DOI:
10.1039/D4RA05984K
(Paper)
RSC Adv., 2024,
14, 33602-33618
Mechanistic insights into carbonic anhydrase IX inhibition by coumarins from Calendula officinalis: in vitro and in silico approaches†
Received
18th August 2024
, Accepted 14th October 2024
First published on 23rd October 2024
Abstract
Given the critical role of carbonic anhydrase IX (CA IX) in various pathological conditions, there is a significant demand for new inhibitors to enhance patient outcomes and clinical management. In this study, we examined the inhibitory effectiveness of five coumarins derived from Calendula officinalis against CA IX using in vitro assays and computational modeling. Among the coumarins tested, xeroboside and isobaisseoside were identified as the most potent inhibitors. Kinetic studies indicated that xeroboside and isobaisseoside exhibit a mixed inhibition mode. Molecular docking analysis showed that the tested coumarins exhibit binding affinities and extensive polar interactions with CA IX. These coumarins demonstrated significant hydrophobic interactions and occupied the same binding site as acetazolamide (AAZ). Molecular dynamics (MD) indicated that xeroboside and isobaisseoside exhibited consistent trajectories and notable energy stabilization during their interaction with CA IX. MM/PBSA calculations showed that xeroboside displayed the lowest binding free energy (−27.26 ± 2.48 kJ mol−1). Potential Energy Landscape (PEL) analysis revealed distinct and stable conformational states for the CA IX–ligand complexes, with xeroboside exhibiting the most stable and lowest energy configuration. These computational findings are consistent with the experimental results, highlighting the potential efficacy of xeroboside and isobaisseoside as CA IX inhibitors. In conclusion, Calendula officinalis-derived coumarins are promising candidates as effective CA IX inhibitors.
1. Introduction
Carbonic anhydrases (CAs) are a family of zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate and a proton, a reaction crucial for maintaining acid–base balance in various tissues and organs.1 These enzymes are implicated in numerous physiological processes including respiration, acid-base homeostasis, and ion transport.1 Among the known isoforms in humans, CA IX is of particular interest due to its overexpression in several types of tumors, particularly under hypoxic conditions.2 CA IX plays a vital role in the regulation of intracellular and extracellular pH in cancer cells, facilitating their survival, proliferation, and invasion.3 The inhibition of CA IX has thus gained significant attention as a therapeutic strategy, aiming to disrupt the pH regulatory mechanism of cancer cells, thereby inhibiting tumor growth and metastasis.4 CA IX is remarkably expressed in cells with high growth and glycolysis rates and cells frequently exposed to acidic and hypoxic environments, such as cancer and endothelial cells (ECs).5 In the endothelium, the role of CA IX extends beyond pH regulation to aerobic glycolysis, EC migration and network formation.6 Targeting CA IX with specific inhibitors could lead to the development of novel anticancer therapies with reduced side effects compared to conventional treatments. In addition, CA IX inhibition might influence ECs and integrity of barriers in different tissues.
Calendula officinalis (C. officinalis), commonly known as marigold, is a well-known medicinal plant with diverse phytochemical composition and broad spectrum of biological activities.7 The plant is rich in secondary metabolites, including flavonoids, triterpenoids, carotenoids, and essential oils, which contribute to its anti-inflammatory, antioxidant, antimicrobial, and anticancer properties.8–11 Among these compounds, coumarins are notable for their therapeutic potential.12 C. officinalis has been documented to exhibit different pharmacological effects,11 making its phytoconstituents, including coumarins prime candidates for further investigation in enzyme inhibition studies. The selection of coumarins from C. officinalis for studying CA IX inhibition is justified by the plant established use in traditional medicine,11 and previous studies indicating the effectiveness of coumarins in inhibiting various enzymes and pathways involved in cancer progression.13 Therefore, exploring the inhibitory effects of these natural compounds on CA IX could provide valuable insights into novel anticancer strategies and enhance the therapeutic applications of C. officinalis.
To comprehensively assess the inhibitory activity of coumarins on CA IX, we employed an integrated approach combining in vitro assays and in silico studies. The in vitro assays provide direct evidence of the potency of the isolated phytoconstituents. Complementing these experiments, molecular docking and molecular dynamics (MD) simulations offer a detailed understanding of the interaction mechanisms at the molecular level.14 Docking studies help predict the binding affinity and orientation of coumarins within the active site of CA IX, while MD simulations reveal the stability and dynamic behavior of the enzyme–inhibitor complexes over time. This integrated approach not only validates the inhibitory activity observed experimentally but also elucidates the underlying molecular interactions, paving the way for the development of more effective CA IX inhibitors.
2. Materials and methods
2.1. Phytochemical investigation
2.1.1. General. NMR spectral analysis, including 1H NMR (500 MHz) and (125 MHz) 13C NMR, of the isolated coumarins was performed using Bruker AM-500 spectrometer. The optical density polarimeter was Rudolph Autopol III. UV-vis spectral analysis was assessed using the Shimadzu UV–Vis 160i spectrophotometer. HREIMS EIMS spectra were obtained using the Finnigan MAT TSQ 700 mass spectrometer. FTIR spectra of the isolated compounds were obtained utilizing the Shimadzu FTIR-8400 instrument with KBr pellets.
2.1.2. Plant collection and isolation of phytochemicals. The aerial parts of C. officinalis were obtained from local private gardens where it is commercially grown. The plant species was identified by expert taxonomists. The powdered dried aerial parts of the plant (4.5 kg) was exhaustively extracted by cold maceration using methanol (MeOH) for three days. A portion of the obtained extract (250 g) was suspended in water and successively extracted with petroleum ether, ethyl acetate (EtOAc), and n-butanol (1.1 L × 3, each). The EtOAc soluble fraction (70 g) underwent chromatographic fractionations using a silica gel column, elution started with the solvent system dichloromethane (DCM)-EtOAc-toluene (7.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.51:1) followed by the DCM-EtOAc (8:2), and MeOH to produce 56 fractions. These fractions were collected and combined based on their TLC profile into six main fractions (A1–A6). Fraction A3 was further chromatographed over a silica gel column eluted with dioxane-hexane (4.5:5.5) to afford five main subfractions after TLC comparison and combinations (A3.1–A3.5). Subfraction A3.1 underwent spontaneous crystallization to afford the pure form of compound 2 (26 mg). Subfraction A3.3 was subjected to chromatographic purification over Sephadex LH-20 column eluted with MeOH to produce the purified compounds 4 (31 mg) and 5 (19 mg). The fraction A4 revealed an interesting TLC profile and was subjected to fractionation using a silica gel column using chloroform–MeOH solvent (90:10) of increasing polarity. A total of six main subfractions were obtained after TLC investigation and combination (A4.1–A4.6). Subfractions A4.2 and A4.3 were purified over the Sephadex LH-20 column eluted with MeOH to afford compound 1 (28 mg) and compound 3 (32 mg), respectively. The purity of the isolated compounds was confirmed by spectroscopic analysis.
:1.51:1) followed by the DCM-EtOAc (8:2), and MeOH to produce 56 fractions. These fractions were collected and combined based on their TLC profile into six main fractions (A1–A6). Fraction A3 was further chromatographed over a silica gel column eluted with dioxane-hexane (4.5:5.5) to afford five main subfractions after TLC comparison and combinations (A3.1–A3.5). Subfraction A3.1 underwent spontaneous crystallization to afford the pure form of compound 2 (26 mg). Subfraction A3.3 was subjected to chromatographic purification over Sephadex LH-20 column eluted with MeOH to produce the purified compounds 4 (31 mg) and 5 (19 mg). The fraction A4 revealed an interesting TLC profile and was subjected to fractionation using a silica gel column using chloroform–MeOH solvent (90:10) of increasing polarity. A total of six main subfractions were obtained after TLC investigation and combination (A4.1–A4.6). Subfractions A4.2 and A4.3 were purified over the Sephadex LH-20 column eluted with MeOH to afford compound 1 (28 mg) and compound 3 (32 mg), respectively. The purity of the isolated compounds was confirmed by spectroscopic analysis.
2.2. In vitro CA IX inhibition assay
The effect of coumarins on CA IX activity conducted as previously described with minor modification,15,16 using 4-nitrophenyl acetate (4-NPA) as a substrate. A buffer solution containing HEPES and Tris–HCl (20 mM) was used as the reaction medium. Acetazolamide (AAZ), a clinically used CA inhibitor, was selected as the standard drug for this study. Compounds 1–5 and AAZ were prepared in DMSO, diluted in the assay buffer to different concentrations, and then utilized in the experiments. The reaction mixture consisted of 150 μL of the prepared reaction medium, 20 μL of the aqueous CA IX solution (0.1 mg mL−1 in deionized water), 20 μL of 4-NPA (0.7 mM in ethanol), and 20 μL of the tested compounds. The production of the yellow-colored 4-nitrophenolate was monitored at 400 nm and the experiment was performed in triplicate. The kinetic analysis of the CA IX inhibition was performed using varying concentrations of the tested compounds and 4-NPA. Following substrate addition, the change in absorbance was monitored every minute for 30 minutes at 25 °C. To elucidate the inhibition mechanism, a Lineweaver–Burk plot was constructed by plotting the reciprocal of the substrate concentration (1/[S]) against the reciprocal of the reaction velocity (1/Vmax). The inhibition constant (Ki) was determined by creating a double reciprocal plot of the inhibitor concentration ([I]) against the slope of the Lineweaver–Burk plot.
2.3. Statistical analysis
The results are presented as the mean ± standard deviation (SD), with each data point reflecting the average of three independent experiments. Comparison of the IC50 was conducted using one-way ANOVA followed by Tukey's test on GraphPad Prism 8.0. A P value <0.05 was considered significant.
2.4. In silico investigations
2.4.1. System preparation. The 3D crystal structure of human CA IX (PDB ID: 5FL4) was obtained from the Protein Data Bank (PDB). The enzyme was checked for missing residues using the Swiss-PdbViewer software.17 UCSF Chimera 1.17.1 was utilized for molecular visualization, removal of nonstandard residues and water molecules, and stripping out the native ligand.18 The geometrical structures of investigated coumarins were fully optimized using the DFT approach at the B3LYP level using the 6-311G (d, p) basis set.19–21 Frequency calculations were also implemented to confirm the absence of imaginary frequencies for the obtained energy minima states. The Gaussian 16 suite was employed for executing all DFT calculations in this investigation.22
2.4.2. Molecular docking analysis. The 3D structure of CA XI was prepared for docking using Autodock Tools (ADT) v1.5.6 software.23 This preparation involves the addition of polar hydrogens and adjusting the grid box size and dimension to involve the native ligand active site amino acid residues.24 The open-source AutoDock Vina v1.5.6 package was utilized to perform the molecular docking calculations of coumarins–CA IX complexes.23
2.4.3. MD simulations. By employing molecular docking calculations, we selected the C. officinalis coumarins complexes with CA IX (PDB ID: 5FL4) that demonstrated the highest binding affinities. These complexes were then subjected to further analysis using 100 ns MD simulations conducted with GROMACS 2022.4,25,26 utilizing GPU acceleration to enhance performance and efficiency. Specifically, the simulations were performed on a high-performance computing cluster equipped with Intel Xeon E5-2680 v3 CPUs running at 2.5 GHz, along with NVIDIA Tesla K80 GPUs (2 GPUs per node) to speed up the calculations and running on the CentOS Linux 7 operating system. The process was initiated by extracting various coumarin molecules from their complex structures with CA IX. These structures were prepared for subsequent simulations using the pdb2gmx tool. For these simulations, all-atom CHARMM36m force field and the CHARMM-modified TIP3P water model were selected.27,28 Geometric parameters and topologies for the various coumarins were generated using the CGenFF online server (https://cgenff.com/). These parameters were then integrated into the full topology of the processed CA IX. Following the formation of the complexes, both the unbound CA IX and the inhibitor–CA IX complexes were solvated by placing them in a dodecahedron-shaped box with a final volume of 289.24 nm3. To ensure electrical neutrality, seven sodium counter ions were added.28 An energy minimization step using the steepest descent method was performed for 10 ps to eliminate any unfavorable interactions.29 The system was equilibrated in two phases using both NVT (constant volume, constant temperature) and NPT (constant pressure, constant temperature) ensembles for 100 ps at 300 K.30 Finally, the production phase of the MD simulation was conducted for 100 ns at a pressure of 1 bar and a temperature of 300 K.
2.4.4. MM/PBSA analysis. The Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) approach was employed to gain comprehensive insight into ligand–receptor interactions. This method integrates molecular mechanics (MM), Poisson–Boltzmann (PB) electrostatics, and Solvent Accessibility (SA) models to estimate binding energies. Renowned for its precision and effectiveness, MM/PBSA is frequently utilized to calculate the binding free energies of noncovalently bound complexes, as corroborated by numerous studies.31,32 In our research, we utilized the gmx_MPBSA tool to calculate the binding free energies derived from the outputs of MD simulations.33 This tool enabled us to assess the energetic contributions of different interactions within the enzyme–inhibitor complexes, providing a detailed understanding of the binding mechanisms involved.
3. Results and discussion
3.1. Phytochemical study
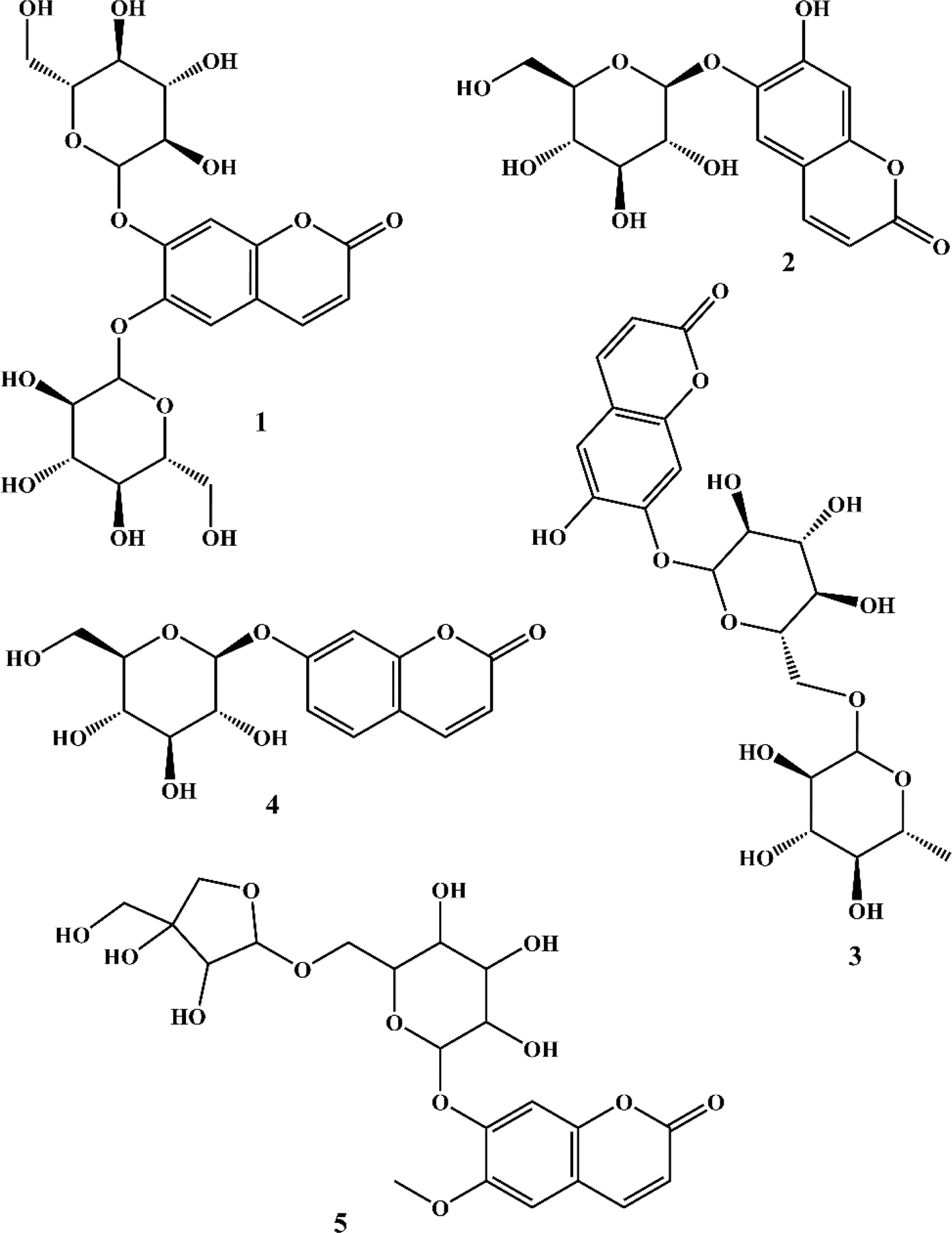
The phytochemical investigation of the aerial parts of C. officinalis led to the isolation and identification of five coumarins. The chemical structures of these isolated compounds (Fig. 1) were elucidated through spectroscopic analysis, comparison of TLC profiles with known standards, and referencing existing scientific literature. Using these methods, the coumarins were identified as 6,7-di-O-glucopyranosyl esculetin (1),34 aesculin (2),35 isobaisseoside (3),36 skimmin (4),37 and xeroboside (5).38
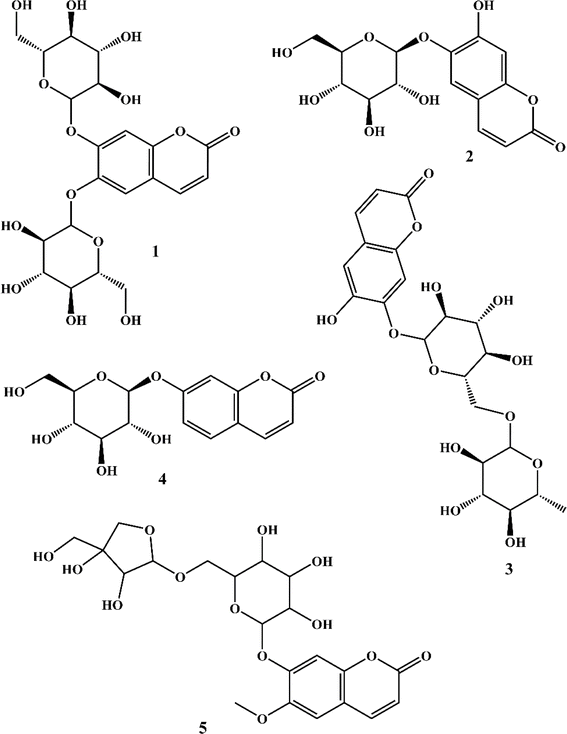 |
| | Fig. 1 Structures of isolated coumarins. | |
3.2. Inhibitory activity of C. officinalis-derived coumarins on CA IX
Given the involvement of CA IX in different physiological and pathological processes, including its overexpression in hypoxic tumor environments and its contribution to tumor growth and survival,1,2 there is a pressing need to identify effective inhibitors to modulate its activity. The dose–response curves and IC50 values for the isolated coumarins and AAZ on CA IX activity are presented in Fig. 2. As shown in Fig. 2A, the compounds and AAZ showed a concentration-dependent inhibition of CA IX activity. The IC50 values (Fig. 2B), indicating the concentration at which 50% inhibition of CA IX activity is achieved, varied among the compounds. Xeroboside demonstrated the most potent inhibition with an IC50 value of 85.34 ± 3.01 nM, following AAZ (IC50 = 42.87 ± 1.39 nM). Isobaisseoside also showed potent inhibition with an IC50 of 247.61 ± 7.27 nM, and aesculin and skimmin displayed moderate inhibition with IC50 values of 327.05 ± 11.46 nM and 547.93 ± 35.66 nM, respectively. The least potent inhibitor was 6,7-di-O-glucopyranosyl esculetin, with an IC50 value of 1.01 ± 0.12 μM. Statistical analysis revealed significant differences between the IC50 value of compounds 1–4 when compared with either AAZ or compound 5. Also, a non-significant difference was observed between IC50 value AAZ and compound 5 (Fig. 2B).
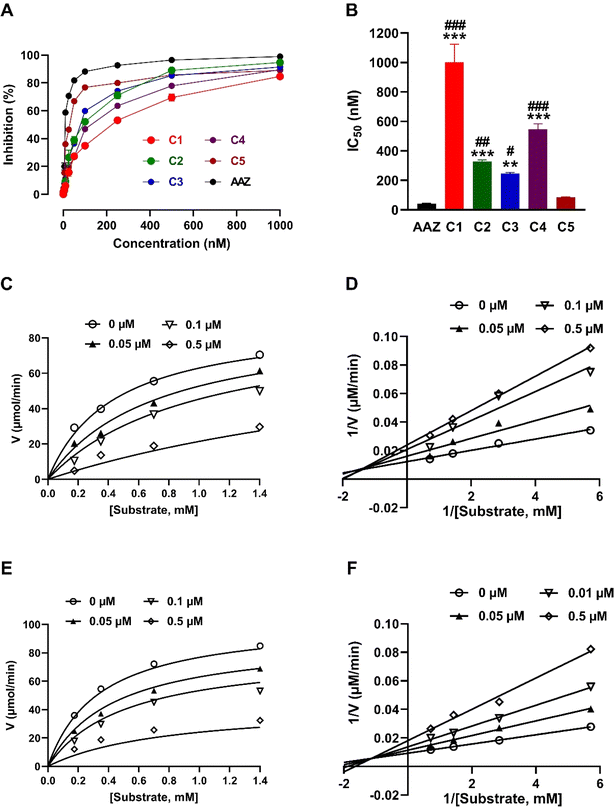 |
| | Fig. 2 CA IX inhibitory activity of C. officinalis-derived coumarins. (A) Dose–response curve of compounds 1–5 and AAZ, and (B) IC50 values of the inhibitory activity of the tested compounds. Data are mean ± SD, N = 3. **P < 0.01 and ***P < 0.001 vs. AAZ. #P < 0.5, ##P < 0.01, and ###P < 0.001 vs. C5 (xeroboside). (C–F) Michaelis–Menten (C and E) and Lineweaver–Burk (D and F) plots of CA IX inhibitory activity of compounds 5 and 3, respectively. | |
To further characterize the inhibitory effects of isobaisseoside and xeroboside on CA IX, kinetic analyses were performed, and the Michaelis–Menten (Fig. 2C and E) and Lineweaver–Burk (Fig. 2D and F) plots were constructed. The Michaelis–Menten plot for isobaisseoside (Fig. 2E) showed a decrease in the maximum reaction velocity (Vmax) with increasing concentration (Ki = 126 nM), suggesting a mixed-type inhibition. The Lineweaver–Burk plot (Fig. 2F) corroborated this, as the lines intersect to the left of the y-axis, indicating changes in both Vmax and Km. Similar to isobaisseoside, xeroboside exhibited a mixed-type inhibition pattern (Fig. 2C and D). The decrease in Vmax with increasing concentration was evident from the Michaelis–Menten plot (Ki = 77.2 nM). The Lineweaver–Burk plot showed intersecting lines, confirming the mixed inhibition. The reference inhibitor AAZ demonstrated non-competitive inhibition.
The efficacy of the tested coumarins against CA IX demonstrates the potential of these natural products as therapeutic agents. Xeroboside and isobaisseoside exhibited the highest inhibition activity among the tested coumarins and the superior activity of xeroboside can be attributed to its lower IC50 and higher inhibition rate compared to other coumarins. The structural features of xeroboside, including the presence of specific functional groups, may contribute to its enhanced binding affinity and inhibitory potency. The kinetic analysis revealed that isobaisseoside and xeroboside exhibited mixed-type inhibition, which suggests that they can bind to both the enzyme active site and an allosteric site, altering both Vmax and Km. This mode of inhibition may provide a therapeutic advantage by offering multiple binding interactions that stabilize the inhibitor–enzyme complex. However, it is noteworthy to highlight that the two-site model of mixed inhibition might not be mechanistically relevant as reported by Pesaresi.39 Using a statistical approach and by combining statistical analysis of enzyme inhibition cases with a theoretical investigation of inhibition models, Pesaresi concluded that mixed inhibitors bind exclusively to the active site, ruling out allosteric involvement.39 Consistently, we propose that our inhibitors interact primarily with the active site, causing a conformational change that affects substrate binding and enzyme activity. This would explain the mixed inhibition observed in our kinetic data without necessarily invoking an allosteric site. While the inhibitors do not completely block substrate binding as in pure competitive inhibition, they still influence the enzyme activity, likely through modulation of the active site conformation. The outcomes of the computational studies conducted in this work support the idea that the inhibitors bind within or near the catalytic region of CA IX, potentially altering the active site structure and leading to mixed inhibition behavior. AAZ, a well-known CA inhibitor, displayed non-competitive inhibition, consistent with its mechanism of action that involves binding to an allosteric site, leading to a decrease in Vmax without affecting Km. The comparison with AAZ highlights the efficacy of the natural coumarins, particularly xeroboside and isobaisseoside, as potent CA IX inhibitors.
3.3. Molecular docking analysis
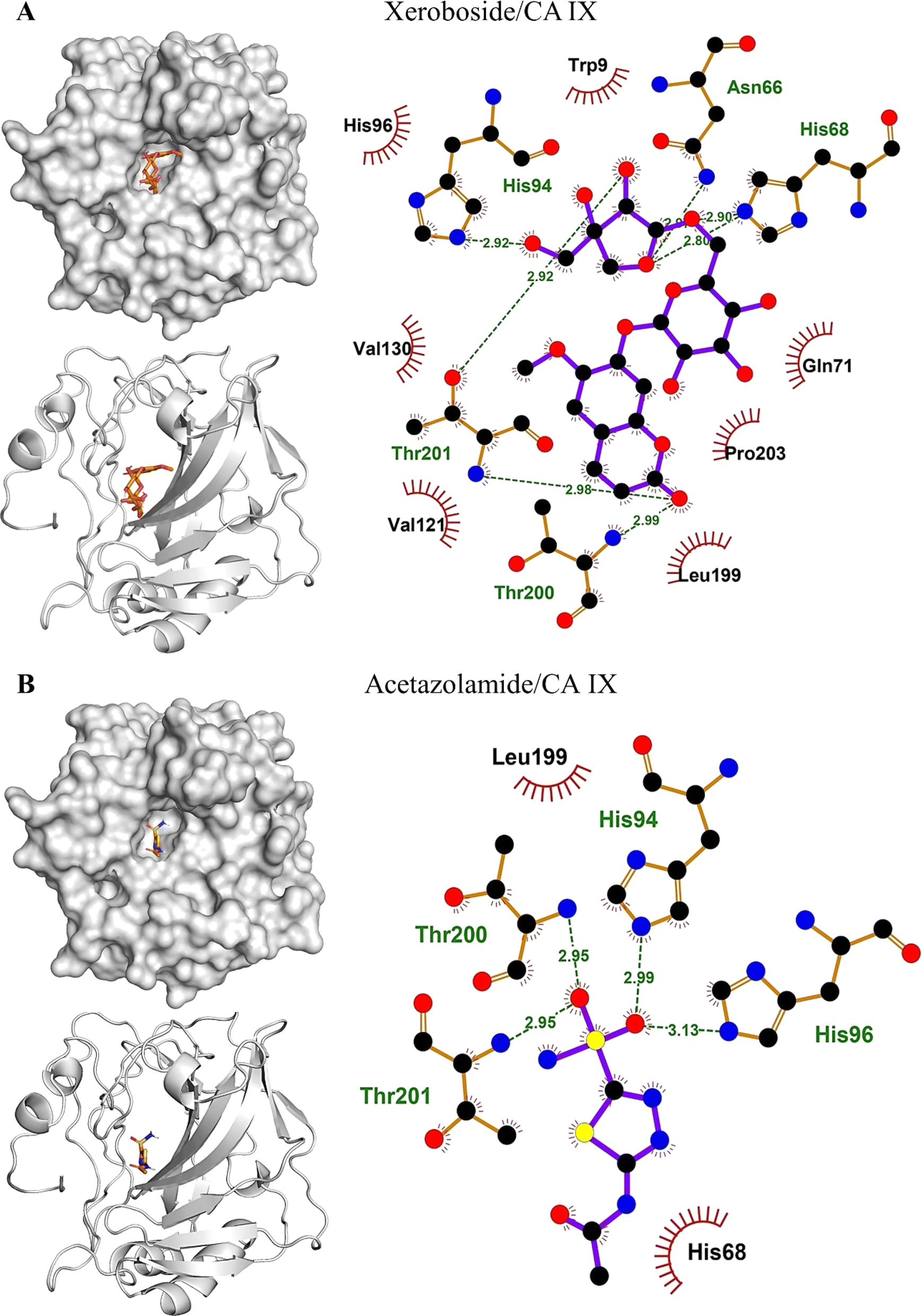
The binding affinities and interacting residues of the isolated coumarins with CA IX were evaluated using molecular docking. Fig. 3, 4, and 5 represent the results of docking simulations, depicting the positioning of different ligands in the binding site of the target enzyme and showcasing residues involved in polar and hydrophobic interactions. Among tested coumarins, 6,7-di-O-glucopyranosyl esculetin and xeroboside demonstrated the lowest binding energies (−8.1 and −8.3 kcal mol−1), respectively (Table 1). However, the remaining coumarins displayed comparable binding energies ranging from −7.3 to −7.7 kcal mol−1, and AAZ had a binding energy of −6.5 kcal mol−1 (Table 1). These findings highlight the potential of these coumarins as effective CA IX inhibitors. The binding site of CA IX displayed a dense network of hydrophobic interactions for all the tested coumarins. These hydrophobic interactions were crucial in ligand–enzyme complexes stability, thereby enhancing the binding affinity and inhibitory potential of the tested compounds. The poses revealed docking of all compounds into the binding site of CA IX, which is also occupied by the reference drug AAZ. This consistent docking pattern suggested that the coumarins and the reference drug share a common binding site, highlighting the potential of these coumarins as effective CA IX inhibitors. In addition, isolated compounds showed a high extent of polar interactions, significantly contributing to the stability and efficacy of these inhibitors.
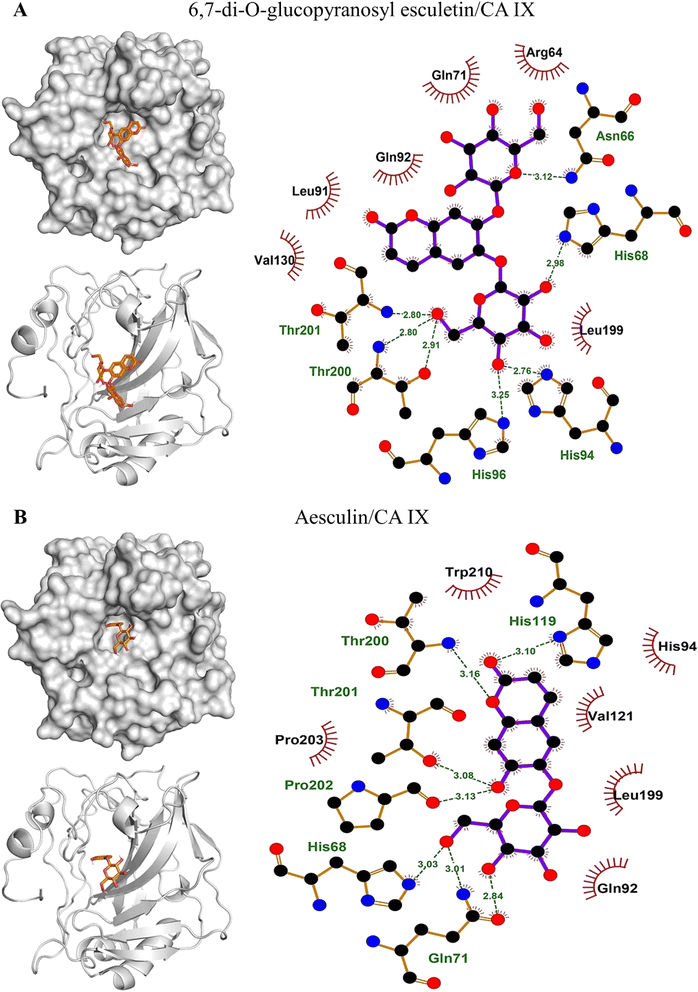 |
| | Fig. 3 Binding site interactions of compounds 1 (A) and 2 (B) with CA IX. | |
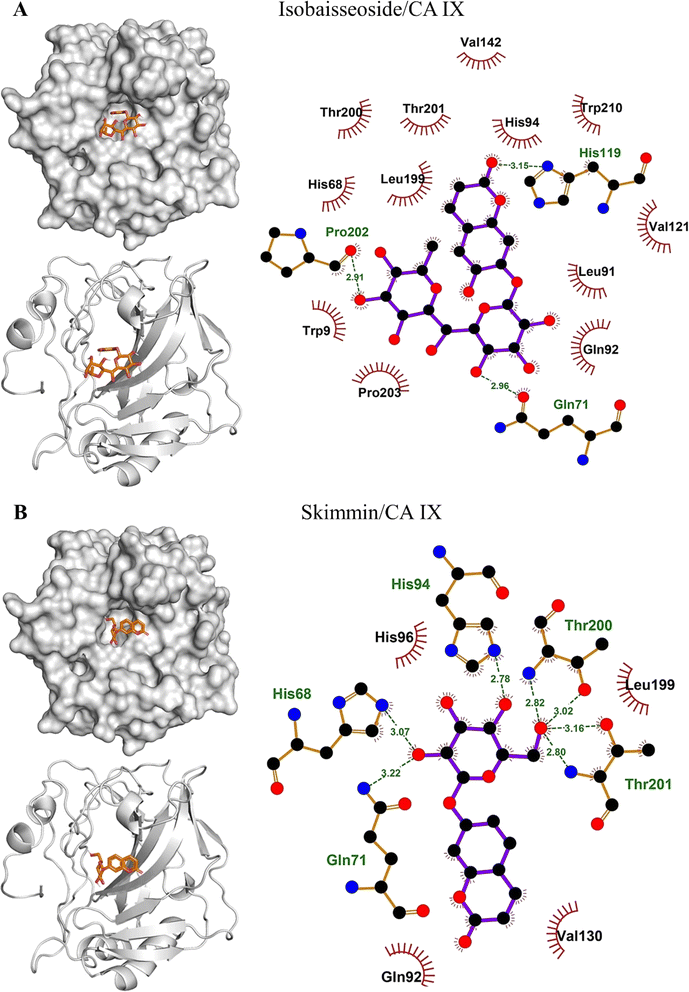 |
| | Fig. 4 Binding site interactions of compounds 3 (A) and 4 (B) with CA IX. | |
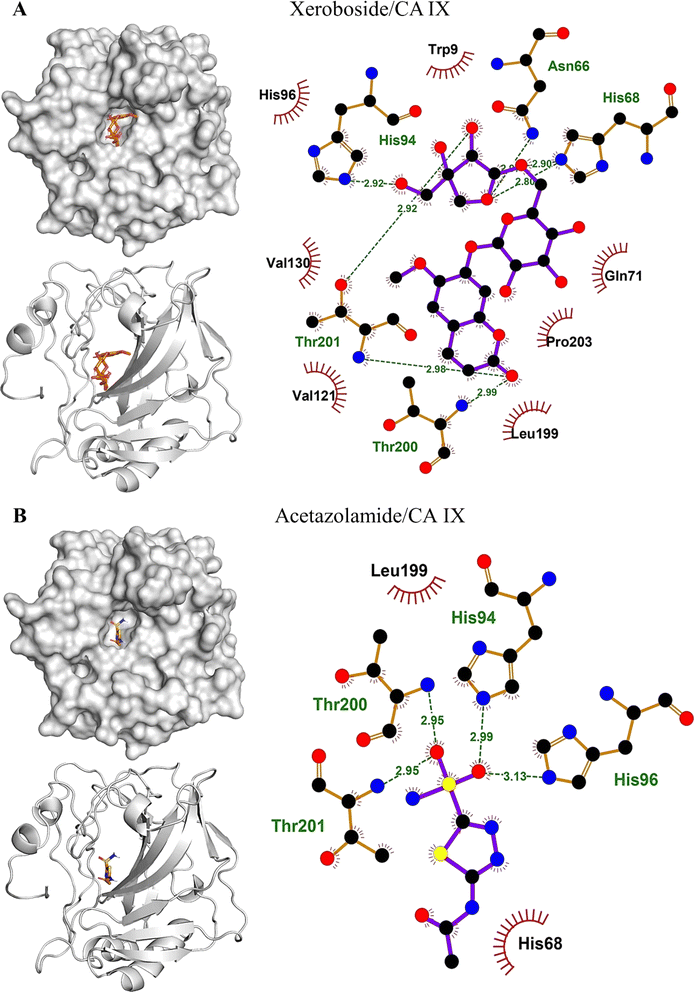 |
| | Fig. 5 Binding site interactions of compound 5 (A) and acetozolamide (B) with CA IX. | |
Table 1 Molecular docking analysis of the binding affinity of C. officinalis-derived coumarins towards CA IX
| |
Lowest binding energy (kcal mol−1) |
Polar interacting residues |
Hydrophobic interacting residues |
| 6,7-di-O-glucopyranosyl esculetin |
−8.1 |
Thr201, Thr200, His96, His94, His68, and Asn66 |
Val130, Leu91, Gln92, Gln71, Arg64, and Leu199 |
| Aesculin |
−7.7 |
Thr200, Thr201, Pro202, His86, Gln71, and His119 |
Trp210, His94, Val121, Leu199, Gln92, and Pro203 |
| Isobaisseoside |
−7.3 |
Pro202, His119, and Gln71 |
Thr200, His68, Thr201, Leu199, His94, Val142, Trp210, Val121, Leu91, Gln91, Trp9, and Pro203 |
| Skimmin |
−7.4 |
His68, Gln71, Thr201, His94, and Thr200 |
His96, Leu199, Val130, and Gln92 |
| Xeroboside |
−8.3 |
His94, Asn66, Hi68, Thr201, and Thr200 |
His96, Trp9, Gln71, Pro203, Leu199, Val130, and Val121 |
| AAZ |
−6.5 |
His94, His96, Thr200, and Thr201 |
His68 and Leu199 |
3.4. Molecular dynamics (MD) simulations
The objective of MD simulations was to elucidate the long-term stability and interactions within the coumarins–CA IX complexes, thereby illuminating the inhibition mechanisms and various binding modes. By carefully examining the simulation trajectories, we aimed to gain comprehensive insights into how the structural dynamics of the compounds influence their CA IX inhibitory potential. Understanding these molecular mechanisms is critical for the strategic design of novel CA IX-targeting therapeutics. We focused on complexes identified through initial docking analyses that exhibited the most favorable binding affinities and conducted an in-depth evaluation using 100 ns MD simulations. This rigorous analysis included key parameters such as interaction energies, MM/PBSA binding free energies, root mean square deviations (RMSD), radius of gyration (Rg), potential energy landscape (PEL), root mean square fluctuations (RMSF), solvent accessible surface area (SASA), and hydrogen bonding profile. Our examination covered both CA IX and its complexes with coumarins, aiming at gaining understanding of their interaction characteristics and dynamic behavior. These insights are essential for elucidating the inhibitory effects of the compounds and guiding the development of effective CA IX inhibitors.
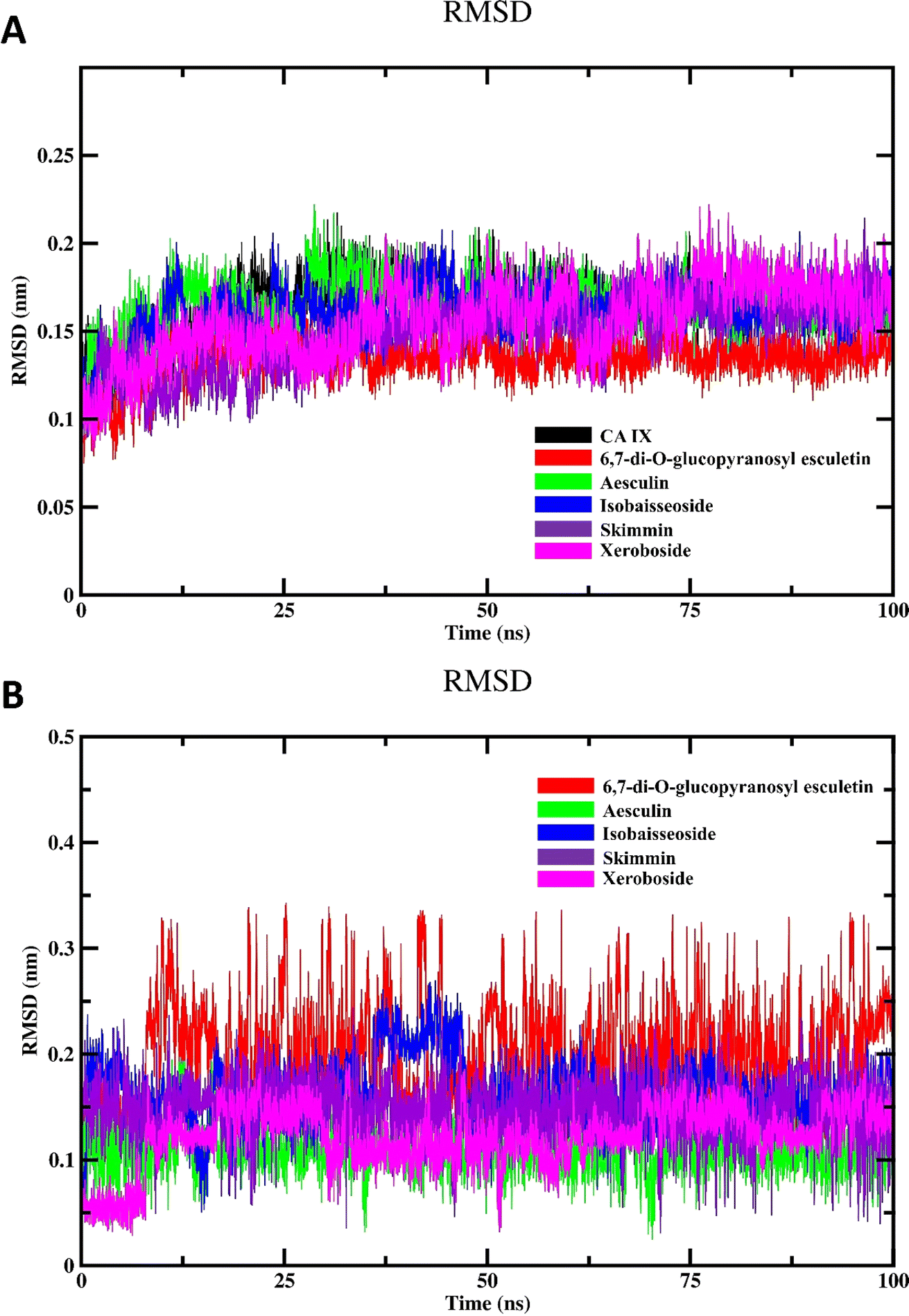
3.4.1. Structural stability and dynamics. The results of the 100 ns RMSD calculation of the CA IX backbone are depicted in Fig. 6A. The RMSD profile of investigated systems initially displayed a rising trend indicative of the system adjusting and transitioning from its initial conformation. This phase of adaptation is common as the system moves towards a more energetically favorable state. Following this initial adjustment, the RMSD profile exhibited a highly fluctuating trend, reflecting the dynamic nature of the enzyme and its interactions with the bound inhibitors. Throughout most of the simulation, these fluctuations persisted, suggesting ongoing conformational changes and adjustments within the enzyme–inhibitor complexes. It wasn't until the last quarter of the simulation that the RMSD values began to stabilize, indicating that the system had reached a state of equilibrium. This period of equilibrium is crucial as it signifies that the conformational changes have settled, and the system is now in a stable state suitable for detailed analysis.
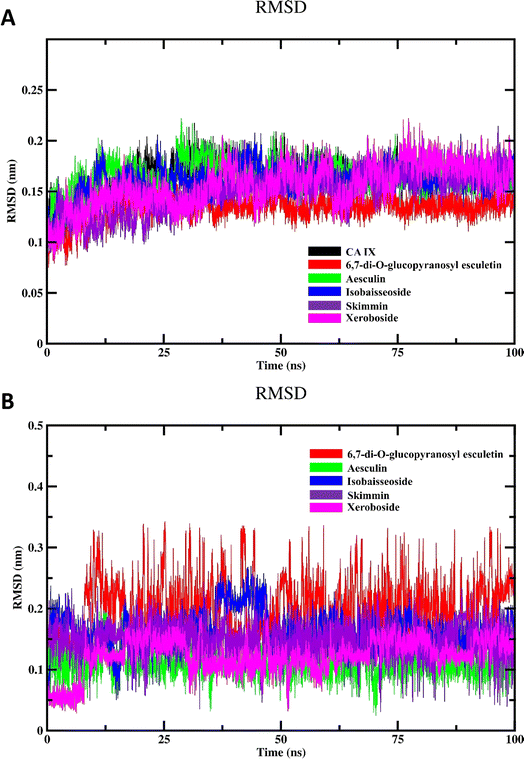 |
| | Fig. 6 MD simulation of CA IX and its complexes with C. officinalis-derived coumarins; (A) backbone RMSD of the unbound CA IX and CA IX–coumarin complexes and (B) RMSD of isolated compounds. | |
To investigate the inherent movement and conformational changes of the coumarin molecules independent of their interaction with CA IX, we investigated their RMSD values (Fig. 6B). The RMSD values were obtained by aligning the trajectory frames to the initial structure of each drug and calculating the deviation of the coordinates. This method enables an assessment of how much each drug deviates from its initial conformation over the 100 ns simulation period. Such an analysis is crucial in drug design research to evaluate the stability and dynamic behavior of drug molecules. As shown in Fig. 6B, the RMSD analysis of different coumarins revealed notable variations in their RMSD profiles. The compounds exhibited fluctuation patterns in their RMSD values indicating that they maintained relatively stable conformations throughout the simulation. This stability suggests the presence of strong and consistent interactions between isolated coumarins and CA IX.
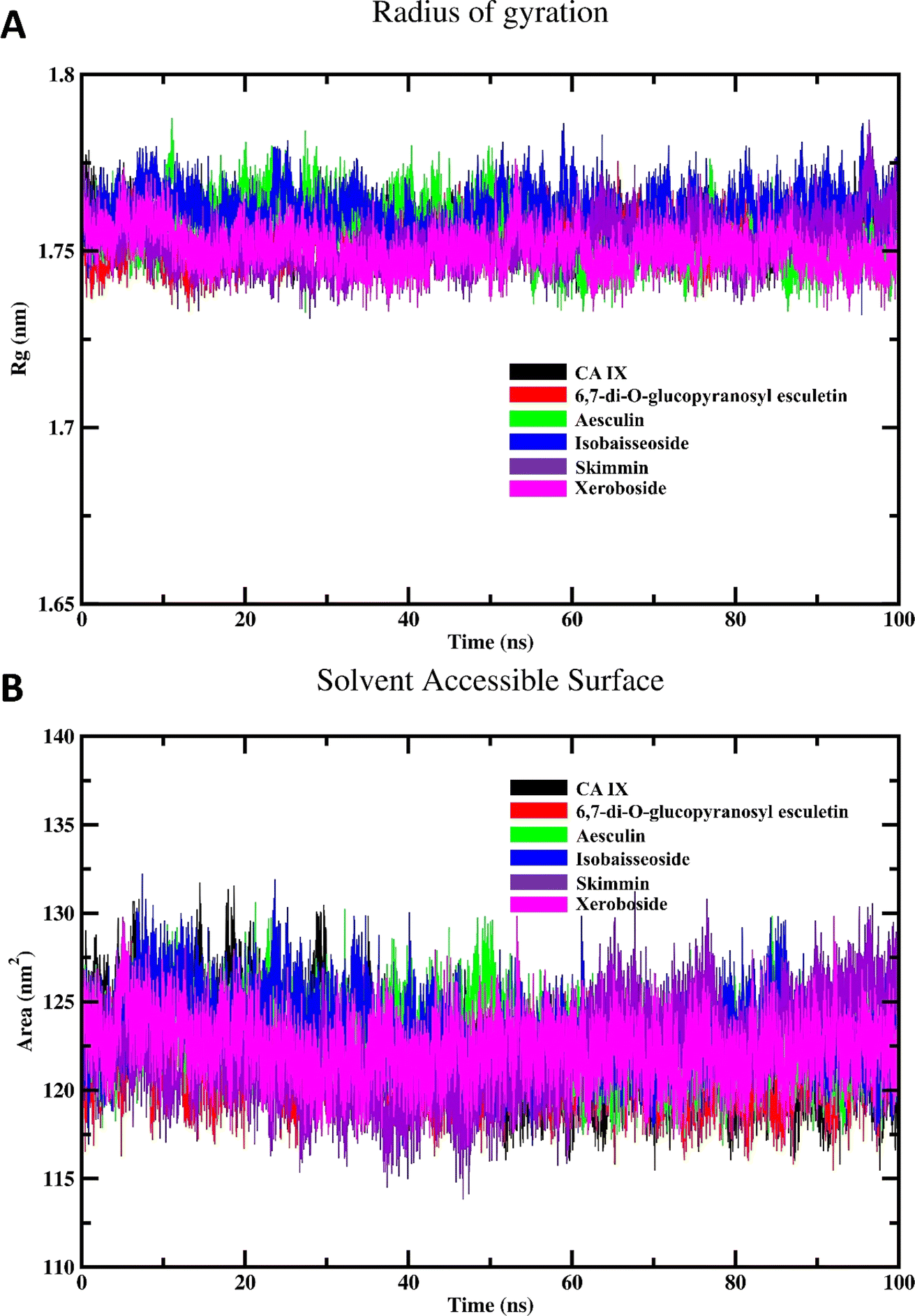
The results of the Rg calculations for the unbound CA IX and its complexes with various coumarins are shown in Fig. 7A. The Rg profile provides insights into structural stability and compactness of the enzyme and/or its complexes over the course of the simulation.40 Throughout the majority of the simulation time, the Rg profiles of the different coumarin–CA IX complexes demonstrated a consistent and stabilized behavior, comparable to that observed for the unbound enzyme. This indicates that the binding of coumarins does not disrupt the overall compactness of the enzyme structure. Interestingly, among all the complexes studied, xeroboside (the compound with the lowest IC50 value) exhibited the lowest average Rg value. This suggested that the xeroboside–CA IX complex is more compact compared to the other coumarin–CA IX complexes. A lower Rg value often correlates with a more tightly bound and stable complex, which could contribute to the enhanced inhibitory potency of xeroboside. The compact nature of the xeroboside–CA IX complex might facilitate stronger interaction with the active site, thus explaining its superior inhibitory activity.
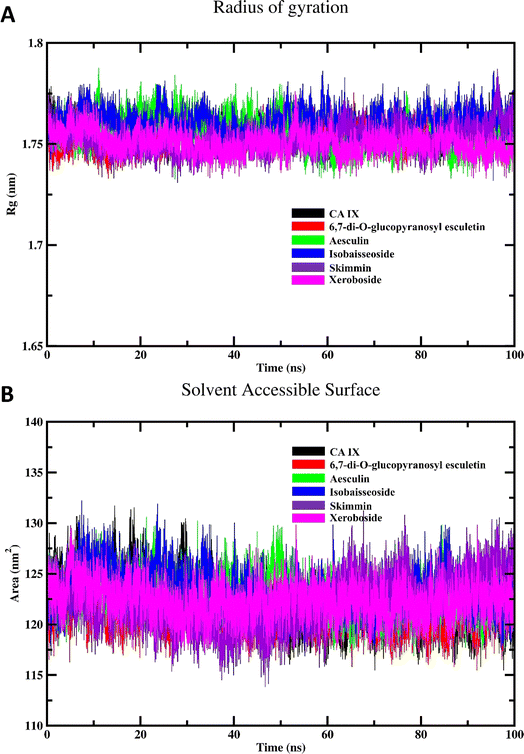 |
| | Fig. 7 MD simulation of CA IX and its complexes with C. officinalis-derived coumarins; (A) radius of gyration of CA IX and CA IX–coumarin complexes and (B) SASA of CA IX and various complexes. | |
The results of the SASA calculations for the free CA IX enzyme and its complexes with various coumarins are depicted in Fig. 7B. The SASA profile provides insights into the surface exposure of the enzyme and its complexes to the solvent, which is a critical factor in understanding the interaction and stability of the complexes.14 Throughout the majority of the simulation span, the SASA profiles of different coumarin–CA IX complexes demonstrated an equilibrated behavior similar to that observed for the unbound enzyme. This indicates that the binding of coumarins does not significantly alter the overall solvent exposure of the enzyme structure. During the simulation, a noticeable decrease in SASA values for all systems was observed. This reduction in SASA values suggested that the enzyme and its complexes become more compact over time, with less surface area exposed to the solvent. Such a decrease in SASA is indicative of a stable binding interaction where the enzyme–inhibitor complex achieves a more energetically favorable conformation by minimizing its solvent-exposed surface area. The equilibrated and reduced SASA profiles across the different complexes suggested that the coumarins contribute to the stabilization of the enzyme by promoting a more compact structure. This stabilization is crucial for the inhibitory activity, as it indicates that the enzyme–inhibitor complex achieves a conformation that is less prone to destabilizing interactions with the solvent.
The SASA findings are consistent with the Rg results, reinforcing the notion that the binding of coumarins leads to a stabilized and compact enzyme–inhibitor complex. The observed decrease in SASA values further supports the idea that these complexes are energetically favorable and structurally stable. This stability is essential for the effective inhibition of CA IX by the coumarins, as a stable complex is less likely to dissociate and more likely to maintain its inhibitory function.
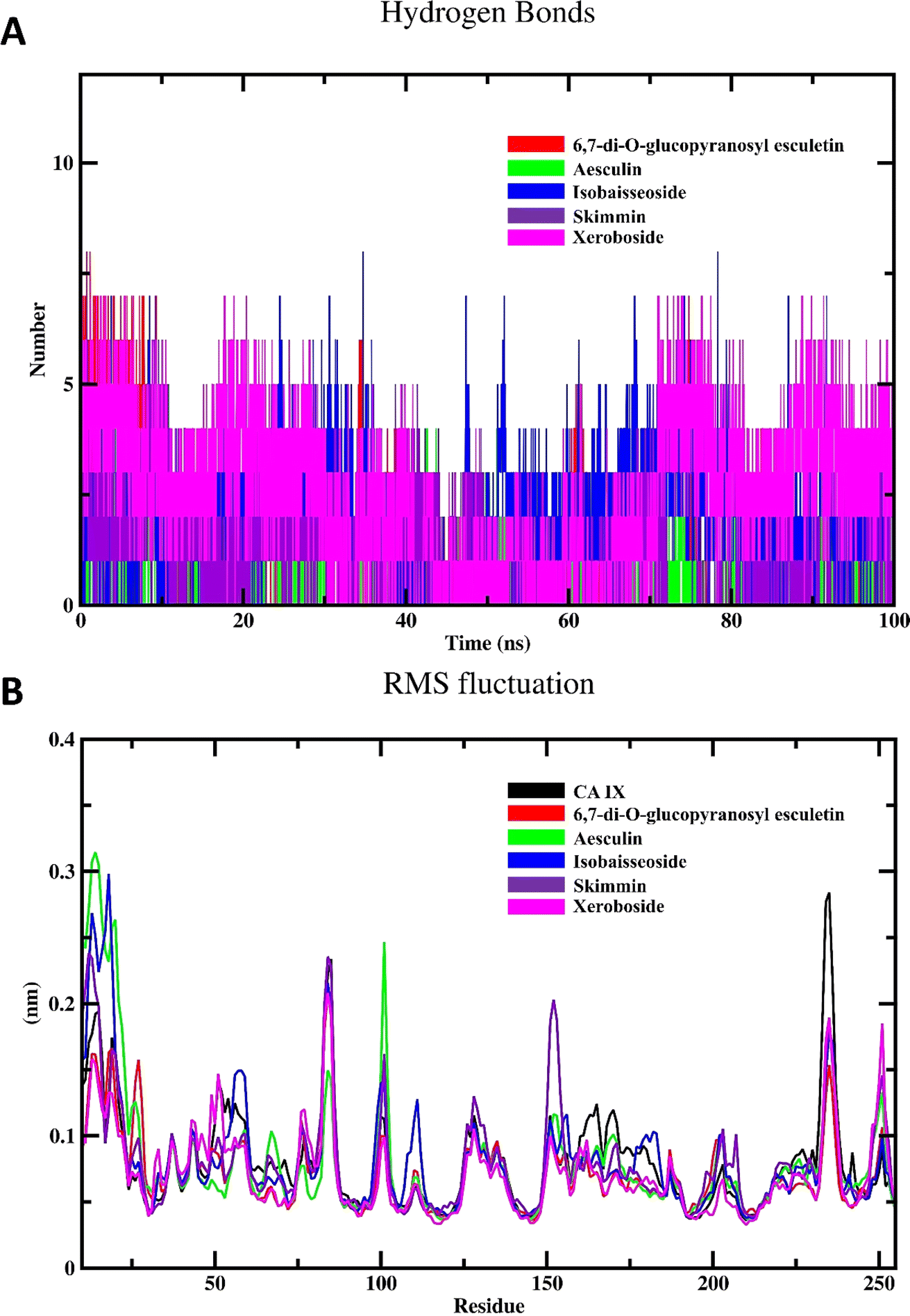
3.4.2. Hydrogen bonding dynamics and flexibility. The hydrogen bonding profile of coumarin–CA IX complexes is represented in Fig. 8A. Hydrogen bonds are critical interactions that contribute significantly to the stability and specificity of enzyme–inhibitor complexes. They play a crucial role in maintaining the structural integrity of the enzyme–ligand complex and in facilitating the correct positioning of the inhibitor within the enzyme active site.41 Our analysis revealed that the coumarin–CA IX complexes exhibited varying extents of hydrogen bonding interactions throughout the simulation period. Notably, xeroboside showed the highest number of hydrogen bonds among the tested coumarins. This extensive hydrogen bonding network suggests a strong and stable interaction between xeroboside and CA IX, which likely contributes to its superior inhibitory activity. The formation of a high number of hydrogen bonds indicates that xeroboside is well accommodated within the enzyme active site, forming stable interactions that enhance its binding affinity. Following xeroboside, isobaisseoside also displayed a significant number of hydrogen bonds, correlating with its relatively low IC50 value. The robust hydrogen bonding interactions observed for isobaisseoside further underline its effective binding and stabilization within the CA IX active site.
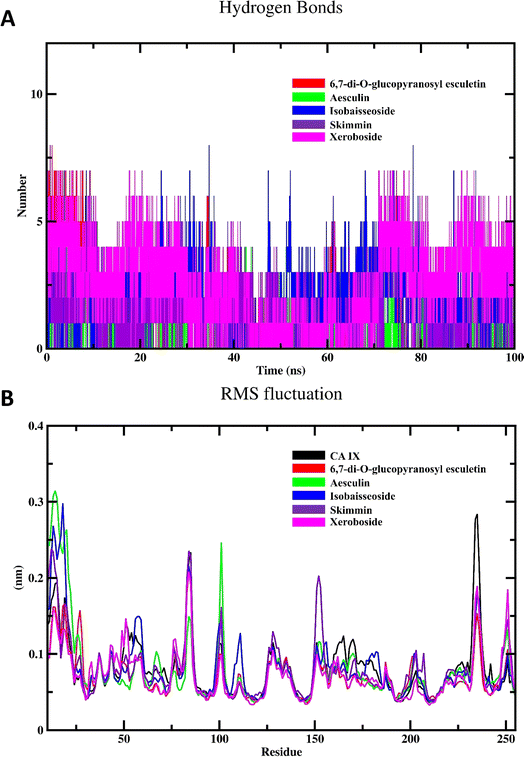 |
| | Fig. 8 MD simulation of CA IX and its complexes with C. officinalis-derived coumarins; (A) hydrogen bonding profile of CA IX–coumarin complexes, and (B) backbone RMSF per residue number for CA IX and CA IX–coumarin complexes. | |
The RMSF profile of the free CA IX and its complexes with different coumarins is depicted in Fig. 8B. RMSF analysis provides insights into the flexibility of individual residues within the protein structure over the course of the MD simulation. High RMSF values indicate regions of the protein that exhibit significant flexibility, while low RMSF values denote more rigid regions. The RMSF profiles of the CA IX–coumarin complexes were found to be similar to that of the free enzyme. This observation suggested that the binding of coumarins with CA IX does not induce significant conformational changes or alter the flexibility of the enzyme residues in a substantial manner. Despite the similarity in RMSF profiles, subtle differences were observed in specific regions, which may reflect localized interactions between the enzyme and coumarins. These localized interactions can stabilize certain regions of the enzyme, potentially enhancing the inhibitory effects of the coumarins without causing widespread conformational changes.
3.4.3. Interaction energy. The analysis of interaction energies highlights the significance of both electrostatic and van der Waals forces in the binding of coumarins to CA IX. The results of the Coulomb short-range (Coul-SR) and Lennard-Jones short-range (LJ-SR) interaction energies for the CA IX–coumarin complexes are presented in Fig. 9 and summarized in Table 2. These interaction energies help evaluate the binding affinities and enzyme–inhibitor complexes stability, with lower values indicating stronger interactions and potentially higher inhibitory efficacy. Among the studied coumarins, xeroboside exhibited the lowest average Coul-SR and LJ-SR interaction energies. This finding correlates with its highest inhibitory activity, as indicated by its low IC50 value. The strong electrostatic (Coul-SR) and van der Waals (LJ-SR) interactions observed for xeroboside suggested a robust binding affinity to the active site of CA IX, contributing to its effective inhibition. Isobaisseoside, the coumarin with the second lowest IC50 value, also demonstrated favorable interaction energies, with average Coul-SR and LJ-SR values slightly higher than those of xeroboside. This trend further supported the correlation between low interaction energies and high inhibitory activity. Aesculin, although not as potent as xeroboside or isobaisseoside, showed LJ-SR interaction energies comparable to those of isobaisseoside, indicating a similar degree of van der Waals interactions.
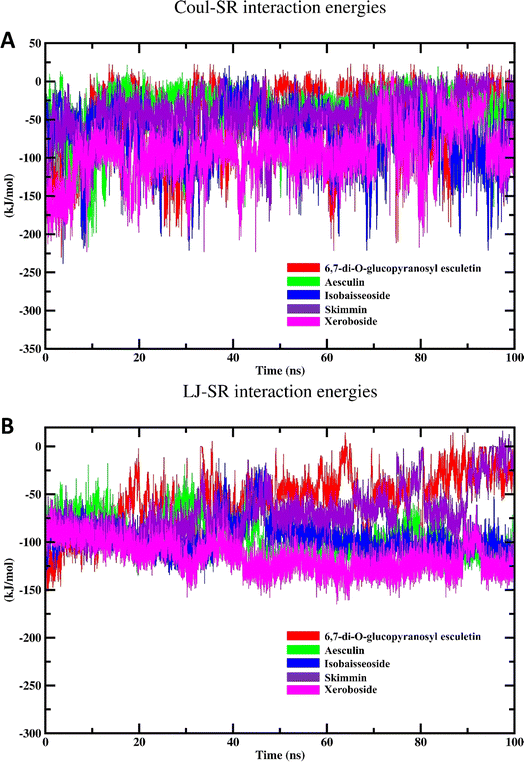 |
| | Fig. 9 MD simulation of CA IX and its complexes with C. officinalis-derived coumarins; (A) Coulomb-SR interactions energies and (B) Lennard-Jones-SR interactions energies of the enzyme amino acid residues with isolated compounds. | |
Table 2 Average Coul-SR and LJ-SR interaction energies for the complexes formed between C. officinalis-derived coumarins and CA IX
| |
Coul-SR interaction energy |
LJ-SR interaction energy |
| Average (kJ mol−1) |
RMSD (nm) |
Average (kJ mol−1) |
RMSD (nm) |
| 6,7-di-O-glucopyranosyl esculetin |
−46.27 ± 4.7 |
45.69 |
−58.32 ± 3.1 |
37.69 |
| Aesculin |
−37.90 ± 3.0 |
7.80 |
−95.40 ± 1.9 |
17.74 |
| Isobaisseoside |
−72.47 ± 3.8 |
36.79 |
−96.67 ± 1.73 |
12.66 |
| Skimmin |
−42.43 ± 3.1 |
32.14 |
−68.74 ± 2.5 |
11.46 |
| Xeroboside |
−94.09 ± 3.6 |
32.45 |
−115.83 ± 1.7 |
11.20 |
3.4.4. MM/PBSA analysis. The MM/PBSA calculations provide an in-depth quantitative analysis of the binding free energies of different coumarins to CA IX. The results summarized in Table 3 highlight the contributions of van der Waals (ΔEvdw), electrostatic (ΔEele), solvation (ΔGsolv), and gas phase (ΔGgas) interactions to the total binding free energy (ΔGtotal) for each complex. Xeroboside exhibited the lowest total binding free energy, indicating the strongest binding affinity among the studied coumarins. This strong binding is reflected in both its substantial van der Waals and electrostatic interaction energies, which are the most favorable among all the compounds. This aligns with the experimental data where xeroboside displayed the lowest IC50 value, underscoring its potent inhibitory effect. Isobaisseoside also demonstrated a strong binding affinity. Similar to xeroboside, the binding of isobaisseoside is characterized by favorable van der Waals and electrostatic interactions, though these interactions are less pronounced than those observed for xeroboside. The strong binding affinity of isobaisseoside is also corroborated by its low IC50 value, making it the second most potent inhibitor among the tested coumarins. The solvation energies (ΔGsolv) for all complexes are positive, indicating an unfavorable desolvation process upon binding. However, the strong negative contributions from van der Waals and electrostatic interactions outweigh these unfavorable solvation effects, resulting in overall negative total binding free energies. Overall, the MM/PBSA analysis underscores xeroboside superior binding affinity towards CA IX, followed by isobaisseoside and aesculin. The strong van der Waals and electrostatic interactions primarily drive the binding affinities of these coumarins. These findings align with the inhibitory activities observed in experimental assays.
Table 3 The results of MM/PBSA calculations (kJ mol−1)
| |
ΔEvdw |
ΔEele |
ΔGsolv |
ΔGgas |
ΔGtotal |
| 6,7-di-O-glucopyranosyl esculetin |
−11.71 ± 0.79 |
−13.42 ± 1.93 |
19.46 ± 1.31 |
−25.13 ± 2.11 |
−5.67 ± 2.35 |
| Aesculin |
−25.08 ± 0.64 |
−7.94 ± 1.64 |
20.86 ± 1.11 |
−33.02 ± 1.77 |
−12.16 ± 2.06 |
| Isobaisseoside |
−25.13 ± 1.76 |
−15.16 ± 1.38 |
26.30 ± 1.30 |
−40.29 ± 2.20 |
−14.00 ± 2.67 |
| Skimmin |
−17.96 ± 1.65 |
−11.66 ± 1.53 |
18.76 ± 3.19 |
−29.62 ± 2.25 |
−10.86 ± 3.72 |
| Xeroboside |
−34.62 ± 1.78 |
−31.58 ± 1.96 |
38.94 ± 0.19 |
−66.20 ± 2.63 |
−27.26 ± 2.48 |
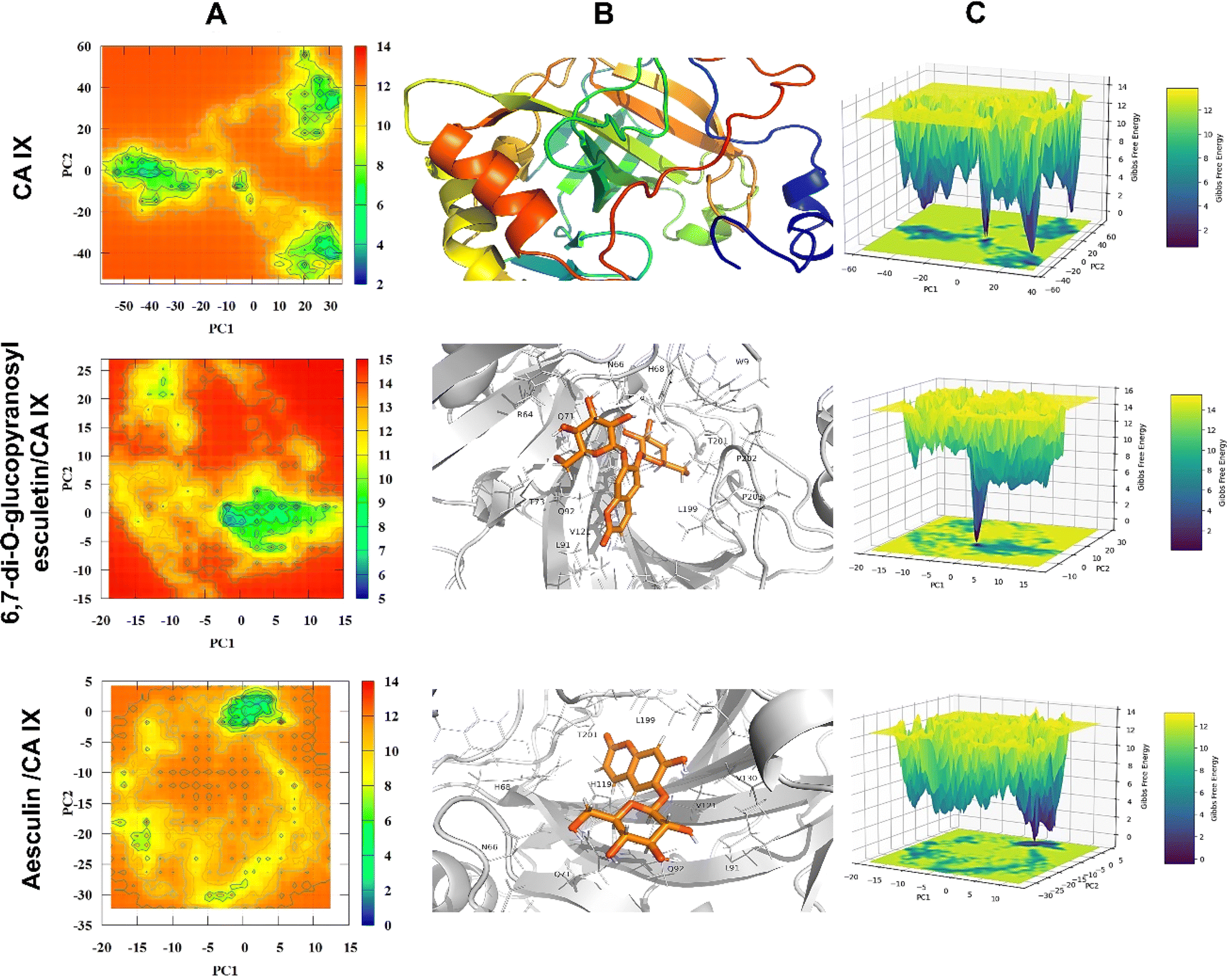
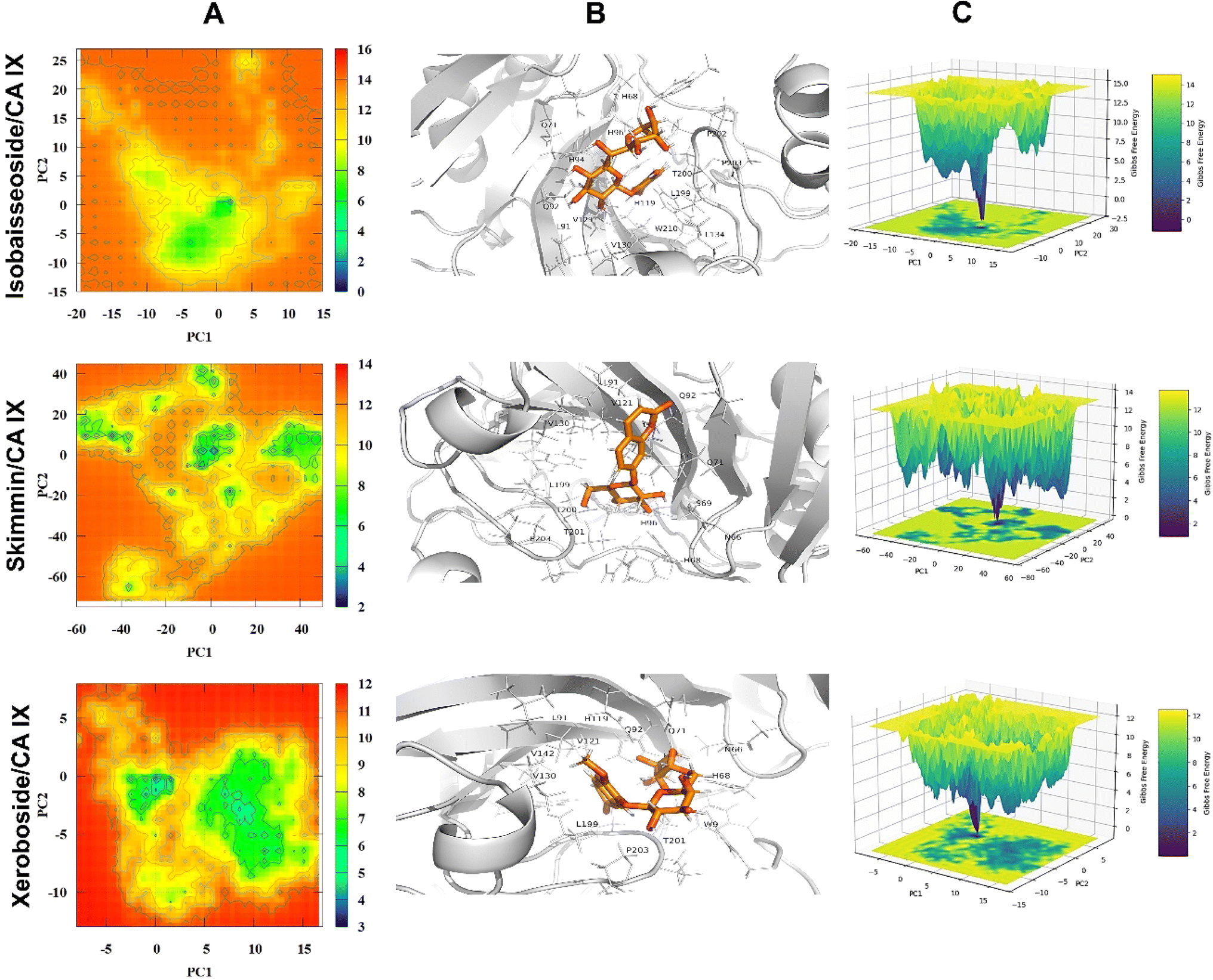
3.4.5. Potential energy landscape. Principal component analysis (PCA) was implemented to assess the conformational dynamics of the unbound CA IX and its complexes with target coumarins. The potential energy landscape (PEL) of the unbound CA IX and its complexes with various coumarins is depicted in Fig. 10 and 11. Panel A in both figures illustrates the 2D PEL of free CA IX and different complexes, showcasing distinct energy minima, which indicate stable conformational states throughout the simulation trajectory. The lowest energy basins in the PEL correspond to the most stable conformations of the enzyme, which are critical for understanding its inherent stability and functional states. Panel B highlights the minimum energy conformations of CA IX in complexes with different coumarins. The CA IX structure serves as a reference for comparing the structural adaptations upon ligand binding. For 6,7-di-O-glucopyranosyl esculetin and aesculin, the snapshots show significant interactions within the enzyme active site, including hydrogen bonding and hydrophobic interactions, which likely contribute to their inhibitory effects. The binding conformations revealed how these compounds fit into the active site, inducing minor or major conformational changes to optimize binding. Panel C presents the three-dimensional potential energy surfaces for these complexes. The stable binding interactions and minimal energy fluctuations throughout the simulations indicated that coumarin–CA IX complexes achieve stable binding conformations essential for their inhibitory function. Notably, the PELs of xeroboside displayed more pronounced energy basins, indicating highly stable and energetically favorable binding conformations. This suggested a robust interaction and a potent inhibitory effect on CA IX. Overall, the PEL analysis underscores the importance of stable and energetically favorable binding conformations in the inhibitory activity of these coumarins against CA IX. The distinct energy minima and stable binding interactions highlight their potential as effective CA IX inhibitors, paving the way for further development and optimization of these compounds for therapeutic applications.
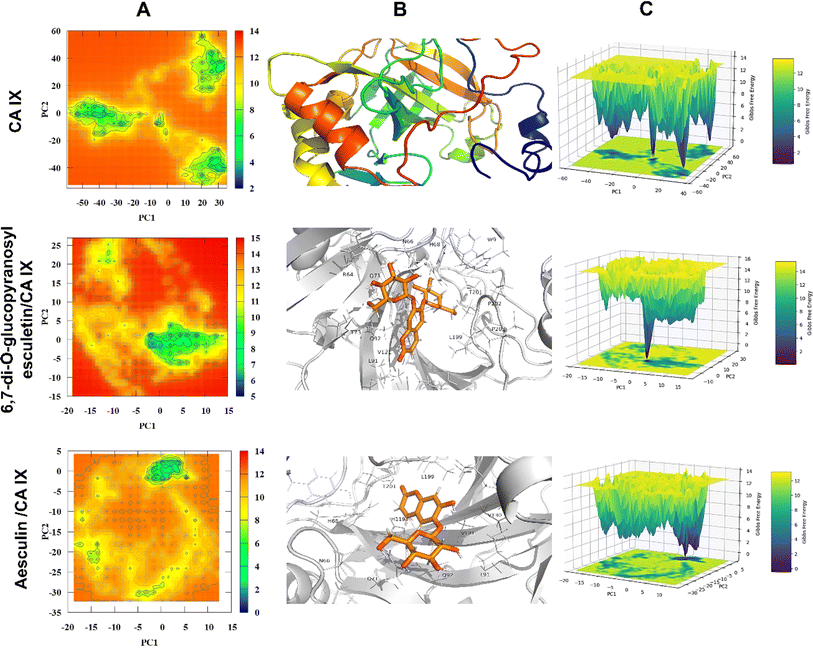 |
| | Fig. 10 The potential energy landscape (PEL) analysis of free CA IX, 6,7-di-O-glucopyranosyl esculetin, and aesculin. (A) The 2D heat map of PEL, (B) the lowest energy conformation of investigated coumarins within the binding site of CA IX, and the key residues involved in the binding mechanism. (C) A 3D representation of PEL. | |
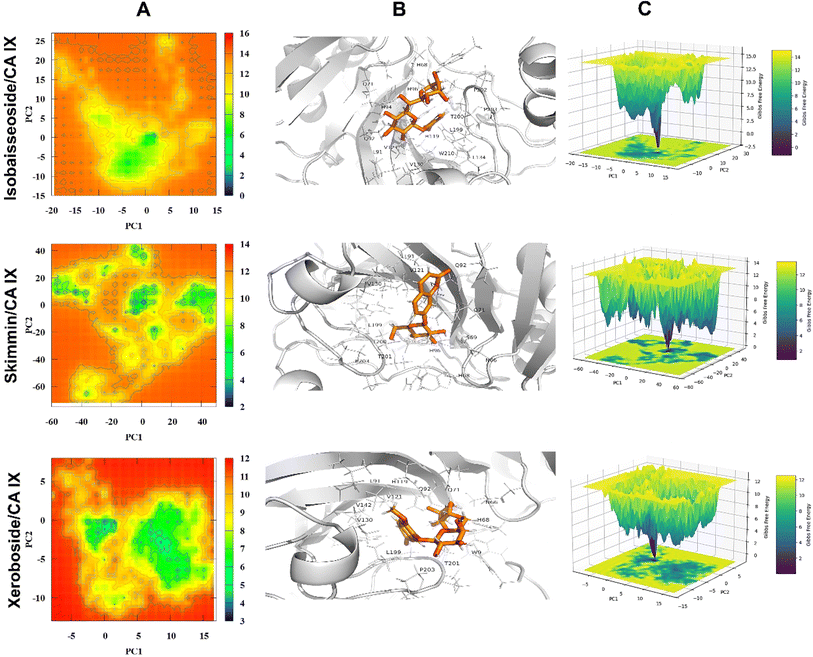 |
| | Fig. 11 The potential energy landscape (PEL) analysis of free isobaisseoside, skimmin, and xeroboside. (A) The 2D heat map of PEL, (B) the lowest energy conformation of investigated coumarins within the binding site of CA IX, and the key residues involved in the binding mechanism. (C) A 3D representation of PEL. | |
4. Conclusion
This study examined the inhibitory capabilities of C. officinalis-derived coumarins against CA IX using an integrated approach, including in vitro and in silico investigations. The results identified xeroboside as the most effective CA IX inhibitor followed by isobaisseoside and both compounds displayed a mixed inhibition mode. Molecular docking revealed polar and hydrophobic interactions between the isolated coumarins and CA IX. MD simulations offered insights into the dynamic behavior of the coumarins within the CA IX binding site. Xeroboside and isobaisseoside exhibited consistent trajectories, with lower Coul-SR and LJ-SR interaction energy values, indicating favorable electrostatic and van der Waals interactions. Xeroboside showed extensive hydrogen bonding with CA IX, affirming its high inhibitory potential. Additionally, xeroboside exhibited lower average Rg and SASA values, suggesting a more compact structure and reduced solvent exposure of the xeroboside–CA IX complex. The MM/PBSA analysis indicated that xeroboside had the most favorable binding free energies when interacting with CA IX. Furthermore, the PEL analysis showed distinct and stable conformational states for the CA IX–ligand complexes, with xeroboside presenting the most stable and lowest energy configuration. These computational results are consistent with our experimental findings, highlighting the potential of xeroboside and isobaisseoside as potent CA IX inhibitors. Therefore, C. officinalis-derived coumarins are promising CA IX inhibitors, pending in vitro, in vivo, and clinical investigations to determine their exact mechanism(s) of action.
Data availability
The manuscript and ESI† contain all data supporting the reported results.
Conflicts of interest
All authors declare no conflicts of interest in relation to the manuscript.
Acknowledgements
Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R381), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. This work was carried out with the support from the project PID2023-150717NB-I00 from Ministerio de Ciencia, Innovacion y Universidades in Spain and the PRIES-CM project Ref: Y2020/EMT-6290 from the Comunidad Autónoma de Madrid. The authors express their gratitude to the Centro de Computación Científica of the UAM (CCC-UAM) for providing the computing time.
References
- M. Imtaiyaz Hassan, B. Shajee, A. Waheed, F. Ahmad and W. S. Sly, Bioorg. Med. Chem., 2013, 21, 1570–1582 Search PubMed.
- A. Queen, H. N. Bhutto, M. Yousuf, M. A. Syed and M. I. Hassan, Semin. Cancer Biol., 2022, 86, 899–913 CrossRef CAS PubMed.
- S.-H. Lee and J. R. Griffiths, Cancers, 2020, 12, 1616 CrossRef CAS PubMed.
- M. Logozzi, C. Capasso, R. Di Raimo, S. Del Prete, D. Mizzoni, M. Falchi, C. T. Supuran and S. Fais, J. Enzyme Inhib. Med. Chem., 2019, 34, 272–278 CrossRef CAS PubMed.
- J. K. Rasmussen and E. Boedtkjer, J. Cereb. Blood Flow Metab., 2018, 38, 492–505 CrossRef CAS PubMed.
- J. Y. Lee, M. Onanyan, I. Garrison, R. White, M. Crook, M. F. Alexeyev, N. Kozhukhar, V. Pastukh, E. R. Swenson, C. T. Supuran and T. Stevens, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2019, 317, L188–L201 CrossRef CAS PubMed.
- D. N. Olennikov and N. I. Kashchenko, Molecules, 2022, 27, 8626 CrossRef CAS PubMed.
- N. F. Komissarenko, V. T. Chernobai and A. I. Derkach, Chem. Nat. Compd., 1988, 24, 675–680 CrossRef.
- A. Raal and K. Kirsipuu, Nat. Prod. Res., 2011, 25, 658–662 CrossRef CAS PubMed.
- H. Neukirch, M. D'Ambrosio, S. Sosa, G. Altinier, R. Della Loggia and A. Guerriero, Chem. Biodiversity, 2005, 2, 657–671 CrossRef CAS PubMed.
- K. Shahane, M. Kshirsagar, S. Tambe, D. Jain, S. Rout, M. K. M. Ferreira, S. Mali, P. Amin, P. P. Srivastav, J. Cruz and R. R. Lima, Pharmaceuticals, 2023, 16, 611 CrossRef CAS PubMed.
- E. H. M. Hassanein, A. M. Sayed, O. E. Hussein and A. M. Mahmoud, Oxid. Med. Cell. Longevity, 2020, 2020, 1675957 Search PubMed.
- D. Pal and S. Saha, in Plant-derived Bioactives: Chemistry and Mode of Action, ed. M. K. Swamy, Springer Singapore, Singapore, 2020, pp. 205–222, DOI:10.1007/978-981-15-2361-8_9.
- R. S. Alruhaimi, A. M. Mahmoud, I. Elbagory, A. F. Ahmeda, A. A. El-Bassuony, A. M. Lamsabhi and E. M. Kamel, Bioorg. Chem., 2024, 147, 107397 CrossRef CAS PubMed.
- J. A. Verpoorte, S. Mehta and J. T. Edsall, J. Biol. Chem., 1967, 242, 4221–4229 CrossRef CAS PubMed.
- S. Iqbal, M. Saleem, M. K. Azim, M. Taha, U. Salar, K. M. Khan, S. Perveen and M. I. Choudhary, Bioorg. Chem., 2017, 72, 89–101 CrossRef CAS PubMed.
- N. Guex and M. C. Peitsch, Electrophoresis, 1997, 18, 2714–2723 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley New York, 1986 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GaussView 5.0. Wallingford, E.U.A., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- S. A. Ramadan, E. M. Kamel, M. A. Ewais, A. A. Khowailed, E. H. M. Hassanein and A. M. Mahmoud, Environ. Sci. Pollut. Res., 2023, 30, 49197–49214 CrossRef CAS PubMed.
- P. Bauer, B. Hess and E. Lindahl, November, 2022, 16, 2022 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell Jr, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. H. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- S. Lin, M. Liu, S. Wang, S. Li, Y. Yang and J. Shi, Chinese Journal of Traditional Chinese Medicine, 2008, 33, 1708–1710 CAS.
- R. Liu, Q. Sun, A. Sun and J. Cui, J. Chromatogr. A, 2005, 1072, 195–199 CrossRef CAS PubMed.
- S. D. Sarker, P. G. Waterman and J. A. Armstrong, J. Nat. Prod., 1995, 58, 1109–1115 CrossRef CAS.
- T. S. Wu, Phytochemistry, 1987, 26, 873–875 CrossRef CAS.
- S. Sibanda, B. Ndengu, G. Multari, V. Pompi and C. Galeffi, Phytochemistry, 1989, 28, 1550–1552 CrossRef CAS.
- A. Pesaresi, J. Enzyme Inhib. Med. Chem., 2023, 38, 2245168 CrossRef PubMed.
- E. M. Kamel, H. A. Alqhtani, M. Bin-Jumah, H. A. Rudayni, A. A. El-Bassuony and A. Mokhtar Lamsabhi, Bioorg. Chem., 2024, 150, 107609 CrossRef CAS PubMed.
- E. M. Kamel, A. Bin-Ammar, A. A. El-Bassuony, M. M. Alanazi, A. Altharawi, A. F. Ahmeda, A. S. Alanazi, A. M. Lamsabhi and A. M. Mahmoud, RSC Adv., 2023, 13, 12361–12374 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Sulaiman M. Alnasserc,
Mohammed A. Alzoghaibid,
Al Mokhtar Lamsabhi
b,
Sulaiman M. Alnasserc,
Mohammed A. Alzoghaibid,
Al Mokhtar Lamsabhi