. The rest of the procedure followed was as discussed above. The proposed analytical methodology is presented as a scheme in Fig. 1.
Table 2 Optimization of the microwave assisted digestion parameters of Gd2Zr2O7
| Sample amount (mg) |
Power |
Ramp (min) |
Hold time |
Reagents |
Remarks |
| Conc. H2SO4 |
Conc. HCl |
Conc. HF |
Milli-Q water |
| ∼100 |
First at 700 W and then at 1400 W |
15 |
30 min at 700 W and 60 min at 1400 W |
1 |
9 |
0 |
0 |
Not dissolved |
| 1 |
8 |
0.5 |
0.5 |
Not dissolved |
| 1 |
7 |
1 |
1 |
Not dissolved |
| 2 |
6 |
1 |
1 |
Partially dissolved |
| 3 |
5 |
1 |
1 |
Completely dissolved |
| 4 |
4 |
1 |
1 |
Completely dissolved |
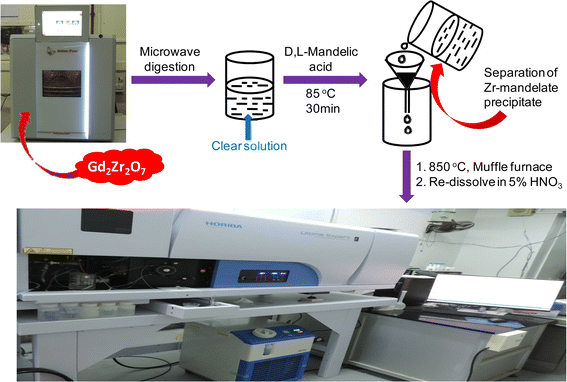 |
| | Fig. 1 Schematic representation of the proposed analytical methodology for trace rare earth determination in Gd2Zr2O7. | |
3. Results and discussion
3.1. Optimization of microwave digestion procedure
The highly refractory Gd2Zr2O7 needs an extreme environment to get completely dissolved. One such method is the fusion method, but this has the possibility of impurity ingression from a large excess of fusion mixture and loss of sample due to sputtering. On the other hand, a microwave digestion system using Suprapur acids provides a clean and closed environment with high temperature and high pressure for the digestion of materials. Hence, we chose the microwave digestion procedure for digesting our sample. A combination of conc. H2SO4, conc. HCl and conc. HF was used to digest the sample. Sulphuric acid was chosen because of its high boiling point which enabled us to reach either a high temperature of 240 °C (maximum pressure 45 bar) or high pressure of 60 bar (maximum temperature 180 °C) inside the Teflon vessels. Both hydrochloric and hydrofluoric acids are highly corrosive and hence they break the metal-oxide bonds to dissolve the material. The volumes of these acids were optimized by following these points: (i) minimum volume of H2SO4 was used so that maximum volumes of corrosive acids can be used; (ii) maximum volume of HCl was utilized because in this medium zirconium was precipitated; (iii) minimum volume of HF was added so that its removal became easy. Since the presence of fluoride ion in the solution could hamper zirconium precipitation, its complete removal was absolutely necessary. The low boiling point of HF (b.p. is near RT of pure HF) compared to H2SO4 (b.p. ∼ 337 °C) allowed the partial removal of the former acid at 275 °C.30 The heating was continued for prolonged time (1 h) to ensure the complete removal of fluoride ions as HF. Using all this information we optimized the proportion of acids as shown in Table 2. The power input was kept constant in all the trials.
3.2. Selection of emission lines
The rare earth elements are rich in atomic and ionic emission lines. However, the ionic lines have the highest theoretical intensities compared to the atomic ones. Hence, we selected the ionic lines for all thirteen rare earth elements. The lines were selected based on the criteria that they should have the maximum intensity irrespective of interference from Zr emission lines but have negligible interference from Gd emission lines. The selected lines are tabulated in Table T2 (ESI†) along with the interference information. Each selected line also has minimum interference of the remaining lanthanide emission lines. This has been observed by plotting the calibration spectra of all thirteen rare earth elements in the range 0.1–5.0 mg L−1 in the presence of each other, as shown in Fig. S1 (ESI†). All thirteen rare earths had a regression coefficient of ≥0.99.
3.3. Optimization of analyte and matrix concentrations
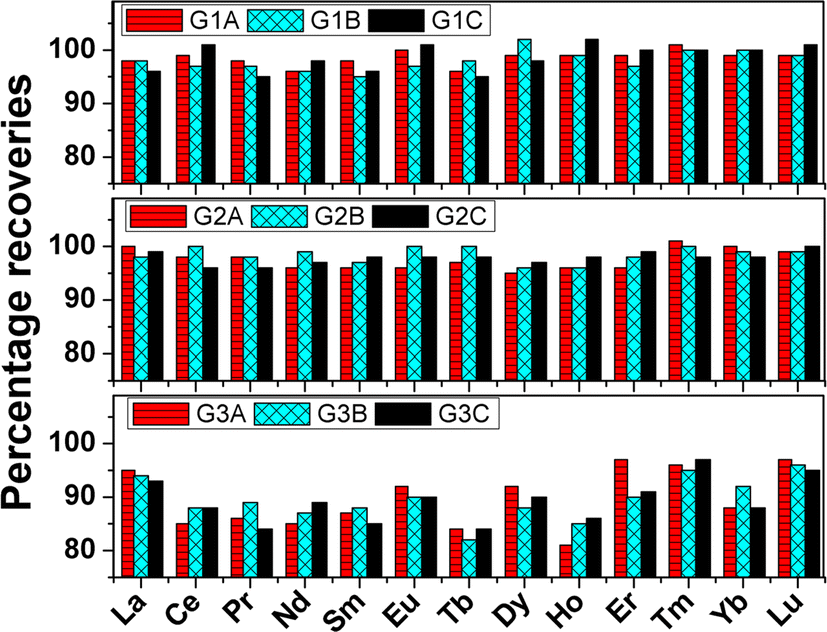
To overcome the hurdle of separating thirteen trace rare earth elements from gadolinium we thought of quantifying the analytes in the presence of the matrix element. However, one of the matrix elements i.e., zirconium was separated by using D,L-mandelic acid in the digested sample solution following the same procedure as Kumins et al.31 The effect of zirconium(IV) separation on the quantification of analytes was first examined by carrying out recovery studies on synthetic standards in Table T1 (see ESI†). The matrix concentrations in all the synthetic standards were maintained by calculating the mass fraction of every element and the dilution factor. The calculations are given in ESI.† The gadolinium and zirconium concentrations in all three synthetic standard sets viz., G1, G2, and G3 were 258 & 150, 516 & 300, and 1032 & 600 mg L−1 respectively. The analyte concentrations were varied in the range of 0.25 to 2.5 mg L−1 in all three sets. Once Zr(IV) was precipitated and filtered out, the raffinate was analyzed for the concentration of thirteen rare earths in the presence of gadolinium. The percentage recoveries of all thirteen rare earth elements in the nine synthetic standards are pictorially represented in Fig. 2. The recovered amounts of the individual elements in all nine synthetic standards are represented in Table T3 (ESI†). In the case of both sets G1 and G2, recoveries of ≥95% were obtained for all the thirteen rare earth elements. The raffinate was also treated with oxalic acid to precipitate Gd(III). Both the precipitate viz., Zr(IV)-mandelate and Gd(III)-oxalate were converted to their respective oxides in a Muffle furnace at 850 °C. The WDXRF spectra of both ZrO2 and Gd2O3 are shown in Fig. S2 and S3 (ESI†) respectively. The ZrO2 spectrum is free from any rare earth elements whereas the Gd2O3 spectrum shows the presence of all other rare earth elements of interest. Hence both the recovery and XRF studies confirmed that the precipitation of Zr(IV) has no adverse effect on the recovery of analytes.
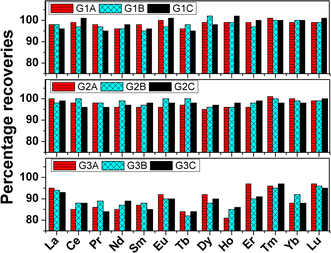 |
| | Fig. 2 Pictorial representation of percentage recoveries of thirteen rare earth elements in nine synthetic standards as prepared in Table T1 (ESI†). The three sets G1, G2, and G3 contain 258, 516, and 1032 mg L−1 gadolinium as matrix during ICP-OES determination. The sub-sets under these three sets were named as A, B, and C which contain thirteen trace rare earth elements according to Table T1 (ESI†). | |
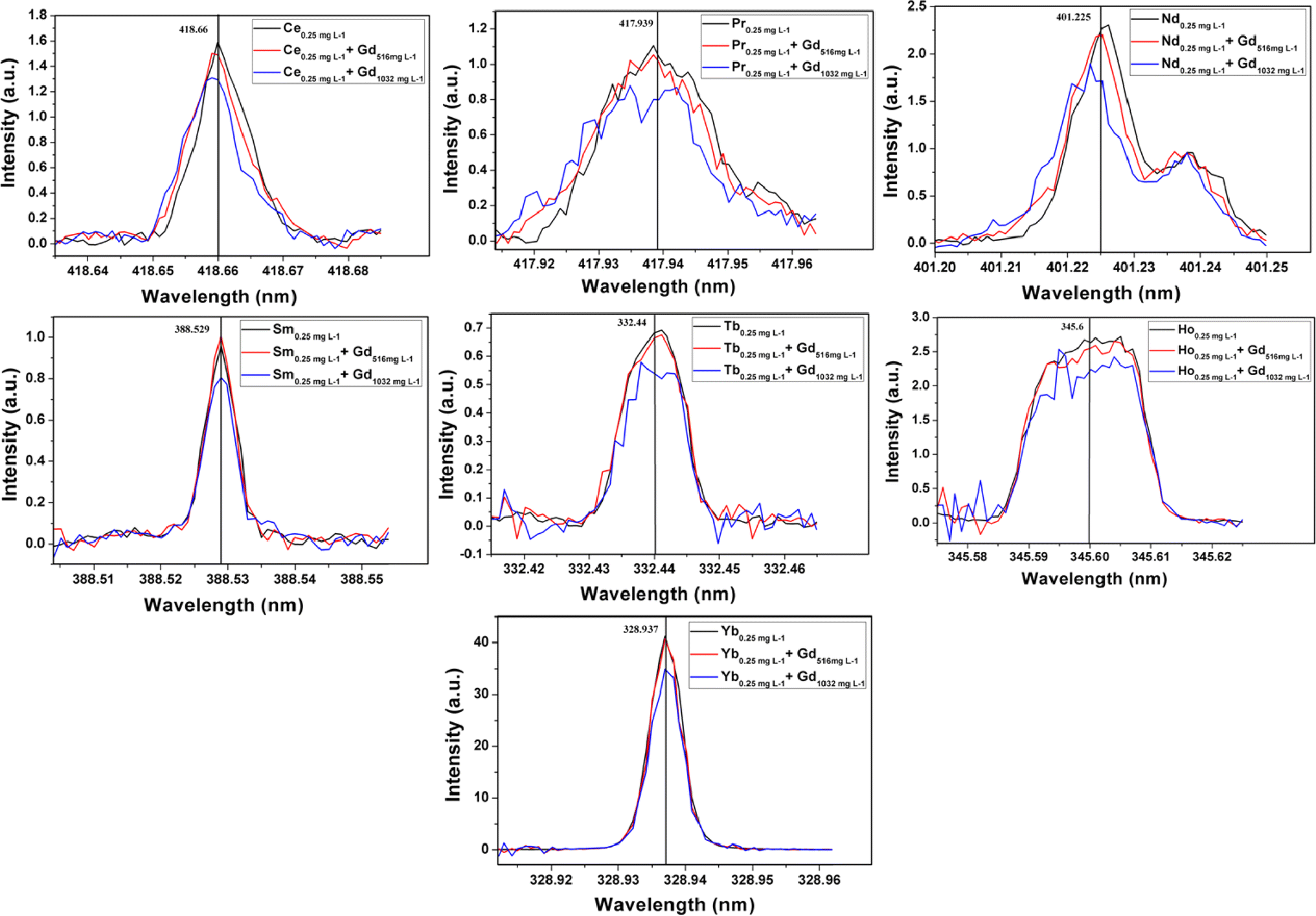
The emission spectra of all thirteen elements at 0.25 mg L−1 level in the absence and presence of gadolinium matrix are given in Fig. S4 and S5 (ESI†). From these figures, it can be seen that elements like Ce, Pr, Nd, Sm, Tb, Ho, and Yb need background correction. The background count for each element in the presence of a matrix was determined by analyzing pure synthetic solutions of 516 and 1032 mg L−1 of gadolinium. The gadolinium emission lines responsible for these blank counts are mentioned in Table T4 (ESI†). In the case of elements like Ce, Pr, Nd, and Sm the nearby single emission line of gadolinium increases the blank intensity. However, the emission lines of Tb and Ho are affected by two or more gadolinium emission lines. Only the emission intensities of Tm and Yb have direct interference from gadolinium emission lines (see Table T2 in ESI†). However, the emission spectra of Tm in the presence or absence of gadolinium matrix were found to be the same at 95% confidence interval, as shown in Fig. S4 (ESI†). The heavy rare earth elements have nearly filled or filled 4f orbitals which results in their highly intense emission lines. The intensity of Tm 313.125 nm line is so high that it remains unaltered by Gd 313.081 line at 0.25 mg L−1 level in the presence of 516 and 1032 mg L−1 of gadolinium. Hence Tm needs no background correction due to the presence of gadolinium matrix. The background corrected spectra of Ce, Pr, Nd, Sm, Tb, Ho, and Yb in the presence of 516 and 1032 mg L−1 of gadolinium are represented in Fig. 3. In the case of all thirteen rare earth elements the minimum analyte concentration of 0.25 mg L−1 was recovered up to ≥95% in presence of 516 mg L−1 of gadolinium. The higher matrix concentration of 1032 mg L−1 resulted in recoveries in between 80–97% for all thirteen analytes. Since the background intensities of Nd, Pr, Tb, and Ho in the presence of 1032 mg L−1 of Gd is nearly equal to or more than the matrix-free analyte intensities at 0.25 mg L−1 level, their recoveries were severely affected. Other elements like Ce, Sm, and Yb also resulted in poor recoveries at 1032 mg L−1 Gd concentration level due to high background intensities. Thus the optimization procedure revealed that a minimum of 0.25 mg L−1 of all thirteen rare earth elements can be precisely determined in the presence of 516 mg L−1 of Gd. This corresponded to the determination of 0.25 mg analyte per g of Gd2Zr2O7 by the proposed methodology.
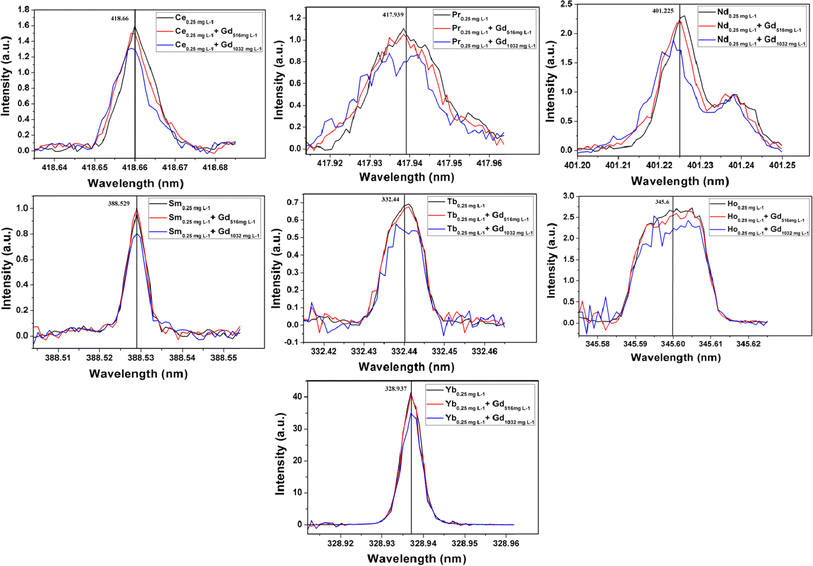 |
| | Fig. 3 Background intensity corrected emission spectra of Ce, Pr, Nd, Sm, Tb, Ho and Yb at 0.25 mg mL−1 level in absence and presence of gadolinium matrix. | |
3.4. Challenges of the procedure
The precise ICP-OES determination of trace rare earth elements in such a refractory matrix needs an efficient and clean dissolution. This difficulty was resolved by microwave assisted digestion of Gd2Zr2O7 using Suprapur acids. The separation of the matrix elements viz., Gd and Zr would nullify the spectroscopic and non-spectroscopic matrix interferences on the impurity analysis. Zr(IV) was separated by mandelate precipitation whereas Gd(III) separation was not performed due to its chemical similarities with trace rare earth impurities. This can be attributed as the limitation of the proposed methodology. The ICP-OES determination of trace rare earth impurities in presence of gadolinium matrix was done by optimizing the matrix concentration, suitable choice of analyte emission lines, and background correction.
3.5. Detection limit of the analysis
The detection limit was determined for both the 5% (v/v) HNO3 solution and sample blank known as instrument detection limit (IDL) and method detection limit (MDL) respectively. A 516 mg L−1 Gd solution was used as the sample blank. The 3σ values of both the blank solutions and the sensitivities (s) of the calibration curves in Fig. S1† were used to calculate the detection limits (3σ/s). The values of both IDL and MDL are mentioned in Table 3.
Table 3 IDL and MDL values
| Elements |
IDL (×10−3) (mg L−1) |
MDL (×10−3) (mg g−1) |
| La |
1.1 |
40 |
| Ce |
3.2 |
65 |
| Pr |
3.8 |
75 |
| Nd |
2.1 |
56 |
| Sm |
2.5 |
60 |
| Eu |
0.5 |
25 |
| Tb |
2.0 |
52 |
| Dy |
1.0 |
35 |
| Ho |
0.8 |
30 |
| Er |
1.5 |
47 |
| Tm |
0.4 |
20 |
| Yb |
0.2 |
12 |
| Lu |
0.2 |
10 |
3.6. Analysis of real sample
Two Gd2Zr2O7 samples were digested in duplicate using a microwave digestion procedure. In the case of each sample, one of the two sets contained only sample and the other set was spiked with the analytes of interest before digestion. All digested samples were subjected to the zirconium precipitation and then subsequent sample treatment. The analysis results are presented in Table 4. The recoveries of all thirteen analytes were found to be ≥95% in both samples. The percentage relative standard deviation (RSD) on the determined values was found to vary between 1 and 3%. These results validate the proposed methodology for its further use in routine analysis. The individual trace rare earth concentrations result in cumulative concentrations of 4.18 and 8.13 mg g−1 in Gd2Zr2O7-1 and Gd2Zr2O7-2 samples respectively. Such information is very much helpful in ascertaining the purity of the compound. The individual concentrations of the trace rare earths having high σa values viz., Sm, Eu, Tb, Dy, Ho, Er, Yb etc. will be helpful in calculating the total negative reactivity imposed by the two compounds.
Table 4 Analysis of real samples (N = 5)a
| Elements |
Gd2Zr2O7-1 |
Gd2Zr2O7-2 |
| Added (mg g−1) |
Found (mg g−1) |
Added (mg g−1) |
Found (mg g−1) |
| The percentage recoveries are given in parenthesis. BDL = below detection limit. |
| La |
0 |
BDL |
0 |
0.277 ± 0.006 |
| 0.25 |
0.248 ± 0.008 (99) |
0.25 |
0.517 ± 0.010 (96) |
| Ce |
0 |
0.824 ± 0.009 |
0 |
1.214 ± 0.016 |
| 0.25 |
1.068 ± 0.012 (98) |
0.25 |
1.456 ± 0.015 (97) |
| Pr |
0 |
BDL |
0 |
0.312 ± 0.008 |
| 0.25 |
0.242 ± 0.007 (97) |
0.25 |
0.559 ± 0.009 (99) |
| Nd |
0 |
0.343 ± 0.012 |
0 |
0.547 ± 0.009 |
| 0.25 |
0.588 ± 0.008 (98) |
0.25 |
0.786 ± 0.013 (96) |
| Sm |
0 |
0.408 ± 0.015 |
0 |
0.573 ± 0.009 |
| 0.25 |
0.648 ± 0.010 (96) |
0.25 |
0.814 ± 0.014 (96) |
| Eu |
0 |
0.377 ± 0.011 |
0 |
0.622 ± 0.015 |
| 0.25 |
0.615 ± 0.015 (95) |
0.25 |
0.873 ± 0.017 (100) |
| Tb |
0 |
0.766 ± 0.016 |
0 |
1.445 ± 0.022 |
| 0.25 |
1.011 ± 0.015 (98) |
0.25 |
1.687 ± 0.020 (97) |
| Dy |
0 |
0.654 ± 0.011 |
0 |
0.844 ± 0.018 |
| 0.25 |
0.897 ± 0.018 (97) |
0.25 |
1.088 ± 0.020 (98) |
| Ho |
0 |
0.542 ± 0.010 |
0 |
0.776 ± 0.016 |
| 0.25 |
0.780 ± 0.016 (95) |
0.25 |
1.021 ± 0.018 (98) |
| Er |
0 |
0.288 ± 0.008 |
0 |
0.463 ± 0.009 |
| 0.25 |
0.536 ± 0.013 (99) |
0.25 |
0.701 ± 0.011 (95) |
| Tm |
0 |
BDL |
0 |
0.357 ± 0.008 |
| 0.25 |
0.246 ± 0.007 (98) |
0.25 |
0.609 ± 0.009 (101) |
| Yb |
0 |
BDL |
0 |
0.313 ± 0.007 |
| 0.25 |
0.252 ± 0.010 (101) |
0.25 |
0.562 ± 0.009 (100) |
| Lu |
0 |
BDL |
0 |
0.386 ± 0.009 |
| 0.25 |
0.251 ± 0.008 (100) |
0.25 |
0.632 ± 0.011 (98) |
4. Conclusion
The determination of thirteen trace rare earth impurities in Gd2Zr2O7 has been reported for the first time in literature. Since the measurement was carried out by separating only zirconium, the presence of gadolinium matrix was optimized to obtain accurate results. The use of ICP-OES was found to provide precise analyte concentrations even in the presence of high matrix levels. The measurement is necessary to calculate the exact negative reactivity of the BA material before putting it into the reactor. This developed methodology can be applied to characterize any A2Zr2O7 type of compounds (where A = rare earth element) which have applications in the fields of solid oxide fuel cells, nuclear waste storage, thermal barrier coating, and nuclear reactor BA or control rod materials. The microwave-assisted dissolution of this refractory BA material is also reported first time in the literature. The developed digestion procedure may also help in carrying out other studies on this BA material. The methodology is free from generating any waste. To the best of the authors' knowledge, this is the first report on analyzing trace rare earth impurities in Gd2Zr2O7 by ICP-OES.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to Dr P. K. Mohapatra, Associate Director, RC & IG, BARC for his constant encouragement throughout the work.
References
- J. A. Evans, M. D. DeHart, K. D. Weaver and D. D. Keiser Jr, Nucl. Eng. Des., 2022, 391, 111726 CrossRef.
- H. S. Kim, C. Y. Joung, B. H. Lee, S. H. Kim and D. S. Sohn, J. Nucl. Mater., 2008, 372, 340–349 CrossRef CAS.
- M. T. Malachevsky, D. R. Salvador, S. Leiva and C. A. D'Ovidio, J. Ceram., 2015, 2015, 298690 Search PubMed.
- D. Lanning, D. Baldwin, M. Cunningham and S. Marschman, Post irradiation examination report for the tritium-producing burnable absorber rods irradiated in the Watts Bar Nuclear Plant, ed. U. S. Nrc, Pacific Northwest National Laboratory, 2002, p. 72 Search PubMed.
- E. P. Naessens, K. S. Allen and B. E. Moretti, Nucl. Sci. Eng., 2006, 152(3), 306–313 CrossRef CAS.
- I. Shaaban and M. Albarhoum, Appl. Radiat. Isot., 2017, 125, 188–195 CrossRef CAS PubMed.
- T. Cardinaels, J. Hertog, B. Vos, L. de Tollenaere, C. Delafoy and M. Verwerft, J. Nucl. Mater., 2012, 424(1–3), 289–300 CrossRef CAS.
- T. Jevremovic, Nuclear Principles in Engineering, Springer, 2nd edn, 2009 Search PubMed.
- H. van Dam, Nucl. Energy, 2000, 27, 63–69 CrossRef CAS.
- J. Porta, S. Baldi, J. P. Chauvin and Ph. Fougeras, Prog. Nucl. Energy, 2001, 38, 355–358 CrossRef CAS.
- M. Asou and J. Porta, Nucl. Eng. Des., 1997, 168(1–3), 261–270 CrossRef CAS.
- V. D. Risovany, E. E. Varlashova and D. N. Suslov, J. Nucl. Mater., 2000, 281, 84–89 CrossRef CAS.
- G. Panneerselvam, R. Venkata Krishnan, M. P. Antony, K. Nagarajan, T. Vasudevan and P. R. Vasudeva Rao, J. Nucl. Mater., 2004, 327, 220–225 CrossRef CAS.
- H. S. Kim, C. Y. Joung, S. H. Kim, B. H. Lee, Y. W. Lee, D. S. Sohn and S. H. Lee, J. Korean Ceram. Soc., 2002, 39(11), 1108–1112 CrossRef CAS.
- A. Saha, S. B. Deb, B. K. Nagar and M. K. Saxena, Spectrochim. Acta Part B At. Spectrosc., 2014, 94–95, 14–21 CrossRef CAS.
- W. J. Weber, J. W. Wald and Hj. Matzke, J. Nucl. Mater., 1986, 138, 196–209 CrossRef CAS.
- W. L. Gong, W. Lutze and R. C. Ewing, J. Nucl. Mater., 2000, 277, 239–249 CrossRef CAS.
- A. J. Feighery, J. T. S. Irvine and C. Zheng, J. Solid State Chem., 2001, 160, 302–306 CrossRef CAS.
- A. Saha, S. B. Deb and M. K. Saxena, J. Anal. At. Spectrom., 2016, 31, 1480–1489 RSC.
- J. W. Olesik and S. Jiao, J. Anal. At. Spectrom., 2017, 32, 951–966 RSC.
- C. Agatemor and D. Beauchemin, Anal. Chim. Acta, 2001, 706(1), 66–83 CrossRef PubMed.
- A. Saha, S. B. Deb, B. K. Nagar and M. K. Saxena, At. Spectrosc., 2013, 34(4), 125–132 CrossRef CAS.
- K. Sanyal, A. Saha, A. Sarkar, S. B. Deb, R. V. Pai and M. K. Saxena, ACS Omega, 2023, 8(44), 41402–41410 CrossRef CAS PubMed.
- C. Bowers, J. Chem. Educ., 2019, 96(11), 2597–2599 CrossRef CAS.
- B. Ticová, K. Novotný and V. Kanický, Chem. Pap., 2019, 73, 2913–2921 CrossRef.
- J. C. Fariñas, I. Rucandio, M. S. Pamarer-Alfonso, M. E. Villanueva-Tagle and M. T. Larrea, Talanta, 2016, 154, 53–62 CrossRef PubMed.
- F. Ardini, F. Soggia, F. Rugi, R. Udisti and M. Grotti, Anal. Chim. Acta, 2010, 678(1), 18–25 CrossRef CAS PubMed.
- J. A. Gásquez, E. DeLima, R. A. Olsina, L. D. Martinez and M. de la Guardia, Talanta, 2005, 67(4), 824–828 CrossRef PubMed.
- L. Bendakovská, A. Krejčová, T. Černohorský and J. Zelenková, Chem. Pap., 2016, 70(9), 1155–1165 Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of elements, Pergamon Press, Oxford, 1984, p. 921 Search PubMed.
- C. A. Kumins, Anal. Chem., 1947, 19(6), 376–377 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Khushboo Kumari,
Sadhan Bijoy Deb* and
Manoj Kumar Saxena
*,
Khushboo Kumari,
Sadhan Bijoy Deb* and
Manoj Kumar Saxena