
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization and quantification of the phytochemical constituents and anti-inflammatory properties of Lindera aggregata†
Song-Song Wenab,
Yong-Jun Liua,
Yu-Lin Liuc,
Yan Zhaob,
Xiao-Xi Xud,
Chang-Chuan Guob,
Chong Niub,
Wei-Jian Wangb,
Yu-Wen Xu*b and
Na Zhang *a
*a
aDepartment of Pharmaceutics, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, 250012, China. E-mail: zhangnancy9@sdu.edu.cn
bNMPA Key Laboratory for Research and Evaluation of Generic Drugs, Shandong Research Center of Engineering and Technology for Consistency Evaluation of Generic Drugs, Shandong Institute for Food and Drug Control, Jinan, 250101, China. E-mail: 13553158409@163.com
cDepartment of Marine Pharmacology, College of Food Science and Technology, Shanghai Ocean University, Shanghai, 201306, China
dBeijing Normal University, Beijing, 100875, China
First published on 11th November 2024
Abstract
The dry roots of Lindera aggregata (Sims) Kosterm have a long-standing history in traditional Chinese medicine, renowned for their ability to regulate vital energy, relieve pain, warm the kidney, and dissipate cold. Recently, L. aggregata has been approved as a new food resource. To gain insights into the bioactive phytochemicals in L. aggregata, an ultrahigh-performance liquid chromatography coupled with high-resolution electrospray ionization quadrupole orbitrap spectrometry method was developed to investigate the chemical profiles of the ethanol extract of L. aggregata. This approach identified 80 compounds, predominantly alkaloids and sesquiterpenoids. Furthermore, 16 selected compounds were simultaneously quantified using the parallel reaction monitoring mode. The quantification method was validated and showed good linearity, sensitivity, and accuracy. The anti-inflammatory activities of the ethanol extract and selected compounds were assessed in vitro using lipopolysaccharide-stimulated RAW 264.7 macrophages. The results revealed that the ethanol extract of L. aggregata and norisoboldine, isolinderalactone, methyllinderone, and linderin B inhibited the production or expression of nitric oxide, inducible nitric oxide synthase (iNOS), tumor necrosis factor-α, and interleukin-6. Molecular docking of iNOS with isolinderalactone, methyllinderone, and linderin B showed that hydrogen bonds, π–π interactions, and hydrophobic interactions contributed to their iNOS inhibitory effects. The results offer insights that may be instrumental in enhancing the quality control for L. aggregata.
1. Introduction
Medicaments used in traditional medicine (TM) are predominantly derived from natural sources. In TM, the equivalent of “clinical trials” has been practiced since ancient times. Specifically, in Traditional Chinese Medicine (TCM), extensive experience and advancements have been accumulated and refined over millennia. These include methods of preparation, herb selection, identification of medicinal materials, and determining the optimal time for harvesting various plants. The dry roots of Lindera aggregata, which belong to the Lauaceae family and are known as Radix Linderae, hold a significant place in traditional Chinese medicine for treating numerous ailments over a long historical span. In line with traditional Chinese medicine principles, L. aggregata, commonly referred to “Wuyao”, is believed to invigorate and harmonize bodily metabolism, offering effects that promote Qi, alleviate pain, warm the kidney, and dispel coldness.1 Characterized by its pungent flavor, mild aroma, and association with the kidney and stomach meridians, L. aggregata has been used to enhance kidney functionality. Recognized for its substantial medicinal value and wide-ranging pharmacological effects, L. aggregata has garnered increasing attention in recent years. Over the past few decades, researchers have explored L. aggregata from diverse perspectives, encompassing the profiling of phytochemical constituents, understanding pharmacological mechanisms, and establishing methods for quality control.2 So far, more than 250 compounds, including flavonoids, alkaloids, terpenes, volatile compounds, and tannins, have been isolated and identified from L. aggregata.2–7 Ongoing pharmacological studies have demonstrated its potential in various areas, such as anti-cancer, anti-inflammatory, anti-arthritis, anti-bacterial, anti-oxidation, anti-diabetic nephropathy, hepatoprotective, and lipid-lowering effects.2 These pharmacological properties have been extensively investigated, with a focus on the crude extract of the root tuber, leaf extract, as well as the chemical compounds.Recognized both as a medicinal herb and a food-related plant, L. aggregata gained approval as a new food resource in China in 2012, signifying its vast development prospects. Consequently, the herb market has witnessed a significant surge in demand for L. aggregata. Its cultivation and utilization in the realms of food and healthcare harbor immense potential for growth and profitability. The growing enthusiasm for the supply of L. aggregata necessitates comprehensive analytical characterization of its bioactive constituents. This approach aims to comprehensively understand their collective impact on both food properties and human health.
While numerous monomeric compounds from L. aggregata have been successfully isolated and identified, research investigations have predominantly focused on the pharmacological effects of the crude extract or primary chemical components. Consequently, there is limited information regarding quality control research, especially concerning the swift identification and quantification of active constituents.7,8 To date, only a few representative sesquiterpenoids and alkaloids have been quantified using LC-MS method. Furthermore, only two active constituents, linderane and norisoboldine, have been designated as the chemical markers for quality control of L. aggregata in China Pharmacopeia 2020. However, it is widely acknowledged that the therapeutic efficacy of L. aggregata relies on the intricate interactions among numerous ingredients in combination, which are different from typical pharmaceutical chemicals. Relying on the determination of only two compounds may not adequately represent the overall clinical therapeutic effects. Therefore, there is a pressing need for a rapid and reliable method to comprehensively determine the chemical profiles of L. aggregata.
Ultra-high performance liquid chromatography (UHPLC)-MS/MS, renowned for its superior speed, enhanced sensitivity, and specificity compared to HPLC-UV analysis, has gained increased attention in the analysis of traditional Chinese medicines. Hence, the objective of this study is to establish a rapid, sensitive, and efficient ultrahigh-performance liquid chromatography coupled with high-resolution electrospray ionization quadrupole orbitrap mass spectrometry (UHPLC-HR-ESI-Q-Orbitrap) method for the qualitative analysis, followed by parallel reaction monitoring (PRM) mode, for the simultaneous quantification of phytochemicals in L. aggregata. The anti-inflammatory effects of the ethanol extract of L. aggregata and selected compounds were measured as the ability to suppress nitril oxide (NO), tumor necrosis factor α (TNF-α), interleukin (IL)-6, and nitric oxide synthase (iNOS) expression in lipopolysaccharide (LPS)-stimulated RAW 264.7 mouse macrophages. In addition, a molecular docking method was carried out to elucidate the protein–ligand interactions between iNOS and the potential bioactive compounds.
2. Materials and methods
2.1. Reagents, chemicals, and plant materials
Ethanol used for extraction was analytical grade, and purchased from Fisher Scientific (Waltham, MA, USA). LC/MS-grade acetonitrile, methanol, water, and trifluoroacetic acid purchased from Merck (Darmstadt, Germany) were used in the sample preparation and UHPLC-MS analysis. LC-MS-grade formic acid was obtained from Fisher Scientific Co. (Waltham, MA, USA). Norisoboldine (P/N, 111![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 825-201802) and linderane (P/N, 111568-201906) were purchased from the National Institute of Food and Drug Control (Beijing, China). Boldine (P/N, X23N9Y73113), isolinderalactone (P/N, S30HB196732), reticuline (P/N, N28HB202502), linderone (P/N, D0HB03066), and methyllinderone (P/N, D0HB03065) were purchase from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). Higenamine (P/N, MUST-22032110), lindenenol (P/N, MUST-22032904), and linderene acetate (P/N, MUST-22111917) were purchased from Chengdu MUST Biotechnology Co., Ltd (Chengdu, China). Coclaruine (P/N, 220511) was obtained from Chengdu Herb Substance Company (Chengdu, China). The standards of linderin B, lindechunisin A, 1-acetyl-4-methoxyl-denudaquinol, (2E,3R,4S)-2-tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide, and (2E,3R,4S)-2-dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide were obtained from our laboratory. Their structures were unambiguously characterized by 1H NMR, 13C NMR, 2D NMR and HR-ESIMS techniques.
825-201802) and linderane (P/N, 111568-201906) were purchased from the National Institute of Food and Drug Control (Beijing, China). Boldine (P/N, X23N9Y73113), isolinderalactone (P/N, S30HB196732), reticuline (P/N, N28HB202502), linderone (P/N, D0HB03066), and methyllinderone (P/N, D0HB03065) were purchase from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). Higenamine (P/N, MUST-22032110), lindenenol (P/N, MUST-22032904), and linderene acetate (P/N, MUST-22111917) were purchased from Chengdu MUST Biotechnology Co., Ltd (Chengdu, China). Coclaruine (P/N, 220511) was obtained from Chengdu Herb Substance Company (Chengdu, China). The standards of linderin B, lindechunisin A, 1-acetyl-4-methoxyl-denudaquinol, (2E,3R,4S)-2-tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide, and (2E,3R,4S)-2-dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide were obtained from our laboratory. Their structures were unambiguously characterized by 1H NMR, 13C NMR, 2D NMR and HR-ESIMS techniques.
Dulbecco's modified Eagle medium (DMEM), penicillin and streptomycin were purchased from Gibco BRL (Grand Island, NY, USA); new-born calf serum (NBCS) was purchased from PAA Laboratories GmbH, Austria; 3-[4,5-dimetylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT), Tween 20, bovine serum albumin (BSA), sodium dodecyl sulfate (SDS), dithiotheitol (DTT), phenylmethylsulfonyl fluoride (PMSF), and LPS were purchased from Sigma Chemical Co. (St. Louis, MO, USA). TNF-α, IL-6, and nitric oxide detection kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
The dried roots of L. aggregata were purchased from Bozhou Hu Herb Company, China, in March 2023, and were authenticated by Dr Qiyan Li, Shandong Institute for Food and Drug Control. The voucher specimen was deposited at the Shandong Institute for Food and Drug Control, Jinan, Shandong Province, China.
2.2. Extraction and sample preparation
The dried roots of L. aggregata underwent comminution using a mill to pass through a 40-mesh sieve. The L.aggregata root powder (150.0 g) was extracted with 80% ethanol (500 mL) under reflux for three times (2 h for each time). The ethanol extract was filtered and combined, and the solvent was evaporated under vacuum to obtain a crude extract. The crude extract was successively lyophilized for subsequent analysis. The extraction yield was 13.6 g of crude extract from 150 g raw herb material. The solution of the freeze-dried L. aggregata ethanolic extract (LAE, 0.5 mg mL−1) was prepared in methanol under sonication, and filtered through a 0.22 μm polyvinylidene difluoride membrane prior to LC-MS detection.2.3. Preparation of standard stock solutions
Based on the compound identification results, individual stock solutions (1.0 mg mL−1) of higenamine, coclaruine, norisoboldine, boldine, reticuline, linderane, isolinderalactone, lindenenol, linderene acetate, linderone, methyllinderone, linderin B, lindechunisin A, 1-acetyl-4-methoxyl-denudaquinol, (2E,3R,4S)-2-tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide, and (2E,3R,4S)-2-dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide were individually prepared in LC-MS-grade methanol. Subsequently, stock solutions containing a mixture of these 16 analytes were prepared and further diluted in the appropriate concentration using methanol to yield a series of concentrations from 1.0 ng mL−1 to 1500 ng mL−1. All the prepared stock solutions were stored in the refrigerator at −20 °C until subsequent analysis.2.4. UHPLC-MS/MS analysis for qualitative study
The quantitative analysis was performed using a Vanquish Flex Binary UHPLC (Thermo Fisher Scientific, Waltham, MA, USA) with a Waters Acquity CSH C18 column (150 × 2.1 mm, 1.7 μm). The column temperature was maintained at 40 °C. The mobile phase A consisted of water and 0.1% formic acid. Mobile phase B comprised methanol and 0.1% formic acid. The flow rate was 0.5 mL min−1. The gradient elution conditions were as follows: 20% B (0–2 min); linear gradient from 20% B to 60% B (2–20 min); 60% B to 80% B (20–21 min); 80% B to 100% B (21–31 min); 100% B for 5 min (31–36 min); back to 20% B at 37 min; 20% B for 6 min balance (37–43 min). The injection volume was 2.00 μL.The detection was carried out using a Q Exactive Plus mass spectrometer system (Thermo Fisher Scientific, Waltham, MA, USA). The parameters for the HRESI source were set as follows: capillary temperature at 275 °C; heater temperature at 300 °C; sheath gas flow, 50 arb; auxiliary gas flow, 10 arb; purge gas flow, 0 arb; spray voltage, 3.5 kV; S-lens RF level, 55%. The mass spectrometer adopted the Full-MS/ddMS2 scan in positive mode. Mass spectra were acquired in the range of 100 to 1200 m/z, and the resolution was set to 70000. The automatic gain control (AGC) was 3 × 106 and the injection time (IT) was 100 ms. For the MS/MS scan, the step-normalized collision energy was set to 20, 40, and 60 N with a resolution of 17500. AGC is 1 × 105 and IT is 50 ms. A data-dependent analysis scan was applied to trigger the second stage fragmentation, whereby the 20 most intense precursor ions at each scan point of the MS were selected as target precursor ions for subsequent MS/MS fragmentation.9
The raw data files obtained from UHPLC-Q-Orbitrap HRESI-MS analysis were processed using the Compound Discoverer 3.3 software (Thermo Fisher Scientific Inc. Waltham, USA). A chromatographic signal/noise (S/N) threshold of 3, mass tolerance of 5 ppm, and a minimum peak intensity of 2 × 103 were used for compound detection. Compound identification was conducted by comparing the accurate mass, MS/MS fragmentation patterns, MzCloud, online metabolite databases of ChemSpider, the in-house compound library, and authentic standards. The in-house compound library on L. aggregata was established based on the reported literature. Approximately 600 compounds were collected from SciFinder and converted to individual structure files (.mol), forming the basis for our in-house library. Compound Discoverer 3.3 utilized exact mass, isotope pattern matching, as well as the MS and MS2 spectra, to conduct the structural identification. The compound database search parameters were adjusted according to the manufacturer's instructions. The collision energy tolerance was set at ±20%, with a match factor threshold of 75% and a maximum of 5 matching results for each compound. The best ion and related fragmentation data (highest resolution and intensity) of each compound were used to predict the elemental composition. Full-MS scans or predicted formulas, when available, were compared with the ChemSpider and in-house library. Fragmentation data (MS2) or predicted formulas, when available, were compared with the MzCloud database.
2.5. UHPLC-MS/MS quantification analysis
Sixteen selected compounds, including higenamine, coclaruine, norisoboldine, boldine, reticuline, linderane, isolinderalactone, lindenenol, linderene acetate, linderone, methyllinderone, linderin B, lindechunisin A, 1-acetyl-4-methoxyl-denudaquinol, (2E,3R,4S)-2-tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide, and (2E,3R,4S)-2-dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide for quantitative study, were accomplished in parallel reaction mode (PRM). Chromatographic separation was performed on a Vanquish Flex Binary UHPLC (Thermo Fisher Scientific, Waltham, MA) with an ACE® Excel® C18-PFP column (2.1 × 100 mm, 3 μm, ACE, UK). The column temperature was set at 40 °C. The mobile phases consisted of 0.1% formic acid aqueous solution (A) and 0.1% formic acid dissolved in methanol (B), and the gradient elution program was as follows: 0–2 min, 40% B; 2–5 min, 40–100% B; 5–16 min, 100% B; 16–17 min, 100–40% B; 17–20 min, 40% B. The flow rate was maintained at 0.5 mL min−1, and the injection volume was 2 μL. The Q-orbitrap mass spectrometer was operated in positive mode. The settings used in HRESI were as follows: spray voltage, 3.5 kV; ion transfer tube temperature, 350 °C; vaporizer temperature, 400 °C sheath gas flow rate, 60 arb; auxiliary gas flow rate, 20 arb. Precursor ion scan mode was used for screening and PRM acquisition mode for quantification of the 16 compounds in LAE. Optimization of the MS/MS conditions for each compound was accomplished using standards through flow injection analysis.2.6. Quantification method validation
The developed quantification method underwent validation in accordance with the International Conferences on Harmonization (ICH, Q2R1) guidelines, encompassing assessments for linearity, limits of detection (LOD), limits of quantification (LOQ), precisions, and recovery studies.10 Linearity was evaluated by constructing the calibration curves, correlating the peak areas against the nominal concentrations of calibration standards using weighted least-square linear regression. Each reference compound was tested at a minimum of five different concentrations to establish the correlation coefficient (r), slope, and intercept. The LOD and LOQ were defined as a S/N equal to 3 and 10, respectively. Precision and reproducibility were assessed by calculating the relative standard deviation (RSD) of the peak areas acquired from six replicates at a medium standard concentration. Accuracy was determined by measuring the mean recovery after adding the standard to actual samples at a medium spiked concentration with six replicates.2.7. Anti-inflammatory activity assay
000 rpm for 20 min. Supernatants were collected, and iNOS protein concentration was determined using a mouse iNOS ELISA kit (Abcam, Cambridge, USA). Dex (25 μM) was used as the positive control.2.8. Molecular docking
The chemical structures of compounds with anti-inflammatory activities were selected as ligands for further molecular docking investigation. Ligands were prepared (minimization of energy done, hydrogen atoms added, and charges added where required) using the UCSF Chimera software (version 1.16) structure build module. Ligand binding site prediction was conducted by PrankWeb (http://prankweb.cz). The X-ray crystal structure of the iNOS with detailed resolution was obtained from Protein Data Bank (PDB) with PDB ID 1R35. The protein was docked with compounds using AutoDock Vina and UCSF Chimera, and the binding energies were calculated. The docking complexes were visualized using the ProteinPlus web server.2.9. Statistical analysis
Statistical significances were determined by the one-way analysis of variance (ANOVA) and the Student's t-test. Data were expressed as mean ± SD of replicated experiments. The values of P < 0.05 were statistically significant.3. Results and discussion
3.1. Characterization and identification of chemical constituents in LAE
In this study, UHPLC-HRESI-Q-orbitrap method was adopted to identify the chemical profiles in LAE, and the total ion chromatogram (TIC) under positive mode is shown in Fig. S1.† The compounds with available standards were identified by comparing the retention time and high-resolution accurate mass. Moreover, the MS fragmentation behaviors of the reference compounds have been previously reported in the literature, which was helpful for structural elucidation of the relative derivatives with the same skeleton.11 For compounds lacking available standards, the structures were tentatively identified by comparing with an in-house compounds database, according to the accurate mass, chromatographic behavior, MS/MS data, and fragmentation patterns. The mass errors for all the precursor ions of the identified compounds were set within ±5 ppm. Ultimately, a total of 80 compounds were unambiguously or tentatively identified, with sesquiterpenes and alkaloids comprising 60% of the total identified compounds. In the positive mode, the quasi-molecular ion peaks of alkaloids and sesquiterpenoids always appeared as [M + H]+ ions, and a series of fragmentation peaks such as [M + H − H2O]+, [M + H − CO]+, and [M + H − NH3]+ were observed in MS/MS spectra. A detailed information of these identified compounds is listed in Table 1, and the MS and MS/MS spectra of the identified compounds are provided in ESI.†| Peak no. | tR (min) | Molecular formula | Calculated mass (m/z) | Measured mass (m/z) | Reference ion | Error (ppm) | MS/MS fragments (m/z) | Identification | Class of compounds |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 7.94 | C17H17NO3 | 283.1198 | 284.1271 | [M + H]+ | −3.58 | 145.0598, 178.0863, 223.0746, 255.1010 | Norcinnamolaurine | Alkaloid |
| 2 | 8.39 | C16H17NO3 | 271.1199 | 272.1272 | [M + H]+ | −3.40 | 107.0495, 123.0445, 161.0603, 255.1022 | Higenamine | Alkaloid |
| 3 | 9.42 | C15H14O6 | 290.0781 | 291.0853 | [M + H]+ | −3.40 | 123.0438, 139.0387, 147.0438, 165.0543 | Catechin | Flavanol |
| 4 | 9.57 | C11H9NO2 | 187.0629 | 188.0702 | [M + H]+ | −2.33 | 118.0649, 146.0598, 170.0596 | Indole-3-acrylic acid | Alkaloid |
| 5 | 10.27 | C11H13NO2 | 191.0943 | 192.1016 | [M + H]+ | −1.71 | 133.0646, 148.0754, 149.0833, 177.0781 | Streptopyrrolidine | Alkaloid |
| 6 | 10.43 | C6H11NO | 113.0839 | 114.0912 | [M + H]+ | −1.24 | 69.0698, 79.0541, 96.0806 | Caprolactam | Amide |
| 7 | 10.80 | C19H21NO4 | 327.1462 | 328.1534 | [M + H]+ | −2.73 | 190.0859, 265.0849, 297.1109 | Laurotetanine | Alkaloid |
| 8 | 10.82 | C18H19NO3 | 297.1356 | 298.1428 | [M + H]+ | −3.19 | 176.0701, 190.0853, 254.1168, 283.1195 | Diolmycin A1 | Alkaloid |
| 9 | 11.53 | C18H19NO4 | 313.1304 | 314.1377 | [M + H]+ | 3.26 | 268.1323, 298.1054, 299.1142 | Norbracteoline | Alkaloid |
| 10 | 12.19 | C17H19NO3 | 285.1357 | 286.1403 | [M + H]+ | −2.54 | 107.0489, 160.0754, 192.1016, 243.1009 | Coclaurine | Alkaloid |
| 11 | 12.83 | C10H11NO3 | 193.0736 | 194.0810 | [M + H]+ | −1.39 | 107.0491, 119.0492, 135.0439, 151.0754 | Northalifoline | Alkaloid |
| 12 | 12.87 | C7H6O3 | 138.0314 | 139.0387 | [M + H]+ | −1.88 | 93.0334, 111.0439, 139.0388 | 3-Hydroxy-2,5-toluquinone | Quinone |
| 13 | 12.99 | C19H21NO4 | 327.1460 | 328.1533 | [M + H]+ | −3.19 | 176.0701, 190.0855, 281.1040, 312.1221, 313.1300 | N-Methylhernovine | Alkaloid |
| 14 | 13.01 | C19H21NO3 | 311.1512 | 312.1585 | [M + H]+ | −3.12 | 177.0906, 204.1015, 206.1171, 269.1163, 283.1318 | Pronuciferine | Alkaloid |
| 15 | 13.37 | C18H19NO4 | 313.1305 | 314.1377 | [M + H]+ | −2.57 | 237.0904, 265.0851, 282.0877, 297.1112 | Norisoboldine | Alkaloid |
| 16 | 14.00 | C19H21NO4 | 327.1460 | 328.1533 | [M + H]+ | 3.20 | 237.0904, 265.0851, 297.1112 | Boldine | Alkaloid |
| 17 | 14.14 | C19H23NO4 | 329.1617 | 330.1690 | [M + H]+ | −3.16 | 137.0595, 143.0489, 192.1015, 299.1267 | Reticuline | Alkaloid |
| 18 | 14.24 | C18H19NO4 | 313.1305 | 314.1378 | [M + H]+ | −2.77 | 107.0489, 237.0903, 265.0851, 297.1112 | Norboldine | Alkaloid |
| 19 | 14.82 | C13H20O2 | 208.1459 | 209.1532 | [M + H]+ | −1.89 | 95.0541, 133.1009, 135.0799, 153.0908, 191.1431 | 4,6,10,12-Tridecatetraene-2,8-diol | Aliphatic alcohol |
| 20 | 15.92 | C19H21NO4 | 327.1460 | 328.1533 | [M + H]+ | −3.23 | 192.1016, 233.0594, 265.0851, 282.0878 | Isoboldine | Alkaloid |
| 21 | 17.33 | C14H16O4 | 248.1040 | 249.1113 | [M + H]+ | −3.44 | 105.0696, 159.0801, 187.0751, 204.0775 | 1-(5-Oxotetrahydrofuran-2-yl)ethyl-2-phenylacetate | Fruanone |
| 22 | 17.40 | C20H23NO4 | 341.1616 | 342.1689 | [M + H]+ | −3.23 | 248.0821, 279.1003, 296.1036, 311.1263 | Isocorydine | Alkaloid |
| 23 | 18.01 | C14H16O4 | 248.1041 | 271.0933 | [M + H]+ | −2.92 | 91.0545, 105.0333 | Epipyriculol | Phenolic derivative |
| 24 | 18.58 | C20H23NO4 | 341.1616 | 342.1689 | [M + H]+ | −3.23 | 237.0903, 265.0851, 280.1085, 296.1033 | N-Methyllaurotetanine | Alkaloid |
| 25 | 18.92 | C15H18O4 | 262.1197 | 263.1270 | [M + H]+ | −3.15 | 105.0333, 107.0490 | Linderolide S | Sesquiterpenoid |
| 26 | 20.64 | C19H15NO5 | 337.0940 | 338.1013 | [M + H]+ | −3.00 | 252.0648, 280.0593, 295.0827, 323.0778 | 9-Hydroxy-1,2,10-trimethoxy-7H-dibenzo[de,g]quinolin-7-one | Alkaloid |
| 27 | 20.76 | C25H27NO5 | 421.1883 | 422.1956 | [M + H]+ | −1.38 | 137.0595, 143.0489, 175.0750, 404.1843 | Karakoramine | Alkaloid |
| 28 | 20.81 | C18H21NO3 | 299.1511 | 300.1584 | [M + H]+ | −3.35 | 107.0489, 151.0749, 174.0673, 189.0907, 283.1317 | Norarmepavine | Alkaloid |
| 29 | 20.88 | C15H18O4 | 262.1196 | 285.1088 | [M + Na]+ | −3.35 | 105.0332, 107.0490, 165.0907, 241.1194 | Linderolide D | Sesquiterpenoid |
| 30 | 20.89 | C15H16O3 | 244.1093 | 245.1166 | [M + H]+ | −2.57 | 107.0488, 189.1272, 199.1114, 217.1218 | Neolinderalactone | Sesquiterpenoid |
| 31 | 21.93 | C18H19NO4 | 313.1303 | 314.1377 | [M + H]+ | −3.41 | 91.0539, 117.0333, 121.0646, 177.0543 | N-Feruloyltyramine | Amide |
| 32 | 22.13 | C15H18O5 | 278.1145 | 279.1218 | [M + H]+ | −3.26 | 123.0829, 233.1167, 243.1009, 261.1124 | Linderolide B | Sesquiterpenoid |
| 33 | 22.90 | C15H18O3 | 246.1247 | 247.1322 | [M + H]+ | −3.62 | 105.0336, 117.0697, 131.0855, 183.1165, 229.1219 | Peniophoral | Aldehyde |
| 34 | 24.67 | C15H18O3 | 246.1249 | 247.1322 | [M + H]+ | −3.02 | 183.1165, 201.1271, 229.1217 | 8-Hydroxylindestenolide | Sesquiterpenoid |
| 35 | 25.09 | C15H16O4 | 260.1040 | 261.1113 | [M + H]+ | −3.51 | 95.0489, 215.1061, 233.1169, 243.1007 | Parvigemone | Sesquiterpenoid |
| 36 | 25.79 | C14H16O3 | 232.1093 | 233.1166 | [M + H]+ | −2.77 | 105.0334, 145.1010, 173.0958, 215.1062 | Citreoviripyrone B | Pyrone |
| 37 | 26.11 | C15H18O4 | 262.1197 | 285.1089 | [M + Na]+ | 2.97 | 105.0333, 133.0644, 227.1066, 241.1192, 267.0984 | Linderolide I | Sesquiterpenoid |
| 38 | 26.40 | C15H20O4 | 264.1353 | 287.1245 | [M + Na]+ | −3.32 | 105.0334, 133.0644, 149.0959, 269.1143 | 1-Methylabscisic-6-acid | Sesquiterpenoid |
| 39 | 26.42 | C16H22O4 | 278.1509 | 301.1401 | [M + Na]+ | −3.39 | 137.0571, 269.1142, 283.1295 | 2-Methyl butyl propyl phthalate | Phthalate esters |
| 40 | 26.62 | C13H20O | 192.1511 | 193.1584 | [M + H]+ | −1.67 | 95.0853, 119.0853, 133.1010, 175.1478 | β-Ionone | Terpenoid |
| 41 | 26.73 | C15H18O2 | 230.1300 | 231.1372 | [M + H]+ | −3.12 | 107.0853, 119.0853, 185.1322, 213.1269 | Dehydrocostus lactone | Sesquiterpenoid |
| 42 | 27.19 | C15H16O2 | 228.1144 | 229.1217 | [M + H]+ | −2.57 | 119.0853, 131.0853, 159.0802, 201.1270 | Lactarioline A | Sesquiterpenoid |
| 43 | 27.64 | C15H24 | 204.1873 | 205.1946 | [M + H]+ | −2.36 | 107.0853, 121.1166, 135.1167, 149.1323 | α-Farnesene | Sesquiterpenoid |
| 44 | 28.00 | C15H18O2 | 230.1301 | 231.1373 | [M + H]+ | −2.73 | 93.0697, 133.0646, 203.1430, 213.1270 | Lindestrenolide | Sesquiterpenoid |
| 45 | 28.05 | C15H22 | 202.1718 | 203.1791 | [M + H]+ | −1.88 | 105.0697, 119.0853, 133.1009, 147.1166 | Calamenene | Sesquiterpenoid |
| 46 | 28.16 | C13H16O | 188.1197 | 189.1270 | [M + H]+ | −2.09 | 105.0697, 131.0854, 143.0853, 174.1035 | 1-Phenylhept-3-en-4-one | Diarylheptanoid |
| 47 | 28.29 | C14H14O2 | 214.0989 | 215.1062 | [M + H]+ | −2.13 | 169.1010, 173.0968, 187.1114, 197.0958 | 2,4,6,8,10,12-Tetradeca-1,14-hexenedial | Aldehyde |
| 48 | 28.30 | C15H16O4 | 260.1040 | 261.1113 | [M + H]+ | −3.20 | 95.0490, 173.0958, 215.1063, 243.1009 | Linderane | Sesquiterpenoid |
| 49 | 28.45 | C13H22O2 | 210.1614 | 228.1952 | [M + NH4]+ | −2.78 | 95.0854, 109.1010, 175.1478, 193.1584 | 10-Methyldodec-3-en-4-olide | Furanone |
| 50 | 28.70 | C15H16O3 | 244.1092 | 245.1165 | [M + H]+ | 3.30 | 157.0654, 171.1173, 199.1124, 227.1072 | Isolinderalactone | Sesquiterpenoid |
| 51 | 28.74 | C15H18O2 | 230.1301 | 231.1374 | [M + H]+ | −2.53 | 95.0490, 107.0853, 119.0854, 147.0801 | Lindenenol | Sesquiterpenoid |
| 52 | 28.74 | C16H20O2 | 244.1456 | 245.1529 | [M + H]+ | −3.05 | 105.0697, 199.1113, 213.1269 | Linderoxide | Sesquiterpenoid |
| 53 | 28.76 | C14H20O2 | 220.1456 | 243.1348 | [M + Na]+ | 3.32 | 93.0697, 105.0697, 109.1010, 161.0595 | Benquoine | Lactone |
| 54 | 28.81 | C15H22O | 218.1666 | 219.1738 | [M + H]+ | −2.35 | 91.0541, 121.0642, 163.1113, 203.1427 | Anaephene A | Alkylphenol |
| 55 | 28.96 | C15H16O2 | 228.1145 | 229.1217 | [M + H]+ | −2.51 | 105.0699, 131.0854, 201.0909, 211.1115 | Dehydrolindestrenolide | Sesquiterpenoid |
| 56 | 29.20 | C17H20O4 | 288.1352 | 289.1425 | [M + H]+ | −3.19 | 105.0697, 183.1165, 211.1113, 229.1218 | Lindenanolide A | Sesquiterpenoid |
| 57 | 29.58 | C15H22O2 | 234.1615 | 235.1688 | [M + H]+ | 2.11 | 123.0441, 179.1063, 217.1579 | Nigriterpene E | Sesquiterpenoid |
| 58 | 30.52 | C15H18O2 | 230.1299 | 253.1190 | [M + Na]+ | −3.45 | 147.0803, 159.1166, 175.0751, 185.1322 | Shizukanolide A | Sesquiterpenoid |
| 59 | 30.57 | C15H22 | 202.1718 | 203.1791 | [M + H]+ | −1.88 | 105.0597, 119.0853, 133.1009, 147.1166 | Rulepidadiene B | Sesquiterpenoid |
| 60 | 31.03 | C17H16O5 | 300.0997 | 301.1066 | [M + H]+ | −1.98 | 155.0488, 241.0856, 269.0804 | Methyllinderone | Cyclopentenedione |
| 61 | 31.27 | C15H18O2 | 230.1301 | 231.1374 | [M + H]+ | −2.71 | 91.0841, 129.0694, 131.0854, 157.1010 | Lorneic acid J | Acid |
| 62 | 31.30 | C15H22O | 218.1666 | 219.1739 | [M + H]+ | −2.15 | 107.0855, 137.0956, 163.1113, 201.1426 | α-Cyperone | Sesquiterpenoid |
| 63 | 32.09 | C17H20O3 | 272.1404 | 273.1478 | [M + H]+ | −3.07 | 105.0697, 157.1010, 185.0957, 213.1270 | Lindenenyl acetate | Sesquiterpenoid |
| 64 | 32.09 | C16H20O2 | 244.1456 | 245.1529 | [M + H]+ | −2.86 | 105.0697, 157.1010, 185.1321, 213.1270 | Isolinderoxide | Sesquiterpenoid |
| 65 | 32.92 | C18H32O4 | 312.2301 | 313.2369 | [M + H]+ | −2.50 | 183.0289, 245.0806, 269.0804 | (2E,3R,4S)-2-Dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide | Butanolide |
| 66 | 32.97 | C15H18O | 214.1352 | 215.1425 | [M + H]+ | −2.45 | 95.0489, 109.0646, 159.0802, 173.1324 | (4aS,8aS)-4,4a,5,6,8a,9-Hexahydro-3,8a-dimethyl-5-methylenenaphtho[2,3-b]furan | Furan |
| 67 | 32.99 | C22H30O5 | 374.2093 | 375.2162 | [M + H]+ | 2.57 | 209.0809, 301.1795, 343.1895 | 1-Acetyl-4-methoxyl-denudaquinol | Phenolic derivative |
| 68 | 33.03 | C16H14O5 | 286.0841 | 287.0911 | [M + H]+ | −2.79 | 183.0289, 245.0806, 269.0804 | Linderone | Cyclopentenedione |
| 69 | 33.29 | C34H42O6 | 546.2981 | 547.3034 | [M + H]+ | −1.57 | 201.1646, 329.1355, 370.8308, 424.9020 | Linderin B | Sesquiterpenoid |
| 70 | 33.63 | C16H33NO | 255.2554 | 256.2627 | [M + H]+ | −3.18 | 88.0755, 102.0911, 115.1057, 130.1225 | Hexadecanamide | Amide |
| 71 | 34.49 | C21H38O4 | 354.2770 | 355.2838 | [M + H]+ | −1.27 | 109.0284, 127.0389, 268.9768 | (2E,3R,4S)-2-Tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide | Butanolide |
| 72 | 34.56 | C10H10O3 | 178.0627 | 179.0700 | [M + H]+ | 1.65 | 118.0412, 133.0645, 161.0595 | Coniferyl aldehyde | Aldehyde |
| 73 | 34.56 | C10H8O2 | 160.0521 | 161.0594 | [M + H]+ | −1.80 | 103.0540, 105.0697, 118.0411, 133.0646 | Ralfuranone A | Furanone |
| 74 | 34.83 | C32H38O5 | 502.2719 | 503.2793 | [M + H]+ | −2.48 | 273.1482, 379.1536, 393.1692 | Lindechunisin A | Sesquiterpenoid |
| 75 | 35.13 | C22H43NO | 337.3335 | 338.3407 | [M + H]+ | −3.00 | 163.1483, 177.1627, 303.3031, 321.3144 | Erucamide | Amide |
| 76 | 36.03 | C5H10O | 86.0730 | 87.0803 | [M + H]+ | −1.65 | 69.0699 | Prenol | Terpenoid |
| 77 | 36.26 | C10H16O | 152.1198 | 153.1271 | [M + H]+ | −1.88 | 97.0646, 107.0653, 109.1010, 135.1156 | Isothujone | Terpenoid |
| 78 | 36.49 | C16H32O | 240.2446 | 241.2519 | [M + H]+ | −2.91 | 69.0699, 83.0854, 97.1010, 111.1168 | Hexadecan-2-one | Ketone |
| 79 | 36.86 | C18H26O2 | 274.1925 | 275.1998 | [M + H]+ | −2.76 | 91.0540, 117.0697131.0853, 157.1009 |
Solwaric acid A | Aromatic acid |
| 80 | 36.87 | C5H10O | 86.0731 | 87.0803 | [M + H]+ | −1.39 | 69.0699 | Isoprenol | Terpenoid |
Previous phytochemical investigations have demonstrated that isoquinoline alkaloids are one of the major components in L. aggregata. In this study, a total of 21 alkaloids were identified from LAE under the positive mode. Notably, the major ion peak 15 displayed a protonated molecular ion at m/z 314. Subsequent examination of the MS/MS spectra revealed two major product ion peaks at m/z 297 and 265 (Fig. 1A). The fragment peak at m/z 297 possibly resulted from the loss of an amino group, while the other fragment peak at m/z 265 arose from the loss of a methanol molecule from the ion m/z 297. Moreover, the product ion at m/z 297 was further fragmented to produce ions at m/z 282 and 237 due to consecutive losses of CH3 and CO, respectively. Based on these spectral characteristics, peak 15 was identified as norisoboldine and further confirmed by comparison with its authentic standard.12 Peaks 16, 18, and 21, exhibiting similar fragment patterns, were identified as boldine, norboldine, and isoboldine, respectively. Peak 17 showed a precursor ion at m/z 330, generating fragment ions at m/z 299 due to the loss of CH3NH2, alongside m/z 192 and 137, corresponding to the isoquinoline and the benzylic cleavage fragment, respectively. Additionally, the product ion peak at m/z 299 further produced ions at m/z 267 and 175, attributed to the loss of methanol and benzene moiety, respectively. Hence, peak 17 was confidently identified as reticuline and confirmed with the standard compound (Fig. 1B).13 Peak 10 displayed a protonated ion at m/z 286, generating a highly abundant product ion at m/z 269 through the loss of an amino group. Consecutive losses of CH4OH and CO resulted in fragment ions at m/z 237 and 209, respectively. Furthermore, a series of fragment ions at m/z 107, 137, 145, and 175, corresponding to β-cleavage of the skeleton, were detected. These characteristic fragment ions supported the identification of peak 10 as coclaurine, further confirmed by comparison with the standard compound (Fig. 1C).13 Similarly, peak 2 showed a comparable MS fragmentation pattern to peak 10. With a molecular weight that was 14 Da lower than that of peak 10, peak 2 was characterized as higenamine according to the proposed fragmentation pathway (Fig. 1D).14
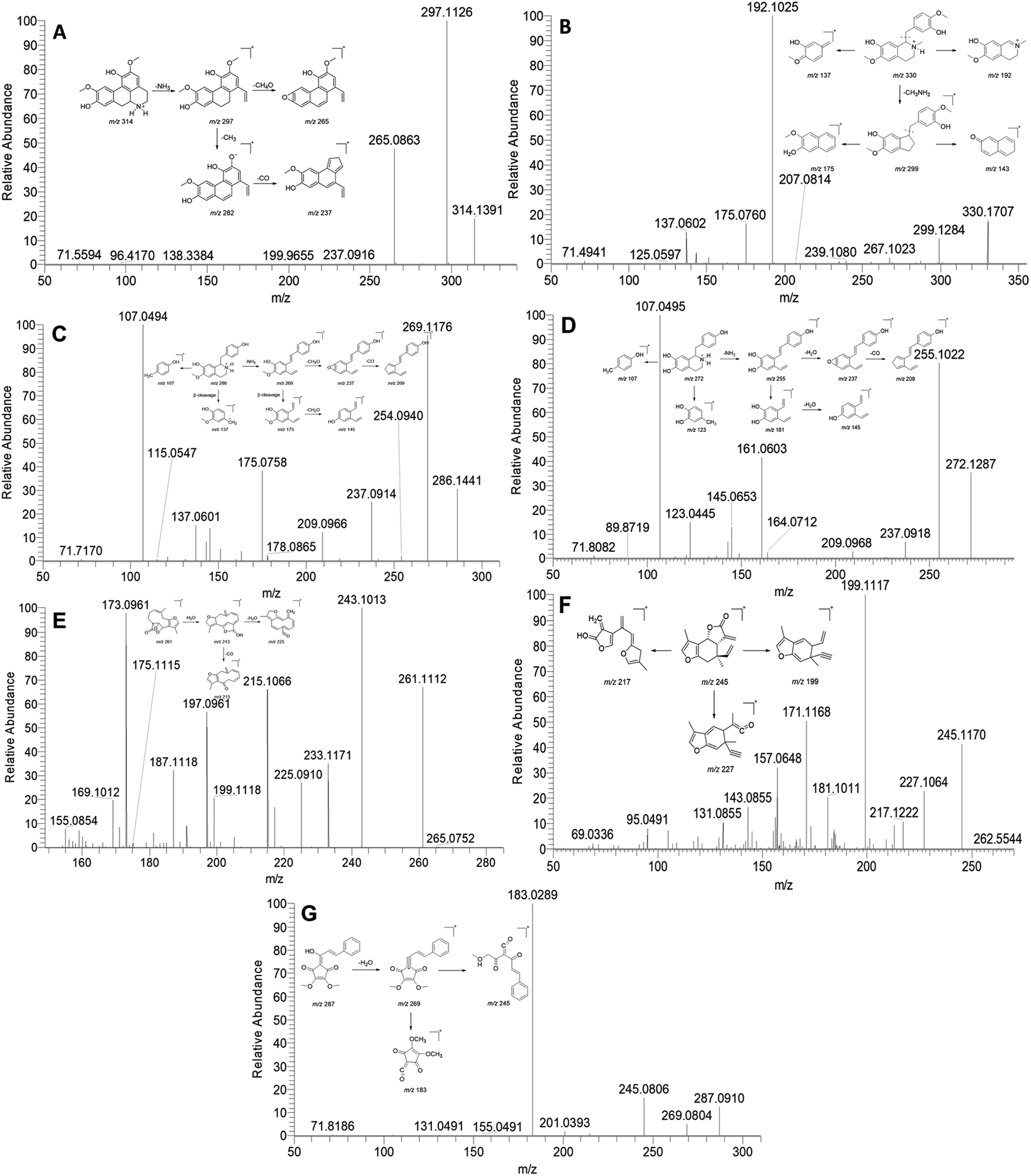 | ||
| Fig. 1 MS/MS spectra and the proposed fragmentation pathway of norisoboldine (A), reticuline (B), coclaurine (C), higenamine (D), linderane (E), isolinderalactone (F), and linderone (G). | ||
Sesquiterpenoid is another major constituent in L. aggregata. A total of 27 sesquiterpenoids were either unambiguously or tentatively identified in LAE. The MS/MS spectra behaviors of sesquiterpenoids in L. aggregata are notably complex due to the presence of multiple sesquiterpenoid skeletons. Taking peak 48 as an example, it yielded a [M + H]+ ion at m/z 261. The MS/MS spectra showed main fragment ion peaks at m/z 243, 225, 215, and 197, which are attributed to the consecutive losses of H2O and CO. Consequently, peak 48 was unambiguously identified as linderane, and further confirmed using the standard compound (Fig. 1E). Peak 50 showed similar MS fragmentation characteristics to peak 48, and it was characterized as isolinderalactone according to the proposed fragmentation pathway (Fig. 1F). Based on these similar fragmentation patterns, peak 51 was identified as lindenenol.15
Peak 68 exhibited a precursor ion at m/z 287. The characteristic fragment ions observed at m/z 269, 245, and 183 were attributed to the loss of H2O and the cleavage of the skeleton, leading to the identification of this peak as linderone (Fig. 1G). Peak 60 displayed a similar MS fragmentation pattern with peak 68, with a molecular weight that is 14 Da higher than that of peak 68. Thus, peak 60 was characterized as methyllinderone.16 Both the above cylcyclopentenediones were confirmed with standard compounds. Additionally, some interesting compounds (including peaks 67, 69, 71, and 74) were first identified from L. aggregata and confirmed with standard compounds. Based on the above results, the 16 compounds (including higenamine (2), coclaurine (10), norisoboldine (15), boldine (16), reticuline (17), linderane (48), isolinderalactone (50), lindeneol (51), methyllinderone (60), lindenenyl acetate (63), (2E,3R,4S)-2-dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide (65), 1-acetyl-4-methoxyl-denudaquinol (67), linderone (68), linderin B (69), (2E,3R,4S)-2-tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide (71), and lindechunisin A (74)) were selected for further quantification analysis.
3.2. Quantitative analysis of the selected compounds
Based on the qualitative results, the aforementioned 16 compounds were selected for quantitative analysis due to their predominance in the chemical profile of L. aggregata and their established or potential bioactivities. These compounds, including some key alkaloids and sesquiterpenoids, are critical for assessing the plant's quality. Herein, an UHPLC-MS/MS quantification method using PRM mode was established. The MS/MS detection parameters, such as ion pairs and collision energy, were optimized by directly injecting each analyte to achieve the most sensitive and stable reaction monitoring transitions. Fig. 2 displayed the PRM extracted ion chromatogram for each compound. | ||
| Fig. 2 Extracted ion chromatograms of the quantified analytes in PRM mode. | ||
The stock solution was diluted with LC-MS-grade solvent for different working concentrations to establish the calibration curves. The calibration curves were constructed using a series of different concentrations by least-square linear regression analysis. The linearity of each compound's calibration curve was assessed through repeated experiments, employing the regression coefficient (r2) within the tested concentration ranges (Table 2). Results demonstrated that good linearity was achievable within the tested concentration ranges. The LODs and LOQs fell within the range of 0.033–0.33 ng mL−1 and 0.11–1.1 ng mL−1, respectively. The RSD for precision and reproducibility were in the range of 1.0–4.2%, and 0.9–5.6%, respectively. The recovery rate of the analytes ranged from 85.4% to 114.7%. All the above results indicated that the analysis method was valid and reliable. Based on the established analysis method, the average content of each compound was determined as follows: higenamine was 0.39 ± 0.02 mg g−1, coclaurine was 0.95 ± 0.15 mg g−1, norisoboldine was 50.34 ± 4.27 mg g−1, boldine was 6.53 ± 0.24 mg g−1, reticuline was 3.30 ± 0.16 mg g−1, linderane was 23.47 ± 0.18 mg g−1, isolinderalactone was 10.45 ± 0.88 mg g−1, lindeneol was 16.70 ± 1.17 mg g−1, methyllinderone was 0.10 ± 0.01 mg g−1, lindenenyl acetate was 11.57 ± 0.08 mg g−1, (2E,3R,4S)-2-dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide was 0.09 ± 0.01 mg g−1, 1-acetyl-4-methoxyl-denudaquinol was 0.28 ± 0.02 mg g−1, linderone was 0.14 ± 0.01 mg g−1, linderin B was 0.55 ± 0.03 mg g−1, (2E,3R,4S)-2-tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide was 0.17 ± 0.01 mg g−1, and lindechunisin A was 0.15 ± 0.01 mg g−1.
| No. | Compounds | Linearity | LOD (ng mL−1) | LOQ (ng mL−1) | Precision RSD (%) | Reproducibility RSD (%) | Recovery (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| Linear range (ng mL−1) | r2 | Regression equation | |||||||
| 2 | Higenamine | 2.65–133.00 | 0.9997 | Y = −115619 + 321943x |
0.086 | 0.273 | 1.5 | 2.8 | 94.1–114.7 |
| 10 | Coclaurine | 2.50–125.00 | 0.9998 | Y = 466122 + 480791x |
0.033 | 0.109 | 1.3 | 3.5 | 91.0–103.6 |
| 15 | Norisoboldine | 2.45–245.00 | 0.9999 | Y = 371228 + 633145x |
0.079 | 0.235 | 1.9 | 3.0 | 85.4–98.5 |
| 16 | Boldine | 2.50–125.00 | 0.9997 | Y = 753908 + 839067x |
0.040 | 0.133 | 1.1 | 3.7 | 94.6–107.3 |
| 17 | Reticuline | 2.38–119.00 | 0.9995 | Y = 84052 + 1123580x |
0.032 | 0.110 | 1.0 | 3.7 | 95.2–114.7 |
| 48 | Linderane | 10.56–1056.00 | 0.9995 | Y = 1745.7 + 2941.5x | 0.308 | 1.062 | 1.6 | 5.6 | 96.7–108.5 |
| 50 | Isolinderalactone | 9.82–982.00 | 0.9994 | Y = −824931 + 100049x |
0.291 | 0.975 | 2.0 | 3.7 | 90.1–109.8 |
| 51 | Lindeneol | 10.80–540.00 | 0.9994 | Y = −59915.2 + 8850.63x |
0.317 | 1.078 | 1.9 | 4.1 | 85.9–97.9 |
| 60 | Methyllinderone | 2.31–116.00 | 0.9998 | Y = −2148.54 + 229864x |
0.077 | 0.228 | 4.2 | 1.7 | 95.3–98.9 |
| 63 | Lindenenyl acetate | 10.61–1061.00 | 0.9999 | Y = −4263.81 + 881.66x | 0.152 | 0.527 | 2.6 | 3.5 | 91.5–113.9 |
| 65 | (2E,3R,4S)-2-Dodecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide | 10.61–1061.00 | 0.9999 | Y = −21702.4 + 644.61x |
0.346 | 1.125 | 4.1 | 2.2 | 96.0–101.5 |
| 67 | 1-Acetyl-4-methoxyl-denudaquinol | 2.50–125.00 | 0.9998 | Y = −79756 + 3264.71x |
0.084 | 0.253 | 1.8 | 4.2 | 97.3–103.7 |
| 68 | Linderone | 2.34–117.00 | 0.9994 | Y = −100140 + 148063x |
0.078 | 0.228 | 2.4 | 0.9 | 97.5–103.0 |
| 69 | Linderin B | 2.48–245.00 | 0.9999 | Y = 27886.34 + 1974.69x |
0.082 | 0.251 | 1.2 | 2.1 | 93.2–98.5 |
| 71 | (2E,3R,4S)-2-Tetradecylinene-3-hydroxy-4-ethoxy-4-methylbutanolide | 9.82–982.00 | 0.9994 | Y = −32503.2 + 617.441x |
0.334 | 0.980 | 1.2 | 1.1 | 91.1–100.1 |
| 74 | Lindechunisin A | 2.65–133.00 | 0.9998 | Y = 17787.3 + 1585.95x |
0.088 | 0.272 | 2.2 | 3.0 | 96.3–102.5 |
3.3. Anti-inflammatory effects of LAE and selected compounds
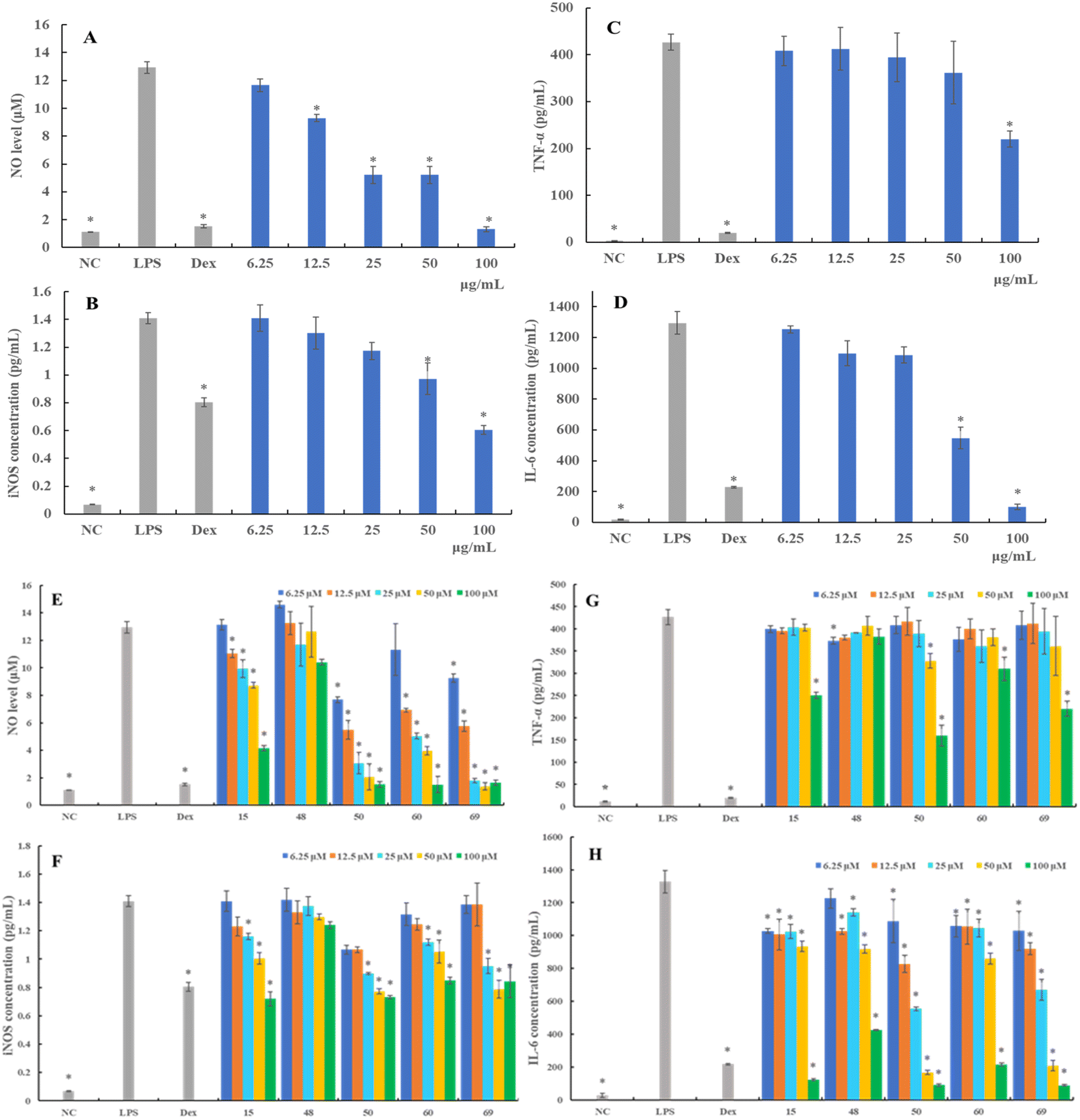 | ||
| Fig. 3 The effects of LAE on the production of NO (A), iNOS (B), TNF-α (C), and IL-6 (D) in LPS-induced RAW264.7 cells. The effects of norisoboldine (15), linderane (48), isolinderalactone (50), methyllinderone (60), and linderin B (69) on the production of NO (E), iNOS (F), TNF-α (G), and IL-6 (H) in LPS-induced RAW264.7 cells. RAW 264.7 cells were treated with various concentrations of LAE and selected compounds in the presence of LPS (1 μg mL−1) for 24 h. Protein expression of iNOS, TNF-α, and IL-6 in the culture medium was assayed by ELISA. NC: negative control group, LPS: LPS-treated group, Dex: dexamethasone-treated group. Data are presented as mean ± SD (n = 3). *p > 0.05, compared with the LPS-treated group. | ||
iNOS mediates inflammatory reactions and catalyzes the synthesis of NO.17 Therefore, we further examined the alternation of iNOS protein production following different treatments. As shown in Fig. 3F, the expression of levels of iNOS in RAW 264.7 cells substantially increased upon LPS stimulation. Compounds 15, 50, 60, and 69 demonstrated a concentration-dependent attenuation of LPS-induced iNOS expression, mirroring its effects on NO production. More specifically, compounds 50, 60, and 69 showed stronger inhibitory activity against iNOS production than compound 15.
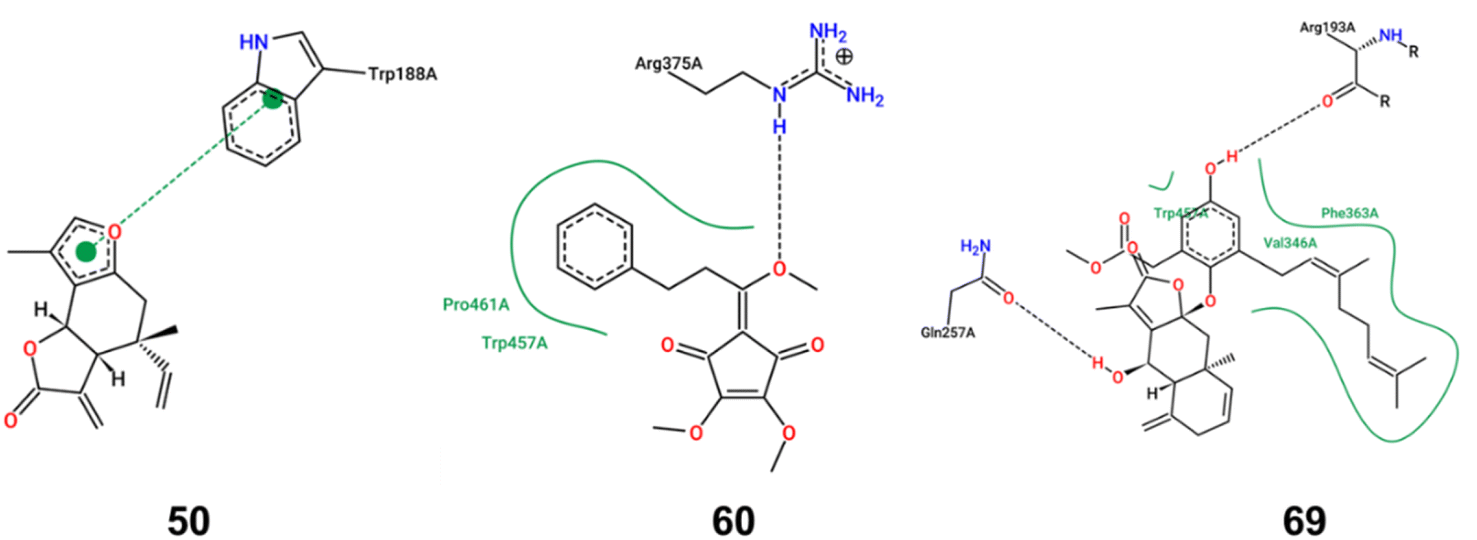 | ||
| Fig. 4 Docking model of compounds 50, 60, and 69 with iNOS. The interaction patterns are composed of hydrogen bonds, displayed as black dashed lines; π interactions, shown as green dashed lines with dots denoting the participating π systems; and hydrophobic contacts, which are represented by the residue labels and spline segments along the contacting hydrophobic ligand's part. | ||
Regarding compound evaluation, compounds 15, 50, 60, and 69 were found to significantly inhibit the expression of TNF-α at the concentration of 100 μM, and the TNF-α level decreased by 41.23%, 62.48%, 27.29%, and 48.43%, respectively (Fig. 3G). However, compound 48 showed a relatively weaker inhibitory effect compared to compounds 15, 50, 60 and 69, and did not exhibit a significant dose–response relationship. Further, the expression of IL-6 decreased dose-dependently following treatment with compound 50, which was as effective as compound 69, and even more effective than compounds 15, 48, and 60 (Fig. 3H). These results suggested that LAE and compounds 15, 48, 50, 60, and 69 suppressed the production of pro-inflammatory cytokines in the LPS-stimulated RAW 264.7 cells, which might be associated with the anti-inflammatory activity.
3.4. Molecular docking studies
iNOS can be activated by cytokines or LPS, leading to substantial NO secretion and subsequent inflammation. To elucidate the inhibitory mechanisms of the bioactive compounds, molecular docking studies were performed with iNOS. Compounds 50, 60, and 69, which demonstrated a significant inhibition effect on iNOS expression, were selected as the ligands. As shown in Fig. 4, compound 50 primarily exhibited π–π stacking interactions with iNOS, suggesting a strong affinity and potential specificity towards the enzyme's active site. In contrast, compounds 60 and 69 primarily engaged in hydrogen bond and hydrophobic interactions with iNOS. The binding energy score value was −7.7, −6.1, and −9.2, respectively. Owing to the lack of hydroxyl groups in compound 50, it is incapable of forming hydrogen bonds with iNOS. Consequently, the unsaturated lactone moiety formed a π–π interaction with the Trp188 residue of iNOS. Such π–π interaction, recognized as the most prevalent noncovalent interaction, manifests as favorable forces between the aromatic subunits of the biochemical molecules.18 The aromatic side chains of the amino acids, tryptophan and phenylalanine, are commonly modeled with indole and benzene, respectively. The π–π interaction, characterized by its broad and large surface area of contact, typically results in high binding energy. This implies that a protein–ligand complex can exhibit a high binding affinity even in the absence of hydrogen bonds. In the complex where iNOS interacted with compound 60, a single hydrogen bond was formed with Arg375, accompanied by hydrophobic interactions involving Pro461 and Trp457. Similarly, the iNOS complex with compound 69 established two hydrogen bonds with Arg193 and Gln257 and hydrophobic interactions with Trp450, Phe363 and Val346. While hydrogen bonds are relatively weaker than covalent or ionic bonds, their collective contribution can be substantial in terms of the overall binding energy between a ligand and its receptor. The oxygen atoms in methoxy group (compound 60) and hydroxy groups (compound 69) acted as hydrogen bond acceptors, significantly influencing the iNOS inhibitory activity. Moreover, the benzene rings and geranyl group in compounds 60 and 69 were likely to engage in robust interactions with the hydrophobic amino acid residue. These interactions were pivotal in enhancing the stability of the ligand–enzyme complex. Collectively, these findings provided a theoretical rationale for the effective binding of compounds 50, 60, and 69 to the inflammatory target iNOS, thereby inhibiting the expression of inflammatory markers.L. aggregata is a widely used traditional Chinese medicine and new food resource with reported curative effects in various aspects, including anti-cancer, anti-arthritis, anti-bacterial, anti-oxidation, anti-diabetic nephropathy, hepatoprotective, and lipid-lowering effects. Owing to its significant medicinal value and wide-ranging pharmacological applications, L. aggregata has attracted increasing attention in China. This herb is found across various regions in China, with specimens from Zhejiang Province being particularly esteemed for their quality.2 However, a notable challenge in the cultivation and utilization of L. aggregata is the genetic diversity and variation in the medicinal component content across different geographical locations. This variability poses a significant challenge to the consistency and stability of L. aggregata's quality, representing a crucial bottleneck in the standardization of its cultivation. This highlights the need for more focused research and development efforts to standardize and optimize the cultivation practices for L. aggregata, ensuring uniformity in the quality of this important medicinal plant. Although over 250 compounds have been isolated from L. aggregata to date, quantitative studies on its roots are relatively limited. Only nine compounds, including five sesquiterpenes and four alkaloids, have been quantified using LC-MS method.7,8 In this study, 80 compounds were identified, and 16 of them were quantified using LC-MS/MS method. Besides expanding the quantitative analysis of alkaloids and sesquiterpenoids in L. aggregata, butanolides and acyclopentendiones were quantified in L. aggregata for the first time, further advancing the quantitative study of L. aggregata. Given the comprehensive nature of the research on the anti-inflammatory effects of L. aggregata, we employed an approach that integrates both qualitative and quantitative methodologies. Our emphasis was on expeditiously identifying and quantifying anti-inflammatory compounds. The findings revealed that compounds 15, 50, 60, and 69 exhibited notable anti-inflammatory properties in LPS-induced RAW 264.7 cells, specifically evidenced by its ability to inhibit the production of NO and iNOS, alongside a marked suppression of cytokines including TNF-α, and IL-6. Extensive previous research has reported norisoboldine (15) as an anti-inflammatory property, demonstrating its capability to reduce systemic inflammation. Studies have demonstrated that norisoboldine possessed the capability to inhibit the production of pro-inflammatory factors and down-regulate the activation of MAPKs in LPS-induced RAW 264.7 cells.19 As a result of these established properties, norisoboldine has been designated as the chemical marker for quality evaluation in China Pharmacopoeia. Compound 50, isolinderalactone, is a representative elemane-type sesquiterpene lactone from L. aggregata. Molecular docking results suggested the unsaturated lactone moiety in compound 50 probably contributed significantly to its anti-inflammatory effect. This observation was echoed in the findings of Shen et al., who also identified the unsaturated lactone moiety as critical for the compound's anti-inflammatory effects.20 Notably, they observed that the anti-inflammatory efficacy of isolinderalactone was almost completely diminished when the unsaturated double bond was reduced, further underscoring the importance of this structural feature.20 Compound 60 (methyllinderone) is a rare natural acyclopentendione. Its anti-inflammatory activity was intimately associated with the presence of the methoxy group based on the molecular docking results, potentially elucidating its better activity compared with linderone. Compound 69 (linderin B), comprising a sesquiterpenoid lactone and a methyl geranylhomogentisate moiety, was identified as a new compound in our previous study.21 Although characterized by a complex conjugate, the presence of a long-chain geranyl group probably enhanced its hydrophobic interactions with iNOS. Building upon the results, we conducted quantitative studies on these compounds, aiming to provide a basis for quality control enhancement. The anti-inflammatory compounds in L. aggregata are crucial to its medicinal properties, traditionally used for treating inflammation-related ailments like arthritis and gastrointestinal disorders. These phytochemicals, particularly sesquiterpenes and alkaloids, could inhibit pro-inflammatory mediators such as cytokines and enzymes, reducing inflammation. This makes L.aggregata effective in treating chronic inflammatory conditions and supports its therapeutic potential in modern medicine. Scientific studies have validated these effects, reinforcing the plant's relevance as a treatment for inflammation in both traditional and contemporary pharmacology. In summary, L. aggregata's diverse bioactive compounds work synergistically, offering a broad therapeutic spectrum through multi-ingredient interactions. Future research will focus on standardizing extracts, exploring additional bioactivities, and elucidating the molecular mechanisms underlying its effects.
4. Conclusions
In conclusion, the chemical profiling and identification of 80 compounds in LAE, followed by quantification of 16 compounds, based on LC-MS/MS method were introduced herein. The quantification results might be used as chemical markers for the quality control of L. aggregata. This study further showcased the capability of LAE and some specific constituents in effectively suppressing NO, iNOS, TNF-α, and IL-6 in LPS-induced RAW 264.7 cells. Molecular docking investigations have provided insights into the mechanisms underlying the iNOS inhibitory activities of specific compounds. These results will hold significance for the enhancement of quality control and assurance processes for L. aggregata and related functional products, contributing to a better understanding of their potential health-promoting properties.Data availability
The authors confirm that the data supporting the results of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 81573606 and 81973486), and the Taishan Industrial Experts Program (No. tscx202306073).References
- Y. Tao, Y. Deng and P. Wang, J. Ethnopharmacol., 2023,(Part A), 116954 Search PubMed.
- Q. Huang, K. Liu, L. Qin and B. Zhu, Med. Plant Biol., 2023, 2, 11, DOI:10.48130/MPB-2023-0011.
- X. Liu, J. Yang, X. J. Yao, X. Yang, J. Fu, L. P. Bai, L. Liu, Z. H. Jiang and G. Y. Zhu, J. Org. Chem., 2019, 84(12), 8242–8247 CrossRef PubMed.
- X. Liu, J. Yang, J. Fu, X. J. Yao, J. R. Wang, L. Liu, Z. H. Jiang and G. Y. Zhu, Org. Lett., 2019, 21(14), 5753–5756 CrossRef PubMed.
- X. Liu, J. Fu, J. Yang, A. C. Huang, R. F. Li, L. P. Bai, Z. H. Jiang and G. Y. Zhu, ACS Omega, 2021, 6(8), 5898–5909 CrossRef PubMed.
- X. Liu, J. Fu, R. S. Shen, X. J. Wu, J. Yang, L. P. Bai, Z. H. Jiang and G. Y. Zhu, Phytochemistry, 2021, 191, 112924 CrossRef PubMed.
- Z. Han, Y. Zheng, N. Chen, L. Luan, C. Zhou, L. Gan and Y. Wu, J. Chromatogr. A, 2008, 1212(1–2), 76–81 CrossRef PubMed.
- Y. Wu, Y. Zheng, X. Liu, Z. Han, Y. Ren, L. Gan, C. Zhou and L. Luan, J. Sep. Sci., 2010, 33(8), 1072–1078 CrossRef PubMed.
- X. Liu, Z. Wang, F. G. Gmitter Jr, J. W. Grosser and Y. Wang, J. Agric. Food Chem., 2023, 71(2), 1246–1257 CrossRef CAS PubMed.
- K. Woertz, C. Tissen, P. Kleinebudde and J. Breitkreutz, J. Pharm. Anal., 2010, 51(3), 497–506 CrossRef CAS PubMed.
- F. Chen, H. L. Li, Y. H. Li, Y. F. Tan and J. Q. Zhang, Chem. Cent. J., 2013, 7, 1–9 CrossRef PubMed.
- X. Wu, H. Long, F. Li, W. Wu, J. Zhou, C. Liu, J. Hou, W. Wu and D. Guo, J. Sep. Sci., 2021, 44(20), 3810–3821 CrossRef CAS PubMed.
- J. Schmidt, K. Raith, C. Boettcher and M. H. Zenk, Eur. J. Mass Spectrom., 2005, 11(3), 325–333 CrossRef CAS PubMed.
- X. Zhao, Y. Yuan, H. Wei, Q. Fei, Z. Luan, X. Wang, Y. Xu and J. Lu, J. Pharm. Biomed. Anal., 2022, 214, 114732 CrossRef CAS.
- H. Cai, J. Wang, Y. Luo, F. Wang, G. He, G. Zhou and X. Peng, Biomed. Pharmacother., 2021, 134, 111098 CrossRef CAS.
- S. M. Lee, S. H. Baek, C. H. Lee, H. B. Lee and Y. H. Kho, Nat. Prod. Sci., 2002, 8(3), 100–102 CAS.
- F. Aktan, Life Sci., 2004, 75(6), 639–653 CrossRef CAS.
- S. Jena, J. Dutta, K. D. Tulsiyan, A. K. Sahu, S. S. Choudhury and H. S. Biswal, Chem. Soc. Rev., 2022, 51(11), 4261–4286 RSC.
- Z. F. Wei, B. Tong, Y. F. Xia, Q. Lu, G. X. Chou, Z. T. Wang and Y. Dai, PLoS One, 2013, 8(3), e59171 CrossRef CAS PubMed.
- X. Shen, H. Chen, H. Zhang, L. Luo, T. Wen, L. Liu, Q. Hu and L. Wang, Int. Immunopharmacol., 2023, 124, 110965 CrossRef.
- S. S. Wen, Y. Wang, J. P. Xu, Q. Liu, J. Zhang, Z. L. Li, N. Zhang, X. Liu, Y. W. Xu and Z. L. Sun, Nat. Prod. Res., 2022, 36(21), 5407–5415 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra05643d |
| This journal is © The Royal Society of Chemistry 2024 |