
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAminodiols, aminotetraols and 1,2,3-triazoles based on allo-gibberic acid: stereoselective syntheses and antiproliferative activities†
Zein Alabdeen Khdara,
Tam Minh Leab,
Zsuzsanna Schelz c,
István Zupkóc and
Zsolt Szakonyi*a
c,
István Zupkóc and
Zsolt Szakonyi*a
aInstitute of Pharmaceutical Chemistry, Interdisciplinary Excellent Center, University of Szeged, Eötvös utca 6, H-6720 Szeged, Hungary. E-mail: szakonyi.zsolt@szte.hu; Fax: +36-62-545705; Tel: +36-62-546809
bHUN-REN-SZTE Stereochemistry Research Group, University of Szeged, H-6720 Szeged, Eötvös utca 6, Hungary
cInstitute of Pharmacodynamics and Biopharmacy, Interdisciplinary Excellent Center, University of Szeged, Eötvös utca 6, H-6720 Szeged, Hungary
First published on 18th November 2024
Abstract
A new series of aminodiols, aminotetraols and 1,2,3-triazoles based on allo-gibberic acid were synthesized in a stereoselective manner, starting from commercially available gibberellic acid. allo-Gibberic acid, prepared from gibberellic acid according to a literature method, was applied to SeO2/t-BuOOH-mediated allylic oxidation, yielding the triol, which is a key intermediate. After protecting the 1,4-diol functionality as acetonide, epoxidation was performed using either m-CPBA or t-BuOOH/VO(acac)2 to produce the epoxy alcohol. Then, the oxirane ring was opened with either primary amines to provide aminodiols or sodium azide to afford azido diols. The latter was subjected to the CuAAC reaction to obtain dihydroxy 1,2,3-triazoles. HCl-mediated acetonide deprotection of the prepared derivatives furnished aminotetraols and tetrahydroxy 1,2,3-triazoles. The antiproliferative effects of the prepared compounds were studied by the in vitro MTT method against a panel of human cancer cell lines (HeLa, SiHa, A2780, MCF-7 and MDA-MB-231) and fibroblasts, and the structure–activity relationships for the prepared compounds were explored.
1 Introduction
In 2022, according to the IARC (International Agency for Research on Cancer) estimates, 20 million new cancer cases and 9.7 million deaths were recorded. Global concerns have been expressed about the escalating burden of cancer as the cases are expected to grow by 77% in 2035.1 Subsequently, efforts in drug discovery alongside other fields, such as genomics, proteomics, molecular biology and pharmaceutical sciences, have been employed to reach the most effective and selective cancer therapy.2–6 Nevertheless, existing cancer treatments often result in significant adverse effects with a high possibility of drug resistance.7 Through the last decades, nature has remained the richest, most sustainable and regenerative source of new promising lead compounds with not only anticancer activity but also a variety of biological properties, for instance, antihypertension, antimalarial and antimicrobial properties.8,9 This was emphasised by Newman and Cragg in their review on drug sources covering the period from 1981 to 2014.10 The main obstacles faced during the development of leads from natural sources have been the structural complexity of natural products and their production in minor quantities.11 However, the enormous progress in genomics and biosynthetic methods has overcome these challenges.12 In the near past, the FDA granted approval for several natural products currently available in the global market, such as doxorubicin13 and vincristine,14 to be used for anticancer therapy. Moreover, the discovery of new lead compounds has been highly accelerated by the application of high throughput screening (HTS) that triggered the need to generate extensive compound libraries by utilising pharmaceutical chemistry aspects.15The ent-kaurene diterpenoids, containing opposite stereochemistry compared to kaurene diterpenoids,16 first isolated from New Zealand Kauri (Agathis australis Salisb.),17 form a distinctive group of the diterpenoid family, and they have attracted the attention of researchers owing to their diverse and complex structures as well as their broad range of bioactive properties, in particular, antitumour18 and antimicrobial activities.19,20 Gibberellic acid (GA3) 1, a famous C19 ent-kaurene diterpenoid, is known for its agricultural applications as a plant growth hormone responsible for seed germination and stem elongation.21,22 Over the past decades, it has captured the interest of synthetic scientists due to its commercial availability and extensive possibilities for chemical transformations.23,24 These efforts brought to light the promising biological applications of 1 and its derivatives, most notably for anticancer activity. The early aspiring report announced in 2009 by Koehler showed the ability of gibberellic acid 1 and 9α-H allo-gibberic acid 2 to modulate the NF-κB pathway activity.25 Lately, Y. Zhang et al. demonstrated the in vitro and in vivo antiangiogenic activity of compound I (GA-13315),26 which was prepared using the synthetic pathway of GA3-based α,β-unsaturated diketone derivatives accomplished by Chen et al.27 This work inspired Jang Wu et al. to engage a 1,2,3-triazole functionality with hydroxy-functionalised epiallo-gibberic acid with an α,β-unsaturated ketone moiety that expressed the ability to induce cell cycle arrest in the S phase and apoptosis.28 Furthermore, a potent anticancer activity was also observed with a group of allo-gibberic acid meta-substituted benzyl esters prepared by Zhu et al.29
In our previous work, we successfully prepared two libraries of aminodiols and aminoalcohols based on N-substituted allo-gibberic acid with potent and selective antiproliferative activity (compounds II, III and IV presented in Fig. 1).30,31 Furthermore, a new series of steviol-based aminotriols (V presented in Fig. 1) was prepared by our research group and these were evaluated through an antiproliferative activity assay (MTT) that showed the importance of the N-benzyl substituents for potent and selective cytotoxicity.32 Integrating these results with our ongoing effort to develop more powerful and selective anticancer agents has guided us to prepare a new library of aminodiols, aminotetraols and 1,2,3-triazoles based on allo-gibberic acid and evaluate their in vitro antiproliferative activity against different human cancer cell lines.
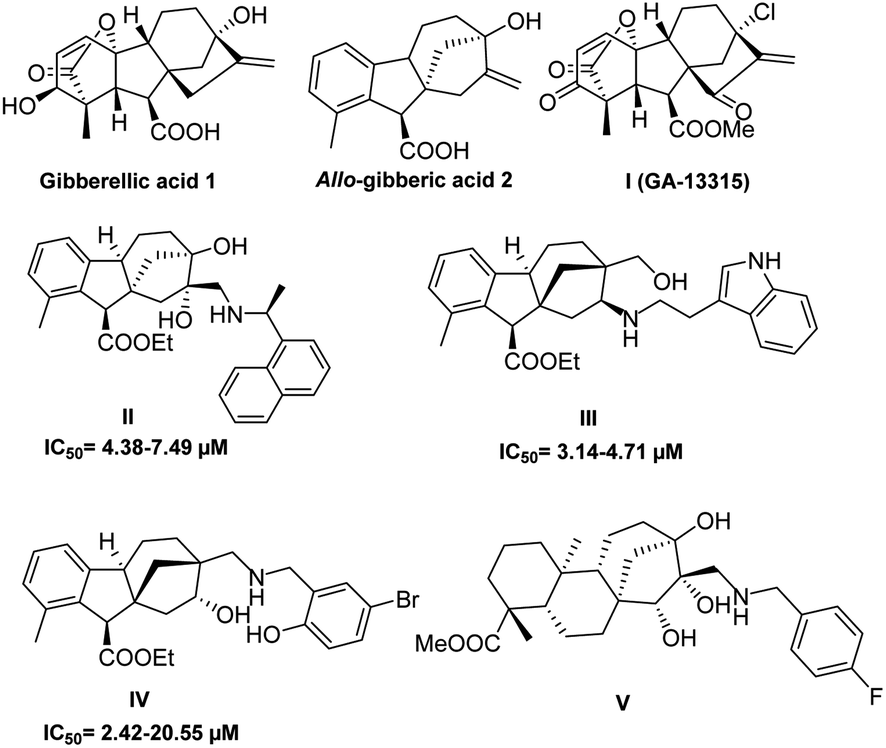 | ||
| Fig. 1 Structures of bioactive gibberellic acid and steviol derivatives. | ||
2 Results and discussion
2.1 Synthetic procedure
2.1.1.1 Stereoselective synthesis of key intermediates (triol 6 and epoxyalcohol 8). Starting from commercially available gibberellic acid 1, allo-gibberic acid 2 was derived by HCl-mediated hydrolysis, in which lactone opening, decarboxylation and then ring A aromatisation occur gradually.29,33 Diol 5 was productively prepared by LiAlH4-mediated reduction of the ester group of either ethyl30 or methyl allo-giberate.28 Allylic diol 5 was oxidised with selenium(IV) dioxide and tert-butyl hydroperoxide (t-BuOOH), yielding triol 6 in a stereospecific manner (16(S)-OH) (de ≥ 95%, NMR determination) (Scheme 1).34 The first attempt to obtain the spiro-epoxide was performed by the direct epoxidation of 6 with t-BuOOH in the presence of vanadyl acetylacetonate (VO(acac)2) as the catalyst. However, it was dismissed considering the low yield and long reaction time, mainly due to the low solubility of triol 6 in organic solvents. To overcome this obstacle, the 1,4-diol functionality was protected as acetonide using dry acetone in the presence of p-toluene sulfonic acid (pTSA).35 Subsequently, the synthesized compound 7 was subjected to the epoxidation of the terminal alkene by utilising either t-BuOOH/VO(acac)2 in dry toluene or m-CPBA in dry DCM. Under the former conditions, the reaction proceeded at a slow rate and heating was needed to obtain the desired product. When using m-CPBA, the reaction was endowed with a higher yield and shorter time at room temperature. In both cases, the reaction took place in a stereospecific manner and cis-epoxy alcohol 8 was observed as a single product (de ≥ 95%, NMR determination) (Scheme 2). Allylic alcohol 7 played a promoting and directing role in epoxidation through the formation of a hydrogen bond with the peracid oxygen of m-CPBA in the transition state.36
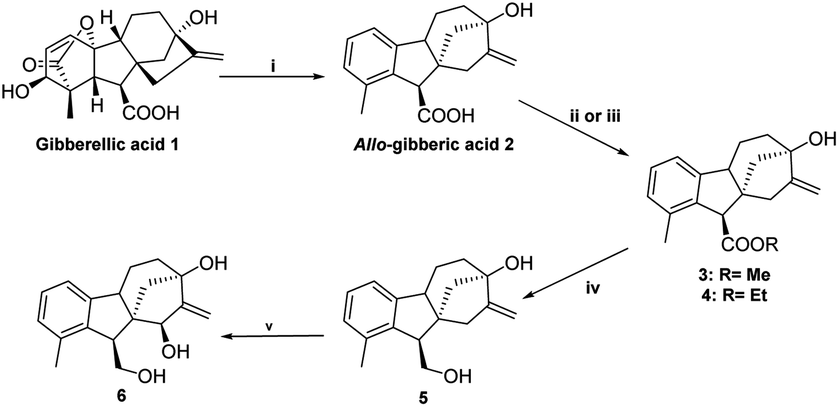 | ||
| Scheme 1 Stereoselective synthesis of triol 6. (i) HCl 1.2 M, 65 °C, 3 h, 70%; (ii) CH2N2, Et2O, 25 °C, 2 min, 85%; (iii) C2H5I (2.5 equ.), Cs2CO3 (2 equ.), MeCN, reflux, 1 h, 99%; (iv) LiAlH4 (2 equ.), dry THF, 40 °C, 12 h, 80%; (v) SeO2 (0.5 equ.), t-BuOOH (3 equ.), dry THF, 40 °C, 12 h, 60%. | ||
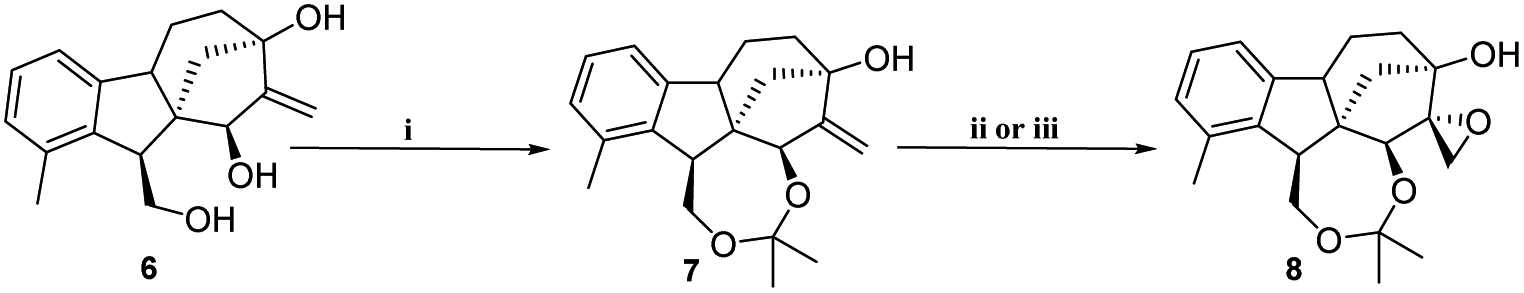 | ||
| Scheme 2 Stereoselective synthesis of the intermediate spiro-epoxide 8. (i) pTSA, dry acetone, 25 °C, 12 h, 80%; (ii) m-CPBA (2.5 equ.), dry DCM, 25 °C, 3 h, 65%; (iii) VO(acac)2, t-BuOOH (1.5 equ.), dry toluene, 50 °C, 12 h, 55%. | ||
2.1.1.2 Synthesis of the desired aminotetraols (aminolysis of epoxyalcohol 8). As described earlier by our research group, nucleophilic addition of amines to epoxides is the optimal approach to obtain aminodiols and aminotriols.37–39 Hence, a library of aminodiols 9–18 based on N-substituted allo-gibberic acid was effectively prepared by the ring opening of oxirane 8 with a variety of primary amines due to the importance of the secondary amine functionality for antiproliferative activity according to our previous work.30 This step was preferably catalysed by LiClO4, where Li+ coordinates with the epoxide oxygen, thereby making the epoxide more prone to nucleophilic attack by amines.40 Moreover, LiClO4 also improves the stereoselectivity of ring opening.41 Aminodiols 9–18 were then deprotected in acidic conditions to provide the desired aminotetraols 19–28 as hydrochloride salts. Nevertheless, in the case of compound 28, a mixture of mono-, di- and tri-hydrochloride salts was obtained; therefore, in order to access the pure compounds, an alkaline liberation was achieved through a chemical extraction applying aqueous Na2CO3 (Scheme 3 and Table 1).
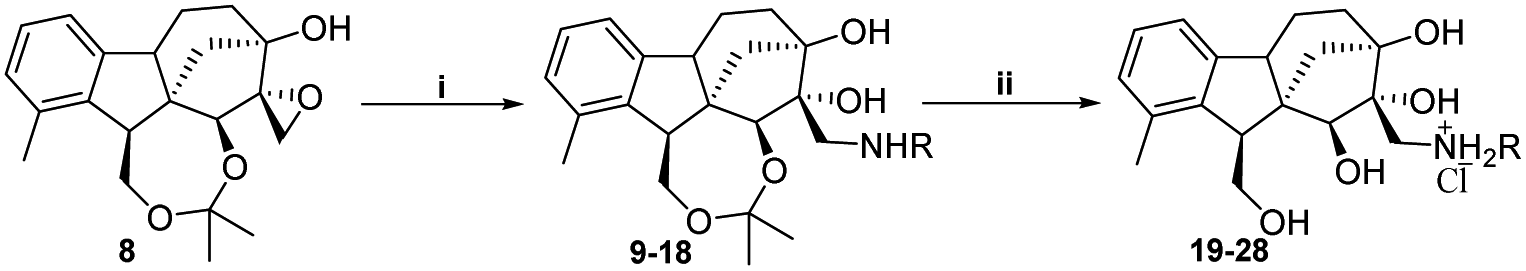 | ||
| Scheme 3 Synthesis of aminodiols 9–18 and aminotetraols 19–28. (i) RNH2 (2.25 equ.), LiClO4 (2.25 equ.), MeCN, 80–90 °C, 24–48 h, 45–85%; (ii) HCl 10%, EtOH, 25 °C, 3 h, 63–91%, then (compound 28) Na2CO3 10%, EtOAc. | ||
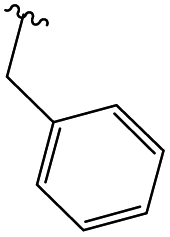
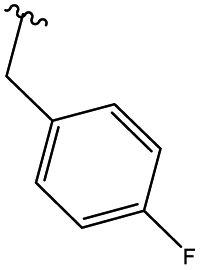
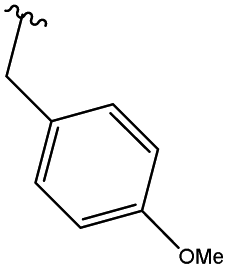
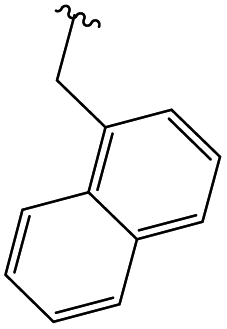
In order to obtain the desired triazoles, azido diol 29 was first prepared by epoxide ring-opening with sodium azide (NaN3) in the presence of NH4Cl. A new series of dihydroxy allo-gibberic acid-based 1,2,3-triazoles 30–35 was again generated by the reaction between dihydroxy azide 29 and pyrimidine-type alkynes48 under Sharpless click chemistry conditions. Acetonide deprotection and subsequent extraction of the resulting hydrochloride salts with aqueous solution of Na2CO3 produced the corresponding tetrahydroxy 1,2,3-triazoles 36–41 (Scheme 4 and Table 2).
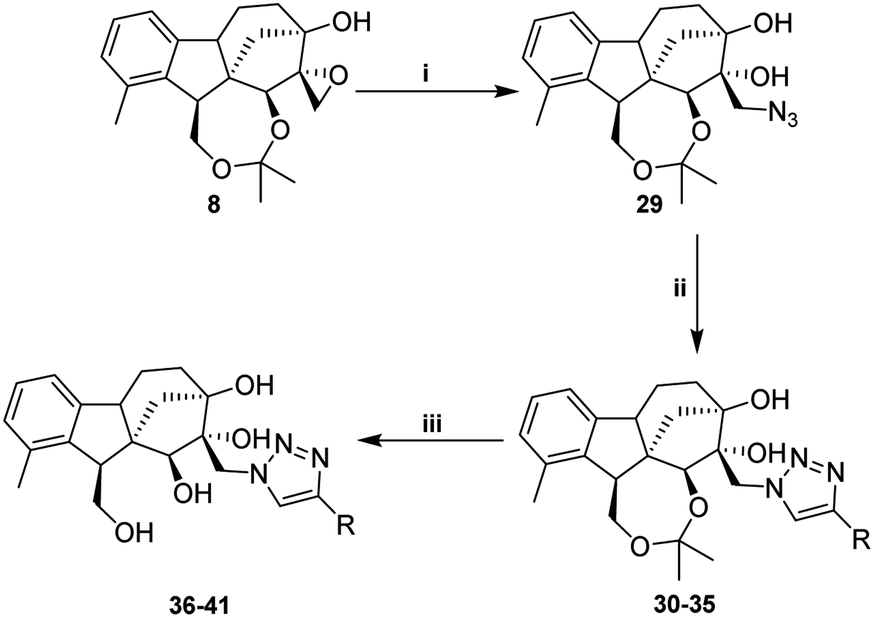 | ||
Scheme 4 Synthesis of dihydroxy and tetrahydroxy 1,2,3-triazoles. (i) NaN3 (5 equ.), NH4Cl (2.5 equ.), EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 8:2, reflux, 48 h, 71%; (ii) acetylenes (1.1 equ.), Cu(OAc)2·H2O (0.1 equ.), sodium ascorbate (0.3 equ.), t-BuOH:H2O (2:1), 40 °C, 12 h, 51–83%; (iii) HCl 10%, EtOH, 25 °C, 3 h, 63–85%. :H2O = 8:2, reflux, 48 h, 71%; (ii) acetylenes (1.1 equ.), Cu(OAc)2·H2O (0.1 equ.), sodium ascorbate (0.3 equ.), t-BuOH:H2O (2:1), 40 °C, 12 h, 51–83%; (iii) HCl 10%, EtOH, 25 °C, 3 h, 63–85%. | ||
| Entry | Compound | R | Yield (%) |
|---|---|---|---|
| 1 | 30 |  |
70 |
| 2 | 31 | 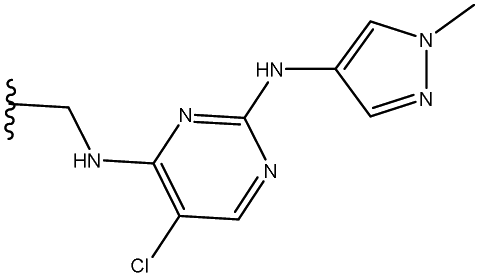 |
83 |
| 3 | 32 | 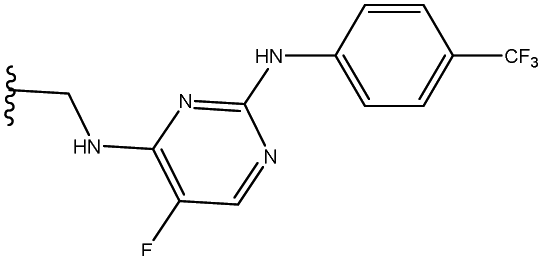 |
83 |
| 4 | 33 | 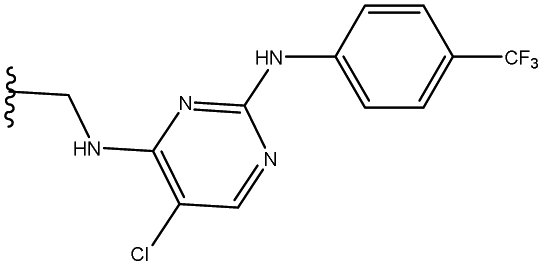 |
66 |
| 5 | 34 | 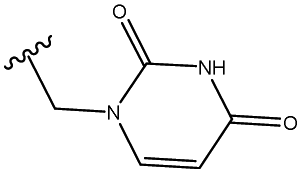 |
51 |
| 6 | 35 | 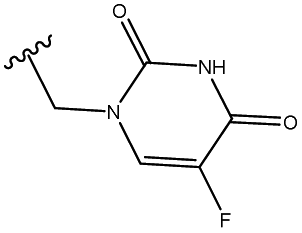 |
56 |
2.1.3.1 Relative configuration of triol 6. As shown in Fig. 2 (compound 6), two clear NOE signals were observed between H-16 and H-13 as well as between OH-16 and H-15. Moreover, a clear COSY signal was observed between H-16 and H-10. In addition, an extra NOE signal was noticed between H-16 and H-9 in compound 7, emphasising the suggested configuration.
 | ||
| Fig. 2 Relative configuration of compounds 6 and 7. | ||
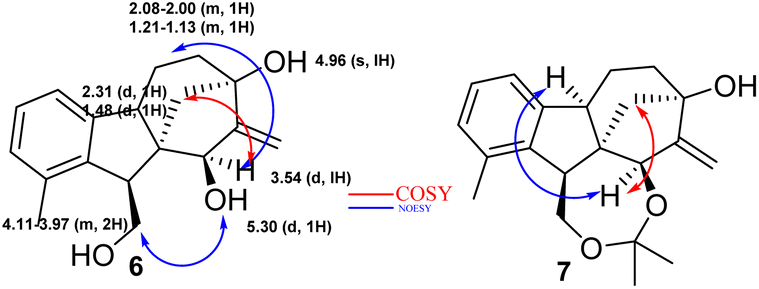
2.1.3.2 Relative configuration of epoxyalcohol 8. The configuration of cis-epoxide was confirmed by two clear NOE signals recorded between H-25 and H-12, along with H-25 and H-13. Since the aminolysis of spiro-epoxide 8 does not affect the absolute configuration, the relative configuration of the chiral centres of 9–41 is known to be the same as that of epoxide 8 (Fig. 3).
 | ||
| Fig. 3 Relative configuration of epoxyalcohol 8. | ||
2.2 Antiproliferative activity
The in vitro antiproliferative activities of the synthesized aminodiols 9–18, aminotetraols 19–28, and dihydroxy and tetrahydroxy substituted 1,2,3-triazoles 30–41 against a panel of different human cancer cell lines, including cervical (SiHa and HeLa), breast (MCF-7 and MDA-MB-231) and ovary (A2780) cancers as well as non-cancerous fibroblast cells (NIH/3T3) were assayed by the MTT method.49 Cisplatin, one of the most widely used anticancer agents, was used as positive control. Results are summarised in Fig. 4. | ||
| Fig. 4 Antiproliferative activities of selected compounds. | ||
2.3 Structure–activity relationship (SAR)
Based on the obtained results, the SARs of these novel derivatives were analysed and summarised (detailed data on antiproliferative effects of all investigated compounds are available in Table S1 in the ESI†).The nitrogen substituent seems to be crucial in determining the cancer-cell-inhibiting properties of the prepared compounds. N-Benzyl-substituted aminodiol 9 displayed modest inhibition values against all tested cancer cell lines (less than 50%). Deprotection of the acetonide moiety, forming aminotetraol 19, led to a moderate increase in the activity. The introduction of either an electron-withdrawing (F) or an electron-donating group (OCH3) at the para position did not show any significant change in the antiproliferative effects of either aminodiols (10 and 11) or aminotetraols (20 and 21).
In the case of N-1-naphthylmethyl-substituted aminodiol 12, the inhibition values were exceedingly low at a concentration of 10 μM, while its related aminotetraol 22 showed more considerable growth in activity with notable selectivity towards MCF-7 and MDA-MDB 231 cell lines (Table 3).
| Compounds | Calculated IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| HeLa | SiHa | MCF-7 | MDA-MB | A2780 | NIH/3T3 | |
| 18 | 3.08 | 3.44 | 2.12 | 1.98 | 1.61 | 4.36 |
| 23 | 3.31 | 3.52 | 3.32 | 5.05 | 6.26 | 4.54 |
| 24 | 3.95 | 3.86 | 3.94 | 5.57 | 6.48 | 4.92 |
| 25 | 3.44 | 2.99 | 3.46 | 4.45 | 5.11 | 4.31 |
| 26 | 3.82 | 4.64 | 3.92 | 5.04 | 5.99 | 5.14 |
| 28 | 2.68 | 3.09 | 1.49 | 3.48 | 3.31 | 4.16 |
| 32 | 4.10 | 5.68 | 3.70 | 5.51 | 5.42 | 4.48 |
| 33 | 4.37 | 6.04 | 3.53 | 6.26 | 4.90 | 5.53 |
| 38 | 4.48 | 6.13 | 4.65 | 7.74 | 5.65 | 7.00 |
| 39 | 5.31 | 8.23 | 5.80 | 8.05 | 5.99 | 8.31 |
| Cisplatin | 12.14 | 4.29 | 8.34 | 25.56 | 5.27 | 5.50 |
In the case of naphthyl derivatives, the tetraol skeleton consistently favoured against their diol analogues, indicating a possible interaction of the 1,4-diol scaffold with the molecular target. The introduction of a methyl group at the α position failed to yield any remarkable increase in the activity of aminodiols 15 and 16. However, S-diastereomer 15 showed moderate activity towards MCF-7, A2780 and HeLa cell lines. Interestingly, their relevant aminotetraols 25 and 26 presented a noteworthy incline in cell growth suppression. In turn, they displayed low selectivity and elevated cytotoxicity towards the fibroblast cell line NIH/3T3. Similar situations were observed with S- and R-α-methyl-2-naphthylmethyl-substituted aminodiols and aminotetraols (13, 14, 23 and 24).
Furthermore, the importance of the “linker” property between allo-gibberic acid and the 2,4-diaminopyrimidine motif was also explored. Based on the results, 2,4-diaminopyrimidine-ethylamine conjugates (compounds 18 and 28) exhibited potent antiproliferative activity, whereas they showed low selectivity and high cytotoxicity towards NIH/3T3. This can be justified by the high flexibility of the ethylamine linker, allowing the rise of various conformations that can mimic a higher number of active sites. Replacement of ethylamine by the 1,2,3-triazole linker (compounds 32, 33, 38 and 39) resulted in similar potent compounds with a slight change in the selectivity profile, which is especially true for the aminotetraol analogues (38, 39). The ability of the triazole moiety to create a restricted number of conformations reduced the possibility of fitting several active sites and probably led to enhanced selectivity. On the other hand, the effect of substituents on the 2,4-pyrimidine ring with respect to antiproliferative properties was investigated. When 5-fluoro substitution (32) was next compared to the 5-chloro analogue (33), the results suggested that 5-chloro-2,4-diaminopyrimidine showed a slightly better selectivity than the 5-fluoro-substituted analogue. Furthermore, the replacement of the N2-(p-trifluoromethyl)phenyl substituent (compound 32 and 33) by the 1-methyl-1H-pyrazol-4-yl group (compound 30 and 31), as expected, the pyrazole analogue became inactive against the cancer cell lines. Additionally, molecules with pyrimidinedione coupled to a triazole linker (34, 35, 40, 41) did not expose any considerable action. The results clearly revealed that the type of substituents at position 4 had a significant effect on the anticancer activity of the 2,4-diaminopyrimidine ring (Table 3).
2.4 Docking study
A docking study was employed to predict the possible target of the most promising compounds (mentioned in Table 1). According to our previous work,30 a group of protein kinases, such as ALK, Pim-1, AKT, and Pak-1, were used as templates for the docking study using the C-Docker method (Table S2†). The findings suggest that all the tested compounds could have a high affinity towards the ATP-binding site of both ALK and Pim-1 kinases by forming strong bonds with several amino acids, with the exception of compound 18, which shows higher affinity to Pim-1 kinase (all the 2D interaction diagrams are provided in the ESI, Table S2†). The CDocker interaction energy of the tested compounds varied between −41.1687 and −58.5523 for ALK and between −47.3708 and −77.4483 for Pim-1. The docking results of the studied compounds show the key role of aminodiol, pyrimidine and triazole in forming hydrogen bonding and ionic interactions with the key amino acids.3 Experimental
Reagents were obtained from commercial suppliers and used without further purification (Molar Chemicals Ltd, Halásztelek, Hungary; Merck Ltd, Budapest, Hungary, and VWR International Ltd, Debrecen, Hungary), while solvents were distilled and dried with several different drying agents following the standard procedures before use (Williams and Lawton 2010). Optical rotations were measured in MeOH at 20 °C with a PerkinElmer 341 polarimeter (PerkinElmer Inc., Shelton, CT, USA). Chromatographic separations and monitoring of reactions were carried out on Merck Kieselgel 60 (Merck Ltd, Budapest, Hungary). The HR-MS flow injection analysis was performed with a Thermo Scientific Q Exactive Plus hybrid quadrupole-Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC™ (Waters, Manchester, UK). Melting points were determined on a Kofler apparatus (Nagema, Dresden, Germany) and were uncorrected. 1H- and 13C J-MOD NMR spectra were recorded on a Brucker Avance DRX 500 spectrometer (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) [500 MHz (1H) and 125 MHz (13C J-MOD), δ = 0 (TMS)]. Chemical shifts are expressed in ppm (δ) relative to TMS as the internal reference. J values are given by Hz. The 1H, 13C J-MOD, 19F J-MOD, COSY, NOESY, HSQC and HMBC NMR spectra of the new compounds are available in the ESI.†Gibberellic acid 1 is commercially available from Merck Co with ee% > 98%, chemical purity >90% ([α]20D = +78.0, c 2.0, MeOH). Since none of the applied transformations reach all the chiral centers at the same time, we believe that the enantiomer purity of all prepared compounds can be defined as ee ≥ 98%, similar to commercial gibberellic acid.
allo-Gibberic acid 2 was prepared from gibberellic acid 1 according to the literature.29,33 Compounds 3–5 were prepared as mentioned in the literature. All spectroscopic data were similar to those described therein.
3.1 Preparation of the reagents
3.1.1.1 Synthesis of R1. Benzyl (2-aminoethyl)carbamate (1.38 g, 7.11 mmol) was dissolved in EtOH (60 mL), then 2,4-dichloro-5-fluoropyrimidine (1.18 g, 7.11 mmol, 1 equ.) and triethylamine (4.9 mL, 4.9 equ.) were added. After being stirred under reflux for 2 hours, the mixture was evaporated to dryness. The residue was diluted with water (50 mL) and extracted with EtOAc (3 × 50 mL). The organic phase was washed with brine solution (50 mL), dried over Na2SO4, filtered and evaporated. The crude product was purified by column chromatography using n-hexane
:EtOAc (1:1) as eluent (Scheme 5).
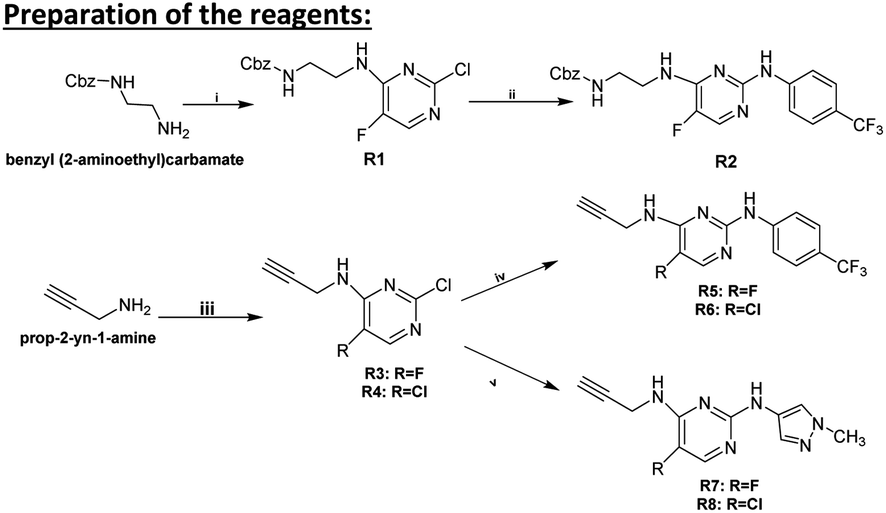 | ||
| Scheme 5 Synthesis of reagents R1–R8. (i) 2,4-Dichloro-5-fluoropyrimidine (1 equ.), triethylamine (4.9 equ.), EtOH, reflux, 2 h, 54%; (ii) 4-trifluoromethylaniline (1 equ.), EtOH, reflux, 24 h, 51%; (iii) 2,4-dichloro-5-fluoropyrimidine or 2,4,5-trichloropyrimidine (0.6 equ.), triethylamine (4.8 equ.), MeCN, 25 °C, 24 h, R3 (65%), R4 (88%); (iv) 4-trifluoromethylaniline (1.1 equ.), EtOH, MW, 150 °C, 200 W, 19 bar, 2 h, R5 (58%), R6 (52%); (iv) 1-methyl-1H-pyrazol-4-amine (1.1 equ.), EtOH, MW, 150 °C, 200 W, 19 bar, 2 h, R7 (91%), R8 (50%). | ||
3.1.1.2 Benzyl (2-((2-chloro-5-fluoropyrimidin-4-yl)amino)ethyl)carbamate R1. Yield: 54%; white crystals; m.p.: 116–118 °C; 1H NMR (500 MHz, DMSO-d6): δ = 8.13 (s, 1H), 8.05 (d, J = 2.7 Hz, 1H), 7.38–7.28 (m, 6H), 5.02 (s, 2H), 3.42 (q, J = 6.0 Hz, 2H), 3.22 ppm (q, J = 6.1 Hz, 2H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 156.7, 154.2 (d, JF,C = 14.9 Hz), 153.9 (d, JF,C = 2.2 Hz), 145.7 (d, JF,C = 255.9 Hz), 139.9 (d, JF,C = 20.1 Hz), 137.6, 128.8 (3C), 128.2 (2C), 65.8, 40.51, 40.31 ppm; 19F J-MOD NMR (470 MHz, DMSO-d6): δ = −157.4 (Cq-F); HR-MS (ESI): m/z calcd for C14H15ClFN4O2 [M + H]+: 325.0867; found: 325.0858.
3.1.1.3 Synthesis of R2. Compound R1 (1.00 g, 3.08 mmol) was dissolved in EtOH (20 mL) then 4-trifluoromethylaniline (0.49 g, 3.08 mmol, 1 equ.) was added. The mixture was stirred under reflux for 24 hours. After that, the mixture was evaporated to dryness, and the crude product was purified by column chromatography using n-hexane
:EtOAc (1:1) as eluent.
3.1.1.4 Benzyl (2-((5-fluoro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)ethyl)carbamate R2. Yield: 51%; white crystals; m.p.: 160–161 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.48 (s, 1H), 7.95–7.91 (m, 3H), 7.57 (d, J = 8.4 Hz, 2H), 7.53 (s, 1H), 7.35–7.27 (m, 6H), 5.01 (s, 2H), 3.50 (q, J = 6.0 Hz, 2H), 3.27 ppm (q, J = 5.8 Hz, 2H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 156.8, 155.8 (d, JF,C = 2.7 Hz), 152.7 (d, JF,C = 12.4 Hz), 145.3, 141.8 (d, JF,C = 248.1 Hz), 138.0 (d, JF,C = 19.0 Hz), 137.5, 128.8 (3C), 128.2 (2C), 126.4 (q, JF,C = 272.3 Hz), 126.1 (q, JF,C = 4.1 Hz, 2C), 120.6 (q, JF,C = 30.8 Hz), 117.9 (2C), 65.7, 40.6, 40.3 ppm; 19F J-MOD NMR (470 MHz, DMSO-d6): δ = −59.7 (Cq-F), −165.9 (Cq-F); HR-MS (ESI): m/z calcd for C21H20F4N5O2 [M + H]+: 450.1553; found: 450.1540.
:EtOAc (4:1) as eluent.
3.1.2.1 2-Chloro-5-fluoro-N-(prop-2-yn-1-yl)pyrimidin-4-amine R3. Prepared with 2,4-dichloro-5-fluoropyrimidine, yield: 65%; white crystals; m.p.: 102–104 °C; 1H NMR (500 MHz, DMSO-d6): δ = 8.59 (t, J = 5.0 Hz, 1H), 8.14 (d, J = 3.2 Hz, 1H), 4.14 (dd, J = 2.2, 5.6 Hz, 2H), 3.14 ppm (t, J = 2.3 Hz, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 153.8 (d, JF,C = 2.8 Hz), 153.4 (d, JF,C = 13.2 Hz), 145.8 (d, JF,C = 256 Hz), 140.8 (d, JF,C = 20.2 Hz), 80.7, 73.8, 29.9 ppm; 19F J-MOD NMR (470 MHz, DMSO-d6): δ = −157.0 (Cq-F); HR-MS (ESI): m/z calcd for C7H6ClFN3 [M + H]+: 186.0234; found: 186.0227.
3.1.2.2 2,5-Dichloro-N-(prop-2-yn-1-yl)pyrimidin-4-amine R4. Prepared with 2,4,5-trichloropyrimidine, yield: 88%; white crystals; m.p.: 100.2–102.9 °C; 1H NMR (500 MHz, DMSO-d6): δ = 8.33 (t, J = 4.8 Hz, 1H), 8.24 (s, 1H), 4.14 (dd, J = 2.7, 2.7 Hz, 2H), 3.11 ppm (t, J = 2.4 Hz, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 158.8, 157.8, 154.7, 113.6, 80.8, 73.5, 30.5 ppm; HR-MS (ESI): m/z calcd for C7H6Cl2N3 [M + H]+: 201.9938; found: 201.9932.
:EtOAc (4:1) for R5, n-hexane:EtOAc (2:1) for R6, and CHCl3:MeOH (19:1) for R7 and R8.
3.1.3.1 5-Fluoro-N4-(prop-2-yn-1-yl)-N2-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine R5. Prepared with R3 and 4-trifluoromethylaniline, yield: 58%; white crystals; m.p.: 188–192 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.59 (s, 1H), 8.00–7.96 (m, 4H), 7.55 (d, J = 8.5 Hz, 2H), 4.16 (d, J = 4.0 Hz, 2H), 3.14 ppm (s, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 155.7 (d, JF,C = 3.1 Hz), 152.0 (d, JF,C = 12.4 Hz), 144.0 (d, JF,C = 301.2 Hz), 140.8, 139.7 (d, JF,C = 18.9 Hz), 126.1 (q, JF,C = 6.1 Hz, 2C), 123.3 (q, JF,C = 270.8 Hz), 120.6 (q, JF,C = 33.2 Hz), 118.0 (2C), 81.7, 73.2, 28.9 ppm; 19F J-MOD NMR (470 MHz, DMSO-d6): δ = −59.7 (Cq-F), −165.9 (Cq-F); HR-MS (ESI): m/z calcd for C14H11F4N4 [M + H]+: 311.0919; found: 311.0908.
3.1.3.2 5-Chloro-N4-(prop-2-yn-1-yl)-N2-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine R6. Prepared with R4 and 4-trifluoromethylaniline, yield: 52%; white crystals; m.p.: 158–160 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.73 (s, 1H), 8.06 (s, 1H), 8.01 (d, J = 8.5 Hz, 2H), 7.71 (t, J = 5.5 Hz, 1H), 7.57 (d, J = 8.5 Hz, 2H), 4.16 (d, J = 4.0 Hz, 2H), 3.12 ppm (s, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 158.0, 157.6, 153.9, 144.8, 126.0 (q, JF,C = 4.1 Hz, 2C), 124.1 (q, JF,C = 276.8 Hz), 121.2 (q, JF,C = 32.2 Hz), 118.6 (2C), 105.2, 81.9, 73.0, 43.4 ppm; 19F J-MOD NMR (470 MHz, DMSO-d6): δ = −59.8 (Cq-F); HR-MS (ESI): m/z calcd for C14H11ClF3N4 [M + H]+: 327.0624; found: 327.0613.
3.1.3.3 5-Fluoro-N2-(1-methyl-1H-pyrazol-4-yl)-N4-(prop-2-yn-1-yl)pyrimidine-2,4-diamine R7. Prepared with R3 and 1-methyl-1H-pyrazol-4-amine, yield: 91%; white crystals; m.p.: 166–169 °C; 1H NMR (500 MHz, DMSO-d6): δ = 8.89 (s, 1H), 7.86 (d, J = 5.0 Hz, 2H), 7.72 (t, J = 5.3 Hz, 1H), 7.45 (s, 1H), 4.15–4.12 (m, 2H), 3.76 (s, 3H), 3.10 ppm (s, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 156.0 (d, JF,C = 1.7 Hz), 152.1 (d, JF,C = 12.4 Hz), 141.8, 139.8 (d, JF,C = 27.1 Hz), 129.9, 124.5, 120.2, 82.1, 73.1, 39.0, 29.7 ppm; 19F J-MOD NMR (470 MHz, DMSO-d6): δ = −169.9 (Cq-F); HR-MS (ESI): m/z calcd for C11H12FN6 [M + H]+: 247.1107; found: 247.1098.
3.1.3.4 5-Chloro-N2-(1-methyl-1H-pyrazol-4-yl)-N4-(prop-2-yn-1-yl)pyrimidine-2,4-diamine R8. Prepared with R4 and 1-methyl-1H-pyrazol-4-amine, yield: 50%; white crystals; m.p.: 194–196 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.07 (s, 1H), 7.93–7.89 (m, 2H), 7.50–7.45 (m, 2H), 4.14 (d, J = 3.8 Hz, 2H), 3.77 (s, 3H), 3.09 ppm (s, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 158.1, 158.0, 157.7, 154.0, 130.2, 124.0, 120.6, 82.2, 72.9, 39.1, 30.3 ppm; HR-MS (ESI): m/z calcd for C11H12ClN6 [M + H]+: 263.0811; found: 263.0802.
:MeOH (9:1) as eluent to obtain compound 6 as a white solid (1.40 g, 66%).
3.1.4.1 (4bR,7S,9S,9aR,10S)-10-(Hydroxymethyl)-1-methyl-8-methylene-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,9-diol 6. Yield: 66%; Rf = 0.4, white crystals; m.p.: 165.6–168.2 °C; [α]20D = −72.5 (c 0.18, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 7.00 (t, J = 7.1 Hz, 1H), 6.89 (d, J = 7.2 Hz, 1H), 6.84 (d, J = 7.2 Hz, 1H), 5.30 (d, J = 5.2 Hz, 1H), 5.06 (s, 1H), 5.04 (t, J = 4.6 Hz, 1H), 5.00 (s, 1H), 4.96 (s, 1H), 4.11–3.97 (m, 2H), 3.54 (d, J = 4.3 Hz, 1H), 3.04 (t, J = 3.9 Hz, 1H), 2.49–2.46 (m, 1H), 2.45 (s, 3H), 2.31 (d, J = 10.5 Hz, 1H), 2.08–2.00 (m, 1H), 1.81–1.73 (m, 1H), 1.56–1.44 (m, 2H), 1.21–1.13 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 161.4, 145.2, 142.1, 134.3, 129.4, 126.1, 119.4, 106.8, 78.1, 78.7, 58.8, 58.3, 54.4, 51.7, 45.1, 40.09, 21.7, 19.8; HR-MS (ESI): m/z calcd for C18H21O2 [M − H2O]+: 269.1541; found: 269.1537.
:EtOAc (2:1) as an eluent to get compound 7 as a white solid (1.00 g, 87.7%).
3.1.5.1 (4aR,4a1R,6S,12bS)-3,3,12-Trimethyl-5-methylene-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepin-6(1H)-ol 7. Yield: 87.7%; Rf = 0.6, white crystals; m.p.: 188.1–190.9 °C; [α]20D = −106.0 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.07 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 6.88 (d, J = 7.5 Hz, 1H), 5.29 (s, 1H), 5.06 (s, 1H), 4.26–4.16 (m, 2H), 4.03 (s, 1H), 3.12 (s, 1H), 2.74 (dd, J = 5.1, 13.2 Hz, 1H), 2.44 (s, 4H), 2.20–2.14 (m, 1H), 1.98–1.91 (m, 1H), 1.79–1.74 (m, 1H), 1.68 (d, J = 10.1 Hz, 1H), 1.45–1.35 (m, 1H), 1.29 (s, 3H), 1.08 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 158.6, 143.9, 140.9, 135.3, 129.2, 126.2, 118.8, 108.7, 101.0, 79.2, 74.9, 56.8, 55.4, 52.5, 50.9, 46.5, 39.1, 26.8, 24.4, 21.7, 20.6; HR-MS (ESI): m/z calcd for C21H27O3 [M + H]+: 327.1960; found: 327.1956.
3.1.6.1 Method A. To a solution of 7 (2.00 g, 6.12 mmol) in DCM (100 mL), m-CPBA (3.16 g, 18.36 mmol, 3 equ., 70% purity) was added. After stirring for 4 hours at room temperature (indicated by TLC), the mixture was washed with a saturated solution of Na2SO3 (3 × 50 mL). The organic phase was then dried over Na2SO4, filtered, and evaporated. The crude product was purified by column chromatography using n-hexane
:EtOAc (1:1) as eluent.
3.1.6.2 Method B. To a solution of 7 (2.00 g, 6.12 mmol) in dry toluene (200 mL), VO(acac)2 (20 mg) was added, and the green solution was stirred at 0 °C for 30 minutes. Then, t-BuOOH (extracted from its 70% H2O solution with toluene and dried over Na2SO4, 9.18 mmol, 1.5 equ.) was added dropwise to the former solution and the mixture turned to dark red and then faded to orange as the reaction proceeded. The reaction mixture was stirred at 70 °C overnight. A saturated solution of NaHCO3 (30 mL) was added, followed by extraction with toluene (3 × 30 mL). The organic phase was washed with brine (3 × 30 mL), dried over Na2SO4 and then evaporated at low pressure. The crude product was purified by column chromatography using n-hexane
:EtOAc (1:1) as eluent.
3.1.6.3 (2R,4a′S,4a1′R,6′S,12b′S)-3′,3′,12′-Trimethyl-7′,8′,8a′,12b′-tetrahydro-4a′H-spiro[oxirane-2,5′-[4a1,6]methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepin]-6′(1′H)-ol 8. Yield: A: 65%, B: 55%; Rf = 0.5, white crystals; m.p.: 173.2–175.9 °C; [α]20D = −90.5 (c 0.17, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.08 (t, J = 7.5 Hz, 1H), 6.96 (d, J = 7.4 Hz, 1H), 6.90 (d, J = 7.4 Hz, 1H), 4.24–4.22 (m, 2H), 3.75 (s, 1H), 3.18 (s, 1H), 2.89 (s, 2H), 2.77 (dd, J = 5.1, 13.2 Hz, 1H), 2.52 (dd, J = 2.1, 10.5 Hz, 1H), 2.44 (s, 3H), 2.41 (s, 1H), 2.33–2.26 (m, 1H), 2.04–1.96 (m, 1H), 1.77 (dd, J = 1.3, 10.5 Hz, 1H), 1.71–1.65 (m, 1H), 1.57–1.50 (m, 1H), 1.32 (s, 3H), 0.93 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.4, 140.8, 135.4, 129.4, 126.3, 118.8, 101.0, 74.0, 73.8, 65.3, 56.8, 56.5, 52.8, 50.5, 48.2, 45.9, 36.1, 26.3, 23.8, 21.1, 20.6; HR-MS (ESI): m/z calcd for C18H21O3 [M − C3H4–H2O]+: 285.1490; found: 285.1483.
:2 (100 mL), NaN3 (0.95 g, 14.6 mmol, 5 equ.) and NH4Cl (0.39 g, 7.29 mmol, 2.49 equ.) were added. After treatment under reflux conditions for 48 h, the reaction mixture was cooled to room temperature, and then ethanol was evaporated. The residue was dissolved in water (50 mL) and extracted with Et2O (3 × 50 mL). The organic phase was washed with a brine solution, dried over Na2SO4, and then evaporated. The compound was purified by flash column chromatography on silica gel (eluted with n-nexane:EtOAc = 1:1) to provide azido diol.
3.1.7.1 (4aR,4a1R,5R,6S,12bS)-5-(Azidomethyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 29. Yield: 71%; Rf = 0.7, white crystals; m.p.: 175.1–176.9 °C; [α]20D = +64.5 (c 0.15, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.09 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.88 (d, J = 7.3 Hz, 1H), 4.61 (s, 1H), 4.24–4.22 (m, 2H), 3.84 (s, 1H), 3.54 (d, J = 12.8 Hz, 1H), 3.24–3.17 (m, 2H), 3.15 (d, J = 12.8 Hz, 1H), 2.76 (dd, J = 5.6, 13.1 Hz, 1H), 2.46 (d, J = 11.2 Hz, 1H), 2.41 (s, 3H), 2.34–2.26 (m, 1H), 2.05–1.93 (m, 1H), 1.77 (dd, J = 5.3, 13.7 Hz, 1H), 1.51 (dd, J = 2.3, 10.9 Hz, 1H), 1.48–1.40 (m, 1H), 1.38 (s, 3H), 0.94 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.0, 140.6, 135.6, 129.5, 126.5, 118.8, 101.3, 80.3, 79.0, 75.4, 57.4, 55.3, 52.9, 52.6, 51.3, 46.0, 31.3, 27.5, 23.6, 21.1, 20.6; HR-MS (ESI): m/z calcd for C21H27N3O4Na [M + Na]+: 408.1899; found: 408.1889.
:MeOH = 19:1), followed by the second column chromatography using n-hexane:EtOAc = 1:2 as eluent.
3.1.8.1 (4aS,4a1R,5R,6S,12bS)-5-((Benzylamino)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 9. Prepared with benzylamine, yield: 85%; Rf = 0.8 (CHCl3
:MeOH = 19:1), white wax; m.p.: 127–130 °C; [α]20D = −50.9 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.29–7.22 (m, 5H), 7.09 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 7.4 Hz, 1H), 6.87 (d, J = 7.4 Hz, 1H), 4.39 (s, 1H), 4.22 (s, 2H), 3.83 (s, 1H), 3.77 (s, 2H), 3.17 (s, 1H), 2.84 (d, J = 11.4 Hz, 1H), 2.75–2.67 (m, 2H), 2.47 (d, J = 10.8 Hz, 1H), 2.41 (s, 3H), 2.24–2.15 (m, 1H), 1.94–1.86 (m, 1H), 1.80 (dd, J = 5.1, 13.5 Hz, 1H), 1.50–1.37 (m, 2H), 1.31 (s, 3H), 0.92 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.6, 140.9, 140.6, 135.5, 129.3, 128.3, 128.2, 128.0, 127.9, 126.8, 126.4, 118.9, 101.0, 79.1, 78.9, 76.1, 57.5, 55.5, 54.3, 53.0, 51.3, 50.6, 45.9, 31.9, 27.9, 24.0, 21.3, 20.7; HR-MS (ESI): m/z calcd for C28H36NO4 [M + H]+: 450.2644; found: 450.2634.
3.1.8.2 (4aS,4a1R,5R,6S,12bS)-5-(((4-Fluorobenzyl)amino)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 10. Prepared with 4-fluorobenzylamine, yield: 45%; Rf = 0.7 (CHCl3
:MeOH = 19:1), white wax; m.p.: 58–60 °C; [α]20D = −55.9 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.23–7.19 (m, 2H), 7.10 (t, J = 7.4 Hz, 1H), 6.98–6.92 (m, 3H), 6.86 (d, J = 7.3 Hz, 1H), 4.38 (s, 1H), 4.22 (d, J = 2.3 Hz, 2H), 3.81 (d, J = 1.4 Hz, 3H), 3.73–3.68 (m, 2H), 3.16 (s, 1H), 2.82 (d, J = 11.7 Hz, 1H), 2.72 (dd, J = 5.6, 12.9 Hz, 1H), 2.66 (d, J = 11.7 Hz, 1H), 2.46 (dd, J = 1.9, 10.5 Hz, 1H), 2.41 (s, 3H), 2.24–2.17 (m, 1H), 1.94–1.86 (m, 1H), 1.78 (dd, J = 5.5, 13.6 Hz, 1H), 1.49–1.36 (m, 2H), 1.31 (s, 3H), 0.91 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 161.8 (d, JF,C = 243.6 Hz), 143.5, 140.8, 136.3 (d, JF,C = 3.6 Hz), 135.5, 129.4, 129.3 (d, JF,C = 4.1 Hz, 2C), 126.4, 118.8, 115.0 (d, JF,C = 20.3 Hz, 2C), 101.0, 79.0, 78.9, 76.2, 57.5, 55.5, 53.8, 53.0, 51.3, 50.5, 45.9, 31.8, 27.8, 23.9, 21.3, 20.6; 19F J-MOD NMR (470 MHz, CDCl3): δ = −116.3 (Cq-F) ppm; HR-MS (ESI): m/z calcd for C28H35FNO4 [M + H]+: 468.2550; found: 468.2533.
3.1.8.3 (4aS,4a1R,5R,6S,12bS)-5-(((4-Methoxybenzyl)amino)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 11. Prepared with 4-methoxybenzylamine, yield: 46%; Rf = 0.7 (CHCl3
:MeOH = 19:1), white wax; m.p.: 133–134 °C; [α]20D = −57.3 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.18 (d, J = 7.8 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H), 6.81 (d, J = 7.8 Hz, 2H), 4.22 (s, 2H), 3.80–3.71 (m, 6H), 3.16 (s, 1H), 2.85 (d, J = 12.0 Hz, 1H), 2.76–2.67 (m, 2H), 2.47 (d, J = 10.1 Hz, 1H), 2.41 (s, 3H), 2.23–2.17 (m, 1H), 1.94–1.85 (m, 1H), 1.82–1.77 (m, 1H), 1.49–1.38 (m, 2H), 1.31 (s, 3H), 0.90 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 158.6, 143.5, 140.9, 135.5, 129.3, 129.2, 129.2 126.4, 118.8, 113.7, 113.7, 101.04, 79.1, 78.8, 76.2, 57.5, 55.5, 55.2, 53.7, 53.0, 51.4, 50.4, 45.9, 31.9, 27.9, 23.9, 21.3, 20.6; HR-MS (ESI): m/z calcd for C29H38NO5 [M + H]+: 480.2749; found: 480.2735.
3.1.8.4 (4aS,4a1R,5R,6S,12bS)-3,3,12-Trimethyl-5-(((naphthalen-1-ylmethyl)amino)methyl)-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 12. Prepared with 1-naphthylmethylamine, yield: 62%; Rf = 0.6 (CHCl3
:MeOH = 19:1), colourless oily compound; [α]20D = −31.0 (c 0.19, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.09 (d, J = 8.2 Hz, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.74 (d, J = 7.9 Hz, 1H), 7.46–7.35 (m, 4H), 7.10 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 7.2 Hz, 1H), 4.25–4.17 (m, 4H), 3.84 (s, 1H), 3.15 (s, 1H), 2.96 (d, J = 11.7 Hz, 1H), 2.82 (d, J = 11.8 Hz, 1H), 2.72 (dd, J = 6.3, 13.5 Hz, 1H), 2.47 (d, J = 10.1 Hz, 1H), 2.41 (s, 3H), 2.24–2.15 (m, 1H), 1.93–1.84 (m, 1H), 1.78 (dd, J = 6.1, 14.2 Hz, 1H), 1.51–1.41 (m, 2H), 1.25 (s, 3H), 0.82 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.5, 140.9, 136.0, 135.5, 133.7, 131.8, 129.3, 128.5, 127.6, 126.4, 125.8, 125.6, 125.5, 125.2, 123.9, 118.8, 101.0, 79.1, 79.0, 76.2, 57.5, 55.5, 53.0, 52.3, 51.4, 51.2, 45.9, 31.9, 27.8, 23.8, 21.3, 20.6; HR-MS (ESI): m/z calcd for C32H38NO4 [M + H]+: 500.2800; found: 500.2789.
3.1.8.5 (4aS,4a1R,5R,6S,12bS)-3,3,12-Trimethyl-5-((((S)-1-(naphthalen-2-yl)ethyl)amino)methyl)-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 13. Prepared with (S)-(−)-1-(2-naphthyl)ethylamine, yield: 63%; Rf = 0.5 (CHCl3
:MeOH = 19:1), white crystals; m.p.: 126–129 °C; [α]20D = −102.1 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.84–7.76 (m, 3H), 7.68 (s, 1H), 7.47–7.43 (m, 3H), 7.06 (t, J = 7.4 Hz, 1H), 6.95 (d, J = 7.5 Hz, 1H), 6.82 (d, J = 7.2 Hz, 1H), 4.32 (s, 1H), 4.24–4.21 (m, 2H), 3.82 (q, J = 6.5 Hz, 1H), 3.75 (s, 1H), 3.16 (s, 1H), 2.73–2.67 (m, 2H), 2.61 (d, J = 11.6 Hz, 1H), 2.49 (d, J = 10.5 Hz, 1H), 2.41 (s, 3H), 2.16–2.08 (m, 1H), 1.88–1.80 (m, 1H), 1.76 (dd, J = 5.4, 13.2 Hz, 1H), 1.44 (d, J = 10.8 Hz, 1H), 1.39–1.34 (m, 4H), 1.33 (s, 3H), 0.91 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.5, 143.2, 140.9, 135.5, 133.4, 132.8, 129.3, 128.2, 127.69, 127.66, 126.3, 125.9, 125.4, 125.1, 124.7, 118.8, 100.9, 78.2, 78.9, 76.4, 59.0, 57.5, 55.4, 53.0, 51.5, 49.5, 46.0, 32.0, 27.9, 24.1, 23.9, 21.3, 20.6; HR-MS (ESI): m/z calcd for C33H40NO4 [M + H]+: 514.2957; found: 514.2942.
3.1.8.6 (4aS,4a1R,5R,6S,12bS)-3,3,12-Trimethyl-5-((((R)-1-(naphthalen-2-yl)ethyl)amino)methyl)-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 14. Prepared with (R)-(−)-1-(2-naphthyl)ethylamine, yield: 68%; Rf = 0.5 (CHCl3
:MeOH = 19:1), white crystals; m.p.: 158–160 °C; [α]20D = −60.8 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.84–7.80 (m, 1H), 7.70 (d, J = 8.7 Hz, 1H), 7.66 (s, 1H), 7.63–7.59 (m, 1H), 7.47–7.44 (m, 2H), 7.34 (d, J = 8.4 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 7.01 (d, J = 7.5 Hz, 1H), 6.65 (d, J = 7.2 Hz, 1H), 4.50 (s, 1H), 4.24 (d, J = 2.1 Hz, 2H), 3.95 (s, 1H), 3.87 (q, J = 6.7 Hz, 1H), 3.15 (s, 1H), 2.72 (d, J = 11.7 Hz, 1H), 2.61 (dd, J = 5.9, 13.0 Hz, 1H), 2.54 (d, J = 12.8 Hz, 1H), 2.44 (s, 3H), 2.44–2.40 (m, 1H), 1.93–1.85 (m, 1H), 1.81–1.72 (m, 1H), 1.59–1.56 (m, 1H), 1.42 (dd, J = 1.8, 10.3 Hz, 1H), 1.38 (s, 3H), 1.32 (d, J = 6.6 Hz, 3H), 1.10 (s, 3H), 1.08–0.98 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.6, 143.4, 140.8, 135.4, 133.4, 132.7, 129.3, 128.1, 127.9, 127.5, 126.3, 125.8, 125.3, 125.0, 1224.8, 118.9, 101.1, 79.3, 78.8, 76.1, 58.4, 57.5, 55.5, 53.0, 51.0, 48.3, 45.9, 31.6, 27.9, 25.1, 24.2, 21.0, 20.6; HR-MS (ESI): m/z calcd for C33H40NO4 [M + H]+: 514.2957; found: 514.2941.
3.1.8.7 (4aS,4a1R,5R,6S,12bS)-3,3,12-Trimethyl-5-((((S)-1-(naphthalen-1-yl)ethyl)amino)methyl)-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 15. Prepared with (S)-(−)-1-(1-naphthyl)ethylamine, yield: 59%; Rf = 0.5 (CHCl3
:MeOH = 19:1), white crystals; m.p.: 177–179 °C; [α]20D = −68.2 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.15 (d, J = 8.5 Hz, 1H), 7.87–7.84 (m, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.57 (d, J = 7.0 Hz, 1H), 7.48–7.43 (m, 3H), 7.07 (t, J = 7.4 Hz, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.84 (d, J = 7.3 Hz, 1H), 4.53 (q, J = 6.6 Hz, 1H), 4.37 (s, 1H), 4.24–4.21 (m, 2H), 3.81 (s, 1H), 3.16 (s, 1H), 2.83 (d, J = 11.7 Hz, 1H), 2.71 (dd, J = 5.6, 13.5 Hz, 1H), 2.64 (d, J = 11.4 Hz, 1H), 2.49 (d, J = 10.5 Hz, 1H), 2.41 (s, 3H), 2.21–2.14 (m, 1H), 1.91–1.77 (m, 2H), 1.48–1.43 (m, 2H), 1.42 (d, J = 6.5 Hz, 3H), 1.31 (s, 3H), 0.91 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.6, 141.4, 140.9, 135.5, 133.9, 131.2, 129.3, 128.9, 127.1, 126.3, 125.69, 125.64, 125.2, 123.0, 122.6, 118.8, 101.0, 79.1, 79.0, 76.4, 57.5, 55.4, 54.3, 53.0, 51.5, 49.6, 46.1, 31.9, 27.9, 23.9, 23.6, 21.3, 20.6; HR-MS (ESI): m/z calcd for C33H40NO4 [M + H]+: 514.2957; found: 514.2943.
3.1.8.8 (4aS,4a1R,5R,6S,12bS)-3,3,12-Trimethyl-5-((((R)-1-(naphthalen-1-yl)ethyl)amino)methyl)-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 16. Prepared with (R)-(+)-1-(1-naphthyl)ethylamine, yield: 54%; Rf = 0.5 (CHCl3
:MeOH = 19:1), white crystals; m.p.: 83–86 °C; [α]20D = −49.9 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.21 (d, J = 8.5 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.53 (d, J = 7.2 Hz, 1H), 7.44 (t, J = 7.1 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 7.00 (d, J = 7.5 Hz, 1H), 6.77 (d, J = 7.2 Hz, 1H), 4.57–4.50 (m, 2H), 4.24 (dd, J = 11.8, 13.4 Hz, 2H), 4.00 (s, 1H), 3.15 (s, 1H), 2.72 (d, J = 11.7 Hz, 1H), 2.64 (dd, J = 5.0, 12.9 Hz, 1H), 2.58 (d, J = 11.7 Hz, 1H), 2.44 (s, 3H), 2.43–2.41 (m, 1H) 1.98–1.91 (m, 1H), 1.80–1.71 (m, 1H), 1.52–1.52 (m, 1H), 1.44–1.42 (m, 1H), 1.42 (d, J = 6.4 Hz, 3H), 1.38 (s, 3H), 1.15–1.07 (m, 1H), 1.05 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.7, 141.2, 140.8, 135.4, 134.0, 131.3, 129.3, 128.8, 127.1, 126.3, 125.6, 125.5, 125.2, 123.0, 122.9, 118.8, 101.1, 79.3, 78.8, 76.0, 57.5, 55.6, 54.8, 53.0, 51.1, 48.7, 45.9, 31.7, 27.9, 24.1, 24.0, 21.0, 20.6; HR-MS (ESI): m/z calcd for C33H40NO4 [M + H]+: 514.2957; found: 514.2943.
3.1.8.9 (4aS,4a1R,5R,6S,12bS)-5-(((3-(1H-Imidazol-1-yl)propyl)amino)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 17. Prepared with 1-(3-aminopropyl) imidazole, yield: 70%; Rf = 0.6 (CHCl3
:MeOH = 19:1), white crystals; m.p.: 68–70 °C; [α]20D = −56.5 (c 0.2, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.42 (s, 1H), 7.10 (t, J = 7.4 Hz, 1H), 7.04 (s, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 7.5 Hz, 1H), 6.85 (s, 1H), 4.23 (d, J = 1.8 Hz, 2H), 3.97 (t, J = 7.0 Hz, 2H), 3.78 (s, 1H), 3.18 (s, 1H), 2.81 (d, J = 11.5 Hz, 1H), 2.75 (dd, J = 5.7, 13.1 Hz, 1H), 2.67–2.59 (m, 2H), 2.53–2.45 (m, 2H), 2.41 (s, 3H), 2.33–2.26 (m, 1H), 1.98–1.81 (m, 4H), 1.59–1.46 (m, 2H), 1.33 (s, 3H), 0.89 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 143.4, 140.8, 137.0, 135.5, 129.44, 129.45, 126.5, 118.8, 118.9, 101.0, 79.1, 78.7, 76.2, 57.5, 55.5, 53.0, 51.45, 51.40, 46.9, 45.9, 44.7, 31.9, 31.4, 27.8, 23.9, 21.4, 20.6; HR-MS (ESI): m/z calcd for C27H38N3O4 [M + H]+: 468.2862; found: 468.2852.
3.1.8.10 (4aS,4a1R,5R,6S,12bS)-5-(((2-((5-Fluoro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)ethyl)amino)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 18. Prior to the aminolysis, the reagent R2 was subjected to a Cbz-deprotection reaction to obtain the primary amine as follows:
An amount of the reagent R2 (0.15 g) was dissolved in MeOH (40 mL), then Pd/C (37 mg, 10% weight) was added to the solution and the mixture was stirred under an H2 atmosphere at room temperature overnight. After that, the mixture was filtered through Celite and evaporated to dryness. The obtained crude product was used for the aminolysis reaction.
Prepared with R2, yield: 47%; Rf = 0.6 (CHCl3:MeOH = 9:1), white wax; m.p.: 90–93 °C; [α]20D = −26.0 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.76 (d, J = 3.1 Hz, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.5 Hz, 2H), 7.17 (s, 1H), 7.08 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 6.87 (d, J = 7.2 Hz, 1H), 5.48 (s, 1H), 4.26–4.18 (m, 2H), 3.79 (s, 1H), 3.65–3.49 (m, 2H), 3.17 (s, 1H), 2.99–2.89 (m, 2H), 2.87–2.80 (m, 1H), 2.78–2.71 (m, 2H), 2.48 (d, J = 10.7 Hz, 1H), 2.41 (s, 3H), 2.31–2.24 (m, 1H), 1.99–1.85 (m, 2H), 1.57–1.46 (m, 2H), 1.31 (s, 3H), 0.88 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 155.1 (d, JF,C = 3.1 Hz), 152.8 (d, JF,C = 12.5 Hz), 143.4, 143.3, 141.8 (d, JF,C = 246 Hz), 140.7, 138.5 (d, JF,C = 20.1 Hz), 135.5, 129.4, 126.4, 126.0 (q, JF,C = 3.6 Hz, 2C), 125.5 (q, JF,C = 293.2 Hz), 123.2 (q, JF,C = 32.1 Hz), 118.8, 117.5 (2C), 101.06, 79.1, 78.7, 76.1, 57.5, 55.5, 53.0, 51.4, 50.9, 49.1, 46.0, 40.2, 31.7, 27.7, 23.8, 21.4, 20.6; 19F J-MOD NMR (470 MHz, CDCl3): δ = −62.5 (Cq-F), −165.1 (Cq-F) ppm; HR-MS (ESI): m/z calcd for C34H40F4N5O4 [M + H]+: 658.3016; found: 658.3005.
:1), Cu(OAc)2·H2O (0.1 equ.), sodium ascorbate (0.3 equ.) and acetylene derivative (0.28 mmol, 1.1 equ.) were added. The mixture was stirred at 40 °C for 48 hours, then t-BuOH was evaporated. The residue was dissolved in water (10 mL) and extracted with DCM (3 × 10 mL). The organic phase was washed with brine (3 × 10 mL), dried over Na2SO4, and then evaporated at low pressure. The crude product was then purified by column chromatography on silica gel using CHCl3:MeOH (19:1) as eluent, followed by recrystallisation using a mixture of n-hexane:Et2O to obtain pure products as white crystals.
3.1.9.1 (4aS,4a1R,5R,6S,12bS)-5-((4-(((5-Fluoro-2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 30. Prepared with R7, yield: 70%; Rf = 0.6 (CHCl3
:MeOH = 9:1), white crystals; m.p.: 139–142 °C; [α]20D = +8.1 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.74 (s, 1H), 7.71 (s, 1H), 7.65 (s, 1H), 7.46 (s, 1H), 7.11 (t, J = 7.4 Hz, 1H), 6.97–6.93 (m, 2H), 6.55 (s, 1H), 5.47 (s, 1H), 4.79 (d, J = 14,2 Hz, 1H), 4.76–4.64 (m, 2H), 4.41 (s, 1H), 4.25 (d, J = 14.0 Hz, 1H), 4.14 (dd, J = 4.9, 13.8 Hz, 1H), 4.05 (d, J = 13.1 Hz, 1H), 3.92 (s, 1H), 3.86 (s, 3H), 3.13 (d, J = 3.5 Hz, 1H), 2.79 (dd, J = 5.3, 13.0 Hz, 1H), 2.47–2.39 (m, 2H), 2.35 (s, 3H), 2.11–2.02 (m, 1H), 1.93 (dd, J = 5.2, 13.8 Hz, 1H), 1.82–1.71 (m, 1H), 1.54 (d, J = 10.7 Hz, 1H), 1.31–1.24 (m, 1H), 0.90 (s, 3H), 0.34 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 155.8 (d, JF,C = 3.1 Hz), 152.3 (d, JF,C = 11.9 Hz), 144.2, 142.7, 141.1 (d, JF,C = 244.9 Hz), 140.3, 139.3, 139.1 (d, JF,C = 19.7 Hz), 135.5, 131.0, 129.6, 126.7, 124.0, 123.3, 121.2, 119.2, 101.2, 79.0, 78.4, 74.8, 57.4, 55.3, 52.7, 51.6, 51.4, 45.8, 39.2, 35.8, 31.3, 27.4, 23.0, 21.1, 20.4 ppm; 19F J-MOD NMR (470 MHz, CDCl3): δ = −170.5 (Cq-F) ppm; HR-MS (ESI): m/z calcd for C32H39FN9O4 [M + H]+: 632.3109; found: 632.3092.
3.1.9.2 (4aS,4a1R,5R,6S,12bS)-5-((4-(((5-Chloro-2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 31. Prepared with R8, yield: 83%; Rf = 0.6 (CHCl3
:MeOH = 9:1), white crystals; m.p.: 237–239 °C; [α]20D = +14.8 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.86 (s, 1H), 7.71 (s, 1H), 7.64 (s, 1H), 7.48 (s, 1H), 7.11 (t, J = 7.4 Hz, 1H), 6.97–6.93 (m, 2H), 6.71 (s, 1H), 5.75 (t, J = 5.0 Hz, 1H), 4.82–4.64 (m, 3H), 4.41 (s, 1H), 4.25 (d, J = 14.0 Hz, 1H), 4.14 (dd, J = 4.1, 13.3 Hz, 1H), 4.05 (d, J = 13.3 Hz, 1H), 3.91 (d, J = 17.8 Hz, 1H), 3.88 (s, 3H), 3.13 (d, J = 3.4 Hz, 2H), 2.79 (dd, J = 5.3, 12.9 Hz, 1H), 2.47–2.39 (m, 2H), 2.35 (s, 3H), 2.06 (dt, J = 5.8, 13.5 Hz, 1H), 1.93 (dd, J = 4.3, 13.6 Hz, 1H), 1.84–1.73 (m, 1H), 1.54 (d, J = 10.8 Hz, 1H), 0.89 (s, 3H), 0.33 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 158.0, 157.6, 153.2, 144.2, 142.7, 140.3, 135.5, 131.2, 129.6, 126.7, 124.0, 122.8, 121.5, 119.2, 101.2, 79.0, 78.6, 74.8, 57.4, 55.3, 52.7, 51.6, 51.4, 45.8, 39.2, 36.2, 31.3, 27.4, 23.0, 21.1, 20.4 ppm; HR-MS (ESI): m/z calcd for C32H39ClN9O4 [M + H]+: 648.2813; found: 648.2800.
3.1.9.3 (4aS,4a1R,5R,6S,12bS)-5-((4-(((5-Fluoro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 32. Prepared with R5, yield: 83%; Rf = 0.5 (CHCl3
:MeOH = 19:1), white crystals; m.p.: 134–136 °C; [α]20D = +7.4 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.81 (d, J = 2.3 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.67 (s, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.20 (s, 1H), 7.10 (t, J = 7.4 Hz, 1H), 6.97–6.92 (m, 2H), 5.65 (s, 1H), 4.80–4.71 (m, 3H), 4.35 (s, 1H), 4.24 (d, J = 14.0 Hz, 1H), 4.14 (dd, J = 4.1, 13.3 Hz, 1H), 4.05 (d, J = 13.3 Hz, 1H), 3.92 (s, 1H), 3.14 (d, J = 3.4 Hz, 1H), 3.08 (s, 1H), 2.78 (dd, J = 5.3, 13.0 Hz, 1H), 2.45–2.38 (m, 2H), 2.35 (s, 3H), 2.08–2.00 (m, 1H), 1.92 (dd, J = 4.2, 13.7 Hz, 1H), 1.82–1.70 (m, 1H), 1.53 (d, J = 10.8 Hz, 1H), 0.90 (s, 3H), 0.35 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 155.1 (d, JF,C = 3.1 Hz), 152.2 (d, JF,C = 12.3 Hz), 143.8, 143.1, 142.7, 140.7, 140.3, 139.0 (d, JF,C = 21.2 Hz), 135.5, 129.6, 126.7, 126.1 (q, JF,C = 3.9 Hz, 2C), 124.0, 125.5 (q, JF,C = 243.6 Hz), 123.3 (q, JF,C = 16.3 Hz), 119.2, 117.92 (2C), 101.23, 79.0, 78.4, 74.8, 57.4, 55.3, 52.7, 51.6, 51.4, 45.8, 35.8, 31.3, 27.4, 23.0, 23.1, 20.4 ppm; 19FNMR (470 MHz, CDCl3): δ = −61.6 (Cq-F), −167.3 (Cq-F) ppm; HR-MS (ESI): m/z calcd for C35H38F4N7O4 [M + H]+: 696.2921; found: 696.2899.
3.1.9.4 (4aS,4a1R,5R,6S,12bS)-5-((4-(((5-Chloro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-3,3,12-trimethyl-4a,5,7,8,8a,12b-hexahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepine-5,6(1H)-diol 33. Prepared with R6, yield: 66%; Rf = 0.5 (CHCl3
:MeOH = 19:1), white crystals; m.p.: 132–134 °C; [α]20D = +5.1 (c 0.18, MeOH); 1H NMR (500 MHz, CDCl3): δ = 7.93 (s, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.65 (s, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.23 (s, 1H), 7.11 (t, J = 7.4 Hz, 1H), 6.97–6.92 (m, 2H), 5.87 (t, J = 5.3 Hz, 1H), 4.81–4.69 (m, 3H), 4.35 (s, 1H), 4.23 (d, J = 14.0 Hz, 1H), 4.14 (dd, J = 4.1, 13.3 Hz, 1H), 4.05 (d, J = 13.4 Hz, 1H), 3.92 (s, 1H), 3.14 (d, J = 3.4 Hz, 1H), 3.10 (s, 1H), 2.79 (dd, J = 5.3, 12.9 Hz, 1H), 2.47–2.40 (m, 2H), 2.35 (s, 3H), 2.10–2.01 (m, 1H), 1.92 (dd, J = 4.2, 13.7 Hz, 1H), 1.83–1.71 (m, 1H), 1.53 (d, J = 10.7 Hz, 1H), 0.90 (s, 3H), 0.35 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 157.5, 157.4, 153.2, 143.8, 142.8, 142.7, 140.2, 135.5, 129.6, 126.7, 126.1 (q, JF,C = 3.5 Hz, 2C), 125.4 (q, JF,C = 161.9 Hz), 124.0, 123.6 (q, JF,C = 25.4 Hz), 119.2, 118.4 (2C), 106.0, 101.2, 79.0, 78.4, 74.8, 57.4, 55.3, 52.7, 51.6, 51.4, 45.8, 36.3, 31.3, 27.5, 23.0, 21.1, 20.4 ppm; 19F J-MOD NMR (470 MHz, CDCl3): δ = −61.7 (Cq-F) ppm; HR-MS (ESI): m/z calcd for C35H38ClF3N7O4 [M + H]+: 712.2625; found: 712.2610.
3.1.9.5 1-((1-(((4aS,4a1R,5R,6S,12bS)-5,6-Dihydroxy-3,3,12-trimethyl-1,4a,5,6,7,8,8a,12b-octahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)pyrimidine-2,4(1H,3H)-dione 34. Prepared with N-1-propargyluracil, yield: 51%; white crystals; m.p.: 260–264 °C; [α]20D = +13.1 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.69 (s, 1H), 7.91 (s, 1H), 7.47 (d, J = 7.9 Hz, 1H), 7.10 (t, J = 7.4 Hz, 1H), 6.97–6.93 (m, 2H), 5.65 (d, J = 7.9 Hz, 1H), 5.00 (d, J = 15.1 Hz, 1H), 4.88 (d, J = 15.4 Hz, 1H), 4.81 (d, J = 13.8 Hz, 1H), 4.49 (s, 1H), 4.29 (d, J = 14.0 Hz, 1H), 4.15 (dd, J = 4.1, 13.3 Hz, 1H), 4.06 (d, J = 13.3 Hz, 1H), 3.91 (s, 1H), 3.29 (s, 1H), 3.15 (d, J = 3.5 Hz, 1H), 2.80 (dd, J = 5.3, 13.0 Hz, 1H), 2.47–2.40 (m, 2H), 2.35 (s, 3H), 2.11–2.02 (m, 1H), 1.94 (dd, J = 4.6, 13.9 Hz, 1H), 1.82–1.70 (m, 1H), 1.55 (d, J = 10.8 Hz, 1H), 0.95 (s, 3H), 0.29 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 163.1, 150.4, 144.1, 142.7, 141.3, 140.2, 135.5, 129.6, 126.7, 125.6, 119.1, 102.5, 101.1, 79.0, 78.5, 74.8, 57.4, 55.3, 52.7, 51.8, 51.4, 45.7, 42.9, 31.4, 27.4, 23.0, 21.1, 20.4 ppm; HR-MS (ESI): m/z calcd for C28H34N5O6 [M + H]+: 536.2509; found: 536.2501.
3.1.9.6 1-((1-(((4aS,4a1R,5R,6S,12bS)-5,6-Dihydroxy-3,3,12-trimethyl-1,4a,5,6,7,8,8a,12b-octahydro-4a1,6-methanobenzo[2,3]azuleno[8,1-de][1,3]dioxepin-5-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)-5-fluoropyrimidine-2,4(1H,3H)-dione 35. Prepared with N-1-propargyl-5-fluorouracil, yield: 56%; white crystals; m.p.: 141–144 °C; [α]20D = +16.4 (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3): δ = 8.61 (s, 1H), 7.88 (s, 1H), 7.57 (d, J = 5.3 Hz, 1H), 7.13–7.09 (m, 1H), 6.98–6.93 (m, 2H), 4.99–4.95 (m, 1H), 4.87 (d, J = 15.1 Hz, 1H), 4.81 (d, J = 14.0 Hz, 1H), 4.46 (s, 1H), 4.30 (d, J = 14.0 Hz, 1H), 4.16 (dd, J = 4.1, 13.3 Hz, 1H), 4.06 (d, J = 13.3 Hz, 1H), 3.92 (s, 1H), 3.20 (s, 1H), 3.16 (d, J = 3.8 Hz, 1H), 2.80 (dd, J = 5.3, 13.0 Hz, 1H), 2.48–2.40 (m, 2H), 2.36 (s, 3H), 2.07 (td, J = 6.8, 19.3 Hz, 1H), 1.94 (dd, J = 4.1, 13.9 Hz, 1H), 1.76 (dq, J = 5.3, 13.4 Hz, 1H), 1.54 (d, J = 5.8 Hz, 1H), 0.96 (s, 3H), 0.32 ppm (s, 3H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 156.5, 148.9, 142.7, 141.3–139.4 (d, JF,C = 242.1 Hz), 140.8, 140.2, 135.6, 129.6, 128.3 (d, JF,C = 32.6 Hz), 126.7, 125.5, 119.1, 101.18, 79.0, 78.4, 74.8, 57.4, 55.4, 52.7, 51.8, 51.4, 45.7, 42.8, 31.3, 27.4, 23.0, 21.1, 20.4 ppm; 19F J-MOD NMR (470 MHz, CDCl3): δ = −164.9 (Cq-F) ppm; HR-MS (ESI): m/z calcd for C28H33FN5O6 [M + H]+: 554.2414; found: 554.2401.
:n-hexane. In the case of compounds (28, 36–40), the crude product was dissolved in 10% Na2CO3 solution (5 mL) and then the free base was extracted with EtOAc (3 × 5 mL). The organic phase was dried over Na2SO4, filtered and then evaporated at low pressure. The crude product was purified by short column chromatography on silica gel using CHCl3:MeOH (9:1) as eluent.
3.1.10.1 N-Benzyl-1-((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methanaminium chloride 19. Yield: 88%; white crystals; m.p.: 245–246 °C; [α]20D = −10.6 (c 0.18, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 8.71 (s, 1H), 8.46 (s, 1H), 7.55–7.52 (m, 2H), 7.41–7.38 (m, 3H), 7.02 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 7.4 Hz, 1H), 6.88 (d, J = 7.4 Hz, 1H), 5.33 (d, J = 6.1 Hz, 1H), 4.81 (s, 1H), 4.66 (s, 1H), 4.20–4.08 (m, 3H), 3.92 (dd, J = 2.4, 11.5 Hz, 1H), 3.40 (d, J = 5.0 Hz, 1H), 3.01–2.86 (m, 3H), 2.56–2.51 (m, 2H), 2.44 (s, 3H), 2.16–2.08 (m, 1H), 1.79–1.69 (m, 1H), 1.59–1.54 (m, 1H), 1.36–1.29 ppm (m, 2H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 144.6, 143.0, 133.8, 132.0, 130.86, 130.84, 129.7, 129.4, 129.01, 129.01, 126.2, 120.0, 78.8, 76.9, 73.0, 59.3, 58.9, 55.0, 51.8, 51.2, 48.6, 43.6, 33.2, 21.3, 19.5; HR-MS (ESI): m/z calcd for C25H32NO4 [M + H]+: 410.2331; found: 410.2318.
3.1.10.2 N-(4-Fluorobenzyl)-1-((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methanaminium 20. Yield: 81%; white crystals; m.p.: 236–238 °C; [α]20D = −2.8 (c 0.17, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 8.72 (s, 1H), 8.46 (s, 1H), 7.61–7.56 (m, 2H), 7.23 (t, J = 8.6 Hz, 2H), 7.02 (t, J = 7.5 Hz, 1H), 6.92–6.87 (m, 2H), 4.17–4.07 (m, 3H), 3.92 (dd, J = 2.6, 11.5 Hz, 1H), 3.40 (s, 1H) 3.01–2.86 (m, 3H), 2.56–2.50 (m, 2H), 2.44 (s, 3H), 2.16–2.9 (m, 1H), 1.78–1.70 (m, 1H), 1.59 (dd, J = 4.7, 13.7 Hz, 1H), 1.38–1.29 ppm (m, 2H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 162.8 (d, JF,C = 246.6 Hz), 144.6, 143.0, 133.8, 133.3 (d, JF,C = 8.2 Hz, 2C), 129.7, 128.3 (d, JF,C = 3.4 Hz), 126.2, 120.0, 115.8 (d, JF,C = 21.2 Hz, 2C), 78.8, 76.9, 73.1, 59.3, 58.9, 55.0, 51.8, 50.4, 48.5, 43.6, 33.2, 21.3, 19.5; 19F J-MOD NMR (470 MHz, DMSO-d6): δ = −112.7 (Cq-F) ppm; HR-MS (ESI): m/z calcd for C25H31FNO4 [M + H]+: 428.2237; found: 428.2224.
3.1.10.3 N-(4-Methoxybenzyl)-1-((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methanaminium chloride 21. Yield: 71%; white crystals; m.p.: 163–165 °C; [α]20D = −18.7 (c 0.18, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 8.58 (s, 1H), 8.47 (s, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.14 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 7.9 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 8.4 Hz, 2H), 4.39 (dd, J = 5.9, 8.8 Hz, 1H), 4.32 (d, J = 8.7 Hz, 1H), 4.12–4.00 (m, 2H), 3.76 (s, 3H), 3.73 (s, 1H), 3.55 (d, J = 5.6 Hz, 1H), 3.00–2.82 (m, 3H), 2.37 (d, J = 11.3 Hz, 1H), 2.30–2.25 (m, 1H), 2.24 (s, 3H), 1.79 (dt, J = 6.1, 13.5 Hz, 1H), 1.61 (dd, J = 4.7, 13.8 Hz, 1H), 1.47 (d, J = 10.5 Hz, 1H), 1.42–1.31 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 160.2, 164.6, 143.1, 134.7, 132.4, 132.3, 129.0, 127.9, 123.8, 121.3, 114.4, 114.3, 84.6, 82.8, 76.2, 74.3, 59.8, 55.9, 50.5, 49.0, 48.2, 48.1, 41.9, 33.6, 23.8, 19.1; HR-MS (ESI): m/z calcd for C26H34NO5 [M + H]+: 440.2436; found: 440.2422.
3.1.10.4 1-(Naphthalen-1-yl)-N-(((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)methanaminium chloride 22. Yield: 71%; white crystals; m.p.: 215–217 °C; [α]20D = −17.7 (c 0.21, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 8.73 (s, 2H), 8.23 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 7.9 Hz, 2H), 7.72 (d, J = 7.4 Hz, 1H), 7.65–7.53 (m, 3H), 7.14 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 7.5 Hz, 2H), 4.71–4.61 (m, 2H), 4.41 (dd, J = 6.0, 8.7 Hz, 1H), 4.33 (d, J = 8.5 Hz, 1H), 3.81 (s, 1H), 3.56 (d, J = 5.6 Hz, 1H), 3.24–3.17 (m, 1H), 3.12–3.04 (m, 1H), 2.98 (dd, J = 5.7, 11.7 Hz, 1H), 2.41 (d, J = 10.4 Hz, 1H), 2.31–2.24 (m, 1H), 2.23 (s, 3H), 1.81–1.74 (m, 1H), 1.66–1.59 (m, 1H), 1.52–1.40 (m, 2H), 1.24 ppm (s, 2H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 144.6, 143.1, 134.8, 133.7, 131.7, 130.1, 129.8, 129.1, 129.0, 128.4, 127.9, 127.2, 126.6, 125.7, 124.1, 121.3, 84.9, 82.9, 76.2, 74.2, 59.8, 49.1, 49.0, 48.3, 47.5, 41.8, 33.5, 23.7, 19.1; HR-MS (ESI): m/z calcd for C29H32NO3 [M − H2O]+: 442.2382; found: 442.2367.
3.1.10.5 (1S)-1-(Naphthalen-2-yl)-N-(((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)ethan-1-aminium chloride 23. Yield: 91%; white crystals; m.p.: 219–222 °C; [α]20D = −56.8 (c 0.18, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 8.93 (s, 1H), 8.59 (s, 1H), 8.01–7.92 (m, 3H), 7.87–7.83 (m, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.59–7.55 (m, 2H), 7.14 (t, J = 7.4 Hz, 1H), 7.03 (d, J = 7.5 Hz, 1H), 6.96 (d, J = 7.3 Hz, 1H), 4.54 (t, J = 6.2 Hz, 1H), 4.44–4.34 (m, 3H), 3.83 (s, 1H), 3.55 (d, J = 5.7 Hz, 1H), 3.00–2.90 (m, 2H), 2.64–2.58 (m, 1H), 2.32 (d, J = 11.0 Hz, 1H), 2.26 (s, 3H), 2.24–2.17 (m, 1H), 1.74–1.67 (m, 1H), 1.66 (d, J = 6.9 Hz, 3H), 1.54–1.47 (m, 1H), 1.41 (d, J = 10.4 Hz, 1H), 1.36–1.22 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 144.6, 143.0, 134.79, 134.73, 133.3, 133.0, 129.2, 129.0, 128.4, 128.15, 128.12, 127.9, 127.2, 127.1, 125.2, 121.3, 84.2, 82.7, 76.3, 74.3, 59.7, 58.7, 49.0, 48.1, 47.9, 41.8, 33.5, 23.8, 20.1, 19.1; HR-MS (ESI): m/z calcd for C30H36NO4 [M + H]+: 474.2644; found: 474.2632.
3.1.10.6 (1R)-1-(Naphthalen-2-yl)-N-(((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)ethan-1-aminium chloride 24. Yield: 82%; white crystals; m.p.: 159–161 °C; [α]20D = −30.5 (c 0.14, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 8.82 (s, 1H), 8.72 (s, 1H), 8.05 (s, 1H), 7.96–7.87 (m, 3H), 7.72 (d, J = 8.5 Hz, 1H), 7.59–7.55 (m, 2H), 7.12 (t, J = 7.4 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 7.3 Hz, 1H), 4.58 (q, J = 6.05 Hz, 1H), 4.37 (dd, J = 5.6, 8.7 Hz, 1H), 4.29 (d, J = 8.6 Hz, 1H), 3.61 (s, 1H), 3.53 (d, J = 5.6 Hz, 1H), 3.03–2.88 (m, 2H), 2.73–2.62 (m, 1H), 2.36 (d, J = 9.5 Hz, 1H), 2.22 (s, 3H), 2.18–2.10 (m, 1H), 1.76–1.69 (m, 1H), 1.68 (d, J = 6.7 Hz, 3H), 1.58–1.51 (m, 1H), 1.44 (d, J = 10.5 Hz, 1H), 1.16–1.08 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 144.5, 142.9, 134.8, 134.7, 133.3, 132.9, 129.0, 128.9, 128.4, 128.2, 128.1, 127.9, 127.2, 127.1, 125.5, 121.2, 84.3, 82.8, 76.3, 74.2, 59.7, 58.8, 48.9, 48.0, 47.9, 41.8, 33.6, 33.7, 19.0, 18.6; HR-MS (ESI): m/z calcd for C30H36NO4 [M + H]+: 474.2644; found: 474.2631.
3.1.10.7 (1S)-1-(Naphthalen-1-yl)-N-(((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)ethan-1-aminium chloride 25. Yield: 80%; white crystals; m.p.: 160–163 °C; [α]20D = −5.3 (c 0.18, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 9.14 (s, 1H), 8.61 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.98–7.91 (m, 3H), 7.61–7.54 (m, 3H), 7.13 (t, J = 7.5 Hz, 1H), 7.02 (d, J = 7.5 Hz, 1H), 6.93 (d, J = 7.3 Hz, 1H), 5.34 (s, 1H), 4.78 (s, 1H), 4.43–4.39 (m, 2H), 4.34 (d, J = 8.7 Hz, 1H), 3.77 (s, 1H), 3.55 (d, J = 5.6 Hz, 1H), 3.08 (t, J = 8.1 Hz, 1H), 2.91 (dd, J = 5.6, 11.8 Hz, 1H), 2.74–2.67 (m, 1H), 2.33 (d, J = 10.1 Hz, 1H), 2.24 (s, 3H), 2.22–2.15 (m, 1H), 1.72–1.66 (m, 1H), 1.66 (d, J = 6.7 Hz, 3H), 1.47–1.39 (m, 2H), 1.32–1.20 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 144.5, 143.0, 134.7, 133.8, 130.8, 129.4, 129.3, 129.0, 127.9, 127.3, 126.6, 126.1, 125.9, 124.4, 122.9, 121.2, 84.1, 82.7, 76.2, 74.2, 59.9, 53.5, 48.9, 48.1, 47.9, 41.8, 33.4, 23.7, 20.6, 19.12; HR-MS (ESI): m/z calcd for C30H36NO4 [M + H]+: 474.2644; found: 474.2629.
3.1.10.8 (1R)-1-(Naphthalen-1-yl)-N-(((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)ethan-1-aminium chloride 26. Yield: 79%; white crystals; m.p.: 161–163 °C; [α]20D = −53.2 (c 0.20, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 9.21 (s, 1H), 8.52 (s, 1H), 8.22 (d, J = 8.5 Hz, 1H), 7.98 (d, J = 7.9 Hz, 1H), 7.95–7.91 (m, 2H), 7.64–7.52 (m, 3H), 7.12 (t, J = 7.4 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.93 (d, J = 7.3 Hz, 1H), 5.30 (s, 1H), 5.04 (s, 1H), 4.59 (s, 1H), 4.37 (dd, J = 7.2, 7.2 Hz, 1H), 4.32 (d, J = 8.5 Hz, 1H), 3.61 (s, 1H), 3.52 (d, J = 5.3 Hz, 1H), 3.17 (dd, J = 9.5, 9.5 Hz, 1H), 2.98–2.90 (m, 2H), 2.39 (d, J = 10.5 Hz, 1H), 2.22 (s, 3H), 2.21–2.13 (m, 1H), 1.75 (td, J = 6.7, 20.6 Hz, 1H), 1.68 (d, J = 6.6 Hz, 3H), 1.62 (dd, J = 5.1, 14.9 Hz, 1H), 1.45 (d, J = 10.5 Hz, 1H), 1.26–1.15 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 144.6, 143.0, 134.8, 134.7, 133.7, 130.7, 129.4, 128.3, 129.0, 127.9, 127.4, 126.6, 125.9, 124.9, 123.2, 121.3, 84.4, 82.9, 76.3, 74.2, 59.8, 53.8, 49.0, 48.7, 48.2, 41.9, 33.7, 23.7, 19.2, 19.0; HR-MS (ESI): m/z calcd for C30H36NO4 [M + H]+: 474.2644; found: 474.2632.
3.1.10.9 3-(1H-Imidazol-1-yl)-N-(((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)propan-1-aminium chloride 27. Yield: 80%; white crystals; m.p.: 207–208 °C; [α]20D = −29.1 (c 0.18, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 9.19 (s, 1H), 8.74 (s, 1H), 8.41 (s, 1H), 7.76 (s, 1H), 7.67 (s, 1H), 7.13 (t, J = 7.4 Hz, 1H), 7.02–6.96 (m, 2H), 4.50–4.36 (m, 2H), 4.33–4.28 (m, 3H), 3.69 (s, 1H), 3.55 (d, J = 5.6 Hz, 1H), 3.17 (s, 1H), 3.00–2.93 (m, 3H), 2.89 (s, 2H), 2.38 (d, J = 10.6 Hz, 1H), 2.29–2.24 (m, 1H), 2.23 (s, 3H), 2.22–2.16 (m, 2H), 1.86–1.76 (m, 1H), 1.74–1.66 (m, 1H), 1.50–1.39 ppm (m, 2H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 144.5, 143.1, 135.51, 134.7, 129.0, 127.7, 122.3, 121.3, 120.4, 84.9, 82.7, 76.3, 74.3, 59.8, 49.2, 49.0, 48.3, 46.2, 44.7, 41.9, 33.5, 26.2, 23.8, 19.1; HR-MS (ESI): m/z calcd for C24H32N3O3 [M − H2O]+: 410.2443; found: 410.2432.
3.1.10.10 (7S,8R,9S,9aR,10S)-8-(((2-((5-Fluoro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)ethyl)amino)methyl)-10-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8,9-triol 28. Yield: 63%; white crystals; m.p.: 92–94 °C; [α]20D = −11.6 (c 0.11, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 9.41 (s, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 3.4 Hz, 1H), 7.55 (d, J = 8.5 Hz, 2H), 7.41 (t, J = 4.8 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 7.3 Hz, 1H), 4.30 (dd, J = 7.2, 7.2 Hz, 1H), 4.26 (d, J = 8.5 Hz, 1H), 2.91 (dd, J = 5.8, 11.7 Hz, 1H), 2.78 (t, J = 6.2 Hz, 2H), 2.64 (d, J = 11.4 Hz, 1H), 2.56 (t, J = 6.2 Hz, 1H), 2.38–2.32 (m, 1H), 2.21 (s, 3H), 2.20–2.16 (m, 1H), 1.73 (td, J = 6.6, 19.2 Hz, 1H), 1.64 (dd, J = 4.1, 13.1 Hz, 1H), 1.41 (d, J = 10.2 Hz, 1H), 1.38–1.30 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 155.7 (d, JF,C = 3.1 Hz), 152.8 (d, JF,C = 12.2 Hz), 145.3, 144.8, 143.3, 142.7–140.7 (d, JF,C = 246.4 Hz), 138.7 (d, JF,C = 18.4 Hz), 134.6, 128.9, 127.7, 126.1 (q, JF,C = 3.5 Hz, 2C), 124.2 (q, JF,C = 274.5 Hz), 120.6 (q, JF,C = 31.2 Hz), 121.14, 117.9 (2C), 85.9, 82.6, 78.0, 73.9, 59.5, 57.5, 50.7, 49.1, 48.9, 48.3, 42.7, 33.6, 24.0, 19.0 ppm; 19F NMR (470 MHz, MeOD): δ = −59.7 (Cq-F), −165.8 (Cq-F); HR-MS (ESI): m/z calcd for C31H36F4N5O4 [M + H]+: 618.2703; found: 618.2685.
3.1.10.11 (7S,8R,9S,9aR,10S)-8-((4-(((5-Fluoro-2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-10-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8,9-triol 36. Yield: 63%; colourless oily compound; [α]20D = +8.2 (c 0.16, MeOH); 1H NMR (500 MHz, MeOD): δ = 7.81 (s, 1H), 7.68 (d, J = 2.7 Hz, 1H), 7.48 (s, 1H), 7.34 (s, 1H), 7.14 (t, J = 7.9 Hz, 1H), 7.02–6.98 (m, 2H), 4.71 (d, J = 15.6 Hz, 1H), 4.63–4.52 (m, 2H), 4.36 (d, J = 14.2 Hz, 1H), 4.21 (dd, J = 6.6, 8.3 Hz, 1H), 4.02 (d, J = 8.7 Hz, 1H), 3.74 (s, 1H), 3.61 (s, 3H), 3.48 (d, J = 5.6 Hz, 1H), 3.03 (dd, J = 5.4, 11.8 Hz, 1H), 2.49–2.39 (m, 2H), 2.15 (s, 3H), 2.05–1.98 (m, 1H), 1.56 (d, J = 10.4 Hz, 1H), 1.42–1.33 (m, 1H), 0.97–0.89 ppm (m, 1H); 13C{1H} J-MOD NMR (125 MHz, MeOD): δ = 159.8, 156.7 (d, JF,C = 12.2 Hz), 149.4, 147.5, 146.4, 145.7–143.8 (d, JF,C = 242.5 Hz), 142.1 (d, JF,C = 18.8 Hz), 138.3, 133.7, 132.5, 131.3, 128.0, 127.8, 125.4, 124.4, 87.9, 86.5, 81.2, 78.1, 63.6, 55.4, 52.7, 52.0, 45.9, 41.3, 38.6, 36.5, 27.4, 21.5; 19F J-MOD NMR (470 MHz, MeOD): δ = −168.3 (Cq-F); HR-MS (ESI): m/z calcd for C29H33FN9O3 [M − H2O]+: 574.2690; found: 574.2676.
3.1.10.12 (7S,8R,9S,9aR,10S)-8-((4-(((5-Chloro-2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-10-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8,9-triol 37. Yield: 64%; white crystals; m.p.: 251–253 °C; [α]20D = +15.7 (c 0.18, MeOH); 1H NMR (500 MHz, MeOD): δ = 7.81 (s, 1H), 7.79 (s, 1H), 7.46 (s, 1H), 7.36 (s, 1H), 7.16 (t, J = 7.4 Hz, 1H), 7.02 (d, J = 7.3 Hz, 2H), 4.75 (d, J = 17.2 Hz, 1H), 4.63 (d, J = 15.2 Hz, 1H), 4.58 (d, J = 14.4 Hz, 1H), 4.38 (d, J = 14.2 Hz, 1H), 4.25–4.19 (m, 1H), 4.01 (d, J = 8.4 Hz, 1H), 3.75 (s, 1H), 3.60 (s, 3H), 3.50 (d, J = 5.8 Hz, 1H), 3.06 (dd, J = 5.6, 11.7 Hz, 1H), 2.50–2.44 (m, 1H), 2.42 (d, J = 10.4 Hz, 1H), 2.17 (s, 3H), 2.04–1.97 (m, 2H), 1.75–1.68 (m, 1H), 1.58 ppm (d, J = 10.7 Hz, 1H); 13C{1H} J-MOD NMR (125 MHz, MeOD): δ = 167.14, 158.2, 157.9, 152.5, 145.4, 142.6, 142.4, 134.3, 129.8, 128.6, 127.4, 123.9, 121.8, 120.5, 84.0, 82.6, 77.3, 74.1, 59.7, 51.5, 48.8, 48.1, 42.0, 37.3, 36.2, 32.8, 23.4, 17.6; HR-MS (ESI): m/z calcd for C29H33ClN9O3 [M − H2O]+: 590.2394; found: 590.2379.
3.1.10.13 (7S,8R,9S,9aR,10S)-8-((4-(((5-Fluoro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-10-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8,9-triol 38. Yield: 85%; colourless oily compound; [α]20D = +9.8 (c 0.14, MeOH); 1H NMR (500 MHz, MeOD): δ = 7.77 (s, 1H), 7.68 (d, J = 3.5 Hz, 1H), 7.57 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 8.5 Hz, 2H), 7.01 (t, J = 7.5 Hz, 1H), 6.90–6.85 (m, 2H), 4.58 (dd, J = 15.6, 27.2 Hz, 2H), 4.44 (d, J = 14.2 Hz, 1H), 4.27 (d, J = 14.2 Hz, 1H), 4.15 (dd, J = 6.3, 8.8 Hz, 1H), 4.03 (d, J = 8.9 Hz, 1H), 3.65 (s, 1H), 3.39 (d, J = 6.3 Hz, 1H), 2.93 (dd, J = 5.6, 12.1 Hz, 1H), 2.36–2.30 (m, 2H), 2.05 (s, 3H), 1.91–1.85 (m, 2H), 1.63–1.52 (m, 1H), 1.45 ppm (d, J = 10.7 Hz, 2H); 13C{1H} J-MOD NMR (125 MHz, MeOD): δ = 155.5 (d, JF,C = 3.1 Hz), 152.5 (d, JF,C = 10.5 Hz), 145.1, 144.2, 143.5, 142.7–140.7 (d, JF,C = 246.7 Hz), 142.4, 137.9 (d, JF,C = 20.2 Hz), 134.2, 130.0 (q, JF,C = 196.7 Hz), 128.5, 127.4, 125.3 (d, JF,C = 3.7 Hz, 2C), 124.2, 122.1 (q, JF,C = 33.1 Hz), 120.5, 117.6 (2C), 84.1, 82.6, 77.3, 74.3, 59.7, 51.5, 48.8, 48.1, 42.0, 35.4, 32.6, 23.4, 17.5 ppm; 19F J-MOD NMR (470 MHz, MeOD): δ = −62.9 (Cq-F), −169.0 (Cq-F); HR-MS (ESI): m/z calcd for C32H33F4N7O3 [M − H2O]+: 638.2581; found: 638.2485.
3.1.10.14 (7S,8R,9S,9aR,10S)-8-((4-(((5-chloro-2-((4-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)methyl)-10-(hydroxymethyl)-1-methyl-4b,5,6,8,9,10-hexahydro-7H-7,9a-methanobenzo[a]azulene-7,8,9-triol 39. Yield: 85%; white crystals; m.p.: 216–219 °C; [α]20D = +30.5 (c 0.17, MeCN); 1H NMR (500 MHz, CDCl3): δ = 7.94 (s, 1H), 7.69–7.64 (m, 3H), 7.54 (d, J = 8.5 Hz, 2H), 7.18 (dd, J = 7.2, 14.7 Hz, 2H), 7.03 (d, J = 7.6 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 5.89 (t, J = 5.1 Hz, 1H), 4.82–4.69 (m, 2H), 4.54 (d, J = 14.2 Hz, 1H), 4.37–4.31 (m, 2H), 4.20 (d, J = 8.9 Hz, 1H), 3.84 (s, 1H), 3.73 (s, 1H), 3.55 (d, J = 6.4 Hz, 1H), 3.04 (dd, J = 5.6, 12.2 Hz, 1H), 2.93 (s, 1H), 2.52–2.44 (m, 1H), 2.30 (d, J = 10.5 Hz, 1H), 2.18 (s, 3H), 2.12–2.03 (m, 1H), 1.97 (dd, J = 4.7, 14.0 Hz, 1H), 1.74–1.65 (m, 1H), 1.62 ppm (d, J = 10.9 Hz, 1H); 13C{1H} J-MOD NMR (125 MHz, CDCl3): δ = 157.7, 157.4, 153.1, 143.9, 143.0, 142.8, 142.4, 134.5, 129.2, 128.0, 126.1 (q, JF,C = 4.1 Hz, 2C), 124.8 (q, JF,C = 163.8 Hz), 123.8, 121.5 (q, JF,C = 32.8 Hz), 120.9, 118.2 (2C), 106.0, 83.7, 83.2, 74.8, 59.9, 51.2, 48.8, 48.3, 43.2, 36.9, 31.8, 29.6, 23.5, 18.9 ppm; 19F J-MOD NMR (470 MHz, CDCl3): δ = −61.7 (Cq-F); HR-MS (ESI): m/z calcd for C32H32ClF3N7O3 [M − H2O]+: 654.2207; found: 654.2194.
3.1.10.15 1-((1-(((7S,8R,9S,9aR,10S)-7,8,9-Trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)pyrimidine-2,4(1H,3H)-dione 40. Yield: 63%; colourless oily compound; [α]20D = +11.6 (c 0.16, MeOH); 1H NMR (500 MHz, DMSO-d6): δ = 11.2 (s, 1H), 7.92 (s, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.14 (t, J = 7.5 Hz, 1H), 7.03–6.99 (m, 2H), 5.53 (d, J = 7.9 Hz, 1H), 4.87 (d, J = 3.5 Hz, 2H), 4.69 (s, 1H), 4.40 (d, J = 14.0 Hz, 1H), 4.30 (d, J = 14.2 Hz, 1H), 4.24 (dd, J = 7.3, 7.3 Hz, 1H), 4.18 (d, J = 8.7 Hz, 1H), 4.08 (s, 1H), 3.70 (s, 1H), 3.52–3.47 (m, 1H), 2.99 (dd, J = 5.7, 11.8 Hz, 1H), 2.35 (d, J = 10.7 Hz, 2H), 2.20 (s, 3H), 1.86 (d, J = 6.3 Hz, 2H), 1.63–1.52 (m, 1H), 1.47 ppm (d, J = 10.5 Hz, 1H); 13C{1H} J-MOD NMR (125 MHz, DMSO-d6): δ = 164.0, 151.2, 145.7, 144.6, 143.1, 141.8, 134.8, 129.0, 127.9, 125.5, 121.3, 101.6, 84.7, 82.7, 77.9, 74.2, 59.7, 52.1, 48.9, 48.3, 42.6, 42.5, 33.5, 24.0, 19.1 ppm; HR-MS (ESI): m/z calcd for C25H28N5O5 [M − H2O]+: 478.2090; found: 478.2077.
3.1.10.16 5-Fluoro-1-((1-(((7S,8R,9S,9aR,10S)-7,8,9-trihydroxy-10-(hydroxymethyl)-1-methyl-4b,6,7,8,9,10-hexahydro-5H-7,9a-methanobenzo[a]azulen-8-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)pyrimidine-2,4(1H,3H)-dione 41. Yield: 65%; white crystals; m.p.: 156–158 °C; [α]20D = +32.1 (c 0.13, MeOH); 1H NMR (500 MHz, MeOD): δ = 8.01 (s, 1H), 7.79 (d, J = 6.3 Hz, 1H), 7.16 (t, J = 7.5 Hz, 1H), 7.04 (d, J = 2.1 Hz, 1H), 7.02 (d, J = 2.4 Hz, 1H), 4.95 (dd, J = 15.7, 19.1 Hz, 2H), 4.59 (d, J = 14.0 Hz, 1H), 4.52 (s, 1H), 4.45 (d, J = 14.2 Hz, 1H), 4.36 (dd, J = 6.3, 8.7 Hz, 1H), 4.28 (d, J = 8.7 Hz, 1H), 3.81 (s, 1H), 3.57 (d, J = 6.0 Hz, 1H), 3.09 (dd, J = 5.7, 12.0 Hz, 1H), 2.52–2.45 (m, 2H), 2.24 (s, 3H), 2.06–2.00 (m, 2H), 1.78–1.67 (m, 1H), 1.61 ppm (d, J = 10.5 Hz, 1H); 13C{1H} J-MOD NMR (125 MHz, MeOD): δ = 158.2, 149.9 (d, JF,C = 12.9 Hz), 143.6, 142.4, 141.3, 141.2–139.4 (d, JF,C = 233.9 Hz), 134.3, 129.1 (d, JF,C = 33.4 Hz), 128.6, 127.4, 125.5, 120.6, 84.2, 82.6, 77.3, 74.4, 58.7, 51.8, 48.9, 48.1, 42.4, 42.0, 32.6, 23.4, 17.6 ppm; 19FNMR (470 MHz, MeOD): δ = −169.8 (Cq-F); HR-MS (ESI): m/z calcd for C25H27FN5O5 [M − H2O]+: 496.1996; found: 496.1985.
3.2 Determination of the antiproliferative properties
A panel of human adherent cancer cell lines and fibroblast cells were utilised to determine the antiproliferative action of the prepared compounds by the MTT assay.49 The applied cell lines and conditions were the same as published earlier.313.3 Docking study
The protein crystal structures were obtained from PDB (protein data bank). ChemBioDraw Ultra 11.0 was used to draw the tested structures for the docking study. The docking study was performed using Accelrys Discovery Studio 2.5 software.3.4 Preparation of the crystal structures
Since the extracted crystal structure from PDB does not have hydrogen atoms, hydrogen atoms must be added first by applying several force fields (CHARMm). The addition of hydrogen atoms leads to steric hindrance, and subsequently, to a high-energy and unstable molecule, which should be minimized. Energy minimization of the complex was performed in 2000 steps of the adopted basis minimization method at finding the most stable and less energy structure and reducing H–H interactions without affecting the basic protein skeleton atoms. Then, the active site was determined by a 10 Å radius sphere.503.5 Docking study (CDocker)
By using the CDocker method, all possible conformations of the compound in the protein active site could be generated. Then, the results can be evaluated by both the CDocker energy and the number of interactions between the ligand and active site. This method requires preparing the crystal structure (as mentioned before) and the designed compounds using the Accelrys Discovery Studio protocol and applying a force field.Before starting this study, it is important to emphasize that the method used is valid by comparing the conformation of the reference compound with its conformations generated by the applied docking method, where RMSD (Root Mean Square Deviation) should not exceed 2 Å.
4 Conclusions
A new series of allo-gibberic acid-based aminodiols, aminotetraols and 1,2,3-triazoles were synthesized in a stereoselective manner starting from commercially available gibberellic acid. The SeO2/t-BuOOH-mediated allylic oxidation yielded a key intermediate triol, which, after the protection of the 1,4-diol functionality and epoxidation, was transformed into epoxide alcohol. The oxirane ring was then opened with either primary amines to provide aminodiols or sodium azide to afford an azido diol. The azido diol was then subjected to the CuAAC reaction to obtain dihydroxy 1,2,3-triazoles. The HCl-mediated acetonide deprotection of the prepared derivatives furnished aminotetraols and tetrahydroxy 1,2,3-triazoles. The antiproliferative effects of the prepared compounds were studied by the in vitro MTT method against a panel of human cancer cell lines (HeLa, SiHa, A2780, MCF-7 and MDA-MB-231) and fibroblasts, and the structure–activity relationship for the prepared compounds was explored. A significant difference was observed in the antiproliferative activity between N-substituted aminotriols. The importance of the “linker” property between allo-gibberic acid and 2,4-diaminopyrimidine motif was also explored. Based on the results, 2,4-diaminopyrimidine-ethylamine conjugates exhibited potent antiproliferative activity; conversely, they were endowed with low selectivity and high cytotoxicity towards the NIH/3T3 regioisomers. These agents seem to be superior to clinically utilised cisplatin. Consequently, some selected compounds could be regarded as potential hit compounds and they may be subjected to further investigation.Data availability
I declare that the data supporting this article have been included as part of the ESI.†Author contributions
Z. S. (Zsolt Szakonyi) and I. Z. conceived and designed the experiments; Z. A. K. and Z. S. (Zsuzsanna Schelz) performed the experiments, analysed the data and wrote the experimental part; Z. S. (Zsolt Szakonyi), T. M. L. and I. Z. discussed the results and contributed to writing the paper. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data in the writing of the manuscript, or in the decision to publish the results.Acknowledgements
We are grateful for financial support from the Hungarian Research Foundation (NKFI K138871). Project no. TKP2021-EGA-32 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme and also by the University of Szeged Open Access Fund (grant no. 7314). The high-resolution mass spectrometric analysis was performed by Robert Berkecz.Notes and references
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef.
- M. F. Braña and A. Sánchez-Migallón, Clin. Transl. Oncol., 2006, 8, 717–728 CrossRef PubMed.
- M.-J. R. Howes, Lancet Oncol., 2018, 19, 293–294 CrossRef PubMed.
- K. K. Jain, Technol. Cancer Res. Treat., 2002, 1, 231–236 CrossRef CAS PubMed.
- A. Sehgal, Curr. Med. Chem., 2003, 10, 749–755 CrossRef CAS.
- A. I. Alqosaibi, Saudi J. Biol. Sci., 2022, 29, 103298 CrossRef CAS PubMed.
- D. Basak, S. Arrighi, Y. Darwiche and S. Deb, Life, 2022, 12, 48 CrossRef CAS PubMed.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- E. J. Barreiro, C. A. M. Fraga and L. M. Lima, in Plant Bioactives and Drug Discovery, ed. V. Cechinel-Filho, Wiley, 1st edn, 2012, pp. 81–126 Search PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS.
- J. Rollinger, T. Langer and H. Stuppner, Curr. Med. Chem., 2006, 13, 1491–1507 CrossRef CAS PubMed.
- F.-M. Zhang, S.-Y. Zhang and Y.-Q. Tu, Nat. Prod. Rep., 2018, 35, 75–104 RSC.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440–446 CrossRef CAS.
- T. Ollila, J. Butera, P. Egan, J. Reagan, A. Thomas, I. Yakirevich, K. MacKinnon, J. Margolis, J. McMahon, V. Rosati and A. J. Olszewski, Oncologist, 2022, 27, e532–e542 CrossRef.
- L. M. Mayr and D. Bojanic, Curr. Opin. Pharmacol., 2009, 9, 580–588 CrossRef CAS PubMed.
- P. Quitt, E. Mosettig, R. C. Cambie, P. S. Rutledge and L. H. Briggs, J. Am. Chem. Soc., 1961, 83, 3720–3722 CrossRef.
- C. Ding, Y. Ding, H. Chen and J. Zhou, in Studies in Natural Products Chemistry, Elsevier, 2017, vol. 54, pp. 141–197 Search PubMed.
- L. Wang, D. Li, C. Wang, Y. Zhang and J. Xu, Mini-Rev. Med. Chem., 2011, 11, 910–919 CrossRef CAS.
- M. O. Fatope, G. B. Varma, N. M. Alzri, R. G. Marwah and R. S. Nair, Chem. Biodiversity, 2010, 7, 1862–1870 CrossRef CAS PubMed.
- I. Kubo, Y. Xu and K. Shimizu, Phytother. Res., 2004, 18, 180–183 CrossRef CAS.
- S.-Y. Chen, S.-R. Kuo and C.-T. Chien, Tree Physiol., 2008, 28, 1431–1439 CrossRef CAS.
- J. Dayan, N. Voronin, F. Gong, T. Sun, P. Hedden, H. Fromm and R. Aloni, Plant Cell, 2012, 24, 66–79 CrossRef CAS PubMed.
- L. N. Mander, Chem. Rev., 1992, 92, 573–612 CrossRef CAS.
- J. R. Hanson, Nat. Prod. Rep., 1990, 7, 41–59 RSC.
- A. N. Koehler, World Intellectual Property Organization, Methods for regulating NF-KB using gibberellins, international application number: WO2010062372A2, 2010.
- Y. Zhang, H. Zhang, J. Chen, H. Zhao, X. Zeng, H. Zhang and C. Qing, Invest. New Drugs, 2012, 30, 8–16 CrossRef CAS PubMed.
- J. Chen, Z. Sun, Y. Zhang, X. Zeng, C. Qing, J. Liu, L. Li and H. Zhang, Bioorg. Med. Chem. Lett., 2009, 19, 5496–5499 CrossRef CAS.
- M.-J. Wu, D.-M. Wu, J.-B. Chen, J.-F. Zhao, L. Gong, Y.-X. Gong, Y. Li, X.-D. Yang and H. Zhang, Bioorg. Med. Chem. Lett., 2018, 28, 2543–2549 CrossRef CAS.
- S. Zhu, F. Luo and P.-H. Sun, Med. Chem. Res., 2020, 29, 1341–1354 CrossRef CAS.
- Z. A. Khdar, T. M. Le, Z. Schelz, I. Zupkó and Z. Szakonyi, Int. J. Mol. Sci., 2022, 23, 10366 CrossRef CAS PubMed.
- Z. Alabdeen Khdar, T. Minh Le, Z. Schelz, I. Zupkó and Z. Szakonyi, RSC Med. Chem., 2024, 15, 874–887 RSC.
- D. Bai, Z. Schelz, D. Erdős, A. K. Kis, V. Nagy, I. Zupkó, G. T. Balogh and Z. Szakonyi, Int. J. Mol. Sci., 2023, 24, 1121 CrossRef CAS PubMed.
- L. Kutschabsky and G. Adam, J. Chem. Soc., Perkin Trans. 1, 1983, 1653 RSC.
- J. R. Annand, A. R. Henderson, K. S. Cole, A. J. Maurais, J. Becerra, Y. Liu, E. Weerapana, A. N. Koehler, A. K. Mapp and C. S. Schindler, ACS Med. Chem. Lett., 2020, 11, 1913–1918 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, C. A. Acaru, J. F. Malone, C. R. O'Dowd, C. C. R. Allen and P. J. Stevenson, Org. Lett., 2010, 12, 2206–2209 CrossRef CAS.
- H. B. Henbest and R. A. L. Wilson, J. Chem. Soc., 1957, 1958–1965 RSC.
- D. Ozsvár, V. Nagy, I. Zupkó and Z. Szakonyi, Int. J. Mol. Sci., 2020, 21, 184 CrossRef.
- T. M. Le, A. Csámpai, F. Fülöp and Z. Szakonyi, Chem.–Eur. J., 2018, 24, 13607–13615 CrossRef CAS.
- Z. Szakonyi, K. Csillag and F. Fülöp, Tetrahedron: Asymmetry, 2011, 22, 1021–1027 CrossRef CAS.
- N. Azizi, B. Mirmashhori and M. R. Saidi, Catal. Commun., 2007, 8, 2198–2203 CrossRef CAS.
- F. A. Saddique, A. F. Zahoor, S. Faiz, S. A. R. Naqvi, M. Usman and M. Ahmad, Synth. Commun., 2016, 46, 831–868 CrossRef CAS.
- M. Albratty and H. A. Alhazmi, Arabian J. Chem., 2022, 15, 103846 CrossRef CAS.
- R. Kaur, P. Kaur, S. Sharma, G. Singh, S. Mehndiratta, P. Bedi and K. Nepali, Recent Pat. Anti-Cancer Drug Discovery, 2014, 10, 23–71 CrossRef.
- K. Bozorov, J. Zhao and H. A. Aisa, Bioorg. Med. Chem., 2019, 27, 3511–3531 CrossRef CAS PubMed.
- S. Zhao, J. Liu, Z. Lv, G. Zhang and Z. Xu, Eur. J. Med. Chem., 2023, 251, 115254 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- L. Zhu, C. J. Brassard, X. Zhang, P. M. Guha and R. J. Clark, Chem. Rec., 2016, 16, 1501–1517 CrossRef CAS PubMed.
- H. B. Lazrek, M. Taourirte, T. Oulih, J. L. Barascut, J. L. Imbach, C. Pannecouque, M. Witrouw and E. De Clercq, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 1949–1960 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- S. Rampogu, A. Baek, M. Son, C. Park, S. Yoon, S. Parate and K. W. Lee, ACS Omega, 2020, 5, 1773–1781 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07334g |
| This journal is © The Royal Society of Chemistry 2024 |