
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time control of distillation process to improve the recovery efficiency of ThF4–LiCl–KCl molten salt
Yujiao Wangab,
Yan Luo a,
Qiang Doua,
Wenxin Lia,
Qingnuan Lia and
Haiying Fu*a
a,
Qiang Doua,
Wenxin Lia,
Qingnuan Lia and
Haiying Fu*a
aShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: fuhaiying@sinap.ac.cn
bShanghai Jiaotong University, Shanghai 200240, China
First published on 28th October 2024
Abstract
The purification and recovery of chloride electrolyte molten salts are vital to reuse this valuable species and reduce the waste. Vacuum distillation method was used to investigate the recovery efficiency of LiCl–KCl mixed molten salt containing 20 wt% ThF4. Rapid mass loss in the initial stage and the subsequent lower evaporation rate were significantly observed under 1173 K and 20 Pa due to the coordination species such as Li3ThF7 and LiTh2F9. To achieve the recovery efficiency of chloride salts, a real-time control distillation was proposed. The distillation was terminated when about 80% mixture salt was evaporated according to the transient weight loss curve. The evaporation ratio of LiCl–KCl reached 91% and the decontamination factors for Th and rare earth elements Nd and Sm were more than 103 and 102, respectively. The results provided a simple and effective scheme to separate ThF4 and recover molten salts from waste electrolyte salts.
Introduction
Maximizing the use of nuclear fuel and minimizing radioactive waste are essential for the sustainable development of nuclear energy. To achieve these goals, chemical treatment of spent fuel from reactors is necessary.1 Pyroprocessing technology employs a molten salt medium mainly composed of alkali and alkaline earth metal fluorides or chlorides. Under high-temperature melting conditions, chemical methods are employed to separate and recover nuclear fuels, such as uranium and plutonium, while removing the majority of the fission products. Compared to traditional aqueous reprocessing technology, pyroprocessing technology has multiple advantages, such as radiation resistance, compact facilities, low critical risk, non-proliferation, and minimal radioactive waste. The treatment was found to be particularly effective in dealing with highly irradiated and extensively burned-out spent fuel, attracting significant attention within the field of nuclear energy.2,3 In recent decades, many countries have developed various dry reprocessing schemes for spent nuclear fuel, for example, the electrolysis and refining of uranium and plutonium, fluorination and volatilization of uranium, liquid–liquid extraction, layer-melt crystallization, and vacuum distillation.4–7 After fuel extraction, vacuum distillation is typically used to purify and recover the molten salt medium, which can be reconstituted for reuse or disposed, according to low-level radioactive waste standards.Vacuum distillation can separate fission products from mixture salts based on the vapor pressure differences of substances under high temperature and low pressure. It has advantages such as simple process, easy control, and no generation of new radioactive waste.8,9 In the 1950s, extensive research on the Molten-Salt Reactor Experiment (MSRE) was conducted at the Oak Ridge National Laboratory (ORNL), and LiF–BeF2 molten salt was recovered successfully from the actual nuclear fuel.10 The experimental results indicated that the removal ratio of rare earth fission products, such as Ce, Pm, Nd, and Sm, exceeded 90% when recovering 7LiF.
In the 1980s, the Argonne National Laboratory (ANL) and the Idaho National Laboratory (INL) applied molten salt electrolytic technology according to the development of the Integral Fast Reactor.11,12 Uranium, plutonium, and other transuranic elements were separated from the fission products. The successful application of electrochemical technology has attracted widespread attention in Russia, France, Japan, South Korea and so on. In recent years, the application scope has expanded from the metal fuel or oxide spent fuel of fast reactors to the spent molten salt fuel of MSR.13–15 Electrochemical separation is typically carried out in chloride salt systems represented by LiCl–KCl (58.5–41.5 mol%).15,16 However, after multiple electrolysis processes, a large amount of rare earth, alkaline earth, and alkali metal fission products accumulate in the electrolytic waste salt.17 Vacuum distillation technology is further employed to purify and recover the electrolyte waste salt, leaving behind highly radioactive fission products for disposal as high-level radioactive waste. The recovered salt is easily contaminated by rare-earth fission products due to the similar vapor pressures of rare-earth chlorides and chloride salts.18 The purification effect of the recovered salt can be effectively improved by first performing a chemical precipitation reaction of rare earths, followed by vacuum distillation.19,20 Therefore, vacuum distillation technology can effectively achieve the separation of molten salt from rare-earth and alkaline-earth fission products in the dry treatment of spent fuel based on the uranium–plutonium cycle.
The thorium–uranium cycle facilitates nuclear fuel breeding by converting 232Th into the 233U isotope through neutron capture and two beta decays. The thorium–uranium fuel cycle has garnered significant interest as a focal point in nuclear energy development due to its advantages of abundant thorium resources, high conversion efficiency, less radioactive waste generation, and easy prevention of nuclear proliferation. Following the substantial separation of nuclear fuel via dry reprocessing, thorium and rare earth elements coexist in the molten salt.21 It is necessary to remove as much thorium and rare-earth fission products as possible to achieve the purification and recovery of the molten salt.
The removal of thorium from LiF–BeF2 molten salt via vacuum distillation technology remain challenging. Smith22 found that the volatility of ThF4 is very low in LiF–BeF2–ThF4 molten salt at 1273 K. However, to achieve a sufficient distillation ratio, the feasibility of distillation separation needs to be re-evaluated at temperatures above 1273 K. Luo et al.23 studied the vacuum distillation of LiF–BeF2 molten salt containing 50 wt% ThF4 and found that the formation of complexes between ThF4 and LiF inhibited the separation of ThF4 and extended the evaporation time by more than 5 times. Therefore, the precipitation process for thorium should be done before vacuum distillation to improve the separation efficiency.
There are fewer reports on the vacuum distillation of LiCl–KCl molten salts containing ThF4. The presence of thorium in the form of fluorides may form complexes that hinder evaporation and the recovery process, while its chloride form may contaminate the recovered salt. The combination of precipitation and vacuum distillation technology can effectively separate thorium from the molten salt while it will introduce new impurities and increase energy consumption. Therefore, the purification and recovery of LiCl–KCl molten salts containing ThF4 using the vacuum distillation technology faces significant challenges. It is crucial to find a solution that can purify and recover the molten salt without the additional chemical precipitation. Therefore, this study conducted a detailed investigation of the vacuum distillation of 20 wt% ThF4–LiCl–KCl mixed molten salt.
Experimental
Reagents
LiCl and KCl (purity ≥ 99%) were purchased from Sigma-Aldrich. NdF3 and SmF3 (purity ≥ 99.99%) were purchased from Alfa Aesar. ThF4 (purity 99.99%) was provided by Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. LiF (purity 99%) was purchased from Shanghai Zhongli Industry Co., Ltd. Argon gas with 99.999% purity was purchased from Shanghai Xiangkun Gas Co., Ltd.Equipment
The preparation of mixed salts was carried out in a resistance furnace connected with a glove box. The resistance furnace can be programmed to raise the temperature up to 1473 K. The furnace chamber is made of 310 S stainless steel and has an inner diameter of 120 mm. The furnace was connected with a glove box with an argon atmosphere (99.99%, H2O < 1.0 × 10−6 g mL−1, O2 < 1.0 × 10−6 g mL−1). A Microwave Digestion System (MARS 6, USA) was utilized to dissolved samples, and the chemical composition of the samples was determined by an inductively-coupled plasma optical emission spectrometer (ICP-OES, DY3614, USA) with a wavelength range of 165–900 nm. X-ray powder diffraction (XRD, X'Pert Pro, Netherlands) was utilized to collect the crystalline phase structure data within a 2θ angle range of 5–90° with a step size of 0.02°.The evaporation behavior of mixed salts was investigated using a self-made gram-level molten salt vacuum distillation apparatus.24 As shown in Fig. 1, the weighing sensor has an accuracy of 0.001 g in the range of 0–100 g. The temperature range of the electric heater is 293–1373 K. The nickel crucible used for distillation has a diameter of 0.9 cm and a height of 3 cm.
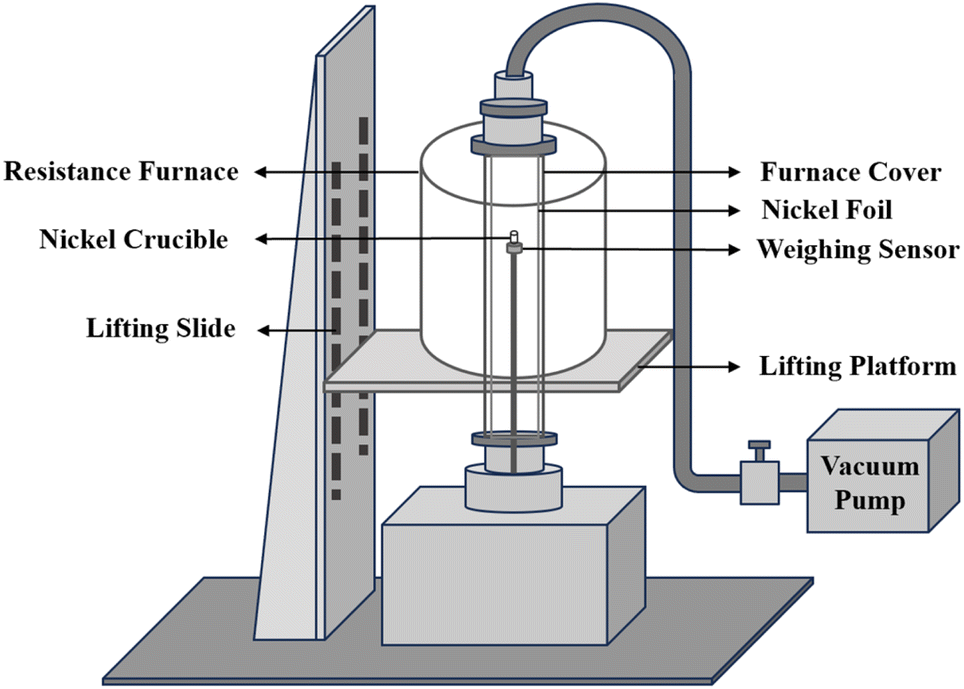 | ||
| Fig. 1 A Schematic diagram of vacuum distillation apparatus. | ||
Preparation of molten salt
About 17.6 g of LiCl and 22.4 g of KCl powder were thoroughly mixed in a nickel crucible. The mixture was loaded into a resistance furnace at 823 K for 6 h to prepare the LiCl–KCl (58.5–41.5 mol%) eutectic salt. The salt was finely ground into powder after cooling to room temperature and subsequently stored in an argon atmosphere glove box.Since the melting point of ThF4 is about 1373 K, it is very difficult to melt ThF4 into LiCl–KCl. However, LiF–ThF4 was more easily dissolved in molten salts than ThF4, as reported in our previous work.23 First, about 2.8 g of LiF was mixed with 10.2 g of ThF4 in a nickel crucible and then the mixture was loaded into an electrical furnace at 973 K for 5 h to prepare the LiF–ThF4 (77–23 mol%) molten salt.
The 20 wt% ThF4–LiCl–KCl mixed salt was prepared by melting a uniform mixture of 3 g LiF–ThF4 and 12 g LiCl–KCl salt under the same conditions. About 4 g LiF–ThF4 (77–23 mol%), 0.2 g NdF3, 0.2 g SmF3, and 15.6 g LiCl–KCl salt were mixed thoroughly in a crucible. 20 wt% ThF4-1 wt% SmF3-1 wt% NdF3–LiCl–KCl mixed salt was prepared at 973 K for 5 h.
Vacuum distillation
A certain amount of mixed salt was loaded in a nickel crucible with a diameter of 0.9 cm and a surface area of approximately 0.63 cm2. The nickel crucible was then put on the weighing sensor of the distillation apparatus. The whole heating process was conducted under argon atmosphere to minimize the salt evaporation. When the temperature reached 1173 K, the pump was turned on and the pressure was sharply decreased. The pressure was maintained at 20 Pa throughout the entire distillation procedure. The real-time monitoring of the mass change was achieved via a mass sensor, namely, a load cell. When the weight no longer showed a change, it indicated that the distillation was completed and the weight loss curve could be obtained. A specific quantity of mixed salt was added to the vacuum distillation experiments under different evaporation times. When the weight loss reached a required time point, the distillation was stopped when argon gas was purged to inhibit further distillation, and the residual molten salt in the crucible was transferred to a glove box and ground to a powder. A small fraction of the sample was used to analyse the relative contents of Li, Be, Th, or rare-earth. Then, the same operation was repeated on gradually increasing the evaporation ratio of the sample.The nickel foil was replaced to collect the condensate salt before each experiment. After the segmented experiments were completed, the salt adhering to the nickel foil was collected and mixed evenly, and a small sample was taken for analysis.
Sample analysis
A microwave digestion method was employed to dissolve the samples with high thorium concentrations, facilitating the dissolution of thorium and rare earth elements at high temperature and pressure.25 Generally, aluminium nitrate was used to improve the solubility of thorium and rare-earth as reported in the previous report.26 0.05 g of salt sample and 0.3 g of aluminium nitrate were first added to 10 mL of 68% nitric acid, followed by microwave heating at 463 K for 1 hour. ICP-OES analysis was then conducted after dilution to measure the elemental concentrations. Three parallel determinations were performed for each sample with a relative deviation below 5%. The results were the average of three tests, and the decontamination factor of elements in the collected salt after distillation was calculated using eqn (1).
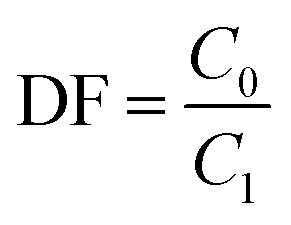 | (1) |
X-ray diffraction (XRD) was utilized for qualitative determination and phase analysis of the sample. The diffraction pattern of the sample was obtained within the range of 10–90°.
Results and discussion
Evaporation behavior of 20 wt% ThF4–LiCl–KCl molten salt
About 1.8 g of LiCl–KCl salt, 1.8 g of 20 wt% ThF4–LiCl–KCl mixed salt, and 2.25 g of 20 wt% ThF4–LiCl–KCl mixed salt were respectively weighed to study the effect of ThF4 on the distillation process of chlorine salt. The samples were placed in a vacuum distillation apparatus and the experiment was conducted at 1173 K and 20 Pa. Real-time weight sensing recorded the mass of the remaining molten salt, producing the distillation weight loss curves for these three mixed salts, as shown in Fig. 2.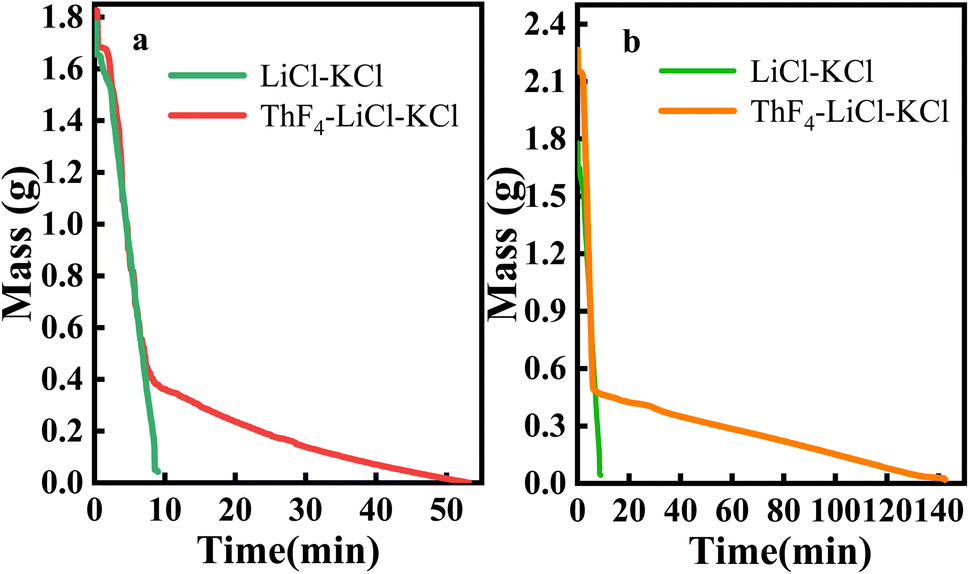 | ||
| Fig. 2 Mass loss curves under vacuum distillation conditions at 1173 K and 20 Pa: (a) 1.8 g of 20 wt% ThF4–LiCl–KCl and 1.8 g of LiCl–KCl; (b) 2.25 g of 20 wt% ThF4–LiCl–KCl and 1.8 g of LiCl–KCl. | ||
The weight loss curves showed that the mass of the LiCl–KCl salt decreased rapidly after the initiation of vacuum distillation. The complete evaporation of 1.8 g of LiCl–KCl salt was only 8 minutes, indicating that LiCl–KCl salt was easily evaporated. For ThF4, the melting point was 1383 K. The vapor pressure of ThF4 at 1273 K was only 0.0688 mmHg,27 which is much lower than that of LiCl and KCl. Therefore, the evaporation of ThF4 is relatively difficult. Research has shown that 0.42 g of ThF4 powder requires 160 minutes to evaporate completely at 1223 K and 10 Pa.19 Fig. 2a shows that the 1.8 g 20 wt% ThF4–LiCl–KCl mixed salt containing 0.36 g of ThF4 takes 50 minutes to completely evaporate. However, Fig. 2b indicates that the 2.25 g 20 wt% ThF4–LiCl–KCl mixed salt containing 0.45 g of ThF4 and 1.8 g of LiCl–KCl takes 140 minutes to completely evaporate, which is significantly longer than the evaporation time of 1.8 g LiCl–KCl salt with the same mass. This indicated that increasing the ThF4 content significantly prolongs the evaporation time. Therefore, the distillation of 20 wt% ThF4LiCl–KCl mixed salt is not a simple superposition of the distillation behaviors of each component. There are interactions between the mixed salts that alter the distillation process.
Fig. 2a showed a distinct turning point in the weight loss curve of the mixed salt, indicating two evaporation modes. Before this point, the average evaporation rate of LiCl–KCl was 0.19 g min−1, while the evaporation rate of 20 wt% ThF4–LiCl–KCl was 0.18 g min−1. Similar rates indicate that the rapid mass decrease was primarily attributed to the evaporation of LiCl–KCl. After the turning point, the mass of 20 wt% ThF4–LiCl–KCl continued to slowly decrease at an average rate of less than 0.01 g min−1. This indicated a significant change in the evaporation process.
The evaporation behavior of the mixed salt cannot be simply attributed to the simple superposition of the LiCl–KCl and ThF4 components. 1.8 g and 2.25 g samples of 20 wt% ThF4–LiCl–KCl mixed salt contained 0.36 g and 0.45 g of ThF4, respectively. As shown in Fig. 2, the weight of the remaining salt at the turning point is 0.4 g and 0.5 g, respectively, both of which are greater than the initial ThF4 content in the molten salt. This indicates that during the distillation process, LiCl–KCl and ThF4 were not completely separated. Further research is required to investigate the mass changes of each component during distillation.
Component analysis
1.8 g of 20 wt% ThF4–LiCl–KCl salt was placed in a vacuum distillation apparatus, and the experiment was conducted at 1173 K and 20 Pa. Distillation was stopped with argon after 3 minutes, and the remaining salt was weighed and analyzed by ICP-OES. The same mass of the mixed salt was subjected to the same conditions with the distillation time extended to 7, 20, and 50 minutes, respectively. The related results are listed in Table 1 and Fig. 3.| Evaporation time (min) | Residual salt mass (g) | Evaporation ratio (%) | Concentration of element (wt%) | Mass of component (g) | ||||
|---|---|---|---|---|---|---|---|---|
| Li | K | Th | LiCl | KCl | ThF4 | |||
| 0 | 1.81 | 0 | 7.23 | 19.91 | 14.17 | 0.79 | 0.69 | 0.33 |
| 3 | 1.52 | 16.04 | 7.08 | 19.78 | 16.51 | 0.65 | 0.57 | 0.33 |
| 7 | 0.37 | 79.76 | 4.95 | 2.66 | 62.82 | 0.11 | 0.018 | 0.31 |
| 20 | 0.30 | 83.34 | 4.74 | 2.18 | 63.93 | 0.08 | 0.013 | 0.26 |
| 50 | 0.18 | 90.24 | 3.96 | 1.73 | 76.37 | 0.04 | 0.005 | 0.18 |
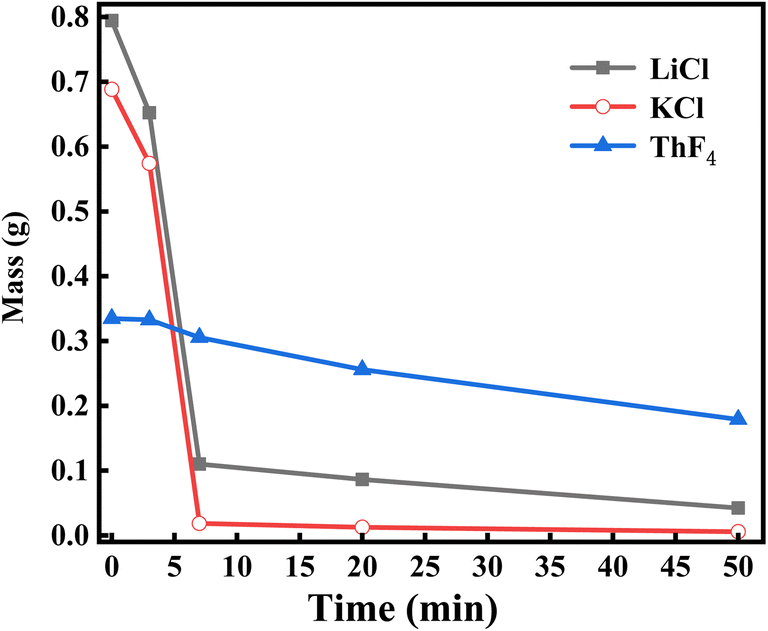 | ||
| Fig. 3 Mass change of the components in the remaining salt with different evaporation times. | ||
The evaporation ratio of each component at various times was calculated by the initial and remaining mass of the components. Specific results are listed in Table 2, while the evaporation ratio changes for the mixed salt and individual components over time are shown in Fig. 4.
| Evaporation time (min) | Residual salt mass (g) | Evaporation ratio (%) | ||||
|---|---|---|---|---|---|---|
| Mixed salt | LiCl | KCl | ThF4 | LiCl–KCl | ||
| 0 | 1.81 | 0 | 0 | 0 | 0 | 0 |
| 3 | 1.52 | 16.04 | 17.90 | 16.59 | 0.55 | 17.25 |
| 7 | 0.37 | 79.76 | 86.14 | 96.76 | 8.71 | 91.45 |
| 20 | 0.30 | 83.34 | 89.10 | 98.18 | 23.55 | 93.64 |
| 50 | 0.18 | 90.24 | 94.67 | 99.15 | 46.48 | 96.91 |
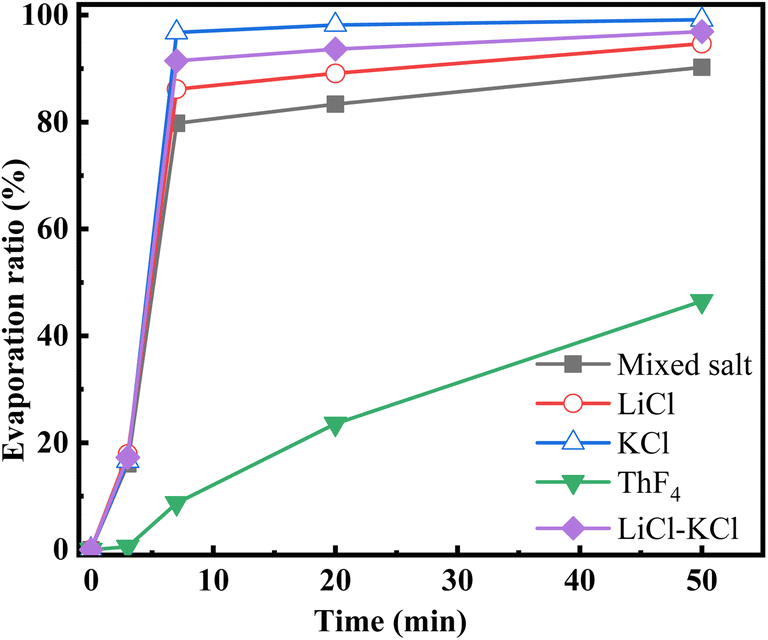 | ||
| Fig. 4 Evaporation ratio of various components at different times. | ||
Fig. 3 and 4 illustrate that LiCl and KCl evaporate quickly at the beginning of distillation, leading to a rapid decrease in the molten salt mass. Table 1 reveals that the mass of ThF4 remains stable during the initial 3 minutes, indicating that the weight loss during this period is due to the evaporation of LiCl and KCl. Between 3 and 7 minutes, as a significant amount of LiCl and KCl decrease, ThF4 begins to evaporate. After 7 minutes, the evaporation ratio drops notably, indicating that LiCl and KCl have been largely depleted, resulting in a slower evaporation stage where mass loss is mainly due to ThF4. Table 2 shows that only 14% and 3% of the initial LiCl and KCl remain after this turning point, respectively. Notably, these residual chlorides do not fully evaporate but co-evaporate with ThF4. This deviation from their characteristic of independent and volatile presence suggests a potential interaction that could lead to the formation of new substances with distinct compositions and evaporation behaviors. XRD analysis was performed on the remaining molten salt at different time points to verify this speculation.
XRD analysis was conducted on salt samples collected at evaporation times of 0, 3, 7, 20, and 50 minutes. As shown in Fig. 5, LiCl and KCl remain in their original chloride compound form in the initial salt of 20 wt% ThF4–LiCl–KCl. Notably, the XRD pattern displayed a peak for the Li3ThF7 coordination compound, indicating the interaction of ThF4 and Li+ during salt preparation.
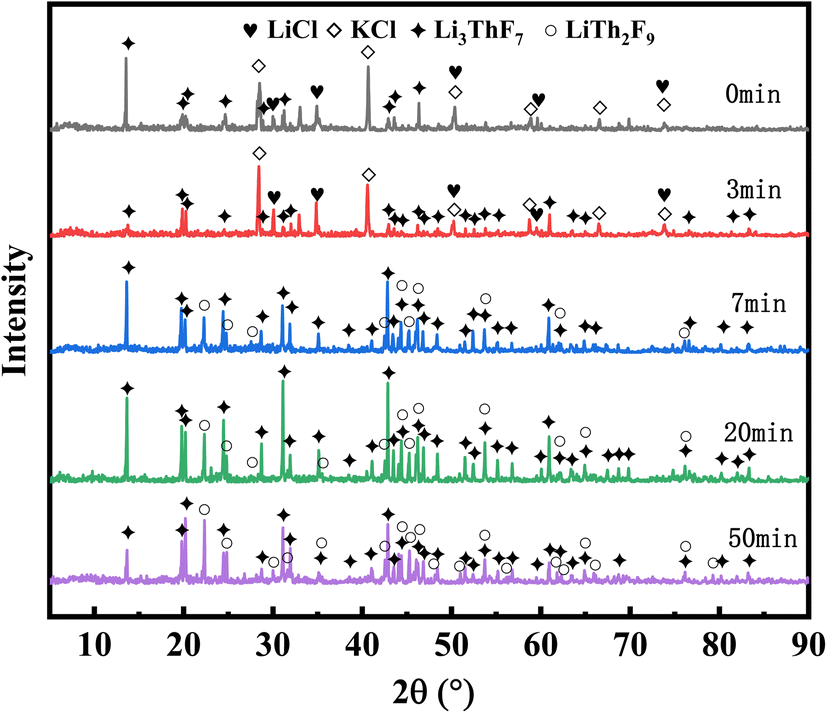 | ||
| Fig. 5 XRD patterns of the remaining salt at different evaporation times. | ||
After 3 minutes of vacuum distillation, the evaporation ratio of the mixed salt was only 16%, with XRD confirming the presence of LiCl, KCl, and Li3ThF7 in the remaining salt. After evaporating for 7 minutes, KCl and most of LiCl were evaporated, resulting in the disappearance of the chloride diffraction peak in the residual salt, which coincided with the evaporation ratio of 91.45%, as shown in Table 2. XRD analysis indicated that the remaining salt primarily consisted of the Li3ThF7 and LiTh2F9 phase. Similarly, the results of the evaporation ratio in Table 2 indicated a stark difference between each component; KCl reached 96.76% at 7 min, while LiCl only reached 86.14%. With combined XRD analysis, it was assumed that while melting, ThF4 easily trapped Li+ and formed the complex such as Li3ThF7 and LiTh2F9. This phenomenon restrained the volatile Li component within the remaining salt, causing it to co-evaporate with thorium at a little lower rate, as reported in our previous work.23 The XRD analysis of the residual salts after the turning point confirms only Li3ThF7 and LiTh2F9, explaining the residual weights of 0.4 g and 0.5 g, which exceed the initial ThF4 amount added. The formation of coordination compounds between Li and ThF4 effectively reduces the volatility of Li, explaining the difference in the evaporation ratio of KCl and LiCl.
The 20 wt% ThF4–LiCl–KCl mixed salt contains more chloride ions, with a molar ratio of about 5.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 compared to fluorine ions. However, Fig. 5 shows that the XRD patterns exhibit the presence of fluorine-coordinated compounds such as Li3ThF7 and LiTh2F9 throughout the distillation process, with no chlorine-coordinated compounds detected. The literature reports that the bond length of Th–F is 2.25 Å, whereas the Th–Cl bond length is 2.66 Å.28 The smaller ionic radius and higher electronegativity of fluoride ions make thorium more inclined to form stable complexes with them in the chloride salt. This is further substantiated by the vapor pressure data from the literature: at 900 °C, the vapor pressures of LiCl, KCl, KF, and LiF were recorded as 6.433 mmHg, 3.605 mmHg, 1.275 mmHg, and 0.06824 mmHg, respectively.29 Compared to other components, LiF exhibits a lower vapor pressure at high temperatures, making it harder to evaporate.
:1 compared to fluorine ions. However, Fig. 5 shows that the XRD patterns exhibit the presence of fluorine-coordinated compounds such as Li3ThF7 and LiTh2F9 throughout the distillation process, with no chlorine-coordinated compounds detected. The literature reports that the bond length of Th–F is 2.25 Å, whereas the Th–Cl bond length is 2.66 Å.28 The smaller ionic radius and higher electronegativity of fluoride ions make thorium more inclined to form stable complexes with them in the chloride salt. This is further substantiated by the vapor pressure data from the literature: at 900 °C, the vapor pressures of LiCl, KCl, KF, and LiF were recorded as 6.433 mmHg, 3.605 mmHg, 1.275 mmHg, and 0.06824 mmHg, respectively.29 Compared to other components, LiF exhibits a lower vapor pressure at high temperatures, making it harder to evaporate.
Consequently, it is more likely to form complexes with ThF4 in the residual salts. The presence of fluorides is critical for addressing thorium fuel solubility issues in the thorium–uranium cycle. Consequently, even trace amounts of fluorides can significantly affect the salt collection process during distillation.
Decontamination of thorium
With the extension of the evaporation time, the majority of the LiCl–KCl salts evaporated rapidly, while the evaporation ratio of ThF4 also gradually increased, which may lead to the contamination of the recovered chlorine salt. Therefore, it is essential to further investigate the decontamination factor of thorium in the collected salts after the vacuum distillation of the 20 wt% ThF4–LiCl–KCl mixed salt. To determine the decontamination effect of Th in the collected salts, a cylindrical nickel foil was positioned in the distillation furnace to collect the evaporated molten salts. The evaporation time was controlled at 3, 7, 20, and 50 minutes, respectively. After cooling to room temperature, the salt on the nickel foil was collected and ground into a powder, and the concentration of Th in the molten salt was analyzed using the same method as previously described. The decontamination factor (DF) of Th was calculated using eqn (1). As shown in Fig. 6, the evaporation ratio of LiCl–KCl salts and the decontamination factor of Th exhibit the opposite trend with the extension of evaporation time. The evaporation ratio of LiCl–KCl rapidly increases to 90% in the initial stage and then slows down. In contrast, the decontamination factor of Th is relatively high in the initial evaporation stage, reaching 1.7 × 103 at 3 minutes and 1.36 × 103 at 7 minutes, but it rapidly decreased with the increase in evaporation time and dropped to 40 with 50- minute evaporation time.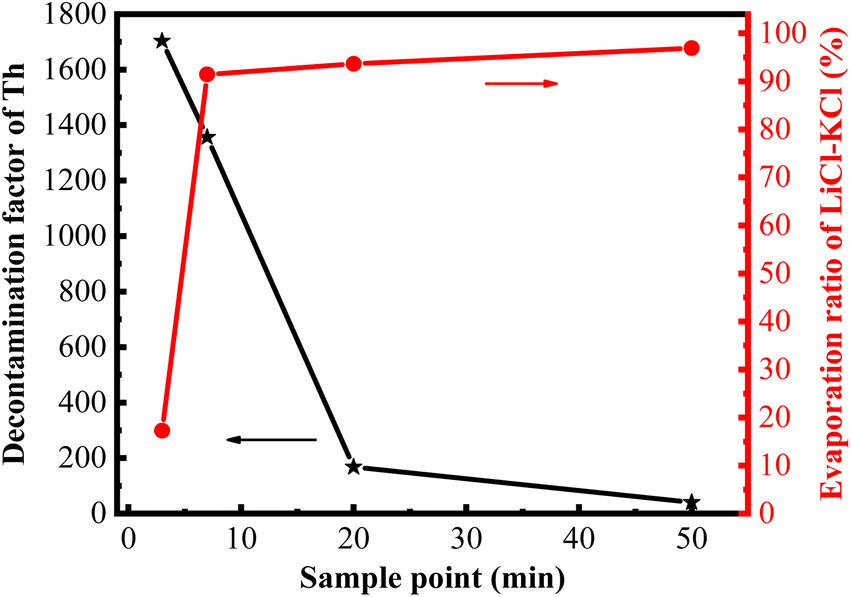 | ||
| Fig. 6 Evaporation ratio of LiCl–KCl and decontamination factor of Th at different evaporation times. | ||
Based on the data from Fig. 6 and Table 2, when the distillation was conducted for 3 minutes, the evaporation ratio of the mixed molten salt was 16%, with the evaporation ratio of the LiCl–KCl salt reaching 17%. At this time, the experimentally determined decontamination factor of Th is 1.70 × 103. When the distillation reached the turning point, the evaporation ratio of the mixed salt and LiCl–KCl salts were 80% and 91%, respectively, and the decontamination factor of Th decreased slightly to 1.36 × 103. This is consistent with the research results of Luo et al., which showed that the decontamination factor of Th in the collected salt was 2.20 × 103 after the distillation of 5 wt% ThF4–LiCl–KCl mixed salt at 1090 K.30 As the distillation time further increased, the evaporation ratio of molten salt continued to increase, and the decontamination factor of Th decreased rapidly. When the evaporation ratio of LiCl–KCl reached 94% and 97%, the DF of Th decreased significantly to 1.68 × 102 and 40, respectively. Similar trends have been observed multiple times in vacuum distillation. Kelly et al.10 reported that the decontamination factor of Eu exceeded 1000 after 80% of the molten salt was distilled, while it decreased to 50 when the evaporation ratio of the mixture salt was up to 98%.
The decrease in the decontamination factor with increasing evaporation ratio during vacuum distillation can be explained by the composition changes of the molten salt during evaporation. According to the kinetic theory of gases,29 the evaporation ratio is proportional to the vapor pressure of the substance and can be expressed as eqn (2).
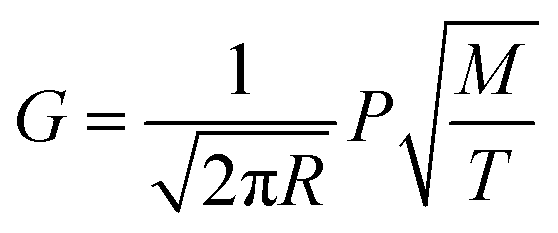 | (2) |
| PA = PA0 × XA × γA | (3) |
Based on the research findings, it can be concluded that during the vacuum distillation process, the evaporation primarily consists of LiCl and KCl salts accompanied by a small amount of co-evaporated thorium before the turning point. After the turning point, co-evaporation mainly occurs in the form of Li3ThF7 and LiTh2F9 coordination compounds. Co-evaporation makes it difficult to purify and recover the LiCl–KCl salt containing high concentrations of ThF4. Therefore, it is necessary to find an appropriate balance between the recovery ratio and purity of the molten salt. This study suggests controlling the distillation at the turning point, where the evaporation ratio of the mixed salt is 80% for 20 wt% ThF4–LiCl–KCl, and the evaporation ratio of the LiCl–KCl salt can reach 91%, which basically meets the requirements for salt recovery. Meanwhile, the evaporation of Th is low, and the DF for Th in the collected salt reaches 1.36 × 102. Therefore, it is feasible to achieve salt recovery by controlling the distillation process in real-time.
Decontamination of thorium and rare earths
When we controlled the evaporation time of the mixed salt and the distillation was terminal at the turning point, the evaporation ratio of LiCl–KCl salt was about 91%. During the electrochemical treatment of spent nuclear fuel, the LiClKCl waste salt often contains rare-earth fission products and thorium. Due to the high yield of rare-earth fission products and their large neutron capture cross-sections, they have a significant impact on the reactivity of the reactor. Therefore, further research is needed to study the recovery efficiency of the molten salt when thorium and rare-earths coexist under this scheme.A gram-scale molten salt vacuum distillation device was used to conduct experiments under conditions of 1173 K and 20 Pa. 1.8 g of 1 wt% SmF3-1 wt% NdF3–LiCl–KCl and 20 wt% ThF4-1 wt% SmF3-1 wt% NdF3–LiCl–KCl mixed salts were respectively put into the furnace. As shown in Fig. 7, it took 10 minutes for 1.8 grams of 1 wt% SmF3-1 wt% NdF3–LiCl–KCl salt to completely evaporate, which was 2 minutes longer than the same mass of LiCl–KCl salt. This is attributed to the addition of SmF3 and NdF3 to LiCl–KCl. The evaporation process of the 20 wt% ThF4-1 wt% SmF3-1 wt% NdF3–LiCl–KCl mixed salt needed 62 minutes, 12 minutes longer than that of the 20 wt% ThF4–LiCl–KCl mixed salt. Comparing Fig. 2 and 7, the weight loss curves of the two mixed salts are similar, both showing a turning point at 7 minutes into the distillation. Before the turning point, the mixed salts lose weight rapidly mainly due to the rapid evaporation of LiCl and KCl. Therefore, the majority of chloride salts can be effectively recovered from the mixed salts by controlling the distillation process in real-time and stopping it at the turning point.
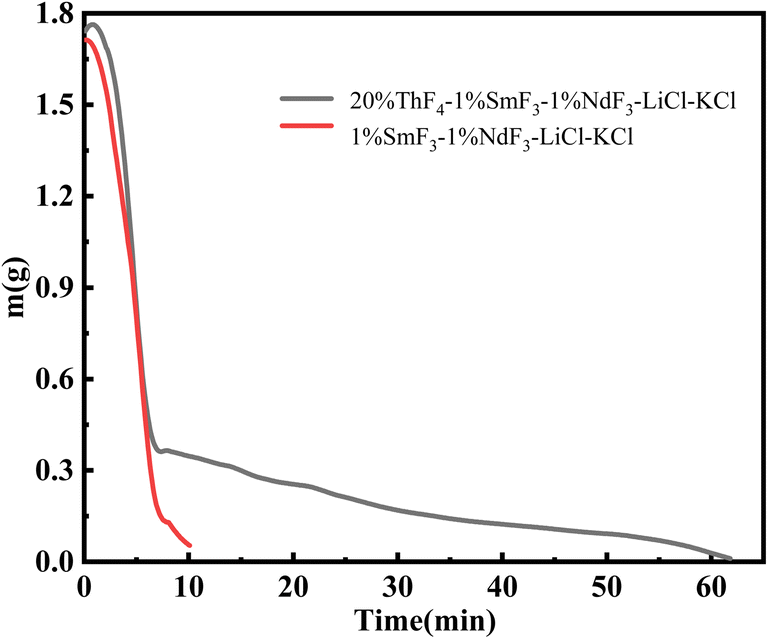 | ||
| Fig. 7 Weight loss curves of 20 wt% ThF4-1 wt% SmF3-1 wt% NdF3–LiCl–KCl mixed salt. | ||
To study the impact of real-time controlled distillation on the decontamination effect of thorium and rare-earth elements in the recovered molten salt, a distillation experiment was conducted on 1.8 g of 20 wt% ThF4-1 wt% SmF3-1 wt% NdF3–LiCl–KCl mixed salt. After the distillation, the cooled salt was uniformly ground into powder and analyzed by ICP-OES. The calculated decontamination factors for Nd, Sm, and Th were 1.23 × 102, 1.06 × 102, and 1.72 × 103, respectively. The decontamination factors for rare-earth elements fluctuate significantly during the vacuum distillation process, which is related to the distillation apparatus, experimental conditions, and sampling analysis methods used in different laboratories.10,31,32 Considering the high evaporation ratio of the molten salt, a decontamination factor slightly above 102 is acceptable in practical applications. On the other hand, the main component of the mixed salt was LiCl–KCl, and thorium fluoride was added, which meant that chloride-fluorine coexisted. The content of F element in the collected salt was further investigated using ion chromatography.
For 20 wt% ThF4-1 wt% SmF3-1 wt% NdF3–LiCl–KCl salt, the content of fluorine was 1.47 × 103 ppm at 3 min, which increased to 8.45 × 103 ppm at 7 min as the evaporation ratio was about 80%. With evaporation for 50 minutes, the content of fluorine was up to 5.69 × 104 ppm in the collected salt. The results indicated that the fluoride mainly remained in the evaporation crucible due to its lower volatility during the evaporation in the first 7 minutes. Moreover, the decontamination factors for thorium and rare-earth elements were high.
Analyzing the relationship between different ThF4 concentrations and the turning point, it was estimated that a better purification will be available when terminating the distillation process at the turning point. For the mixed salt with less than 20 wt% ThF4, it is a reasonable scheme to stop the distillation when 80% of the mixed salt was evaporated. The scheme avoids the chemical precipitation process that is necessary before vacuum distillation, which not only improves the recovery efficiency of the molten salt but also reduces the amount of radioactive waste. To further implement this plan, some process control issues may be introduced for the development of artificial intelligence technology.
Conclusions
In the present work, we investigated the vacuum distillation process of 20 wt% ThF4–LiCl–KCl mixture salt. The results showed that ThF4 cannot be completely separated from the chloride salts due to the formation of coordination compounds such as Li3ThF7 and LiTh2F9. A real-time controlled distillation scheme was proposed based on the evaporation behavior of the components during the vacuum distillation process. Terminating the distillation when 80% of the mixed salt was evaporated can achieve an evaporation ratio of over 91% for LiCl–KCl salt, with the decontamination factor for Th reaching 1.72 × 103, and the decontamination factors of Nd and Sm were 1.23 × 102 and 1.06 × 102, respectively. The actual recovery ratio of the molten salt can reach 90%, and the content of fluoride ions in the recovered salt is relatively low. The study essentially achieved the separation of Th and the purification of LiCl–KCl salt from the 20 wt% ThF4–LiCl–KCl mixed salt without the additional use of precipitants. This research shows the significant reference value for the recovery of molten salts in the dry reprocessing of thorium–uranium cycles.Data availability
The data supporting this article have been included within the manuscript.Author contributions
Yujiao Wang: writing – original draft, methodology, data curation. Yan Luo: formal analysis, data curation. Qiang Dou: writing – review & editing, validation. Wenxin Li: methodology. Qingnuan Li: resources, supervision. Haiying Fu: writing – review & editing, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This study was supported by the Natural Science Foundation of China (No. 12275349).Notes and references
- A. O. Adeola, K. O. Iwuozor, K. G. Akpomie, K. A. Adegoke, K. O. Oyedotun, J. O. Ighalo, J. F. Amaku, C. Olisah and J. Conradie, Environ. Geochem. Health, 2023, 45, 2663–2689 CrossRef CAS PubMed.
- K. M. Goff, J. C. Wass, K. C. Marsden and G. M. Teske, Nucl. Eng. Technol., 2011, 43, 335–342 CrossRef CAS.
- S. Park and R. C. Ewing, Annu. Rev. Environ. Resour., 2023, 48, 713–736 CrossRef.
- A. Y. Galashev, Int. J. Energy Res., 2022, 46, 3891–3905 CrossRef CAS.
- L. X. Sun, Y. S. Niu, C. W. Hu, X. H. Wang, Z. Q. Zhao, J. G. Chen, X. Z. Cai, H. Y. Fu, Q. Dou and Q. N. Li, J. Fluorine Chem., 2022, 261, 110016 CrossRef.
- M. Shim, H. G. Choi, J. H. Choi, K. W. Yi and J. H. Lee, J. Nucl. Mater., 2017, 491, 9–17 CrossRef CAS.
- R. Pierce, T. Caldwell and D. Pak, Sep. Sci. Technol., 2012, 47, 2065–2073 CAS.
- H. Y. Fu, J. X. Geng, Y. Yang, Y. Luo, Q. Dou, W. X. Li and Q. N. Li, Nucl. Tech., 2018, 41, 1–8 Search PubMed.
- C. D. Scott and W. L. Carter, ORNL-3830, Oak Ridge National Laboratory, 1965 Search PubMed.
- M. J. Kelly, ORNL-3789, Oak Ridge National Laboratory, 1965 Search PubMed.
- Y. I. Chang, Fission Energy: The Integral Fast Reactor, 1989 Search PubMed.
- J. P. Ackerman, Ind.Eng.Chem.Res, 1991, 30, 141–145 CrossRef CAS.
- F. Jiang, N. Ji, W. Huang and H. Y. Fu, J. Electrochem. Soc., 2022, 169, 102505 CrossRef CAS.
- Y. Zuo, X. J. Li, F. Jiang, C. F. She, W. Huang and Y. Gong, Sep. Purif. Technol., 2023, 315, 123717 CrossRef.
- T. J. Zhu, W. Huang and Y. Gong, Sep. Purif. Technol., 2020, 235, 116227 CrossRef CAS.
- F. Jiang, W. Huang, C. F. She, Y. Gong and H. Y. Fu, Nucl. Technol., 2024, 47, 060301 Search PubMed.
- B. J. Riley, Ind. Eng. Chem. Res., 2020, 59, 9760–9774 CrossRef CAS.
- B. R. Westphal, K. C. Marsden, J. C. Price and D. V. Laug, Nucl. Eng. Technol., 2008, 40, 163–174 CrossRef CAS.
- H. C. Eun, Y. Z. Cho, H. Park, I. Kim and H. Lee, J. Radioanal. Nucl. Chem., 2012, 292, 531–535 CrossRef CAS.
- H. C. Eun, J. H. Choi, I. H. Cho, T. K. Lee, T. J. Kim, J. S. Shin, H. S. Park and D. H. Ahn, J. Radioanal. Nucl. Chem., 2016, 307, 1419–1427 CrossRef CAS.
- X. Z. Cai, Z. M. Dai and H. J. Xu, Physics, 2016, 45, 578–590 Search PubMed.
- F. J. Smith, L. M. Ferris and C. T. Thompson, ORNL-4415, Oak Ridge National Laboratory, 1969 Search PubMed.
- Y. Luo, J. X. Dai, Q. Dou, H. Y. Fu and Q. N. Li, RSC Adv., 2023, 13, 6637–6642 RSC.
- L. Xu, H. Y. Fu and Q. Dou, Utility model Pat., CN201921846080.7, 2019 Search PubMed.
- Y. Luo, H. X. Cong, Z. Q. Zhao, W. Hu, W. Zhou and S. He, J. Nucl. Radiochem., 2015, 37, 37–40 CAS.
- R. A. Edge, Anal. Chim. Acta, 1963, 28, 278–281 CrossRef CAS.
- A. J. Darnell and F. J. Keneshea, J. Phys. Chem., 1958, 62, 1143–1145 CrossRef CAS.
- J. B. Liu, X. Chen, J. B. Lu, H. Q. Cui and J. Li, J. Comput. Chem., 2018, 39, 2432–2438 CrossRef CAS PubMed.
- C. L. Yaws, Handbook of vapor pressure, Gulf Professional Publishing, 1995, vol 4 Search PubMed.
- Y. Luo, T. H. Yang, J. X. Geng, H. Y. Fu and Q. Dou, J. Radioanal. Nucl. Chem., 2020, 324, 1365–1371 CrossRef CAS.
- J. R. Hightower and L. E. NcNeese, Ind. Eng. Chem. Process Des. Dev., 1973, 12, 232–236 CrossRef CAS.
- J. X. Geng, Y. Luo, H. Y. Fu, Q. Dou, H. He, G. A. Ye and Q. N. Li, Prog. Nucl. Energy, 2022, 147, 104212 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |