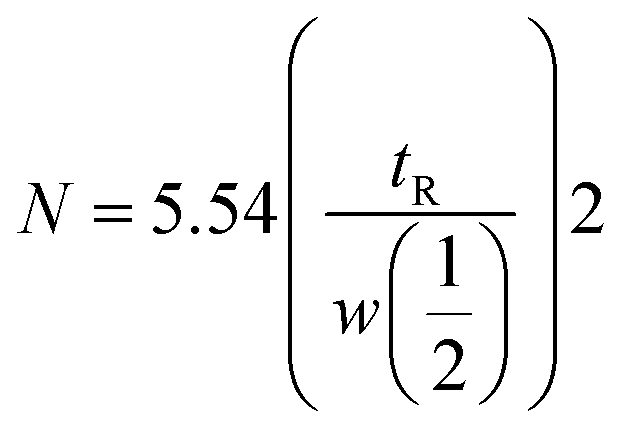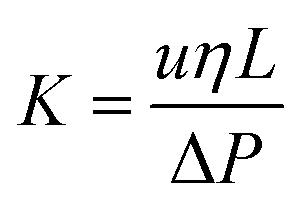DOI:
10.1039/D4RA06215A
(Paper)
RSC Adv., 2024,
14, 34486-34497
A simple and cost-effective hybrid stationary phase for the separation of peptides, proteins and benzene derivatives through liquid chromatography
Received
28th August 2024
, Accepted 6th October 2024
First published on 29th October 2024
Abstract
A very simple and cost-effective silica-based hybrid stationary phase was synthesized for the separation of five synthetic peptides, five proteins, and benzene derivatives. Silica monolith was synthesized via sol–gel process. Particles obtained through the grinding of silica monolith were suspended in methanol and sedimented under gravity to obtain sub-2 μm particles. A hybrid LC stationary phase was obtained by coating an ethylene glycol dimethacrylate co-polymer onto the particles via RAFT polymerization. The resultant stationary phase was characterized using SEM, FTIR, BET, and EDX analysis. A stainless steel column (200 mm long × 0.2 mm ID) was packed with the resultant stationary phase via pressure tapering protocol using a slurry packing machine. The column resulted in the average number of theoretical plates/meter (287![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 for synthetic peptides, 276800 for proteins, and 226100 for benzene derivatives) under the elution conditions of acetonitrile/50 mM ammonium format (68/32 v/v%) with a flow rate of 0.785 μL min−1. Since the column resulted in the separation of synthetic peptides, proteins, and benzene derivatives with very good chromatographic performance, it could possibly separate any complex mixture of peptides and proteins with better chromatographic performance.
500 for synthetic peptides, 276800 for proteins, and 226100 for benzene derivatives) under the elution conditions of acetonitrile/50 mM ammonium format (68/32 v/v%) with a flow rate of 0.785 μL min−1. Since the column resulted in the separation of synthetic peptides, proteins, and benzene derivatives with very good chromatographic performance, it could possibly separate any complex mixture of peptides and proteins with better chromatographic performance.
Introduction
Peptides and proteins are considered gene coded translational products in nature,1 and they are essential building blocks that regulate various biological activities.2 These essential blocks play a vital role in biological processes, including cell signaling, proliferation, immune response, and metabolic processes.3,4 Proteins are made of amino acids, which are essential to the human health.5 Peptides and proteins are often applied as reagents in biopharmaceutical industries.6 Therefore, purification is necessary to achieve peptides of required purity.7
Various parameters are considered in the descriptive study of diseases, including the identification of proteins that help in establishing a relationship between genes and their variants.8 Such relationships play a significant role in the diagnosis of certain diseases through the identification of different biomarkers.9 The sensitivity and selectivity of analytical technique has a significant impact on biomarker discovery. For studying peptides and proteins in terms of their activities, expression, interaction, and behavioral changes linked to protein alterations, a simplified proteomic analytical technique is important.10 Peptidome methods and processes are an effective way to determine novel peptide biomarkers for diagnostic and therapeutic applications.11 This process can be divided into the following steps: (1) discovery and identification of potential biomarker applicants, (2) confirmation of their expression in the targeted disease state, and (3) assessment of their specificity for the targeted disease in comparison to other conditions.12 However, peptide and protein separation is a challenging task owing to the complex physiochemical properties and presence of many detectable species in a single sample.13
Various laboratory-based chromatographic techniques are available for peptide and protein purification, including reverse-phase liquid chromatography (RPLC),14 affinity chromatography,15 size-exclusion chromatography (SEC),16 ion-exchange chromatography (IEC),17 hydrophilic interaction liquid chromatography (HILIC),18 and hydrophobic interaction chromatography (HIC).19 RPLC, IEC, and HIC are considered better chromatographic modes for the separation of peptides and proteins.20 However, RPLC stationary packing materials do not provide satisfactory results in the separation of peptides, proteins, and their derivatives.21
Organic/inorganic hybrid stationary phases synthesized by chemical binding of an initiator to the porous and non-porous surfaces have been documented in many research publications22 for various applications. Hydroxyapatite, graphite, silica, and metal oxides are widely used as the inorganic packing materials in various chromatographic research areas.23 Owing to its characteristic features, silica monolith particles have been used as the packing media in separation science.24 The particles obtained by crushing the silica monolith are calcined to eliminate the organic contents, and chemicals were modified with different organic ligands for a variety of chromatographic applications.25,26
Hybrid stationary phases are considered as one of the attractive packing materials to achieve the enhanced separation selectivity of complex biological analytes, such as peptides, proteins, and their derivatives. Hybrid packing materials provide multimodal interactions of analytes with the stationary phase and mobile phases.27 Hence, different single-mode conventional columns can be replaced by a single hybrid column with enhanced chromatographic characteristics.28,29 A hybrid chromatography column combines several retention/separation mechanisms in a single column for the analysis of complex samples.30 To prevent the irreversible adsorption of peptides/proteins, a polymer-based stationary bed is more biocompatible.31 Nevertheless, a silica-based stationary bed provides high column efficiency for the separation of peptides and proteins compared to polymer-based stationary beds.28 A total of N ∼ 224000 plates per m has been reported in a recently published article for a mixture of Trp-Gly, Thr-Tyr-Ser, angiotensin I, isotocin, and bradykinin,32 where the packing is somewhat similar to mixed/multimodal packing modes. The authors of another article33 reported on the separation efficiency of 110000 plates per m for the targeted vasopressin. Some of the research articles reported N-values from chromatograms under isocratic elution mode, which are inferior to those of the previous studies.34 Separation efficiency under gradient elution mode can be considered for comparison purposes. N-values of 197,000 m−1, 14,000 m−1, and 111,000 m−1, were reported for lysozyme, bovine insulin, and metenkephalin, respectively.35 A comparison of the EGDMA polymer-bound silica monolith stationary phase with other silica-based mixed mode packing materials is summarized in Table 1. The modification of silica particles with the polymer using EGDMA as the monomer, their packing in the column, and their evaluation for the separation of peptides, proteins and benzene derivatives, has never been reported prior to this study.
Table 1 Comparison of the EGDMA-bound silica monolith stationary phase with previously reported stationary phases for the separation of peptides, proteins, and benzene derivatives
| Stationary phase |
Analytes |
Separation efficiency (plates per meter) |
References |
| Poly(styrene-methacrylic acid-N-phenylacrylamide) stationary phase |
Peptides and proteins |
351000 |
36 |
| N-Phenylmaleimide embedded polystyrene (PMP) stationary phases |
Peptides and proteins |
200000 |
27 |
| Chlorodimethyl octadecyl silane (C18) modified silica monolith |
Peptides and proteins |
400000 |
37 |
| TSPU/C18 phase |
Peptides |
174000 |
38 |
| Proteins |
11800 |
| C18-modified silica monolith |
Benzene derivatives |
157000–195000 |
25 |
| Polystyrene-immobilized silica monolith stationary phase |
Benzene derivatives |
197000 |
39 |
| Polystyrene-modified sub-1 μm particles originating from the silica monolith stationary phase |
Benzene derivatives |
256200 |
40 |
| Polystyrene-bound stationary phase |
Benzene derivatives |
165000 |
41 |
| EGDMA-embedded silica |
Peptides |
288000 |
Current study |
| Proteins |
277500 |
| Benzene derivatives |
227500 |
Monolith silica particles were synthesized and modified with EGDMA, resulting in the polymer-bound silica monolith hybrid stationary phase. After packing the stationary phase in a stainless steel column (20 cm long × 0.2 mm ID), very good chromatographic performances were achieved for the separation of synthetic peptides, proteins, and benzene derivatives in liquid chromatography. The synthesis steps of the EGDMA polymer-bound silica monolith particles are very easy and cost-effective with very good yield of the final product. The chromatographic performance of the newly designed stationary phase in terms of separation efficiency is very good, i.e., N-value of 288000, 277500, and 227500 plates per m were obtained for the mixture of synthetic peptides, proteins, and benzene derivatives, respectively.
Materials and methods
Chemicals and materials
Glacial acetic acid, urea, polyethylene glycol (PEG 1500) of molecular weight in the range of 1350–1650 g mol−1, trimethylorthosilicate (TMOS), 3-aminopropyl triethoxysilane, ethylene glycol dimethylacrylate (EGDMA), and sodium diethyldithiocarbamate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Analytical grade ammonia was supplied by Scharlau (Barcelona, Spain). Toluene and anhydrous THF were obtained from Guangfu Co., Ltd (Tianjin, China). Anhydrous DMF was purchased from Heowns Biochemical Technology Co., LTD. (Tianjin, China). HPLC-grade ethanol, acetone and distilled water were obtained from Mallinckrodt Baker (Phillipsburg, NJ, USA). Glass-lined stainless steel tubing (20 cm long × 0.2 mm) and a silica capillary column of 50 μm ID, 365 μm OD were obtained from Grace (Deerfield, IL, USA). Screen frits (diameter 1.6 mm, thickness 0.08 mm, and pore size 0.5 mm) were bought from Valco (Houston, TX, USA).
Instrumentations
Setup of the HPLC. A liquid chromatography system consisting of a pressure pump, a detector, injector, and a computer installed with data processing software, as reported in ref. 40 and 41, was used for checking the separation performance of the hybrid packed column. A Shimadzu LC-10AD pump (Japan), UV-2075 capillary window detector (Jasco, Japan), C14W.05 injector with a 50 nL injection loop from Valco (Houston, TX, USA), and Shimadzu DGU-14A membrane degasser were connected to construct the μLC system. The Lab solution (Shimadzu, Japan) software was installed for processing the chromatographic data. The resultant chromatographic data were analyzed by Origin Pro8 (Northampton, MA, USA).In the current study, SEM (JSM 5910, JEOL Japan) was used for analyzing the surface morphology, and FTIR (PerkinElmer, spectrum version 10.5.1) was used for the investigation of the surface functionalities. Furthermore, a particle size analyzer (Micromeritics, USA) and an EDX (JSM-6390LV) for elemental composition were used. A digital balance (Shimadzu ATY 224), Lc oven (Model 3511) at 40 °C, and GC oven (model 6850B) at 120 °C were used for heating purposes. Furthermore, a muffle furnace (Model TMF-4-13, Shanghai, China) was used for calcination, a shaker (Taiwan) was used to agitate the samples, and a magnetic stirrer (Irmico, Germany) was used for stirring purposes. A Mettler-Toledo EL20 (Mettler-Toledo, Switzerland) was used for pH measurement.
Synthesis of silica particles obtained by the crushing of silica monolith
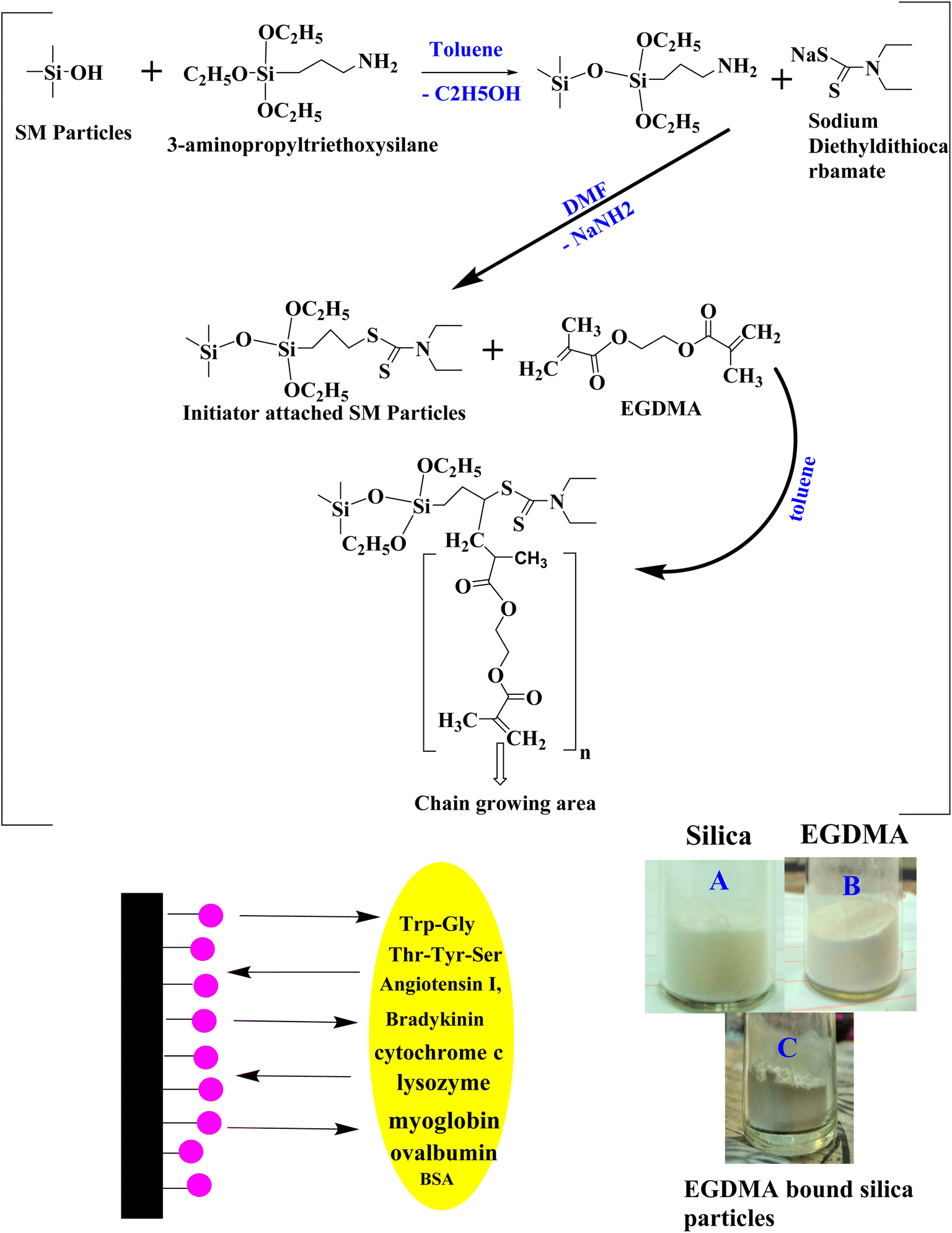
Silica monolith was synthesized by adopting some modifications in the basic sol–gel process. The monolithic body was crushed into silica particles of small dimensions, followed by calcination.42,43 The production scale was improved by using increased amounts of the reacting species. Thus, 3.3 gram PEG and 3.375 gram urea were dissolved in 15 mL, 0.01 M acetic acid solution. The reacting species were taken in a Teflon vial. The solution was magnetically stirred for 15 min under ice cold conditions. During the stirring process, 15 mL TMOS was slowly added to the reaction mixture, and the solution was continuously stirred for 40 min. The Teflon vial containing the reaction mixture was placed at 40 °C in an oven for 48 h (gel formation process). The aging process of the gel was carried out at 120 °C for 48 h. The residual water was dried at 55 °C for 20 h under vacuum conditions. The solid monolithic body was ground with a mortar and pestle. The scheme of the reaction is given in Fig. 1.
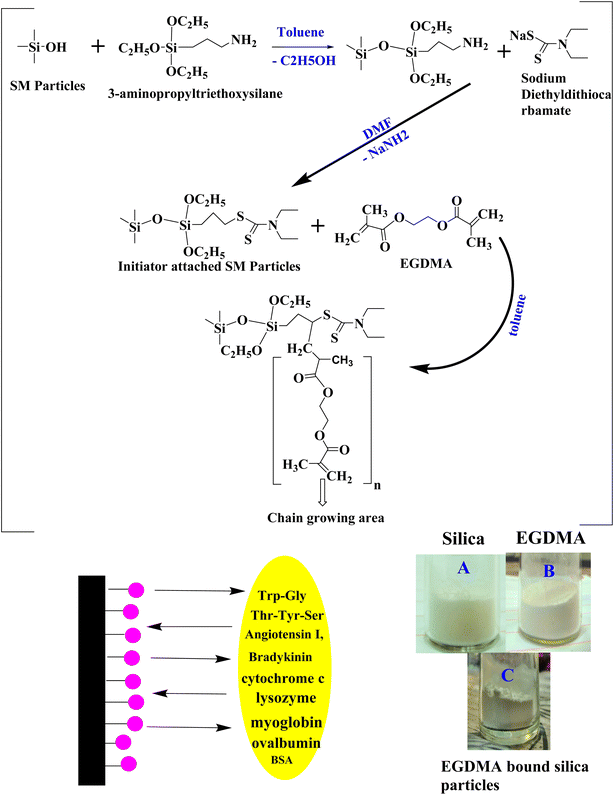 |
| | Fig. 1 Reaction scheme for EGDMA-embedded silica particles, and images of silica (A), EGDMA (B) and EGDMA-bound silica particles (C). | |
Chemical reaction of the initiator with silica particles
The silica monolith-originated particles (0.9 g) were dried at 80 °C for 2 h in a vacuum-LC oven and suspended in 60 mL anhydrous toluene with continuous stirring in a 3-neck round bottom flask of 500 mL capacity. 3-Aminopropyl triethoxysilane (3 mL) dissolved in anhydrous 10 toluene was added dropwise to the reaction mixture under reflux conditions at 104 °C for 10 h under vigorous stirring and a nitrogen-saturated environment. The product was kept undisturbed for 10 min. The upper portion containing fine suspended particles was decanted off. The ligand-bound particles present in the lower portion of the flask were washed with toluene and acetone, filtered and vacuum-dried at 50 °C for 1–2 h, and shifted to a 3-neck round-bottom flask of 100 mL capacity. A volume of 25 mL DMF was added to the flask, and 0.5 g sodium dithiocarbamate dissolved in 5 mL DMF was added dropwise using the dropping funnel. The reaction mixture was heat-treated under a nitrogen environment at 100 °C overnight with vigorous shaking. The resultant initiator-bound silica particles were washed with DMF, toluene and acetone. The product was vacuum-dried in a desiccator at room temperature overnight.
Polymer coating on silica particle surface
A solution of 2 mL EGDMA and 30 mL anhydrous toluene was sonicated for 10 min. Then, 2.2 g initiator-bound silica particles were dispersed in the solution. RAFT polymerization was carried out under reflux conditions for the immobilization of the polymer coating at the particle surface. The EGDMA solution was added dropwise to the reacting mixture under vigorous stirring and nitrogen purging for 20 h. The EGDMA polymer-bound silica particles were filtered, washed using 2-propanol, toluene, and acetone. The polymer-bound silica particles were dried under vacuum conditions. Fig. 1 illustrates the reaction scheme for the synthesis of the polymer-bound silica particles.
Characterization
The BET/BJH nitrogen adsorption/desorption analysis of silica particles before and after modification with polymer were carried out using BEL-Japan (Osaka, Japan) BELSORP-Max. A stable pressure of less than 10−3 Torr was obtained by degassing the sample at 393 K for 10 h. The pore volume was determined using adsorbed N2 at a relative pressure of P/P0 = 0.99. SEM SU 8010 (Hitachi High Technologies, Tokyo, Japan) was used to examine the morphological appearance of the bare and polymer-bound silica particles. The dried samples (modified and bare silica particles) were placed on aluminum stubs using carbon tape. To create a thin coating, the particles were evenly distributed across the carbon tape. Elemental analysis was performed using a Thermo Electron (Waltham, MA, USA) Flash EA1112 elemental analyzer. Particle size distribution was carried out using a Mastersizer 2000 particle size analyzer (Malvern, Worcestershire, UK).
Column packing
A home-assembled stainless-steel column (20 cm long × 0.2 mm ID) was packed by the pressure tapering procedure using a slurry packing setup in accordance with the procedure reported in ref. 44. The reservoir of the slurry packing machine was fed with a stationary phase slurry. The column with an outlet union containing a 1 μm screen frit (Alltech Deerfield, IL, USA) was connected to the reservoir of the slurry packer. The slurry was prepared by suspending the polymer-bound silica particles in methanol. 800 mg of the EGDMA polymer-bound silica particles were suspended in 10 mL methanol. The quality of packing was improved by vigorously vibrating the reservoir and column during packing. Nitrogen gas was used at a pressure of 20000 psi for 3 min, at 15000 psi for 15 min, and then the column was left in position until the pressure was down to zero, which took about 1 h. Two vibrators (Alltech, Deerfield, IL, USA) provided mechanical agitation to ensure the homogenous packing of the column. The capillaries of the UV-detector and outlet union of the column were connected via Teflon tubing. Peptides, proteins, and benzene derivatives were chosen for evaluating the separation performance of the column. The stock sample solution was stored below 4 °C.
Column evaluation
Peptides (Trp-Gly, Thr-Tyr-Ser, angiotensin-I, isotocin, bradykinin), proteins (cytochrome c, lysozyme, myoglobin, ovalbumin, and BSA), and benzene derivatives (acetophenone, phenol, 4-methyl-2-nitroaniline, toluene) were analyzed under isocratic elusion mode using the newly developed column. The results of the column from the current study were compared with those of the previously reported columns. Numbers of theoretical plates (N) were calculated by the following equation:| |
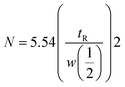 | (1) |
where tR is the retention time and w1/2 is the bandwidth at half height.
Results and discussion
Morphological appearance of the stationary phase
Morphology of the cross-linked EGDMA polymer-bound monolith silica particles is visualized in the form of SEM images given in Fig. 2. The modified silica particles are smooth enough due to the EGDMA polymer immobilization. The monolithic architecture was developed by packing the irregular-shaped silica particles inside the stainless-steel column, which offers flow through channels, leading to enhanced permeability of the column. The irregular shape and nature of the particles can be seen in the figure. The microscopic image of the EGDMA polymer-bound silica particles given in Fig. 2 suggests that the particles are well spread with no agglomeration. Good dispersity of the stationary phase particles is crucial for achieving a good packing bed inside the column.
 |
| | Fig. 2 SEM images (A: wide view, B: close view) and microscopic image of the EGDMA-modified silica particles (C). | |
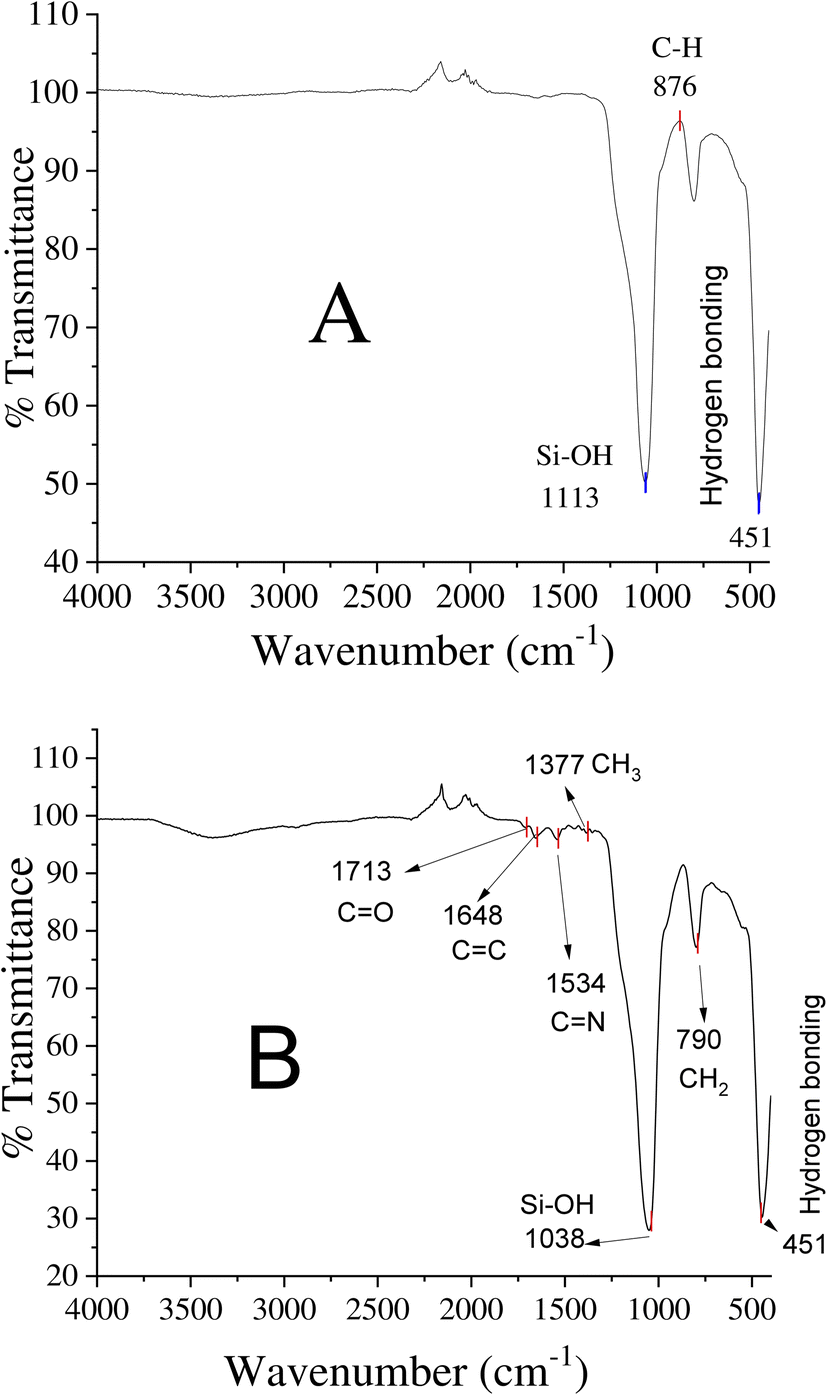
FT-IR studies
Surface functionalities of the unmodified and EGDMA-modified silica particles were observed via FTIR scanning in the range of 4000–500 cm−1 (Fig. 3). The peaks observed at 1113 cm−1, 875 cm−1, and 451 cm−1 are the peaks due to Si–OH, C–C, and hydrogen bonding, corresponding to the unmodified silica particles (Fig. 3A). The peak at 1713 cm−1 corresponds to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O present in EGDMA, while the peaks at 1648 cm−1, 1377 cm−1, and 790 cm−1 are due to the presence of CC, –CH3, and –CH2 groups in EGDMA, respectively. The band around 1534 cm−1 of the EGDMA-modified silica belongs to the stretching vibration of the C–N groups, and indicates the presence of dithiocarbamate in the product.
O present in EGDMA, while the peaks at 1648 cm−1, 1377 cm−1, and 790 cm−1 are due to the presence of CC, –CH3, and –CH2 groups in EGDMA, respectively. The band around 1534 cm−1 of the EGDMA-modified silica belongs to the stretching vibration of the C–N groups, and indicates the presence of dithiocarbamate in the product.
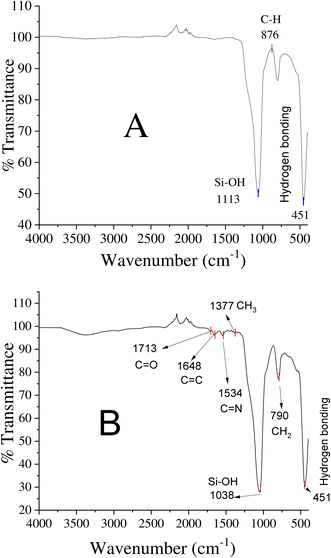 |
| | Fig. 3 FTIR spectra: (A) unmodified silica and (B) EGDMA polymer-modified silica particles. | |
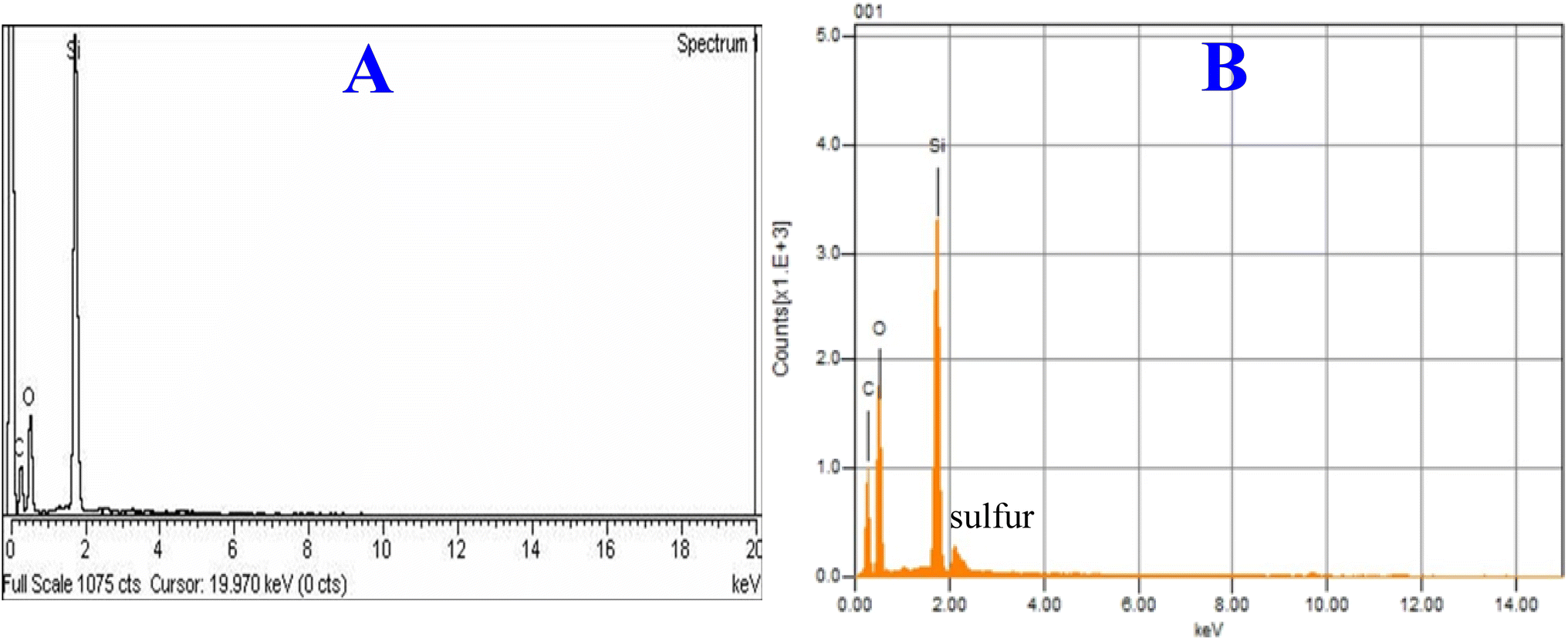
Elemental analysis by EDX
Energy dispersive X-ray analysis of the new hybrid stationary phase was carried out before and after modification with EGDMA (Fig. 4). The EDX plot of the silica monolith contains C – 31.13%, O – 41.99%, and Si – 14.99%, and that of the EGDMA-modified silica was C – 36.61%, O – 48.30%, Si – 16.10%, and S – 6%, respectively. The increase in the percentage composition of carbon and oxygen, and the presence of sulfur in the modified silica monolith particles provide clues of EGDMA polymerization at the surface of the silica monolith particles.
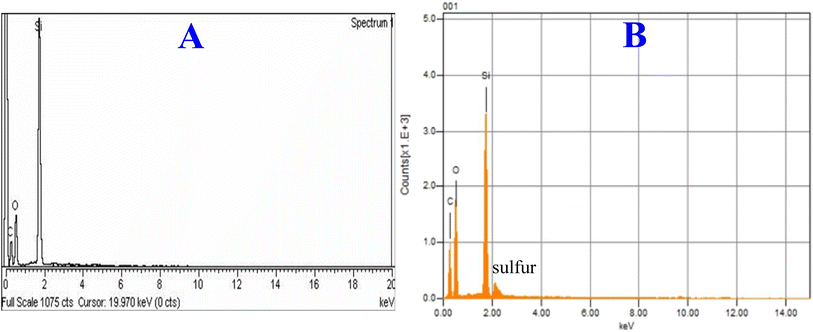 |
| | Fig. 4 EDX plots of the silica monolith particles before (A) and after (B) modification with EGDMA. | |
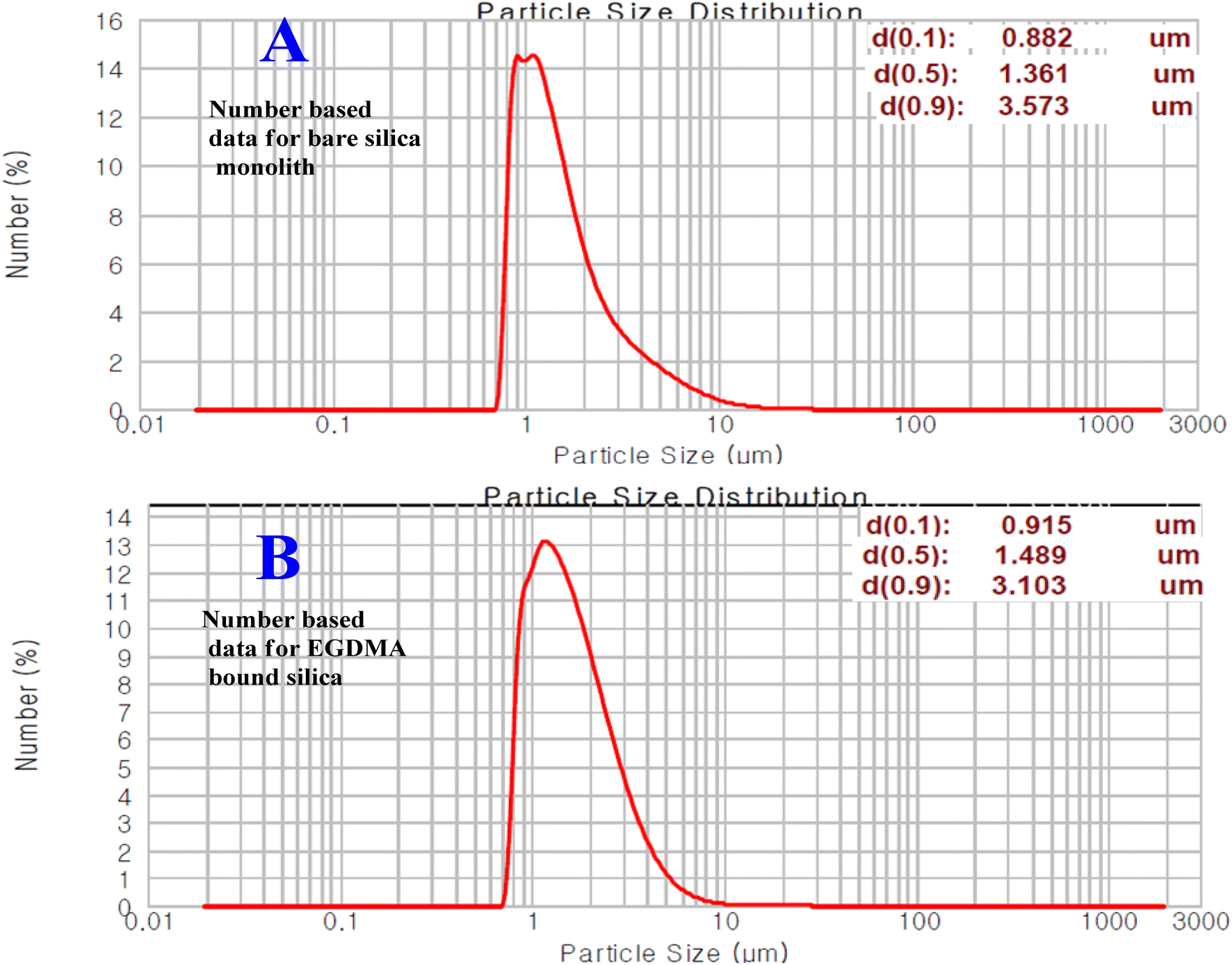
Particle size of the silica particles
The particle size distribution data and plots for the bare and modified silica particles originating from silica monolith are given in Fig. 5. The silica particle size distribution data of the current and previous studies are comparatively summarized in Table 2. The number-based particle size of the monolithic silica was in the range of 1.4 μm. After modification with the EGDMA copolymer, the number-based particle size has been increased to 1.5 μm. The increasing particle size with EGDMA modification is in accordance with ref. 41.
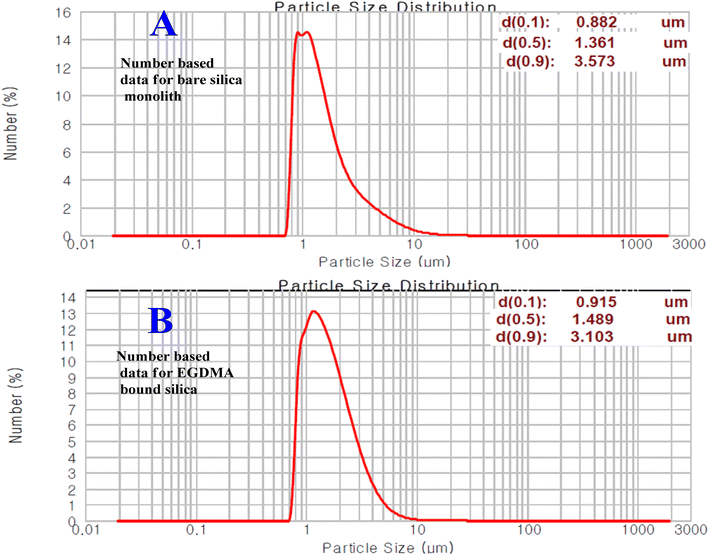 |
| | Fig. 5 Number-based particle size distribution plots of silica particles before (A) and after (B) modification. | |
Table 2 The BET/BJH analysis data of silica monolith particles and EGDMA-bound silica monolith particles
| Parameters |
Bare silica monolithic particles |
Polystyrene-bound silica particles |
C18-bound silica particles |
EGDMA-bound silica particles |
| Previous studies |
Current study |
Previous studies |
Current study |
| 41 |
45 |
42 |
37 |
41 |
45 |
42 |
37 |
| Pore size (Å) |
343 |
212 |
298 |
310 |
337 |
252 |
146 |
204 |
241 |
241 |
| Pore volume (cm3 g−1) |
1.06 |
0.83 |
0.61 |
0.67 |
0.99 |
0.84 |
0.53 |
0.49 |
0.58 |
0.58 |
| Surface area (m2 g−1) |
1.36 |
283 |
122 |
116 |
381 |
131 |
161 |
105 |
105 |
405 |
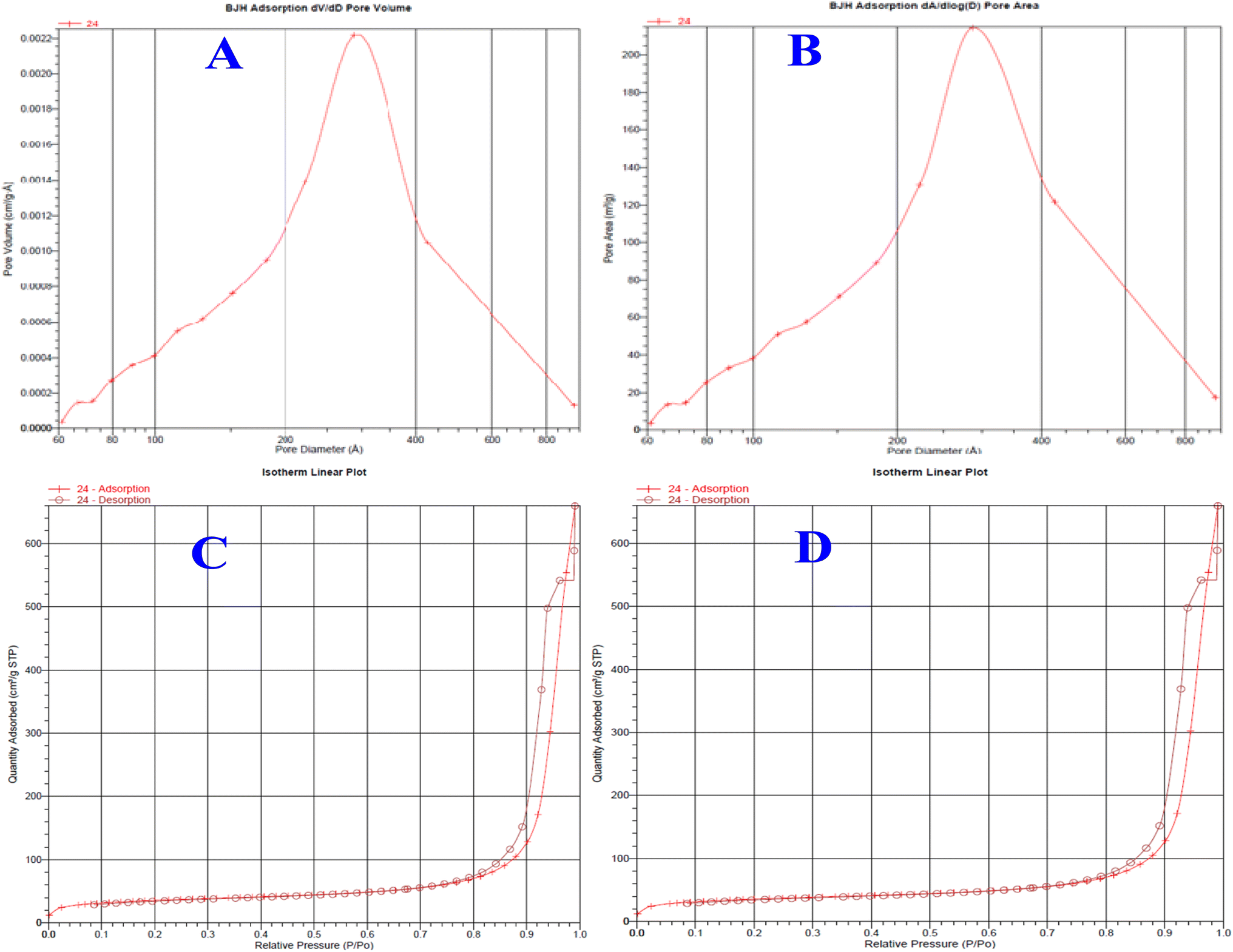
Nitrogen adsorption desorption analysis
The BJH adsorption-based pore size of the bare silica and EGDMA-modified silica particles can be seen in the plots given in Fig. 6. The average pore size of the bare silica particle is 337 Å, while that of the EGDMA-bound silica particle is 241 Å. The polymer formation at the pore surface has resulted in a diminished pore size and corresponding pore volume. The extent of the decrease in the pore size is roughly comparable to those of the previous studies summarized in Table 2. The average pore volume of monolithic silica is 0.996 cm3 g−1, while that of the EGDMA-bound silica is 1.204 cm3 g−1. The BET specific surface area of the silica monolith particles is 118 m2 g−1, while that of the EGDMA-bound silica is 157 m2 g−1. The changes in the pore size, pore volume, and surface area of the current study are comparable to those of the previous study.46
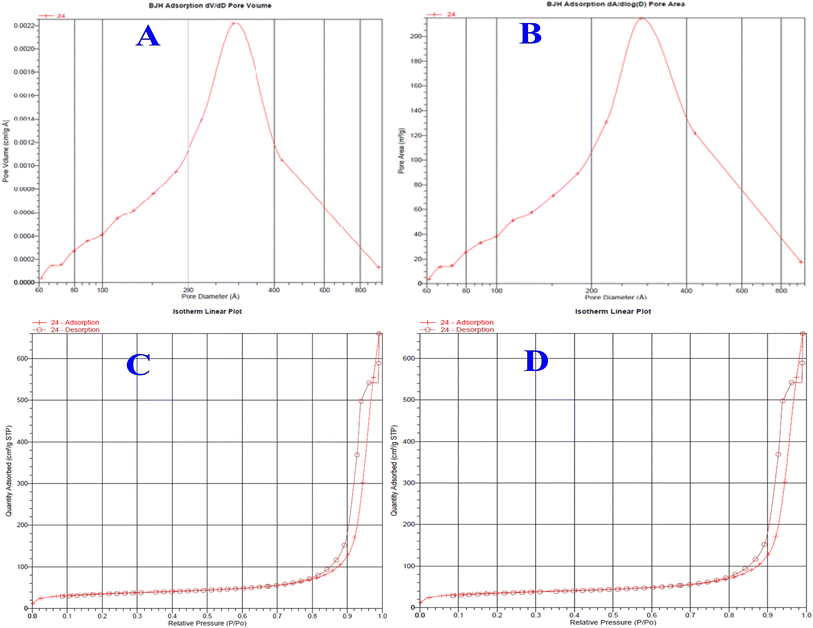 |
| | Fig. 6 Pore size distribution plots of silica monolith particles (A), EGDMA-bound silica particles (B), and isotherm linear plots of the silica monolith (C) and EGDMA polymer-immobilized stationary phase (D). | |
Optimization of the elution conditions for better separation of peptides and proteins
The effects of the salt ionic strength and pH value on the separation of analytes with complex properties, such as peptides and proteins, were critically studied. When a dual-retention mechanism is in action in any chromatographic set up, then parameters (such as pH, ionic strength of the mobile phase, etc.) affect the overall separation of complex analytes, such as peptides and proteins. The retention time of the polar analytes increases by increasing the salt concentration. A greater cohesive energy density of the mobile phase was observed at elevated ionic strength. The polar analytes were more readily driven by the mobile phase of the higher salt content. Peptides possess complex physicochemical characteristics, causing various trends of retention times at different salt concentrations. In the current study, the optimized elution conditions were found to be 62/38 acetonitrile/30 mM, ammonium formate buffer (adjusted at pH 6.4) for better separation of the synthetic peptides and proteins. The same elution condition was used for the separation of small organic molecules such as benzene derivatives, which resulted in a very good chromatographic performance.
Retention mechanism of peptides/proteins
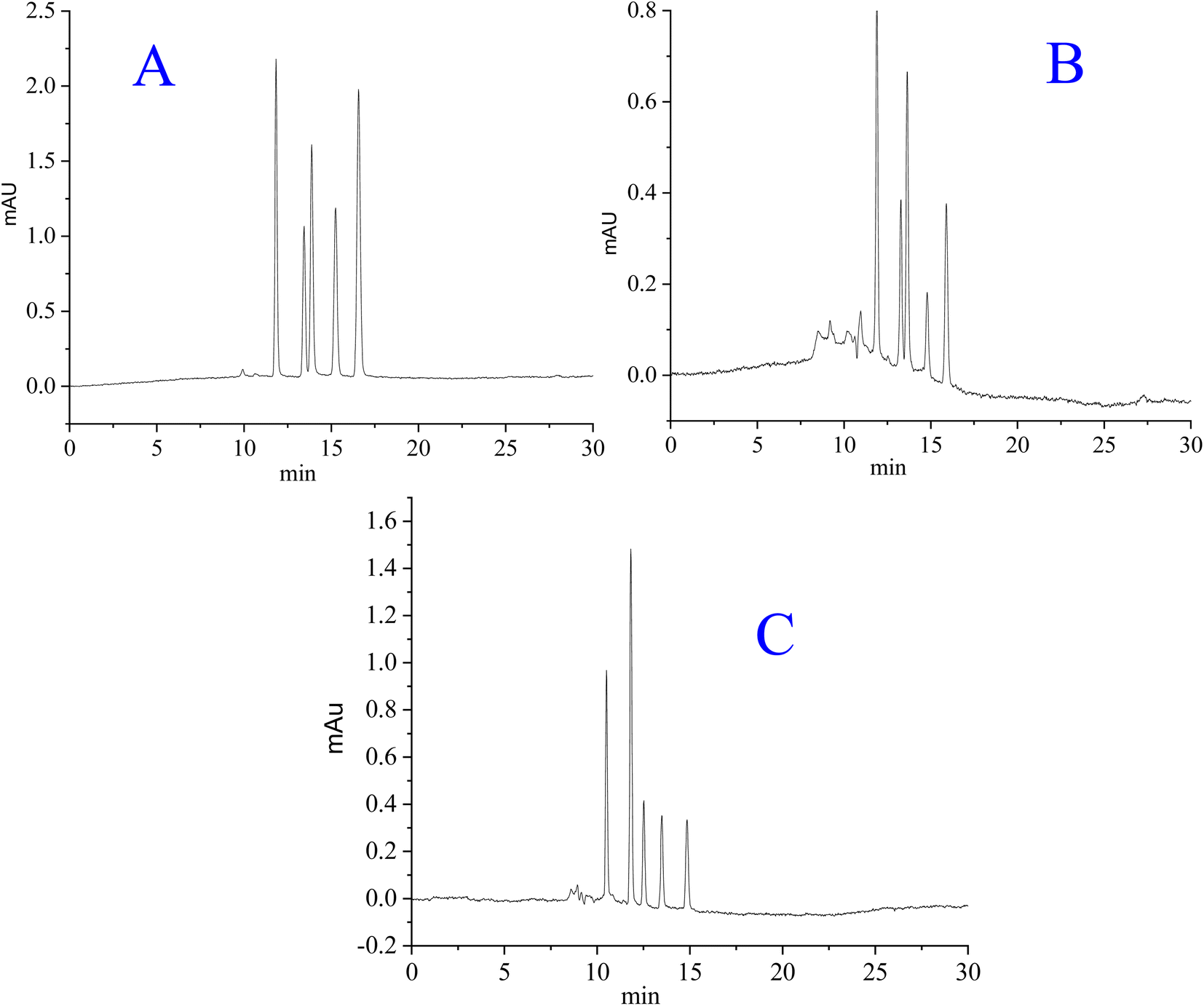
The magnitude of hydrogen bonding is different for different peptides,45 resulting in different retention times of individual peptides. Peptides/proteins are retained by the hydrophilic and (to a lesser extent) hydrophobic interactions between the analytes and hybrid packings.38 A thin layer of water is formed at the surface of silica hybrid stationary phases having polar moieties.47 Thus, the liquid–liquid partitioning effect is established between the water layer at the stationary bed and the mobile phase, whereby peptides and proteins are separated under HILIC mode.48 The separation/retention of peptides, proteins, and benzene derivatives on polar silica-based stationary phases is a complicated process, in which the quadruple interactions among analytes, mobile phase, and silica particles may not be excluded.45 For the separation of polar species such as peptides and proteins, the HILIC mode might be very effective.49 The chromatograms for the separation of selected analytes are shown in Fig. 7. Because of their reduced mass transfer kinetics, proteins have poorer separation efficiency than peptides. The less retained myoglobin is close to neutral, exhibiting less interaction with the polar stationary phase in comparison to albumin and insulin, which are more polar (slightly charged). Beyond the preview of this work, there may be geometry–selective interactions of certain analytes with the stationary phase, regulating their retentions. In the current project, the advantageous effects of the RPLC and HILIC modes have been successfully availed in the form of hybrid stationary phases.
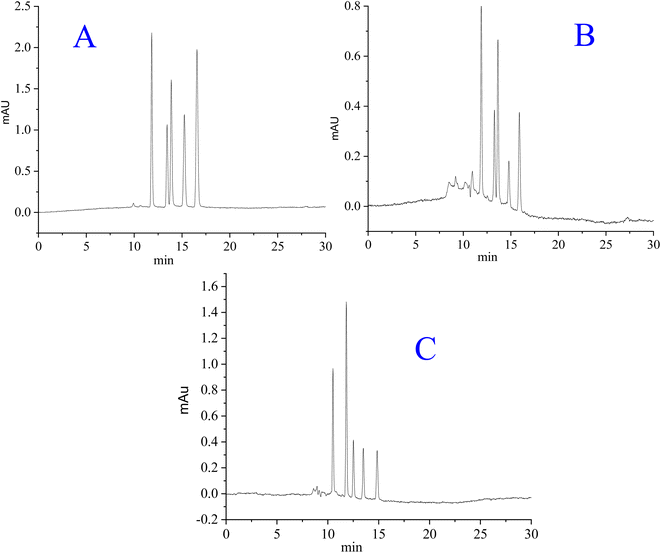 |
| | Fig. 7 Separation of (A): synthetic peptides (Thr-Tyr-Ser, angiotensin I, bradykinin, isotocin, Trp-Gly), (B) proteins (myoglobin, ovalbumin, cytochrome c, lysozyme, and BSA), and (C) benzene derivatives (phenol, acetophenone, 4-methyl-2-nitroaniline, benzene and toluene). Mobile phase: acetonitrile/30 mM ammonium formate (62/38 v/v %) at pH 6.4, flow rate = 0.785 μL min−1, column dimensions = 20 cm long × 0.2 mm ID. | |
Permeability of column
The permeability of the packed hybrid column was calculated using the following equation:
where “η” is the viscosity of the mobile phase, “ΔP” is the back-pressure of the column, “L” is the length of the column, and “u” is the mobile phase linear velocity. The permeability of the column was computed as 1.024 × 10−14 m2 at 0.785 μL min−1 using a 60/40 v/v% ACN/water. The phase permeability of the current study is comparable to those of the monolith stationary phases reported in the literature.41
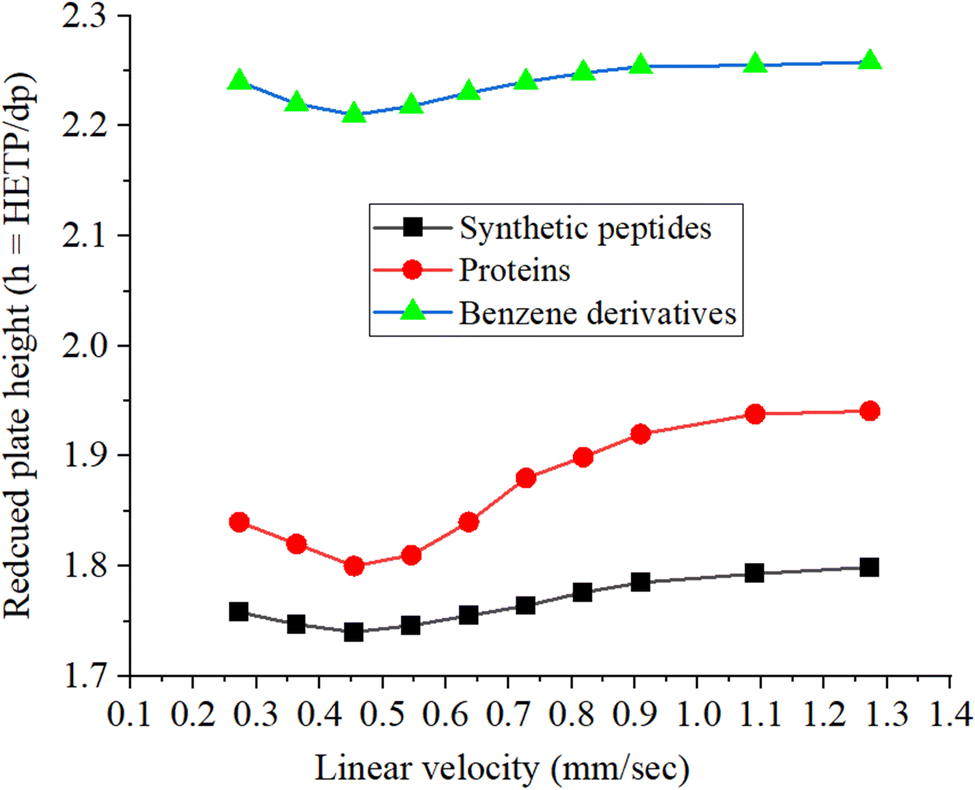
Van Deemter plot
Fig. 8 shows the Van Deemter plots for the synthetic peptides, proteins, and benzene derivatives. The reduced plate height is calculated by dividing the height equivalent to the theoretical plate (HETP) by the particle size of the stationary phase (dp), where dp is the average particle size of the stationary phase that is computed from the particle size distribution data. A lower value of the reduced plate height higher will be the separation efficiency of the column. The reduced plate height values (h = HETP/dp) used in the construction of the Van Deemter plot are the average values of the reduced plate heights for peptides, proteins, and test analytes of benzene derivatives. The lowest values of the reduced plate height (h = HETP/dp) corresponding to the highest separation efficiency calculated under optimized elution conditions for synthetic peptides, proteins, and benzene derivatives are 1.74, 1.80, and 2.21, respectively, as can be seen in the plot (Fig. 8). The optimized flow rate of the mobile phase (60/40 v/v% ACN/water) is 0.785 μL min−1, corresponding to a linear velocity of 0.45 mm s−1.27,41,50
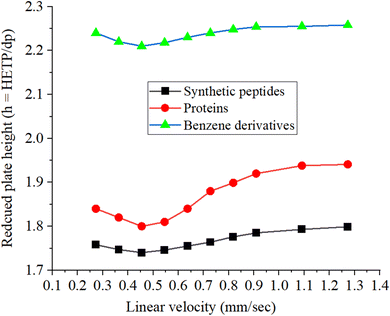 |
| | Fig. 8 Van Deemter plots in terms of the reduced plate height vs. linear velocity for the synthetic peptides, proteins, and small molecules of benzene derivatives. The reduced plate heights are the average values calculated for each class. | |
The retention time of the unretained analytes on the column from the current study (Rt) is 8 min. The following calculations were used to determine the flow rate and linear velocity of the column. When the units of the column length and radius (column internal diameter/2) are taken in millimeter, the unit of volume will be in μL.
| Volume of the column = column length × πr2 |
| = 200 mm × 3.14 (0.2/2)2 = 6.28 μL |
| Volumetric flow rate = Volume of the column/Retention time of the unretained analyte |
| Volumetric flow rate = 6.28 μL/8 min = 0.785 μL min−1. |
| Linear velocity = volume flow rate/column cross-section area = 0.785 μL/3.14 × (0.1 mm)2 = 25 μL mm−2 min−1, |
Since 1 μL = 1 mm3, so
| Linear velocity = 25 mm min−1, = 25/60 = 0.417 mm s.−1 |
The linear velocity was confirmed using the alternate method of calculation. The average linear velocity of the mobile phase is given by the length of the column divided by the retention time of the mobile phase or the dead/void time.
| V (linear velocity) = L (column length in mm)/t0 (Retention time of unretained analyte in sec) |
The dead volume of the column equivalent to the unretained analyte in the unit of time is 8 min.
| = 200/8 × 60 = 0.45 mm s−1. |
The separation efficiency of the hybrid column developed in the current study is better than those of the commercially available columns. The monolith pattern can be observed in the Van Deemter plot obtained in the current study, which is in agreement with previous studies.25
Column separation performance
Separation efficiency. The hybrid column developed in the current study was checked for the separation of synthetic peptides, proteins and benzene derivatives as the test analytes. The chromatograms are given in Fig. 7. The earlier eluting peaks are sharper than the later eluting peaks under isocratic elution mode due to the peak tailing phenomena. In contrast, some of the earlier eluting peaks are less sharp than the later eluting peaks in the chromatograms. The overlap of several peptide peaks with similar retention times to each other may not be excluded when dealing with protein samples, which results in wider peaks. The separation performance of the hybrid column was evaluated by analyzing a mixture of peptides/proteins and benzene derivatives. Synthetic peptides, proteins, and benzene derivatives are separated well with an average number of theoretical plates at 287500 m−1, 276800 m−1, and 226100 m−1, respectively, corresponding to their respective chromatograms (Fig. 7). For high separation efficiency, the confinement of particles to a narrow size range, a proper packing protocol, and a packing setup to get the best packed bed inside the column are very important. The separation efficiency in terms of the plate number/reduced plate height, along with reproducibility in the stationary phase, batch analysis, and column packing are summarized in Table 3. The %RSD values computed in the separation efficiency and retention times for three different columns developed by the same protocol are within the permissible limit of analytical chemistry. The %RSD in the plate count is less than 2%, while the %RSD in the retention time is less than 4%. For evaluating the reproducibility in the synthesis of the stationary phase and packing procedure, three different batches of the same stationary phase were synthesized and packed in three different columns of the same dimensions using the same protocol. The reproducibility was evaluated in terms of checking the columns by injecting the same sample for a single run. The day-to-day repeatability of one of the three columns was checked by injecting the sample for more than 15 runs through the same column on different days, where the average %RSD values were found to be less than 1.5% for different samples under study. Similarly, the intraday repeatability of that column was checked by injecting the same sample within different times of the same day for ten times, where the %RSD was found to be less than 1%. The behavior of the analyte was purely sorption type without any clear evidence of adsorption.
Table 3 Separation efficiency and column-to-column reproducibility in terms of %RSD in the plate count, and the retention time computed for three different batches of the stationary phase synthesized using the same protocola
| |
Name |
Average N-value/meter |
Reduced plate height |
%RSD in plate count |
Retention time |
%RSD in retention time |
| Elution conditions: acetonitrile/30 mM ammonium formate (62/38 v/v%) at pH 6.4, flow rate = 0.785 μL min−1, column dimensions = 20 cm long × 0.2 mm ID. |
| Synthetic peptides |
Thr-Tyr-Ser |
289000 |
1.73 |
2.1 |
11.7 |
3.2 |
| Angiotensin I |
284500 |
1.75 |
1.9 |
13.2 |
3.8 |
| Bradykinin |
290000 |
1.72 |
1.9 |
13.4 |
3.9 |
| Isotocin |
287500 |
1.74 |
1.3 |
15.1 |
4.1 |
| Trp-Gly |
286500 |
1.74 |
1.1 |
16.8 |
3.5 |
| Proteins |
Cytochrome C |
279500 |
1.79 |
2.6 |
11.7 |
3.4 |
| Lysozyme |
276500 |
1.80 |
2.2 |
13.1 |
3.9 |
| Myoglobin |
276000 |
1.81 |
2.9 |
13.2 |
4.0 |
| Ovalbumin |
277000 |
1.80 |
2.8 |
14.8 |
3.8 |
| BSA |
275000 |
1.81 |
3.1 |
16.2 |
3.6 |
| Benzene derivatives |
Phenol |
229000 |
2.18 |
1.5 |
10.7 |
4.0 |
| Acetophenone |
228500 |
2.19 |
2.1 |
12 |
3.9 |
| 4-Methyl-2-nitroaniline |
225500 |
2.22 |
1.8 |
12.4 |
4.0 |
| Benzene |
224500 |
2.23 |
1.6 |
13.4 |
3.6 |
| Toluene |
223000 |
2.24 |
1.4 |
14.9 |
3.5 |
Comparative analysis of the column of current study
The literature reports on efficient and rapid HPLC separation analysis consist of very tiny porous particles,51 core–shell particles,52 and shell-type, sub-2 μm fully porous, and monolith stationary phases.53 A detailed comparative study of the mass-transfer kinetics for the stationary phases comprising a core–shell type, sub-2 μm fully porous, and monolith has been reported in ref. 53. The core–shell type phases were found to be dominant, followed by the fully porous phases, and the monolithic phase in order of minimum HETP and reduced mass-transfer resistance.54 Particularly, compared to completely porous particles of the same size, the core–shell type particles with a 1.7 μm size demonstrated superior separation efficiency. Monolithic columns have a higher separation efficiency that gradually drops with an increase in flow rate, and are known to be very permeable. Thus, they are particularly helpful in many LC applications.41,53 The efficiency of the column from the current study for proteins ∼ 276800 m−1, peptides ∼ 287500 m−1, and benzene derivatives ∼ 226100 m−1 are either better or comparable to the N-value reported in the literature37 for the same biomolecules. Despite the relatively narrow column ID (0.2 mm) and broad range of particle size dispersion, the efficiency of the column in the current study is greater than those of previously reported columns. A significant portion of the particles in the phase of the current study is in the sub-1 μm range, which might disturb the packing quality inside the column, so we removed that portion by simple sedimentation process as reported in the articles published from our laboratory. A low back-pressure due to the monolithic architecture of the column from the current study is one of the advantageous features of the current project. Monolith flow through the channels results in high mass transfer kinetics. So, the current project has made room for the development of highly efficient, low cost, and disposable silica-based HPLC columns for the separation of biomolecules in research laboratories and pharmaceutical companies.
Concluding remarks
The current project aimed to synthesize silica-based hybrid stationary phases for the separation of five synthetic peptides (Trp-Gly, Thr-Tyr-Ser, angiotensin I, isotocin, bradykinin and acetone), proteins (cytochrome c, lysozyme, myoglobin, ovalbumin, and BSA), and benzene derivatives (acetophenone, phenol, 4-methyl-2-nitroaniline, benzene and toluene). The modified sol–gel strategy, along with the sedimentation process, was used to synthesize the confined sized silica particles, and the EGDMA polymer layer was subsequently chemically bound onto the silica particle surface by RAFT polymerization. The Alltech slurry packer was used to pack the hybrid stationary phase in a stainless steel column (20 cm long × 0.2 mm). The separation efficiency of the column is very attractive, where the Navg values for proteins is 276800 m−1, peptides is 287500 m−1, and benzene derivatives is 226100 m−1 when checked for protein samples and a test mixture of benzene derivatives using a mobile phase of acetonitrile/30 mM ammonium formate (62/38 v/v%) at pH 6.4, and a flow rate of 0.785 μL min−1.
Data availability
The data that support the findings of this paper are available from the corresponding author upon request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the University of Malakand through Higher Education Commission (HEC) of Pakistan Project no: 20-14499/NRPU/R&D/HEC/2021. The authors are grateful to the researchers supporting Project No. (RSP2024R1), King Saud University, Riyadh, Saudi Arabia.
References
- N. Budisa, Curr. Opin. Biotechnol., 2013, 24, 591–598 CrossRef CAS.
- J. Xie and P. G. Schultz, Methods, 2005, 36, 227–238 CrossRef CAS PubMed.
- N. P. Möller, K. E. Scholz-Ahrens, N. Roos and J. Schrezenmeir, Eur. J. Nutr., 2008, 47, 171–182 CrossRef PubMed.
- S. R. Hertzler, J. C. Lieblein-Boff, M. Weiler and C. Allgeier, Nutrients, 2020, 12, 3704 CrossRef CAS.
- A. Shen, Z. Guo, X. Cai, X. Xue and X. Liang, J. Chromatogr. A, 2012, 1228, 175–182 CrossRef CAS.
- G. Wu, Amino acids, 2009, 37, 1–17 CrossRef CAS.
- B. Gomes, M. T. Augusto, M. R. Felício, A. Hollmann, O. L. Franco, S. Gonçalves and N. C. Santos, Biotechnol. Adv., 2018, 36, 415–429 CrossRef CAS PubMed.
- J. Mayne, A. E. Starr, Z. Ning, R. Chen, C. K. Chiang and D. Figeys, Anal. Chem., 2014, 86, 176–195 CrossRef.
- F. Leung, N. Musrap, E. P. Diamandis and V. Kulasingam, Transl. Proteomics, 2013, 1, 74–86 CrossRef.
- M. Ye, X. Jiang, S. Feng, R. Tian and H. Zou, TrAC, Trends Anal. Chem., 2007, 26, 80–84 CrossRef.
- E. S. Nakayasu, M. Gritsenko, P. D. Piehowski, Y. Gao, D. J. Orton, A. A. Schepmoes and J. P. Krischer, Nat. Protoc., 2021, 16, 3737–3760 CrossRef PubMed.
- I. Schulte, H. Tammen, H. Selle, H.-D. Zucht and P. Schulz-Knappe, Compr. Anal. Chem., 2005, 46, 385–409 Search PubMed.
- P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Drug Discov. Today, 2010, 15, 40–56 CrossRef.
- N. D. Ostryanina, G. P. Vlasov and T. B. Tennikova, J. Chromatogr. A, 2002, 949, 163–171 CrossRef PubMed.
- J. Carroll, I. M. Fearnley and J. E. Walker, Proc. Natl. Acad. Sci., 2006, 103, 16170–16175 CrossRef PubMed.
- H. Xia, G. Wan, J. Zhao, J. Liu and Q. Bai, J. Chromatogr. A, 2016, 1471, 138–144 CrossRef PubMed.
- A. R. Ivanov, C. Horváth and B. L. Karger, J. Electrophor., 2003, 24, 3663–3673 CrossRef PubMed.
- L. Wang, W. Wei, Z. Xia, X. Jie and Z. Z. Xia, TrAC, Trends Anal. Chem., 2016, 80, 495–506 CrossRef.
- O. Núñez, K. Nakanishi and N. Tanaka, J. Chromatogr. A, 2008, 1191, 231–252 CrossRef.
- K. Zhao, F. Yang, H. Xia, F. Wang, Q. Song and Q. Bai, J. Sep. Sci., 2015, 38, 703–710 CrossRef.
- J. B. Müller, P. E. Geyer, A. R. Colaco, P. V. Treit, M. T. Strauss, M. Oroshi and N. Köhler, Nature, 2020, 582, 592–596 CrossRef.
- E. S. Hosseini and K. T. Heydar, Talanta, 2021, 221, 121445 CrossRef PubMed.
- H. Qiu, X. Liang, M. Sun and S. Jiang, Anal. Bioanal. Chem., 2011, 399, 3307–3322 CrossRef.
- D. F. Moyano, K. Saha, G. Prakash, B. Yan, H. Kong, M. Yazdani and V. M. Rotello, ACS Nano, 2014, 8, 6748–6755 CrossRef PubMed.
- A. Ali, F. Ali and W. J. Cheong, J. Chromatogr. A, 2017, 1525, 79–86 CrossRef PubMed.
- V. Pérez-Fernández, S. Morante-Zarcero, D. Pérez-Quintanilla, M. Á. García, M. L. Marina and I. S. Alonso, J. Electrophor., 2014, 35, 1666–1676 CrossRef PubMed.
- S. Alharthi, A. Ali, M. Iqbal, A. Ibrar, B. Ahmad, S. Nisa and F. Mabood, Sci. Rep., 2022, 12, 4061 CrossRef PubMed.
- Y. Yang and X. Geng, J. Chromatogr. A, 2011, 1218, 8813–8825 Search PubMed.
- U. Andjelković, S. Tufegdžić and M. Popović, J. Electrophor., 2017, 38, 2851–2869 CrossRef PubMed.
- K. Zhang and X. Liu, J. Pharm. Biomed. Anal., 2016, 130, 19–34 CrossRef.
- G. Sun, J. S. Kim, Y. S. Kim, H. J. An and W. J. Cheong, J. Sep. Sci., 2019, 42, 2612–2620 CrossRef.
- J. De Vos, M. De Pra, G. Desmet, R. Swart, T. Edge, F. Steiner and S. Eeltink, J. Chromatogr. A, 2015, 1409, 138–145 CrossRef.
- F. Gritti and G. Guiochon, J. Chromatogr. A, 2012, 1228, 2–19 CrossRef.
- H.-S. Wang, M. Song and T. J. Hang, ACS Appl. Mater. Interfaces, 2016, 8, 2881–2898 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- F. Ali, W. J. Cheong, A. Rafique, Z. A. AlOthman, M. Sadia and M. Muhammad, J. Sep. Sci., 2021, 44, 1430–1439 CrossRef PubMed.
- A. Ali, S. Alharthi, N. H. Al-Shaalan and E. Y. Santali, Polymers, 2022, 14, 2576 CrossRef PubMed.
- G. Sun, A. Ali, Y. S. Kim, J. S. Kim, H. J. An and W. J. Cheong, J. Sep. Sci., 2019, 42, 3621–3630 CrossRef PubMed.
- G. Sun and Y. Lu, J. Chromatogr. Sci., 2021, 59, 949–955 Search PubMed.
- F. Ali, A. R. Malik, W. J. Cheong and N. U. Rehman, J. Liq. Chromatogr. Relat. Technol., 2019, 42, 662–672 CrossRef.
- F. Ali, W. J. Cheong, Z. A. ALOthman and A. M. ALMajid, J. Chromatogr. A, 2013, 1303, 9–17 CrossRef PubMed.
- A. Ali, F. Ali and W. J. Cheong, Bull. Korean Chem. Soc., 2015, 36, 1733–1736 CrossRef.
- A. Ali and W. J. Cheong, Bull. Korean Chem. Soc., 2017, 38, 919–927 CrossRef.
- S.-M. Lee, S. A. Zaidi and W. J. Cheong, Bull. Korean Chem. Soc., 2010, 31, 2943–2948 CrossRef.
- M. Skoczylas, S. Bocian, T. Kowalkowski and B. Buszewski, J. Sep. Sci., 2017, 40, 4152–4159 CrossRef PubMed.
- M. Skoczylas, S. Bocian and B. Buszewski, J. Chromatogr. A, 2020, 1609, 460514 CrossRef.
- D. Yeung, N. Klaassen, B. Mizero, V. Spicer and O. V. Krokhin, J. Chromatogr. A, 2020, 1619, 460909 CrossRef PubMed.
- S. Di Palma, M. L. Hennrich, A. J. Heck and S. Mohammed, J. Proteom., 2012, 75, 3791–3813 CrossRef CAS PubMed.
- S. Fekete, E. Oláh and J. Fekete, J. Chromatogr. A, 2012, 1228, 57–71 CrossRef PubMed.
- A. Ali, G. Sun, J. S. Kim and W. J. Cheong, J. Chromatogr. A, 2019, 1594, 72–81 CrossRef.
- G. Guiochon and F. Gritti, J. Chromatogr. A, 2011, 1218, 1915–1938 CrossRef PubMed.
- G. Guiochon, J. Chromatogr. A, 2007, 1168, 101–168 CrossRef PubMed.
- E. Oláh, S. Fekete, J. Fekete and K. Ganzler, J. Chromatogr. A, 2010, 1217, 3642–3653 CrossRef PubMed.
- S. Fekete, K. Ganzler and J. Fekete, J. Pharm. Biomed. Anal., 2011, 54, 482–490 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Sana Beguma,
Muhammad Ilyasa,
Zeid A. ALOthman
*a,
Sana Beguma,
Muhammad Ilyasa,
Zeid A. ALOthman