
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of bicyclomahanimbine by Cu(II)-promoted photoredox process†
Shilpa Dangar a,
Tiyasa Roya,
Suman Noskara and
Alakesh Bisai*ab
a,
Tiyasa Roya,
Suman Noskara and
Alakesh Bisai*ab
aIndian Institute of Science Education and Research Kolkata, Mohanpur, Nadia, West Bengal 741246, India. E-mail: alakesh@iiserkol.ac.in; alakeshb@gmail.com
bDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhauri, Bhopal 462 066, Madhya Pradesh, India
First published on 23rd September 2024
Abstract
Since the isolation of carbazole alkaloids, the synthetic chemists have witnessed an upsurge in research of them due to their potential pharmacological properties. Our approach shows the total syntheses of five such biorelevant pyrano-[3,2a]-carbazole alkaloids, emphasizing biomimetic and innovative synthetic methodologies such as cascade reactions and strategic bond formations through sustainable electrochemical and photochemical conditions.
Introduction
Carbazole alkaloids, a prominent class of natural compounds, are renowned for their unique and wide biological effects, and have had a great impact on pharmaceutical research and development.1–4 The curry tree (Murraya koenigii), a significant medicinal plant in Asia, is particularly notable for its abundance of biologically active carbazole alkaloids. It was from Murraya koenigii that Chakraborty et al. (1965) isolated the first carbazole (murrayanine, shows antibiotic properties) from a biological source.5The foremost alkaloid component of Murraya koenigii is the monoterpenoid pyrano-[3,2-a]-carbazole, mahanimbine (Fig. 1); first isolated by Chakraborty et al. from the stem bark in 1966.6 The structure elucidation was done by Narasimhan et al. later in 1968.7 The 2H-chromene unit on mahanimbine indicates that its biosynthesis likely involves geranylation and subsequent cyclization of 2-hydroxy-3-methylcarbazole. Further annulations that transpire in vivo construct the bicyclic units that contribute to the structural variety of carbazoles found in this Murraya genus. Among them, curryanin (1969, Dutta et al.),8 murrayazolinine (1973, Chakraborty et al.)9 and feature pentacyclic scaffold whereas bicyclomahanimbine and isocyclomahanimbine (1969, Kapil et al.)10 have hexacyclic heptane [3,2,0] core (Fig. 1).
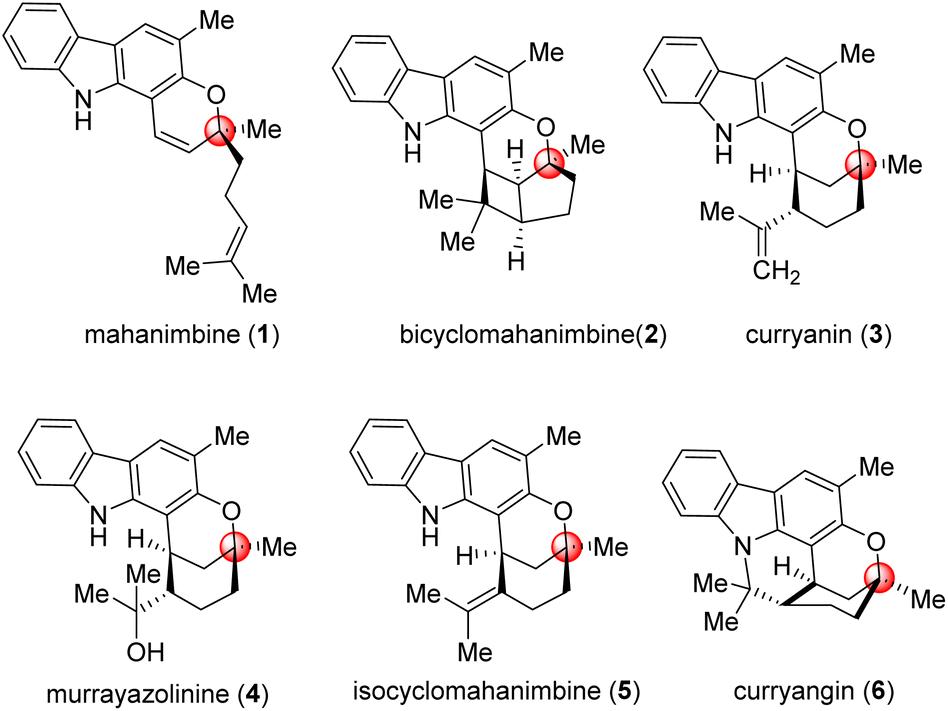 | ||
| Fig. 1 Representatives of pyrano [3,2-a] carbazole alkaloids. | ||
Among these mahanimbine (1) has shown broad spectra of biological effects, primarily cytotoxic and antitumor properties against various cancer cell lines including CEM-SS, HL-60, MCF-7, P388, and HeLa.11,12 It also exhibits anti-plasmodial (IC50 – 9.12 mM),13 anti-HIV,14 antioxidant,3 anti-inflammatory,3 and melanogenesis inhibitory activities,15 highlighting its potential in pharmacology. Additionally, it lowers blood glucose levels by enhancing peripheral glucose uptake or stimulating pancreatic insulin secretion4 underscoring its versatility to become a therapeutic agent and emphasizing its importance for further research and synthetic efforts. It was explored that, due to their parent skeleton (3-methylcarbazole), they exhibit potential biological activities, including antitumor, antibacterial, antiviral, and antifungal properties.3,4 Adding methyl groups to biologically active molecules can alter their conformation, potentially enhancing their activity, as well as improving their hydrophilicity and solubility (‘Magic Methyl’ effect).16
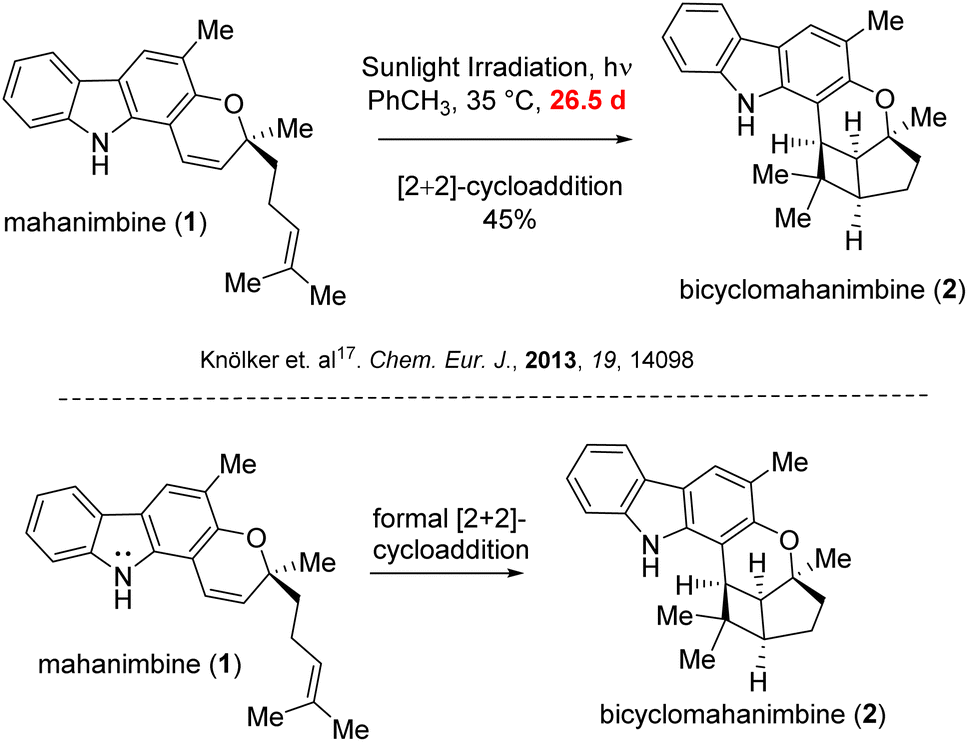
The traditional methods for crafting the carbazole motifs or achieving subsequent annulations often rely on pre-functionalization steps, oxidants, metal catalysis or significantly high temperatures, which come with a number of disadvantages. These include the need for stoichiometric concentrations of oxidizers, and rather costly metal complexes which can be hazardous or poisonous, limits functional group tolerance and also sometimes, the requirement for essential leaving groups, leading to significant reagent waste. Previous studies by various groups predominantly utilized such Pd-catalyzed cross-coupling reactions to synthesize the parent 2-hydroxy-3-methyl carbazole skeleton.17–20 Furthermore, the sole documented method for converting mahanimbine into bicyclohamanimbine required sunlight irradiation over nearly a lunation and resulted only 45% yield.17
Thus, advancements in atom economics and improvement in environmental impact, reaction rate are much required. Hence, in our approach to synthesize pyrano-[3,2-a]-carbazole alkaloids, we reduced the usage of metal complexes wherever possible and utilized photo-redox chemistry as well as electrochemistry, which are ‘green’ substitutes in synthetic chemistry offering exceptional reaction paths and intrinsic safety. Correspondingly, the waste generation will significantly reduce.
The potential of mahanimbine (1) as a versatile intermediate for accessing diverse monoterpenoid pyrano-[3,2-a]-carbazole alkaloids, we developed a synthetic strategy centered on this compound. Inspired by biosynthetic pathways, we explored a sustainable formal [2 + 2]-cycloaddition approach to construct the bicyclomahanimbine (2) skeleton. Here, toxic reagents can be superseded by using photoredox catalysis for ring annulation. Constructing curryanin (3) can also be facilitated by using an Alder-ene type reaction on mahanimbine (1). Subsequent regioselective hydration of curryanin (3) can lead to the formation of murruyazolinine (4). Furthermore, isocyclomahanimbine (5) can be achieved through the isomerization of curryanin (3). Therefore, the synthesis of mahanimbine (1) serves as a crucial starting point in our synthetic strategy (Scheme 1).
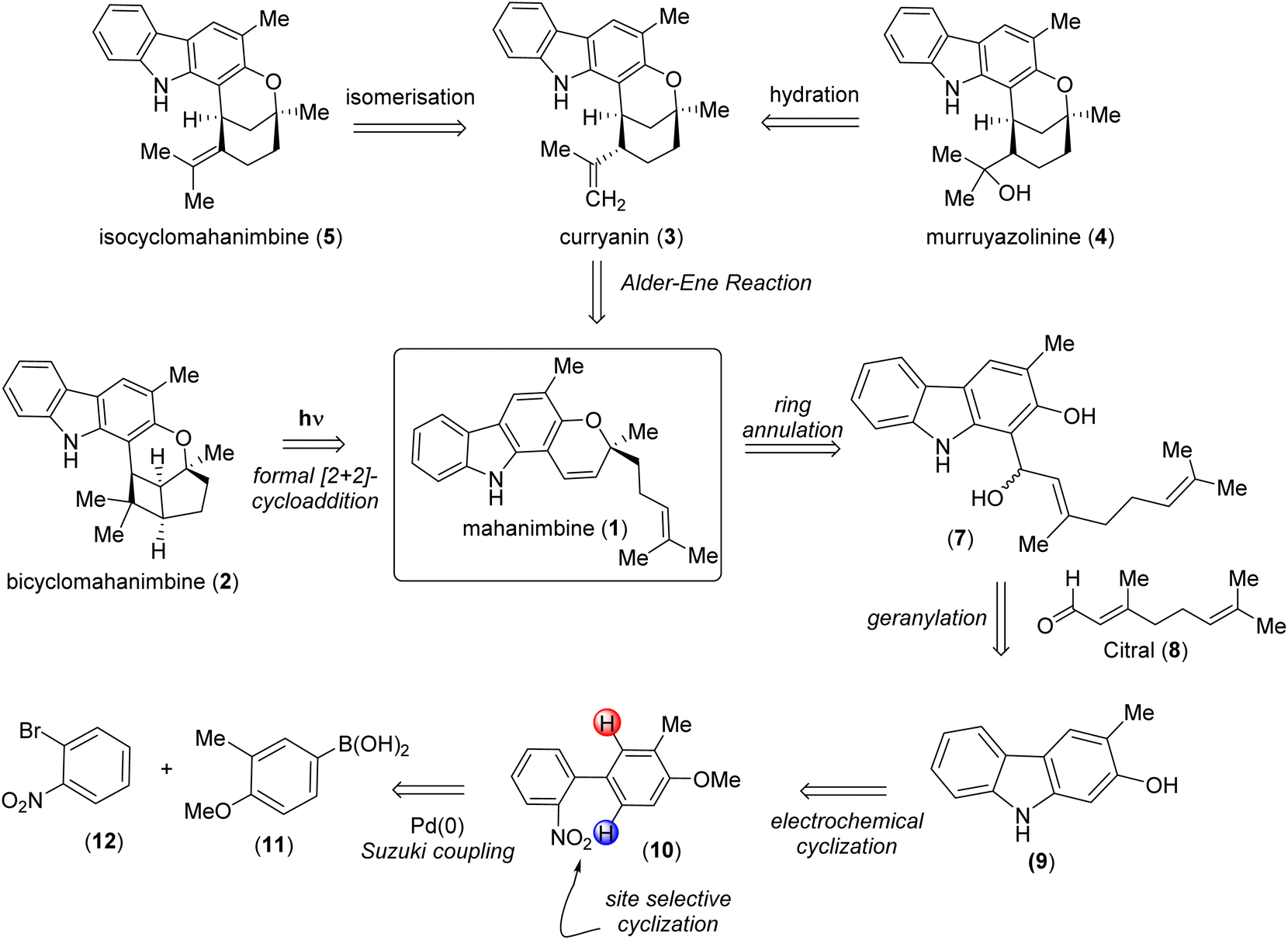 | ||
| Scheme 1 Divergent retrosynthetic analysis of pyrano-[3,2-a] carbazole alkaloids using mahanimbine (1) as the precursor. | ||
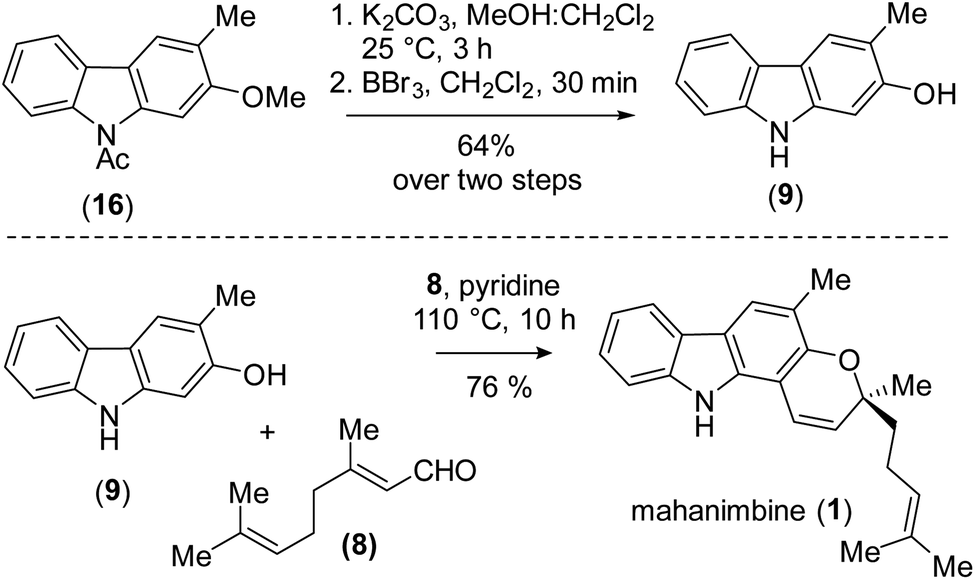
To design the 2H-chromene ring on mahanimbine (1), we envisaged the established biomimetic geranylation protocol for pyran annulation on 2-hydroxy-3-methylcarbazole (9). Sustainable and cost-effective electrochemical C–N bond formation was hypothesized to craft the carbazole motif.21 To construct the C–C bond, Suzuki–Miyaura coupling has been chosen. Therefore, the synthesis begins with commercially available 4-bromo-2-methylanisole (13) (Scheme 2).
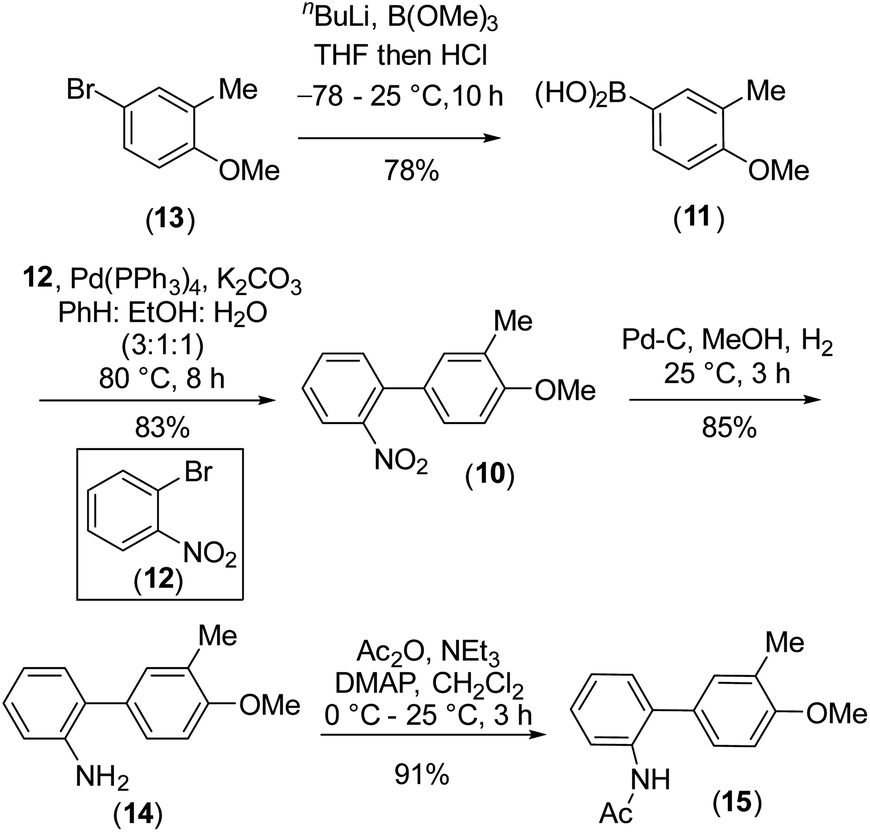 | ||
| Scheme 2 Generation of carbazole precursor biphenyl compound (10) for electrochemical cyclization. | ||
Our experimental approach commenced by treating 4-bromo-2-methylanisole (13) with nBuLi for metal-halogen exchange, followed by quenching of the generated aryl lithium species with B(OMe)3 and subsequent acidic work-up. This process yielded 4-methoxy m-tolyl boronic acid (11), which underwent a Pd(0)-catalyzed Suzuki–Miyaura coupling22 with readily available 2-bromonitrobenzene (12) to form compound (10). After forming the C–C bond, the nitro group on (10) was fully reduced to an amino group with Pd/C-occluded H2 (Scheme 2). Subsequently, we attempted electrochemical oxidation to construct the carbazole scaffold from compound (14). Due to their susceptibility towards side reactions,23 we implemented a constant potential protocol.
However, this approach was unsuccessful. Since the use of amidyl radicals as reactive intermediates has been extensively studied and is closely linked to cyclization reactions,25 we then explored same dehydrogenative electrochemical reaction, protecting the amino group with various electron-withdrawing groups.26,27 Notably, when the amino group was protected with an acetyl group (as in 15), the desired C–N bond formation (see, carbazole 16) occurred with a yield of 73% (r![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :r = 3:1) (Scheme 3) [for detailed optimization, see the ESI† for details].
:r = 3:1) (Scheme 3) [for detailed optimization, see the ESI† for details].
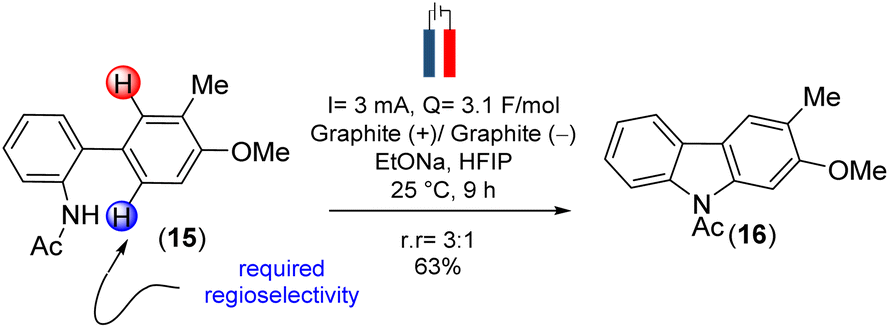 | ||
| Scheme 3 Sustainable dehydrogenative electrochemical C–N bond formation to construct the carbazole scaffold. | ||
Having ample quantities of carbazole (16) available, we proceeded with alkaline hydrolysis of amide followed by phenolic ether cleavage using BBr3. These set of reactions provided the biological carbazole precursor, 2-hydroxy-3-methylcarbazole (9). Now, to construct the 2H-chromene motif, we accomplished base-mediated cascade pyran-ring annulation28 with an established C10-building block such as citral (8), over the previously reported Lewis-acid mediated cyclization [which yielded only 74% using Ti(OiPr)4].15 After exhaustive optimization, we found that refluxing the phenolic compound (9) in pyridine solvent in the presence of 10 equiv. citral (8) yielded mahanimbine (1) with an impressive yield of 76% (Scheme 4). The tentative mechanism for the formation of mahanimbine (1) from a reaction of carbazole 9 with citral (see, ESI for details†).
 | ||
| Scheme 4 Biomimetic synthesis of mahanimbine (1). | ||
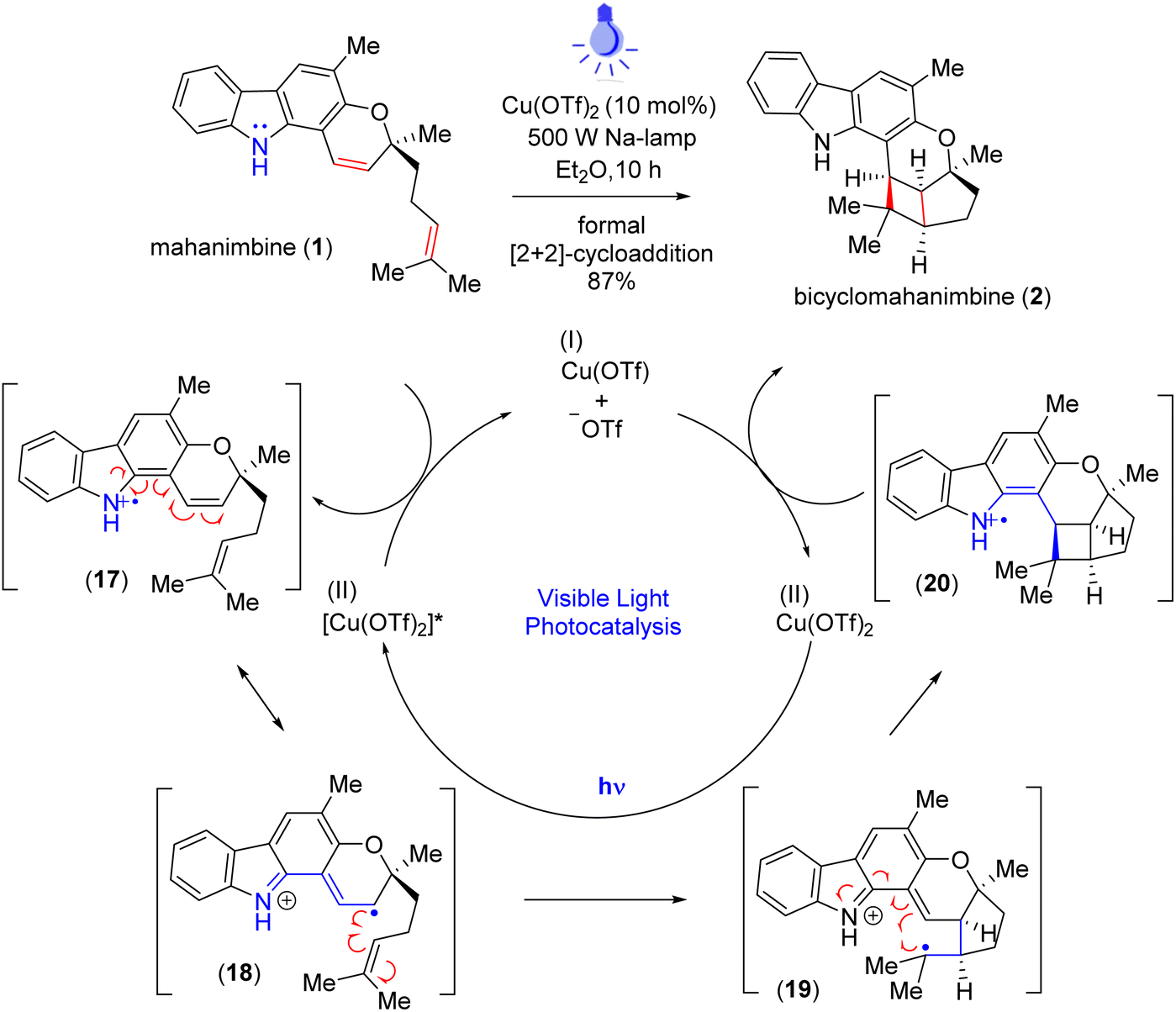
In 2013, Knölker group17 subjected mahanimbine (1) to prolonged exposure to sunlight. After 26.5 days, they isolated bicyclomahanimbine (2) with a yield of 45%. With utilization of 5 mol% FeCl3, Tan group also achieved this conversion but only with 42% yield.29 Hence, our focus was also into expediting the synthesis of bicyclomahanimbine (2) within a significantly shortened timeframe. As hypothesized, we explored various photocatalytic optimization strategies (Table 1) for synthesizing bicyclo-[3,2,0] heptane system in bicyclomahanimbine (2) from mahanimbine (1). The most successful approach involved using Cu(OTf)2 as a catalyst (10 mol%) in diethyl ether solvent under the irradiation of a 500 W Na-lamp, achieving a maximum yield of 87%. Under visible light, Cu(OTf)2 undergoes excitation30 and subsequent abstraction of an electron from the nitrogen atom of mahanimbine (1), forms a cation radical intermediate and ultimately Cu(II) reduces to Cu(I). The intermediate then undergoes electronic rearrangement, resulting in the formation of a five-membered ring, with the radical stabilizing at a tertiary position. Concurrently, a four-membered ring forms adjacent to the fused five-membered ring, with a radical cation generated at the nitrogen center. Subsequently, the Cu(I) species donates an electron, oxidizing the nitrogen radical cation intermediate resulting in the formation of bicyclomahanimbine (2) via a formal [2 + 2]-cycloaddition and regenerating Cu(OTf)2 in the process (Scheme 5).
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Lamp | Time | Yielda,b |
| a Optimization reactions were carried out on 0.33 mmol of substrate.b Yields are isolated after column chromatography. | |||||
| 1 (ref. 24) | LDA | THF | Tungsten bulb | 5 h | ND |
| 2 | fac-Ir(ppy)3 | MeOH | 456 nm LED | 10 h | 32% |
| 3 | — | Et2O | 500 W Na-lamp | 3 d | NR |
| 4 | Cu(OTf)2 (10 mol%) | Et2O | 500 W Na-lamp | 10 h | 87% |
 | ||
| Scheme 5 Proposed mechanism of visible-light photocatalysis for the synthesis of bicyclomahanimbine (2). | ||
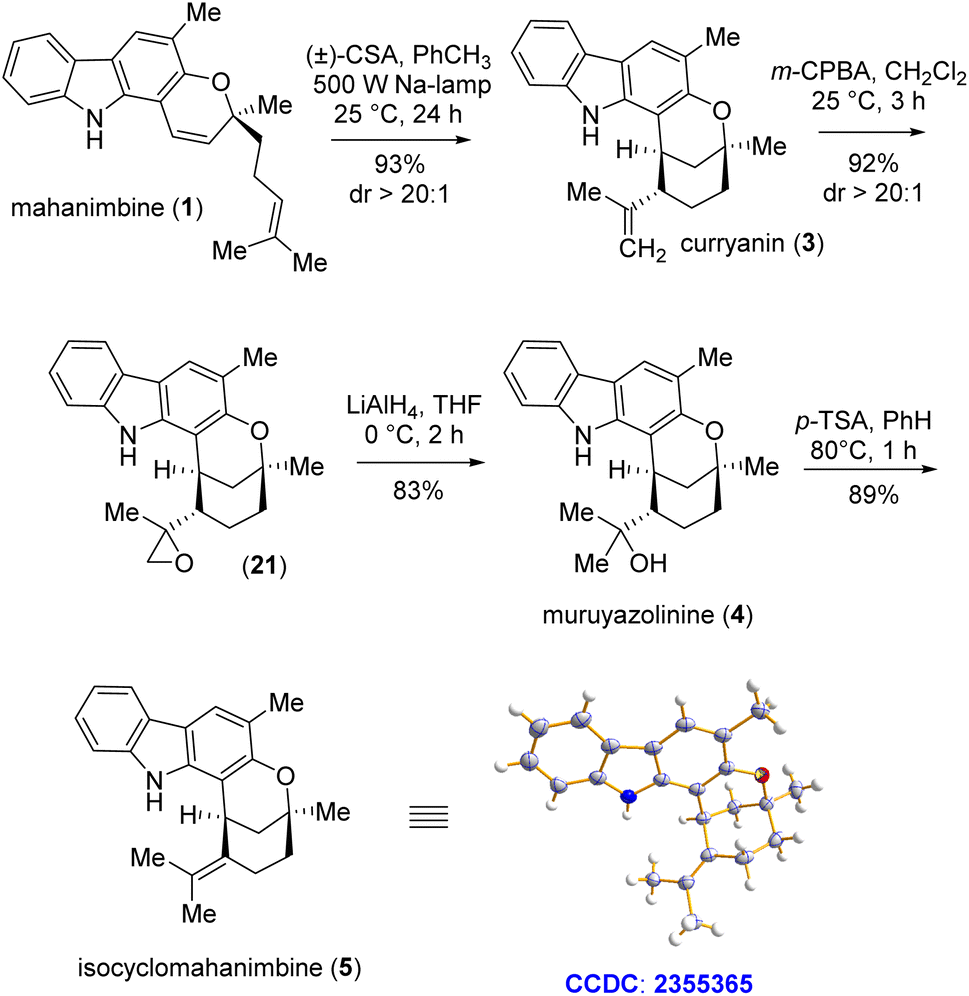
Conversion of mahanimbine (1) into curryanin (3) using camphor sulfonic acid (CSA) as a Brønsted acid has previously been accomplished by the Knölker group.17 However, their procedure had few drawbacks as being time-consuming, nevertheless prone to generating side-products which could have reduced the output.17 In this regard, Sarpong et al. achieved 99% yield in the reaction under the same conditions with a desmethylated mahanimbine analog; however, it still required 41.5 hours.20 To reduce such time-consumption, we employed CSA in toluene medium under sodium light conditions. Within 24 hours of reaction time, we successfully acquired our intended product compound (3) with a striking yield of 93% (Scheme 6). Following the synthesis pathway, to design murrayazolinine (4), we first reacted the olefin on curryanin (3) with m-CPBA. Subsequently, the generated epoxide compound (21) was charged with LiAlH4-mediated SN2-reaction for epoxy-ring opening. These sequential reactions resulted in the regioselective hydration of the olefin, which ultimately led to murrayazolinine (4) with an excellent yield of 76.5% over two steps (Scheme 6). Then, instead of directly attempting the isomerization of curryanin (3), we chose to proceed with dehydration of murrayazolinine (4). This approach aimed to favor the formation of the more substituted olefin, which is the thermodynamically controlled product, namely isocyclomahanimbine (5). Catalytic para-toluenesulfonic acid (20 mol%) in benzene under reflux conditions was employed to carry out this dehydration and after 1 hour of reaction time, we successfully isolated the crystalline form of the natural product isocyclomahanimbine (5) with 89% yield (Scheme 6).
 | ||
| Scheme 6 End-game synthesis of curryanin (3) and conversion to murrayazolinine (4) and isocyclomahanimbine (5). | ||
Conclusions
In summary, to achieve complex monoterpenoid pyrano-[3,2-a]-carbazole alkaloids, this approach not only enhances the efficiency of the synthesis but also aligns with principles of sustainability and environmental responsibility redressing the drawbacks of previous reports. Key highlights include the successful application of photoredox and electrochemical methods for efficient bond formations, as well as the regioselective hydration and dehydration strategies to access different structural motifs. The use of Cu(OTf)2 played crucial roles in the photoredox process controlling reaction outcomes with excellent yields. Overall, the synthetic approach demonstrated a blend of modern sustainable methodologies and traditional organic chemistry techniques, showcasing the importance of innovation and optimization in natural product synthesis.Experimental
Synthetic procedures and full characterization data are provided in the ESI.†Data availability
Experimental details and spectral analysis are available free of charge from the ESI† available with this article.Conflicts of interest
There are no conflicts to declare.Notes and references
- D. P. Chakraborty, The Alkaloids: Chemistry and Pharmacology, 1993, vol. 44, pp. 257–364 Search PubMed.
- H.-J. Knölker and K. R. Reddy, The Alkaloids: Chemistry and Pharmacology, 2008, 65, 181–193 Search PubMed.
- R. S. Ramsewak, M. G. Nair, G. M. Strasburg, D. L. DeWitt and J. L. Nitiss, J. Agric. Food Chem., 1999, 47, 444–447 CrossRef PubMed.
- T. Nagappan, T. C. Segaran, M. E. A. Wahid, P. Ramasamy and C. S. Vairappan, Molecules, 2012, 17, 14449–14463 CrossRef PubMed.
- D. P. Chakraborty, B. K. Barman and P. K. Bose, Tetrahedron, 1965, 21, 681–685 CrossRef.
- K. C. Das, D. P. Chakraborty and P. K. Bose, Sci. Cult., 1966, 32, 83–84 Search PubMed.
- N. S. Narasimhan, M. V. Paradkar and V. P. Chitgupp, Tetrahedron Lett., 1968, 53, 5501–5504 CrossRef PubMed.
- N. L. Dutta, C. Quasim and M. S. Wadia, Indian J. Chem., 1969, 7, 1168–1169 CAS.
- D. P. Chakraborty, S. N. Ganguly, P. N. Majhi, A. R. Mitra, K. C. Das and B. Wesntein, Chem. Ind., 1973, 322–323 CAS.
- S. P. Kureel, R. S. Kapil and S. P. Popli, Tetrahedron Lett., 1969, 10, 3857–3862 CrossRef PubMed.
- S.-P. Tan, A. M. Ali, M. A. Nafiah, K. Awang and K. Ahmad, Tetrahedron, 2015, 71, 3946–3953 CrossRef CAS.
- T. Nagappan, P. Ramasamy, M. E. Abdul Wahid, T. C. Segaran and C. S. Vairappan, Molecules, 2011, 16, 9651–9664 CrossRef CAS PubMed.
- Y. Nali, V. Thakur, A. Mohmmed, V. Gupta and A. Ali, New J. Chem., 2017, 41, 4923–4930 RSC.
- K. M. Meragelman, T. C. McKee and M. R. Boyd, J. Nat. Prod., 2000, 63, 427–428 CrossRef CAS PubMed.
- S. Nakamura, S. Nakashima, Y. Oda, N. Yokota, K. Fujimoto, T. Matsumoto, T. Ohta, K. Ogawa, S. Maeda, S. Nishida, H. Matsuda and M. Yoshikawa, Bioorg. Med. Chem., 2013, 21, 1043–1049 CrossRef CAS PubMed.
- H. Schonherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- R. Hesse, K. K. Gruner, O. Kataeva, A. W. Schmidt and H. J. Knölker, Chem.–Eur. J., 2013, 19, 14098–14111 CrossRef CAS PubMed.
- K. K. Julich-Gruner, O. Kataeva, A. W. Schmidt and H. J. Knölker, Chem.–Eur. J., 2014, 20, 8536–8540 CrossRef CAS PubMed.
- A. Polley, K. Varalaxmi, A. Nandi and R. Jana, Asian J. Org. Chem., 2021, 10, 1207–1215 CrossRef CAS.
- (a) K. Norseeda, V. Gasser and R. Sarpong, J. Org. Chem., 2019, 84, 5965–5973 CrossRef CAS PubMed; (b) For synthesis of mahanimbine via a hexadehydro Diels-Alder reaction, see; T. Wang and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 13870–13873 CrossRef CAS PubMed and references cited therein..
- A. Kehl, N. Schupp, V. M. Breising, D. Schollmeyer and S. R. Waldvogel, Chem.–Eur. J., 2020, 26, 15847–15851 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) J. F. Ambrose and R. F. Nelson, J. Electrochem. Soc., 1968, 115, 1159–1164 CrossRef CAS; (b) J. F. Ambrose, L. L. Carpenter and R. F. Nelson, J. Electrochem. Soc., 1975, 122, 876–894 CrossRef CAS; (c) K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef CAS.
- B. M. Trost and W. Tang, J. Am. Chem. Soc., 2002, 124, 14542–14543 CrossRef CAS.
- (a) G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226–9229 CrossRef CAS PubMed; (b) B. Janza and A. Studer, J. Org. Chem., 2005, 70, 6991–6994 CrossRef CAS; (c) H.-C. Xu, J. M. Campbell and K. D. Moeller, J. Org. Chem., 2014, 79, 379–391 CrossRef; (d) C. Moutrille and S. Z. Zard, Chem. Commun., 2004, 1848–1849 RSC; (e) K. C. Nicolaou, P. S. Baran, Y.-L. Zhong, S. Barluenga, K. W. Hunt, R. Kranich and J. A. Vega, J. Am. Chem. Soc., 2002, 124, 2233–2244 CrossRef; (f) L. Zhu, P. Xiong, Z.-Y. Mao, Y.-H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem. Int. Ed., 2016, 55, 2226–2229 (Angew. Chem., 2016, 128, 2266–2269) CrossRef.
- A. Kehl, V. M. Breising, D. Schollmeyer and S. R. Waldvogel, Chem.–Eur. J., 2018, 24, 17230–17233 CrossRef.
- S. Mallick, T. Mandal, N. Kumari, L. Roy and S. De Sarkar, Chem.–Eur. J., 2024, 30, e202304002 CrossRef CAS PubMed.
- S. P. Kureel, R. S. Kapil and S. P. Popli, Chem. Commun., 1969, 1120–1121 RSC.
- S.-P. Tan, K. Ahmad and M. T. Nafiah, Tetrahedron, 2017, 73, 4805–4810 CrossRef CAS.
- T. Bach, C. Krüger and K. Harms, Synthesis, 2000, 305–320 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure and characterisation of all compounds. CCDC 2355365. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra05863a |
| This journal is © The Royal Society of Chemistry 2024 |