
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient convergent synthesis of 1,3-diazepinone nucleosides by ring-closing metathesis and direct glycosylation†
Adam K. Hedger ab,
Jonathan Findellac,
Dinesh S. Baraka,
Celia A. Schiffera,
Jonathan K. Watts*b and
Akbar Ali*a
ab,
Jonathan Findellac,
Dinesh S. Baraka,
Celia A. Schiffera,
Jonathan K. Watts*b and
Akbar Ali*a
aDepartment of Biochemistry and Molecular Biotechnology, University of Massachusetts Chan Medical School, Worcester, Massachusetts 01605, USA. E-mail: Akbar.Ali@umassmed.edu; Tel: +1 508 856 8873
bRNA Therapeutics Institute, University of Massachusetts Chan Medical School, Worcester, Massachusetts 01605, USA. E-mail: Jonathan.Watts@umassmed.edu; Tel: +1 774 455 3784
cDepartment of Chemistry, School of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 20th November 2024
Abstract
A new and highly efficient ring-closing metathesis-based strategy was developed for the synthesis of the cyclic urea 1,3-diazepinone, presenting significant improvement upon previous methods. Using a direct glycosylation approach, analogues of the potent cytidine deaminase (CDA) inhibitor diazepinone riboside were then synthesized including 2′-deoxyribo-, 2′-deoxy-2′-fluoroarabino-, and 2′-deoxy-2′,2′-difluoro-diazepinone nucleosides, all with moderate to good yield and excellent anomeric selectivity. Crucially, in contrast to the previous multistep linear synthesis of 2′-deoxyribo- and 2′-deoxy-2′-fluoroarabino-diazepinone nucleosides, this is the first report of direct glycosylation to access these nucleosides. Overall, we report efficient convergent routes to multiple 2′-modified-diazepinone nucleosides for further evaluation as CDA and potential APOBEC3 inhibitors.
Introduction
Cytidine deaminase enzyme (CDA) catalyzes the deamination of cytidine and deoxycytidine to uridine and deoxyuridine, respectively,1 as a part of the pyrimidine salvage pathway.2 CDA has been found to readily metabolize and deactivate pyrimidine-based anticancer and antiviral nucleoside drugs such as gemcitabine, azacytidine and decitabine, leading to resistance and poor clinical outcomes.2,3 These anticancer nucleoside drugs show improved therapeutic efficacy when co-administered with a CDA inhibitor (e.g. cedazuridine), culminating in the FDA approval of decitabine/cedazuridine combination therapy. The APOBEC3 family of enzymes also catalyzes the deamination of cytosine (C) to uracil (U), but only in nucleic acids (DNA and RNA) rather than single nucleosides. The APOBEC3 proteins play an important role in innate immunity, but when misregulated cause somatic mutations to the human genome that lead to cancer evolution and drug resistance.4,5 Recent biochemical and structural data has shown that nucleoside-based CDA inhibitors can also inhibit APOBEC3 enzymes when incorporated into short oligonucleotides.6–8 Therefore, there is significant interest in synthetically straightforward routes to modified nucleosides that can inhibit CDA and APOBEC3 enzymes.Several riboside-based CDA inhibitors have been reported including zebularine, tetrahydrouridine, and 1,3-diazepinone riboside (Fig. 1). However, unlike the CDA enzyme, APOBEC3s do not bind single nucleosides, and prefer to bind DNA substrates rather than RNA. Therefore, CDA inhibitors must be incorporated into oligonucleotides as their 2′-deoxy counterparts to have good activity against APOBEC3 enzymes, as is the case for zebularine and 2′-deoxyzebularine (Fig. 1B). The diazepinone riboside is one of the most potent CDA inhibitors reported with a Ki of 25 nM.9 The corresponding 2′-deoxy- and 2′-fluorinated-diazepinone nucleosides also potently inhibit CDA,10,11 and we hypothesize that they could inhibit APOBEC3 enzymes once incorporated into oligonucleotides. Indeed, the 2′-fluorination of CDA inhibitor tetrahydrouridine has also been shown to increase stability while retaining good levels of activity, resulting in the FDA approved cedazuridine (Fig. 1C).12
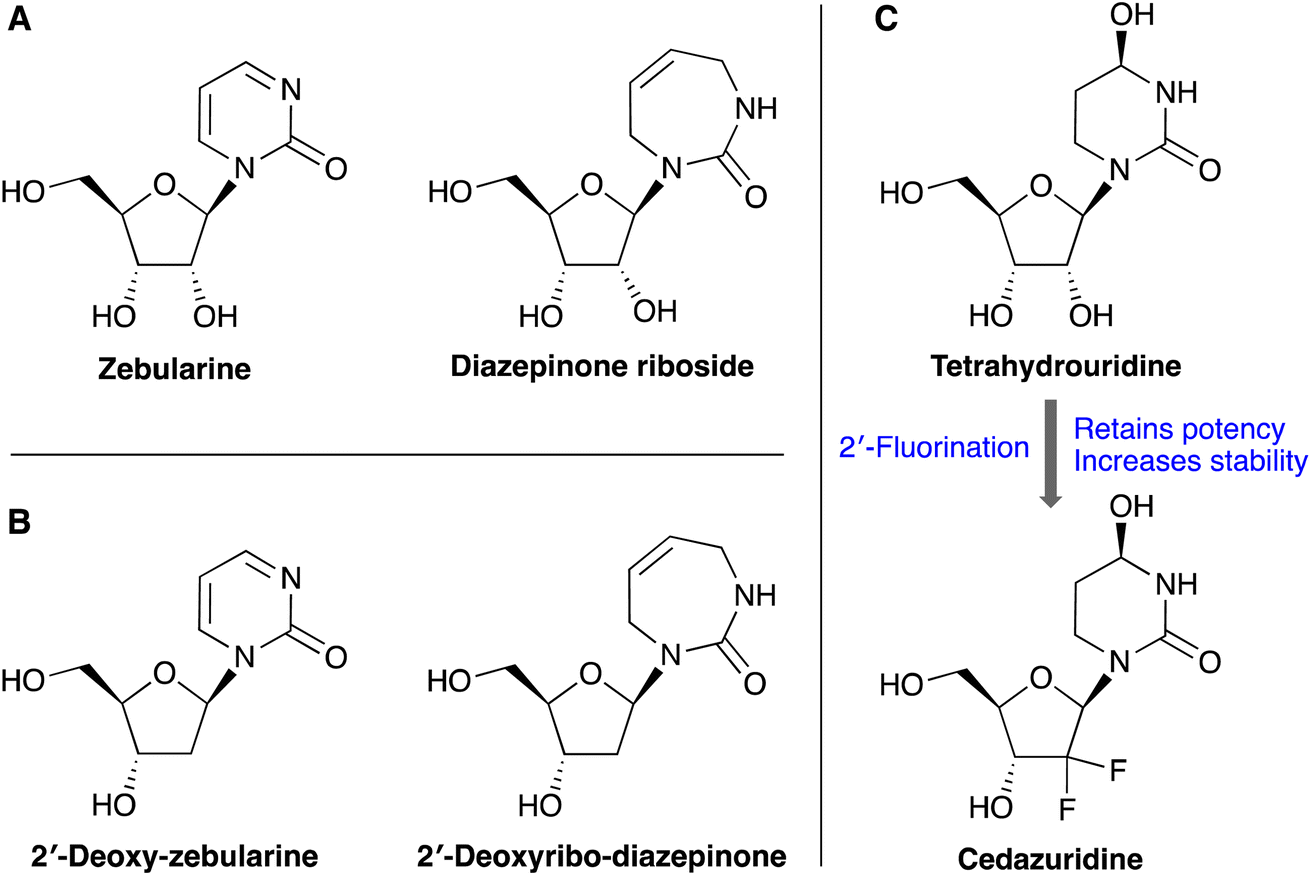 | ||
| Fig. 1 Example of chemically modified cytidine analogues: (A) ribose-based nucleoside inhibitors of cytidine deaminase (CDA). (B) Corresponding 2′-deoxy analogues which inhibit APOBEC3 enzymes when incorporated into oligonucleotides. (C) 2′-Fluorination has successfully been used to increase the stability of CDA inhibitors. | ||
The first reported synthesis of the 7-membered diazepinone nucleobase involved an undesirable and low yielding ring-closing reaction of (Z)-but-2-ene-1,4-diamine using carbonyl sulfide (COS) gas (Scheme 1).13 A recent method reported by Kim et al.14 avoids the use of COS gas, but still requires 5 steps with orthogonal protection and deprotection steps. This improved method utilized a ring-closing metathesis-based strategy originally developed by the Marquez laboratory for the synthesis of carbocyclic analogues of diazepinone riboside.15 To access the diazepinone riboside, both reports employ a direct glycosylation strategy using mercury salts as catalysts, which are commonly used for non-aromatic cyclic-urea nucleobases.9,14,16 However, for 2′-modified diazepinone nucleosides, including the recently reported 2′-deoxy-, and 2′-fluoroarabino diazepinone nucleosides, a linear synthetic strategy of building the nucleobase on the sugar itself is used (Scheme 1).11,17 Starting from the sugar, these multistep linear routes are less efficient as the glycosylation and diazepinone ring-building is carried out early in the synthesis, with the key ring-closing metathesis (RCM) steps resulting in only ∼40% yield. Furthermore, this approach would likely require re-optimization of many steps for each new target nucleoside.
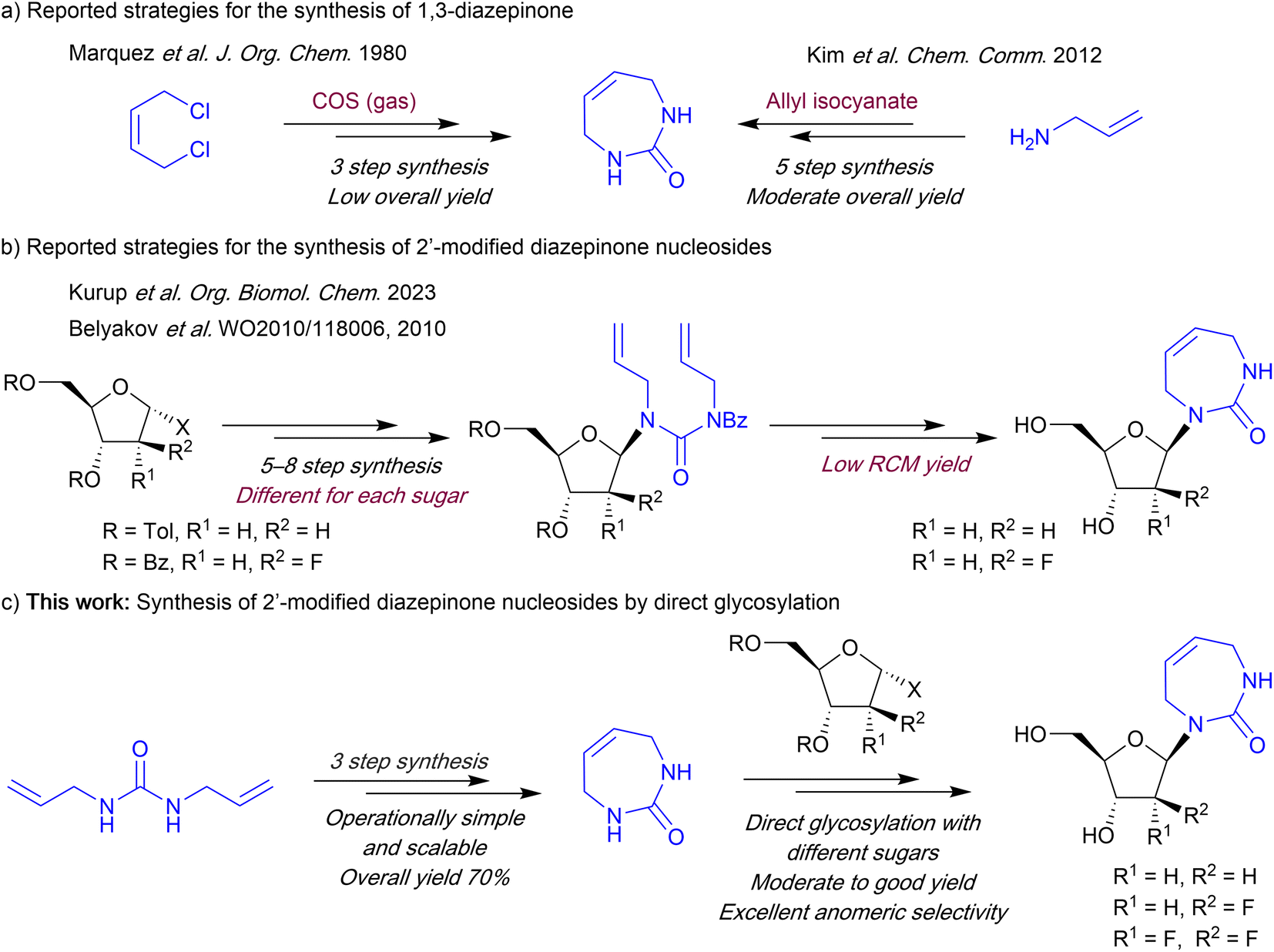 | ||
| Scheme 1 Strategies for the synthesis of 1,3-diazepinone and 2′-deoxyribo and 2′-fluorinated diazepinone nucleosides. | ||
Here we report an efficient convergent route to the 1,3-diazepinone cyclic urea nucleobase and corresponding 2′-modified nucleosides. Using our approach, the 7-membered diazepinone ring can be synthesized in 3 steps utilizing RCM with 70% overall yield. Next, we optimized a direct glycosylation approach to install the diazepinone onto various 2′-substituted sugars to provide easy access to diazepinone nucleosides in a convergent manner. In our hands the 2′-deoxy-diazepinone nucleoside shows limited stability, as reported for other non-aromatic nucleosides.18 We expanded our direct glycosylation approach to access the corresponding 2′-fluoroarabino nucleoside, as well as the 2′-deoxy-2′,2′-difluoro nucleoside. Crucially, 2′-fluorination improves the stability of the 2′-deoxy-nucleosides, suggesting promise for further evaluation as CDA and APOBEC3 inhibitors.
Results and discussion
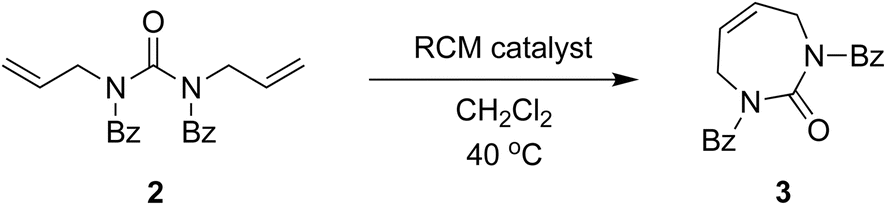
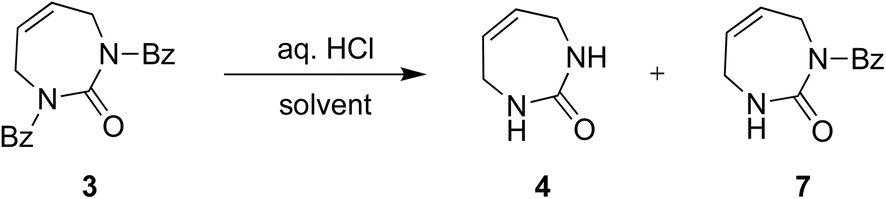
The direct glycosylation approach first involved developing an efficient route to the 7-membered diazepinone nucleobase 4, which we envisaged could then be glycosylated directly to 2′-modified sugars of interest. The direct RCM of unprotected diallylurea derivatives (e.g., 1 to 4, Scheme 2) does not work due to unfavorable orientation of the allyl groups,15,19 as we also found. We hypothesized that a general strategy of bis-N-protection, RCM, and then deprotection would provide an efficient route to the target diazepinone 4 (Scheme 2), and that benzoyl protection would be a good candidate for this strategy.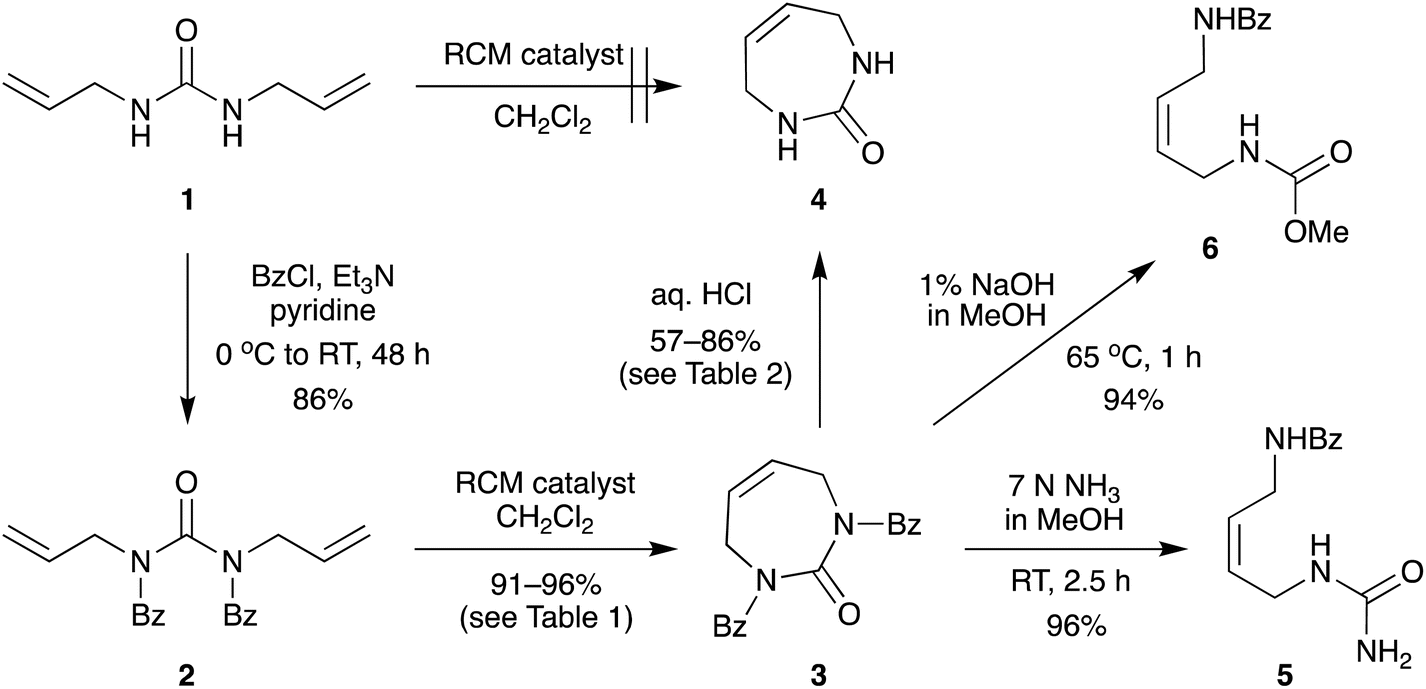 | ||
| Scheme 2 Improved 3-step synthesis of 1,3-diazepinone by ring-closing metathesis. | ||
Therefore, diallylurea 1 was first bis-benzoyl (Bz) protected using excess BzCl in pyridine to form 2. Next, RCM gave the Bz-protected diazepinone 3 in high yield, using as little as 1% RCM catalyst in anhydrous CH2Cl2. Finally, 3 was deprotected under strongly acidic conditions and recrystallized from MeOH to give the free diazepinone 4, which was used in subsequent glycosylation reactions.
We explored optimization of the RCM step using multiple catalysts, catalyst loadings, and repeat reaction cycles in the same vessel (Table 1). We observed that the RCM reaction with the Grubbs first-generation catalyst was slower compared to reactions with second-generation catalysts, although yields were similar. All reactions were high yielding, and we managed to systematically reduce catalyst loading. Optimal RCM conditions (Table 1, entry 10) used just 1 mol% catalyst, and allowed for a substantial reduction in solvent, by performing 5 repeat reaction cycles in the same flask.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Catalyst loading (mol%) | Reaction cyclesa | Substrate conc. (mM) | Time | Yieldb (%) |
| a Repeat cycles of substrate and catalyst addition in same vessel.b Isolated yield. | ||||||
| 1 | Grubbs 1st gen | 10 | 1 | 6 | 2 h | 90 |
| 2 | Grubbs 2nd gen | 10 | 1 | 6 | 1 h | 91 |
| 3 | Zhan-1b | 10 | 1 | 6 | 1 h | 93 |
| 4 | Grubbs 2nd gen | 5 | 1 | 6 | 1 h | 91 |
| 5 | Grubbs 2nd gen | 2 | 1 | 6 | 1 h | 96 |
| 6 | Hoveyda–Grubbs 2nd gen | 2 | 1 | 6 | 1 h | 95 |
| 7 | Zhan-1b | 2 | 1 | 6 | 1 h | 96 |
| 8 | Zhan-1b | 1 | 1 | 6 | 1 h | 95 |
| 9 | Zhan-1b | 1 | 2 | 5 | 2 × 1 h | 94 |
| 10 | Zhan-1b | 1 | 5 | 5 | 5 × 1 h | 96 |
Surprisingly, deprotection of the N-Bz groups to provide the free nucleobase 4 proved to be quite challenging. Attempted deprotection under basic conditions provided only ring opened products (Scheme 2). For example, treatment of bis-N-Bz-diazepinone 3 with 7 N NH3 in MeOH cleanly gave the ring-opened urea derivative 5 in high yield. Similarly, reaction with 1% NaOH in MeOH also provided the ring-opened product, in this case the carbamate derivative 6, in 94% yield. Both these conditions have previously been reported to deprotect a N-Bz group on the diazepinone system, albeit with orthogonal substitution of the second urea nitrogen.15,17
Next, we explored acidic conditions to remove the bis-Bz protection (Table 2). Reaction of 3 with aqueous 6 N HCl in THF or 1,4-dioxane at room temperature (RT) for 24 hours was unsuccessful, giving only small amounts of mono deprotected 7 (Table 2, entries 1 and 2). Extended time, or warming to 30 °C in 6 N HCl in 1,4-dioxane, gave a mixture of 4 and 7, and unreacted 3 (Table 2, entries 3–5). Increasing the reaction temperature further to 50 °C, in either solvent gave predominantly the desired product 4 in good yield (∼60%, Table 2, entries 6 and 7). These encouraging results, and the fact that the diazepinone ring remained intact, prompted us to further optimize the acid-catalyzed deprotection reaction. Next, we tried 8 N HCl in 1,4-dioxane at RT for 24 and 48 hours and observed 7 as the major product (Table 2, entries 8 and 9). Increasing the temperature to 45 °C for 48 hours gave our optimal conditions, producing fully deprotected 4 in 86% yield (Table 2, entry 10).
|
||||
|---|---|---|---|---|
| Entry | [HCl]/solvent | Time (h) | Temp. (°C) | Product/yield (%) |
| a RT = 23 °C.b Monitored by TLC, products not isolated. | ||||
| 1 | 6 N/THF | 24 | RTa | (3 major) + 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
| 2 | 6 N/1,4-dioxane | 24 | RT | (3 major) + 7b |
| 3 | 6 N/1,4-dioxane | 48 | RT | 3 + 4 + (7 major)b |
| 4 | 6 N/1,4-dioxane | 24 | 30 | 3 + (7 major)b |
| 5 | 6 N/1,4-dioxane | 48 | 30 | 3 + 4 + (7 major)b |
| 6 | 6 N/THF | 24 | 50 | 4: 58% |
| 7 | 6 N/1,4-dioxane | 24 | 50 | 4: 57% |
| 8 | 8 N/1,4-dioxane | 24 | RT | 3 + 4 + (7 major)b |
| 9 | 8 N/1,4-dioxane | 48 | RT | 3 + 4 + (7 major)b |
| 10 | 8 N/1,4-dioxane | 48 | 45 | 4: 86% |
Overall, we present a highly efficient 3-step synthesis (70% overall yield) of the 7-membered diazepinone nucleobase. This improved method avoids the use of COS gas,9 and requires fewer steps and significantly less RCM catalyst and solvent than the previous report.14
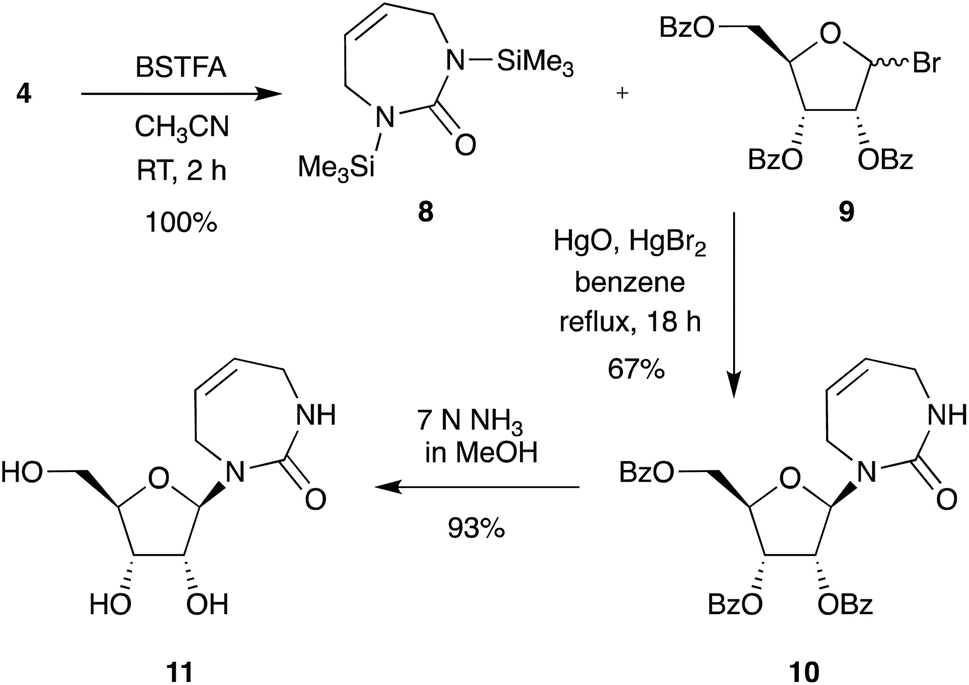
With efficient access to the free diazepinone nucleobase established, we next chose to explore synthesis routes to multiple diazepinone nucleosides. First, we repeated the synthesis of diazepinone riboside following the direct glycosylation conditions developed by Marquez et al. (Scheme 3).9 Silylated diazepinone 8 was coupled to bromosugar 9 via mercury-catalyzed glycosylation in refluxing benzene, providing exclusively the β-anomer 10 in good yield (67%). The nucleoside 10 was deprotected using methanolic ammonia, and lyophilized to give the diazepinone riboside 11. As with previous reports, we found the type and quality of mercury salts can affect the N-/O-linked glycoside ratio and must be carefully controlled.16
 | ||
| Scheme 3 Synthesis of diazepinone riboside. | ||
Next, we tried to obtain the corresponding 2′-deoxy-diazepinone nucleoside. As there is limited literature precedent for the direct glycosylation of non-aromatic nucleobases to 2′-deoxy sugars, and anticipating poor anomeric selectivity, we first opted to pursue 2′-deoxygenation of the diazepinone riboside (Scheme S1†). The Barton–McCombie deoxygenation has found widespread utility in carbohydrate synthesis, accessing novel ribonucleosides through exclusive β-anomer glycosylation selectivity, then removing the 2′-hydroxyl group to provide their 2′-deoxy counterparts.20–23 To attempt 2′-deoxygenation we performed a series of protection/deprotection steps starting from protected riboside 10, followed by deoxygenation under Barton–McCombie conditions to successfully give the fully protected target 2′-deoxy-diazepinone nucleoside 30. However, deprotection proved troublesome and multiple conditions gave a mixture of products (Scheme S1† and associated text). These problems, along with the long synthetic scheme prompted us to explore an alternative strategy.
We therefore attempted direct glycosylation to obtain the target 2′-deoxy-diazepinone nucleoside. Initially, we tried the same conditions as Cristalli et al.10 using excess N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) for silylation and SnCl4 for glycosylation. However, multiple attempts failed to provide any detectable nucleoside product in our hands. Next, we applied controlled silylation conditions,24 similar to those used for coupling the acyclic urea 1 to Hoffer's chlorosugar 13 in the linear synthetic approach to access the 2′-deoxy-diazepinone nucleoside.17 Gratifyingly, using similar silylation and glycosylation conditions with diazepinone 4 and Hoffer's chlorosugar 13 under general Vorbrüggen conditions (SnCl4, 1,2-dichloroethane)25 yielded the desired 2′-deoxy nucleoside in a 9:1 anomeric ratio, providing the β-anomer 14 in 48% isolated yield (Table 3, entry 2). The α-anomer 15 was also characterized. The anomeric identity of the β-anomer 14 and α-anomer 15 was confirmed by 2D NOESY NMR experiments (Fig. S4 and S5†).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Silylation conditions | Solvent/catalyst | Höffer's sugar (eq.) | Temp. (°C) | Anomeric ratio (crude) (β/α)a | Isolated yield (%) (β-anomer) |
| a Entry 1 as per Cristalli et al.10b Crude anomeric ratio's measured by H NMR. | ||||||
| 1 | BSTFA (excess)a acetonitrile | CH2Cl2 | 1.1 | RT | N/A | N/A |
| SnCl4 (1.0 eq.) | ||||||
| 2 | Et3N (1.6 eq.) | ClCH2CH2Cl | 0.85 | −30 | 9:1 |
48 |
| TMS-Cl (1.2 eq.) benzene | SnCl4 (3.0 eq.) | |||||
| 3 | Et3N (2.0 eq.) | ClCH2CH2Cl | 0.85 | −30 | 9:1 |
51 |
| TMS-Cl (1.4 eq.) benzene | SnCl4 (3.0 eq.) | |||||
| 4 | Et3N (1.6 eq.) TMS-Cl (1.2 eq.) benzene | ClCH2CH2Cl | 1.2 | −30 | 5:1 |
28 |
| SnCl4 (3.0 eq.) | ||||||
| 5 | Et3N (1.6 eq.) TMS-Cl (1.2 eq.) benzene | ClCH2CH2Cl | 0.85 | 0 | 10:1 |
60 |
| SnCl4 (3.0 eq.) | ||||||
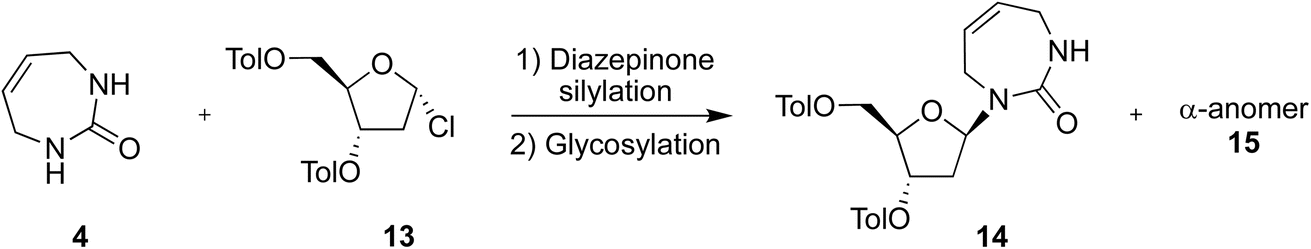
We then tried to optimize this glycosylation reaction (Table 3) by varying reagent amounts and temperature. First, we slightly increased the equivalents of silylation reagent, but this had little effect on crude anomeric ratio or reaction yield (Table 3, entry 3). Next, we increased the amount of Hoffer's chlorosugar to 1.2 eq., but this lowered both anomeric ratio and yield (Table 3, entry 4). Finally, we repeated entry 2 but at 0 °C rather than −30 °C, and saw a marginal increase in selectivity but moderate increase in yield, giving the target 2′-deoxy nucleoside in a 10:1 crude anomeric ratio, and 60% isolated yield of the β-anomer 14 (Table 3, entry 5).
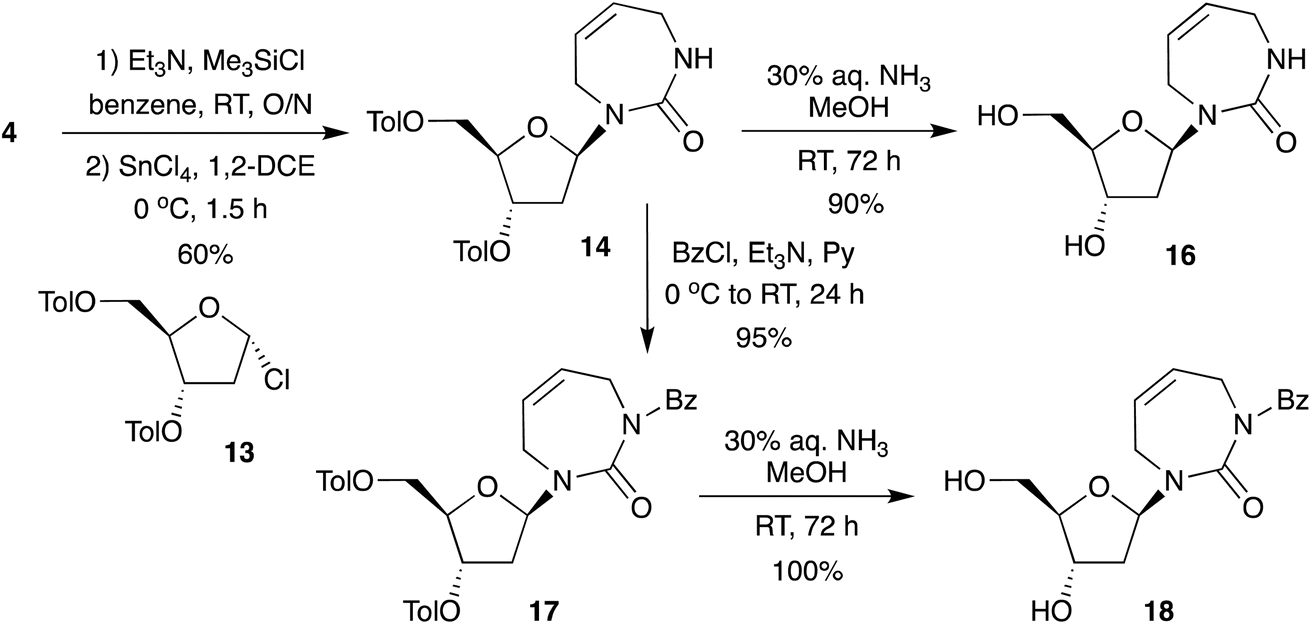
Therefore, under controlled silylation and Vorbrüggen conditions we report the direct glycosylation of the non-aromatic diazepinone nucleobase (Scheme 4), giving access to the protected 2′-deoxy-diazepinone nucleoside 14 in excellent yield and anomeric ratio. Toluoyl-protected 14 was deprotected using aqueous ammonia in MeOH to give nucleoside 16 in 90% yield after lyophilization. However, we noticed this nucleoside appeared to be unstable in solution. 1H NMR samples in D2O showed extra signals form within hours, clearly visible around the anomeric and 2′ proton range (Fig. S1†). In addition, multiple attempts to purify by water/chloroform extraction (to remove deprotection by-products) resulted in a mixture of nucleoside products after lyophilization of the aqueous phase. Indeed, instability of non-aromatic nucleobases has been reported previously, including with 7-membered cyclic urea nucleosides.18 In this process, ring-opening of the sugar via Schiff base formation at the glycosidic bond allows for anomeric interconversion, as well as thermodynamically driven interconversion from furanose to pyranose.11,26,27 We reasoned that blocking of the diazepinone NH with an electron-withdrawing group might help to provide additional stability by reducing electron density on the glycosidic nitrogen, and so 14 was Bz-protected in quantitative yield to a form fully protected nucleoside 17. The 5′- and 3′-toluoyl protecting groups were selectively removed under mild ammonia treatment to give N-Bz-protected 2′-deoxy-diazepinone nucleoside 18 (Scheme 4). While careful handling of 18 was still required, this derivative appeared more stable than 16. Interestingly, N-boc-protected 2′-deoxy-diazepinone nucleoside 31 from the deoxygenation scheme (Scheme S1B†) also appeared to be stable, and the instability and acid sensitivity of 16 likely explains why we couldn't remove the N-boc group in Scheme S1B.†
 | ||
| Scheme 4 Synthesis of 2′-deoxyribo-diazepinone nucleoside by direct glycosylation. | ||
Overall, our method provides a highly efficient route to 2′-deoxy-diazepinone nucleoside 16 in excellent yield through direct glycosylation to the sugar. This convergent synthesis over 5 steps results in overall 38% yield which is significantly improved in comparison to overall 8% yield obtained for the linear strategy recently reported.17
Motivated by the efficient synthesis of the protected 2′-deoxy nucleoside 14 via direct glycosylation, we next explored whether this strategy could also be extended to 2′-fluorinated analogues. We started from commercially available 2-deoxy-2-fluoro-1,3,5-tri-O-benzoyl-α-D-arabinofuranose and 2-deoxy-2,2-difluoro-1-O-methanesulfonyl-3,5-di-O-benzoyl-ribo-furanose as glycosyl donors (Scheme 5).
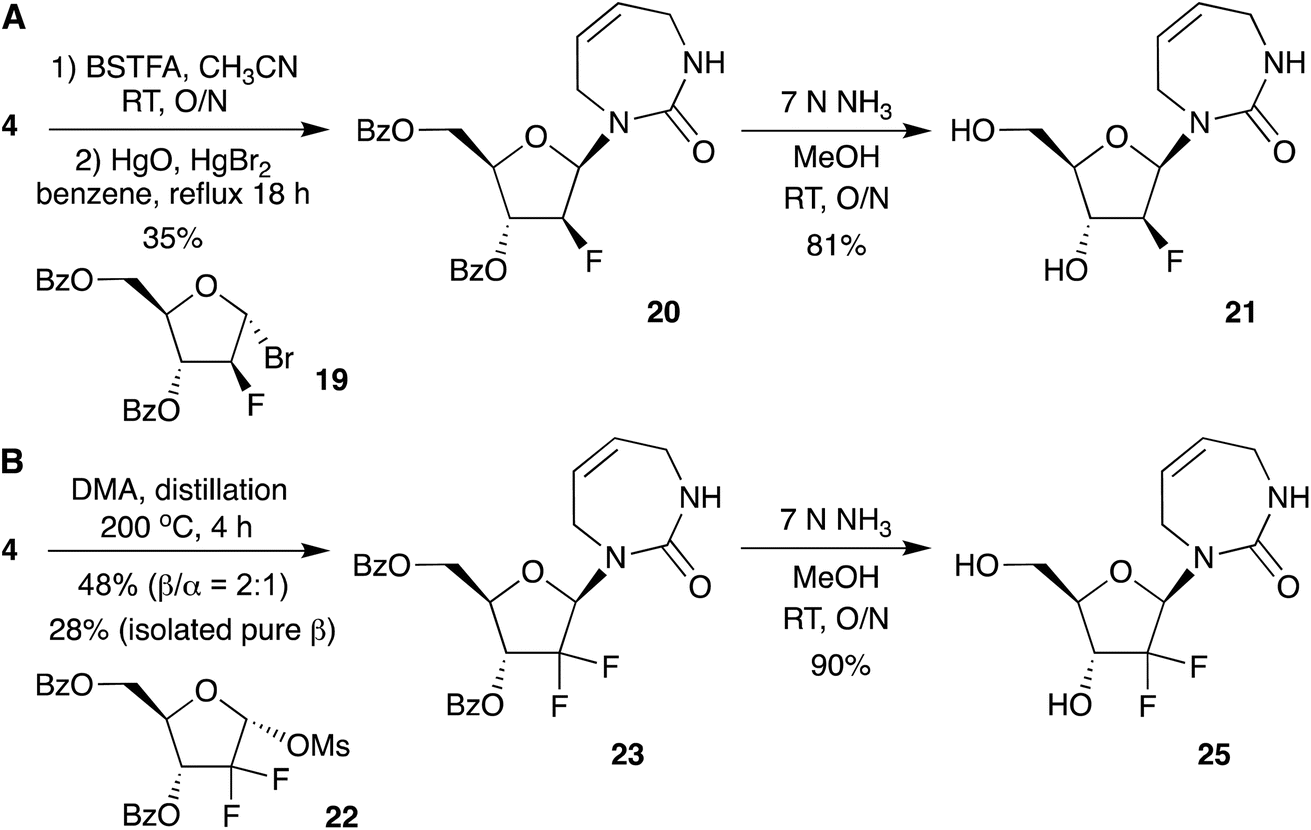 | ||
| Scheme 5 Synthesis of fluorinated diazepinone nucleosides by direct glycosylation. | ||
For the 2-fluoroarabino-1-bromo derivative 19 (Scheme 5A), glycosylation did not proceed under the optimized SnCl4 catalyzed conditions that worked for Hoffer's chlorosugar (13 to 14 in Scheme 4). However, the reaction proceeded well with the mercury-catalyzed conditions employed for glycosylation with the 1-bromo ribofuranose (9 to 10 in Scheme 3). The 2-deoxy-2-fluoro-1,3,5-tri-O-benzoyl-α-D-arabinofuranose was converted to its 1-bromo derivative 19 using 30% HBr in AcOH in quantitative yield.28 Bromosugar 19 was then coupled with 4 via mercury-catalyzed glycosylation in refluxing benzene to afford the protected 2′-fluoroarabino nucleoside 20 as the pure β-anomer in 35% isolated yield. No α-anomer was detected. We also isolated the minor O-glycoside by-product, as observed previously for mercury-catalyzed glycosylation reactions.16 Protected nucleoside 20 was then deprotected in methanolic ammonia to furnish the 2′-fluoroarabino-diazepinone nucleoside 21. 1H NMR studies of 21 in D2O showed complete stability out to at least 48 hours (Fig. S2†), in contrast to 2′-deoxyribo-diazepinone 16, which showed partial degradation within hours. The 2′-fluorination of nucleosides (as well as oligonucleotides containing them) is a known strategy to stabilize them to acidic conditions.12,29,30
Next, to access the 2′-deoxy-2′,2′-difluoro nucleoside analogue, we followed the conditions reported in the patent literature (Scheme 5B).11 Condensation of 4 with 2-deoxy-2,2-difluoro sugar 22 was achieved by vigorous distillation in DMA, without a catalyst. This gave protected 2′-deoxy-2′,2′-difluoro diazepinone nucleoside in 48% yield with 2:1 β selectivity. Separation of anomers was challenging as reported previously, but we isolated the pure β-anomer 23 in 28% yield. Compound 23 was deprotected using 7 N NH3 in MeOH to give nucleoside 25, in 90% yield. Similar to 2′-fluoroarabino nucleoside 21, we also observed that the difluoro analogue 25 was more stable than 2′-deoxyribo-diazepinone 16 (Fig. S3†).
Conclusions
Cyclic ureas are a broadly useful class of synthetic intermediates and bioactive molecules. In particular, diazepinone-based nucleosides, which are attractive deaminase inhibitors, have proved synthetically challenging. We present efficient synthetic methods to access the 7-membered 1,3-diazepinone cyclic urea, as well as direct glycosylation approaches to access diazepinone-based nucleosides. These strategies can be applied to the synthesis of other cyclic urea derivatives, as well as other diverse nucleoside analogues by direct glycosylation with modified sugars.We first developed an improved 3-step synthesis of 1,3-diazepinone 4 starting from readily available 1,3-diallyl urea, via a highly efficient RCM reaction, with 70% overall yield. Next, we pursued a convergent direct glycosylation approach to install the diazepinone on different 2′-substituted sugars. This provided direct access to the ribo- and 2′-deoxyribo-diazepinone nucleosides (11 and 16), as well as the more stable 2′-fluoroarabino-, and 2′-deoxy-2′,2′-difluoro-diazepinone nucleosides (21 and 25), with excellent anomeric selectivity. Overall, our work provides efficient synthetic routes to multiple potent diazepinone-based CDA inhibitors. These compounds are also key synthetic intermediates required to synthesize diazepinone-containing oligonucleotides as potential APOBEC3 inhibitors,6–8 work which is ongoing in our laboratory.
Experimental section
General information
All chemicals, reagents and solvents were purchased from commercial sources (Sigma-Aldrich, Fisher Scientific, Chem-Impex, Biosynth) and were used as received, unless otherwise stated. Anhydrous solvents were purchased from Sigma-Aldrich and were further dried over activated 3 Å molecular sieves. All reactions were performed in oven-dried round bottomed flasks fitted with rubber septa under argon atmosphere, unless otherwise noted. Automated flash column chromatography was performed on an Interchim PuriFlash system equipped with a UV-vis detectors using prepacked silica gel cartridges or was performed manually using silica gel (230–400 mesh, EMD Millipore). Analytical thin-layer chromatography (TLC) was performed using silica gel (60 F254) coated aluminum plates (EMD Millipore), and spots were visualized by exposure to ultraviolet light (UV), and/or stained with 10% H2SO4 in ethanol or ninhydrin, followed by brief heating. 1H, 13C, 19F, DEPT-135, and 2D NMR spectra were acquired on a Bruker Avance III HD 500 MHz NMR instrument. Chemical shifts are reported in ppm (δ scale) relative to the solvent signal and coupling constant (J) values are reported in hertz. Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet of doublets), coupling constant in Hz, and integration. Structural assignments were made using a combination of 1D and 2D NMR spectra, including gradient selected COSY, HSQC, HMBC, and NOESY experiments (particularly for nucleoside anomer assignment) as needed. High-resolution mass spectra (HRMS) were recorded on a Thermo Scientific Orbitrap Velos Pro mass spectrometer coupled with a Thermo Scientific Accela 1250 UPLC and an autosampler using electrospray ionization (ESI) in the positive mode.Synthesis of 1,3-diazepinone
:1) mixture and subjected to flash chromatography, initially eluting with the same mixture and then with CH2Cl2. Fractions containing the product were pooled and evaporated to provide a pale-yellow solid. Recrystallization from EtOAc/hexanes (1:5) mixture provided the bis-benzoylated dialy urea 2 (10.7 g, 86%) as colorless needles. TLC: Rf = 0.65 (25% EtOAc/hexanes).1H NMR (500 MHz, CDCl3): δ 7.55–7.50 (m, 2H, H-Bz), 7.41 (apparent d, J = 4.4 Hz, 8H, H-Bz), 5.89–5.81 (m, 2H, H-5, H-8), 5.20 (dd, J = 17.1, 1.3 Hz, 2H, H-6/H-9), 5.15 (dd, J = 10.2, 1.2 Hz, 2H, H-6/H-9), 3.98 (d, J = 6.1 Hz, 4H, H-4, H-7) ppm; 13C NMR (126 MHz, CDCl3): δ 171.04 (2C, Bz-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 158.39, (CO, C-2), 135.49 (2C, C-Ph), 132.44 (2C, CH-5, CH-8), 132.13 (2C, CH-Ph), 128.75 (4C, CH-Ph), 128.05 (4C, CH-Ph), 118.99 (2C, CH-6, CH-9), 50.10 (2C, CH2-4, CH2-7) ppm; HRMS (ESI): m/z Calcd. for C21H21N2O3 [M + H]+ 349.1547; found 349.1536.
O), 158.39, (CO, C-2), 135.49 (2C, C-Ph), 132.44 (2C, CH-5, CH-8), 132.13 (2C, CH-Ph), 128.75 (4C, CH-Ph), 128.05 (4C, CH-Ph), 118.99 (2C, CH-6, CH-9), 50.10 (2C, CH2-4, CH2-7) ppm; HRMS (ESI): m/z Calcd. for C21H21N2O3 [M + H]+ 349.1547; found 349.1536.
:1) mixture, providing the pure bis-benzoylated 1,3-diazepinone 3 (9.40 g) as a white crystalline solid. The filtrate was evaporated in vacuo and the solid residue was recrystallized, giving additional pure product (1.60 g) as a white crystalline solid. Total yield: 11.0 g, 96%. TLC: Rf = 0.50 (25% EtOAc/hexanes).1H NMR (500 MHz, CDCl3): δ 7.53–7.50 (m, 4H, H-Bz), 7.46–7.43 (m, 2H, H-Bz), 7.38–7.35 (m, 4H, H-Bz), 6.00 (t, J = 2.3 Hz, 2H, H-5, H-6), 4.82 (d, J = 2.2 Hz, 4H, H-4, H-7) ppm; 13C NMR (126 MHz, CDCl3): δ 172.11 (2C, Bz-CO), 159.88 (CO, C-2), 134.94 (2C, C-Ph), 132.03 (2C, CH-5, CH-6), 128.60 (4C, CH-Ph), 127.71 (4C, CH-Ph), 126.20 (2C, CH-Ph), 42.93 (2C, CH2-4, CH2-7) ppm; HRMS (ESI) m/z: Calcd. for C19H17N2O3 [M + H]+ 321.1234; found 321.1227.
:1) mixture (3 × 200 mL) and filtered. The filtrate was passed through a short pad of silica gel (8 × 4 cm) and the pad was further eluted with the same solvent mixture (3 × 100 mL). The combined fractions were evaporated in vacuo to provide the product as a white crystalline solid, which was contaminated with the by-product benzoic acid. Recrystallization from MeOH provided the pure 1,3-diazepinone 4 (0.83 g, 86%) as a white crystalline solid. TLC: Rf = 0.60 (10% MeOH/CH2Cl2).1H NMR (500 MHz, DMSO-d6): δ 5.95 (s, 2H, H-1, H-3), 5.76 (t, J = 2.7 Hz, 2H, H-5, H-6), 3.53–3.51 (m, 4H, H-4, H-7) ppm; 1H NMR (500 MHz, CDCl3): δ 5.80 (t, J = 2.8 Hz, 2H), 4.84 (br s, 2H), 3.74 (s, 4H) ppm; 13C NMR (DMSO-d6, 126 MHz): δ 164.44 (CO, C-2), 127.35 (2 × CH, CH-5, CH-6), 40.36 (2 × CH2, CH2-4, CH2-7) ppm; 13C NMR (126 MHz, CDCl3): δ 165.65, 126.90 (2C), 41.70 (2C) ppm; HRMS (ESI) m/z: Calcd. for C5H9N2O [M + H]+ 113.0709, found 113.0704.
Synthesis of 2′-deoxyribo-diazepinone nucleoside
The above silylated diazepinone compound (1.18 g, 6.42 mmol) was dissolved in 1,2-DCE (45 mL) to give a colorless solution and cooled to 0 °C using an ice bath. Freshly distilled SnCl4 (2.0 mL, 17 mmol) was added dropwise, followed by Hoffer's chlorosugar 13 (2.20 g, 5.60 mmol). This gave a clear yellow solution which was stirred at 0 °C for 1.5 h, at which point the mixture had turned dark brown. Pyridine (7.5 mL) and water (40 mL) were added, and the reaction mixture was stirred at room temperature for 1 h. After diluting with water (50 mL), the reaction mixture was extracted with CH2Cl2 (2 × 150 mL). The combined organic fractions were filtered through a bed of Celite, washed with saturated aqueous NaCl (100 mL), dried (Na2SO4), and evaporated to dryness. The resulting dark brown residue was dissolved in a minimum volume of CH2Cl2 and purified by flash column chromatography using a silica gel column (SiliaSep, 40 g, gradient elution with 0–20% acetone/CH2Cl2) to give the pure β-anomer compound 14 (1.58 g, 60%) as a white foamy solid. TLC: Rf = 0.40 (8% acetone/CH2Cl2).
1H NMR (500 MHz, CDCl3) δ 7.94–7.92 (m, 4H, H-Ph), 7.26–7.23 (m, 4H, H-Ph, overlapping), 6.17 (dd, J = 9.1, 5.8 Hz, 1H, H-1′), 5.70–5.66 (m, 1H, H-5/6), 5.65–5.60 (m, 1H, H-5/6), 5.53–5.50 (m, 1H, H-3′), 4.65 (dd, J = 11.9, 3.6 Hz, 1H, H-5′), 4.54 (dd, J = 11.9, 3.7 Hz, 1H, H-5′), 4.50 (br. s, 1H, H-3), 4.37 (dd, J = 6.2, 3.6 Hz, 1H, H-4′), 3.84–3.77 (m, 2H, H-4, H-7), 3.71–3.63 (m, 2H, H-4, H-7), 2.42 (s, 6H, CH3-Tol), 2.31–2.20 (m, 2H, H-2′) ppm; 13C NMR (126 MHz, CDCl3) δ 166.31 (CO-Tol), 166.30 (CO-Tol), 165.57 (C-2), 144.36 (C-Ph), 144.17 (C-Ph), 129.92 (2 × CH-Ph), 129.72 (2 × CH-Ph), 129.39 (2 × CH-Ph), 129.32 (2 × CH-Ph), 127.27 (CH-5/6), 127.14 (C-Ph), 126.80 (C-Ph), 125.96, (CH-5/6), 86.71 (CH-1′), 80.88 (CH-4′), 75.16 (CH-3′), 64.68 (CH2-5′), 43.58 (CH2-7), 40.11 (CH2-4), 35.20 (CH2-2′), 21.84 (CH3-Tol), 21.83(CH3-Tol) ppm; HRMS (ESI): m/z Calcd. for C26H29N2O6 [M + H]+ 465.2020; found 465.2018.
1H NMR (500 MHz, D2O) δ 6.00–5.95 (m, 1H, H-5/6), 5.97 (dd, J = 8.3, 6.4 Hz, 1H, overlapping, H-1′), 5.93–5.88 (m, 1H, H-5/6), 4.33 (dt, J = 6.7, 3.4 Hz, 1H, H-3′), 3.86–3.72 (m, 6H), 3.69 (dd, J = 12.2, 5.4 Hz, 1H, H-4′), 2.20 (ddd, J = 15.2, 8.2, 7.2 Hz, 1H, H-2′), 2.03 (ddd, J = 14.2, 6.4, 3.2 Hz, 1H, H-2′); 1H NMR (500 MHz, DMSO-d6) δ 6.02 (br s, 1H, NH), 5.82–5.77 (m, 1H, H-5/6), 5.77 (dd, J = 8.6, 6.1 Hz, 1H, overlapping, H-1′), 5.73–5.69 (m, 1H, H-5/6), 5.0 (br s, 1H), 4.73 (br s, 1H), 4.05 (br d, J = 2.8 Hz, 1H, H-3′), 3.67–3.49 (m, 5H), 3.46–3.30 (m, 2H), 1.86 (ddd, J = 15.0, 8.5, 6.5 Hz, 1H, H-2′), 1.71 (ddd, J = 13.2, 6.1, 2.7 Hz, 1H, H-2′); 13C NMR (126 MHz, D2O) δ 166.02 (C-2), 128.05 (CH-5/6), 126.80 (CH-5/6), 86.30 (CH-1′), 84.65 (CH2-4′), 70.92 (CH-3′), 61.68 (CH2-5′), 41.75 (CH2-4/7), 39.89 (CH2-4/7), 36.06 (CH2-2′); HRMS (ESI): m/z Calcd. for C10H17N2O4 [M + H]+ 229.1183; found 229.1180.
1H NMR (500 MHz, CDCl3): δ 7.97 (d, J = 8.0 Hz, 2H, H-Ph), 7.88 (d, J = 8.0 Hz, 2H, H-Ph), 7.56–7.53 (m, 2H, H-Ph), 7.48–7.44 (m, 1H, H-Ph), 7.40–7.35 (m, 2H, H-Ph), 7.29 (d, J = 8.0 Hz, 2H, H-Ph), 7.22 (d, J = 8.0 Hz, 2H, H-Ph), 6.19 (dd, J = 9.4, 5.4 Hz, 1H, H-1′), 5.79 (dt, J = 10.8, 3.3 Hz, 1H, H-5), 5.70–5.65 (m, 1H, H-6), 5.56–5.54 (m, 1H, H-3′), 4.74 (dd, J = 12.0, 3.3 Hz, 1H, H-5′), 4.65 (br d, J = 18.5 Hz, 1H, H-4), 4.57 (dd, J = 12.0, 3.6 Hz, 1H, H-5′), 4.35 (q, J = 3.1 Hz, 1H, H-4′), 4.31 (br d, J = 18.5 Hz, 1H, H-4), 4.10–4.06 (m, 1H, H-7), 3.96 (dd, J = 17.0, 5.0 Hz, 1H, H-7), 2.45 (s, 3H, CH3-Tol), 2.40 (s, 3H, CH3-Tol), 2.31 (ddd, J = 14.0, 5.4, 1.5 Hz, 1H, H-2′), 2.18 (ddd, J = 14.1, 9.4, 6.6 Hz, 1H, H-2′) ppm; 13C NMR (126 MHz, CDCl3): δ 171.05 (CO-Bz), 166.23 (CO-Tol), 166.20 (CO-Tol), 159.71 (CO–C-2), 144.53 (C-Ph), 144.45 (C-Ph), 135.22 (C-Ph), 131.62 (CH-Ph), 129.86 (2C, CH-Ph), 129.68 (2 × CH-Ph), 129.52 (2 × CH-Ph), 129.34 (2 × CH-Ph), 128.57 (2 × CH-Ph), 128.32 (CH-5/6), 127.38 (2 × CH-Ph), 126.98 (C-Ph), 126.52 (C-Ph), 124.03 (CH-5/6), 85.64 (CH-1′), 81.52 (CH-4′), 74.94 (CH-3′), 64.34 (CH2-5′), 43.27 (CH2-4), 39.29 (CH2-7), 35.44 (CH2-2′), 21.85 (CH3-Tol), 21.82 (CH3-Tol) ppm; HRMS (ESI): m/z Calcd. for C33H32N2O7Na [M + Na]+ 591.2102; found 591.2102.
1H NMR (500 MHz, D2O): δ 7.65–7.61 (m, 1H, H-Ph), 7.58–7.56 (m, 2H, H-Ph), 7.54–7.51 (m, 2H, H-Ph), 6.03–5.96 (m, 1H, H-5/6), 5.99 (dd, J = 8.0, 6.5 Hz, 1H, overlapping, H-1′), 5.89–5.85 (m, 1H, H-5/6), 4.48 (br s, 2H, H-4/7), 4.38 (td, J = 6.6, 3.2 Hz, 1H, H-3′), 4.20 (br d, J = 3.7 Hz, 2H, H-4/7), 3.90 (ddd, J = 7.5, 5.1, 3.8 Hz, 1H, H-4′), 3.78 (dd, J = 12.3, 4.0 Hz, 1H, H-5′), 3.72 (dd, J = 12.3, 5.2 Hz, 1H, H-5′), 2.20 (ddd, J = 14.4, 7.8, 6.6 Hz, 1H, H-2′), 2.07 (ddd, J = 14.1, 6.3, 3.2 Hz, 1H, H-2′) ppm; 1H NMR (500 MHz, CDCl3): δ 7.56–7.53 (m, 2H), 7.46–7.44 (m, 1H), 7.40–7.37 (m, 2H), 5.95 (t, J = 7.1 Hz, 1H), 5.91–5.87 (m, 1H), 5.83–5.80 (m, 1H), 4.57–4.53 (m, 1H), 4.38–4.35 (m, 1H), 4.38–4.35 (m, 1H), 4.14–4.09 (m, 1H), 4.07–4.02 (m, 1H), 3.80 (dd, J = 7.5, 3.7 Hz, 1H), 3.75 (m, 1H), 3.71 (dd, J = 11.5, 3.7 Hz, 1H), 2.08 (dt, J = 13.8, 7.3 Hz, 1H), 2.01 (ddd, J = 13.7, 6.6, 3.8 Hz, 1H) ppm; 13C NMR (126 MHz, D2O): δ 172.90 (CO-Bz), 160.29 (C-2), 133.85 (C-Ph), 132.21 (CH-Ph), 128.94 (2 × CH-Ph), 127.02 (CH-Ph), 126.86 (2 × CH-Ph), 123.86 (CH-Ph), 85.79 (CH-1′), 85.74 (CH-4′), 70.84 (CH-3′), 61.50 (CH2-5′), 43.61 (CH2-4), 39.71 (CH2-7), 36.43 (CH2-2′) ppm; 13C NMR (126 MHz, CDCl3): δ 171.05 (CO-Bz), 159.53 (C-2), 135.18 (C-Ph), 131.62 (CH-Ph), 128.57 (2 × CH-Ph), 128.23 (CH-Ph), 127.48 (2 × CH-Ph), 124.26 (CH-Ph), 86.47 (CH-1′), 85.74 (CH-4′), 71.69 (CH-3′), 62.77 (CH2-5′), 43.36 (CH2-4), 41.04 (CH2-7), 38.14 (CH2-2′) ppm; HRMS (ESI): m/z Calcd. for C17H20N2O5Na [M + Na]+ 355.1264; found 335.1265.
Synthesis of 2′-fluorinated diazepinone nucleosides
A solution of the above persilylated cyclic urea 8 in dry benzene (10 mL) was rapidly added to a refluxing mixture of HgO (1.80 g) and HgBr2 (1.80 g) in dry benzene (110 mL) under dry N2 atmosphere. After 10 min, a solution of 2-deoxy-2-fluoro-3,5-di-O-benzoyl-α-D-arabinofuranosyl bromide 19 in dry benzene (10 mL) was added and refluxing continued for 18 h. After cooling to room temperature, the reaction mixture was filtered through a pad of Celite, and the filter cake was washed with EtOAc. The combined filtrates were washed with saturated aqueous NaHCO3 (2 × 200 mL) and water (200 mL), dried (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified by flash column chromatography using a silica gel column (SiliaSep, 80 g, gradient elution with 0–100% EtOAc/hexanes) to provide the protected nucleoside 20 (0.72 g, 35%) as a white foamy solid. TLC: Rf = 0.35 (60% EtOAc/hexanes).
1H NMR (500 MHz, CDCl3): δ 8.10–8.03 (m, 4H, Ph), 7.63–7.55 (m, 2H, Ph), 7.48–7.43 (m, 4H, Ph), 5.99 (dd, J = 25.6, 3.3 Hz, 1H, H-1′), 5.99 (dd, J = 25.6, 3.3 Hz, 1H, H-1′), 5.80–5.75 (m, 1H, H-6), 5.73–5.69 (m, 1H, H-5), 5.57 (dd, J = 18.9, 3.3 Hz, 1H, H-3′), 5.23 (dd, J = 50.8, 3.2 Hz, 1H, H-2′), 4.75 (dd, J = 11.9, 4.0 Hz, 1H, H-5′), 4.67 (dd, J = 11.9, 4.7 Hz, 1H, H-5′), 4.48 (t, J = 3.0 Hz, 1H, NH), 4.32 (q, J = 4.1 Hz, 1H, H-4′), 4.01–3.91 (m, 2H, H-7), 3.85–3.75 (m, 2H, H-4) ppm; 13C NMR (126 MHz, CDCl3): δ 166.36 (C), 165.44 (C), 164.72 (C), 133.98 (CH-Ph), 133.36 (CH-Ph), 130.05 (2 × CH-Ph), 129.90 (2 × CH-Ph), 129.81 (C-Ph), 128.75 (2 × CH-Ph), 128.58 (2 × CH-Ph), 127.09 (CH-6), 126.84 (CH-5), 95.0 (d, J = 190.9 Hz, CH-2′), 86.2 (d, J = 16.0 Hz, CH-1′), 79.06 (CH-4′), 77.3 (d, J = 30.3 Hz, CH-3′), 63.76 (CH2-5′), 43.52 (CH2-4), 42.0 (d, J = 7.3 Hz, CH2-7) ppm; 19F NMR (470 MHz, CDCl3): δ −201.05 ppm; HRMS (ESI): m/z Calcd. for C24H24FN2O6 [M + H]+ 455.1613; found 455.1612.
1H NMR (500 MHz, D2O): δ 6.00–5.96 (m, 1H, H-5), 5.93–5.90 (m, 1H, H-4), 5.88 (dd, J = 18.1, 5.2 Hz, 1H, overlapping, H-1′), 5.07 (ddd, J = 53.0, 5.1, 3.7 Hz, 1H, H-2′), 4.33 (ddd, J = 22.8, 6.3, 3.7 Hz, 1H, H-3′), 3.96–3.85 (m, 3H, H-5′, H-7), 3.82–3.76 (m, 4H, H-4′, H-5′, H-3) ppm; 1H NMR (500 MHz, DMSO-d6): δ 6.10 (t, J = 2.9 Hz, 1H), 5.79–5.75 (m, 1H), 5.73–5.69 (m, 1H), 5.63 (dd, J = 21.2, 4.6 Hz, 1H), 5.63 (d, J = 5.1 Hz, 1H, overlapping), 4.88–4.85 (m, 1.5H), 4.77 (dd, J = 4.6, 3.0 Hz, 0.5H), 4.03 (ddd, J = 22.5, 7.9, 5.0 Hz, 1H), 3.76–3.66 (m, 2H), 3.60–3.54 (m, 3H), 3.53–3.47 (m, 2H) ppm; 13C NMR (126 MHz, D2O): δ 165.44 (CO, C-2), 127.88 (CH-4/5), 127.28 (CH-4/5), 96.95 (d, J = 190.5 Hz, CH-2′), 84.36 (d, J = 16.5 Hz, CH-1′), 80.08 (d, J = 6.1 Hz, CH-4′), 73.98 (d, J = 24.7 Hz, CH-3′), 60.46 (CH2-5′), 41.88 (d, J = 6.1 Hz, CH2-7), 41.65 (CH2-4) ppm; 13C NMR (126 MHz, DMSO-d6): δ 163.89 (CO, C-2), 127.27 (CH-4/5), 126.86 (CH-4/5), 97.86 (d, J = 189.4 Hz, CH-2′), 84.18 (d, J = 16.3 Hz, CH-1′), 81.45 (d, J = 5.0 Hz, CH-4′), 74.01 (d, J = 23.7 Hz, CH-3′), 60.60 (CH2, C-5′), 41.90 (CH2-4), 41.69 (d, J = 6.2 Hz, CH2-7), ppm; 19F NMR (470 MHz, DMSO-d6): δ −197.70 ppm; HRMS (ESI): m/z Calcd. for C10H16FN2O4 [M + H]+ 247.1089; found 247.1090.
:1) (0.79 g, 48%) as a pale-yellow viscid solid. This material was further purified by flash column chromatography using a silica gel column (SiliaSep, 40 g, gradient elution with 10–20% CH3CN/toluene) to provide the pure β-nucleoside 23 (0.46 g, 28%) as a clear viscid solid. TLC: Rf = 0.30 (β-anomer), Rf = 0.28 (α-anomer) (12% CH3CN/toluene).1H NMR (500 MHz, CDCl3): δ 8.08–8.03 (m, 4H, Ph), 7.63–7.60 (m, 1H, Ph), 7.58–7.55 (m, 1H, Ph), 7.48–7.45 (m, 2H, Ph), 7.44–7.41 (m, 2H, Ph), 5.99 (dd, J = 11.8, 10.7 Hz, 1H, H-1′), 5.76–5.69 (m, 2H, H4 and H5), 5.55 (ddd, J = 13.7, 6.4, 5.5 Hz, 1H, H-3′), 4.78 (dd, J = 12.2, 3.6 Hz, 1H, H-5′), 4.68 (br s, 1H, NH), 4.57 (dd, J = 12.2, 4.2 Hz, 1H, H-5′), 4.39 (dt, J = 7.1, 3.6 Hz, 1H, H-4′), 3.94–3.80 (m, 3H, H-3/7), 3.73–3.68 (m, 1H, H3/7) ppm; 13C NMR (126 MHz, CDCl3): δ 166.11 (CO-Bz), 165.06 (CO-Bz), 164.86 (C-2), 134.12 (CH-Ph), 133.51 (CH-Ph), 130.22 (2 × CH-Ph), 129.81 (2 × CH-Ph), 129.53 (C-Ph), 128.76 (2 × CH-Ph), 128.64 (2 × CH-Ph), 128.38 (C-Ph), 127.01 (CH-4/5), 126.16 (CH-4/5), 121.66 (t, J = 260.3 Hz, C-2′), 86.25 (dd, J = 37.4, 20.6 Hz, CH-1′), 75.81 (d, J = 5.4 Hz, CH-4′), 72.08 (dd, J = 32.5, 17.7 Hz, CH-3′), 62.91 (CH2-5′), 43.35 (CH2-4), 42.31 (d, J = 5.1 Hz, CH2-7) ppm; 19F NMR (470 MHz, DMSO-d6): δ −115.43 (d, J = 247.0), −116.75 (d, J = 246.9) ppm; HRMS (ESI): m/z Calcd. for C24H23F2N2O6 [M + H]+ 473.1519; found 473.1523.
1H NMR (500 MHz, D2O): δ 5.99–5.92 (m, 1H, H-4, H-5), 5.78 (dd, J = 14.6, 6.1 Hz, 1H, H-1′), 4.25 (ddd, J = 20.3, 11.6, 8.7 Hz, 1H, H-3′), 3.96 (d, J = 11.8 Hz, 1H, H-5′), 3.87–3.77 (m, 6H, H-4′, H5′, H-4, H-7) ppm; 1H NMR (500 MHz, DMSO-d6): δ 6.41 (t, J = 3.1 Hz, 1H), 6.02 (dd, J = 6.6 Hz, 1H), 5.81–5.75 (m, 2H), 5.60 (dd, J = 14.5, 7.3 Hz, 1H), 4.97 (t, J = 5.4 Hz, 1H), 4.02–3.93 (m, 1H), 3.72–3.51 (m, 7H) ppm; 13C NMR (126 MHz, D2O): δ 164.36 (CO, C-2), 127.13 (CH-4/5), 126.02 (CH-4/5), 122.00 (dd, J = 261.0, 252.4 Hz, CH-2′), 85.81 (dd, J = 41.5, 22.3 Hz, CH-1′), 77.23 (d, J = 8.9 Hz, CH-4′), 69.30 (d, J = 27.5, 18.5 Hz, CH-3′), 58.75 (CH2-5′), 40.82 (d, J = 4.0 Hz, CH2-7), 40.59 (CH2-4) ppm; 13C NMR (126 MHz, DMSO-d6): δ 163.45 (CO, C-2), 127.62 (CH-4/5), 126.58 (CH-4/5), 123.70 (dd, J = 259.6, 253.2 Hz, CH-2′), 85.85 (dd, J = 39.8, 22.0 Hz, CH-1′), 78.84 (d, J = 9.0 Hz, CH-4′), 69.87 (d, J = 26.7, 18.5 Hz, CH-3′), 59.48 (CH2-5′), 41.62 (CH2-4), 41.25 (d, J = 3.0 Hz, CH2-7) ppm; 19F NMR (470 MHz, DMSO-d6): δ −113.68 (d, J = 237.0), −115.16 (d, J = 237.2) ppm; HRMS(ESI): m/z Calcd. for C10H15F2N2O4 [M + H]+ 265.0994; found 265.0996.
Data availability
The data underlying this study are available in the published article and its online ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases of the NIH (R01 AI150478). AKH was supported by a fellowship from the PhRMA foundation.References
- T. Cacciamani, A. Vita, G. Cristalli, S. Vincenzetti, P. Natalini, S. Ruggieri, A. Amici and G. Magni, Arch. Biochem. Biophys., 1991, 290, 285–292 CrossRef CAS PubMed.
- A. Frances and P. Cordelier, Mol. Ther., 2020, 28, 357–366 CrossRef CAS PubMed.
- C. Serdjebi, G. Milano and J. Ciccolini, Expert. Opin. Drug Metab. Toxicol., 2015, 11, 665–672 CrossRef CAS PubMed.
- M. Petljak, L. B. Alexandrov, J. S. Brammeld, S. Price, D. C. Wedge, S. Grossmann, K. J. Dawson, Y. S. Ju, F. Iorio, J. M. C. Tubio, C. C. Koh, I. Georgakopoulos-Soares, B. Rodriguez-Martin, B. Otlu, S. O'Meara, A. P. Butler, A. Menzies, S. G. Bhosle, K. Raine, D. R. Jones, J. W. Teague, K. Beal, C. Latimer, L. O'Neill, J. Zamora, E. Anderson, N. Patel, M. Maddison, B. L. Ng, J. Graham, M. J. Garnett, U. McDermott, S. Nik-Zainal, P. J. Campbell and M. R. Stratton, Cell, 2019, 176, 1282–1294 CrossRef CAS PubMed.
- S. Revathidevi, A. K. Murugan, H. Nakaoka, I. Inoue and A. K. Munirajan, Cancer Lett., 2021, 496, 104–116 CrossRef CAS PubMed.
- M. V. Kvach, F. M. Barzak, S. Harjes, H. A. M. Schares, G. B. Jameson, A. M. Ayoub, R. Moorthy, H. Aihara, R. S. Harris, V. V. Filichev, D. A. Harki and E. Harjes, Biochemistry, 2019, 58, 391–400 CrossRef CAS PubMed.
- A. Maiti, A. K. Hedger, W. Myint, V. Balachandran, J. K. Watts, C. A. Schiffer and H. Matsuo, Nat. Commun., 2022, 13, 7117 CrossRef PubMed.
- A. K. Hedger, W. Myint, J. M. Lee, D. Suchenski-Loustaunau, V. Balachandran, A. M. Shaqra, N. Kurt-Yilmaz, J. K. Watts, H. Matsuo and C. A. Schiffer, bioRxiv, 2024, preprint, 2024.2009.2005.611238, DOI:10.1101/2024.09.05.611238.
- P. S. Liu, V. E. Marquez, J. S. Driscoll, R. W. Fuller and J. J. McCormack, J. Med. Chem., 1981, 24, 662–666 CrossRef CAS PubMed.
- G. Cristalli, R. Volpini, S. Vittori, E. Camaioni, G. Rafaiani, S. Potenza and A. Vita, Nucleosides Nucleotides, 1996, 15, 1567–1580 CrossRef CAS.
- S. Belyakov, B. Duvall, D. Ferraris, G. Hamilton and M. Vaal, WO2010/118006, 2010.
- D. Ferraris, B. Duvall, G. Delahanty, B. Mistry, J. Alt, C. Rojas, C. Rowbottom, K. Sanders, E. Schuck, K.-C. Huang, S. Redkar, B. B. Slusher and T. Tsukamoto, J. Med. Chem., 2014, 57, 2582–2588 CrossRef CAS PubMed.
- V. E. Marquez, P. S. Liu, J. A. Kelley and J. S. Driscoll, J. Org. Chem., 1980, 45, 485–489 CrossRef CAS.
- M. Kim, K. Gajulapati, C. Kim, H. Y. Jung, J. Goo, K. Lee, N. Kaur, H. J. Kang, S. J. Chung and Y. Choi, Chem. Commun., 2012, 48, 11443–11445 RSC.
- O. R. Ludek, G. K. Schroeder, C. Liao, P. L. Russ, R. Wolfenden and V. E. Marquez, J. Org. Chem., 2009, 74, 6212–6223 CrossRef CAS PubMed.
- V. E. Marquez, P. S. Liu and J. K. Linevsky, J. Org. Chem., 1982, 47, 1712–1717 CrossRef CAS.
- H. M. Kurup, M. V. Kvach, S. Harjes, G. B. Jameson, E. Harjes and V. V. Filichev, Org. Biomol. Chem., 2023, 21, 5117–5128 RSC.
- J. A. Kelley, J. S. Driscoll, J. J. McCormack, J. S. Roth and V. E. Marquez, J. Med. Chem., 1986, 29, 2351–2358 CrossRef CAS PubMed.
- N. Gimeno, P. Formentín, J. H. G. Steinke and R. Vilar, Eur. J. Org. Chem., 2007, 2007, 918–924 CrossRef.
- G. Vasquez, G. C. Freestone, W. B. Wan, A. Low, C. L. De Hoyos, J. Yu, T. P. Prakash, M. E. Østergaard, X.-h. Liang and S. T. Crooke, Nucleic Acids Res., 2021, 49, 1828–1839 CrossRef CAS PubMed.
- P. M. J. Jung, A. Burger and J.-F. Biellmann, J. Org. Chem., 1997, 62, 8309–8314 CrossRef CAS PubMed.
- H. Ueda and Y. Ueno, Bioorg. Med. Chem., 2022, 60, 116690 CrossRef CAS PubMed.
- H. Liu, J. Gao and E. T. Kool, J. Org. Chem., 2005, 70, 639–647 CrossRef CAS PubMed.
- K. Ebenryter-Olbinska, J. Karolak-Wojciechowska and E. Sochacka, Carbohydr. Res., 2014, 392, 7–15 CrossRef CAS PubMed.
- U. Niedballa and H. Vorbruggen, J. Org. Chem., 1974, 39, 3654–3660 CrossRef CAS PubMed.
- Y. Jian, G. Lin, L. Chomicz and L. Li, J. Am. Chem. Soc., 2015, 137, 3318–3329 CrossRef CAS PubMed.
- T.-X. Xiang, R. Niemi, P. Bummer and B. D. Anderson, J. Pharm. Sci., 2003, 92, 2027–2039 CrossRef CAS PubMed.
- C. J. Wilds and M. J. Damha, Nucleic Acids Res., 2000, 28, 3625–3635 CrossRef CAS PubMed.
- J. K. Watts, A. Katolik, J. Viladoms and M. J. Damha, Org. Biomol. Chem., 2009, 7, 1904–1910 RSC.
- V. E. Marquez, C. K. H. Tseng, H. Mitsuya, S. Aoki, J. A. Kelley, H. Ford Jr, J. S. Roth, S. Broder, D. G. Johns and J. S. Driscoll, J. Med. Chem., 1990, 33, 978–985 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07318e |
| This journal is © The Royal Society of Chemistry 2024 |