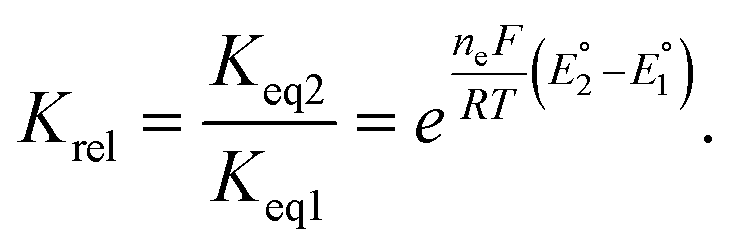DOI:
10.1039/D4RA06738J
(Paper)
RSC Adv., 2024,
14, 37797-37802
Can selenenyl sulfides be a substrate of glutathione reductase enzyme? A theoretical insight†
Received
18th September 2024
, Accepted 6th November 2024
First published on 26th November 2024
Abstract
Glutathione reductase (GR) catalyzes the reduction of glutathione disulfide (GSSG) to glutathione. As selenium is a congener of sulfur, the possibility of reducing selenenyl sulfide (RSeSG) at the catalytic site of GR has been investigated using density functional theory. Calculations on the redox potential and the Se–S bond strength of some studied RSeSG compounds with a phenyl selenide backbone suggested that the unsubstituted and amine-based selenenyl sulfide intermediates could have a promising tendency to be reduced at the catalytic site of GR.
1 Introduction
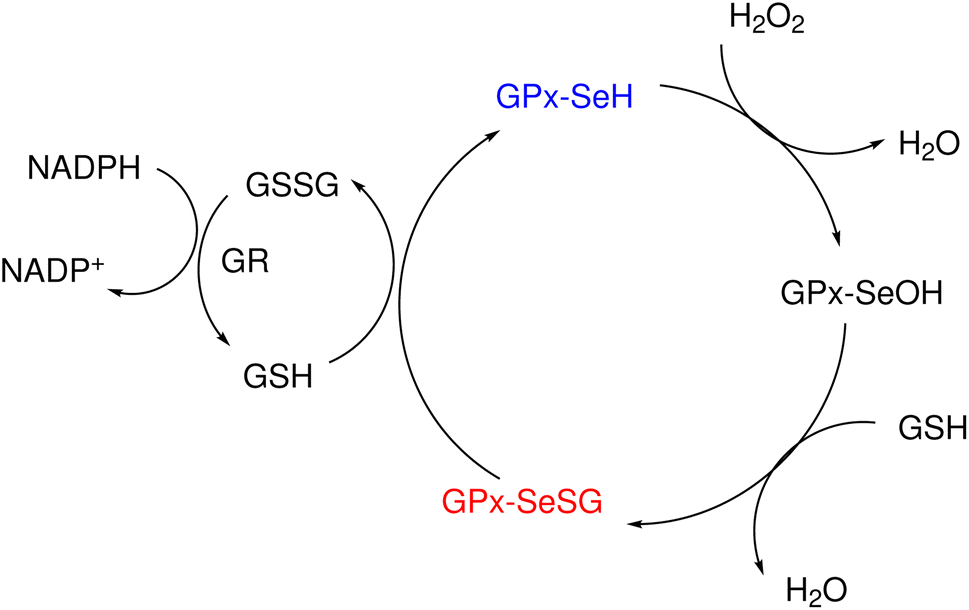
Glutathione peroxidase (GPx) plays an important role in regulating cellular oxidative stress. The enzymes in this family contain selenium in their active sites. In the catalytic cycle of GPx (Fig. 1), selenol (RSeH) is oxidized by reactive oxygen species (ROS), such as hydrogen peroxide or organic peroxides, to form selenenic acid (RSeOH). The elimination of ROS is followed by the regeneration of the catalyst in two essential events: regeneration of RSeH from RSeOH and regeneration of glutathione (GSH). The regeneration of RSeH proceeds through reduction of RSeOH to an important intermediate, selenenyl sulfide (RSeSG). This RSeSG is further reduced to selenol by another GSH molecule, forming glutathione disulfide (GSSG). GSH serves as the reagent necessary for regenerating the selenol in its reduced form (RSeH). In the cellular environment, GSSG is reduced back to GSH by glutathione reductase (GR) in the presence of NADPH1–4 as depicted in eqn (1a). During this redox process, cysteines in the GR are oxidized to disulfides (eqn (1b)), and consequently, GSSG is reduced to two molecules of GSH (eqn (1c)).| |
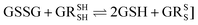 | (1a) |
| |
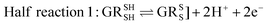 | (1b) |
| | |
Half reaction 2: GSSG + 2H+ + 2e− ⇌ 2GSH
| (1c) |
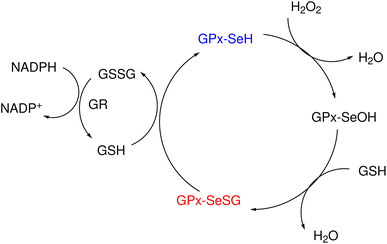 |
| | Fig. 1 Experimentally suggested mechanism for the catalytic cycle of GPx. | |
The Se–S bond appears naturally during the catalytic cycle of GPx when selenocysteine interacts with GSH in the active site of GPx. Besides GPx, the Se–S bond in selenenyl sulfides (RSe–SR′) is crucial in several other biological antioxidant regeneration enzymes such as thioredoxins.5,6 However, in many cases, this bond is inherently unstable under standard atmospheric pressures and temperatures, posing significant challenges in studying its properties.2 Previous studies7,8 have demonstrated that compounds containing Se–S bonds exhibit antioxidant capabilities in animal studies. Moreover, selenenyl sulfides have emerged as promising agrochemical candidates9 and have shown potential to act as antiviral inhibitors.10
After the successful discovery of ebselen as an antioxidant and anti-inflammatory drug, many small organoselenium compounds have been studied which mimic the antioxidant activity of GPx enzymes,11,12 and many of them show a catalytic cycle similar to the (GPx) enzymes.13–15 Their GPx-like activity has been evaluated using chemical, biochemical or electrochemical assays. The chemical assay is based on the rate of oxidation of an organic thiol (e.g. thiophenol (PhSH)) which forms the corresponding selenenyl sulfide RSeSPh, an important intermediate in the GPx-like catalytic cycle. This commonly used assay is prone to a thiol-exchange reaction when the Se atom in the selenenyl sulfide is more electrophilic than the S atom of the incoming thiol,12,16 thus limiting its usefulness. The coupled reductase assay involves the reduction of GSSG by GR in the presence of GSH and NADPH.4,17–20 This biochemical assay mimics the biological conditions in which the GPx enzymes function and is not limited by the thiol-exchange reaction. On the other hand, the electrochemical assay directly measures the rate of peroxide reduction employing a hydrogen peroxide selective electrode, which is capable of working in diverse thiol substrates and is free of any potential artefacts arising due to interaction of the organoselenium compound with any components of the assay.21
While comparing the activity of ebselen and diphenyldiselenide using an electrochemical assay and coupled reductase assay, Giles et al.21 observed a two- to four-fold higher activity in the case of the coupled reductase assay. The observed increase in the activity for the coupled reductase assay compared to that of the electrochemical assay was proposed due to the interaction of the organoselenium compounds with GR. In a recent study,22 it was observed that some of the derivatives of ebselen lacked thiophenol assay activity, while they showed similar or better activity in the coupled reductase assay. A similar observation was recorded by Yu et al.,23 who suggested that the selenenyl sulfide intermediate could be responsible for showing the activity in the coupled reductase assay for the mimics, which lacked the activity in the thiophenol assay. Employing a molecular docking study, they showed that there is a possibility for a RSeSG-type intermediate to react at the catalytic site of GR, leading to the production of GSH and RSeH. This process is similar to the regeneration of two GSH molecules from GSSG at the active site of GR within the enzymatic cycle (eqn (1c)). These findings imply that RSeSG could mimic the behaviour of GSSG at the catalytic site of GR.
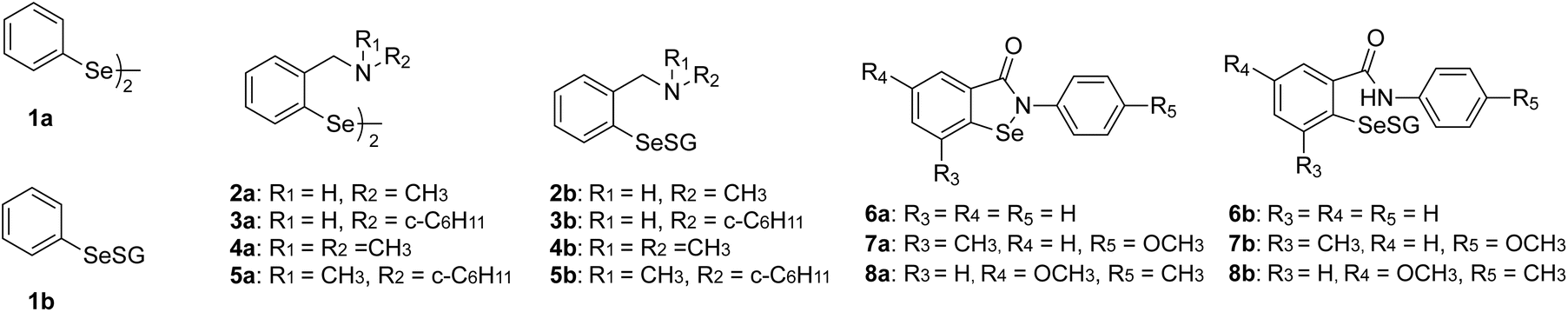
Based on these observations, we hypothesise that selenenyl sulfide can act as a substrate to GR. Accordingly, we consider the plausible redox reactions given in eqn (2a)–(2c) to model the conversion of RSeSG to RSeH (hereafter eqn (2c) will be denoted as RSeSG|(RSeH + GSH)). We have chosen RSeSG-type intermediates belonging to diphenyl diselenide 1a, amine-based diselenides 2a to 5a, and ebselene-based 6a to 8a (amides) mimics for the investigation. The studied RSeSG intermediates, along with the parent GPx mimics, are given in Fig. 2. Factors influencing the catalyst regeneration include the redox potential of RSeSG|(RSeH + GSH) and the ease with which the Se–S bond breaks, etc., have been investigated.
| |
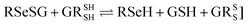 | (2a) |
| |
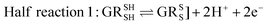 | (2b) |
| | |
Half reaction 2: RSeSG + 2H+ + 2e− ⇌ RSeH + GSH
| (2c) |
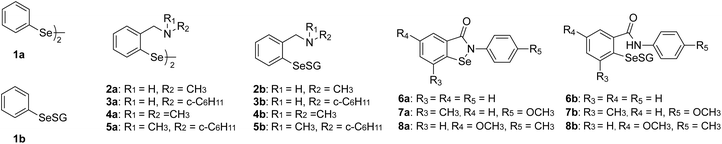 |
| | Fig. 2 Nomenclature of the parent GPx mimics (1a–8a) and the corresponding selenenyl sulfide intermediates (1b–8b) that were investigated. | |
2 Computational methodology
Geometry optimization of all the studied compounds was carried out using Gaussian 16.24 Unless specified, all the calculations were performed at the M06-2X/6-311++g(2df,2pd) level of theory. Frequency calculations were conducted to verify that the optimized geometry has no imaginary frequency and, thus, is a minimum on the potential-energy surface. The solvent effect was modeled using the self-consistent reaction field (SCRF) approach utilizing the polarizable continuum model (PCM) with water as the solvent. The free energy of species in the solvent was calculated from the corresponding gas-phase geometries using a single-point calculation.25 The non-covalent interactions in the intermediates were studied using NBO analysis as implemented in Gaussian 16.
The standard reduction potential 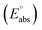 of the half-reaction
of the half-reaction
can be calculated from its standard state free energy (SFE) from the relation
| |
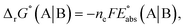 | (4) |
where the oxidized species A gets reduced to B in the reaction by exchanging
ne number of electrons. “s” and “g” in the subscript or parentheses represents the species in solution and the gas phase, respectively.
F is the Faraday constant.
The SFE of a reaction in solution 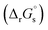 can be calculated from its corresponding gas-phase SFE
can be calculated from its corresponding gas-phase SFE 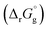 using the thermodynamic (Born–Haber) cycle (Fig. 3) as follows:26–28
using the thermodynamic (Born–Haber) cycle (Fig. 3) as follows:26–28
| |
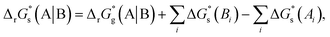 | (5) |
where the 2nd and 3rd terms on the RHS represent the SFE of solvation for the products and the reactants, respectively. The index
i goes over all the reactants or products.
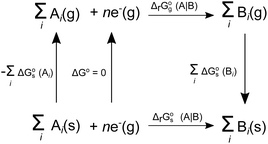 |
| | Fig. 3 Thermodynamic cycle designed to calculate 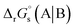 . Here, . Here, 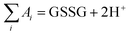 (or RSeSG + 2H+) and (or RSeSG + 2H+) and 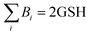 (or RSeH + GSH) for the half-reaction (1c) (or (2c)). (or RSeH + GSH) for the half-reaction (1c) (or (2c)). | |
The difference in SFE between the gas-phase reference state (1 atm, 24.46 L at 298.15 K) and that of the PCM solvation calculation (1 M) is calculated as per eqn (6):27
| |
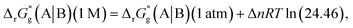 | (6) |
where Δ
n is the net change of moles in
Fig. 3,
R is the gas constant and
T is absolute temperature. The standard redox potential relative to the standard hydrogen electrode (SHE) for (A|B) can be expressed according to
eqn (7).
| |
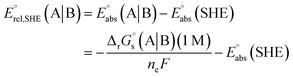 | (7) |
We have used 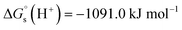 ,26,29
,26,29 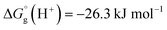
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27,30 and
27,30 and 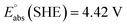 26 at T = 298.15 K in this work.
26 at T = 298.15 K in this work.
The homolytic bond-dissociation energy (BDE) of the X–Y bond is calculated using the following equation:
| | |
BDE = ΔfH(R1X) + ΔfH(R2Y) − ΔfH(R1X–YR2),
| (8) |
in which R1X–YR2 is the neutral molecule, and R1X and R2Y are the corresponding radicals. The geometries of the radicals were optimized at the unrestricted M06-2X/6-311++g(2df,2pd) level of theory.
3 Results and discussion
3.1 Redox potential and feasibility of the reaction of RSeSG at GR
The method for calculating the redox potential of RSeSG (shown in Fig. 2) follows a similar approach to that used for the GSSG, as outlined in the ESI† (Justification of functionals). Similar to the GSSG|2GSH couple, the redox potentials of RSeSG|(RSeH + GSH) couple (eqn (2c)) were calculated from 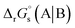 , where
, where 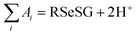 and
and 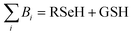 , using the thermodynamic cycle shown in Fig. 3 and are given in Table 1. The calculated redox potentials for amine-based selenenyl sulfides 2b to 5b are about 10–40 mV more positive than that for GSSG, whereas the ebselen-based selenenyl sulfide intermediates exhibit redox potential values that are about 110–150 mV more negative than that for GSSG. The reported values of the redox potentials of selenenyl sulfides depend on various conditions, and range from −0.381 to −0.215 V at pH = 7.31–33
, using the thermodynamic cycle shown in Fig. 3 and are given in Table 1. The calculated redox potentials for amine-based selenenyl sulfides 2b to 5b are about 10–40 mV more positive than that for GSSG, whereas the ebselen-based selenenyl sulfide intermediates exhibit redox potential values that are about 110–150 mV more negative than that for GSSG. The reported values of the redox potentials of selenenyl sulfides depend on various conditions, and range from −0.381 to −0.215 V at pH = 7.31–33
Table 1 Important thermodynamic and structural parametersa of the studied compounds using the M06-2X/6-311++g(2df,2pd) level of theory
| Compd |
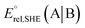
b |
Krelc |
rS–Se/Sd |
rN⋯Sed |
∠N⋯Se–Sd |
BDEe |
E2f |
Illustrative figures (Fig. S1 and S2) showing the structural parameters are included in the ESI.† Calculated redox potentials, 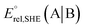 at pH = 7 in volts; here A|B is either the RSeSG|(RSeH + GSH) or GSSG|2GSH couple. Krel represents the relative equilibrium constant with respect to that of the GSSG|2GSH pair. S–Se/S, and N⋯Se, bond distance in Å, bond angle N⋯Se–S in degrees. The gas-phase bond dissociation energy (BDE) for the S–Se/S bond in kcal mol−1. Second-order perturbation energy (E2) from NBO for the Se⋯N non-covalent interactions in kcal mol−1. at pH = 7 in volts; here A|B is either the RSeSG|(RSeH + GSH) or GSSG|2GSH couple. Krel represents the relative equilibrium constant with respect to that of the GSSG|2GSH pair. S–Se/S, and N⋯Se, bond distance in Å, bond angle N⋯Se–S in degrees. The gas-phase bond dissociation energy (BDE) for the S–Se/S bond in kcal mol−1. Second-order perturbation energy (E2) from NBO for the Se⋯N non-covalent interactions in kcal mol−1. |
| 1b |
−0.251 |
1.95 × 101 |
2.178 |
— |
— |
56.8 |
— |
| 2b |
−0.252 |
1.88 × 101 |
2.204 |
2.833 |
173.6 |
53.6 |
8.11 |
| 3b |
−0.257 |
1.24 × 101 |
2.207 |
2.792 |
173.9 |
53.9 |
9.20 |
| 4b |
−0.264 |
6.96 |
2.209 |
2.770 |
174.5 |
53.2 |
9.74 |
| 5b |
−0.280 |
2.02 |
2.212 |
2.726 |
175.4 |
53.5 |
10.56 |
| GSSG |
−0.289 |
1.00 |
2.055 |
— |
— |
65.3 |
— |
| 6b |
−0.396 |
2.50 × 10−4 |
2.194 |
— |
— |
63.6 |
— |
| 7b |
−0.404 |
1.38 × 10−4 |
2.198 |
— |
— |
62.1 |
— |
| 8b |
−0.443 |
6.42 × 10−6 |
2.196 |
— |
— |
69.1 |
— |
| 2′b |
−0.231 |
9.78 × 101 |
2.204 |
2.925 |
159.0 |
52.9 |
4.85 |
More positive redox potentials (Table 1) of the amine-based intermediates (2b to 5b) than those of GSSG or the ebselen-based selenenyl sulfide intermediates (6b to 8b) indicate that the amine-based intermediates have a higher tendency to be reduced to form selenol – the active catalyst in the GPx-like catalytic cycle. For the amine-based intermediates 2b to 5b, the rSe⋯N distances are found to be smaller than the sum of their van der Waals radii34 (rvdW(Se) + rvdW(N)), and also the N⋯Se–S angles are almost linear (173–175°) (see Table 1, and Fig. S1 and S2 in the ESI†) indicating the presence of non-covalent Se⋯N interactions. The strength of the (Se⋯N) interactions (which involve 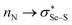 orbitals)35,36 obtained by the NBO second-order perturbation energy (E2) are also given in Table 1. The Se⋯N interaction is relatively weaker in intermediates with secondary amino groups (2b and 3b), compared to intermediates with tertiary amino groups (4b and 5b). Thus, E2 for these intermediates increases as the calculated redox potential decreases, indicating that a stronger Se⋯N interaction results in a weaker reducing ability of the selenenyl sulfide intermediates. Specifically, intermediates with a methyl group as the only substituent on the amino nitrogen, 2b (8.11 kcal mol−1, −0.252 V) and 4b (9.74 kcal mol−1, −0.264 V) exhibit a relatively weaker Se⋯N interaction and, consequently, more positive redox potentials compared to their counterparts with cyclohexyl substitution 3b (9.20 kcal mol−1, −0.257 V) and 5b (10.56 kcal mol−1, −0.280 V). This observation suggests that the presence of a cyclohexyl ring on the amino nitrogen enhances the Se⋯N interaction. The absence of Se⋯N non-covalent interactions in ebselen-based intermediates (6b–8b) could be a reason for showing relatively more negative redox potentials than glutathione.
orbitals)35,36 obtained by the NBO second-order perturbation energy (E2) are also given in Table 1. The Se⋯N interaction is relatively weaker in intermediates with secondary amino groups (2b and 3b), compared to intermediates with tertiary amino groups (4b and 5b). Thus, E2 for these intermediates increases as the calculated redox potential decreases, indicating that a stronger Se⋯N interaction results in a weaker reducing ability of the selenenyl sulfide intermediates. Specifically, intermediates with a methyl group as the only substituent on the amino nitrogen, 2b (8.11 kcal mol−1, −0.252 V) and 4b (9.74 kcal mol−1, −0.264 V) exhibit a relatively weaker Se⋯N interaction and, consequently, more positive redox potentials compared to their counterparts with cyclohexyl substitution 3b (9.20 kcal mol−1, −0.257 V) and 5b (10.56 kcal mol−1, −0.280 V). This observation suggests that the presence of a cyclohexyl ring on the amino nitrogen enhances the Se⋯N interaction. The absence of Se⋯N non-covalent interactions in ebselen-based intermediates (6b–8b) could be a reason for showing relatively more negative redox potentials than glutathione.
To further understand the tendency of RSeSG reduction at the catalytic site of GR, we define a relative equilibrium constant Krel as follows. The thiol–disulfide equilibrium represents a dynamic state where the rates of the forward and reverse reactions are balanced to sustain constant concentrations of reactants and products during the reduction of GSSG at GR as depicted in eqn (1a). The equilibrium constant Keq is related to the standard free-energy change ΔG° and standard electrode potential through eqn (9):
| |
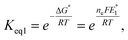 | (9) |
where
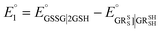
. Similarly, the equilibrium constant for the RSeSG reduction (
eqn (2a)) can be written as
| |
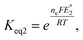 | (10) |
where
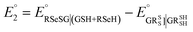
. Assuming that the RSeSG and GSSG interact similarly with the enzyme, the
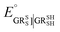
terms may cancel out, and an equilibrium constant for RSeSG conversion relative to that of GSSG conversion may be defined as
| |
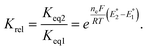 | (11) |
The relative equilibrium constant, Krel depends on the redox potentials of RSeSG and GSSG and indicates the ability of RSeSG to be reduced at the active site of GR in comparison to GSSG. The calculated values of Krel (Table 1) imply that the unsubstituted phenyl selenide intermediate 1b would have a higher tendency to be reduced at the GR, then the secondary-amine-containing analogues 2b and 3b, followed by the tertiary amines 4b and 5b. Conversely, ebselen and its substituted compounds (6b to 8b) show a much lower tendency (Krel) to be reduced compared to GSSG. The compounds having a relatively positive redox potential tend to reach equilibrium rapidly.33
We have carried out a molecular docking study taking GSSG and RSeSG (as ligands) at the active site of GR. This study shows that many of the studied ligands can approach the active site of GR in a range of 5 Å, indicating that there is a possibility of interactions between some of the RSeSG and GR. The details are given in the ESI (Fig. S3, S4, and Table S3).†
3.2 Bond dissociation energy
To facilitate the easy reduction of selenenyl sulfides (RSeSG) to selenol (RSeH) and thiol (GSH) at the catalytic site of GR, the RSeSG intermediates should not only possess a more positive redox potential, but also have a weaker Se–S bond. The calculated value of BDE for the disulfide bond in GSSG is found to be 65.3 kcal mol−1, which is comparable to the experimental value of a typical disulfide bond (e.g. 66.2 kcal mol−1 for dialkyl disulfide37). The calculated Se–S BDE data and bond distances are presented in Table 1. The Se–S BDEs of all the RSeSG compounds (except 8b) are lower than the S–S BDE of GSSG. Notably, all our studied intermediates contain a phenyl selenide backbone. Compared to the parent phenyl selenenyl sulfide 1b, the amine-substituted selenenyl sulfides (2b to 5b) exhibit Se–S BDEs that are about 3 kcal mol−1 lower (ranging from 53.2 to 53.9 kcal mol−1), while those amide-substituted (ebselen-based) selenenyl sulfides (6b to 8b) showed BDEs that are higher by amounts ranging from approximately 5 to 12 kcal mol−1 (with values ranging from 62.1 to 69.1 kcal mol−1).
Among the amide-substituted (ebselen-based) compounds, 6b shows a relatively more positive redox potential (than 7b or 8b), while 7b has the lowest BDE (compared to 6b and 8b). Using a coupled reductase assay it was reported recently that 7a (the parent compound of 7b) showed better activity than those of 6a and 8a (about 1.6 times better than that of 6a, while 6a showed an activity about 1.2 times higher than 8a).22 Since RSeSG is formed during the coupled reductase assay, a lower activity of 8a could be ascribed to its relatively higher Se–S BDE. However, the relatively better activity of 7a compared to 6a indicated that the interplay of redox potential and Se–S BDE would play a crucial role during the reduction of RSeSG at GR.
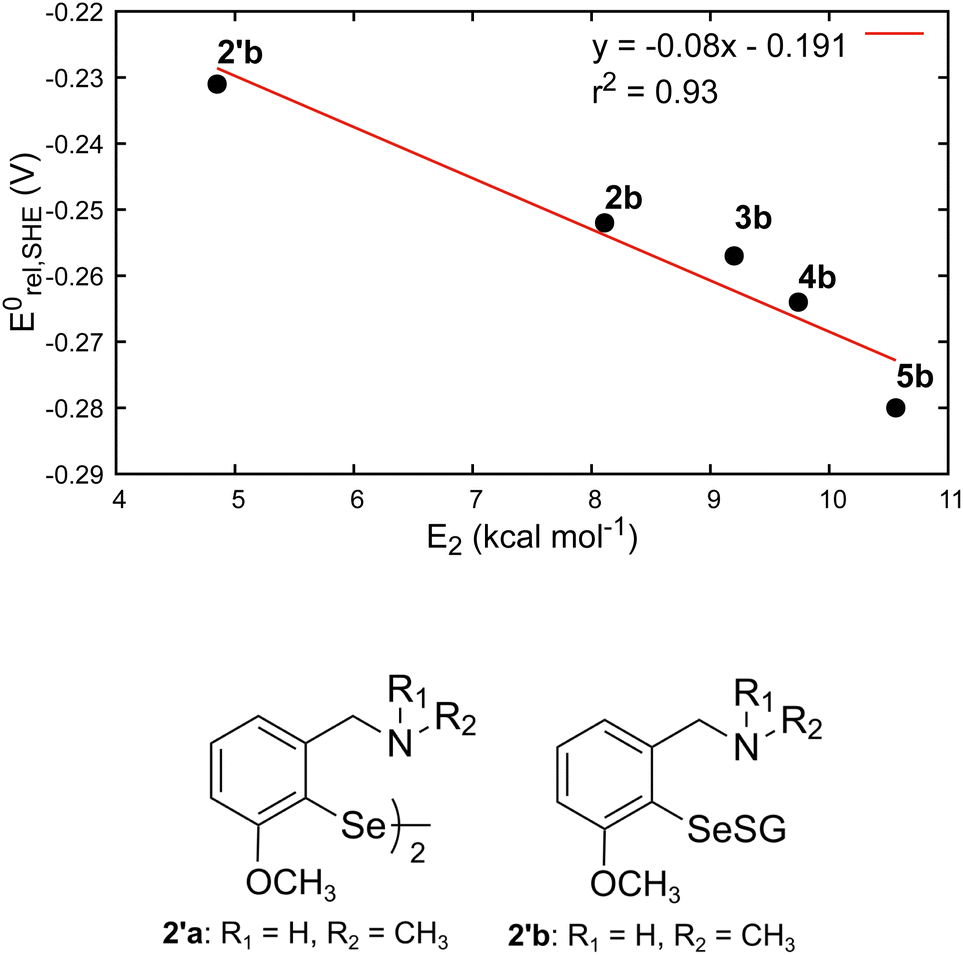
The amine-substituted compounds (2b to 5b) showed the lowest Se–S BDEs, while the parent phenyl selenenyl sulfide (1b) showed the most positive redox potential. The lower BDEs of 2b to 5b may be attributed to the presence of Se⋯N non-covalent interactions, which is known to weaken the Se–S bond.36,38 As the strength of these interactions (E2) decreases, the BDEs change a little, while the redox potential shifts towards more positive values (reaching close to that of 1b). This indicates that the strength of the Se⋯N interactions altered not only the Se–S BDE but also the redox potential of RSeSG. To test this hypothesis, we chose the parent compound 2′a, which was found to give better catalytic activity in a thiophenol assay during previous testing of this family of amine-based GPx mimics.39 The results on its selenenyl sulfide intermediate 2′b are given in Table 1 and Fig. 4. As expected, 2′b showed the weakest Se⋯N interaction (E2 = 4.85 kcal mol−1), the lowest Se–S BDE (52.9 kcal mol−1), and the most positive redox potential (–0.231 V). Also, we find a very good correlation (R2 = 0.93) between the strength of the Se⋯N interaction and the redox potential among the studied amine-substituted RSeSG compounds (Fig. 4). Although it is reported that ebselen-based compounds exhibit Se⋯O interactions,40 NBO analysis of the ebselen-based compounds under study did not show any Se⋯O or Se⋯N interactions. The Se⋯O distances in the present case are in the range of 3.07 to 3.13 Å (Fig. S2†).
 |
| | Fig. 4 Correlations between the strength of the Se⋯N non-covalent interaction (E2) and the calculated redox potential for amine-based RSeSGs (top). Structure of 2′a and 2′b (bottom). | |
Thus, the presence of Se⋯N non-covalent interactions in RSeSG lowers the Se–S BDE, while a moderate (not too strong) value of such interactions results in a higher positive redox potential and may modulate its peroxidase activity. This prediction is consistent with the literature results that some of the most successful GPx mimics contain a selenium–nitrogen interaction (Se–N bond or a close Se⋯N interaction).38 Mimics with weaker Se⋯N interactions (E2 < ∼7 kcal mol−1) showed relatively higher activities, while those with strong Se⋯N interactions (E2 > ∼11 kcal mol−1) showed relatively lower activities.41 The presence of strong Se⋯N interactions in the selenenyl sulfide intermediates has shown a preference for the thiol-exchange reactions over the regeneration of active selenol (RSeH), thereby reducing the GPx activity significantly.12,16
4 Conclusions
In summary, we have explored the possibility of reduction of potential selenenyl sulfide intermediates (RSeSG) at the catalytic site of GR. The diphenyl diselenide-based selenenyl sulfides (1b to 5b, and 2′b) showed more positive redox potentials and lower Se–S BDEs than GSSG. The molecular docking study indicated a closer approach of some of these RSeSG compounds in proximity to the active site of GR (∼5 Å), suggesting that these species can potentially be reduced at the catalytic site of GR. The presence of Se⋯N non-covalent interactions alters both the Se–S BDEs as well as the redox potentials of RSeSG. Thus, fine-tuning the strength of these non-covalent interactions could be a way to modulate the GPx activity and identify more potent antioxidants.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Vishnu Rama Chari: conceptualization, investigation, writing – original draft, visualization. Raghu Nath Behera: conceptualization, investigation, writing – review & editing, funding acquisition, supervision.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Science and Engineering Research Board (Project No. EMR/2016/003210). We acknowledge the DST-FIST supported computational facility to the Department of Chemistry, and the HPC facility of Birla Institute of Technology and Science Pilani, K. K. Birla Goa Campus for this work. VRC is thankful to UGC-SAP and DST-FIST funding to School of Chemical Sciences, Goa University. We thank the anonymous reviewers for their helpful comments and suggestions.
Notes and references
- L. Flohé, Glutathione Peroxidase, in, Ciba Foundation Symposium 65 - Oxygen Free Radicals and Tissue Damage, ed. D. W. Fitzsimons, 1979, pp. 95–122 Search PubMed.
- A. Hamsath and M. Xian, Antioxid. Redox Signaling, 2020, 33, 1143–1157 CrossRef CAS PubMed.
- E. Lubos, J. Loscalzo and D. E. Handy, Antioxid. Redox Signaling, 2011, 15, 1957–1997 CrossRef CAS PubMed.
- M. D. Tiezza, G. Ribaudo and L. Orian, Curr. Org. Chem., 2019, 23, 1381–1402 CrossRef CAS.
- B. K. Sarma and G. Mugesh, Org. Biomol. Chem., 2008, 6, 965–974 RSC.
- C. Gallo-Rodriguez and J. B. Rodriguez, ChemMedChem, 2024, 19, e202400063 CrossRef CAS PubMed.
- C. V. Rao, C. Wang, B. Simi, J. G. Rodriguez, I. Cooma, K. El-Bayoumy and B. S. Reddy, Cancer Res., 2001, 61, 3647–3652 CAS.
- M. Prigol, E. A. Wilhelm, C. C. Schneider and C. W. Nogueira, Chem.-Biol. Interact., 2008, 176, 129–136 CrossRef CAS PubMed.
- M.-H. Shang, X.-W. Sun, H.-L. Wang, H.-R. Li, J.-S. Zhang, L.-Z. Wang, S.-J. Yu, X. Zhang, L.-X. Xiong and Y.-H. Li, et al., Pest Manage. Sci., 2023, 79, 1885–1896 CrossRef CAS PubMed.
- J.-Y. Tang, S. Dai, X. Wang, M. Zhang, J.-R. Shi, Y.-X. Hong, Z.-J. Sun, H.-Q. Dai and J.-G. Wang, Arabian J. Chem., 2024, 17, 105713 CrossRef CAS.
- N. V. Barbosa, C. W. Nogueira, P. A. Nogara, A. F. de Bem, M. Aschner and J. B. Rocha, Metallomics, 2017, 9, 1703–1734 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Acc. Chem. Res., 2010, 43, 1408–1419 CrossRef CAS PubMed.
- M. Iwaoka and S. Tomoda, J. Am. Chem. Soc., 1994, 116, 2557–2561 CrossRef CAS.
- G. S. Heverly-Coulson and R. J. Boyd, J. Phys. Chem. A, 2010, 114, 1996–2000 CrossRef CAS PubMed.
- J. K. Pearson and R. J. Boyd, J. Phys. Chem. A, 2007, 111, 3152–3160 CrossRef CAS PubMed.
- B. K. Sarma and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 11477–11485 CrossRef CAS PubMed.
- V. P. Singh, J.-f. Poon, R. J. Butcher, X. Lu, G. Mestres, M. K. Ott and L. Engman, J. Org. Chem., 2015, 80, 7385–7395 CrossRef CAS PubMed.
- M. Kumar and V. P. Singh, New J. Chem., 2022, 46, 12010–12022 RSC.
- G. Mugesh and H. B. Singh, Chem. Soc. Rev., 2000, 29, 347–357 RSC.
- E. F. Pai and G. E. Schulz, J. Biol. Chem., 1983, 258, 1752–1757 CrossRef CAS PubMed.
- N. M. Giles, S. Kumari, R. A. Stamm, S. Patel and G. I. Giles, Anal. Biochem., 2012, 429, 103–107 CrossRef CAS PubMed.
- M. Kumar, B. Chhillar, D. Verma, S. Nain and V. P. Singh, J. Org. Chem., 2023, 88, 4273–4285 CrossRef CAS PubMed.
- S.-C. Yu, I.-C. Kim, K.-J. Ri, J. Ri and H. Kuehn, J. Inorg. Biochem., 2021, 215, 111276 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian-16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- M.-H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407–7412 CrossRef CAS.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- H. Mohammad-Shiri, M. Ghaemi, S. Riahi and A. Akbari-Sehat, Int. J. Electrochem. Sci., 2011, 6, 317–336 CrossRef CAS.
- O. Hammerich and B. Speiser, Organic Electrochemistry: Revised and Expanded, CRC Press, 2015 Search PubMed.
- M. D. Liptak, K. C. Gross, P. G. Seybold, S. Feldgus and G. C. Shields, J. Am. Chem. Soc., 2002, 124, 6421–6427 CrossRef CAS PubMed.
- J. J. Fifen, Z. Dhaouadi and M. Nsangou, J. Phys. Chem. A, 2014, 118, 11090–11097 CrossRef CAS PubMed.
- D. Besse, F. Siedler, T. Diercks, H. Kessler and L. Moroder, Angew. Chem., Int. Ed. Engl., 1997, 36, 883–885 CrossRef CAS.
- F. Li, P. B. Lutz, Y. Pepelyayeva, E. S. Arnér, C. A. Bayse and S. Rozovsky, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6976–6981 CrossRef CAS PubMed.
- K. Arai, T. Matsunaga, H. Ueno, N. Akahoshi, Y. Sato, G. Chakrabarty, G. Mugesh and M. Iwaoka, Chem.–Eur. J., 2019, 25, 12751–12760 CrossRef CAS PubMed.
- A. v. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- A. Panda and R. N. Behera, J. Hazard. Mater., 2014, 269, 2–8 CrossRef CAS PubMed.
- R. N. Behera and A. Panda, RSC Adv., 2012, 2, 6948–6956 RSC.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357–4416 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2009, 15, 9846–9854 CrossRef CAS PubMed.
- S. Antony and C. A. Bayse, Inorg. Chem., 2011, 50, 12075–12084 CrossRef CAS PubMed.
- K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2008, 14, 8640–8651 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
Raghu Nath Behera
ab and
Raghu Nath Behera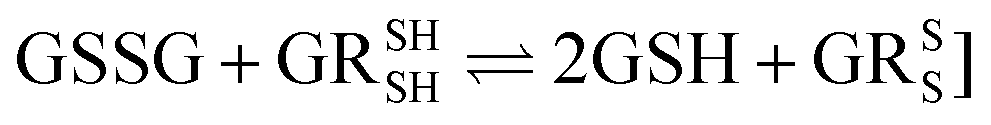
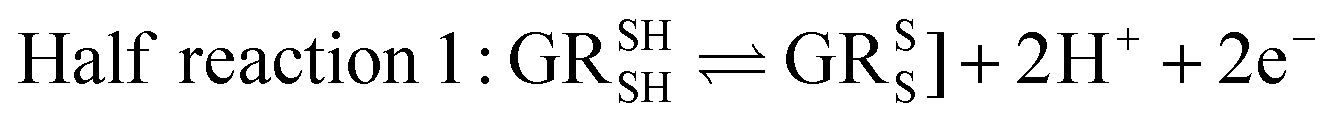
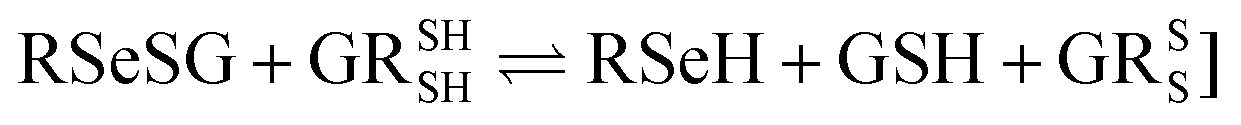
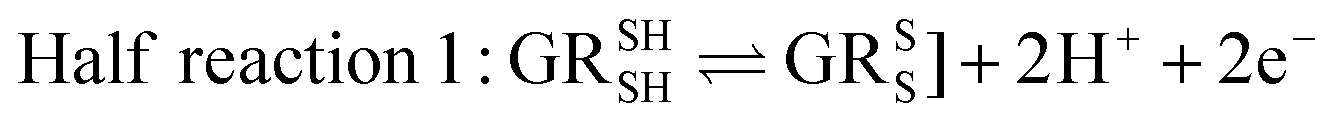
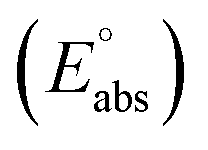 of the half-reaction
of the half-reaction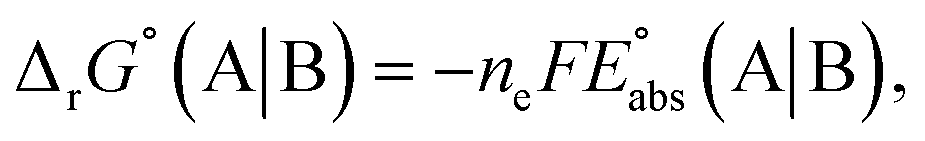
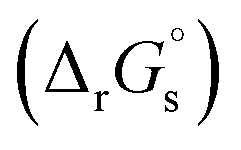 can be calculated from its corresponding gas-phase SFE
can be calculated from its corresponding gas-phase SFE 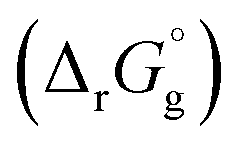 using the thermodynamic (Born–Haber) cycle (Fig. 3) as follows:26–28
using the thermodynamic (Born–Haber) cycle (Fig. 3) as follows:26–28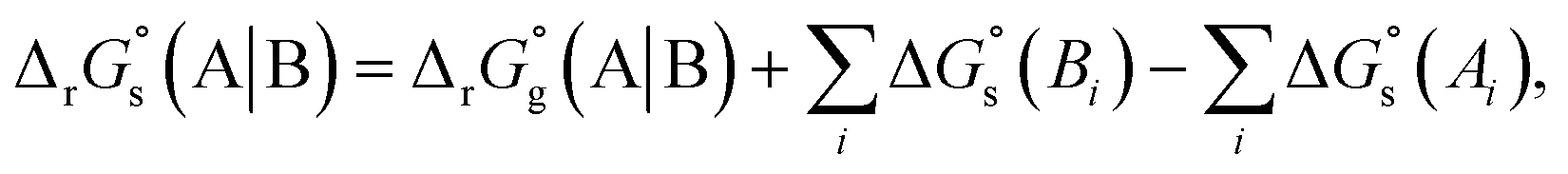
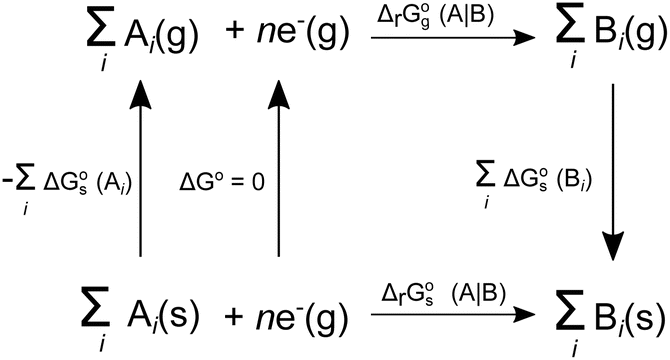
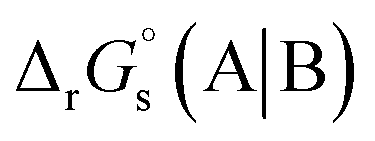 . Here,
. Here, 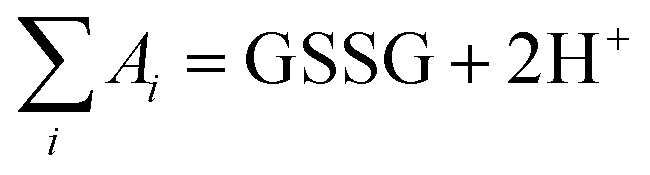 (or RSeSG + 2H+) and
(or RSeSG + 2H+) and 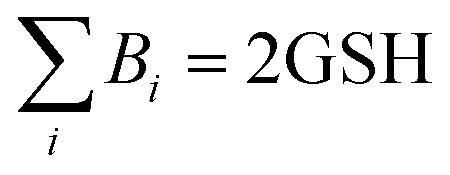 (or RSeH + GSH) for the half-reaction (1c) (or (2c)).
(or RSeH + GSH) for the half-reaction (1c) (or (2c)).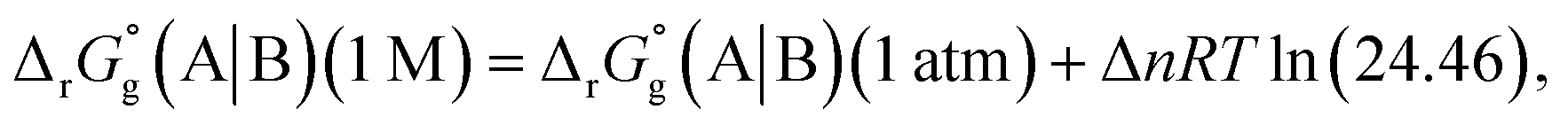
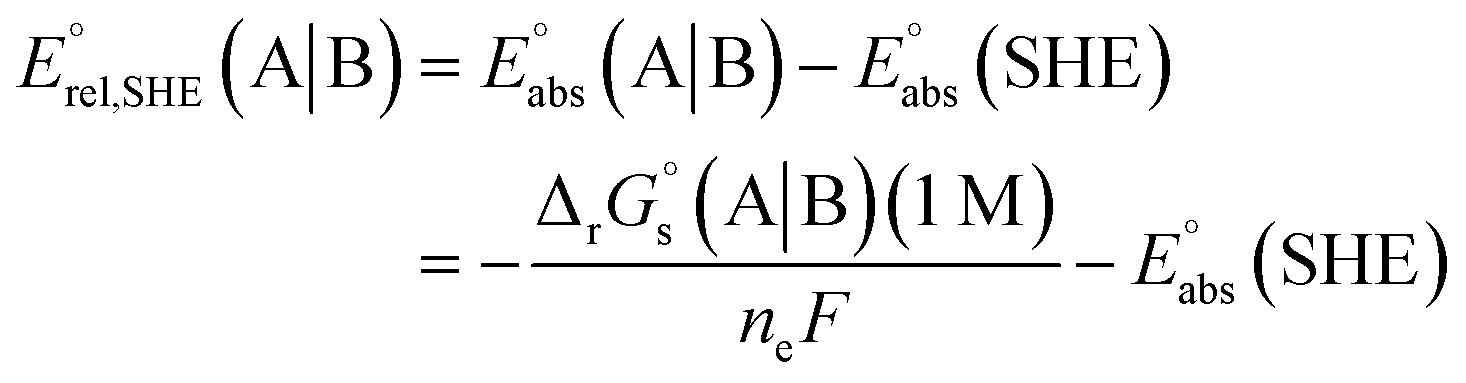
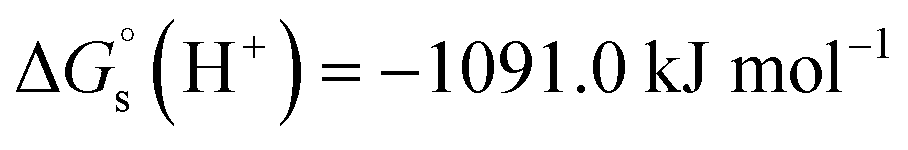 ,26,29
,26,29 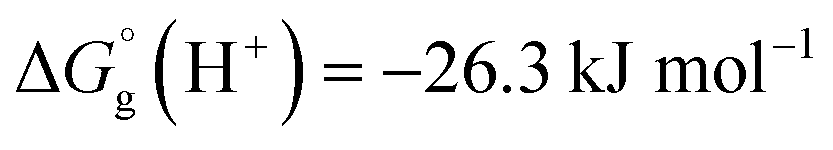
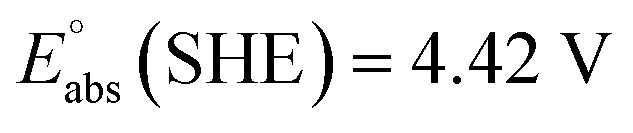
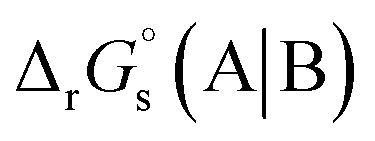 , where
, where 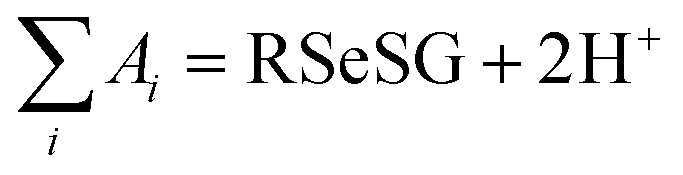 and
and 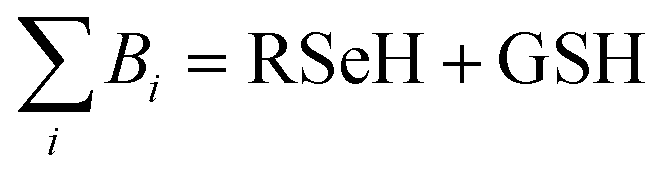 , using the thermodynamic cycle shown in Fig. 3 and are given in Table 1. The calculated redox potentials for amine-based selenenyl sulfides 2b to 5b are about 10–40 mV more positive than that for GSSG, whereas the ebselen-based selenenyl sulfide intermediates exhibit redox potential values that are about 110–150 mV more negative than that for GSSG. The reported values of the redox potentials of selenenyl sulfides depend on various conditions, and range from −0.381 to −0.215 V at pH = 7.31–33
, using the thermodynamic cycle shown in Fig. 3 and are given in Table 1. The calculated redox potentials for amine-based selenenyl sulfides 2b to 5b are about 10–40 mV more positive than that for GSSG, whereas the ebselen-based selenenyl sulfide intermediates exhibit redox potential values that are about 110–150 mV more negative than that for GSSG. The reported values of the redox potentials of selenenyl sulfides depend on various conditions, and range from −0.381 to −0.215 V at pH = 7.31–33
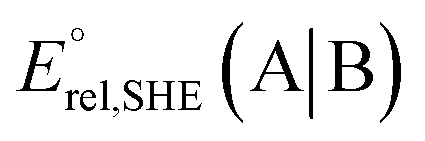 at pH = 7 in volts; here A|B is either the RSeSG|(RSeH + GSH) or GSSG|2GSH couple.c Krel represents the relative equilibrium constant with respect to that of the GSSG|2GSH pair.d S–Se/S, and N⋯Se, bond distance in Å, bond angle N⋯Se–S in degrees.e The gas-phase bond dissociation energy (BDE) for the S–Se/S bond in kcal mol−1.f Second-order perturbation energy (E2) from NBO for the Se⋯N non-covalent interactions in kcal mol−1.
at pH = 7 in volts; here A|B is either the RSeSG|(RSeH + GSH) or GSSG|2GSH couple.c Krel represents the relative equilibrium constant with respect to that of the GSSG|2GSH pair.d S–Se/S, and N⋯Se, bond distance in Å, bond angle N⋯Se–S in degrees.e The gas-phase bond dissociation energy (BDE) for the S–Se/S bond in kcal mol−1.f Second-order perturbation energy (E2) from NBO for the Se⋯N non-covalent interactions in kcal mol−1.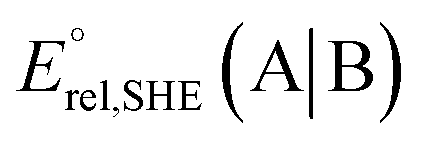
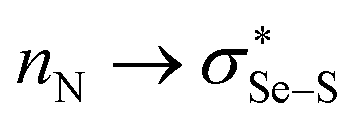 orbitals)35,36 obtained by the NBO second-order perturbation energy (E2) are also given in Table 1. The Se⋯N interaction is relatively weaker in intermediates with secondary amino groups (2b and 3b), compared to intermediates with tertiary amino groups (4b and 5b). Thus, E2 for these intermediates increases as the calculated redox potential decreases, indicating that a stronger Se⋯N interaction results in a weaker reducing ability of the selenenyl sulfide intermediates. Specifically, intermediates with a methyl group as the only substituent on the amino nitrogen, 2b (8.11 kcal mol−1, −0.252 V) and 4b (9.74 kcal mol−1, −0.264 V) exhibit a relatively weaker Se⋯N interaction and, consequently, more positive redox potentials compared to their counterparts with cyclohexyl substitution 3b (9.20 kcal mol−1, −0.257 V) and 5b (10.56 kcal mol−1, −0.280 V). This observation suggests that the presence of a cyclohexyl ring on the amino nitrogen enhances the Se⋯N interaction. The absence of Se⋯N non-covalent interactions in ebselen-based intermediates (6b–8b) could be a reason for showing relatively more negative redox potentials than glutathione.
orbitals)35,36 obtained by the NBO second-order perturbation energy (E2) are also given in Table 1. The Se⋯N interaction is relatively weaker in intermediates with secondary amino groups (2b and 3b), compared to intermediates with tertiary amino groups (4b and 5b). Thus, E2 for these intermediates increases as the calculated redox potential decreases, indicating that a stronger Se⋯N interaction results in a weaker reducing ability of the selenenyl sulfide intermediates. Specifically, intermediates with a methyl group as the only substituent on the amino nitrogen, 2b (8.11 kcal mol−1, −0.252 V) and 4b (9.74 kcal mol−1, −0.264 V) exhibit a relatively weaker Se⋯N interaction and, consequently, more positive redox potentials compared to their counterparts with cyclohexyl substitution 3b (9.20 kcal mol−1, −0.257 V) and 5b (10.56 kcal mol−1, −0.280 V). This observation suggests that the presence of a cyclohexyl ring on the amino nitrogen enhances the Se⋯N interaction. The absence of Se⋯N non-covalent interactions in ebselen-based intermediates (6b–8b) could be a reason for showing relatively more negative redox potentials than glutathione.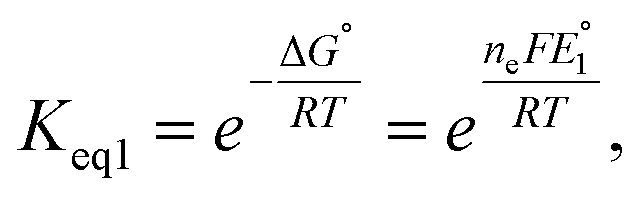
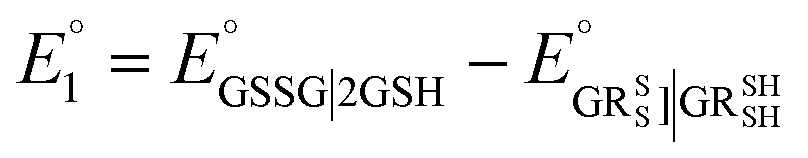 . Similarly, the equilibrium constant for the RSeSG reduction (eqn (2a)) can be written as
. Similarly, the equilibrium constant for the RSeSG reduction (eqn (2a)) can be written as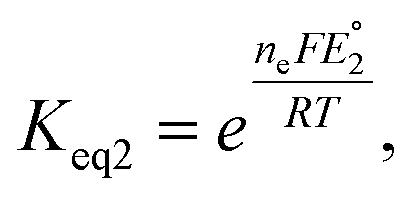
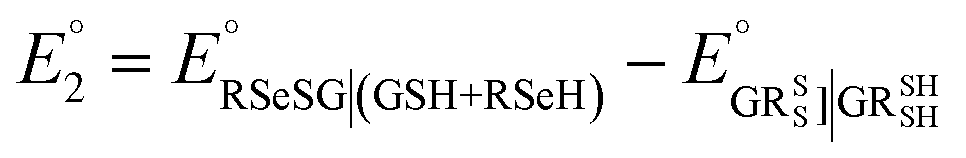 . Assuming that the RSeSG and GSSG interact similarly with the enzyme, the
. Assuming that the RSeSG and GSSG interact similarly with the enzyme, the 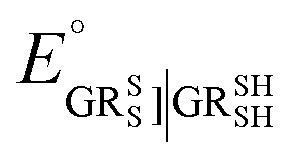 terms may cancel out, and an equilibrium constant for RSeSG conversion relative to that of GSSG conversion may be defined as
terms may cancel out, and an equilibrium constant for RSeSG conversion relative to that of GSSG conversion may be defined as