DOI:
10.1039/D4RA05517A
(Paper)
RSC Adv., 2024,
14, 29789-29799
Discovery of novel SS-31 (D-Arg-dimethylTyr-Lys-Phe-NH2) derivatives as potent agents to ameliorate inflammation and increase mitochondrial ATP synthesis†
Received
30th July 2024
, Accepted 12th September 2024
First published on 18th September 2024
Abstract
Neuroinflammation and mitochondrial function are crucial for neuronal function and survival. SS-31 is a novel mitochondria-targeted peptide antioxidant that reduces mitochondrial reactive oxygen species production, increases ATP generation, protects the integrity of mitochondrial cristae and the mitochondrial respiratory chain, and reduces inflammatory responses. Exploring novel SS-31 derivatives is important for the treatment of neurodegenerative diseases. In this study, nineteen SS-31 derived peptides (5a-5s) were synthesized. Through cellular activity screening, we discovered that 5f and 5g exhibited significantly greater anti-inflammatory activity compared to SS-31, reducing LPS-induced TNF-α levels by 43% and 45%, respectively, at a concentration of 10 μM. Furthermore, treatment with 50 nM of 5f and 5g increased ATP synthesis by 42% and 41% in rotenone-induced HT22 cells and attenuated mitochondrial ROS production by preserving mitochondrial integrity. These findings demonstrate their direct protective effects on neuronal mitochondria. This work highlights the potential of 5f and 5g in the treatment of neurodegenerative diseases associated with inflammation and mitochondrial damage, offering a promising therapeutic avenue for mitochondrial-related conditions such as Alzheimer's disease.
1 Introduction
Neuroinflammation is the brain's defense mechanism and is implicated in the development of various brain disorders. 1–3 The brain comprises nerve cells and glial cells, including microglia, oligodendrocytes, and astrocytes. When harmful pathogens invade, these support cells, particularly astrocytes, and microglia, become activated. 4 Excessive activation of these cells results in the release of various inflammatory mediators, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6). 5 Research indicates that these mediators play a crucial role in the initiation, detection, and treatment of brain disorders. Furthermore, they contribute to the pathogenesis of diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), multiple sclerosis (MS), stroke, and others. 6–10 Therefore, investigating substances that inhibit inflammatory responses is essential for the treatment of neurodegenerative diseases.
The mitochondrion, a membrane-bound organelle, is widespread in eukaryotic cells. It serves as the primary hub for energy transfer between intracellular and extracellular compartments and plays crucial roles in cell function and apoptosis. 11–13 However, mitochondria also significantly contribute to the production of reactive oxygen species (ROS) as a byproduct when there is an electron transport chain (ETC) deficiency. 14,15 Mitochondrial ROS mediates the activation of apoptotic pathways and various inflammatory cytokines, leading to neuronal injury. 16,17 Oxidative damage is implicated in clinical conditions such as neurodegenerative diseases, ischemia-reperfusion injury, diabetes, and heart failure. 18,19 Thus, mitigating mitochondrial oxidative stress and preserving mitochondrial function offer potential therapeutic strategies.
SS-31(D-Arg-Dmt-Lys-Phe-NH2), also known as elamipretide, is a mitochondria-targeted tetrapeptide that has garnered significant attention in biomedical research due to its potential therapeutic effects on mitochondrial dysfunction. SS-31 selectively targets the inner mitochondrial membrane where it binds to cardiolipin, a phospholipid crucial for mitochondrial function. By stabilizing cardiolipin and maintaining mitochondrial membrane integrity, SS-31 improves mitochondrial bioenergetics and reduces the production of ROS. These actions help mitigate mitochondrial dysfunction, which is implicated in a variety of diseases. SS-31 also prevents the opening of the mitochondrial permeability transition pore, which occurs during mitochondrial stress, such as in traumatic brain injury, stroke, and neurodegenerative diseases. 20 Recent studies have demonstrated the potential anti-inflammatory properties of SS-31, which include the inhibition of NLRP3 inflammasome activation, the suppression of lipopolysaccharide (LPS)-stimulated IL-1β production in macrophages, and the prevention of mitochondrial hyperfission. 21–23
Traditional treatments, such as Coenzyme Q10 and MitoQ, while effective in mitigating oxidative stress, do not specifically target mitochondrial membrane components like cardiolipin. Peptides like SS-20 and SS-22, which are also designed to address mitochondrial dysfunction, may lack the refined targeting and enhanced stability exhibited by SS-31. 24 SS-31 is primarily investigated for its role in mitochondrial protection and has potential applications in various conditions involving mitochondrial dysfunction, such as cardiovascular diseases, neurodegenerative disorders, and muscle dystrophies. In cases where resistance to SS-31 might develop, new derivatives can offer alternative options that bypass resistance mechanisms, ensuring continued therapeutic efficacy. Novel SS-31 derivatives are engineered to more precisely interact with cardiolipin, potentially leading to more targeted mitochondrial protection. In order to improve the therapeutic effectiveness of SS-31, we designed and synthesized nineteen novel derivatives of SS-31. Analytical RP-HPLC and ESI-MS were utilized to confirm the correctness of the obtained peptides. The anti-inflammatory activity and the ability to restore ATP synthesis were also investigated.
2 Results and discussions
2.1 Design and synthesis of SS-31-derived peptides
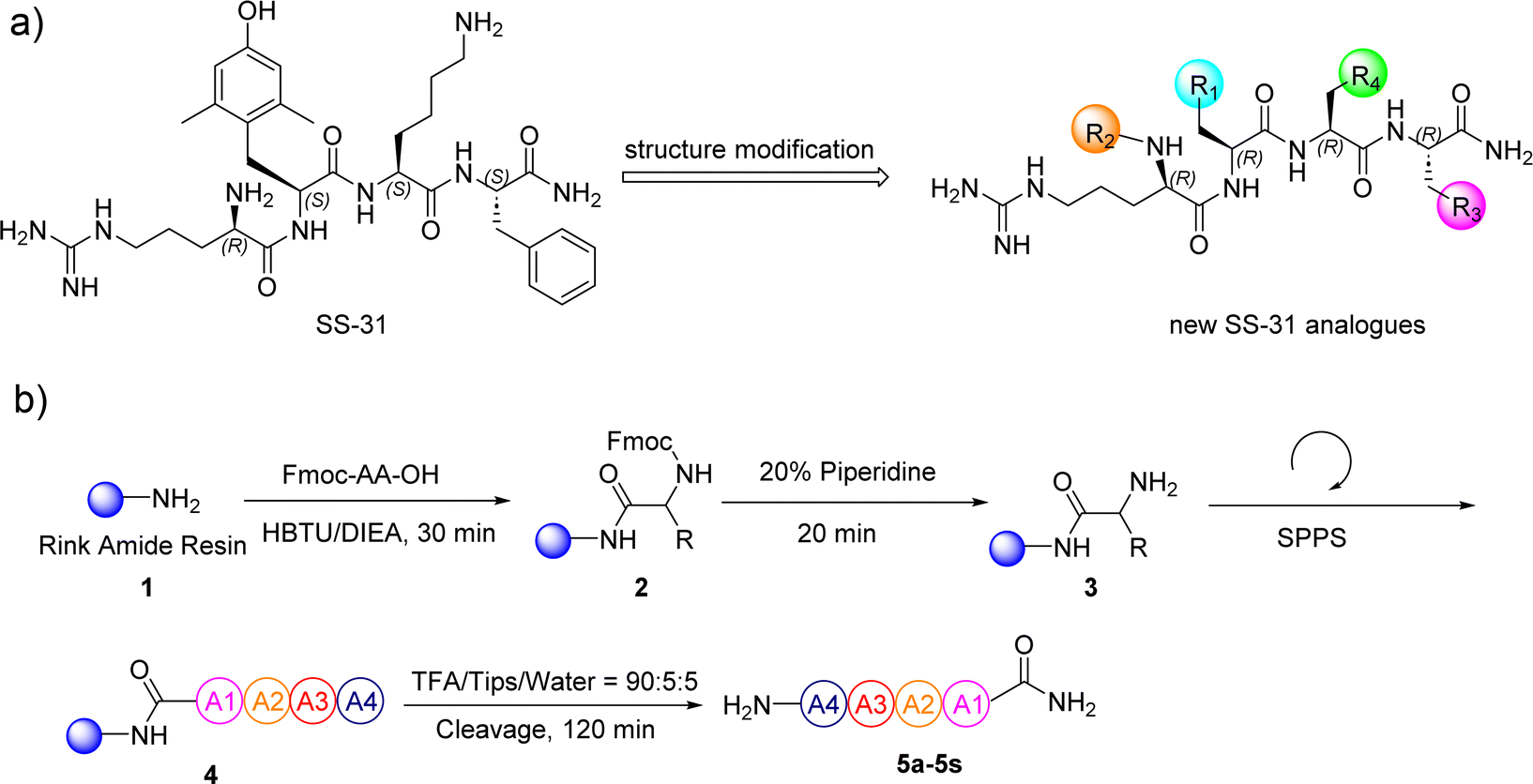
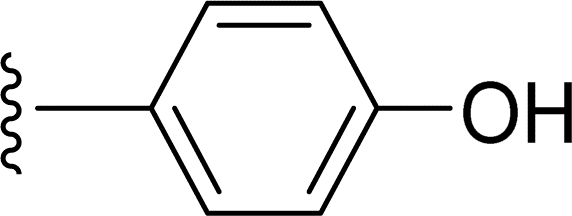
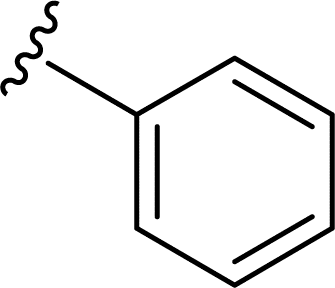
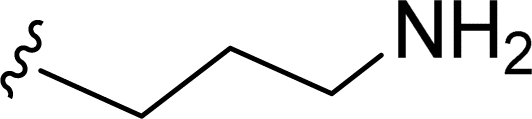
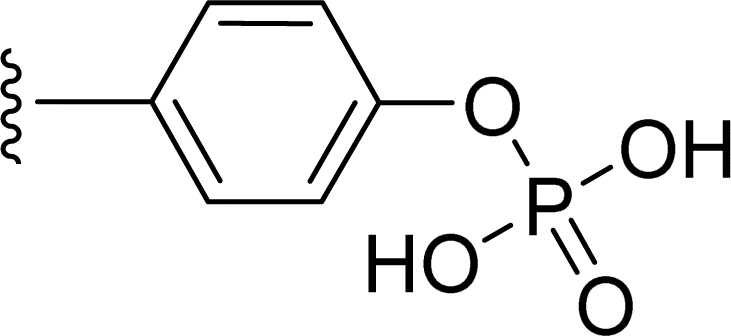
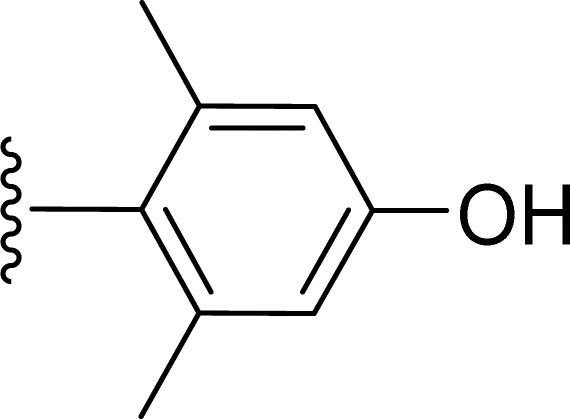
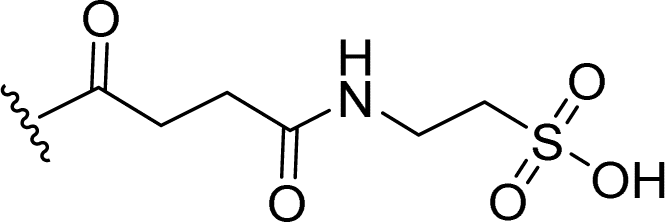
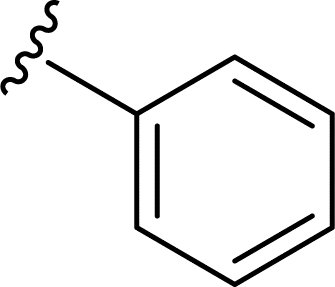
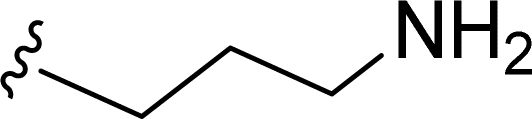
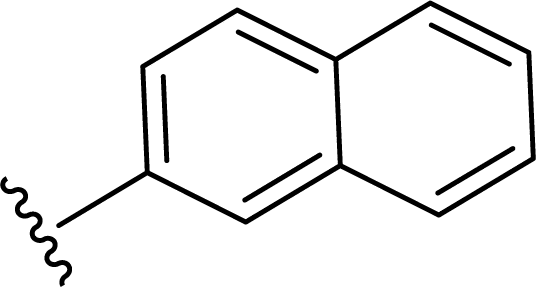
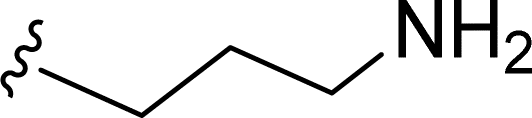
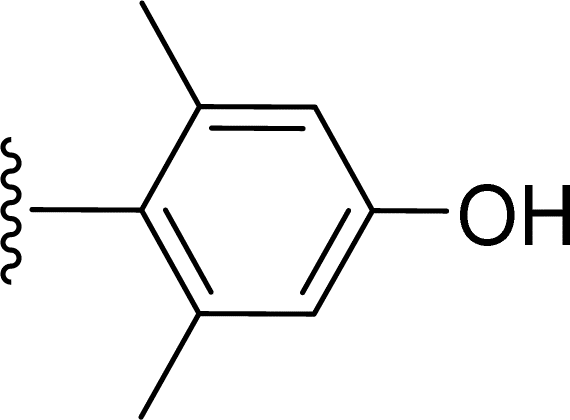
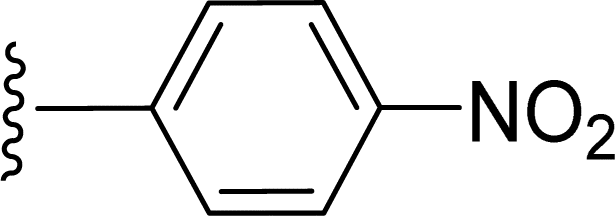
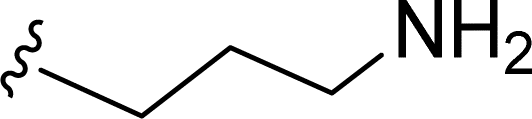
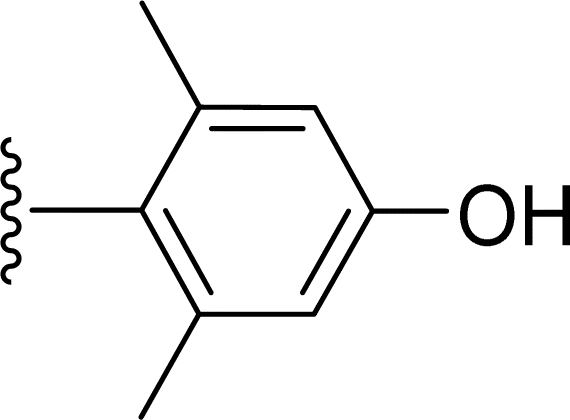
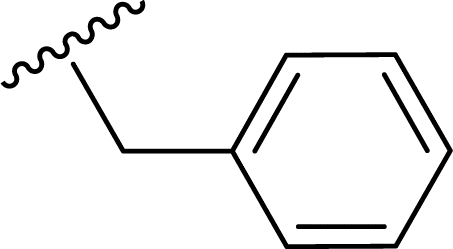
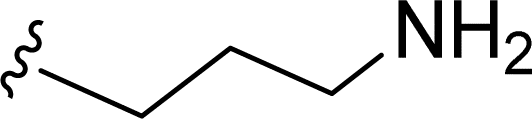
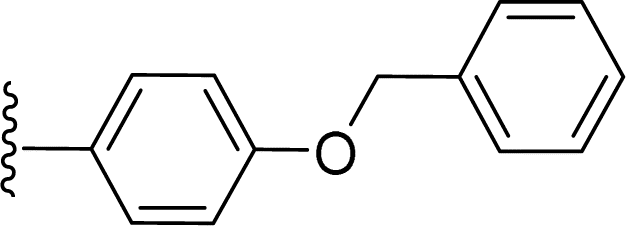
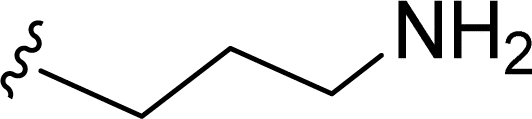
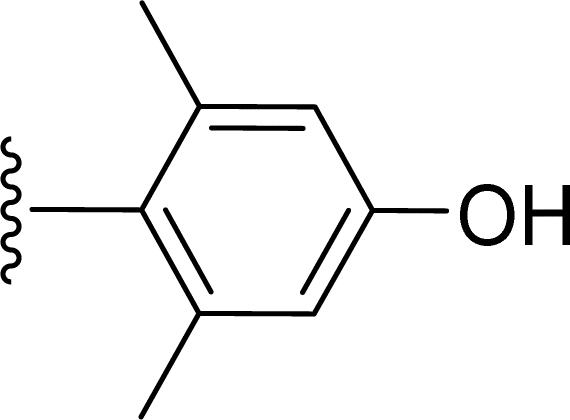
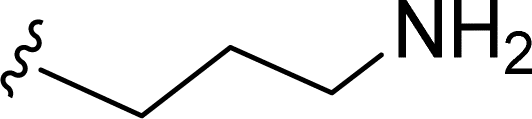
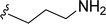
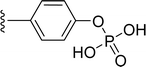
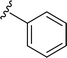
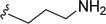
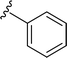
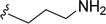
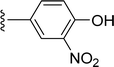
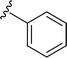
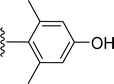
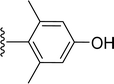
SS-31 possesses a highly polar peptide backbone and a net charge of +3 due to the N-terminal amino group and the side chains of Arg and Lys. Recent studies have shown that SS-31 selectively binds to cardiolipin via both electrostatic and hydrophobic interactions. 24 Dmt, an amino acid residue of SS-31, is known for its ability to scavenge free radicals. 25 SS-31 has been demonstrated to dose-dependently scavenge hydrogen peroxide, hydroxyl radical, and peroxynitrite in vitro.26 Based on these findings, we retained the positive charge and free radical scavenging properties of SS-31 and performed structural modifications on specific amino acids. By modifying the Dmt residue, we synthesized a series of peptides 5a-5e. A taurine-modifying group was introduced to the arginine residue, which regulates mitochondrial function, yielded 5f. Structural modifications of phenylalanine led to the synthesis of 5g-5p, and modifications of lysine produced 5q-5s (Fig. 1A).
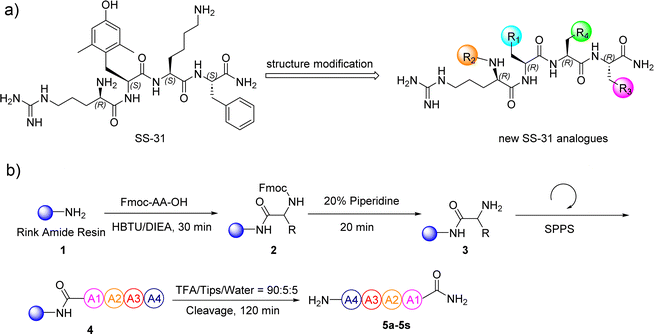 |
| | Fig. 1 (a) The modification strategy for SS-31. (b) The general synthetic procedure for SS-31 analogs. | |
In this study, nineteen SS-31-derived peptides 5a-5s were synthesized using standard 9-fluorenylmethyloxycarbonyl (Fmoc)-based solid-phase peptide synthesis (SPPS) (Fig. 1B and Table 1). Following sequential manual SPPS, peptide cleavage, semi-preparative RP-HPLC purification, and lyophilization, the target peptides were obtained. Each synthetic peptide was produced on a scale of more than twenty milligrams, with RP-HPLC purity above 95%. The synthesized peptides were systematically characterized by analytical RP-HPLC and ESI-MS (Table 2). Detailed spectra of the original analytical RP-HPLC and ESI-MS for each synthetic peptide are provided in Fig. S2–S39.†
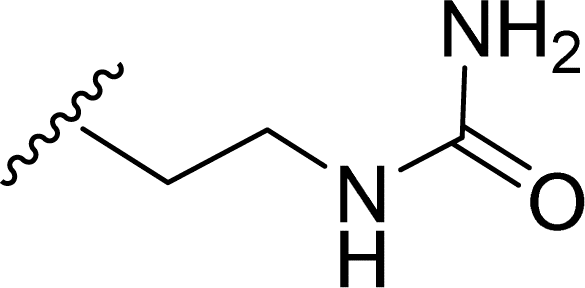
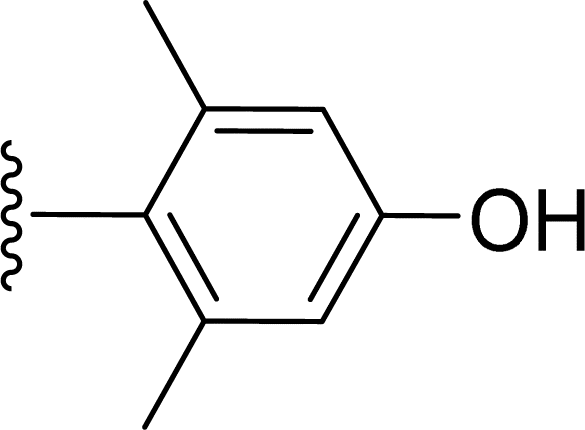
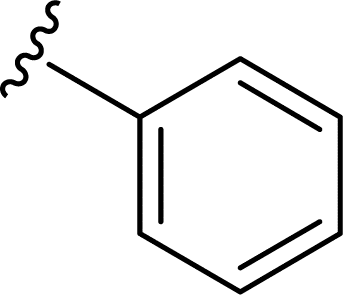
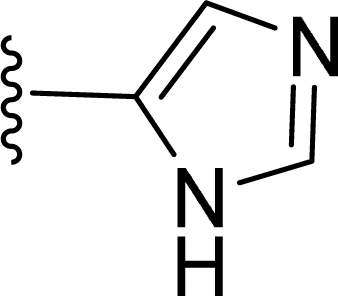
Table 1 Molecular structures of 5a-5s
| Peptides |
R1 |
R2 |
R3 |
R4 |
| 5a |
 |
H |
 |
 |
| 5b |
 |
H |
 |
 |
| 5c |
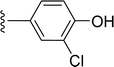
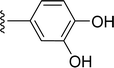 |
H |
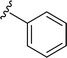 |
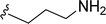 |
| 5d |
 |
H |
 |
 |
| 5e |
 |
H |
 |
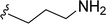 |
| 5f |
 |
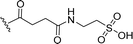 |
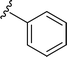 |
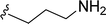 |
| 5g |
 |
H |
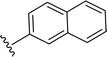 |
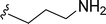 |
| 5h |
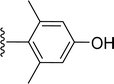 |
H |
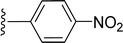 |
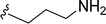 |
| 5i |
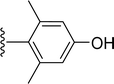 |
H |
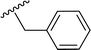 |
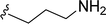 |
| 5j |
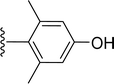 |
H |
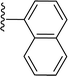 |
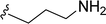 |
| 5k |
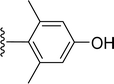 |
H |
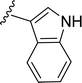 |
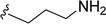 |
| 5l |
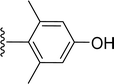 |
H |
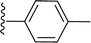 |
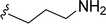 |
| 5m |
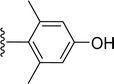 |
H |
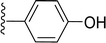 |
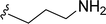 |
| 5n |
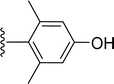 |
H |
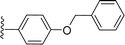 |
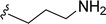 |
| 5o |
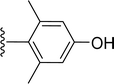 |
H |
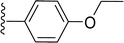 |
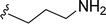 |
| 5p |
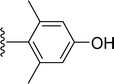 |
H |
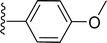 |
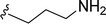 |
| 5q |
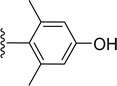 |
H |
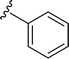 |
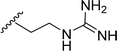 |
| 5r |
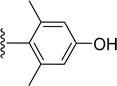 |
H |
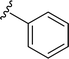 |
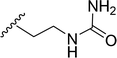 |
| 5s |
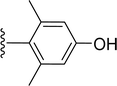 |
H |
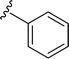 |
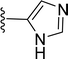 |
Table 2 The synthetic results for SS-31derived peptides
| Peptides |
Rt (min)a |
ESI-MS (Da)b |
| Calculated mass |
Observed mass |
| Rt, retention time in analytical RP-HPLC. RP-HPLC conditions are listed in the ESI. Calculated and observed molecular weight and average isotopes. |
| 5a |
11.198 |
611.748 |
611.495 |
| 5b |
11.764 |
691.727 |
691.598 |
| 5c |
11.098 |
627.747 |
627.519 |
| 5d |
9.412 |
646.190 |
645.421 |
| 5e |
13.988 |
656.745 |
656.939 |
| 5f |
13.077 |
847.002 |
846.205 |
| 5g |
15.884 |
689.862 |
689.634 |
| 5h |
9.722 |
684.799 |
684.590 |
| 5i |
10.261 |
653.829 |
653.620 |
| 5j |
9.735 |
689.862 |
689.656 |
| 5k |
5.702 |
678.839 |
678.695 |
| 5l |
9.928 |
653.829 |
653.649 |
| 5m |
11.075 |
655.801 |
655.664 |
| 5n |
9.790 |
745.926 |
745.666 |
| 5o |
10.380 |
683.855 |
683.676 |
| 5p |
13.448 |
669.828 |
669.665 |
| 5q |
10.570 |
667.816 |
667.661 |
| 5r |
9.752 |
668.800 |
668.680 |
| 5s |
9.722 |
648.769 |
648.627 |
2.2 Inhibition of LPS-induced inflammatory response in BV2 cells by SS-31 derived peptides
Microglia are the primary immune cells of the nervous system, playing a crucial role in the initiation and perpetuation of inflammatory responses. It is known that SS-31 can inhibit the LPS-induced inflammatory response in microglia. 27 To evaluate the activity of SS-31 derivatives, we assessed the effects of 5a-5s on the LPS-induced inflammatory response in the BV2 microglial cell line. Ginsenoside Rb1, a major active component of ginseng, has been previously demonstrated to exhibit anti-inflammatory activity by inhibiting LPS-induced activation of microglia. 28 Prior research by the authors suggests that Rb1 can suppress the LPS-induced inflammatory response in macrophages; 29 therefore, we selected Rb1 as a positive control.
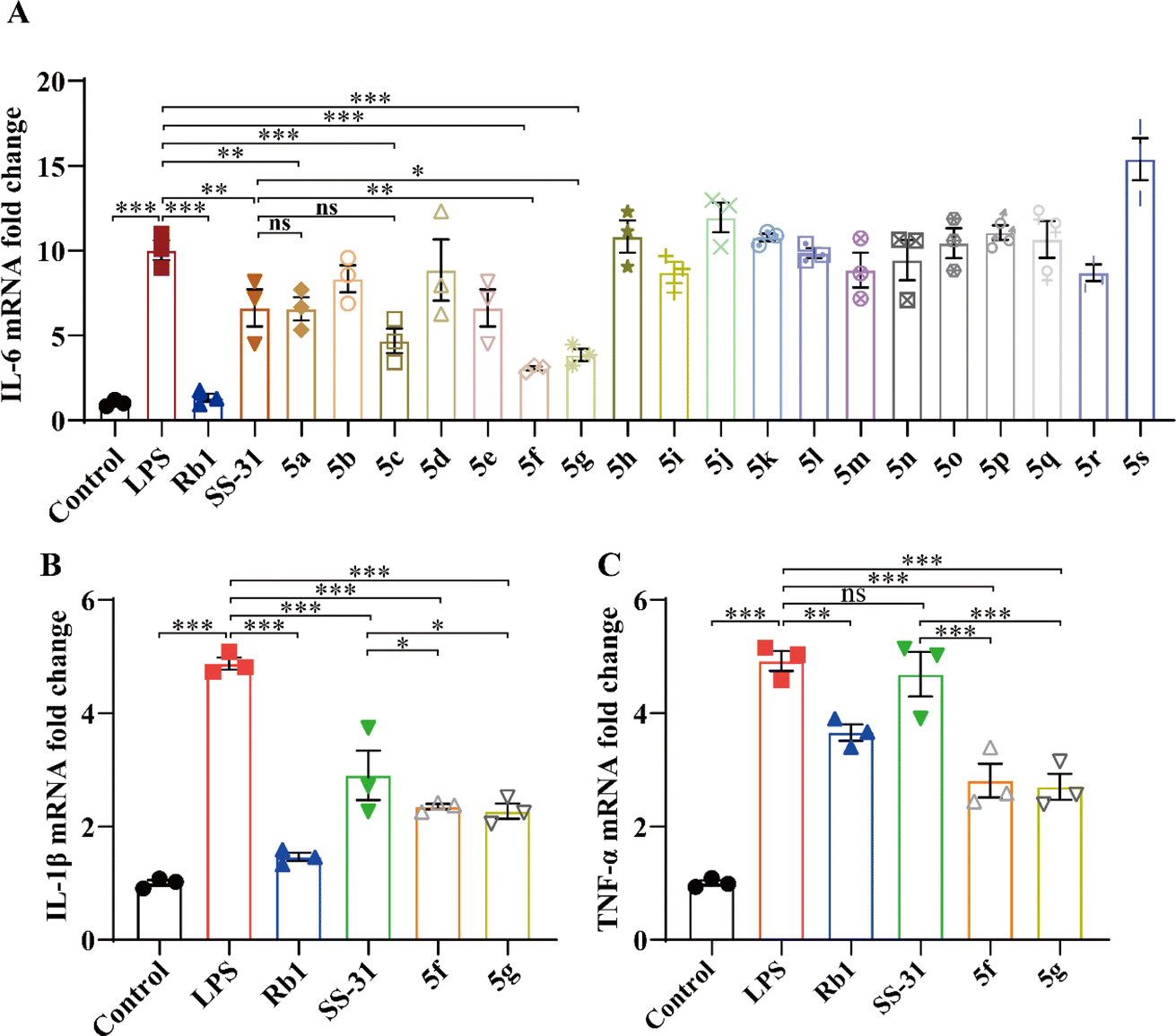
As shown in Fig. 2A, LPS induced an increase in IL-6 mRNA expression, which was significantly inhibited by SS-31 and its derivatives. Notably, 5f and 5g exhibited more pronounced inhibitory effects compared to SS-31. To further validate the anti-inflammatory activity of 5f and 5g, we measured the expression levels of the inflammatory cytokines IL-1β and TNF-α. As depicted in Fig. 2B and C, both SS-31, 5f, and 5g inhibited the LPS-induced upregulation of IL-1β expression. Interestingly, 5f and 5g demonstrated superior anti-inflammatory activity compared to SS-31. Importantly, while SS-31 did not significantly inhibit the LPS-induced increase in TNF-α expression, both 5f and 5g successfully suppressed the LPS-induced upregulation of TNF-α expression. These results collectively indicate that 5f and 5g exhibit more significant anti-inflammatory activity than SS-31. The toxicity of SS-31, 5f, and 5g in HT22 cells was assessed; even at a concentration of 100 μM, 5f and 5g showed no significant toxicity (Fig. S1†).
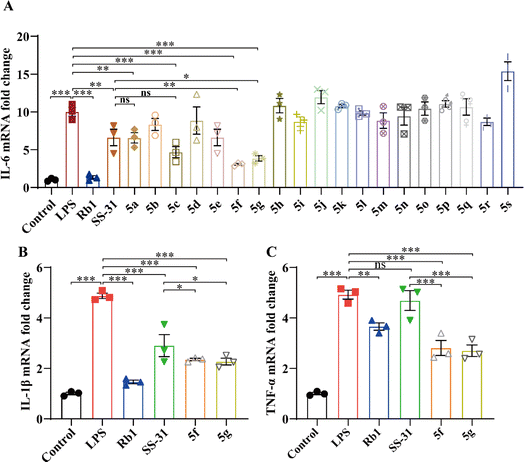 |
| | Fig. 2 BV2 cells were treated with LPS (5 ng mL−1) for 24 hours, followed by intervention with Rb1 (100 μM), SS-31, and its derivatives (10 μM). Real-time qPCR was used to measure changes in mRNA expression of IL-6 (A), IL-1β (B), and TNF-α (C) mRNA. Experimental data are presented as mean ± standard error of the mean (Mean ± S.E.M) (n = 3).***, P < 0.001; **, P < 0.01; *, P < 0.05, ns, no significance. | |
2.3 5f and 5g inhibited mitochondrial damage in LPS-induced BV2 cells
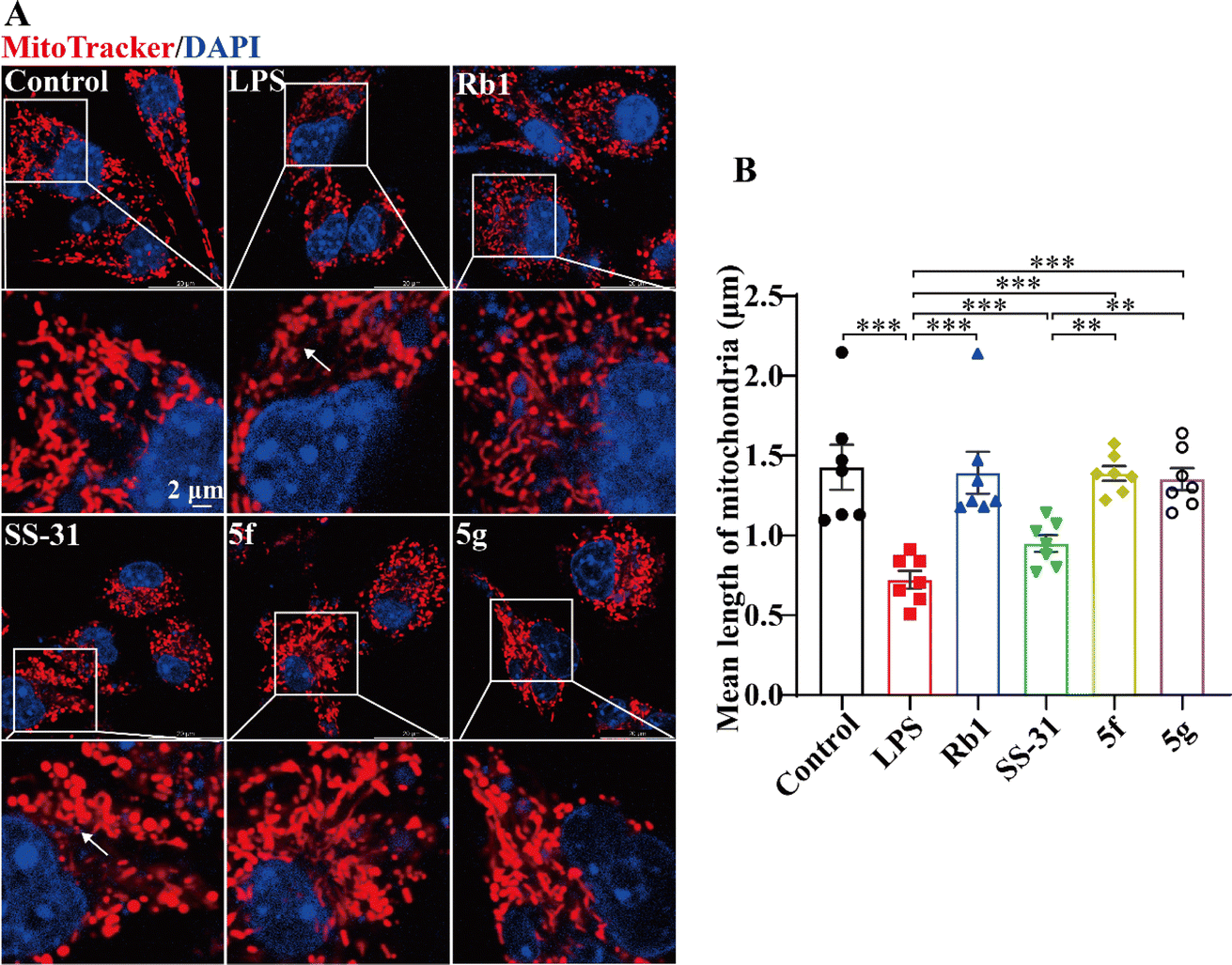
Previous studies have demonstrated that LPS can induce mitochondrial damage in BV2 cells, leading to mitochondrial fragmentation. 30–32 SS-31 has been shown to exert anti-inflammatory activity by protecting mitochondria. 33,34 We analyzed the protective effects of SS-31, 5f, and 5g on mitochondria. Mitochondria were labeled with MitoTracker™. Cells with elongated and highly interconnected mitochondria were classified as tubular, while those with small and spherical mitochondria were classified as fragmented. As shown in Fig. 3A, LPS-treated BV2 cells exhibited a reduction in tubular mitochondrial structures and an increase in fragmented mitochondria. SS-31, 5f, and 5g significantly inhibited LPS-induced mitochondrial fragmentation. As shown in Fig. 3B, statistical analysis of at least 60 mitochondria per group revealed that LPS-treated BV2 cells exhibited a decrease in mitochondrial length, which was mitigated by SS-31, 5f, and 5g. Furthermore, compared to SS-31, 5f and 5g better maintained mitochondrial morphology, showing protective effects comparable to the positive control, Rb1. These results suggest that the superior anti-inflammatory activity of 5f and 5g is based on their enhanced mitochondrial protective effects.
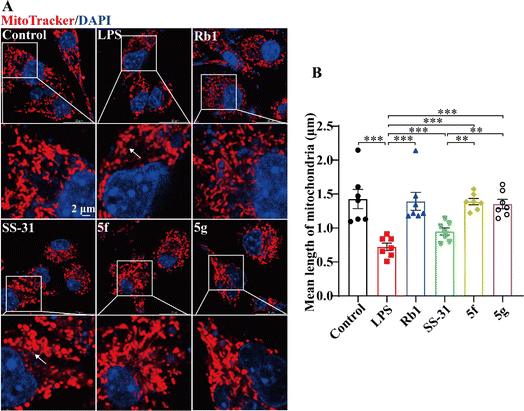 |
| | Fig. 3 5f and 5g inhibited mitochondrial network fragmentation in LPS-induced BV2 cells. BV2 cells were treated with 5 ng mL−1 LPS for 24 hours, followed by intervention with 100 μM Rb1 and 10 μM SS-31, 5f, or 5g. (A) Representative images of mitochondria labeled with MitoTracker™ (red) and nuclei labeled with Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 (blue), with arrows indicating fragmented mitochondria. (B) Statistical graph of average mitochondrial length. Experimental data are presented as mean ± standard error of the mean (Mean ± S.E.M). ***, P < 0.001; **, P < 0.01. 342 (blue), with arrows indicating fragmented mitochondria. (B) Statistical graph of average mitochondrial length. Experimental data are presented as mean ± standard error of the mean (Mean ± S.E.M). ***, P < 0.001; **, P < 0.01. | |
2.4 5f and 5g effectively restored ATP levels depleted by rotenone in HT22 cells
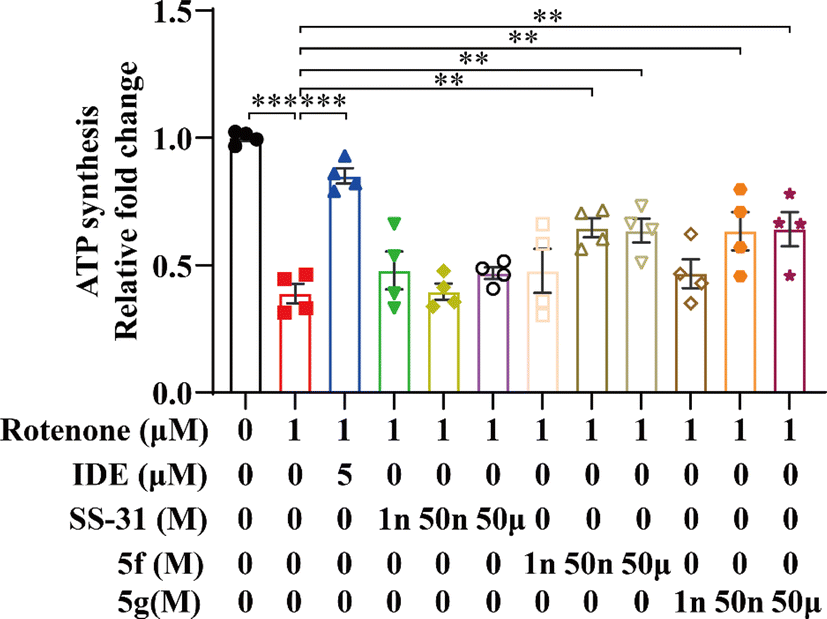
Rotenone, a compound derived from the plant Derris elliptica, selectively inhibits the activity of mitochondrial respiratory complex I, blocking cellular oxidative respiration. This leads to mitochondrial damage, increased generation of mitochondrial ROS, and reduced ATP synthesis. Idebenone is known to inhibit mitochondrial oxidative stress by directly transferring electrons to mitochondrial complex III (bypassing complex I deficiency), thereby restoring ATP production. 35 Idebenone has been shown to significantly restore ATP levels in cells treated with rotenone. 36 To further evaluate the mitochondrial protective effects of 5f and 5g, we employed a rotenone-induced damage model in the mouse hippocampal neuronal cell line HT22, using idebenone as a positive control. Initially, we measured intracellular ATP levels in HT22 cells using an ATP assay kit. As shown in Fig. 4, ATP levels in cells treated with rotenone for 12 hours decreased by approximately 50% compared to the control group. Idebenone significantly restored ATP levels in these cells. In contrast, SS-31 had minimal effect on ATP restoration, whereas 5f and 5g dose-dependently and significantly inhibited the rotenone-induced decrease in ATP production, with notable effects observed at 50 nM. These results further confirm that 5f and 5g possess stronger mitochondrial protective activity compared to SS-31, thereby exerting neuroprotective effects.
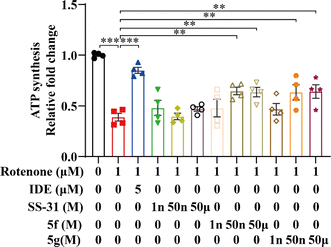 |
| | Fig. 4 5f and 5g restored rotenone-induced ATP depletion in HT22 cells. HT22 cells were treated with 1 μM rotenone for 24 hours, followed by intervention with 5 μM idebenone, and 1 nM, 50 nM, or 50 μM of SS-31, 5f, and 5g. ATP content changes were measured using an ATP assay kit. Experimental data are presented as mean ± standard error of the mean (Mean ± S.E.M) (n = 4). ***, P < 0.001; **, P < 0.01. | |
2.5 5f and 5g inhibited mitochondrial damage induced by rotenone in HT22 cells
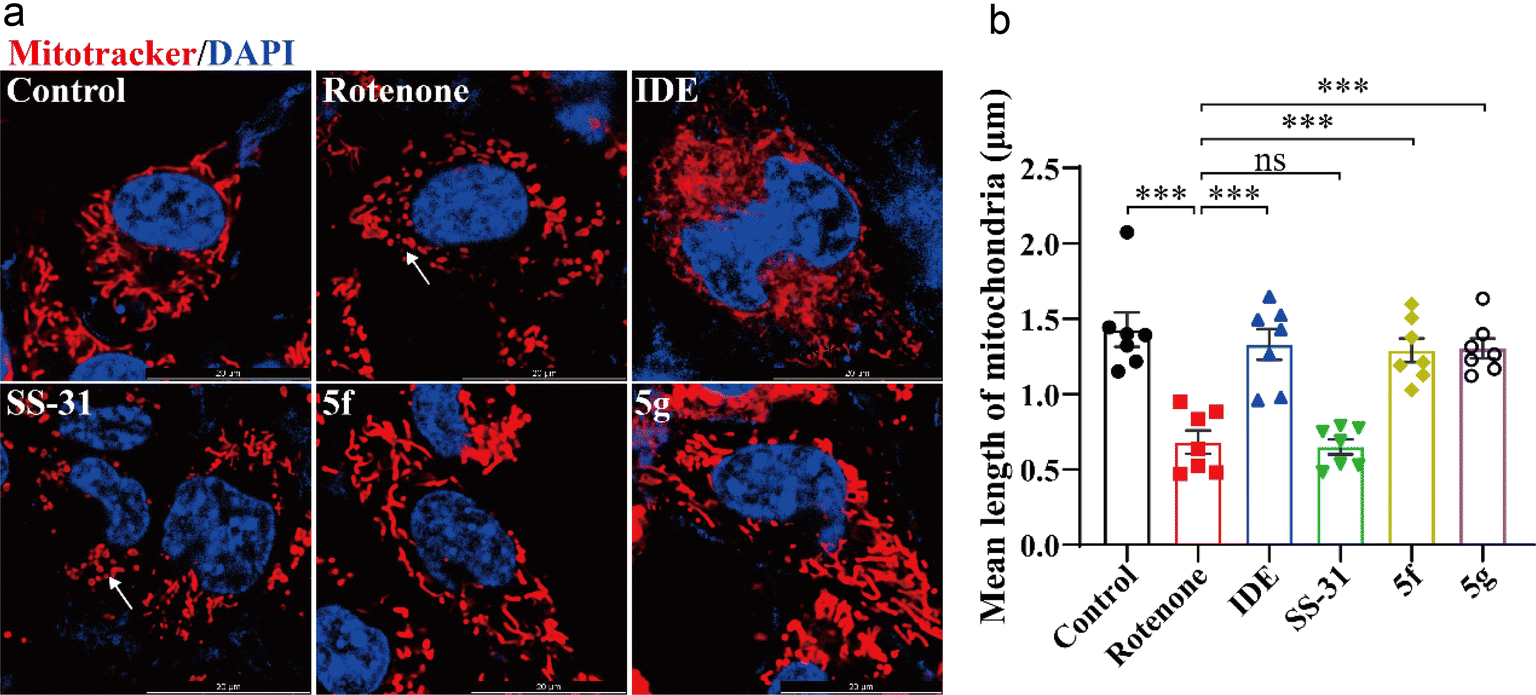
Next, we used MitoTracker™ to label mitochondria and evaluate the direct protective effects of 5f and 5g on HT22 cell mitochondria. As shown in Fig. 5A, rotenone treatment led to a decrease in mitochondrial tubular structure and increased mitochondrial fragmentation in HT22 cells. Idebenone, 5f, and 5g significantly inhibited rotenone-induced mitochondrial fragmentation. As shown in Fig. 5B, statistical analysis of at least 60 mitochondria per group revealed that rotenone treatment reduced mitochondrial length in HT22 cells. Idebenone, 5f, and 5g inhibited the rotenone-induced reduction in mitochondrial length, whereas SS-31 showed similar mitochondrial morphology and length as the model group. These results further confirm that, compared to SS-31, 5f and 5g exert a direct protective effect on neuronal mitochondria, with significantly higher activity. They can directly mitigate the neurotoxic effects of the mitochondrial-damaging agent rotenone.
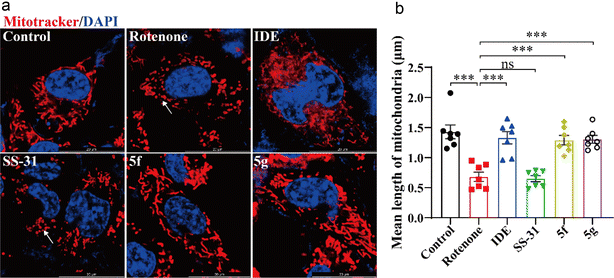 |
| | Fig. 5 5f and 5g inhibited mitochondrial damage induced by rotenone in HT22 cells. HT22 cells were treated with 1 μM rotenone for 24 hours, followed by intervention with 5 μM idebenone, 50 nM SS-31, or 50 nM of 5f and 5g. (a) Representative images of mitochondria labeled with MitoTracker™ (red) and nuclei stained with Hoechst 33342 (blue), with arrows indicating fragmented mitochondria. (b) Statistical analysis of average mitochondrial length. Experimental data are presented as mean ± standard error of the mean (Mean ± S.E.M). ***, P < 0.001; **, P < 0.01, ns, no significance. | |
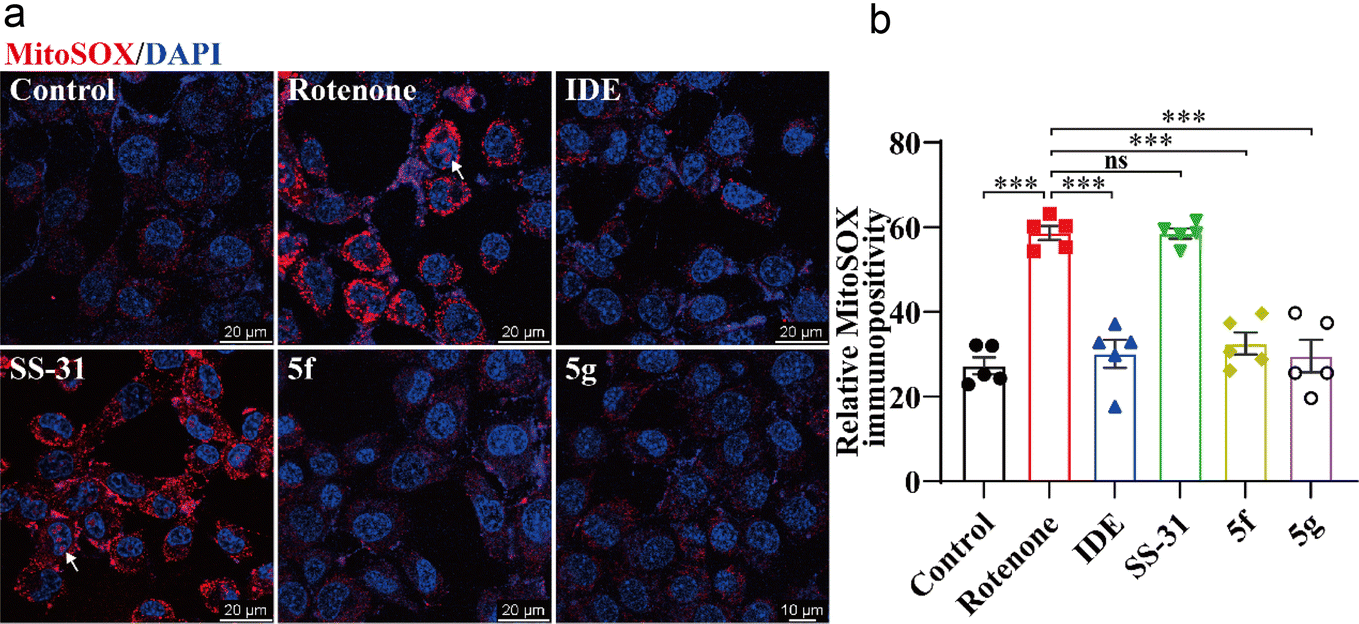
2.6 5f and 5g inhibited mitochondrial oxidative stress induced by rotenone in HT22 cells
Next, we used the mitochondria-specific superoxide indicator MitoSOX™ Red to further evaluate the effects of 5f and 5g on mitochondrial oxidative stress. As shown in Fig. 6, treatment of HT22 cells with 1 μM rotenone for 12 hours significantly increased mitochondrial ROS levels. This increase was evidenced by the enhanced immunofluorescence intensity of MitoSOX™ in perinuclear mitochondria, accompanied by dye entry into the nuclei, indicating nuclear membrane damage. Idebenone, 5f, and 5g significantly inhibited the production of mitochondrial ROS, whereas SS-31 showed no significant inhibition of rotenone-induced mitochondrial ROS production. These results further confirm the direct protective effects of 5f and 5g on neuronal mitochondria, with their efficacy being markedly superior to that of SS-31.
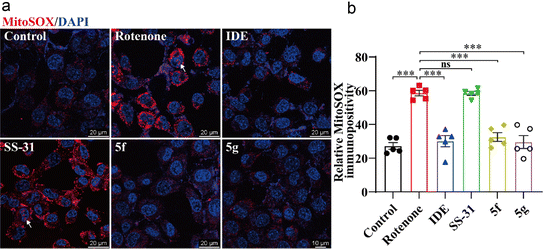 |
| | Fig. 6 5f and 5g inhibited mitochondrial damage induced by rotenone in HT22 cells. HT22 cells were treated with 1 μM rotenone for 24 hours, followed by intervention with 5 μM idebenone, 50 nM SS-31, or 50 nM of 5f and 5g. (a) Representative images of mitochondria labeled with MitoSOX (red) and nuclei stained with Hoechst 33342 (blue), with arrows indicating MitoSOX dye entering the nuclei. (b) Statistical analysis of mitochondrial MitoSOX fluorescence intensity. Experimental data are presented as mean ± standard error of the mean (Mean ± S.E.M) (n = 5). ***, P < 0.001, ns, no significance. | |
3 Conclusions
Mitochondria are organelles responsible for energy metabolism, playing a crucial role in maintaining cellular function. SS-31 is a novel mitochondria-targeted peptide antioxidant that can rapidly cross the blood–brain barrier and cell membranes, accumulating in the inner mitochondrial membrane at concentrations over 5000 times higher than in the cytoplasm. 24,37 SS-31 reduces mitochondrial ROS production, increases ATP generation, maintains mitochondrial membrane potential, and inhibits the formation of the mitochondrial permeability transition pore. 24,38 Additionally, SS-31 protects the integrity of mitochondrial cristae and the mitochondrial respiratory chain, inhibits apoptosis, and reduces inflammatory responses.27 In recent studies, the mitochondrial protective effects of SS-31 have been extensively investigated. It has been reported that SS-31 can alleviate mitochondrial dynamic abnormalities in neurons associated with neurodegenerative diseases such as AD, PD, and HD, thereby improving mitochondrial biogenesis. However, the development of more efficient SS-31 derivatives has not yet been reported in the literature.
In the present study, we synthesized and tested a series of novel SS-31-derived peptides. During cellular analysis of the peptides, we discovered that 5f and 5g exhibit significantly greater anti-neuroinflammatory effects compared to SS-31. Additionally, 5f and 5g counteract the rotenone-induced reduction in neuronal ATP and the increase in mitochondrial ROS by protecting the mitochondria. These findings demonstrate their direct protective effects on neuronal mitochondria. Interestingly, in the 5f, taurine is introduced as a substituent via succinic acid, which serves as a linker on the arginine substituent of SS-31. Taurine is a sulfur-containing conditionally essential amino acid widely distributed in the body's tissues and cells, with the highest concentrations found in excitable tissues such as nerves and muscles. 39,40 Taurine exerts multiple biological functions in the human body, acting as a neurotransmitter, osmotic regulator, and cell membrane stabilizer. It also plays roles in regulating glucose and lipid metabolism, reducing oxidative stress, and inhibiting endoplasmic reticulum stress. Beyond maintaining normal physiological functions, taurine provides protective effects in metabolic and inflammatory diseases and is associated with improved cognition, anti-aging, and anti-damage effects. 41,42 In this study, we introduced taurine substituents into SS-31. The anti-inflammatory effects and mitochondrial protective abilities of 5f were superior to SS-31. This promising peptide holds great potential for practical applications, and we are currently investigating its specific mechanisms. The 5g molecule is derived by replacing the phenyl group of phenylalanine in SS-31 with a 2-substituted naphthyl group, resulting in enhanced anti-inflammatory activity and mitochondrial protection compared to SS-31. However, when the phenyl group of phenylalanine is replaced with a 1-substituted naphthyl group (5j), both anti-inflammatory activity and mitochondrial protection are reduced compared to SS-31. We will further investigate the specific mechanisms underlying these observations.
In summary, this study demonstrates that 5f and 5g exhibit significantly stronger anti-neuroinflammatory effects than SS-31. These effects are attributed to their enhanced mitochondrial protective activity. Furthermore, 5f and 5g mitigate the rotenone-induced reduction in neuronal ATP and the increase in mitochondrial ROS by protecting the mitochondria. These findings demonstrate their direct protective effects on neuronal mitochondria. This work highlights the potential of 5f and 5g in the treatment of neurodegenerative diseases associated with inflammation and mitochondrial damage, warranting further investigation into their neuroprotective efficacy in mammalian systems.
4 Materials and methods
4.1 Peptide synthesis
All peptides were purchased from GL Biochem and prepared using manual solid-phase peptide synthesis with the 9-fluorenylmethyloxycarbonyl (Fmoc) strategy. The C-terminal phenylalanyl residue was coupled to the resin (p-methylbenzhydrylamine polystyrene-1% divinylbenzene) via a Rink amide linker [p-(Fmoc-2,4-dimethoxybenzyl)-phenoxyacetic acid]. The other amino acid residues were incorporated through successive cycles of Fmoc deprotection and amino acid coupling. After the solid-phase assembly of the peptide, the cleavage reaction from the resin and concomitant side-chain deprotection in one step with TFA yielded the crude peptide with C-terminal amides. The crude preparation was then precipitated and dried. Purification was performed by preparative reverse-phase HPLC in the TFA buffer. The selected pools were mixed to form a homogeneous solution in water before freeze-drying and packaging. The detailed peptide synthesis procedures can be found in the ESI.†
4.2 Reagents
Ginsenoside Rb1 (Rb1) (purity > 98%, Cas. 41753-43-9, Cat. no. BD9470) was purchased from Bide Pharmatech Co.,Ltd (China). Idebenone (purity > 98%, Cas. 58186-27-9, Cat. No. BD134310) was purchased from Bide Pharmatech Co.,Ltd (China).
4.3 Cell culture and treatment
BV2 and HT22 cell line was purchased from the Chinese Academy of Sciences Cell Bank of Type Culture Collection (Shanghai, China) and cultured in DMEM/high glucose medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. BV2 cells were seeded in 12-well plates at the number of 5 × 105 cells per well or 8-well Lab-Tek chamber slides at a density of 0.8 × 105 cells per well, then incubated with Rb1 at 100 μM, SS-31 and SS-31-derived peptides at 10 μM or vehicle, followed by LPS (5 ng mL−1) incubation for 24 h. HT22 cells were seeded in 96-well plates at the number of 1 × 104 cells per well or 8-well Lab-Tek chamber slides at a density of 0.5 × 105 cells per well and subject to the indicated experiments. For the experiments, HT22 cells were treated with IDE at 5 μM, SS-31, 5f, and 5g at indicated concentrations followed by rotenone incubation at 1 μM for 24 h.
4.4 Real-time quantitative polymerase chain reaction (qPCR)
The BV2 cells were seeded in 12-well plates as previously described. After the indicated treatments, total RNA was isolated from BV2 cells using 5× All-In-One RT MasterMix (ABM, Canada), followed by reverse transcription using ChamQ Universal SYBR qPCR Master Mix (Vazyme, China). The mRNA expression of Il6, Il1b, and Tnf was analyzed using LightCycler 480 SYBR Green I Master (Roche, Germany) on a LightCycler 480 II system (Roche, USA). GAPDH was analyzed in parallel for normalization purposes. The primer sequences are included in Table 3. The fold change in the gene expression was calculated according to 2−[Ct (candidate) − Ct (GAPDH)].
Table 3 Primer Sequences for Real-Time qPCR Analyses
| Gene name |
Forward primer (5′-3′) |
Reverse Primer(5′-3′) |
| Il6 |
TTCACAAGTCGGAGGCTTA |
CAAGTGCATCATCGTTGTTC |
| Il1b |
TGCCACCTTTTGACAGTGATG |
AAGGTCCACGGGAAAGACAC |
| Tnf |
ACGTCGTAGCAAACCACCAA |
GCAGCCTTGTCCCTTGAAGA |
| Gapdh |
AGAACATCATCCCTGCATCCA |
CTTCACCACCTTCTTGATGTCAT |
4.5 ATP assay
Cellular adenosine triphosphate (ATP) luminescence was assessed in HT22 cells using a CellTiter-Glo Cell Viability Assay kit (Promega, Chilworth, UK). The CellTiter-Glo Assay is used as an indicator of metabolically active cells by determining the number of viable cells in culture based on quantitation of the ATP present. This assay measures free intracellular ATP from viable cells to generate photons of light (bioluminescence). The intensity of the luminescent signal is directly proportional to the cellular ATP concentration. HT22 cells were seeded in opaque, white 96-well plates and treated as previously described. The method was followed according to the manufacturer's instructions. The luminescence was recorded using SpectraMax i3x Multi-Mode Microplate Reader (Molecular Devices, PA, US).
4.6 Mitochondria staining and imaging
BV2 and HT22 cells were seeded in 8-well Lab-Tek chamber slides as previously described. After the indicated treatments, We stained the mitochondria using the MitoTracker™ Dyes for Mitochondria Labeling (Invitrogen, UK) to observe the changes of mitochondria in BV2 and HT22 cells. Hoechst 33342 was used as counterstaining with nuclei of living cells. The red fluorescence of MitoTracker and the blue fluorescence of Hoechst 33342 were observed and recorded using a confocal fluorescence microscope (Leica TCS SP8, Germany) with a 63× oil immersion objective. Leica LAS AF software was used to acquire fluorescence images. The length of mitochondria was analyzed using ImageJ.
4.7 Mitochondrial superoxide measurement
We used the mitochondrial superoxide indicator MitoSOX™ Red (Invitrogen, UK) to assess Mitochondrial superoxide production in HT22 cells. MitoSOX™ Red is a fluorogenic dye used for the highly selective detection of mitochondrial superoxide in live cells and displays red fluorescence upon oxidation of the MitoSOX™ fluorescent probe. The fluorescent intensity of the cells is proportional to the amount of mitochondrial superoxide generated within the cells. HT22 cells were seeded and treated as previously described. After the indicated treatments, HT22 cells were incubated in working solutions of 5 μM MitoSOX™ and 10 μg mL−1 Hoechst 33342 in cell culture media at 37 °C for 10 min. The red fluorescence of MitoSOX™ and the blue fluorescence of Hoechst 33342 were observed and recorded using a confocal fluorescence microscope (Leica TCS SP8, Germany) with a 63× oil immersion objective. Leica LAS AF software was used to acquire fluorescence images. Quantification of immunofluorescence was performed using ImageJ.
4.8 Statistical analysis
The data were presented as mean ± standard error of mean (S.E.M). The statistical analyses were performed by Student's t-test or one-way ANOVA with the Turkey multiple-comparisons test. Statistical significance was defined as P < 0.05.
Data availability
Data will be made available on request. The data supporting this study's findings are available from the corresponding author, G. W., upon reasonable request.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors gratefully acknowledge the financial support from the Young Taishan Scholars tsqn (no. 202306352) and the Qilu Young Scholars Program of Shandong University (SDQLQN2021-01).
References
- J. Suescun, S. Chandra and M. C. Schiess, The role of neuroinflammation in neurodegenerative disorders, in Translational Inflammation, Elsevier, 2019, pp. 241–267, DOI:10.1016/B978-0-12-813832-8.00013-3.
- M. Schain and W. C. Kreisl, Neuroinflammation in neurodegenerative disorders—a review, Curr. Neurol. Neurosci. Rep., 2017, 17, 1–11, DOI:10.1007/s11910-017-0733-2.
- H. Hong, B. S. Kim and H.-I. Im, Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders, Int. Neurourol. J., 2016, 20, S2, DOI:10.5213/inj.1632604.302.
- M. Colonna and O. Butovsky, Microglia function in the central nervous system during health and neurodegeneration, Annu. Rev. Immunol., 2017, 35, 441–468, DOI:10.1146/annurev-immunol-051116-052358.
- A. Rauf, H. Badoni, T. Abu-Izneid, A. Olatunde, M. M. Rahman, S. Painuli, P. Semwal, P. Wilairatana and M. S. Mubarak, Neuroinflammatory markers: key indicators in the pathology of neurodegenerative diseases, Molecules, 2022, 27, 3194, DOI:10.3390/molecules27103194.
- M. T. Heneka, M. J. Carson, J. El Khoury, G. E. Landreth, F. Brosseron, D. L. Feinstein, A. H. Jacobs, T. Wyss-Coray, J. Vitorica and R. M. Ransohoff, Neuroinflammation in Alzheimer's disease, Lancet Neurol., 2015, 14, 388–405, DOI:10.1016/S1474-4422(15)70016-5.
- Q. Wang, Y. Liu and J. Zhou, Neuroinflammation in Parkinson's disease and its potential as therapeutic target, Transl. Neurodegener., 2015, 4, 1–9, DOI:10.1186/s40035-015-0042-0.
- J. Saba, F. L. Couselo, J. Bruno, L. Carniglia, D. Durand, M. Lasaga and C. Caruso, Neuroinflammation in Huntington's disease: a starring role for astrocyte and microglia, Curr. Neuropharmacol., 2022, 20, 1116, DOI:10.1016/S1474-4422(15)70016-5.
- I. Bjelobaba, D. Savic and I. Lavrnja, Multiple sclerosis and neuroinflammation: the overview of current and prospective therapies, Curr. Pharm. Des., 2017, 23, 693–730, DOI:10.2174/1381612822666161214153108.
- R. L. Jayaraj, S. Azimullah, R. Beiram, F. Y. Jalal and G. A. Rosenberg, Neuroinflammation: friend and foe for ischemic stroke, J. Neuroinflammation, 2019, 16, 1–24, DOI:10.1186/s12974-019-1516-2.
- J. S. Bhatti, G. K. Bhatti and P. H. Reddy, Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies, Biochim. Biophys. Acta, Mol. Basis Dis., 2017, 1863, 1066–1077, DOI:10.1016/j.bbadis.2016.11.010.
- P. M. Herst, M. R. Rowe, G. M. Carson and M. V. Berridge, Functional mitochondria in health and disease, Front. Endocrinol., 2017, 8, 296, DOI:10.3389/fendo.2017.00296.
- J. S. Harrington, S. W. Ryter, M. Plataki, D. R. Price and A. M. Choi, Mitochondria in health, disease, and aging, Physiol. Rev., 2023, 103, 2349–2422, DOI:10.1152/physrev.00058.2021.
- D. B. Zorov, M. Juhaszova and S. J. Sollott, Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release, Physiol. Rev., 2014, 94, 909–950, DOI:10.1152/physrev.00026.2013.
- E. Bertero and C. Maack, Calcium signaling and reactive oxygen species in mitochondria, Circ. Res., 2018, 122, 1460–1478, DOI:10.1161/CIRCRESAHA.118.310082.
- T. Chen, J. Zhu, Y.-H. Wang and C.-H. Hang, ROS-mediated mitochondrial dysfunction and ER stress contribute to compression-induced neuronal injury, Neuroscience, 2019, 416, 268–280, DOI:10.1016/j.neuroscience.2019.08.007.
- K. H. Flippo and S. Strack, Mitochondrial dynamics in neuronal injury, development and plasticity, J. Cell Sci., 2017, 130, 671–681, DOI:10.1242/jcs.171017.
- I. Dalle-Donne, R. Rossi, R. Colombo, D. Giustarini and A. Milzani, Biomarkers of oxidative damage in human disease, Clin. Chem., 2006, 52, 601–623, DOI:10.1373/clinchem.2005.061408.
- J. Luo, K. Mills, S. le Cessie, R. Noordam and D. van Heemst, Ageing, age-related diseases and oxidative stress: What to do next?, Ageing Res. Rev., 2020, 57, 100982, DOI:10.1016/j.arr.2019.100982.
- J. Wu, M. Zhang, H. Li, X. Sun, S. Hao, M. Ji, J. Yang and K. Li, BDNF pathway is involved in the protective effects of SS-31 on isoflurane-induced cognitive deficits in aging mice, Behav. Brain Res., 2016, 305, 115–121, DOI:10.1016/j.bbr.2016.02.036.
- L. Zhong, X. Ren, Y. Ai and Z. Liu, SS-31 improves cognitive function in sepsis-associated encephalopathy by inhibiting the drp1-NLRP3 inflammasome activation, NeuroMol. Med., 2023, 25, 230–241, DOI:10.1007/s12017-022-08730-1.
- Y. Nie, J. Li, X. Zhai, Z. Wang, J. Wang, Y. Wu, P. Zhao and G. Yan, Elamipretide (SS-31) attenuates idiopathic pulmonary fibrosis by inhibiting the nrf2-dependent NLRP3 inflammasome in macrophages, Antioxidants, 2023, 12, 2022, DOI:10.3390/antiox12122022.
- X. Peng, K. Wang, C. Zhang, J.-P. Bao, C. Vlf, J.-W. Gao, Z.-M. Zhou and X.-T. Wu, The mitochondrial antioxidant SS-31 attenuated lipopolysaccharide-induced apoptosis and pyroptosis of nucleus pulposus cells via scavenging mitochondrial ROS and maintaining the stability of mitochondrial dynamics, Free Radical Res., 2021, 55, 1080–1093, DOI:10.1080/10715762.2021.2018426.
- H. H. Szeto, First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics, Br. J. Pharmacol., 2014, 171, 2029–2050, DOI:10.1111/bph.12461.
- C. C. Winterbourn, H. N. Parsons-Mair, S. Gebicki, J. M. Gebicki and M. J. Davies, Requirements for superoxide-dependent tyrosine hydroperoxide formation in peptides, Biochem. J., 2004, 381, 241–248, DOI:10.1042/BJ20040259.
- H. H. Szeto, Mitochondria-targeted cytoprotective peptides for ischemia–reperfusion injury, Antioxid. Redox Signaling, 2008, 10, 601–620, DOI:10.1089/ars.2007.1892.
- Y. Mo, S. Deng, L. Zhang, Y. Huang, W. Li, Q. Peng, Z. Liu and Y. Ai, SS-31 reduces inflammation and oxidative stress through the inhibition of Fis1 expression in lipopolysaccharide-stimulated microglia, Biochem. Biophys. Res. Commun., 2019, 520, 171–178, DOI:10.1016/j.bbrc.2019.09.077.
- D. W. Li, F. Z. Zhou, X. C. Sun, S. C. Li, J. B. Yang, H. H. Sun and A. H. Wang, Ginsenoside Rb1 protects dopaminergic neurons from inflammatory injury induced by intranigral lipopolysaccharide injection, Neural Regener. Res., 2019, 14, 1814–1822, DOI:10.4103/1673-5374.257536.
- S. Wang, Y. Cui, M. Xiong, M. Li, P. Wang, J. Cui, X. Du, Y. Chen and T. Zhang, Dual Activity of Ginsenoside Rb1 in Hypertrophic Cardiomyocytes and Activated Macrophages: Implications for the Therapeutic Intervention of Cardiac Hypertrophy, J. Inflammation Res., 2021, 14, 1789–1806, DOI:10.2147/jir.S310633.
- O. R. Pereira Jr., V. M. Ramos, J. V. Cabral-Costa and A. J. Kowaltowski, Changes in mitochondrial morphology modulate LPS-induced loss of calcium homeostasis in BV-2 microglial cells, J. Bioenerg. Biomembr., 2021, 53, 109–118, DOI:10.1007/s10863-021-09878-4.
- J. Park, H. Choi, B. Kim, U. Chae, D. G. Lee, S. R. Lee, S. Lee, H. S. Lee and D. S. Lee, Peroxiredoxin 5 (Prx5) decreases LPS-induced microglial activation through regulation of Ca(2+)/calcineurin-Drp1-dependent mitochondrial fission, Free Radical Biol. Med., 2016, 99, 392–404, DOI:10.1016/j.freeradbiomed.2016.08.030.
- J. Park, H. Choi, J. S. Min, S. J. Park, J. H. Kim, H. J. Park, B. Kim, J. I. Chae, M. Yim and D. S. Lee, Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells, J. Neurochem., 2013, 127, 221–232, DOI:10.1111/jnc.12361.
- X. Peng, K. Wang, C. Zhang, J. P. Bao, C. Vlf, J. W. Gao, Z. M. Zhou and X. T. Wu, The mitochondrial antioxidant SS-31 attenuated lipopolysaccharide-induced apoptosis and pyroptosis of nucleus pulposus cells via scavenging mitochondrial ROS and maintaining the stability of mitochondrial dynamics, Free Radical Res., 2021, 55, 1080–1093, DOI:10.1080/10715762.2021.2018426.
- W. Zhao, Z. Xu, J. Cao, Q. Fu, Y. Wu, X. Zhang, Y. Long, X. Zhang, Y. Yang and Y. Li, et al., Elamipretide (SS-31) improves mitochondrial dysfunction, synaptic and memory impairment induced by lipopolysaccharide in mice, J. Neuroinflammation, 2019, 16, 230, DOI:10.1186/s12974-019-1627-9.
- K. A. Lyseng-Williamson, Idebenone: A Review in Leber's Hereditary Optic Neuropathy, Drugs, 2016, 76, 805–813, DOI:10.1007/s40265-016-0574-3.
- V. Giorgio, M. Schiavone, C. Galber, M. Carini, T. Da Ros, V. Petronilli, F. Argenton, V. Carelli, M. J. Acosta Lopez and L. Salviati, et al., The idebenone metabolite QS10 restores electron transfer in complex I and coenzyme Q defects, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 901–908, DOI:10.1016/j.bbabio.2018.04.006.
- K. Zhao, G. Luo, S. Giannelli and H. H. Szeto, Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines, Biochem. Pharmacol., 2005, 70, 1796–1806, DOI:10.1016/j.bcp.2005.08.022.
- H. Szeto and A. Birk, Serendipity and the discovery of novel compounds that restore mitochondrial plasticity, Clin. Pharmacol. Ther., 2014, 96, 672–683, DOI:10.1038/clpt.2014.174.
- J. A. Kurtz, T. A. VanDusseldorp, J. A. Doyle and J. S. Otis, Taurine in sports and exercise, J. Int. Soc. Sports Nutr., 2021, 18, 39, DOI:10.1186/s12970-021-00438-0.
- G. Wu, Important roles of dietary taurine, creatine, carnosine, anserine and 4-hydroxyproline in human nutrition and health, Amino Acids, 2020, 52, 329–360, DOI:10.1007/s00726-020-02823-6.
- V. Castelli, A. Paladini, M. d'Angelo, M. Allegretti, F. Mantelli, L. Brandolini, P. Cocchiaro, A. Cimini and G. Varrassi, Taurine and oxidative stress in retinal health and disease, CNS Neurosci. Ther., 2021, 27, 403–412, DOI:10.1111/cns.13610.
- J. McGaunn and J. A. Baur, Taurine linked with healthy aging, Science, 2023, 380, 1010–1011, DOI:10.1126/science.adi3025.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc