
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced light-harvesting efficiency in Au metal–NiFe2O4 semiconductor hetero-nanostructures with implications for photoelectrochemical sensors towards the sensitive detection of paracetamol in human urine†
Tuan-Anh Nguyen *a,
Ngo Thi Loanab,
Mai Thi Ngoc Phamc,
Pham Duc Thangd,
Vu Ngoc Phana,
Ngo Xuan Dinha,
Tien Van Manhe,
Van Hoang Ongea,
Vu Dinh Lamf and
Anh-Tuan Le*ab
*a,
Ngo Thi Loanab,
Mai Thi Ngoc Phamc,
Pham Duc Thangd,
Vu Ngoc Phana,
Ngo Xuan Dinha,
Tien Van Manhe,
Van Hoang Ongea,
Vu Dinh Lamf and
Anh-Tuan Le*ab
aPhenikaa University Nano Institute (PHENA), Phenikaa University, Hanoi 12116, Vietnam. E-mail: anh.nguyentuan1@phenikaa-uni.edu.vn; tuan.leanh@phenikaa-uni.edu.vn
bFaculty of Materials Science and Engineering, Phenikaa University, Hanoi 12116, Vietnam
cFaculty of Chemistry, VNU University of Science, Vietnam National University, Hanoi, 19 Le Thanh Tong, Hoan Kiem, Hanoi, 11000, Vietnam
dFaculty of Physics, Center of Materials Science, VNU University of Science, Vietnam National University, Hanoi, 334 Nguyen Trai, Thanh Xuan, Hanoi, 11416, Vietnam
eUniversity of Transport Technology, Trieu Khuc, Thanh Xuan District, Hanoi, Vietnam
fGraduate University of Science and Technology (GUST), Institute for Materials Science (IMS), Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Hanoi 10000, Vietnam
First published on 29th October 2024
Abstract
In this study, Au–NiFe2O4 hetero-nanostructures (Au–NFO HNs) have been successfully developed and proposed as an efficient photoactive material for the construction of a photoelectrochemical sensor for the detection of paracetamol (PCM) under visible light irradiation. When Au NPs were in intimate contact with NFO NFs, a built-in electric field and Schottky barrier at the NFO/Au interface were established and downward band bending occurred. By using monochromatic 532 nm laser excitation, the photo-generated electrons on the NFO conduction band were promptly migrated to the Au Fermi level due to the presence of a built-in electric field and downward band bending, and together with the hot electrons induced by localized surface plasmon resonance characteristics of Au NPs could concurrently contribute to the enhanced photoelectrochemical activity. Furthermore, the Schottky barrier prevents the transfer of photo-generated holes from the valence band of NFO to Au, thereby suppressing the recombination of photo-generated electron–hole pairs and prolonging charge carrier lifetimes. A series of electrochemical kinetic parameters were determined and the results showed that in the presence of visible light irradiation, the interfacial charge transfer ability, electrocatalytic activity, and adsorption/diffusion capacity of Au–NFO HNs-modified electrode were remarkably enhanced compared to the dark environment. As a result, the photoelectrochemical sensing platform based on Au–NFO HNs showed noteworthy analytical performance towards PCM detection in the wide linear ranges from 0.5–200 μM, high electrochemical sensitivity of 1.089 μA μM−1 cm−2, and low detection limit of 0.38 μM. Furthermore, the repeatability, anti-interference ability, and feasibility of the proposed photoelectrochemical sensor were also evaluated. This study will establish a more comprehensive understanding and promote ongoing interest in constructing advanced and efficient plasmonic metal/semiconductor hetero-nanostructures for analytical photoelectrochemistry.
1. Introduction
The photoelectrochemical sensor, as an evolutionary generation of electrochemical sensing technology, has been becoming a fast-developing and high-performance analytical platform due to the advantages of optical and electrochemical analysis.1–3 Compared with traditional analytical techniques such as high-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC/MS), or enzyme-linked immunosorbent assay (ELISA), photoelectrochemical sensing has the merits of operational simplicity, remarkable sensitivity and selectivity, inherent miniaturization, cost effectiveness, and strong real-time monitoring capability.2,4 Since the sensing performance of a photoelectrochemical sensor is closely related to the photoelectric conversion efficiency of the photoactive electrode, the light harvesting and converting ability of photoactive materials play a non-negligible role.2,5,6 Fortunately, the breakthroughs and tremendous advances in nanoscience and nanotechnology have now paved the way for the rational design and engineering strategies employed in the construction of promising photoactive nanomaterials with well-designed configurations, robust structural/electrochemical stability, tailorable morphologies, and improved electrochemical properties to fulfill practical applications.In recent years, semiconductor nanostructures-based photocatalysts have formed the basis for the fast development in a myriad of fields such as photochemical applications,7,8 solar energy conversion and storage,9,10 environmental remediation,11,12 and other areas. As is well known, under visible and/or ultraviolet irradiation, the photo-generated electron–hole pairs at photoactive semiconductor materials can directly interact with molecules in the reaction medium, which in turn act as reducing agent and oxidizing agent and initiate the photochemical reactions.13,14 However, the performance of such semiconductor-based photoelectrochemical systems is restricted by two major intrinsic constraints. Since visible light constitutes ∼43% of the solar spectrum, the core issue is the visible light harvesting ability of photoactive semiconductor nanomaterials, which directly affect its practical applications in normal light conditions.15 Therefore, the semiconductor photocatalyst should first have sufficiently narrow bandgap (1.23 eV < Eg < 3.0 eV, 400 < λ < 1000 nm) to absorb as much visible light as possible, thereby maximizing the photocatalytic activity.13 Another critical issue is the dynamics of photo-generated electron–hole pairs, e.g., charge separation efficiency, mobility, and lifetimes. After the photo-excited charge carriers are generated, the recombination rate and the lifetimes needs to be hindered and prolonged, respectively. In addition, a high migration rate of photo-generated electrons and holes to the catalyst surface is needed, allowing them to participate in photoelectrochemical reactions at the photocatalytic active sites.
Among various types of semiconductors, nickel ferrite (NiFe2O4 – NFO) has recently been recognized as an efficient photoactive material, owing to the inherent merits of its relatively narrow bandgap energy, high photoreactivity, high photosensitivity, easy synthesis methodology, stable physicochemical properties and cost-effectiveness.16,17 Our previous work has verified the feasibility of utilizing NiFe2O4 nanoflakes in improving the analytical sensing performance of visible light-assisted electrochemical sensor for the sensitive detection of azithromycin antibiotic.18 Nevertheless, to further enhance the analytical performance of photoelectrochemical sensors, it is imperative to develop a new class of high-efficiency visible-light sensitive photocatalyst. Noble metal nanomaterials have been attracting great attention for their ability to interact intensely as well as absorb strongly with light in the visible range, thanks to their localized surface plasmon resonance (LSPR).19,20 They can serve as electron traps and active reaction sites to efficiently suppress the recombination of photo-generated electron–hole pairs, prolong charge carrier lifetimes, and improve charge separation due to the Schottky barrier built at the semiconductor–metal interface.21–23
Recently, substantial endeavors have been devoted to developing hetero-nanostructures (HNs) composed of semiconductors and plasmonic metal components, because they exhibit superior optical characteristics in the visible-to-near-infrared regions, which was found to be highly effective in amplifying the efficiency of light harvesting and charge transfer.22,24 The engineering of HNs consisting of plasmonic noble metal nanomaterials (e.g., Au, Ag, Pt, etc.) and low-bandgap semiconductors is considered as an efficient strategy to improve the lifetimes and efficiency of photo-excited in the semiconductors upon light absorption.14,22,23,25 The integration of plasmonic materials and semiconductors with the close conjunction of LSPR effect and low band gap may result in the energy band alignment on both sides of the interface,24,26 thus leading to spatial charge separation across the semiconductor–metal nanojunction under visible light irradiation. The high separation efficiency of photo-generated charge carriers can be harnessed for redox reactions and play a significant role in enhancing the sensing performance of photoelectrochemical reactions. By taking advantage of the combinatorial properties and the synergistic coupling effects, diverse noble metal–semiconductor HNs have been extensively explored in many fields, such as photocatalytic organic pollutant degradation,27 photochemical water splitting,28 surface-enhanced Raman scattering detection,29 and solar-to-chemical energy conversion.30 Nevertheless, to the best of our knowledge, there have been few reports of the application of photoactive noble metal/low-bandgap semiconductor HNs for the detection of antibiotics using photoelectrochemical sensing platform under visible light irradiation.
Motivated by previous pioneering studies, we have developed an innovative approach using Au–NFO HNs as an efficient photoactive nanomaterial for the construction of a photoelectrochemical sensing platform towards the sensitive detection of paracetamol (PCM). The crystalline phase formation, micro/nanostructures, morphological features, and optical properties of the as-prepared Au–NFO HNs were investigated. A greatly enhanced interfacial electron transfer, adsorption/diffusion capacity, and intrinsic electrocatalytic activity of Au–NFO HNs-based photoelectrochemical sensor under visible light irradiation is demonstrated by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), chronoamperometry (CA) and differential pulse voltammetry (DPV) techniques. Furthermore, by investigating the energy band diagram of metal and n-type semiconductor, the underlying mechanism of the interactions between Au and NFO before and after contact was also proposed to clarify the possible electron-transfer pathways across the Au/NFO interfaces under visible light irradiation. According to that, the synergistic coupling effect of the built-in electric field and Schottky barrier for capturing photo-generated electrons, prolonging their lifetimes, and suppressing the recombination of electron–hole pairs during the photoelectrochemical reaction was undoubtedly unraveled. As a result, the Au–NFO-based photoelectrochemical sensing platform presented high sensitivity, satisfactory repeatability, excellent anti-interference ability, and good practical applicability toward the detection of PCM. We hope that this study will provide researchers with a unique approach to improve the further exploration of diverse and advanced plasmonic metal/semiconductor hetero-nanostructure and develop more high-performance photoelectrochemical nanosensors.
2. Experimental procedures
2.1. Chemicals
Nickel dichloride hexahydrate (NiCl2·6H2O), ferric chloride hexahydrate (FeCl3·6H2O), gold(III) chloride hydrate (HAuCl4·H2O), sodium citrate (Na3C6H5O7), and sodium hydroxide (NaOH) were purchased from Xilong Scientific Co., Ltd. Potassium ferricyanide (K3Fe(CN)6), potassium ferrocyanide (K3Fe(CN)6), potassium dihydrogen phosphate (KH2PO4), and dibasic sodium phosphate (Na2HPO4), potassium chloride (KCl), and sodium chloride (NaCl) were purchased from Merck KGaA, Germany. The synthetic standard of PCM analyte was supplied by the National Institute of Drug Quality Control of Vietnam. All chemicals employed in this study were analytical grade and used without additional refinement.2.2. Synthesis of Au–NFO HNs and construction of Au–NFO-modified SPE electrode
The NFO HNs were synthesized by a one-pot hydrothermal method. Typically, 0.594 g of NiCl2·6H2O and 0.676 g of FeCl3·6H2O (molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were dispersed in 80 mL distilled water, followed by stirring for 1 hour until creating a homogeneous solution. Then, 10 mL of 2 M NaOH solution was added to the previous mixture drop by drop and the mixture was stirred for an additional 30 min. After that, the resulting mixture was transferred into a Teflon container, sealed, and heated in an oven at 180 °C for a duration of 2 hours. After cooling down to room temperature, the resulting precipitate was thoroughly washed with deionized water and absolute ethanol to remove impurities, dried in a vacuum oven at 80 °C overnight to obtain NFO HNs. For the preparation of Au–NFO HNs, 100 mg of the obtained NFO HNs was dissolved in 100 mL distilled water containing 10 mg HAuCl4·H2O, followed by ultrasonication in the sonication bath for 30 min until creating a homogeneous solution. Subsequently, 0.1 wt% Na3C6H5O7 was added and this mixture was maintained at 80 °C under stirring for 1 hour and the precipitate was centrifuged for 20 min at 5000 rpm and filtered, washed to remove any residual material. Finally, the obtained product was naturally dried in an oven at 80 °C for a period of 24 h to obtain the Au–NFO HNs.
:1) were dispersed in 80 mL distilled water, followed by stirring for 1 hour until creating a homogeneous solution. Then, 10 mL of 2 M NaOH solution was added to the previous mixture drop by drop and the mixture was stirred for an additional 30 min. After that, the resulting mixture was transferred into a Teflon container, sealed, and heated in an oven at 180 °C for a duration of 2 hours. After cooling down to room temperature, the resulting precipitate was thoroughly washed with deionized water and absolute ethanol to remove impurities, dried in a vacuum oven at 80 °C overnight to obtain NFO HNs. For the preparation of Au–NFO HNs, 100 mg of the obtained NFO HNs was dissolved in 100 mL distilled water containing 10 mg HAuCl4·H2O, followed by ultrasonication in the sonication bath for 30 min until creating a homogeneous solution. Subsequently, 0.1 wt% Na3C6H5O7 was added and this mixture was maintained at 80 °C under stirring for 1 hour and the precipitate was centrifuged for 20 min at 5000 rpm and filtered, washed to remove any residual material. Finally, the obtained product was naturally dried in an oven at 80 °C for a period of 24 h to obtain the Au–NFO HNs.
The simple multiple drop-casting method was used to prepare the Au–NFO-modified SPE electrode. Firstly, 1 mg Au–NFO HNs powder were ultrasonically dispersed in 1 mL of distilled water for 3 h to obtain a homogeneous suspension. Subsequently, a 2 μL amount of the resulting Au–NFO suspension was coated on the working electrode surface of the bare SPE and dried naturally. Eventually, the Au–NFO-modified SPE electrode was rinsed carefully with PBS solution and stored at 4 °C for further use.
2.3. Electrochemical assessments and characterization of Au–NFO HNs
Electrochemical experiments including cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), chronoamperometry (CA) and differential pulse voltammetry (DPV) were all carried out using PSTrace 5.8 software with a PalmSens 4 electrochemical workstation (PS Trace, PalmSens, The Netherlands). The commercially screen-printed carbon electrode was utilized throughout the electrochemical experiments with carbon working electrode, a carbon counter electrode, and a silver/silver chloride (Ag/AgCl) reference electrode.Powder X-ray diffraction (XRD, Bruker D5005 X-ray Diffractometer, Cu Kα radiation (λ = 0.15405 nm)), scanning electron micrographs (SEM, field emission scanning electron microscopy FESEM, S-4800), and UV-vis absorbance spectra (Macherey-Nagel spectrophotometer nanocolor UV-vis II within a wavelength range from 300 to 900 nm) were conducted in order to characterize the structural, morphological, and optical properties of the as-synthesized NFO and Au–NFO HNs.
3. Results and discussion
3.1. Physical characterization of NFO–Au HNs
Firstly, a powder X-ray diffractometer is used to get information about the crystalline structure, phase composition and crystallite size of as-synthesized Au–NFO HNs. XRD patterns of Au–NFO sample is shown in Fig. 1a, revealing the coexistence of two different crystalline phases in the Au–NFO HNs. Six characteristic diffraction peaks at 30.1°, 35.5°, 43.1°, 53.6°, 57.1°, and 62.8° are indexed to (220), (311), (400), (422), (511), and (440) planes can be clearly seen, which was in agreement with the cubic spinel phase of NiFe2O4 with the space group Fd3m. This XRD pattern is well matched with the Joint Committee on Powder Diffraction Standards (JCPDS) card no. 10-0325, illustrating trevorite phase of NiFe2O4. Some secondary peaks at 2θ = 38.1°, 44.1°, and 64.4° coincided with the diffraction planes of (111), (200), and (220) were observed, indicating a typical crystalline face-centered cubic (fcc) structure of Au (JCPDS card no. 04-0784). The result verifies the existence of NFO and Au in the as-prepared Au–NFO HNs. Furthermore, the average grain sizes of NFO and Au were calculated to be 20.4 and 10.8 nm, respectively, obtained by the standard Scherrer equation:31
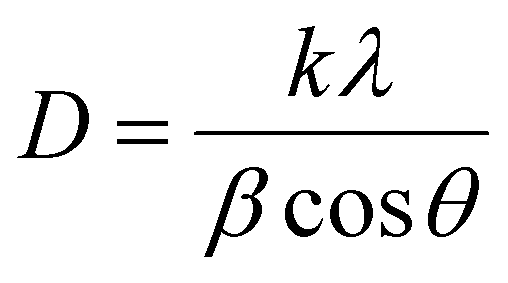 | (1) |
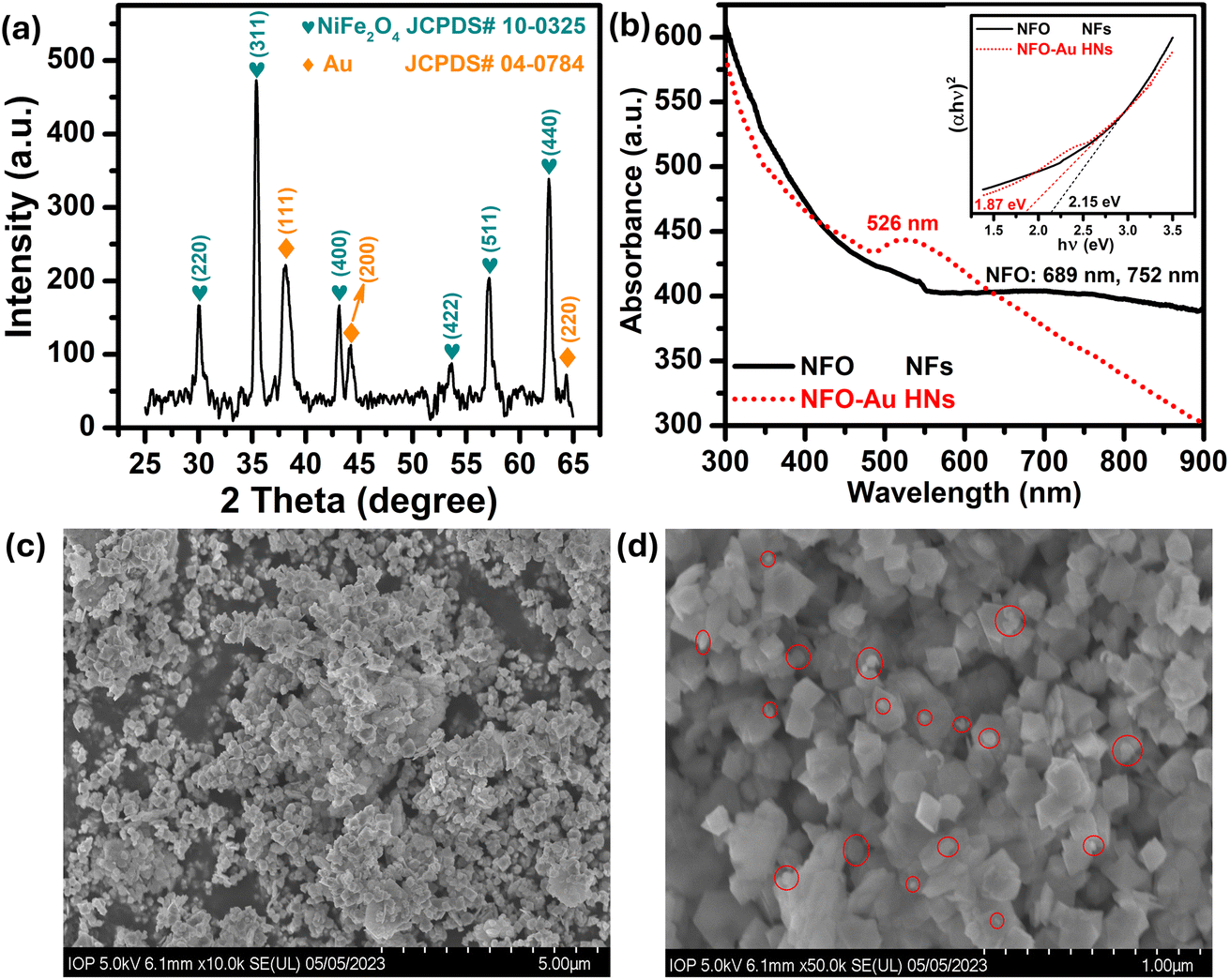 | ||
| Fig. 1 (a) XRD diffractogram and (b) UV-vis absorption spectra of pure NFO and Au–NFO HNs with inset showing the Tauc plot and band gap value of the NFO and Au–NFO HNs; (c and d) FE-SEM images with different magnification of the Au–NFO HNs. | ||
The optical properties of the prepared Au–NFO HNs is an essential factor in the visible light-assisted electrochemical sensor. Therefore, ultraviolet-visible (UV-vis) measurements of pure NFO and Au–NFO HNs through the wavelength range from 300 to 900 nm were conducted to determine light absorption characteristics and band gap properties and the results were shown in Fig. 1b. As for pure NFO, the spectrum displayed a broad absorption band in the wavelength range from 600 nm to 900 nm. To clarify the presence of possible peaks, fitting the Gaussian function was conducted in Origin 9.0 (Fig. S1†). The blue and pink lines represent a correlated Gaussian fit of these data. It can be found that the two Gaussian curves fitted the original curve perfectly with two peaks at 689 and 752 nm which can be assigned to d–d transitions.32 As compared to pure NFO, a new absorption peak at 526 nm was observed in the UV-vis absorption spectra of the as-prepared Au–NFO HNs. This peak can be attributed from the localized surface plasmon resonance (LSPR) characteristics of Au nanoparticles (NPs).32 The appearance of two kinds of characteristic absorption bands further confirms that the Au–NFO sample is composed of NFO nanoflakes and Au nanoparticles. The optical band gap of pure NFO and Au–NFO HNs has been evaluated from the absorption spectrum using the Tauc's equation:
| (αℏν)1/n = C(ℏν − Eg) | (2) |
The morphology, surface characteristics and particle size of the Au–NFO HNs was confirmed using FE-SEM analysis different magnification of 5 and 1 μm; the corresponding images are shown in Fig. 1c and d. The low-magnification FE-SEM image shows that the as-obtained Au–NFO HNs have homogeneous particle size, uniform distribution, and robust network structure. The size distribution analysis of Au and NFO in the Au–NFO HNs from SEM images was performed using the widespread image analysis software ImageJ (National Institutes of Health, Bethesda, Maryland, USA) and their average diameters were determined by Lorentz fitting of size histograms. The high-magnification FE-SEM image revealed that the NFO has a homogeneous population of flake-shaped particles with an average diameter in the range of 65 to 203 nm. Interestingly, it was found that small spherical-like Au NPs (marked by the red circle) with an average size of less than 35 nm were tightly coupled and sparingly scattered over the surface of flake-like NFO. This observation further suggests the successful formation of the heterostructural system of the as-prepared Au–NFO HNs and the strong interactions between two components.
3.2. Electrochemical properties of unmodified and Au–NFO-modified SPE electrodes
In order to obtain insight into the electrochemical redox properties and electrocatalytic activity of the bare SPE and Au–NFO-modified SPE, CV and EIS measurements were conducted in 0.1 M KCl solution containing 5 mM [Fe(CN)6]3–/4– (Fig. 2). As shown in Fig. 2a, two pairs of well-defined redox peaks are observed in the CV curves of the bare SPE and Au–NFO/SPE, corresponding to the one-electron transfer process of Fe2+ reversibly into Fe3+. The rectangular-like shape of CV curves together with reversible peaks verify the combination of pseudo-capacitance behavior and double electric layer effect through reversible redox reaction of [Fe(CN)6]3–/4–.33 For unmodified electrode, the anodic and cathodic peak currents (ΔIpa and ΔIpc) were calculated to be about 129.0 and 138.9 μA, respectively (Fig. 2b). The CVs of Au–NFO-modified SPE showed an augmentation in both ΔIpa and ΔIpc values (166.4 and 167.2 μA), which were 1.29 and 1.20 times larger than unmodified SPE counterpart, respectively. More interestingly, it is noteworthy that the ratio of anodic and cathodic peak currents of the bare SPE was 0.929, and with the introduction of Au–NFO HNs onto working electrode of SPE, ΔIpa/ΔIpc value was almost equal to unity. Furthermore, owing to the presence of Au–NFO HNs on the electrode surface, the onset potential value of Au–NFO/SPE (71.8 mV) was found to shift towards a more negative potential compared to the bare SPE (81.2 mV). The surface area under the CV curves was determined according to the equation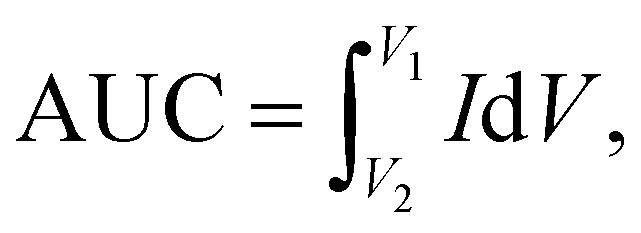 where AUC stands for the absolute area under the curve (AV), V2–V1 stands for the potential range, and I stands for the response current (A).34 The CV absolute area of the bare SPE and Au–NFO/SPE were calculated to be 1 × 10−4 and 1.05 × 10−4 AV. These obtained results indicate the modification of the bare SPE with Au–NFO HNs can accelerate the electrochemical reaction and the electron transfer rate.
where AUC stands for the absolute area under the curve (AV), V2–V1 stands for the potential range, and I stands for the response current (A).34 The CV absolute area of the bare SPE and Au–NFO/SPE were calculated to be 1 × 10−4 and 1.05 × 10−4 AV. These obtained results indicate the modification of the bare SPE with Au–NFO HNs can accelerate the electrochemical reaction and the electron transfer rate.
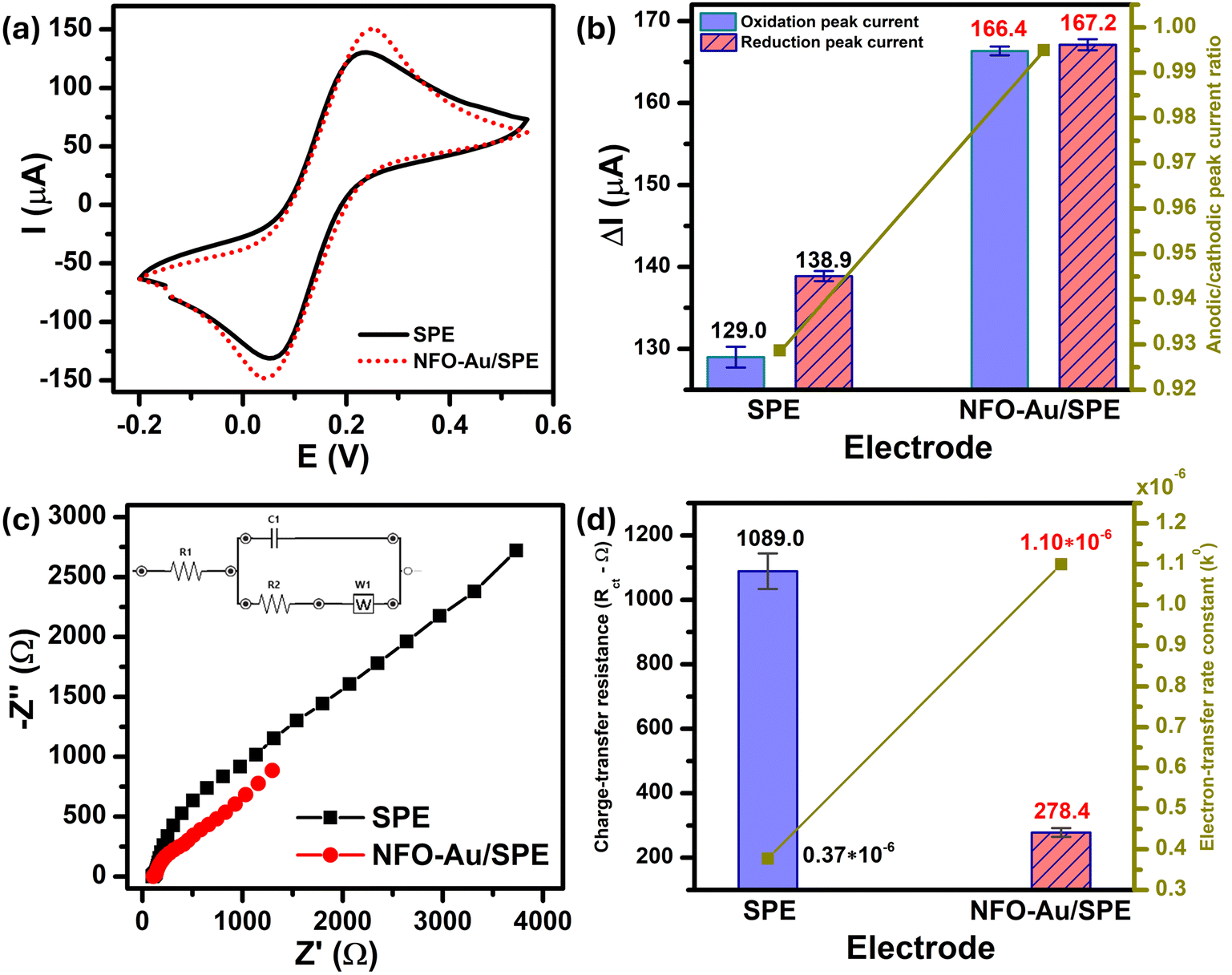 | ||
| Fig. 2 (a) CV curves of the bare SPE and Au–NFO/SPE at a scan rate of 50 mV s−1 in 0.1 M KCl solution containing 5 mM [Fe(CN)6]3–/4– and (b) the corresponding bar chart diagram of the oxidation/reduction peak currents with error bars and peak current ratio; (c) Nyquist plots and (d) the corresponding bar chart diagram of charge-transfer resistance (Rct) and the heterogeneous electron-transfer rate constants (k0) value of the bare SPE and Au–NFO/SPE in 0.1 M KCl solution containing 5 mM [Fe(CN)6]3–/4–. Inset (c): the diagram of Randles equivalent circuit model. | ||
To further confirm the intrinsic electrocatalytic activity of Au–NFO HNs, one of the essential factors governing the electrochemical sensing performance, the electrochemically active surface area (ECSA) was determined. Fig. S2† depicts CV curves of the bare SPE and Au–NFO/SPE in 0.1 M KCl solution containing 5 mM [Fe(CN)6]3–/4– at different scan rates from 10 to 60 mV s−1 and linear plots for square root of scan rate vs. anodic/cathodic peak currents. It can be seen that with an increment in the scan rate, the ΔIpa and ΔIpc values increased in both unmodified and modified electrodes. Furthermore, an excellent linear dependence between the redox current intensity and the square root of the scan rate was observed, demonstrating that the processes at both electrodes are diffusion controlled.35,36 Its linear regression equations were expressed as:
For SPE:
| ΔIpa (μA) = 372.8ν1/2 (V s−1) + 42.0/R2 = 0.97 |
| ΔIpc (μA) = −445.1ν1/2 (V s−1) − 33.4/R2 = 0.98 |
For Au–NFO/SPE:
| ΔIpa (μA) = 582.2ν1/2 (V s−1) + 27.3/R2 = 0.98 |
| ΔIpc (μA) = −595.6ν1/2 (V s−1) − 26.7/R2 = 0.98 |
The ECSA values of the bare SPE and Au–NFO/SPE were determined by utilizing the Randles–Sevcik equation,37 which describes the relationship between anodic/cathodic peak currents (ΔIp, μA), square root of scan rate (ν1/2, V s−1), number of transferred electrons (n), the coefficient of diffusion of [Fe(CN)6]3–/4– redox probe (D, cm2 s−1), the bulk concentration of [Fe(CN)6]3−/4− solution (C, mol cm−3), and electrochemically active surface area (ECSA, cm2). This equation is explained as:
| ΔIp = 2.69 × 105n3/2D1/2ACν1/2 | (3) |
According to that, the ECSA value of Au–NFO-modified electrode was 0.18 cm2, which is much higher than that of unmodified electrode (0.13 cm2). In general, the larger the active surface area, the higher the enrichment efficiency of target analytes.38,39 This finding suggests that the Au–NFO-modified SPE is poised to possess excellent electron and mass transport at the electrode/electrolyte interface, resulting in superior electrocatalytic performance.
EIS is a powerful tool in electrochemical analysis for the determination of the charge transfer resistance, the standard heterogeneous electron transfer rate constant, and the exchange current density as it gives some information about the electron-transfer kinetics for the analyte at the interface of the electrode surface-electrolyte solution during the redox reactions.40 Fig. 2c shows the Nyquist plots of the bare SPE and Au–NFO/SPE in 0.1 M KCl solution containing 5 mM [Fe(CN)6]3–/4– in the frequency range between 100 kHz and 0.01 Hz with an excitation signal of 10 mV amplitude. Evidently, while the bare SPE presents a large semi-circle at high-frequency region, indicative of a high charge transfer resistance, the presence of Au–NFO HNs onto the working electrode markedly reduced the impedance of SPE. Employing the conventional Randles equivalent circuit, the charge transfer resistance (Rct) values of the bare SPE and Au–NFO/SPE were determined (Fig. 2d). As illustrated in Fig. S3a,† the unmodified electrode exhibits the high radius with Rct value of 1089 Ω due to slow heterogeneous electron transfer and indirect electron transfer ability. Upon the incorporation of Au–NFO HNs at the SPE working surface, a notably lower Rct value of 278.4 Ω was obtained, showing obvious enhancement in the electrical conductivity of Au–NFO/SPE (Fig. S3b†). Furthermore, the heterogeneous electron-transfer rate constant (k0) and the exchange current density (j0) values for the bare SPE and Au–NFO/SPE can be calculated from the obtained ECSA and Rct values using the equation:36,39
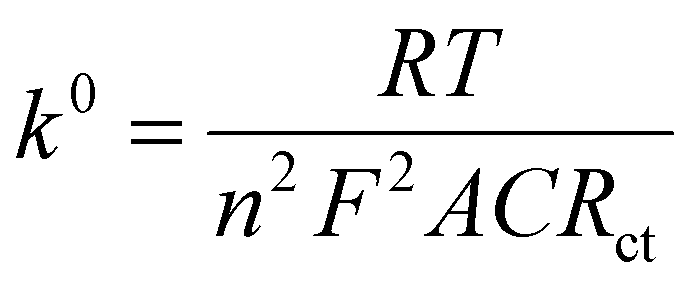 | (4) |
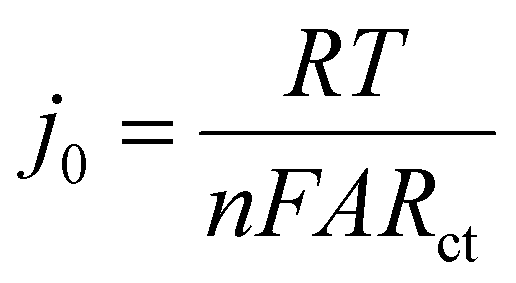 | (5) |
485.33 C mol−1). It was observed that the k0 and j0 values for the Au–NFO/SPE (1.1 × 10−6 cm s−1 and 5.3 × 10−4 A cm−2) were 2.97 and 2.94 times greater than the bare SPE (0.37 × 10−6 cm s−1 and 1.8 × 10−4 A cm−2), respectively.
Based on the visible-light-response property of Au–NFO HNs and the CV and EIS results in [Fe(CN)6]3–/4– redox probe, it is expected that the activation of Au–NFO HNs by visible light irradiation together with large ECSA and predefined electron transfer pathway may provide a rapid charge separation of photogenerated electron–hole pairs and efficient interfacial charge transfer, which is a key factor in the visible light enhanced photoelectrochemical sensing performance.
3.3. Photoelectrochemical redox reaction of PCM at Au–NFO-modified SPE electrodes
In order to investigate the influence of visible light on the electrochemical redox behavior of PCM at Au–NFO/SPE, CV measurement was conducted in 0.1 M PBS (pH 7.0) solution containing 50 μM PCM at a scan rate of 50 mV s−1 under dark environment and visible light irradiation, and the results are presented in Fig. 3. The Au–NFO-modified electrode in the presence of visible light irradiation exhibits a relatively high background current intensity, suggesting that visible light-assisted-Au–NFO/SPE has a higher electrochemical response as compared to under the dark condition (Fig. 3a). Moreover, it is clearly seen that the Au–NFO/SPE possesses electrocatalytic activities toward the redox reaction of PCM with an anodic potential at around 0.2 V vs. Ag/AgCl corresponds to the electro-oxidation of PCM to N-acetyl-p-benzoquinone-imine (NAPQI) and a cathodic peak at around 0.3 V vs. Ag/AgCl corresponds to the electro-reduction of NAPQI to PCM.41,42 The calculated peak-to-peak separation potential value, ΔEp = |Epa − Epc| of 105 mV, was greater than the theoretically predicted (59/n) mV for a reversible electrochemical process. This observation is indicative of a quasi-reversible behavior of the PCM electrochemical redox reaction on Au–NFO-modified electrode.36,41 More interestingly, when the modified electrode is shined by visible light during the process of detecting PCM, the anodic and cathodic peak current is significantly enhanced compared to that without visible light irradiation. As shown in Fig. 3b, the ΔIpa and ΔIpc values in the presence of visible light irradiation were 1.41 and 1.19 times higher than in the dark, respectively. Furthermore, the electron transfer kinetic parameters such as electron transfer coefficient (α) and apparent charge transfer rate constant (ks) can be determined using eqn (5)–(7):43,44
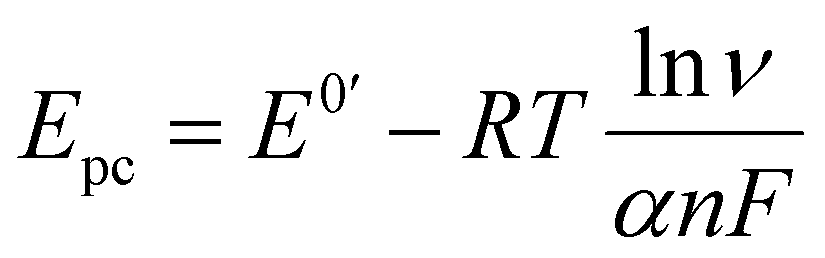 | (6) |
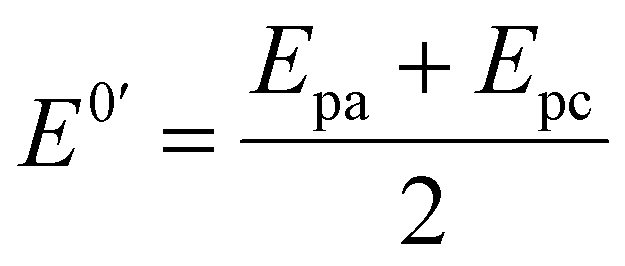 | (7) |
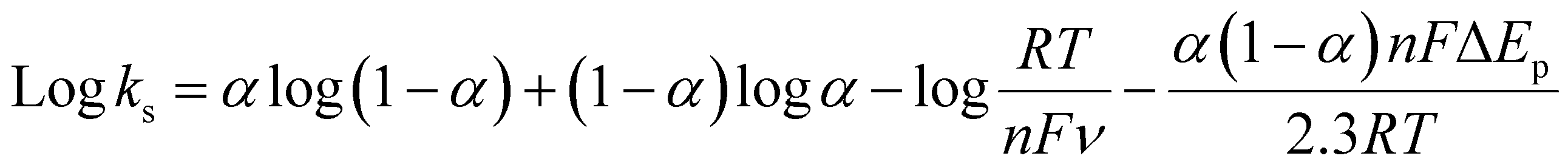 | (8) |
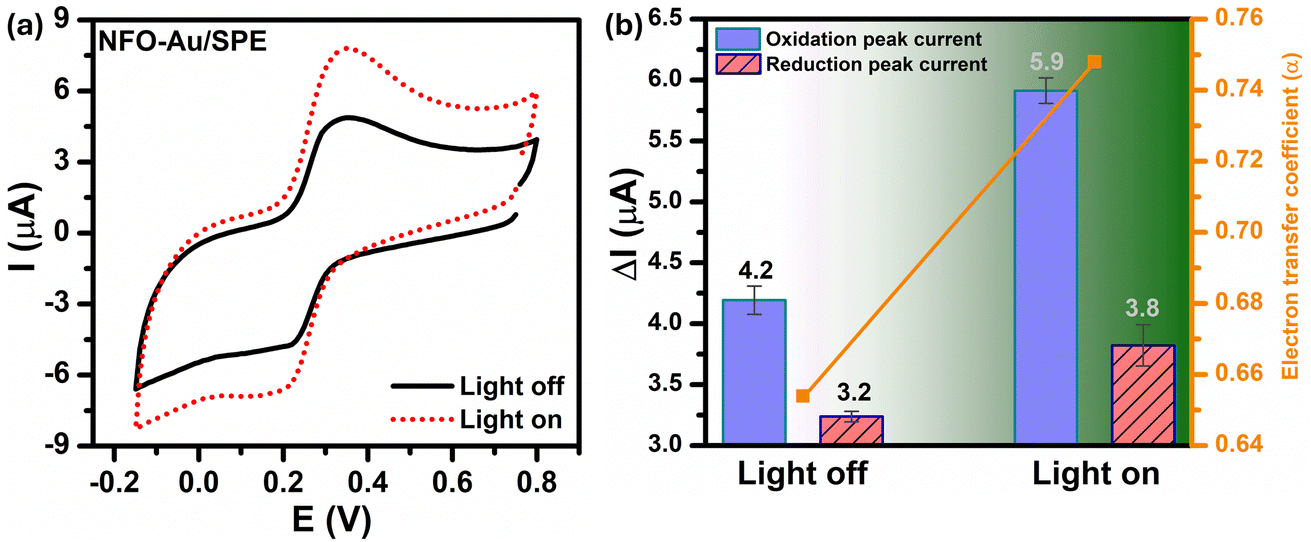 | ||
| Fig. 3 (a) CV curves and (b) the corresponding bar diagram of oxidation/reduction peak currents and the electron transfer coefficient of Au–NFO/SPE in 0.1 M PBS (pH 7.0) solution containing 50 μM PCM in the absence and presence of visible light. The scan rate of CV is 50 mV s−1. | ||
To further quantitatively distinguish the relative contribution of the surface-controlled kinetic and diffusion-controlled processes, CV analyzes of Au–NFO/SPE at various scan rates from 10 to 60 mV s−1 in the absence and presence of visible light were conducted (Fig. 4). As illustrated in Fig. 4a and d, the electrochemical oxidation and reduction processes of PCM at Au–NFO/SPE in the absence and presence of visible light were substantially enhanced upon increasing the scan rate. In both cases, the anodic and cathodic peak current of PCM increased with the increasing scan rate. Surprisingly, linear relationships were observed for both redox peak currents vs. scan rate and vs. square root of scan rate under dark environment (Fig. 4b and c) and visible light irradiation (Fig. 4e and f). This result suggested that the quasi-reversible electron transfer reaction on the Au–NFO-modified electrode is controlled by the mixing of diffusion and adsorption processes, which was consistent with the redox behavior of PCM in the previous studies.36,45 According to Laviron's theory, the calculated slope value between the anodic/cathodic peak currents and the scan rate is equal to 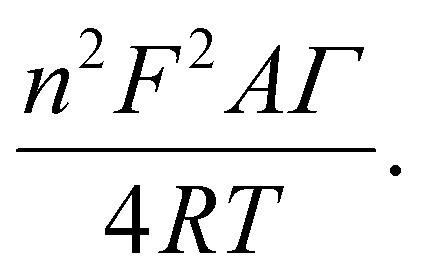 Therefore, the adsorption capacity (Γ) of PCM analyte in the absence and presence of visible light were calculated to be about 2.3 × 10−7 and 4.1 × 10−7 mol cm−2, respectively. The Au–NFO-modified electrode shows the higher electron transfer coefficient, apparent charge transfer rate constant, and adsorption capacity under the visible light irradiation compared to those of in the dark environment, which suggests that the introduction of external visible light provides the improved PCM analytical performance.
Therefore, the adsorption capacity (Γ) of PCM analyte in the absence and presence of visible light were calculated to be about 2.3 × 10−7 and 4.1 × 10−7 mol cm−2, respectively. The Au–NFO-modified electrode shows the higher electron transfer coefficient, apparent charge transfer rate constant, and adsorption capacity under the visible light irradiation compared to those of in the dark environment, which suggests that the introduction of external visible light provides the improved PCM analytical performance.
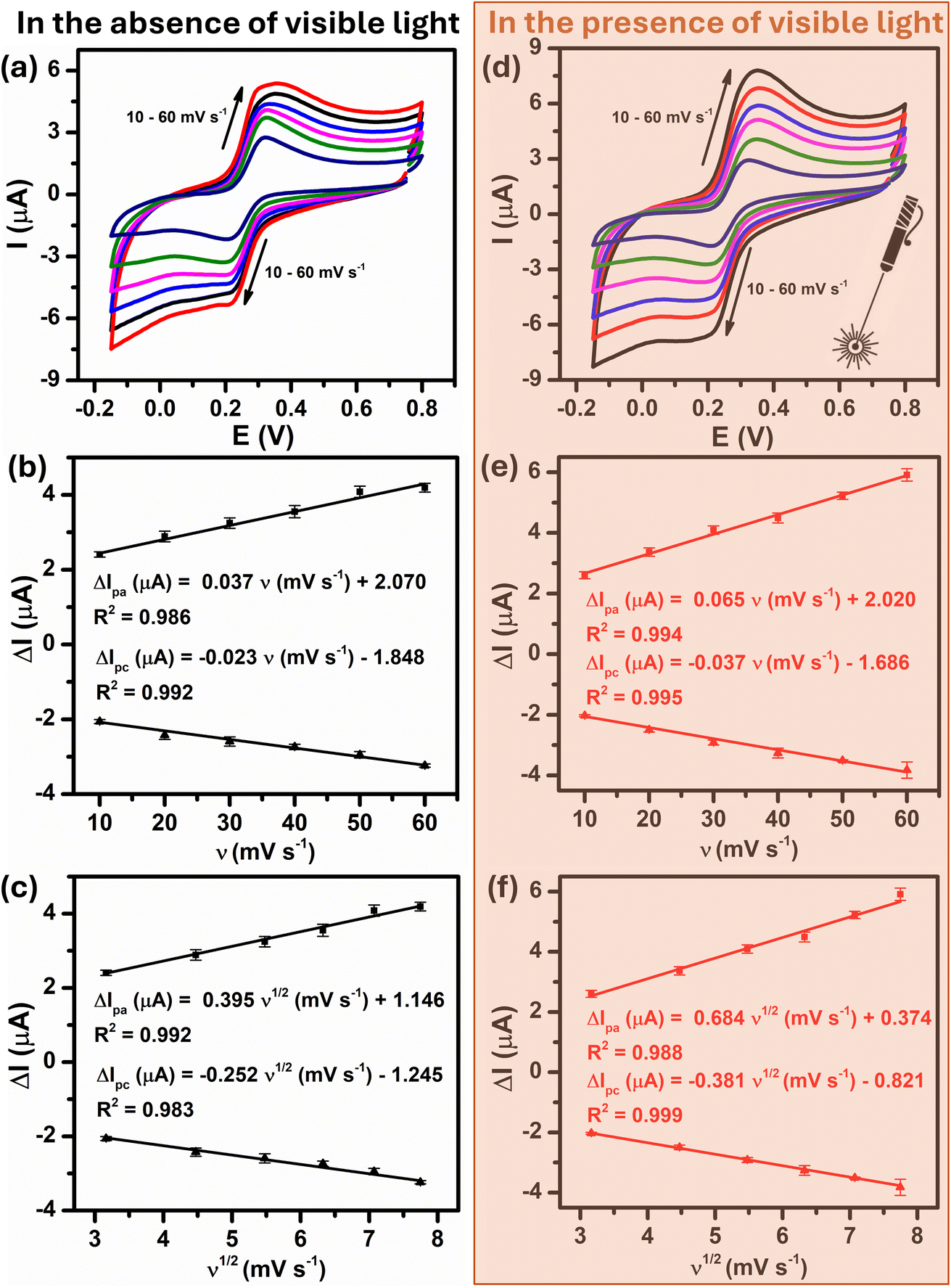 | ||
| Fig. 4 CV curves of Au–NFO/SPE recorded at various scan rates from 10 to 60 mV s−1, the corresponding calibration plots of oxidation/reduction peak current vs. scan rate and the square root of scan rate in the absence (a–c) and in the presence (d–f) of visible light, respectively. | ||
In order to get more information about the electrocatalytic activity of PCM analyte at Au–NFO-modified electrode in the absence and presence of visible light irradiation, chronoamperometry (CA) was used to evaluate the diffusion coefficient (D) and the catalytic rate constant (kcat). For the determination of D values, CA measurements of 25, 50, 100, 150, and 200 μM PCM in 0.1 M PBS (pH 7.0) under dark and visible light conditions were recorded with the potential of the working electrode being set at 250 mV vs. Ag/AgCl, as shown in Fig. 5a and c. As can be observed, the current responses of the PCM electrochemical oxidation increase with the increasing the concentration of PCM, and with the extension of the catalysis time, the transient current gradually decayed. The plot of catalytic current (Icat) to different concentration of PCM vs. negative square root of time (t−1/2) presented a good level of linearity in both dark and visible light conditions (Fig. 5b and d). Meanwhile, an excellent linear dependence was seen between the slopes of the resulting straight lines and the corresponding PCM concentration. The linear regression equations are expressed as follow:
| SlopeI–t−1/2 (μA s−1/2) = 0.011CPCM (μM) + 0.410/R2 = 0.994 (in the absence of visible light irradiation) |
| SlopeI–t−1/2 (μA s−1/2) = 0.016CPCM (μM) + 0.527/R2 = 0.998 (in the presence of visible light irradiation) |
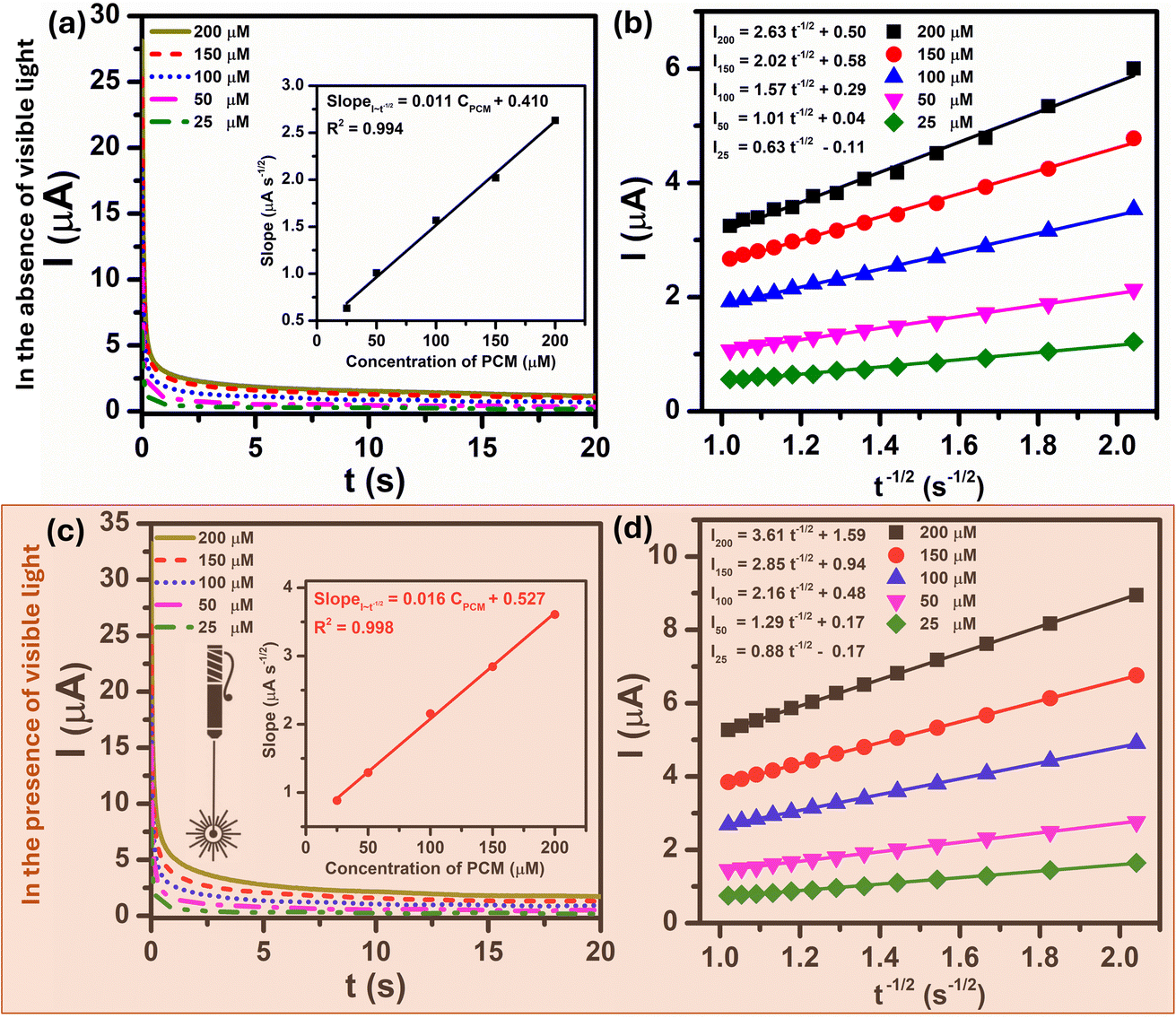 | ||
| Fig. 5 CA curves of Au–NFO/SPE in 0.1 M PBS (pH 7.0) solution containing different concentrations of PCM (25, 50, 100, 150, and 200 μM) and plot of I vs. t−1/2 in the absence (a and b) and presence (c and d) of visible light, respectively. Inset the plot of the slope of the straight lines against PCM concentration. | ||
The diffusion coefficient (D) in the electro-oxidation process of PCM at Au–NFO/SPE under dark environment and visible light irradiation was obtained according to the Cottrell equation:37
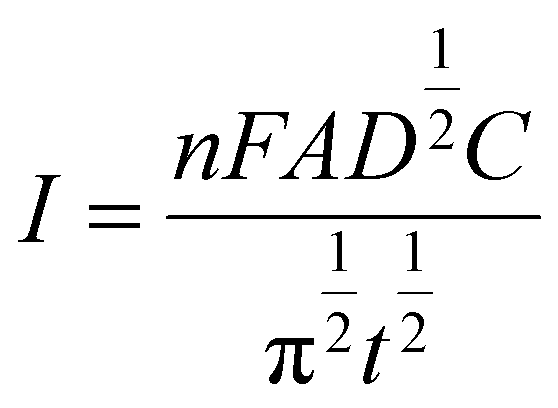 | (9) |
From the Cottrell equation and slope of the straight line shown in inset Fig. 5a and c, the average diffusion coefficient values of PCM in the absence and presence of visible light were calculated as 0.56 × 10−7 and 1.01 × 10−7 cm2 s−1, respectively.
The catalytic rate constant (kcat) for the electro-oxidation reaction between PCM and Au–NFO/SPE in the absence and presence of visible light irradiation can be also determined by using CA technique, according to the Galus method:37
| IC/IL = π1/2(kcatCbt)1/2 | (10) |
Fig. S4a† shows chronoamperograms recorded at Au–NFO-modified electrode in 0.1 M PBS and 0.1 M PBS containing 200 μM PCM under dark environment and visible light condition. At the potential of the working electrode of 250 mV vs. Ag/AgCl, the presence of visible light irradiation led to an increase in the catalytic current of PCM oxidation. Plots of the ratio of catalytic current to blank current (IC/IL) vs. the square root of time (t1/2) without and with the visible light yields two straight lines with slopes equal to π1/2(kcatCb)1/2 (Fig. S4b and c†), which can be expressed as:
| IC/IL = −81.78t1/2 + 344.9/R2 = 0.97 (in the absence of visible light irradiation) |
| IC/IL = −142.3t1/2 + 589.3/R2 = 0.97 (in the presence of visible light irradiation) |
As a result, the mean values of the catalytic rate constant (kcat) of Au–NFO/SPE for the electrocatalytic oxidation reaction of PCM under dark environment and visible light condition were estimated to be 1.06 × 107 and 3.22 × 107 M−1 s−1, respectively. To sum up, during the photoelectrocatalytic reaction, when the working surface of Au–NFO modified electrode is irradiated with visible light, the diffusion coefficient and the catalytic rate constant were 1.8 and 3 times higher than in the dark condition, respectively, indicating a substantial impact and a strong electrocatalytic effect of visible light irradiation on the analytical sensing performance of the proposed Au–NFO-based electrochemical sensor.
3.4. Optimization of electrochemical parameters
The peak current response and peak potential strongly depend on the pH of the supporting electrolyte because of the involvement of protons in the overall electrode reaction. Therefore, to determine the optimal pH value of buffer solution for the PCM electro-oxidation using Au–NFO/SPE, differential pulse voltammograms (DPVs) were recorded in various pH values from acidic to basic values. As shown in Fig. 6a, as there is an increment on pH value from 3.0 to 11.0, the oxidation peak potential of PCM shifted slightly towards less positive potentials, which indicated that protons have taken part in the electrode reaction processes. Fig. 6b summarizes the plots between peak current response vs. pH value (black plot) and peak potential vs. pH value (red plot). With an elevation in pH from acidic to basic values (pH 3.0–7.0), the electro-oxidation peak current of PCM increases. In contrast, the electro-oxidation peak current of PCM decreases when the solution deviated from the neutral condition. Both acidic and alkaline conditions made the electrochemical oxidation reaction of PCM happen on the Au–NFO/SPE less favorable, which suggested that pH 7.0 was the most optimal pH for all subsequent analyses of PCM electrochemical determination.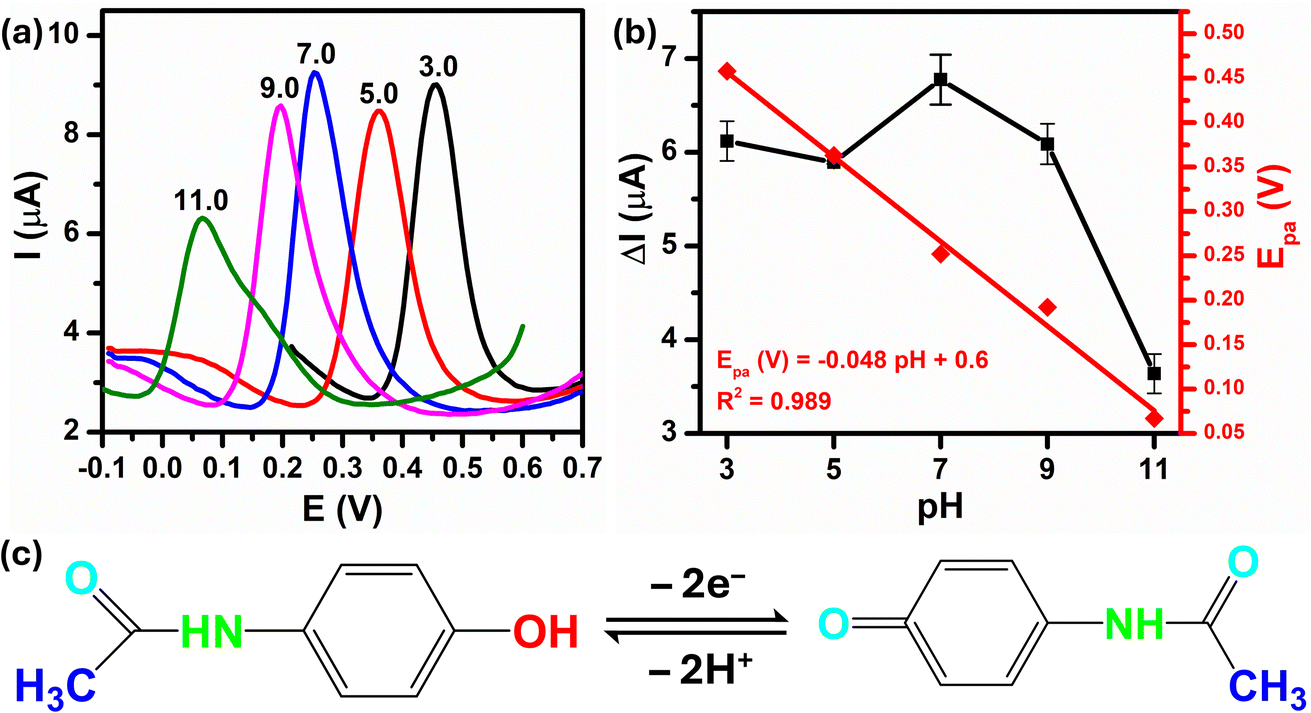 | ||
| Fig. 6 (a) DPV curves of 50 μM PCM at various pH values from 3.0 to 11.0 at Au–NFO/SPE, (b) the corresponding calibration plots of oxidation peak current responses and oxidation peak potentials against pH values, and (c) the electrochemical oxidation mechanism of PCM. | ||
More interestingly, the PCM oxidation peak potential became pH-independent within examined pH range with the linear relationship between the peak potential and the pH of the PBS electrolyte can be expressed as follows:
| Epa (V) = −0.048pH + 0.06/R2 = 0.989 |
The slope obtained was −0.048 V pH−1 which was nearer to the Nernst theoretical value −0.059m/n V pH−1 (m and n stand for number of proton and electron involved in electrochemical reaction), indicating the number of proton transfer is equal to the number of electron transfer. As the electro-oxidation of PCM is a two-electron process, the number of protons taking part in the electrode reaction was found to be two protons. Thus, the possible electrochemical oxidation reaction mechanism of PCM on the Au–NFO-modified electrode were in good agreement with some previously proposed mechanisms,36,42,46 as shown in Fig. 6c.
The influences of crucial factors, including pulse potential and pulse time on the analytical performance were also optimized using DPV technique at different Epluse values from 0.025 to 0.15 V and various tpluse values from 0.01 to 0.15 s (Fig. S5a and c†). The results in Fig. S5b† show that the anodic peak current of AZM increased up to a pulse potential of 0.125 V and substantially decreased for higher pulse potential value. It is clearly noticed from Fig. S5d† that when the pulse time value increased from 0.01 to 0.025 s, the anodic peak current of PCM was improved, and further increase in the pulse time value from 0.025 to 0.15 s resulting in a decrease in the anodic peak current. Therefore, 0.125 V and 0.025 s were selected as the optimum operating conditions for subsequent experiments.
3.5. Photoelectrochemical determination of PCM at Au–NFO-modified SPE electrodes
Under the optimized experimental conditions described above, the influence of visible light irradiation of the analytical sensing performance of Au–NFO/SPE towards PCM including linear range, limit of detection, and electrochemical sensitivity was investigated. Fig. 7a and c present the DPV curves for the PCM concentration ranges of 2.5–200 μM and 0.5–200 μM under the dark environment and visible light condition, respectively. From the figures, it is observed that the anodic peak current increased as the PCM concentration increased in both cases. Comparatively, the introduction of visible light into Au–NFO-based electrochemical sensor made the anodic peak current of the electro-oxidation at each PCM concentration much higher than the anodic peak current under the dark condition. As a result, the minimum detectable concentration of PCM using Au–NFO-modified electrode in the presence of visible light irradiation was 0.5 μM, which was 5 times lower than under the dark environment (2.5 μM). Simultaneously, Fig. 7b and d show that there are good linear correlations between the anodic peak current and the PCM concentration in the linear range from 2.5 to 100 μM in the absence of visible light and from 0.5 to 200 μM in the presence of visible light. The linear equations were described as following:| ΔIpa (μA) = 0.179CPCM (μM) + 0.337/R2 = 0.998 (in the absence of visible light irradiation) |
| ΔIpa (μA) = 0.196CPCM (μM) + 0.307/R2 = 0.999 (in the presence of visible light irradiation) |
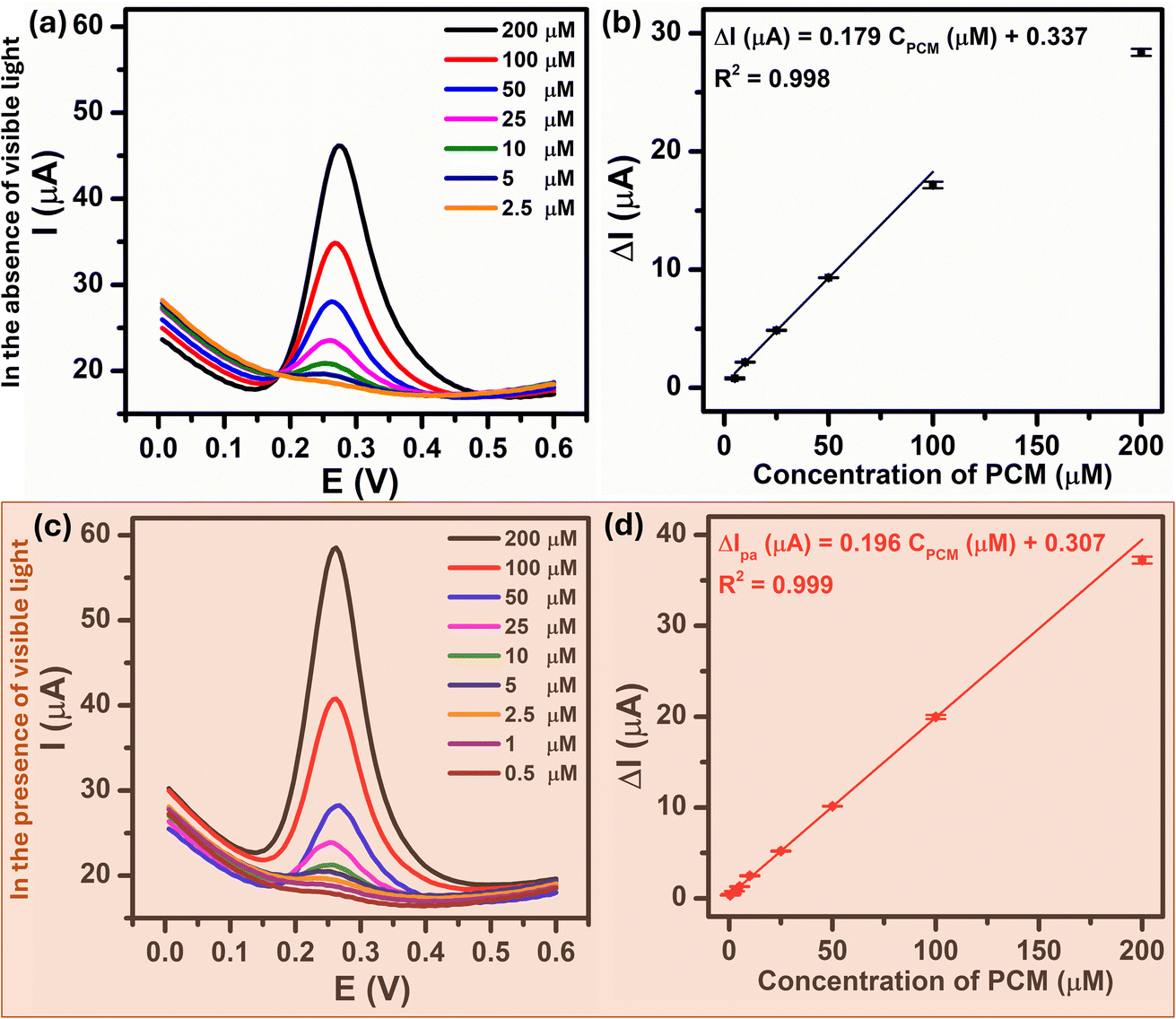 | ||
| Fig. 7 DPV curves of Au–NFO/SPE in 0.1 M PBS (pH 7.0) solution containing different concentrations from 0.5 to 200 μM in 0.1 PBS (pH 7.0) and the calibration plots between oxidation peak current responses vs. various concentrations of PCM in the absence (a and b) and presence (c and d) of visible light, respectively. | ||
By using IUPAC formula as follows 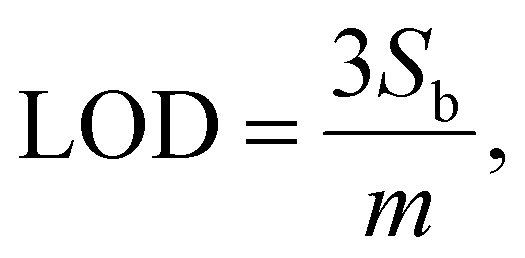 where Sb is the standard deviation of the blank solution m is the slope of the calibration curve, the detection limit of the Au–NFO-based electrochemical sensor for the detection of PCM under the dark environment and visible light condition were determined to be 1.85 and 0.37 μM, respectively. Furthermore, the electrochemical sensitivities were calculated to be 0.994 μA μM−1 cm−2 under the dark condition and 1.089 μA μM−1 cm−2 under visible light irradiation. These obtained values are compared with analytical parameters reported by other research groups for the electrochemical determination of PCM, and the corresponding results were summarized and listened in Table S1.† As expected, the Au–NFO-based electrochemical sensor with the assistance of visible light irradiation possesses comparable electroanalytical performance for the robust and efficient detection of PCM, suggesting that the proposed photoelectrochemical sensing platform has strong potential for the determination of other analytes.
where Sb is the standard deviation of the blank solution m is the slope of the calibration curve, the detection limit of the Au–NFO-based electrochemical sensor for the detection of PCM under the dark environment and visible light condition were determined to be 1.85 and 0.37 μM, respectively. Furthermore, the electrochemical sensitivities were calculated to be 0.994 μA μM−1 cm−2 under the dark condition and 1.089 μA μM−1 cm−2 under visible light irradiation. These obtained values are compared with analytical parameters reported by other research groups for the electrochemical determination of PCM, and the corresponding results were summarized and listened in Table S1.† As expected, the Au–NFO-based electrochemical sensor with the assistance of visible light irradiation possesses comparable electroanalytical performance for the robust and efficient detection of PCM, suggesting that the proposed photoelectrochemical sensing platform has strong potential for the determination of other analytes.
3.6. Mechanisms of Schottky barrier formation and the enhanced mechanism of proposed photoelectrochemical performance under visible light irradiation
Fig. 8a depicts a schematic illustration of the energy-band diagram of Au–NFO nanoheterostructures before and after Au NPs contacting with NFO NFs. According the previous works, the work functions of Au metal and NFO p-type semiconductor are 5.1 and 5.28 eV, respectively.47,48 On the other hand, the electron affinity (χS) in a semiconductor is defined as the energy difference between the lowest energy level in the conduction band and the vacuum level, and it equals 3.22 V for NFO.47,49 The band gap of NFO was calculated to be about 2.15 eV in the Characterization section. The degree of bending of the energy band of the semiconductor at the interface equals the work function difference between metal and semiconductor, VBB = |WM − WS|, where ϕM and ϕS are work functions of metal and semiconductor, respectively. According to that, the VBB value was estimated to be about 0.18 eV. Generally, when metal is intimately integrated with p-type semiconductor with their work functions satisfying ϕM > ϕS, a Schottky barrier will be established at the interface of metal and semiconductor to drive the transfer of electrons from the low work function material to high work function material.49–51 Therefore, in Au–NFO HNs case, when Au and NFO came into contact with each other, electrons flow from Au into NFO and the situation would be reversed for holes, thereby forming an internal electric field (also known as built-in electric field) in the interfacial region at the thermodynamic equilibrium. Meanwhile, after the contact between Au and NFO is made, since the Fermi energy level of NFO is lower than that of Au, the electron transfer process from Au to NFO will continue until the Fermi level on the two sides reach equilibrium and form a new Fermi energy level. Due to the formation of built-in electric field, the energy band edges of NFO are raised up and bent downward toward the interface, forming the Schottky barrier at the metal–semiconductor interface.49,52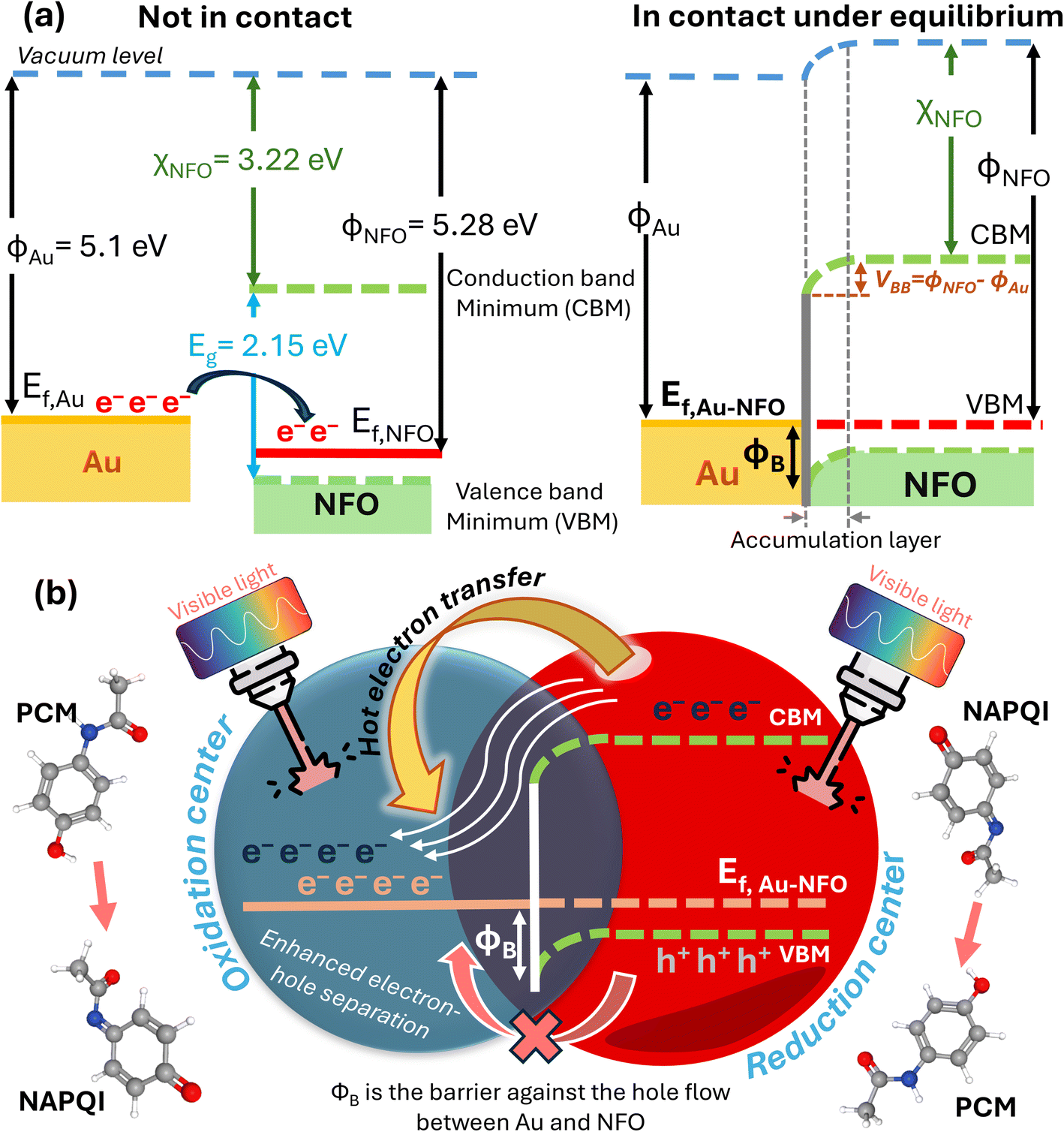 | ||
| Fig. 8 (a) Energy-band diagram of Au and NFO before and after physical contact with each other; (b) mechanism of the enhanced analytical performance of Au–NFO-based photoelectrochemical sensor for the detection of PCM under visible light irradiation. | ||
To propose the mechanism of the enhanced analytical sensing performance of the Au–NFO-based photoelectrochemical sensor under visible light irradiation, it is imperative to clarify the role of each components (metal and semiconductor) in the as-prepared nanoheterostructures. As a semiconductor, upon excitation by visible light irradiation with energy (hν) larger than or equal to bandgap (Eg) of NFO, the photogenerated electrons (e−) will be excited from the valence band (VB) to the conduction band (CB), simultaneously creating the same amount of holes (h+) in the VB, leading to the generation of electron–hole pairs.13,22 Generally, these negatively charged electrons at CB are not physically separate from positively charged holes at VB, most of the photogenerated electrons and holes tend to recombine in just several nanoseconds,22,53,54 thereby lowering the photoelectrochemical activity. For the case of Au NPs, due to the localized surface plasmon resonances (LSPR) of the noble metal NPs induced by the oscillating electric field of the incident light with a specific frequency, the electric polarization results in a large local electro-magnetic fields and enhanced light harvesting ability.20,25,55 Because metals have no band gap, so when Au NPs are irradiated with visible light, the LSPR absorption can couple the light flux to the conducting electrons to excite these electrons to higher energy states from the lower levels.20 The resulting energetic electrons can promote photoelectrochemical reactions through direct interaction with reactant molecules absorbed on the working electrode of modified electrode and activating them for oxidation or reduction reactions.
In this developed electrochemical sensor, judicious integration of Au with NFO at nanoscale for rationally constructing Au–NFO HNs, where both materials can be simultaneously excited by the light in the visible region affords the synergistic effect of internal electric field and Schottky barrier, making them ideal material for enhanced analytical sensing performance. Fig. 8b shows the proposed mechanism for the enhanced photoelectrochemical detection of PCM using Au–NFO HNs-modified electrode under visible light irradiation. Under simultaneous excitation, the photo-generated electrons from VB to CB in NFO are directly transferred to the adjacent Au because the CB edge of NFO lies higher in energy than the Fermi level of Au, allowing fast interfacial electron transfer. In this way, the electron transfer process from semiconductor to metal becomes energetically favorable and remains predominating, in which Au serves as an “electron sink” and an active reaction site for the accumulation of photo-generated electrons. Moreover, at the thermodynamic equilibrium, the established and enhanced internal electric field at the interface of Au and NFO could facilitate direct interfacial electron transfer and make the electron transfer rapidly. As a result, the recombination of photo-generated electrons and holes is suppressed and the lifetime of transferred hot electrons is substantially prolonged. Besides the photo-generated electrons from NFO, the resonant optical excitation of surface plasmons also generated energetic hot electrons.25,55,56 These plasmon-induced hot electrons of Au, together with the transferred photo-generated electrons from NFO to Au could initiate the electrochemical reduction reaction with the PCM analytes absorbed on the working electrode, concurrently contributing to improved photoelectrochemical activity.
For the case of holes, due to the localized Schottky barrier of the Au–NFO interface, it is very difficult for photo-generated holes on the VB of NFO to migrate to Au. Consequently, the photogenerated electron–hole recombination is hindered by the absence of electrons in NFO under visible light irradiation. As a result, the separated holes on the VB of NFO could promote the electrochemical oxidation reaction of PCM to NAPQI. Generally speaking, the coupling of the metal Au plasmon and semiconductor NFO exciton can build up built-in electric field and Schottky barrier at the interface, which could drive the photo-generated electrons and holes to migrate in different directions, thereby limiting the interaction and facilitating the superior separation of photogenerated carriers. The detailed kinetic parameters and analytical parameters of the Au–NFO-based electrochemical sensing platform under dark environment and visible light are shown in Table 1. On the basis of the above discussions, it can be concluded that the synergistic combination of Au NPs and NFO NFs would promote interfacial charge transfer, electrocatalytic activity, and large adsorption/diffusion capacity, which contribute to markedly improved photoelectrochemical performance for the detection of PCM of Au–NFO HNs.
| Analyte: [Fe(CN)6]3–/4– | |||
|---|---|---|---|
| Electrochemical kinetic parameters | Bare SPE | Au–NFO/SPE | Variance (%) |
| Anodic peak current (ΔIpa, μA) | 129.0 | 166.4 | 29.0 |
| Cathodic peak current (ΔIpc, μA) | 138.9 | 167.2 | 20.4 |
| Anodic/cathodic peak current ratio | 0.929 | 0.995 | 7.1 |
| Onset potential (mV) | 81.2 | 71.8 | 11.6 |
| Surface area under the CV curve | 1 × 10−4 | 1.05 × 10−4 | 5 |
| Electrochemically active surface area (ECSA, cm2) | 0.13 | 0.18 | 38.5 |
| Charge-transfer resistance (Rct, Ω) | 1089 | 278.4 | 291.2 |
| Heterogeneous electron-transfer rate constant (k0, cm s−1) | 0.37 × 10−6 | 1.1 × 10−6 | 197.3 |
| Exchange current density (j0, A cm−2) | 1.8 × 10−4 | 5.3 × 10−4 | 194.4 |
| Electrode: Au–NFO/SPE; analyte: PCM | |||
|---|---|---|---|
| Analytical parameters | Without visible light | With visible light | Variance (%) |
| Anodic peak current (ΔIpa, μA) | 4.2 | 5.9 | 40.5 |
| Cathodic peak current (ΔIpc, μA) | 3.2 | 3.8 | 18.8 |
| Electron transfer coefficient (α) | 0.65 | 0.75 | 15.4 |
| Apparent charge transfer rate constant (ks, s−1) | 2.07 | 2.30 | 11.1 |
| Adsorption capacity (Γ, mol cm−2) | 2.3 × 10−7 | 4.1 × 10−7 | 78.3 |
| Diffusion coefficient (D, cm2 s−1) | 0.56 × 10−7 | 1.01 × 10−7 | 80.4 |
| Catalytic rate constant (kcat, M−1 s−1) | 1.06 × 107 | 3.22 × 107 | 203.8 |
| Linear range (μM) | 2.5–100 | 0.5–200 | — |
| Electrochemical sensitivity (μA μM−1 cm−2) | 0.994 | 1.089 | 9.5 |
| Limit of detection (μM) | 1.85 | 0.38 | 386.9 |
3.7. Repeatability, anti-interference ability, and feasibility of the proposed photoelectrochemical sensor
In real-world condition, the presence of non-target metal ions, organic compounds, and many types of antibiotics can interfere with the analytical results of the PCM electrochemical detection. Therefore, the anti-interference ability of the Au–NFO-based photoelectrochemical sensing platform was performed using DPV technique with consecutive additions of 1000 μM of metal ions (K+, Na+, Fe2+, Fe3+, Co2+, and Cr6+) and antibiotic drugs (amoxicillin (AMX), erythromycin (ERM), azithromycin (AZM), furazolidone (FZD), chloramphenicol (CAP), and ofloxacin (OFX)) in the presence of 100 μM PCM (Fig. 9a). It can be clearly seen that the presence of the interfering metal ions and antibiotics caused slightly changes in the DPV background current but did not possess any interference peak within the potential range of 0.0 to 0.6 V. As shown in Fig. 9b, the relative error values, which was calculated by dividing the absolute error of ΔIpa value of 100 μM PCM in the presence of the interferers by the measured ΔIpa value of 100 μM PCM, were less than 8.8%, implying good anti-interfering capability of the Au–NFO-modified electrode for the electrochemical detection of PCM under visible light irradiation.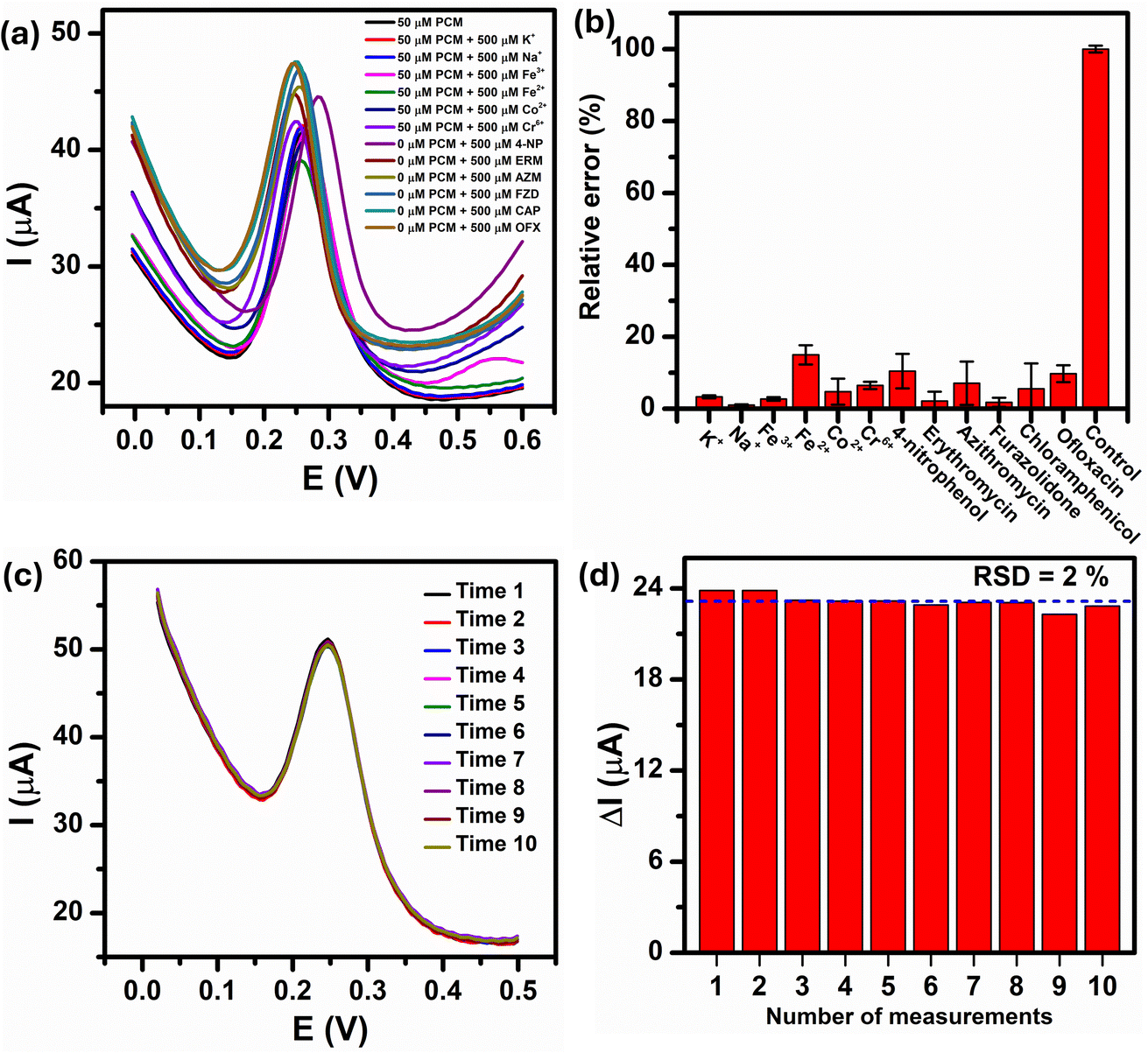 | ||
| Fig. 9 (a and b) Interference investigation of Au–NFO/SPE in 0.1 M PBS solution containing 100 μM PCM with 10-fold concentration of interference substances and (c and d) the repeatability performance of Au–NFO/SPE for 10 consecutive measurements in 0.1 M PBS solution containing 100 μM PCM. | ||
The electrochemical repeatability of the developed sensor was also scrutinized by measuring the anodic peak current response of PCM using DPV technique for 10 consecutive measurements, and the results were presented in Fig. 9c. The Au–NFO/SPE exhibited excellent antifouling properties under the visible light condition with a relative standard deviation (RSD) of 2.0% was obtained for 10 successive DPV measurements (Fig. 9d), indicating the good signal stability and repeatability of the visible light-assisted electrochemical sensor.
To evaluate the feasibility of the developed visible light assisted-electrochemical sensor, the Au–NFO/SPE were employed to determine PCM in human urine samples using the standard addition method. Initially, no anodic or cathodic peak currents were found in urine samples, indicating that the real samples were absolutely free of PCM or the concentrations of PCM were below the limit of detection. Therefore, the standard solutions of 5, 25, and 50 μM PCM were spiked into urine samples, and recorded anodic peak currents were compared with the values from the linear regression. The calculated results are summarized in Table S2.† The mean recoveries obtained from triplicate DPV measurements was in the range of 92.0 and 98.6% with the largest RSD value among all the measurements was 2.0%. The obtained satisfactory recovery and relative standard deviation results confirm the accuracy and feasibility of the proposed electrochemical sensor.
4. Conclusions
In summary, by taking advantage of the suitable energy-level matching between metal Au NPs and low bandgap semiconductor NFO NFs, we have successfully demonstrated the employment of Au–NFO nano-heterostructures-based electrochemical sensor to significantly improve the electrochemical sensing performance towards detection of PCM under visible light irradiation. The intimate contact of Au NPs with NFO NFs led to the formation of built-in electric field and Schottky barrier at the Au–NFO interface. Upon excitation by visible light irradiation, the built-in electric field together with the downward band bending caused the effective migration of photo-generated electrons from the CB of NFO to the Fermi level of Au, while the plasmon-induced hot electrons were also simultaneously generated on the outer surface of Au NPs. In the meantime, the Schottky barrier prevents the transfer of photo-generated holes from the VB of NFO to Au. As results, the recombination between photo-generated electrons, plasmon-induced hot electrons, and photo-generated holes is suppressed and the lifetime of charge carriers is prolonged, making them available for participating and promoting the electrochemical oxidation/reduction reaction. Through optimizing the detection conditions, the Au–NFO-modified electrode exhibited a high electrochemical sensitivity of 1.089 μA μM−1 cm−2, a wide linear sensing range from 0.5 to 200 μM, and a low detection limit of 0.38 μM. Furthermore, the visible light-assisted electrochemical sensor based on Au–NFO HNs showed satisfactory repeatability, high anti-interference ability, and good practical applicability with admire recovery results in human urine samples. This work will provide useful guidance for designing efficient plasmonic metal/semiconductor nano-heterostructures for improving electrochemical sensing performance under visible light irradiation, which has a broad application prospect in the field of environmental and pharmaceutical analysis.Data availability
The data supporting this study's findings are available from the corresponding author, Tuan Anh Nguyen upon reasonable request. Additionally, the data supporting this article have been included as part of the ESI.†Author contributions
N. T. Anh: conceptualization, methodology, validation, investigation, writing-original draft; N. T. Loan: validation, formal analysis; P. T. N. Mai: validation, methodology, investigation; P. D. Thang: methodology, formal analysis, writing-review & editing; V. N. Phan: validation, investigation; N. X. Dinh: methodology, supervision, formal analysis; T. V. Manh: validation, formal analysis; O. V. Hoang: validation, investigation; V. D. Lam: conceptualization, methodology, formal analysis; L. A. Tuan: conceptualization, methodology, supervision, project administration, writing-review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research is partially supported by the Ministry of Science and Technology of Vietnam under grant (ĐTĐL.CN-26/23), Physics Development Program 2020–2025. The authors would like to acknowledge the support for Raman, electrochemical & UV-vis measurements from NEB Lab (Phenikaa University) and SEM supports from IOP-VAST and XRD from VNU-Hanoi.References
- J. Wang and Z. Liu, TrAC, Trends Anal. Chem., 2020, 133, 116089 CrossRef CAS.
- J. Shu and D. Tang, Anal. Chem., 2019, 92, 363–377 CrossRef PubMed.
- W.-W. Zhao, J.-J. Xu and H.-Y. Chen, Chem. Soc. Rev., 2015, 44, 729–741 RSC.
- Y. Chen, W. Gu, C. Zhu and L. Hu, Anal. Chem., 2024, 96(22), 8855–8867 CrossRef CAS PubMed.
- Z. Qiu and D. Tang, J. Mater. Chem. B, 2020, 8, 2541–2561 RSC.
- Y. Zang, J. Lei and H. Ju, Biosens. Bioelectron., 2017, 96, 8–16 CrossRef CAS PubMed.
- C. V. Reddy, K. R. Reddy, N. P. Shetti, J. Shim, T. M. Aminabhavi and D. D. Dionysiou, Int. J. Hydrogen Energy, 2020, 45, 18331–18347 CrossRef CAS.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- B. Fang, Z. Xing, D. Sun, Z. Li and W. Zhou, Adv. Powder Mater., 2022, 1, 100021 CrossRef.
- F. Wang, Q. Li and D. Xu, Adv. Energy Mater., 2017, 7, 1700529 CrossRef.
- K. Rajeshwar and N. R. De Tacconi, Chem. Soc. Rev., 2009, 38, 1984–1998 RSC.
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang and P. Huo, Chin. J. Catal., 2022, 43, 178–214 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- S. K. Dutta, S. K. Mehetor and N. Pradhan, J. Phys. Chem. Lett., 2015, 6, 936–944 CrossRef CAS.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS.
- X. Gao, X. Liu, Z. Zhu, X. Wang and Z. Xie, Sci. Rep., 2016, 6, 30543 CrossRef CAS PubMed.
- B. Palanivel, C. Ayappan, V. Jayaraman, S. Chidambaram, R. Maheswaran and A. Mani, Mater. Sci. Semicond. Process., 2019, 100, 87–97 CrossRef CAS.
- T. A. Nguyen, T. N. M. Pham, D. T. Pham, Q. H. Nguyen, D. N. Xuan and A.-T. Le, J. Electrochem. Soc., 2024, 171, 046502 CrossRef CAS.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- L. Yuan, Z. Geng, J. Xu, F. Guo and C. Han, Adv. Funct. Mater., 2021, 31, 2101103 CrossRef CAS.
- C. Jia, X. Li, N. Xin, Y. Gong, J. Guan, L. Meng, S. Meng and X. Guo, Adv. Energy Mater., 2016, 6, 1600431 CrossRef.
- R. Jiang, B. Li, C. Fang and J. Wang, Adv. Mater., 2014, 26, 5274–5309 CrossRef CAS.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2017, 118, 2927–2954 CrossRef.
- N. Waiskopf, Y. Ben-Shahar and U. Banin, Adv. Mater., 2018, 30, 1706697 CrossRef PubMed.
- N. B. Saber, A. Mezni, A. Alrooqi and T. Altalhi, J. Mater. Res. Technol., 2020, 9, 15233–15262 CrossRef.
- M. Valenti, M. P. Jonsson, G. Biskos, A. Schmidt-Ott and W. A. Smith, J. Mater. Chem. A, 2016, 4, 17891–17912 RSC.
- Y. Liu, H. Ma, X. X. Han and B. Zhao, Mater. Horiz., 2021, 8, 370–382 RSC.
- M. Wang, M. Ye, J. Iocozzia, C. Lin and Z. Lin, Adv. Sci., 2016, 3, 1600024 CrossRef.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- N. Barnawi, S. Allehyani and R. Seoudi, J. Mater. Res. Technol., 2022, 17, 537–545 CrossRef CAS.
- G. Ma, J. Li, K. Sun, H. Peng, J. Mu and Z. Lei, J. Power Sources, 2014, 256, 281–287 CrossRef CAS.
- A. Valipour, N. Hamnabard, S. M. H. Meshkati, M. Pakan and Y.-H. Ahn, Dalton Trans., 2019, 48, 5429–5443 RSC.
- N. T. Anh, N. X. Dinh, T. N. Pham and A.-T. Le, RSC Adv., 2021, 11, 30544–30559 RSC.
- N. T. Anh, N. Van Quy, O. Van Hoang, N. X. Dinh and A.-T. Le, Nanoscale Adv., 2024, 6, 256–267 RSC.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2022 Search PubMed.
- N. T. Anh, N. X. Dinh, N. N. Huyen, P. T. L. Huong, V. N. Phan, P. D. Thang, H. V. Tuan, T. V. Tan and A.-T. Le, J. Electrochem. Soc., 2023, 170, 017510 CrossRef CAS.
- N. T. Anh, N. N. Huyen, N. X. Dinh, N. T. Vinh, N. V. Quy, V. D. Lam and A.-T. Le, New J. Chem., 2022, 46, 7090–7102 RSC.
- E. P. Randviir and C. E. Banks, Anal. Methods, 2013, 5, 1098–1115 RSC.
- T. Kokab, A. Shah, M. A. Khan, M. Arshad, J. Nisar, M. N. Ashiq and M. A. Zia, ACS Appl. Nano Mater., 2021, 4, 4699–4712 CrossRef CAS.
- N. T. Anh, N. X. Dinh, N. N. Huyen, P. T. L. Huong, V. N. Phan, P. D. Thang, H. V. Tuan, T. V. Tan, J. Lee and A.-T. Le, J. Electrochem. Soc., 2023, 170, 077506 CrossRef CAS.
- E. J. J. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 19–28 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1974, 52, 355–393 CrossRef CAS.
- B. Healy, F. Rizzuto, M. de Rose, T. Yu and C. B. Breslin, J. Solid State Electrochem., 2021, 25, 2599–2609 CrossRef CAS.
- D. Nematollahi, H. Shayani-Jam, M. Alimoradi and S. Niroomand, Electrochim. Acta, 2009, 54, 7407–7415 CrossRef CAS.
- B. Thomas and L. K. Alexander, J. Solid State Chem., 2020, 288, 121417 CrossRef CAS.
- N. Miura, T. Nanjo, M. Suita, T. Oishi, Y. Abe, T. Ozeki, H. Ishikawa, T. Egawa and T. Jimbo, Solid-State Electron., 2004, 48, 689–695 CrossRef CAS.
- A. Kahn, Mater. Horiz., 2016, 3, 7–10 RSC.
- W. Zhang, B. Wang, C. Hao, Y. Liang, H. Shi, L. Ao and W. Wang, J. Alloys Compd., 2016, 684, 445–452 CrossRef CAS.
- L. Wang, J. Ge, A. Wang, M. Deng, X. Wang, S. Bai, R. Li, J. Jiang, Q. Zhang and Y. Luo, Angew. Chem., 2014, 126, 5207–5211 CrossRef.
- Z. Zhang and J. T. Yates Jr, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- Z. Zhang, C. Zhang, H. Zheng and H. Xu, Acc. Chem. Res., 2019, 52, 2506–2515 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra06752e |
| This journal is © The Royal Society of Chemistry 2024 |