DOI:
10.1039/D4RA07602H
(Review Article)
RSC Adv., 2024,
14, 39436-39461
Construction, structural modification, and bioactivity evaluation of pentacyclic triterpenoid privileged scaffolds in active natural products
Received
24th October 2024
, Accepted 9th December 2024
First published on 13th December 2024
Abstract
Pentacyclic triterpenoids, as important representatives of natural products, have garnered widespread attention due to their diverse biological activities, including anti-inflammatory, antiviral, and antitumor effects. Oleanolic acid (OA), betulinic acid (BA), ursolic acid (UA), triptolide, and glycyrrhetinic acid (GA) are typical examples of pentacyclic triterpenoids. Despite their significant biological activities, their poor water solubility and low bioavailability have limited further development and application. In recent years, researchers have developed a series of derivatives with enhanced biological activities and improved drug properties through structural modifications of these compounds, particularly achieving notable progress in the field of antitumor therapy. This review summarizes recent advances in the structural modification of pentacyclic triterpenoids and explores their promising applications in the development of antitumor, antiviral, and other therapeutic agents.
Terpenoids are a class of compounds and their derivatives with a molecular scaffold based on isoprene units, which are widely distributed in nature. They are major components of secondary metabolites such as plant essential oils, resins, and pigments. Terpenoids are classified into monoterpenes, sesquiterpenes, diterpenes, sesterterpenes, triterpenes, tetraterpenes, and polyterpenes based on the number of isoprene units in their molecular structure. Triterpenes, composed of a basic skeleton of 30 carbon atoms, are important plant secondary metabolites found in nature either in free form or as glycosides or esters bound to sugars. They exhibit various pharmacological activities, including anti-cancer, antiviral, and cholesterol-lowering effects. Triterpenoids can be further classified into monocyclic, bicyclic, tricyclic, tetracyclic, and pentacyclic triterpenes, based on the number of rings in their structure, with tetracyclic and pentacyclic triterpenes being the most common.
Pentacyclic triterpenoids possess various modification types in their carbon skeletons, resulting in complex and diverse structures, which have garnered significant attention due to their wide range of functions. Based on their aglycone types, pentacyclic triterpenoids are divided into four main categories: oleanane-type [e.g., OA, GA, and hawthorn acid (HA)], lupane-type (e.g., BA, lupeol, and betulin), ursane-type [e.g., UA and asiatic acid (AA)], and friedelane-type (e.g., Tripterygium wilfordii polyglycosides). Oleanane-type triterpenoids exhibit extensive pharmacological activity; OA is commonly used clinically for its hepatoprotective effects,1–3 while glycyrrhizin-based formulations have been marketed for the treatment of hepatitis.4 The oleanane-type, also known as the β-amyrane-type, includes compounds such as β-amyrin, germanicol, OA, and GA.
Lupane-type compounds mainly include lupeol, betulin, BA, and their derivatives. Lupeol is widely distributed in nature and serves as the core skeleton for lupane-type triterpenoids, showing significant anti-inflammatory5 and anti-cancer6 activities. Betulinic acid, formed through three consecutive oxidation steps at the C-28 position of lupeol, not only demonstrates potent anti-HIV activity,7 but also selectively inhibits melanoma cell proliferation,8 making it a promising candidate for anti-HIV and anti-melanoma drug development.9,10 BA is predominantly found in the bark of birch trees.
The ursane-type, also known as the α-amyrane-type, includes compounds such as α-amyrin, UA, and taraxasterol. α-Amyrin is a precursor for ursane-type triterpenoids, whose structural diversity can be enhanced through glycosylation, acylation, and redox reactions. UA, also known as ursane acid or UA, is an oxidation product of α-amyrin at the C-28 position and exhibits anti-cancer, anti-diabetic, anti-ulcer, and lipid-lowering effects.11–13 UA is found in various plants, including fruit trees, aromatic herbs, and traditional Chinese medicinal herbs.
1 Construction of pentacyclic triterpenoid privileged scaffolds
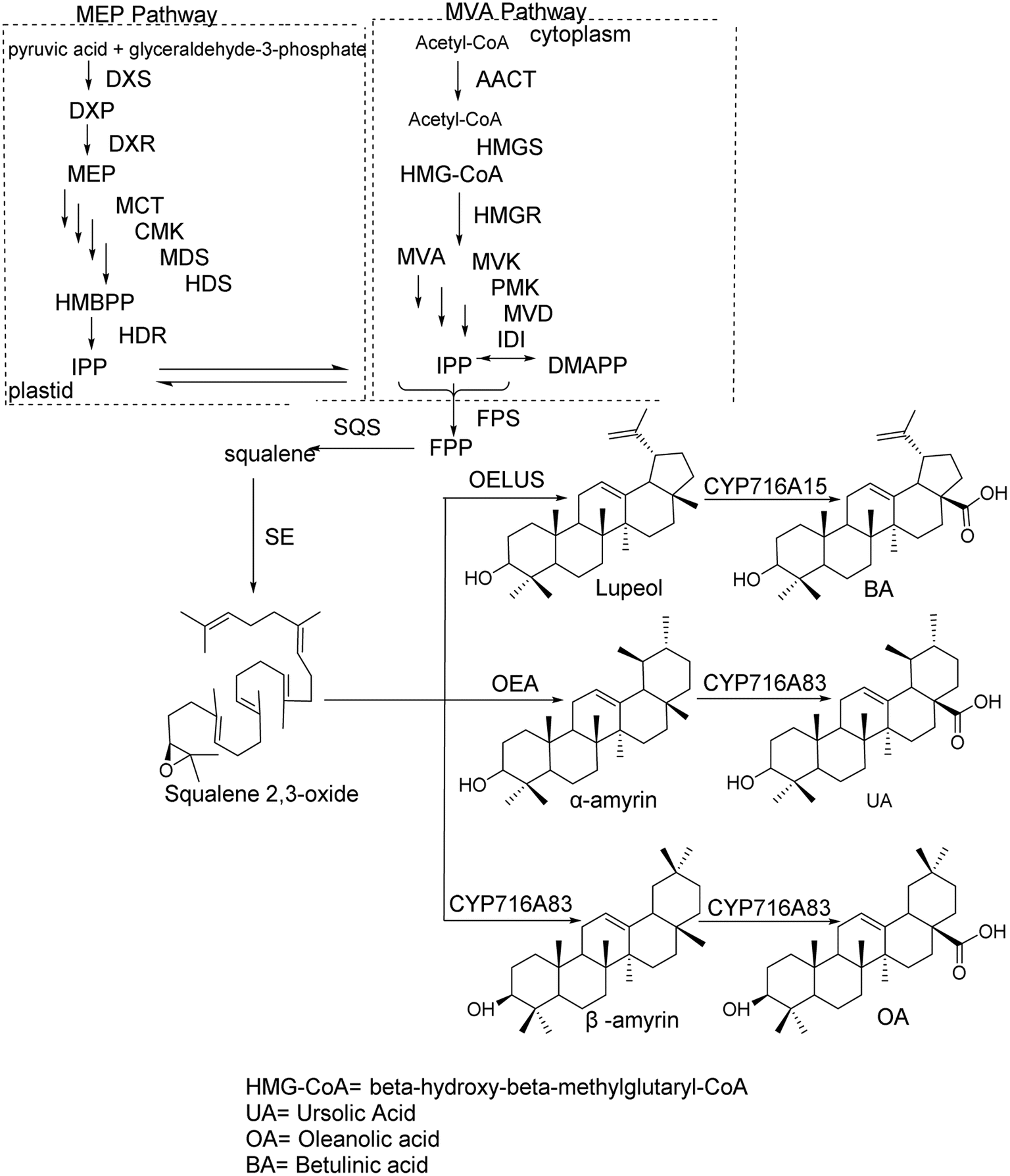
The synthesis of pentacyclic triterpenoid scaffolds primarily occurs through two pathways:14 the methylerythritol phosphate (MEP) pathway in plastids and the mevalonate (MVA) pathway. In plastids, isopentenyl diphosphate (IPP) is synthesized from pyruvate and glyceraldehyde-3-phosphate via seven enzymatic reactions, after which IPP is transported into the cytoplasm to participate in terpenoid synthesis. Under the action of IPP isomerase, IPP and dimethylallyl pyrophosphate (DMAPP) interconvert. In the cytoplasmic MVA pathway, acetyl-CoA acts as the initial donor to produce IPP. IPP and DMAPP are then converted into farnesyl diphosphate under the action of farnesyl diphosphate synthase, which is subsequently converted into squalene by squalene synthase. Squalene is further oxidized into 2,3-oxidosqualene via squalene epoxidase (SE).15 The cyclization of 2,3-oxidosqualene, catalyzed by oxidosqualene cyclase (OSC), produces various tetracyclic and pentacyclic triterpenoid scaffolds. These scaffolds undergo chemical modifications, such as oxidation, substitution, and glycosylation, through the action of cytochrome P450 monooxygenases and glycosyltransferases, with uridine diphosphate (UDP) as the glycosyl donor, ultimately forming various pentacyclic triterpenoid compounds. The synthesis pathways of OA, BA, and UA scaffolds are shown in Fig. 1. OSC, a key enzyme in this process, catalyzes the cyclization of 2,3-oxidosqualene to produce sterol and triterpenoid precursors, representing the critical step in generating the diversity of triterpenoid products.
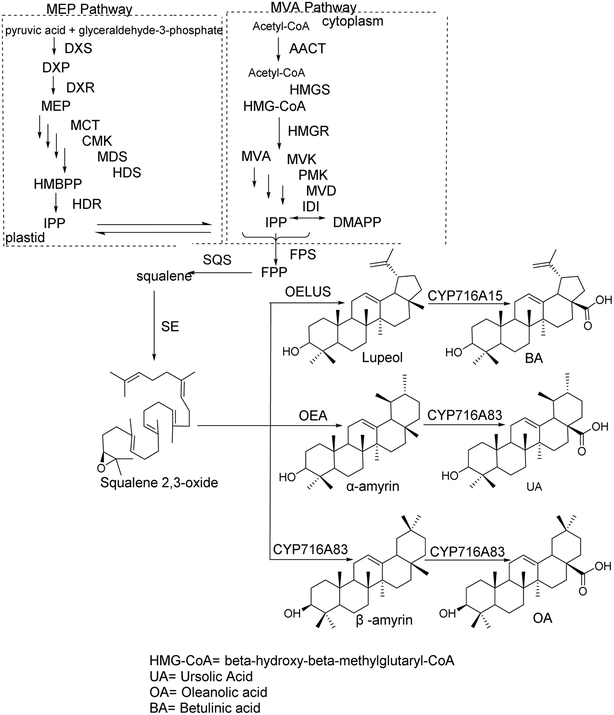 |
| | Fig. 1 Synthesis of the pentacyclic triterpenoid frameworks OA, BA, and UA. | |
2 Structural modification of pentacyclic triterpenoid compounds
In the structure of pentacyclic triterpenoids, the modifications of OA, BA, and UA primarily occur at the hydroxyl group on the A-ring, the carboxyl group at the C-28 position, and the double bond on the C-ring. Rational structural modifications of these active groups can significantly enhance the antitumor activity of these compounds.
2.1 Salt formation modification
Based on the structural characteristics of pentacyclic triterpenoid compounds, there are three direct methods of salt formation: carboxyl groups forming salts with alkali metal ions; introduction of hydrophilic groups followed by salt formation with alkali metal ions; and the introduction of quaternary ammonium salts. Zhao et al.16 and Zhai et al.17 demonstrated that converting OA into its sodium salt significantly increased its solubility in water compared to OA. After administering sodium oleanolate to rats, Li et al.18 observed a marked improvement in subacute liver injury, with the hepatoprotective effect being significantly stronger than that of OA. This indicates that salt formation significantly enhances the water solubility, bioavailability, and bioactivity of OA.
2.2 Amino acid conjugation
Amino acids, as essential nutrients for the human body, are commonly used in the treatment of liver diseases, neurological disorders, and psychiatric conditions. Due to the presence of both carboxyl and hydroxyl polar groups in their structure, coupling amino acids with pentacyclic triterpenoid compounds can enhance the water solubility of the drug and improve its bioavailability. Zhang et al.19 introduced amino acids at the C-28 position of OA, and the resulting OA derivatives exhibited significantly higher antitumor activity compared to the parent compound.
2.3 Glycosylation
Glycosides are polyhydroxy compounds widely distributed in the roots, stems, and leaves of plants, characterized by high solubility and strong polarity. Currently, numerous studies have focused on using OA as a starting material to design and synthesize OA glycoside derivatives with enhanced biological activities. These OA derivatives have shown significantly improved pharmacological activities. Meng et al.20 synthesized 10 OA derivatives through a series of reactions, using natural OA as the lead compound. Pharmacological activity analysis of two glycoside derivatives revealed that one of the derivatives exhibited significantly better antitumor activity than OA, while the other showed even stronger antitumor activity, comparable to the antitumor drug gefitinib.
2.4 Introduction of ester and amide bonds
Based on the structural characteristics of UA, OA, and BA, the hydroxyl group at the C-3 position can undergo esterification reactions with compounds containing carboxyl groups to form ester bonds. Additionally, the carboxyl group at the C-28 position can react with amino or hydroxyl groups through dehydration to form either ester or amide bonds, thereby improving their water solubility and enhancing bioavailability. Zhang et al.21 applied the principle of molecular hybridization in drug synthesis, coupling oxo-oleanolic acid with aminophosphonic acid aryl esters through amide bonds. The resulting novel OA derivatives exhibited better water solubility and higher bioactivity compared to the parent compound.
3 Structural modification and bioactivity of oleanolic acid
OA, also known as Cenoside, is a pentacyclic triterpenoid compound of the oleanane type. It is widely distributed in nature and can be found in various plants such as the fruit of Ligustrum lucidum and the whole herb of Rabdosia rubescens, existing either in its free form or as glycosides. OA exhibits a wide range of biological activities, common to pentacyclic triterpenoids, including antitumor activity,22–24 antiviral properties,25 antihyperlipidemic effects,26,27 anti-inflammatory actions,28,29 and hypoglycemic effects. OA is considered an ideal drug for the treatment of chronic viral hepatitis and acute jaundice hepatitis30 due to its low toxicity and side effects. The anticancer activity of OA and its derivatives is particularly broad, showing inhibitory effects on various human tumor cells. Yang et al.31 reported that OA and its derivatives are well-known for their potent antitumor activity, including inhibiting tumor cell proliferation, inducing cell cycle arrest, apoptosis, and differentiation. These effects have been observed in various cancer cell lines, such as A549, SGC7901, MCF-7, HT1080, and MKN-45 cells. Jannus et al.32 further highlighted that the anticancer properties of OA and its derivatives have been demonstrated in numerous in vitro and in vivo models, showing significant anticancer effects. In cancer, the homeostatic balance between cell proliferation and death is disrupted, and apoptosis, a physiological process of programmed cell death, is one of the key mechanisms for controlling this balance.
However, the major limitation of OA and its derivatives as anticancer agents is their poor water solubility, which leads to low bioavailability in vivo. Therefore, it is necessary to introduce polar groups and perform rational structural modifications to enhance solubility and bioavailability, thus improving the clinical applicability of these compounds.
3.1 Structural modifications of oleanolic acid and antitumor activity
3.1.1 Modification of hydroxyl group at C-3 or carboxyl group at C-28 of oleanolic acid. Spivak et al.33 designed and synthesized derivatives of OA with a C-28 carboxyl group connected to a guanidine group through various linkers to investigate the influence of the guanidine moiety on the anticancer properties of OA. The synthesized guanidine-containing OA derivatives (compound 3, Fig. 2) exhibited significantly higher cytotoxicity in Jurkat cells compared to the original OA. Most of the guanidine derivatives tested demonstrated higher IC50 values than the amine counterparts, while showing lower toxicity towards human fibroblasts.
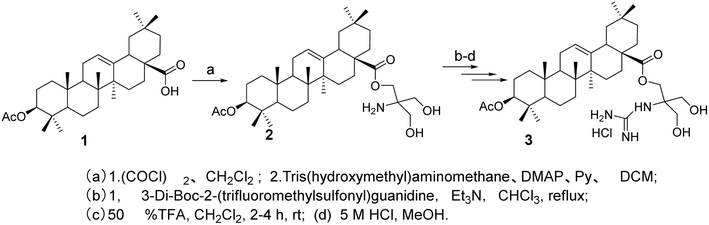 |
| | Fig. 2 Synthesis of OA derivative 3. | |
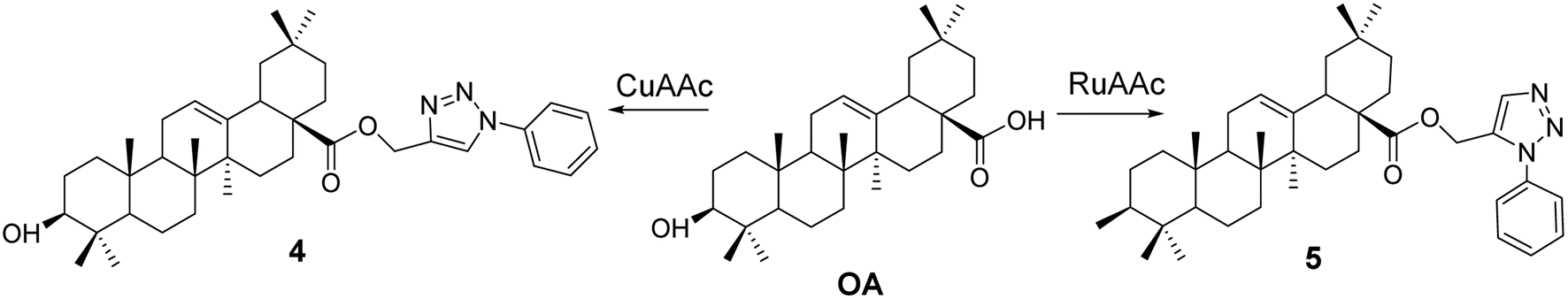
Chouaïb et al.34 synthesized two series of OA 1,2,3-triazole derivatives (compounds 4 and 5, Fig. 3) using CuAAC or RuAAC as catalysts under microwave conditions. The introduction of the 1,2,3-triazole moiety enhanced their activity against mouse mammary cancer (EMT-6) and human colon cancer (SW480) cells.
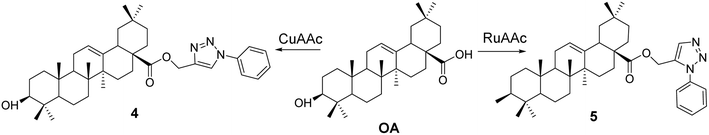 |
| | Fig. 3 Synthesis of OA derivatives 4 and 5. | |
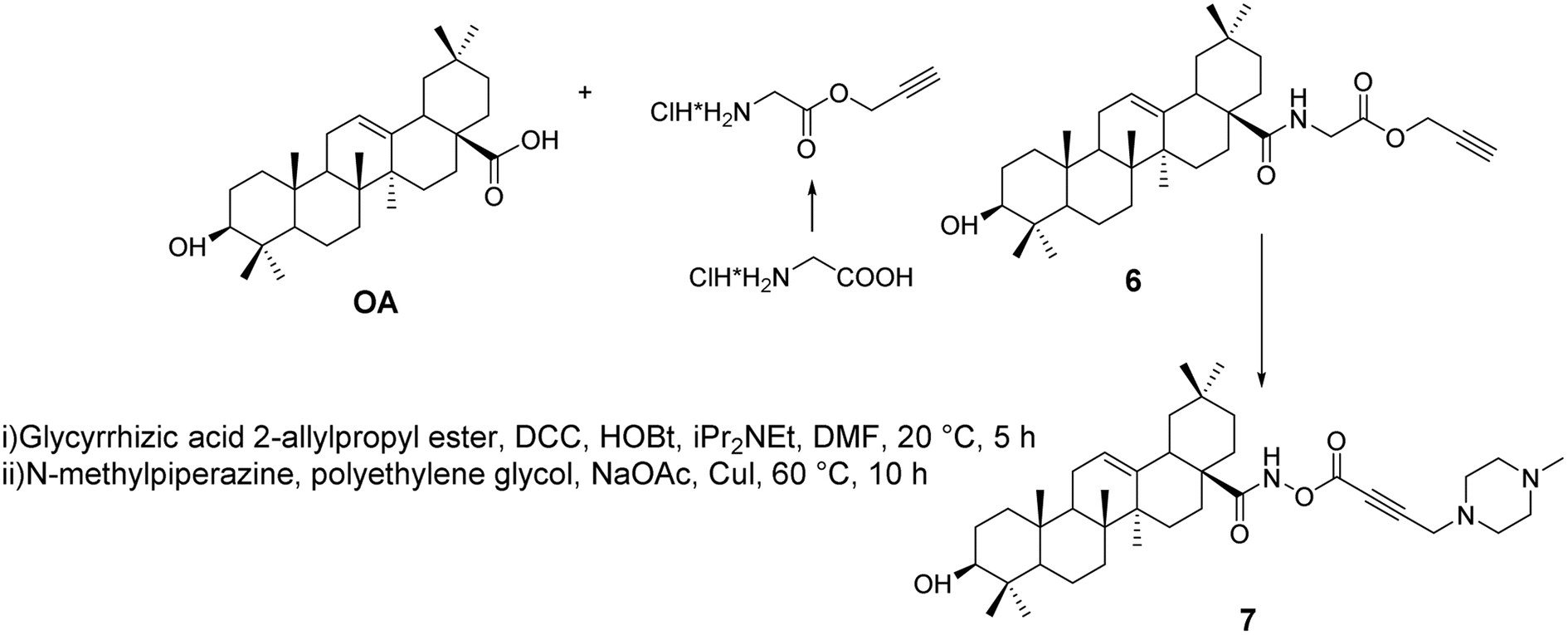
Khusnutdinova et al.35 synthesized alkynyl derivatives 6 and 7 (Fig. 4) through amidation and the Mannich reaction. These derivatives exhibited in vitro anticancer activity against leukemia cells and human colon cancer, acting through their pro-apoptotic activity, stimulating the activity of apoptosis-related caspases 3 and 8, enhancing the activity of NF-κB transcription factors, influencing BAX protein expression, and inhibiting DNA polymerase.
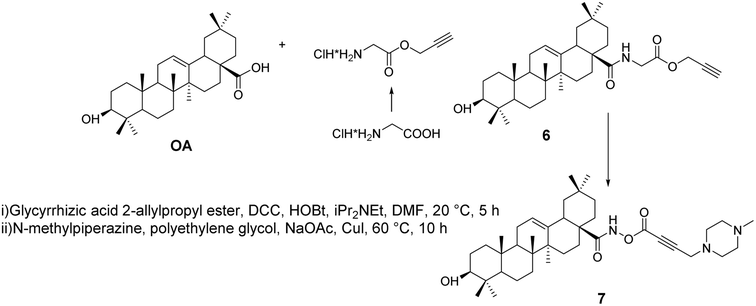 |
| | Fig. 4 Synthesis of OA derivatives 6 and 7. | |
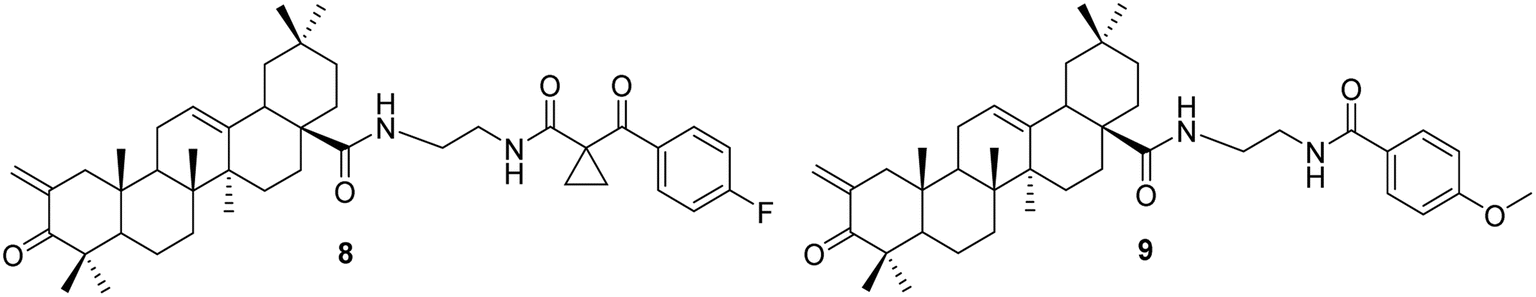
3.1.2 Modification of C-2, ring A, and other positions of oleanolic acid. Liu et al.36 synthesized a variety of OA derivatives (compounds 8 and 9, Fig. 5) involving the condensation of formaldehyde at the C-2 position, oxidation of the C-3 hydroxyl group to a carbonyl, and the attachment of ethylenediamine at C-28. Compounds 8 and 9 demonstrated inhibitory effects on various types of cancer stem cells (CSCs) involved in tumorigenesis, tumor recurrence, invasion, metastasis, and resistance.
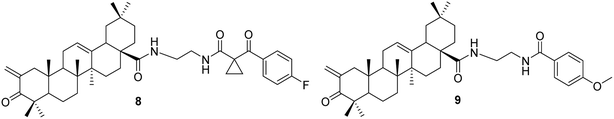 |
| | Fig. 5 The structures of OA derivatives 8 and 9. | |
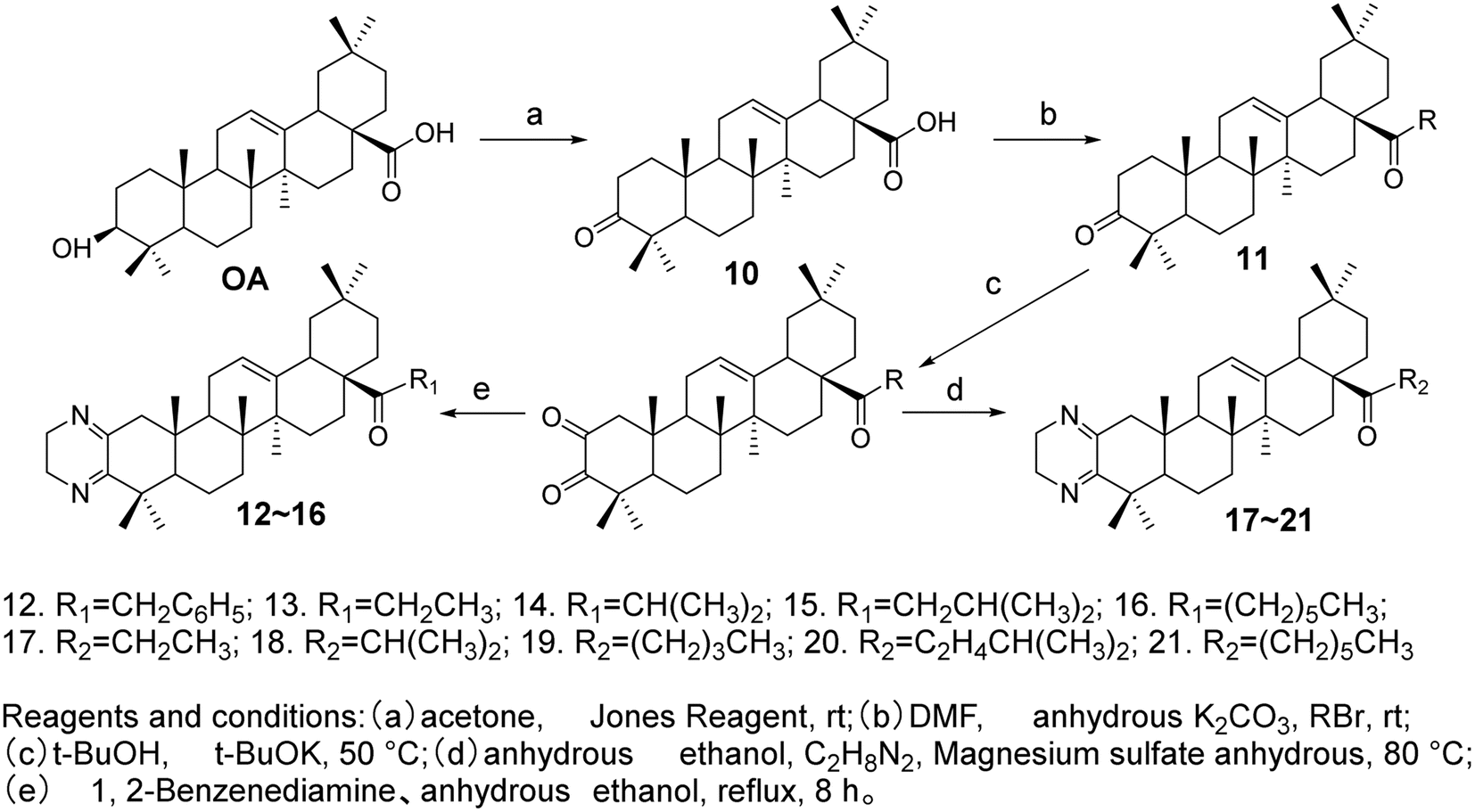
Meng et al.37 modified the A ring and C-28 position of OA to synthesize ten OA derivatives (compounds 12–21, Fig. 6). The inhibitory activities of these derivatives were evaluated against the SGC7901 and A549 cell lines, confirmed through the MTT assay. The experimental results indicated that all compounds exhibited certain antitumor activities against the SGC-7901 and A-549 cell lines. Among them, the OA derivatives 15 and 21 demonstrated superior antitumor activity against both SGC7901 and A549 cells compared to the positive drug gefitinib.
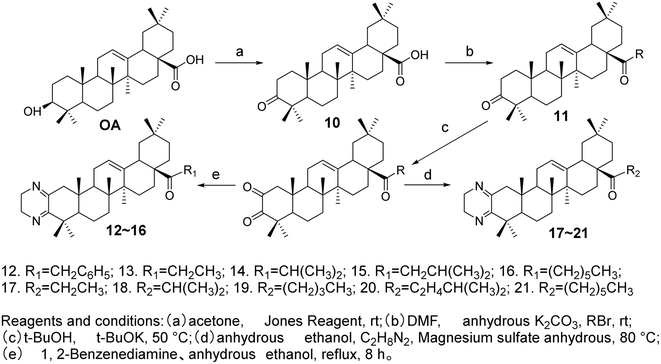 |
| | Fig. 6 Synthesis of OA derivatives 12 to 21. | |
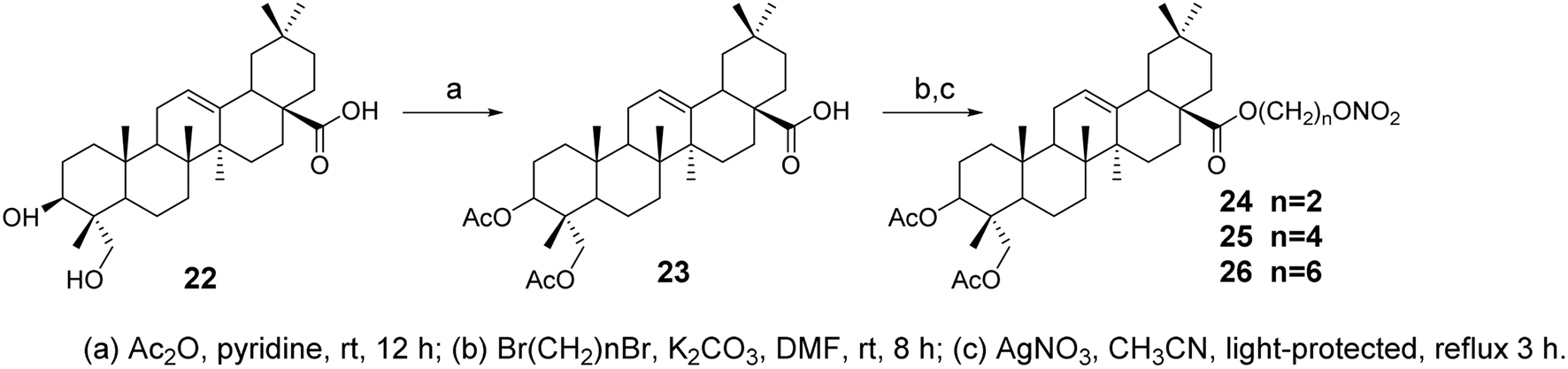
Nitric oxide (NO) is associated with several carcinogenic signaling pathways. Ling's group synthesized several hybrid compounds of OA and ivy saponin NO donors38 (compounds 24–26, Fig. 7) and evaluated their cytotoxicity against various cancer cell lines. The most effective derivative, compound 25, significantly inhibited the proliferation of five cancer cell lines (IC50 = 4.6–5.2 μmol L−1). Furthermore, compound 25 exhibited a stronger inhibitory effect on EGFR-LTC kinase activity (IC50 = 0.01 μmol L−1) compared to ivy saponin (IC50 > 20 μmol L−1) and inhibited the proliferation of gefitinib-resistant H1975 (IC50 = 8.1 μmol L−1) and osimertinib-resistant H1975-LTC (IC50 = 7.6 μmol L−1) non-small cell lung cancer (NSCLC) cells. This suggests that NO may synergistically act with compound 15 (Fig. 6) to induce the maximum production of NO in H1975 cells.
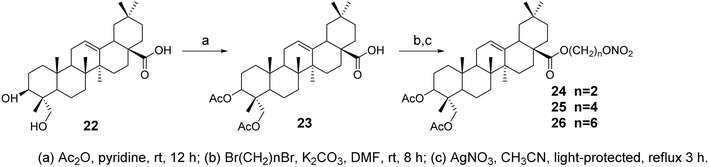 |
| | Fig. 7 Synthesis of OA derivatives 24 to 26. | |
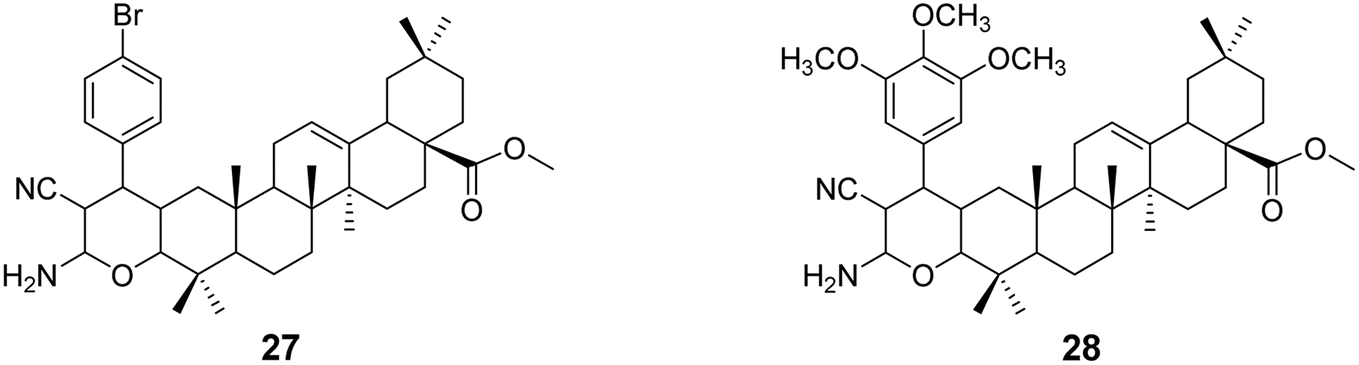
In 2018, Raghuvanshi et al.39 synthesized a series of OA-based chromene derivatives and evaluated their anticancer activity. Among the derivatives with promising activity (compounds 27 and 28, Fig. 8), they further analyzed their ability to trigger apoptosis in A549 and MDA-MB-231 cells and found that these compounds caused cell cycle arrest in the G2/M phase. The most effective compound, 27 (IC50 = 3.6 μmol L−1), exhibited anticancer activity through microtubule destabilization, and it was found to be non-toxic to human red blood cells.
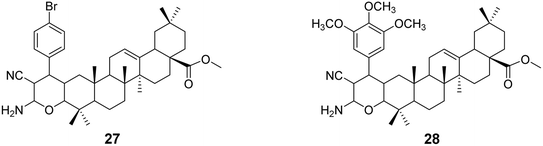 |
| | Fig. 8 The structures of OA derivatives 27 and 28. | |
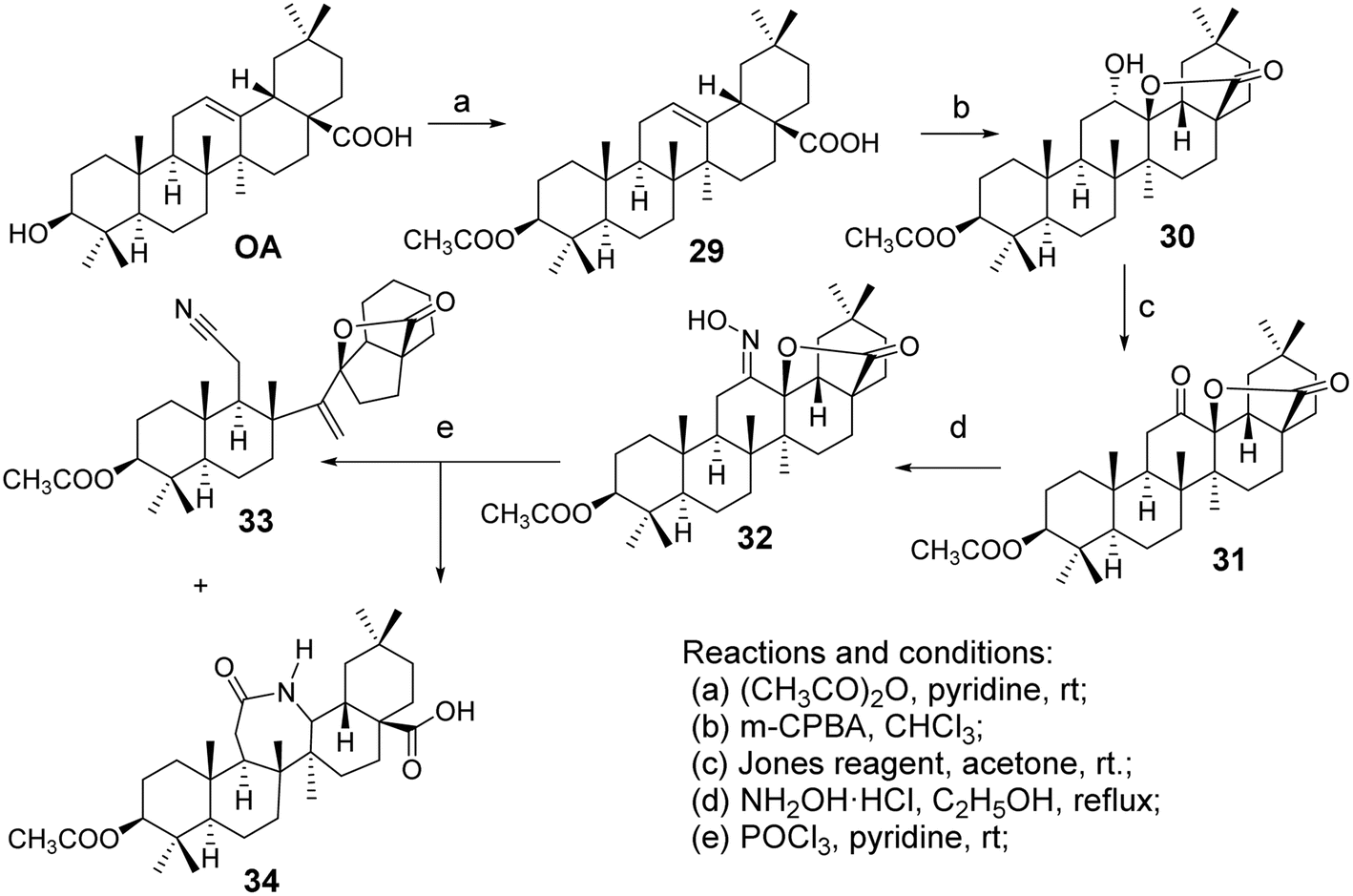
Froelich et al.40 introduced a lactam system into the pentacyclic triterpenoid structure of OA, facilitating the Beckmann rearrangement of the C-ring lactone to form OA derivatives (oxime 32, nitrile 33, and lactam 34, Fig. 9). Zaprutko et al.41 conducted tests on these derivatives as percutaneous permeation enhancers, finding that their activity was comparable to that of the positive control drug Azone.
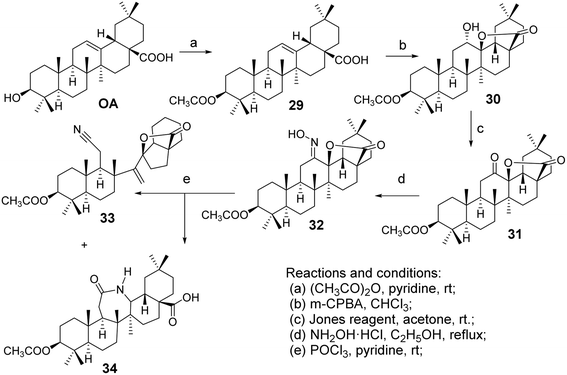 |
| | Fig. 9 Synthesis of OA derivatives 32 to 34. | |
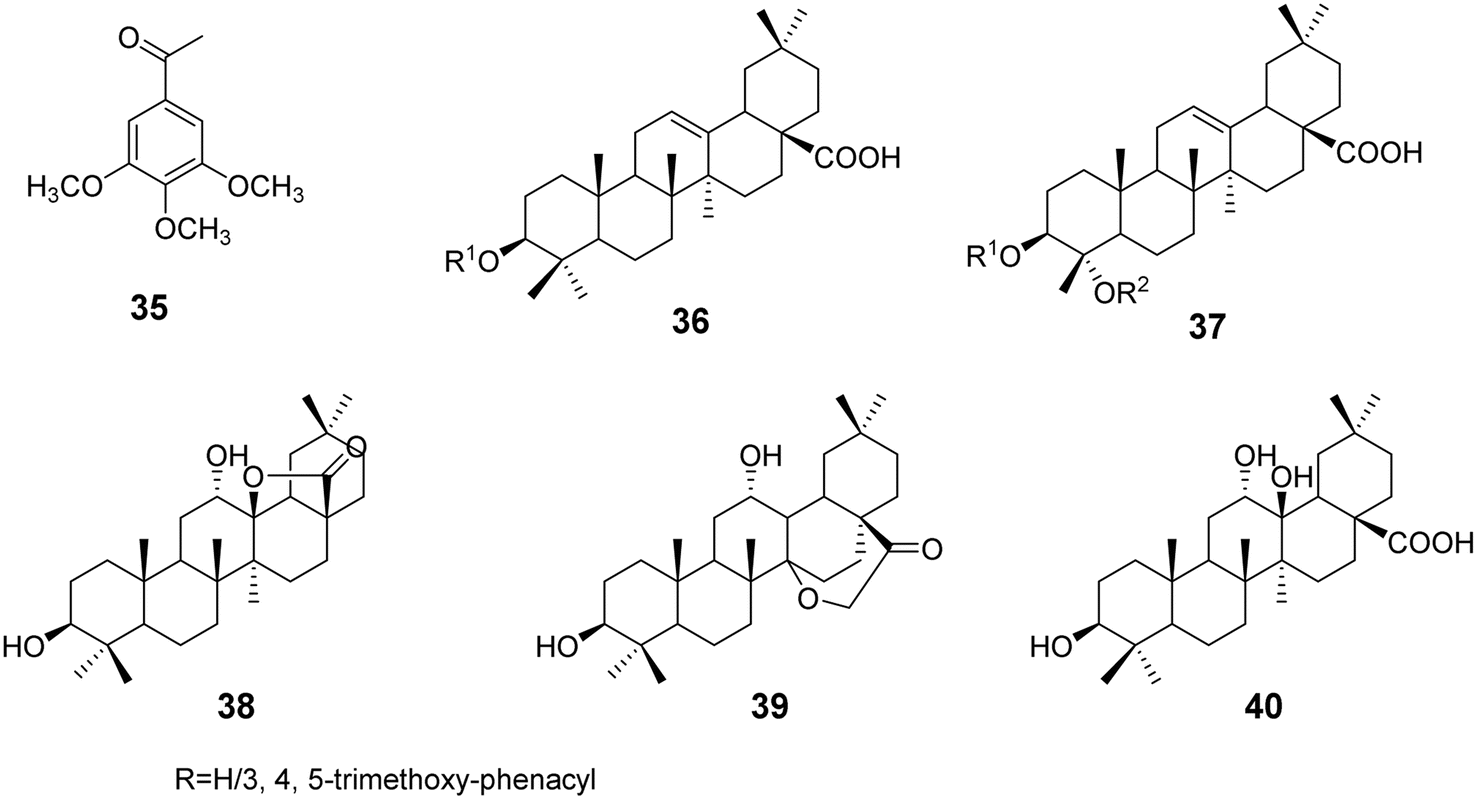
3.1.3 Modifications of C-3, C-4 of the A ring and C-17 of oleanolic acid. Xu et al.42 discovered that methoxy substitutions in triterpene structures exhibit better anticancer activity compared to hydroxyl substitutions. Based on this finding, they modified the pentacyclic triterpene acid through esterification, using compound 35 as the ester moiety to synthesize a series of triterpene derivatives (compounds 36 and 37, Fig. 10), with modifications at the C-3 and C-4 positions of the A ring and the C-17 position of the five-membered ring. Activity assays indicated that most derivatives showed enhanced antifibrotic and antiproliferative effects. The increased lipophilicity improved the ability of the drugs to penetrate cell membranes, demonstrating that the introduction of ester moieties can enhance the anticancer activity of triterpenes. Furthermore, studies have shown that introducing amide bonds at the C-28 position and carbonyl groups at the C-11 position can also enhance the antitumor activity of triterpene acids.43 Heise et al.44 modified OA derivatives to obtain two structurally unique lactone triterpenes (derivatives 38–40, Fig. 10) and a tri-hydroxy OA derivative (compound 40). Among these, derivative 38 exhibited no cytotoxicity against all tested cell lines, while derivative 39 showed a degree of selectivity against MCF-7 and FaDu cancer cells. The tri-hydroxy compound 40 demonstrated the strongest cytotoxic effect on MCF-7 cells, with toxicity approximately 5.5 times greater than that observed in non-malignant fibroblasts. This unique structural modification provides new avenues for enhancing the anticancer activity and selectivity of triterpenes.
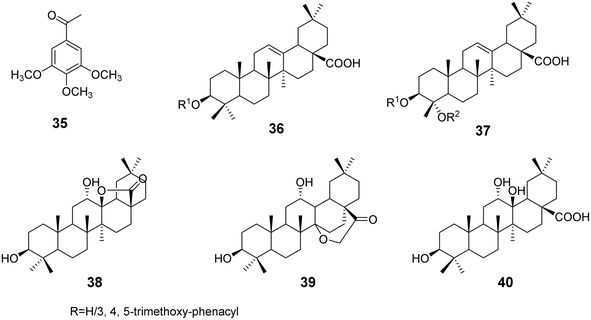 |
| | Fig. 10 The structures of triterpenic acid derivatives 35 to 40. | |
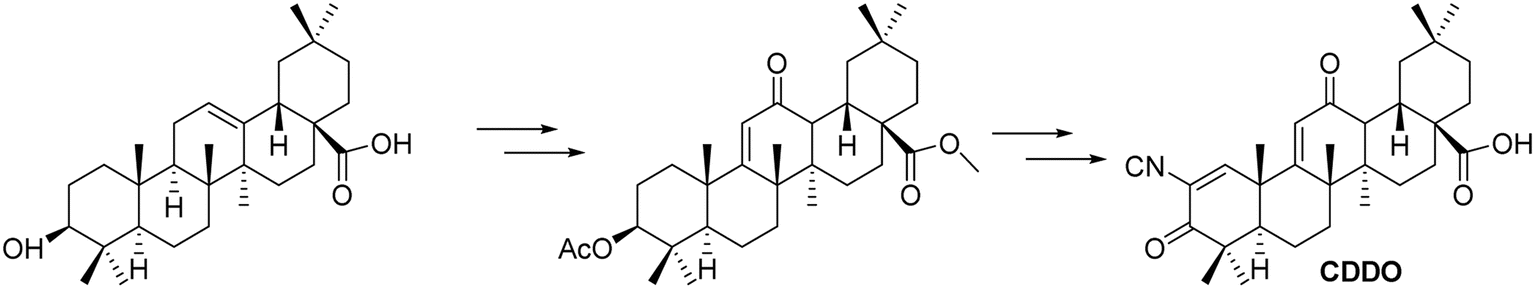
3.1.4 Improvements and antitumor activity of CDDO based on oleanolic acid derivatives. Honda et al.45 synthesized a derivative of OA over two decades ago, known as 2-cyano-3,12-dioxo-olean-1,9(11)-dien-28-oic acid (CDDO, Fig. 11). Their research revealed that CDDO not only prevents somatic cancers but also shows increasing evidence of efficacy against gender-specific cancers. CDDO and its derivatives are relatively non-toxic and serve as promising candidates for testing therapies for various diseases, demonstrating effectiveness against multiple types of cancer. The CDDO compound provides protection against ROS-induced cell death in normal cells by upregulating Nrf2, while simultaneously inducing death in cancerous or abnormal cells. In ovarian cancer models, CDDO directly targets and inhibits hsp90, reducing cell proliferation; it also upregulates NQO1, which contributes to PARP1-mediated programmed necrosis in breast cancer, offering a potential therapeutic approach for this malignancy.
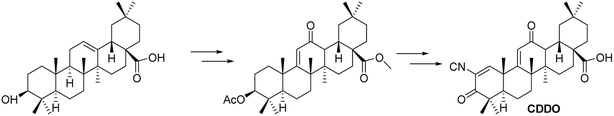 |
| | Fig. 11 Synthesis of OA derivative CDDO. | |
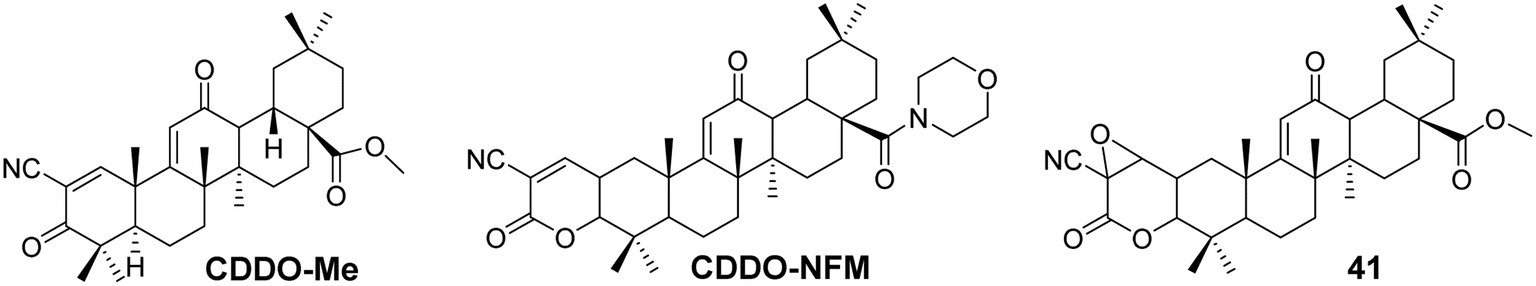
The most common modification site for CDDO is the C-28 carboxyl group. For instance, the introduction of a methyl ester at C-28 yields CDDO-Me (Fig. 12), which exhibits potent anti-inflammatory and anticancer activity.46 CDDO-Me has shown inhibitory effects on prostate cancer cells, ovarian cancer cells, acute myeloid leukemia, pancreatic cancer cells, esophageal cancer cell lines, and triple-negative breast cancer cells. In ovarian cancer, CDDO-Me induces apoptosis by inhibiting AKT, NF-κB, and mTOR signaling pathways, without affecting PDK1 kinase or PP2A activity. It induces prostate cancer cell death by inhibiting Akt, and apoptosis in acute myeloid leukemia by inhibiting ERK 1/2 phosphorylation through activation of p38/MAPK. CDDO-Me can also downregulate telomerase activity (hTERT) and induce cell death in pancreatic cancer cell lines.47 Wong et al.48 synthesized a novel CDDO-Me analog (compound 41, Fig. 12), which can irreversibly react with model thiols, aiding in the identification of pharmacologically relevant target interaction sites. Compound 41 exhibited cytotoxicity in rat liver cancer cells and comparable efficacy to CDDO-Me in activating Nrf2. Molecular modeling supported the stable modification of specific cysteine residues within Keap1 by compound 41. Gao et al.49 explored whether CDDO-NFM (Fig. 12) has potential antitumor effects for cancer treatment, and they found that CDDO-NFM effectively inhibits OS cell growth, and this inhibitory effect is independent of apoptosis-related and cell cycle-related proteins.
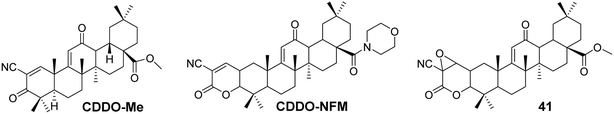 |
| | Fig. 12 The structures of OA derivatives CDDO-Me, CDDO-NFM, and 41. | |
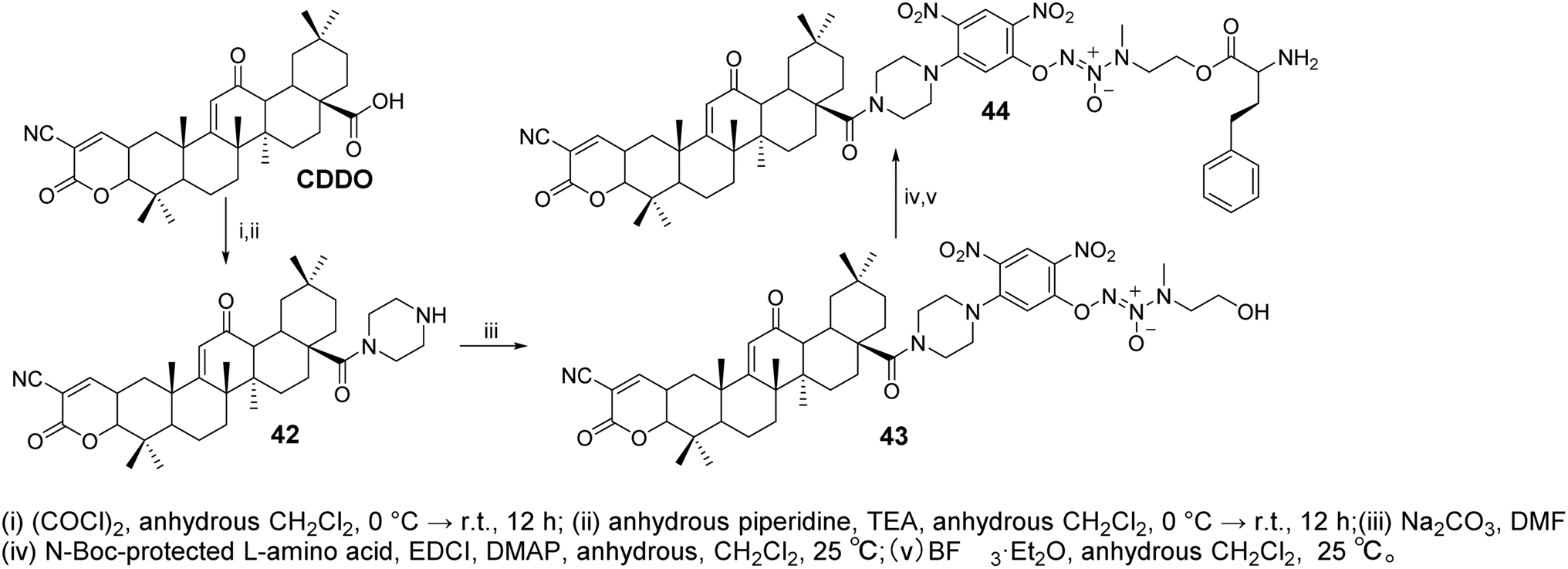
Zhang et al.50 designed and synthesized CDDO derivatives (compounds 42–44, Fig. 13). Their anticancer activity was further evaluated by measuring IC50 values using the MTT assay. These compounds demonstrated potent inhibitory activity against both A549/Taxol (IC50 = 0.349–1.087 μmol L−1) and A549 (IC50 = 1.066–1.438 μmol L−1) cells, showing greater antiproliferative efficacy than CDDO-Me (IC50 = 1.703 and 2.313 μmol L−1) and JS-K (IC50 = 1.987 μmol L−1 and 2.313 μmol L−1). The most active compound, 44, induced higher levels of NO and reactive oxygen species (ROS) in paclitaxel-resistant A549/Taxol cells with overexpression of glutathione S-transferase π (GSTπ), significantly inhibiting cell proliferation (IC50 = 0.349 ± 0.051 μmol L−1). Moreover, compound 44 showed stronger inhibition of Lon protease expression, as well as more pronounced induction of apoptosis and cell cycle arrest in A549/Taxol cells, compared to CDDO-Me.
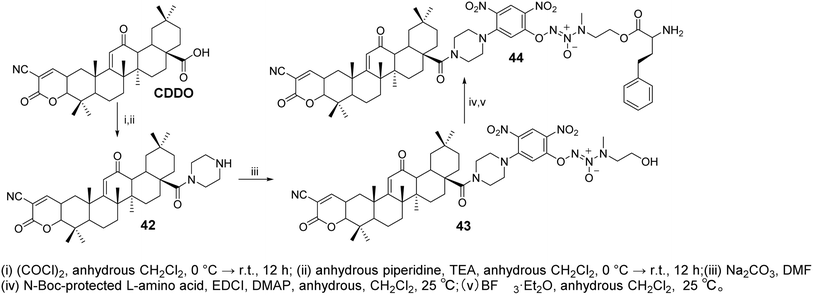 |
| | Fig. 13 Synthesis of OA derivatives 42 to 44. | |
3.2 Structural modifications of oleanolic acid and antiviral activity
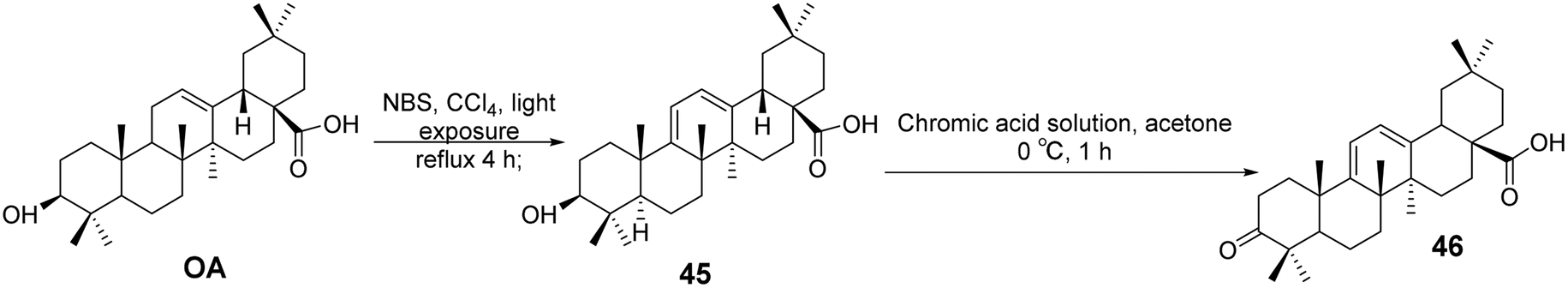
Yan et al.51 synthesized a series of OA derivatives by introducing conjugated dienes and utilizing photosensitized oxidation to form epoxides. The cytotoxicity of these derivatives was evaluated in HepG 2.2.15 cells using the standard MTS assay. Most of the derivatives exhibited activity in inhibiting HBV antigen secretion in HepG 2.2.15 cells. Among the compounds tested, the most active derivative (compound 46, Fig. 14) demonstrated significant activity in inhibiting the secretion of HBsAg, HBeAg, and the replication of HBV DNA. It was more effective than lamivudine in preventing the rebound of viral replication.
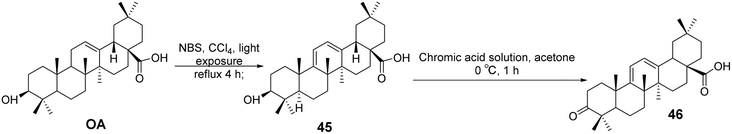 |
| | Fig. 14 Synthesis of OA derivative 46. | |
Li et al.52 introduced amino conjugates at the C-28 position of OA (compounds 48–52, Fig. 15) and evaluated their antiviral activity against influenza A virus A/WSN/33 (H1N1) in Madin–Darby Canine Kidney (MDCK) cells. Based on the biological results, the structure–activity relationship of the compounds was discussed. Compound 52, which contains a six-carbon chain and a terminal hydroxyl group, exhibited the strongest anti-influenza activity, with an IC50 of 2.98 μmol L−1. Hemagglutination inhibition and surface plasmon resonance (SPR) analysis suggested that compound 52 may interfere with viral invasion by interacting with the HA protein of the influenza virus.
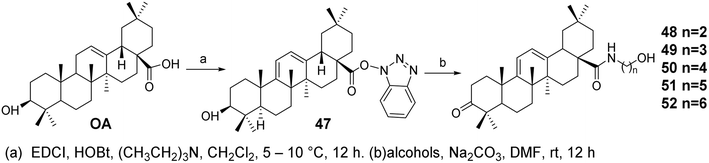 |
| | Fig. 15 Synthesis of OA derivatives 48 to 52. | |
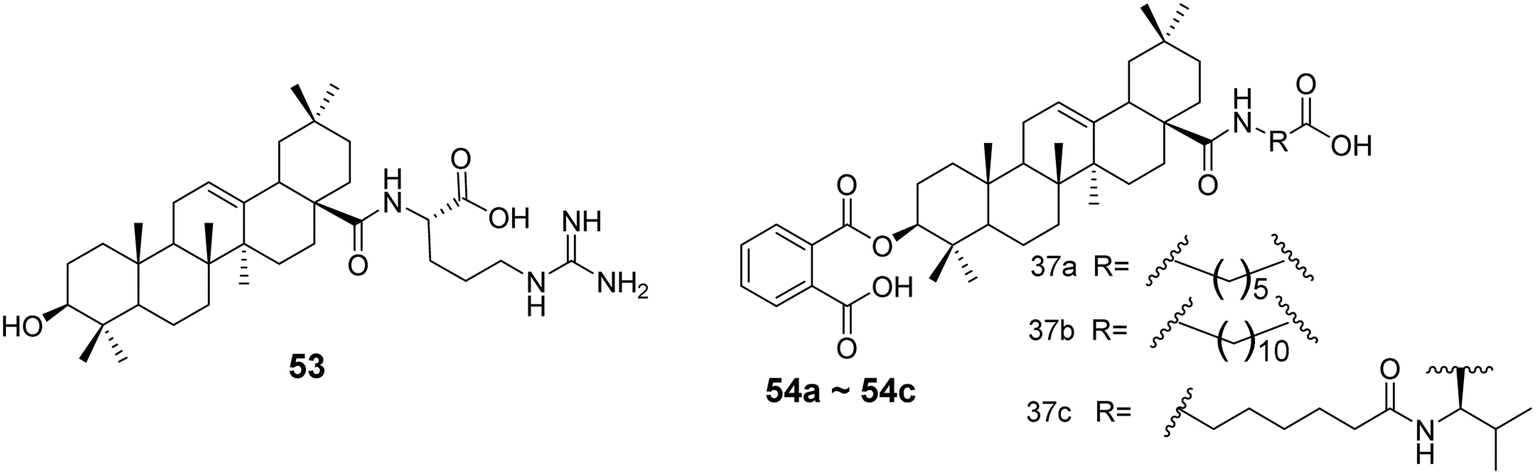
In 2019, Yang's team53 designed and synthesized 32 derivatives by coupling various amino acids with the 28-COOH of OA. The most active derivative (compound 53, Fig. 16) demonstrated robust efficacy and a broad antiviral spectrum against four different influenza strains. Hemagglutination inhibition (HI) assays and docking studies suggested that these derivatives likely share the same mechanism as their parent compound, blocking viral entry by binding to hemagglutinin (HA) and preventing the interaction between the viral HA protein and the sialic acid receptors on host cells. In the same year, Medina-O’Donnell et al.54 esterified one or two amino acids at the C-28 position and substituted phthaloyl groups at the C-3 position to obtain three 3-phthaloyl derivatives (compounds 54a–54c, Fig. 16), which showed enhanced inhibition of HIV-1 protease activity.
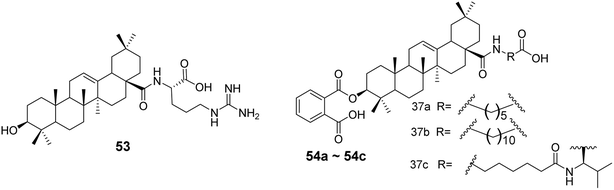 |
| | Fig. 16 Synthesis of OA derivative 53 and derivatives 54a to 54c. | |
4 Structural modifications and biological activities of glycyrrhetinic acid
Glycyrrhiza, derived from the dried roots and rhizomes of Glycyrrhiza uralensis, Glycyrrhiza inflate, or Glycyrrhiza glabra, is a valuable traditional Chinese medicine. The active constituents found in its dried roots and rhizomes primarily include flavonoids such as glycyrrhizin, GA, liquiritin, and isoliquiritin. Current research on the structural modifications of GA mainly focuses on the hydroxyl group at C-3, the carboxyl group at C-30, and the carbonyl group at C-11. Through modifications of the GA skeleton, a series of derivatives with physiological activities have been developed.
For instance, after replacing the hydroxyl group at the C-3 position of GA, the anti-inflammatory and anti-ulcer activities were significantly enhanced. When acyl groups, glycosides, or alkoxy and glycosyl groups were introduced at the C-3 hydroxyl or C-30 carboxyl positions, the anti-ulcer and anti-inflammatory activities were improved. Introducing substituents at both the C-3 hydroxyl and C-30 carboxyl positions further increased anti-inflammatory and antiviral activities. Additionally, the removal of the hydroxyl group at the C-11 position of GA has been shown to reduce or eliminate side effects and enhance pharmacological activity.
4.1 Structural modifications of the C-30 carboxyl group of glycyrrhetinic acid
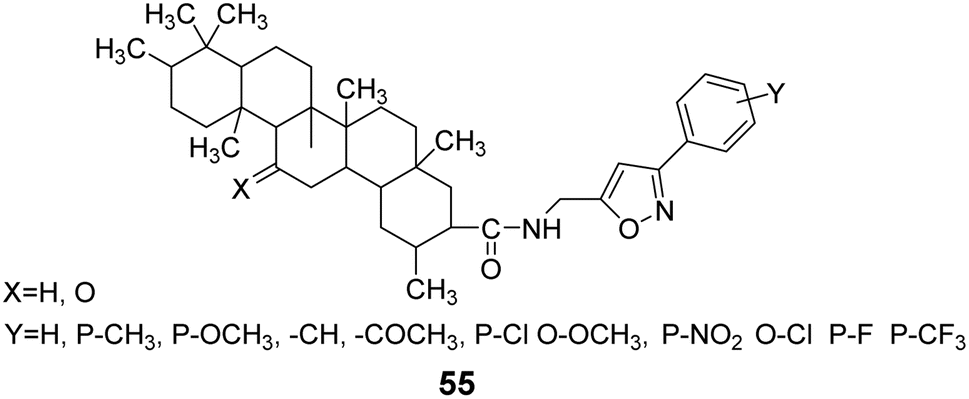
Wang et al.55,56 utilized addition and elimination reactions to modify substituted benzaldehyde, yielding substituted methylamine intermediates. These intermediates were then reacted with GA and deoxyglycyrrhetinic acid (DGA), respectively, to produce GA and DGA derivatives at the C-30 position with amide bonds, forming heterocyclic derivatives (compound 55, Fig. 17).
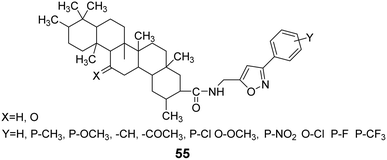 |
| | Fig. 17 The structure of isoxazole heterocyclic derivatives 55 of glycyrrhizic acid and dehydroglycyrrhizic acid. | |
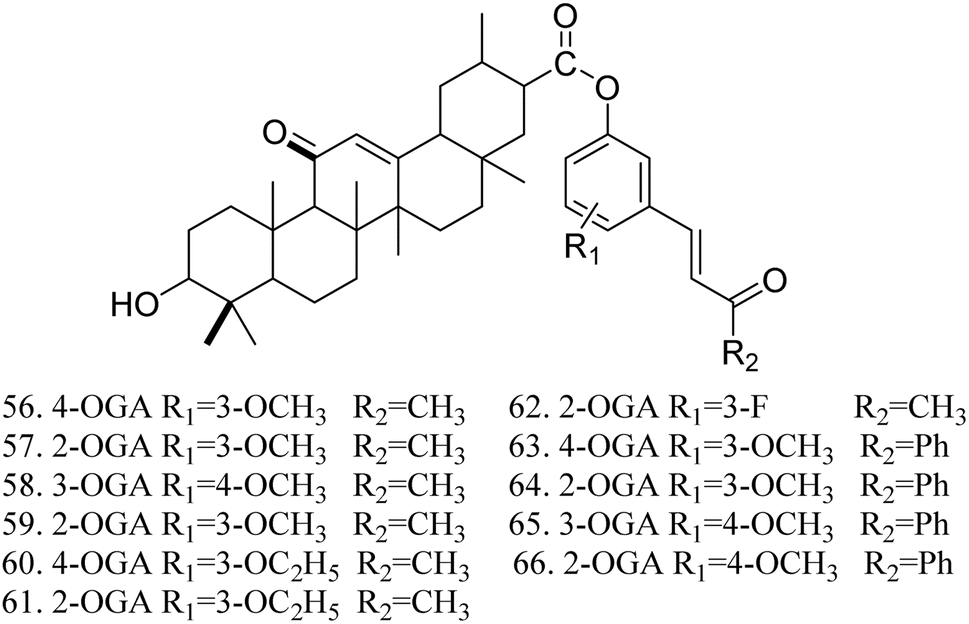
The activity testing results for these compounds indicated that they exhibited high analgesic, anti-inflammatory, and antitussive activities, along with increased safety. The findings also demonstrated that 1-(3-methoxyphenyl)-5-aminomethyl-2-isoxazole DGA possesses certain hepatoprotective effects, effectively reducing the mortality rate of mice infected with the H1N1 influenza virus at a dose of 100 mg kg−1. Lee et al.57 synthesized 11 GA derivatives by introducing dehydrogingerol at the C-30 position. The activity tests revealed that these GA derivatives significantly inhibited tumor cell replication and exhibited anticancer effects. Some derivatives displayed strong cytotoxic activity; however, substituting the methoxy groups in compounds 56 and 57 with ethoxy groups resulted in a reduction in the cytotoxic activity of compounds 60 and 61 (Fig. 18).
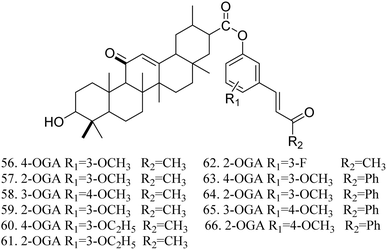 |
| | Fig. 18 The structures of dehydrogingdiolide derivatives of glycyrrhizic acid 56 to 66. | |
Zhang Yan et al.58 synthesized GA methyl ester and GA ethyl ester through esterification reactions between GA and methanol or ethanol. GA and its derivatives reacted with peracetic acid, resulting in the oxidation of the C-3 hydroxyl group to a carbonyl group, yielding oxidized GA and its methyl and ethyl ester derivatives. The catalytic asymmetric epoxidation activity of these compounds was tested, revealing the following activity order: oxidized glycyrrhetinic acid (OGA) > oxidized glycyrrhetinic acid derivatives > GA derivatives. Furthermore, GA derivatives were found to be ineffective in catalyzing the reactions of sterically hindered alkenes.
4.2 Structural modifications of C-3 hydroxyl and C-30 carboxyl groups of glycyrrhetinic acid
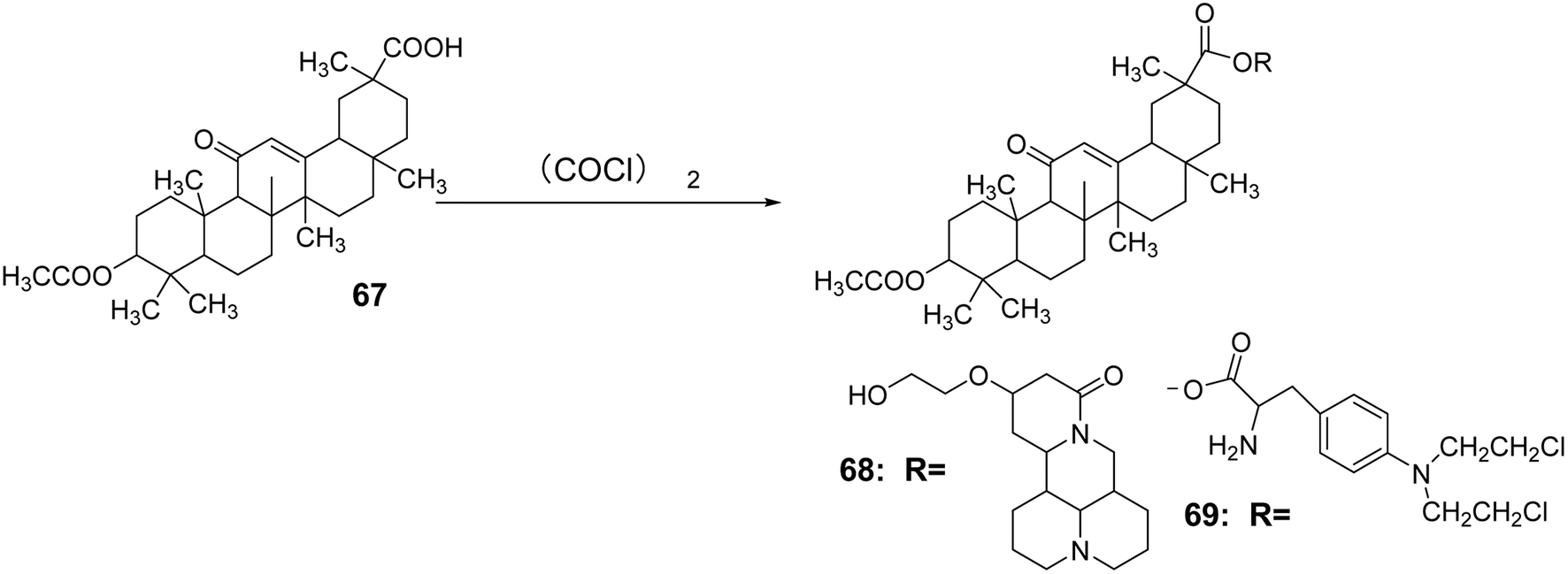
Zhang Na et al.59 introduced an acetyl group at the C-3 position through an acetylation reaction to synthesize compound 67. Subsequently, under the reaction conditions of (COCl)2, compounds 56 and 57 (Fig. 18) were reacted to obtain compounds 68 and 69. Using berberine, 18α-GA, and methotrexate as controls, it was found that compounds a and b exhibited significant inhibitory effects on the liver cancer cell line SMMC-7721, with IC50 values of 85.7 μmol L−1 and 178.2 μmol L−1, respectively. Animal experiments indicated that these compounds were almost non-toxic to normal liver cells (Fig. 19).
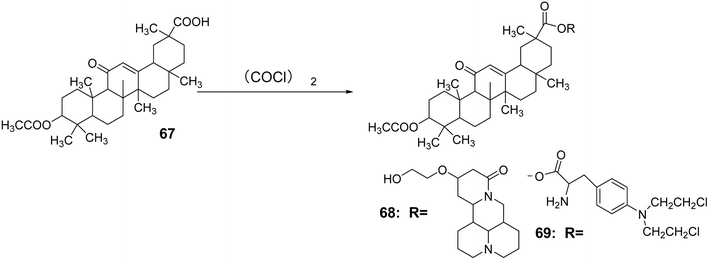 |
| | Fig. 19 Synthesis of 3-acetyl glycyrrhizic acid lipid derivatives 68 to 69. | |
Csuk et al.60 modified GA methyl ester (GA-Me) by reacting it with chloroacetyl chloride, followed by a condensation reaction with various diamines to introduce straight-chain amino groups at the C-3 hydroxyl position, resulting in a series of GA derivatives 70. The results of antitumor activity testing demonstrated that the introduction of straight-chain amino groups at the C-3 position enhanced the activity of GA, with the highest activity observed when the carbon chain length n = 6, yielding an IC50 range of 0.6–3.0 μmol L−1 (Fig. 20).
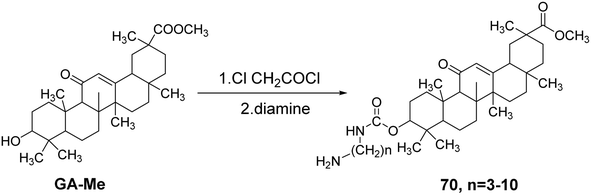 |
| | Fig. 20 Synthesis of 3-O-(linear aminocarbonyl) glycyrrhizic acid lipid derivative 70. | |
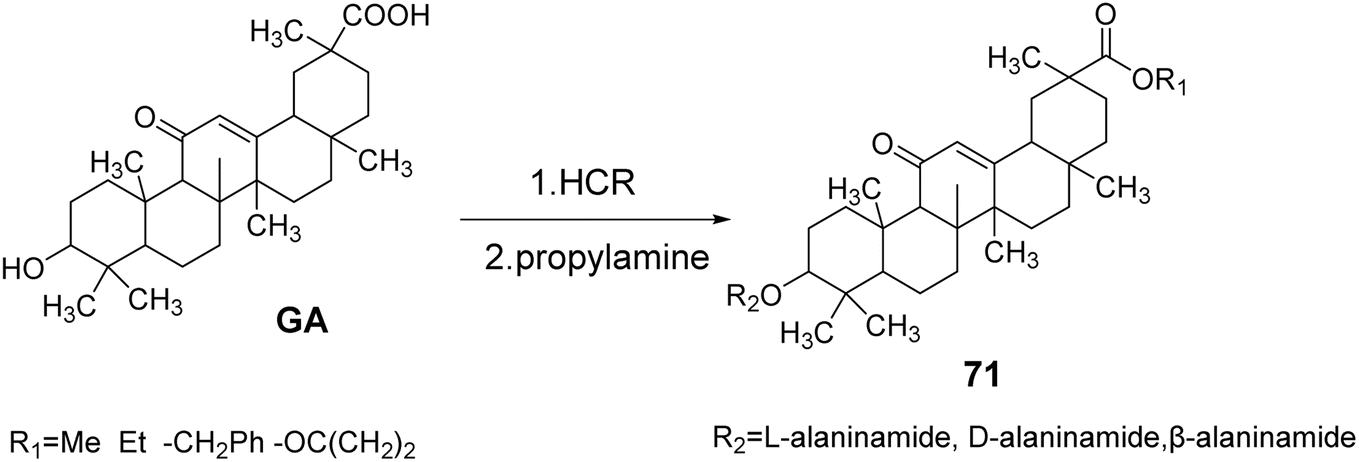
Schwarz et al.61 synthesized GA derivatives by introducing methoxy, ethoxy, and benzyloxy groups at the C-30 position through esterification reactions. They also introduced different configurations of alanine at the C-3 position, resulting in a series of C-3 and C-30 GA derivatives 71. The activity and selectivity of these derivatives against tumor cells were found to be superior to those of the control group, which consisted of GA (Fig. 21).
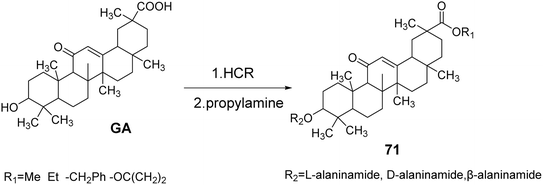 |
| | Fig. 21 Synthesis of 3-alanine type glycyrrhizic acid lipid derivative 71. | |
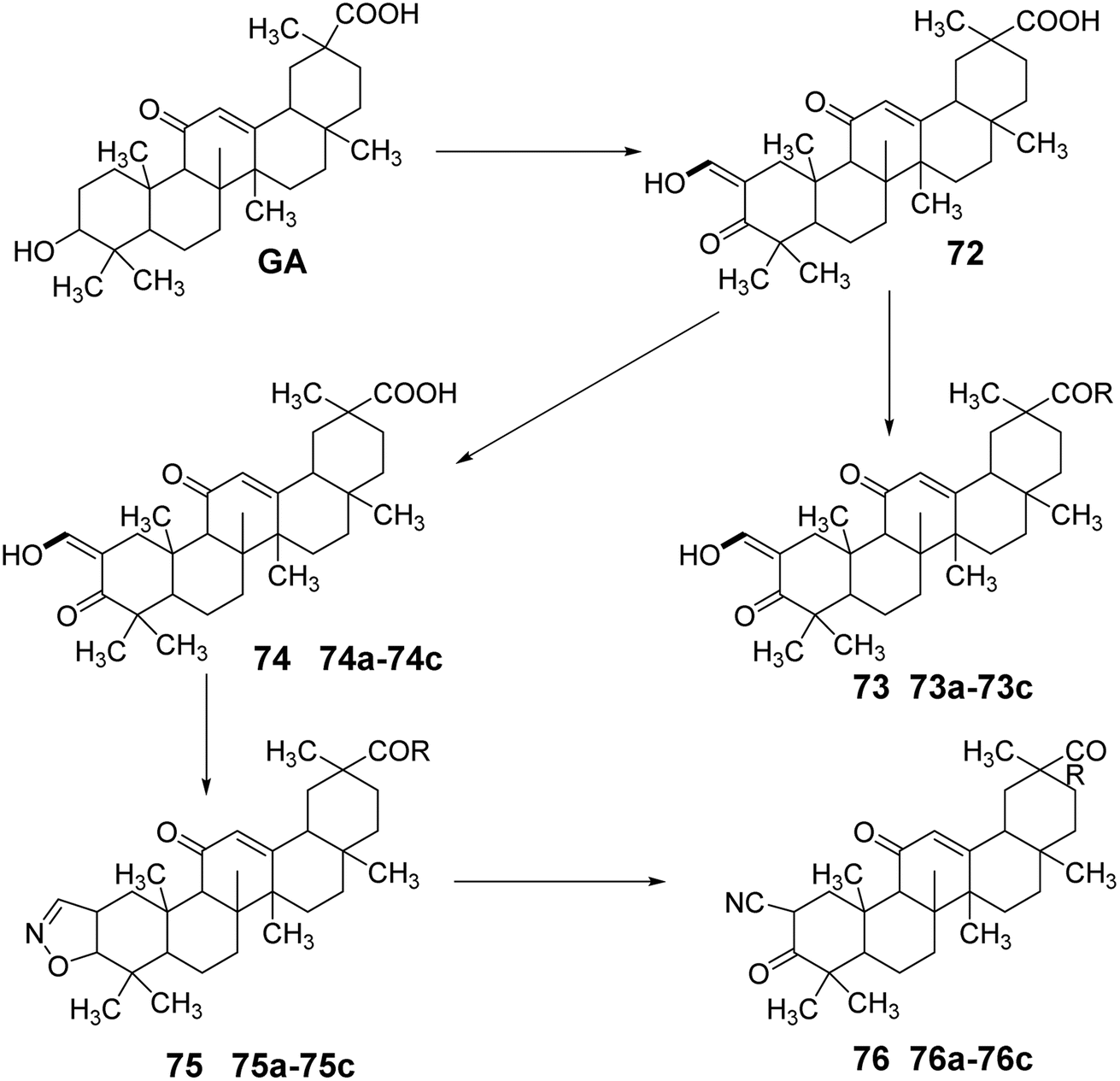
Gao et al.62 simultaneously modified the C-30 carboxyl and C-3 hydroxyl groups of GA through oxidation, esterification, substitution, and hydrolysis reactions to obtain compound 72. Compound 72 underwent nucleophilic substitution to introduce various N-substituents at the carboxyl group, yielding amide derivatives 73a–73c. Compound 2 was synthesized by forming a ring between C-3 and C-2, followed by nucleophilic substitution to obtain compounds 74a–74d. Subsequently, ring-opening elimination and reduction led to various structural derivatives of oxidized GA amides, including 75a–75d and 76a–76d. Activity studies indicated that these compounds exhibited strong inhibitory effects on the growth of HL-60 leukemic cells (Fig. 22).
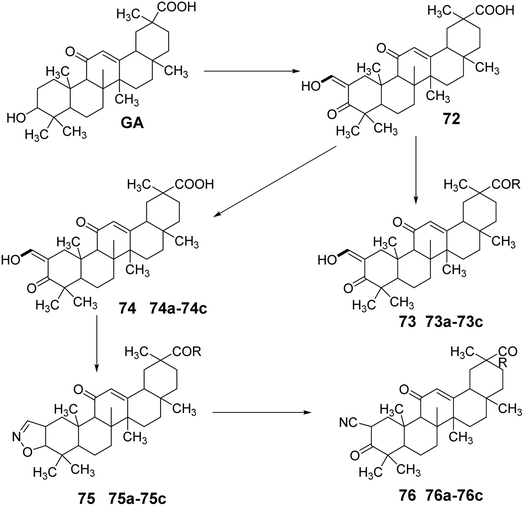 |
| | Fig. 22 Synthetic route of oxidized glycyrrhizic acid amide derivatives. | |
4.3 Structural modification of the C-11 carbonyl group of glycyrrhetinic acid
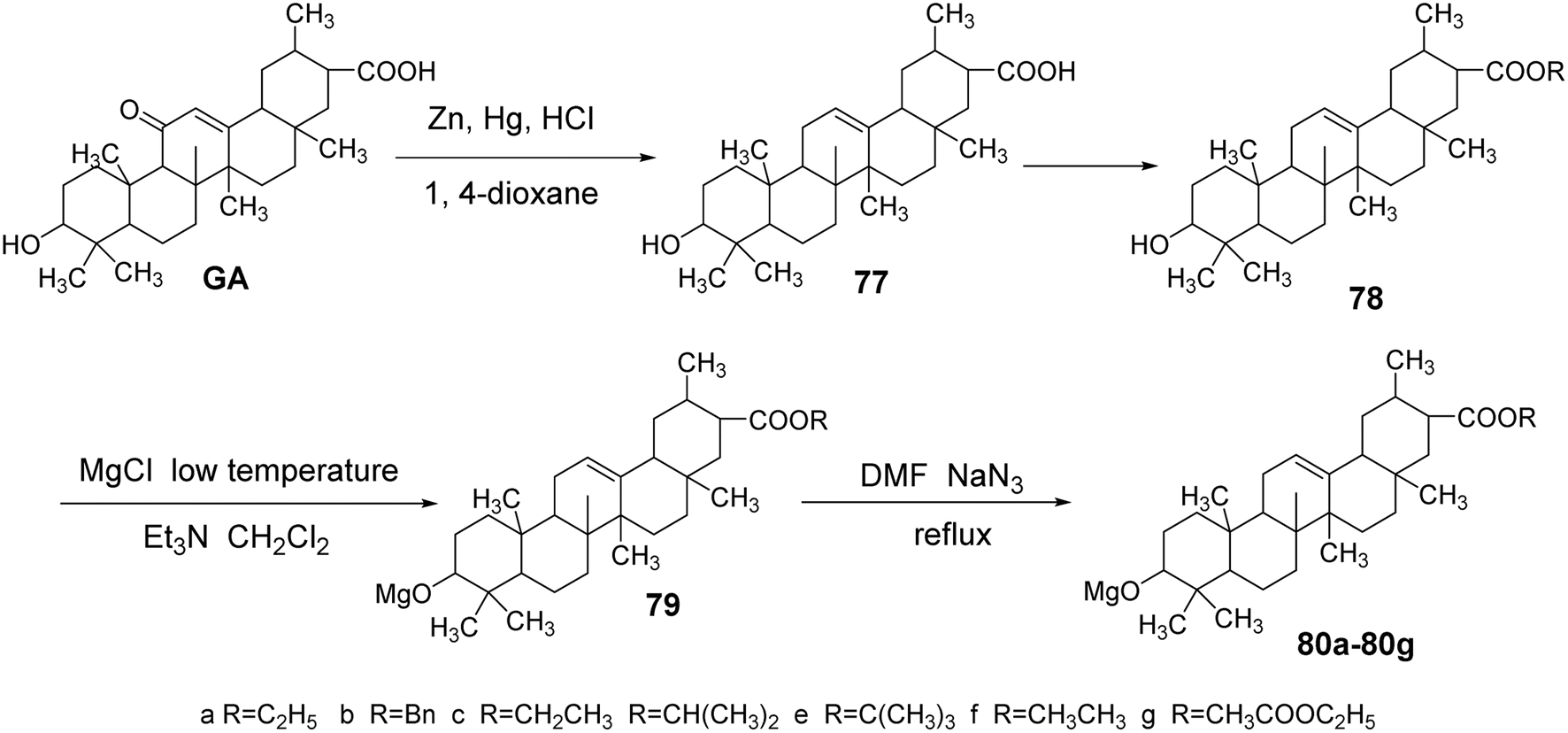
Meng Yanqiu et al.63 reduced the C-11 carbonyl group of GA to obtain DGA (compound 77) using zinc–mercury amalgam. Subsequently, they introduced an ester bond at the C-30 position through an esterification reaction. The reaction with p-toluenesulfonyl chloride in a dichloromethane and triethylamine medium at low temperatures facilitated the introduction of a p-toluenesulfonyl group at the C-3 hydroxyl. Finally, refluxing with sodium azide in tetrahydrofuran yielded compounds 80a–80g.
In vitro antitumor activity tests, using paclitaxel and gefitinib as controls, revealed that compounds 80a, 80c, and 80e exhibited significant inhibitory effects on human breast cancer cells (MCF-7) and lung cancer cells (A549). At a concentration of 10 μmol L−1, the inhibition rates against MCF-7 were 71.57%, 14.53%, and 22.02%, respectively. Notably, compound 80a demonstrated a significantly enhanced inhibitory capacity against breast cancer cells compared to glycyrrhetinic acid, with an IC50 of 4.29 ± 0.07 μmol L−1. The inhibition rates against A549 cells at 10 μmol L−1 were 4.91%, 26.60%, and 19.97%, all exceeding that of glycyrrhetinic acid. The activity testing results of compounds 80a, 80c, and 80e further indicated that the different ester bonds at the C-30 position significantly influenced the inhibition of MCF-7 cells (Fig. 23).
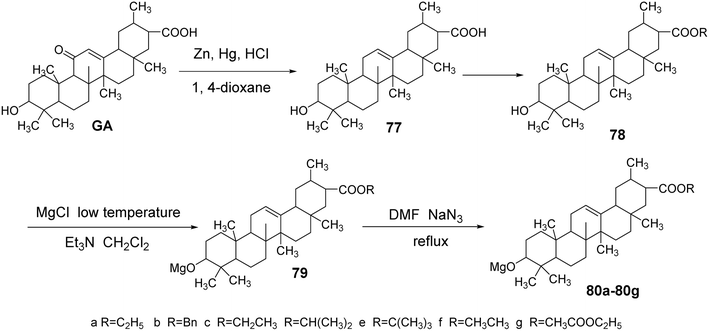 |
| | Fig. 23 Synthesis route of 3-azido glycyrrhizic acid derivatives. | |
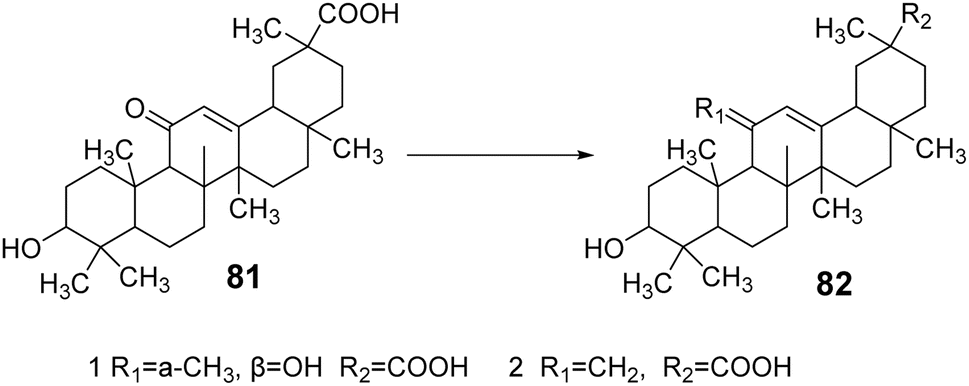
Su et al.64 modified the carbonyl group at the C-11 position and the carboxyl group at the C-30 position of GA, yielding two GA derivatives, 81 and 82. These derivatives exhibited strong inhibitory activity against 11β-HSD1, with IC50 values of 0.4 μmol L−1 and 1.1 μmol L−1, respectively. Notably, the presence of the carboxyl group at the C-30 position contributed to the inhibition of 11β-HSD1 activity (Fig. 24).
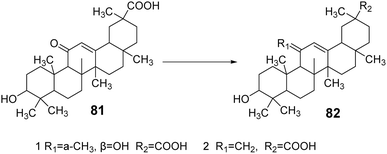 |
| | Fig. 24 The synthesis route of 11-position modified glycyrrhizic acid derivatives 81 and 82. | |
5 Betulinic acid: structural modifications and biological activity
5.1 Structural modifications of betulinic acid and antitumor activity
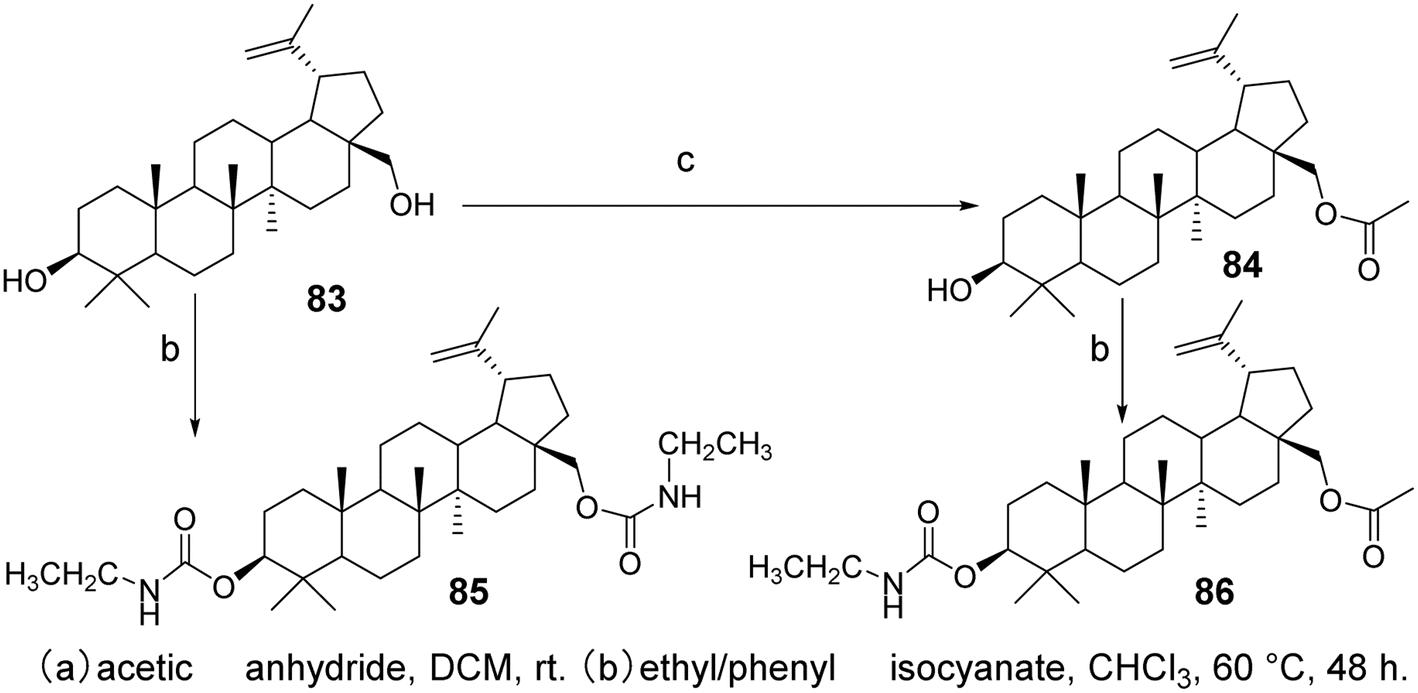
5.1.1 Structural modifications at C-3 and C-28 of betulinic acid. Research has indicated that derivatives with modifications at the C-3 and C-28 positions show promising potential. The introduction of amino acid conjugates at the C-28 position enhances solubility and cytotoxicity, while the incorporation of polar groups such as acids, amines, or hydroxyls increases biological activity.65 Hydroxylation at the C-3 position has yielded encouraging results against murine melanoma cells. Additionally, another chemical modification at C-3, namely dimethylsuccinoyl BA, has transformed BA from a proteasome activator to a proteasome inhibitor. BA derivatives substituted with new carbamate derivatives at C-3 and C-28 (compounds 85 and 86, Fig. 25) exhibit strong cytotoxicity and can induce apoptosis in lung tumor cell lines. Furthermore, compounds 85 and 86 demonstrate higher selectivity for tumor cells compared to normal cells.66
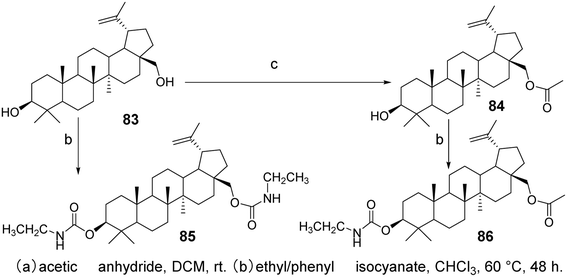 |
| | Fig. 25 Synthesis of BA derivatives 85 and 86. | |
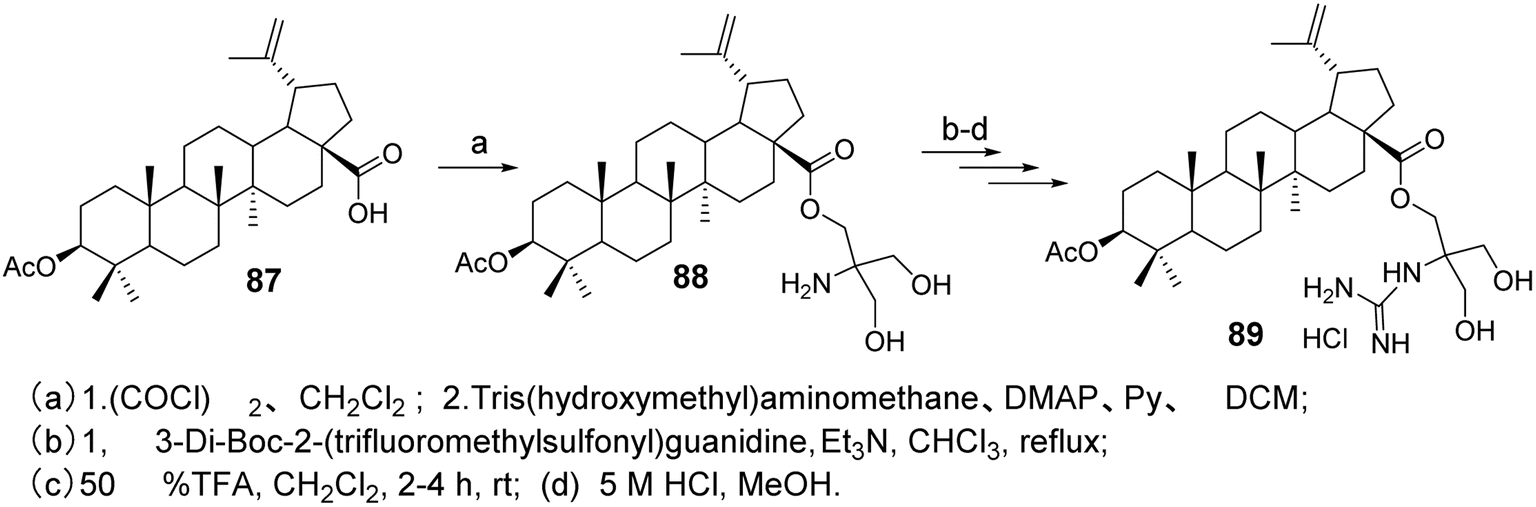
Spivak et al.33 designed and synthesized guanidine-containing BA derivatives (compound 89, Fig. 26), which exhibited higher cytotoxicity in Jurkat cells compared to the original triterpenic acid. Most of the tested guanidine derivatives showed higher IC50 values than amines, while demonstrating lower toxicity to human fibroblasts.
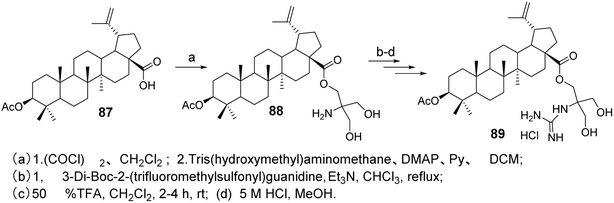 |
| | Fig. 26 Synthesis of BA derivative 89. | |
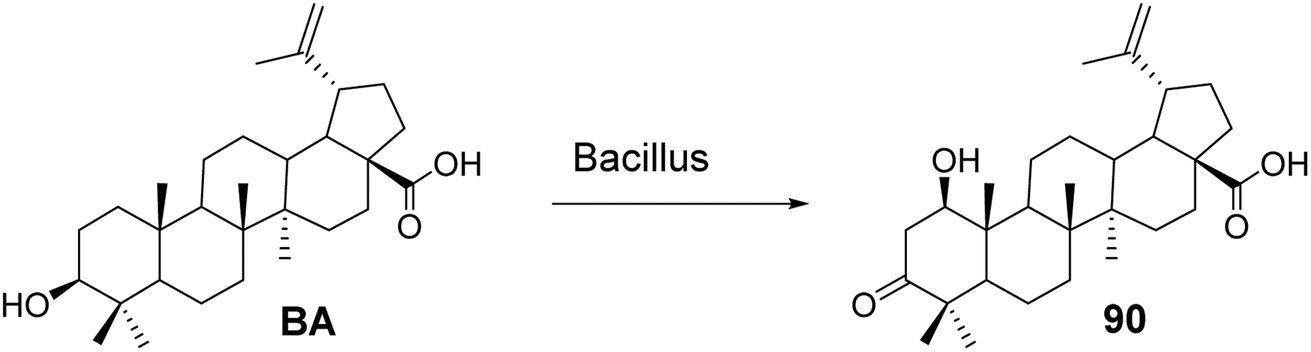
5.1.2 Structural modifications of betulinic acid in the A ring. Chatterjee et al.67 investigated the metabolic conversion of BA using Bacillus subtilis to biosynthesize derivative 90 (Fig. 27). Compared to the activity of BA (ED50 ranging from 2.1 μmol L−1 to 7.0 μmol L−1), compound 90 exhibited cytotoxic activity against human melanoma cell lines (Mel-1 and Mel-2) in the micromolar range (ED50 < 40 μmol L−1).
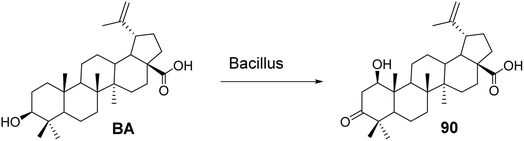 |
| | Fig. 27 Synthesis of BA derivative 90. | |
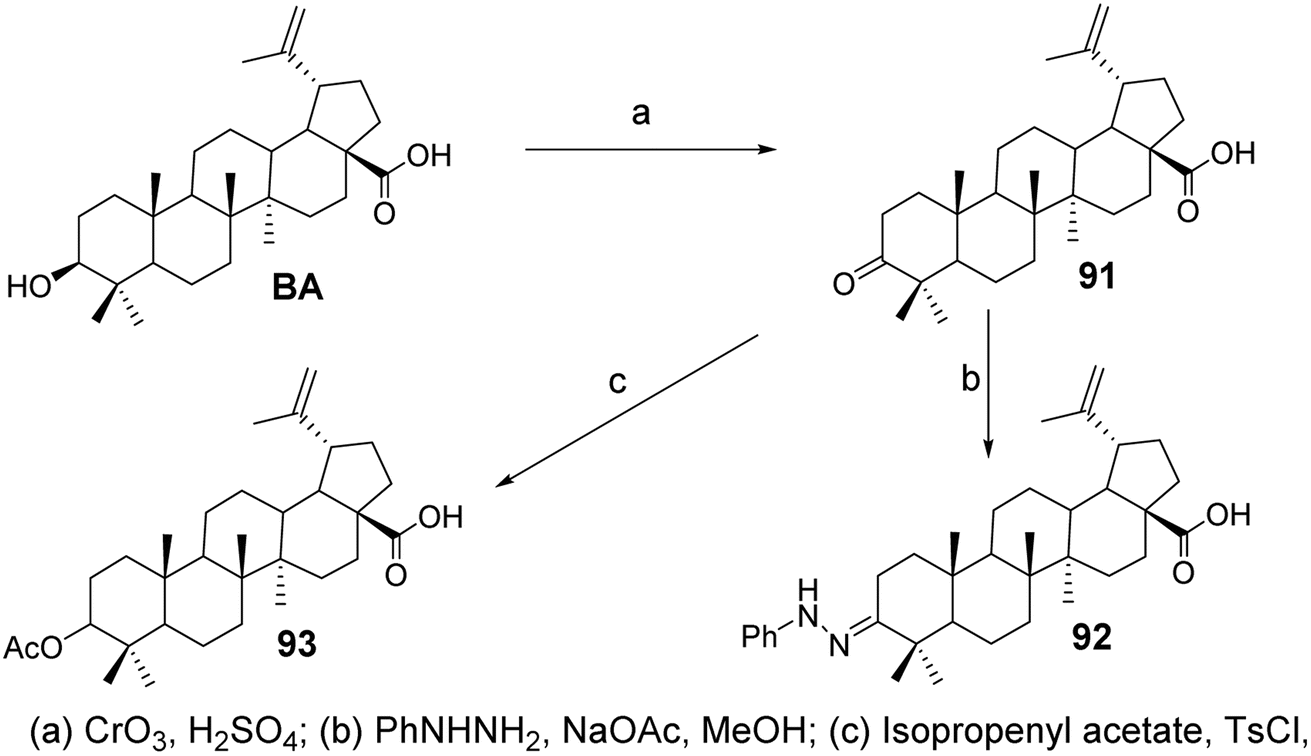
In the experiment by Mukherjee et al.,68 BA was oxidized with Jones reagent to form its 3-oxo derivative, BA ketone (compound 91, Fig. 28), which then reacted with phenylhydrazine and sodium acetate to yield the 3-phenylhydrazone (compound 92, Fig. 28). The results from anti-angiogenesis screening indicated that derivative 91 exhibited lower cytotoxicity compared to BA (with an IC50 of 4.4 μmol L−1 against the ECV304 endothelial cell line) and comparable low endothelial cell specificity (with an EC50 of 1.9–4.2 μmol L−1 across four cancer cell lines). Compound 91 was then reacted with isopropenyl acetate and p-toluenesulfonyl chloride under reflux for 9 hours, resulting in the enol acetate (compound 93, Fig. 28). Compound 93 was screened against a panel of five cancer cell lines and demonstrated low cytotoxic activity (IC50 ranging from 40 μmol L−1 to 100 μmol L−1).
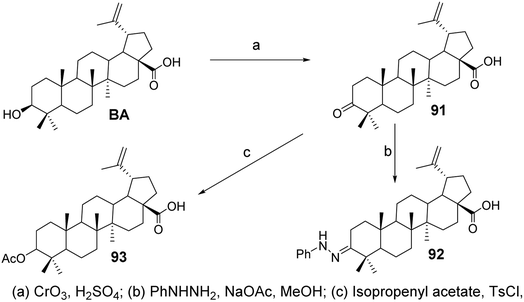 |
| | Fig. 28 Synthesis of BA derivatives 91 to 93. | |
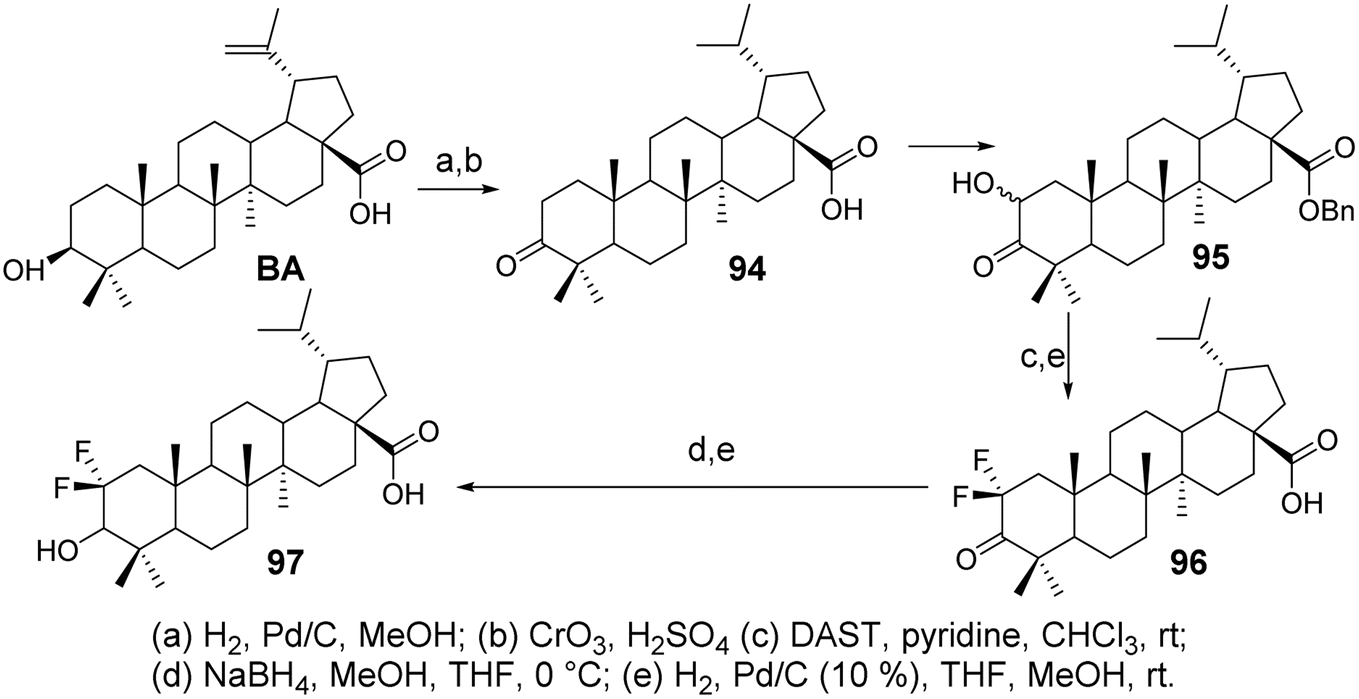
Li et al.69 synthesized difluorinated derivatives (compounds 96 and 97, Fig. 29) starting from dihydrobetulinic acid (DHBA) through a five-step or six-step reaction pathway. They investigated the cytotoxic activity of all compounds on the CCRF-CEM cell line. Compared to DHBA, the difluorinated derivatives 96 and 97 exhibited higher cytotoxicity, with IC50 values of 2.2 μmol L−1 and 4.0 μmol L−1, respectively.
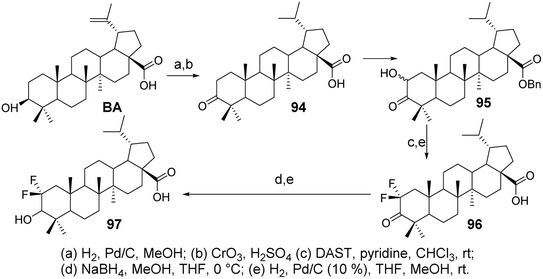 |
| | Fig. 29 Synthesis of BA derivatives 96 and 97. | |
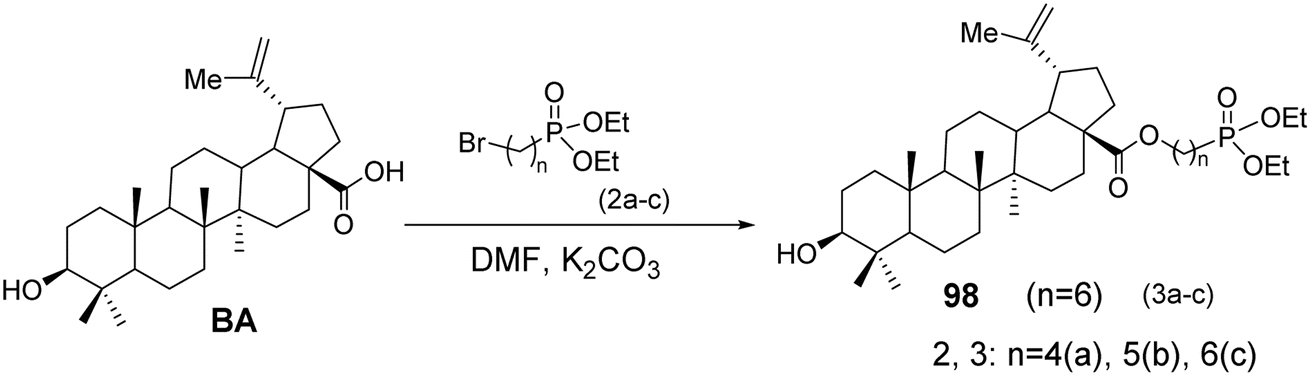
5.1.3 Structural modifications of betulinic acid at the C-3 and C-28 positions with phosphorus. Tsepaeva et al.70 used BA as the starting material and, in the presence of K2CO3, reacted it with phosphoric acid esters in DMF solvent to obtain derivative 98 (Fig. 30). The compounds containing triphenylphosphonium cations are promising ionic molecules for the targeted delivery of neutral bioactive compounds to the mitochondria of cancer cells, including anticancer agents that possess pro-oxidant properties and destabilize the mitochondrial membrane.
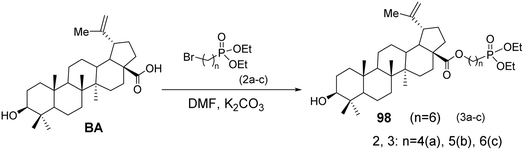 |
| | Fig. 30 Synthesis of BA derivative 98. | |
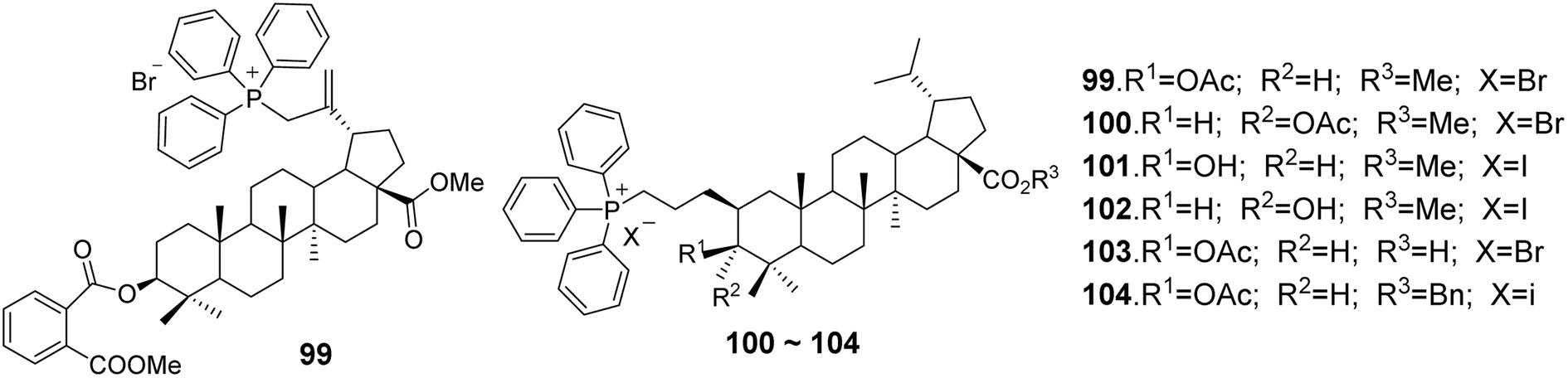
In the study by Spivak et al.,71 BA derivatives (compounds 99–104, Fig. 31) containing triphenylphosphonium cations featured various functional groups at the C-3 and C-28 positions (3β-OAc, 3α-OAc, 3β-OH, 3α-OH, 3β-O-phthalate, 28-COOMe, 28-COOH). Despite the differences in their chemical structures, the cytotoxic effects exhibited against EAC cells and mastocytoma P-815 cells were not significantly different, with IC50 values in Ehrlich cells ranging from 1.2 to 1.9 μmol L−1 and in P-815 cells from 0.9 to 1.1 μmol L−1. However, the benzyl ester of DHBA (compound 99) demonstrated reduced antitumor activity against both cell types, with IC50 values of 4.80 μmol L−1 for Ehrlich cells and 4.89 μmol L−1 for P-815 cells.
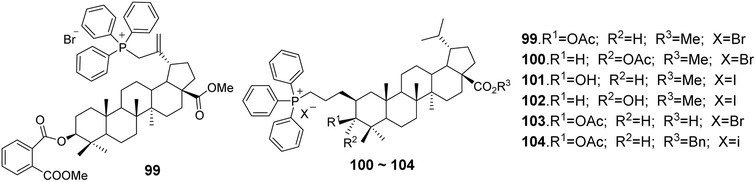 |
| | Fig. 31 The structures of BA derivatives 99 to 104. | |
5.2 Structural modifications of betulinic acid and their antiviral activity against HIV-1
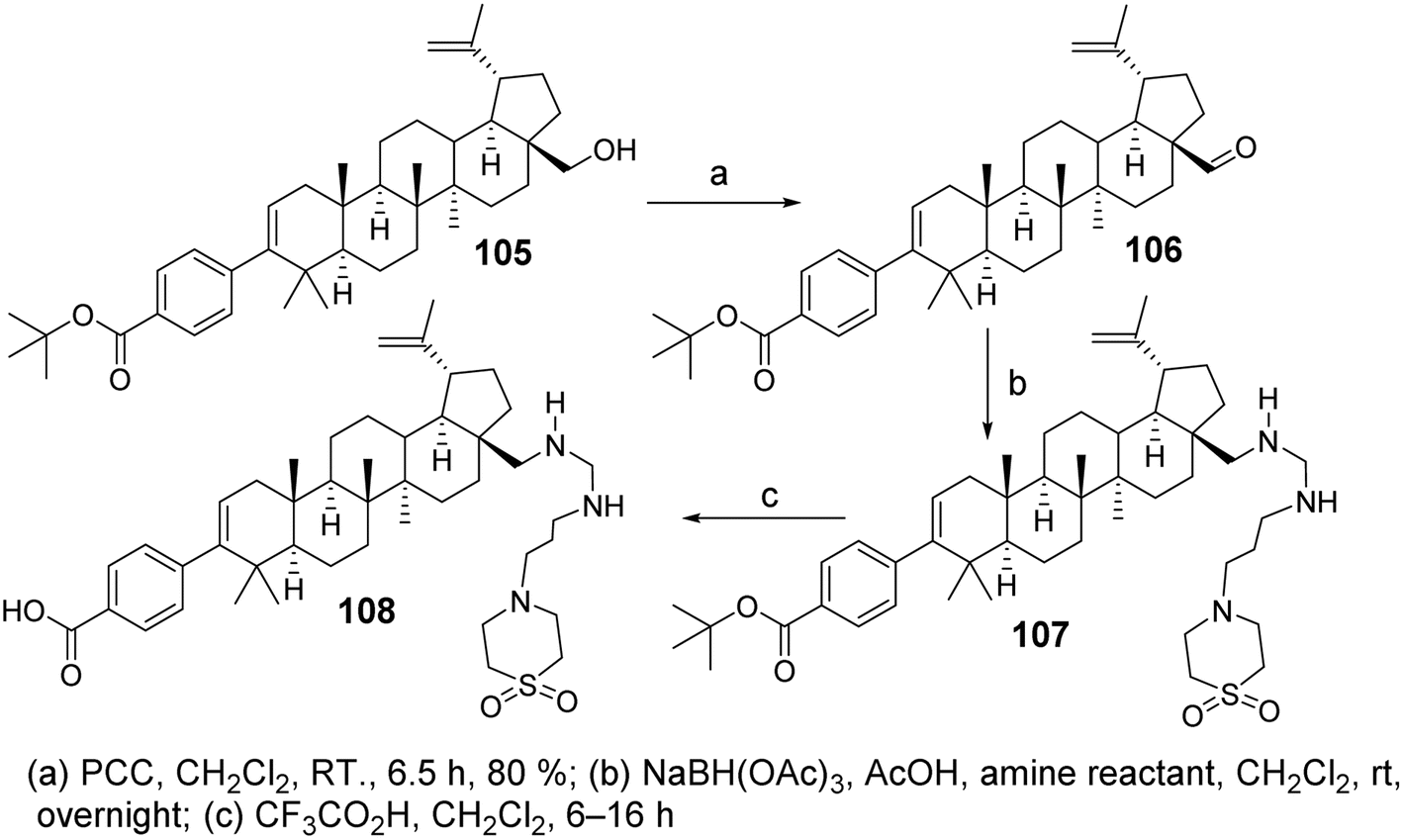
Chen et al.72 designed and synthesized a series of C-28 amine derivatives of BA (107) based on C-3 benzoyl-substituted BA derivatives (105, 106) as HIV-1 inhibitors. Compared to the C-28 amide series, the C-28 amine derivatives (107) showed enhanced inhibitory activity against HIV-1, specifically targeting Gag polyprotein polymorphisms. Their activity was further improved in the presence of human serum, even though basic amines generally exhibit lower plasma exposure after oral administration in rats. Among the C-28 amine series, the derivative with the best antiviral activity and acceptable pharmacokinetic properties (compound 108, Fig. 32) demonstrated a 2- to 4-fold improvement in antiviral potency compared to the C-28 amide. It exhibited low EC50 shifts in the presence of human serum or human serum albumin against the V370A and DV370 viral strains and showed promising efficacy against polymorphic variants T371A and V362I.
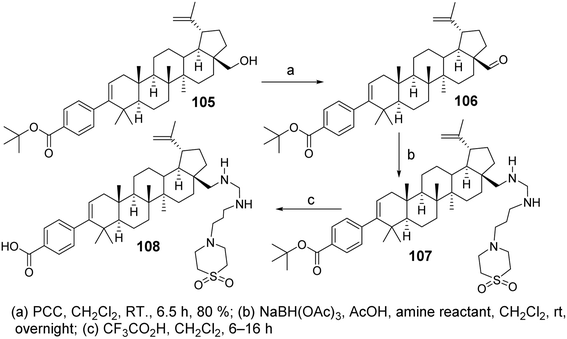 |
| | Fig. 32 Synthesis of BA derivative 108. | |
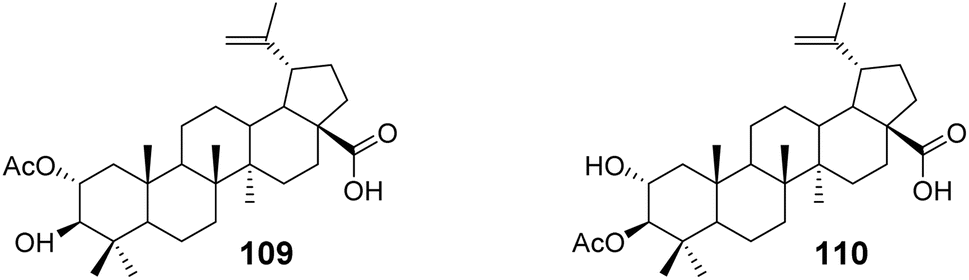
Reutrakul et al.73 isolated two partially acetylated 2α-hydroxy BA derivatives (compounds 109 and 110, Fig. 33) from the leaves, branches, and resin of Garcinia species. Both acetylated derivatives, 109 and 110, exhibited anti-HIV-1 activity in HIV-1 reverse transcriptase and syncytium formation assays. In the syncytium formation assay, compounds 109 and 110 demonstrated efficacy with IC50 values of 30.9 μmol L−1 and 38.6 μmol L−1, respectively, which is 2–3 times more potent than the activity of BA.
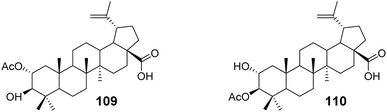 |
| | Fig. 33 Structure of BA derivatives 109 and 110. | |
6 Structural modifications of ursolic acid and its biological activities
Ursolic acid (UA), also known as UA or ursolic acid, is a representative compound of the ursane-type pentacyclic triterpenoids. It is widely distributed in many plants, including medicinal herbs and edible plants such as the leaves of Rosaceae plants like loquat and species from the Ericaceae family. Significant progress has been made in recent years in studying the antitumor activities of UA. Li et al.74 found that UA can exert anticancer effects by inhibiting cancer initiation and progression as well as DNA mutations. Young et al.75 reported that UA can induce the differentiation of teratoma stem cells, demonstrating its anti-carcinogenic properties, and has been recognized as one of the most promising natural compounds for cancer prevention. Additionally, UA shows potential therapeutic applications for prostate cancer by controlling the proliferation and apoptosis of PC-3 and LNCaP cells.76 UA exerts its anticancer activity by inhibiting kinases involved in cancer cell signaling, showing antitumor effects in various cell types.
6.1 Structural modifications of ursolic acid and antitumor activity
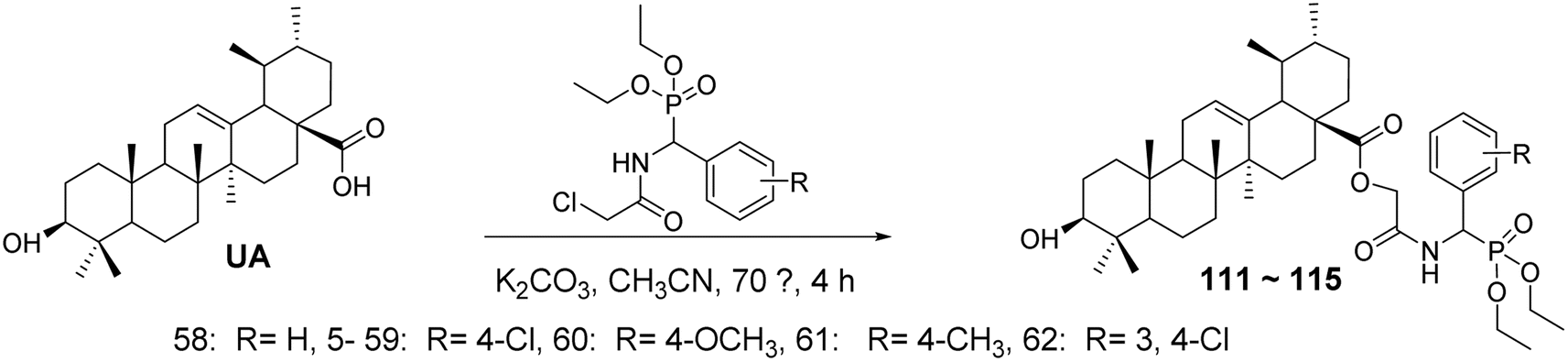
6.1.1 Structural modifications of ursolic acid at the C-3 and C-28 positions. Zhang Dajun et al.77 synthesized benzyl phosphonic acid diethyl ester acetamide derivatives of UA (compounds 111–115, Fig. 34) through a substitution reaction between UA and various substituted diethyl benzyl phosphonic acids. The in vitro anti-hepatocellular activity of the target compounds was evaluated using the MTT assay. All target compounds exhibited low cytotoxicity against normal human liver cells (L02), while compound 115 demonstrated significant antiproliferative activity against HepG2 cells, with an IC50 value of 2.69 μmol L−1, showing good selectivity between tumor cells and normal cells.
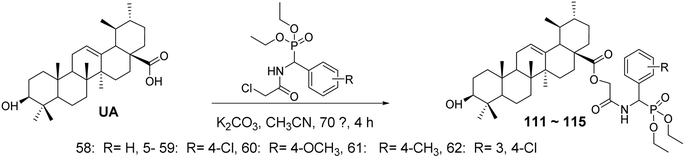 |
| | Fig. 34 Synthesis of UA derivatives 111–115. | |
Spivak et al.33 designed and synthesized guanidine-containing UA derivatives (compound 118, Fig. 35). Compared to the original UA, these guanidine-containing derivatives exhibited lower toxicity toward human fibroblasts while demonstrating higher cytotoxicity in Jurkat cells. Most of the guanidine derivatives tested showed higher IC50 values compared to their amine counterparts.
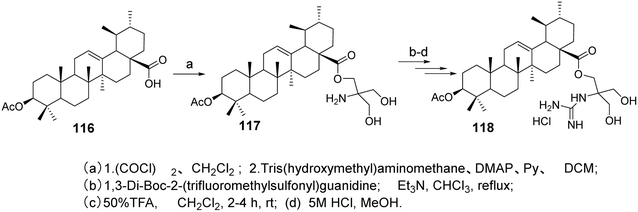 |
| | Fig. 35 Synthesis of UA derivative 118. | |
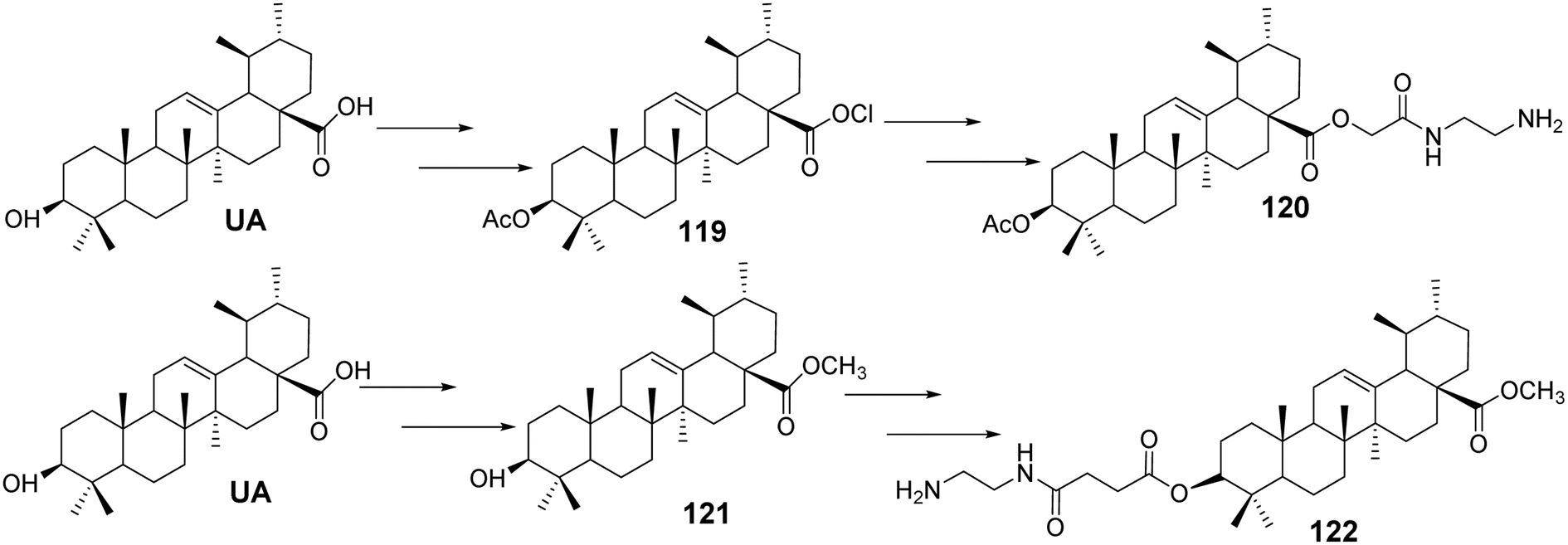
Tian et al.78 designed and synthesized a series of novel nitrogen-containing UA derivatives by introducing diamine fragments at the C-3 and C-28 positions. The derivatives with the highest activity (compounds 120 and 122, Fig. 36) were tested for their cytotoxicity against MCF-7, HeLa, and A549 cell lines. UA-4a and UA-8a showed significant inhibitory activity against all three human cancer cell lines, with IC50 values of less than 10 μmol L−1. Analyzing the antitumor activity of the synthesized compounds revealed no significant difference in activity between the introduction of diamine compounds at the C-3 and C-28 positions, both displaying strong antitumor properties.
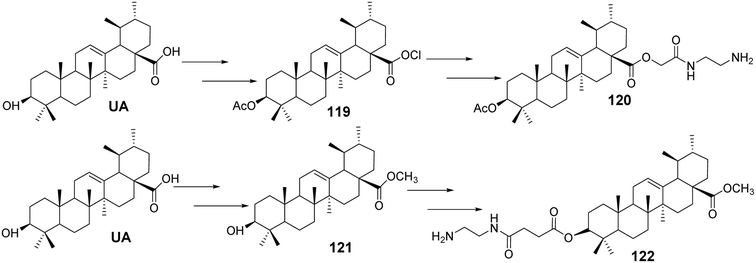 |
| | Fig. 36 Synthesis of UA derivatives 120 and 122. | |
Furthermore, analysis of the synthesized series suggested that the carbon chain length of the diamine compounds had little impact on antitumor activity, and no linear correlation was observed. However, a slight decrease in activity was noted when the carbon chain length reached six carbons. Considering factors such as ease of isolation, purification, yield, and activity during synthesis, the diamine compounds with a two-carbon chain were deemed the most valuable for further research. These compounds were characterized by low synthesis cost, easy purification, simple synthetic steps, and high cytotoxicity against cancer cell lines, making them promising candidates for further study as novel antitumor drugs. Additionally, they provide the potential for further salt formation modifications.
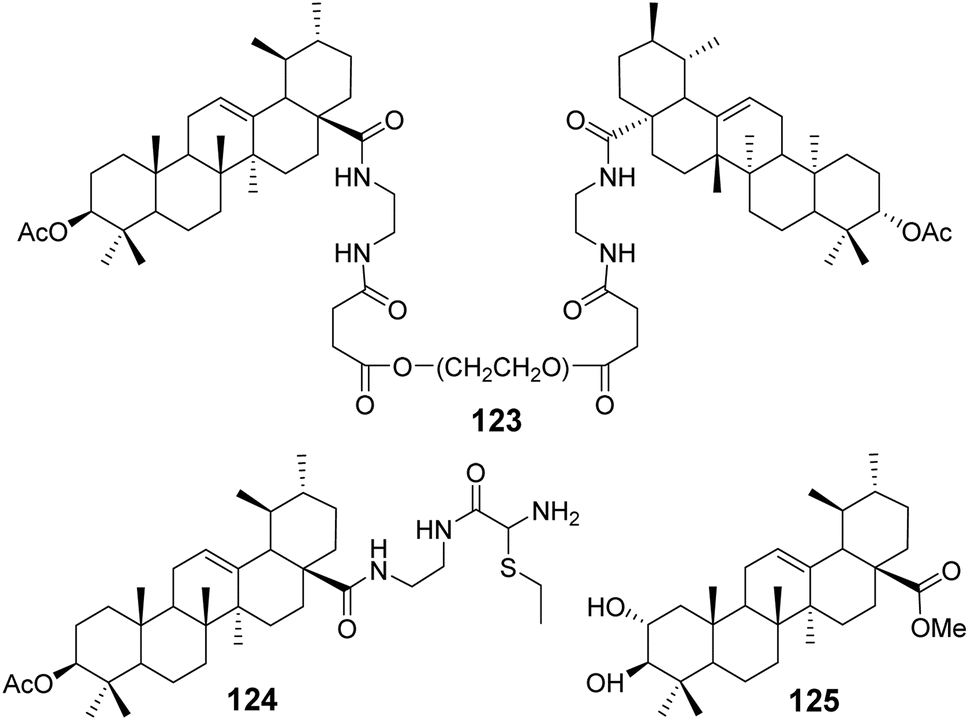
Bai et al.79–81 conducted comprehensive studies on modifications at the C-3 and C-28 positions of UA. The introduction of hydrophilic polyethylene glycol (PEG) significantly enhanced the solubility of the drug. Conjugating PEG at the C-28 position of the UA scaffold notably improved the in vitro antitumor activity. For example, compound 123 (Fig. 37) exhibited a solubility between 0.01 g mL−1 and 0.033 g mL−1 in water and demonstrated potent anticancer activity with IC50 values below 10 μmol L−1 against five tumor cell lines: HepG2, BGC-823, AGS, HT-29, and PC-3.
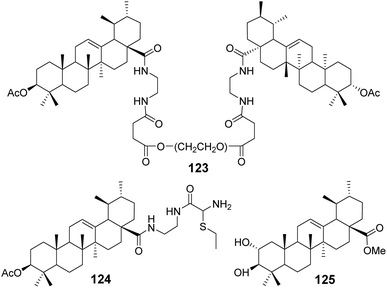 |
| | Fig. 37 Structure of UA derivatives 123 to 125. | |
Furthermore, the attachment of groups with varying electronegativities to UA effectively improved its water solubility, particularly the positively charged UA derivatives, which exhibited significantly enhanced anticancer activity. Compound 124 (Fig. 37) displayed a 70-fold increase in water solubility and an IC50 value below 10 μmol L−1 against PC-3 cancer cells. Additionally, it was found that introducing amino acid derivatives with primary amino groups at the C-28 position of UA substantially enhanced its anticancer activity compared to UA alone, with the mechanism of action involving the induction of apoptosis.
Zhao et al.82 carried out structural modifications on the A and C rings of UA, as exemplified by compound 125 (Fig. 37). These modifications involved multiple oxygen substitutions on the A ring and oxidation at the 11th position of the C ring. The resulting compounds exhibited cytotoxicity against cancer cells such as KB and PC-3, comparable to cisplatin, and demonstrated an inhibitory effect on α-glucosidase that was an order of magnitude stronger than that of acarbose.
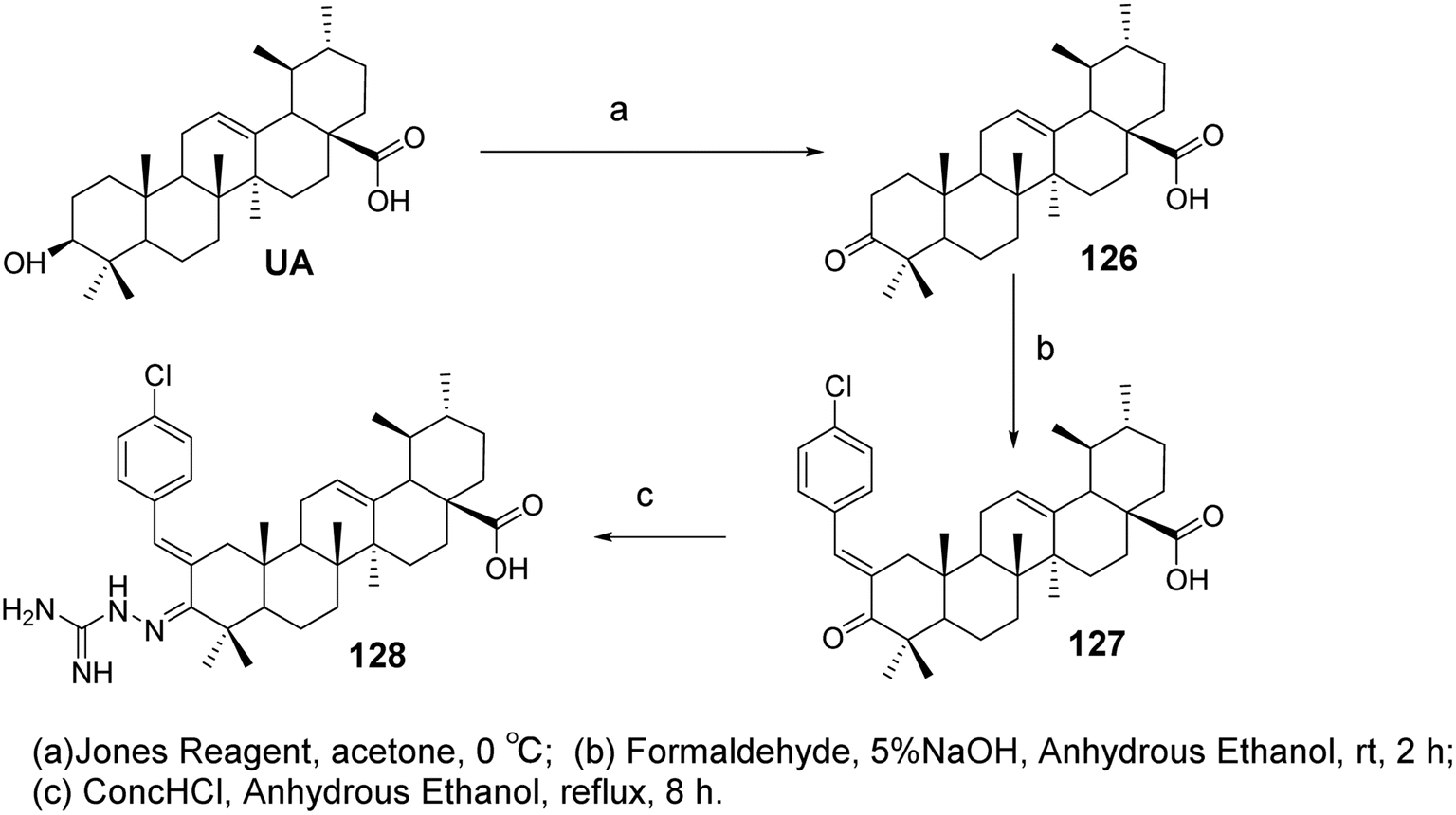
6.1.2 Structural modifications of ursolic acid at the C-2 and C-3 positions of the A ring. Research on the structural modification of UA at the C-2 and C-3 positions of the A ring has provided insights into enhancing its biological activity. Modifications in these positions have been shown to impact the compound's pharmacological properties, particularly its anticancer effects. Wu et al.83 designed and synthesized a series of UA derivatives containing an aminoguanidine structure and evaluated their activity as HIF-1α inhibitors and anticancer agents. Using a luciferase reporter gene assay based on Hep3B cells, most compounds exhibited good HIF-1α inhibitory activity. Among them, the most active derivative (compound 128, Fig. 38) demonstrated the strongest HIF-1α inhibition under hypoxic conditions, with an IC50 value of 4.0 μmol L−1, and showed no significant cytotoxicity in the tested cells. The mechanism of action may involve the downregulation of HIF-1α protein expression through inhibition of HIF-1α protein synthesis, as indicated by western blotting, RT-PCR assays, and cell aggregation experiments. This reduction in HIF-1α expression subsequently leads to decreased production of vascular endothelial growth factor (VEGF), thereby inhibiting cancer cell proliferation.
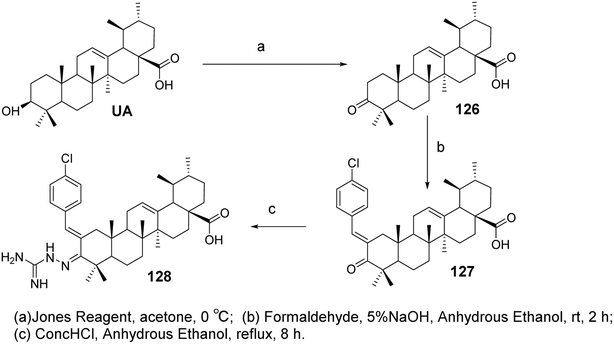 |
| | Fig. 38 Synthesis of UA derivative 128. | |
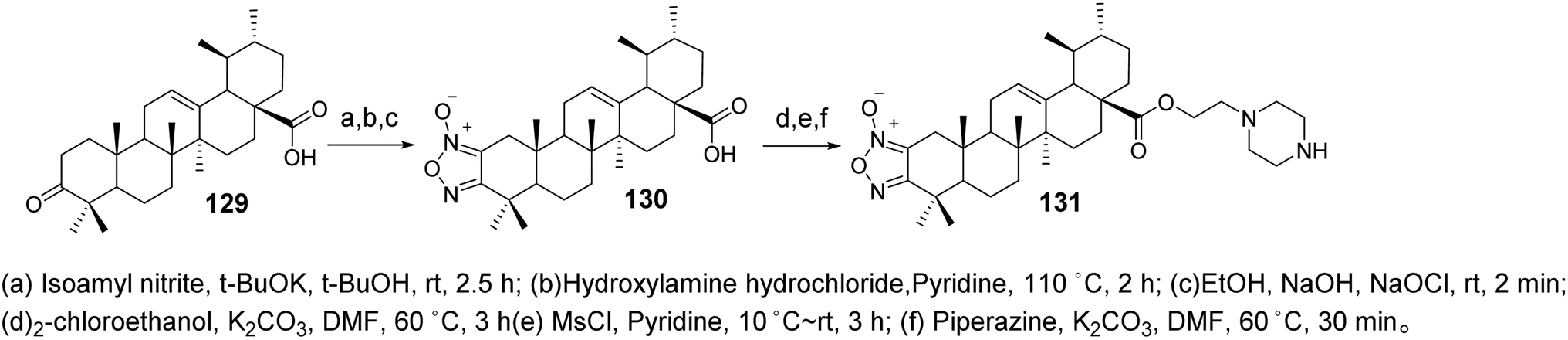
Wang et al.84 synthesized derivatives of UA with a furan moiety at the C-2 and C-3 positions based on the method of Khan (compound 130, Fig. 31). These derivatives were further modified by conjugation with ethyl-piperazine to obtain piperidine-containing derivatives (compound 131, Fig. 39). The anti-proliferative activities of the target compounds against the HeLa and MKN45 cancer cell lines were evaluated using the MTT assay, with cisplatin and UA used as positive controls. Derivative 130 exhibited significantly higher activity than UA, with an IC50 value below 10 μmol L−1, while derivative 131 displayed moderate to low activity against both cancer cell lines. Structure–activity relationship studies indicated that the introduction of thiazole on the A ring and the incorporation of triazole or tetrazole structures at C-28 had minimal impact on enhancing antitumor activity. However, the inclusion of piperazine or higher piperazine derivatives significantly improved the antitumor activity.
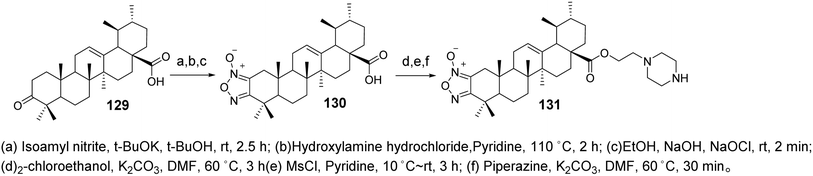 |
| | Fig. 39 Synthesis of UA derivatives 130 and 131. | |
6.2 Structural modifications of ursolic acid and antiviral activity
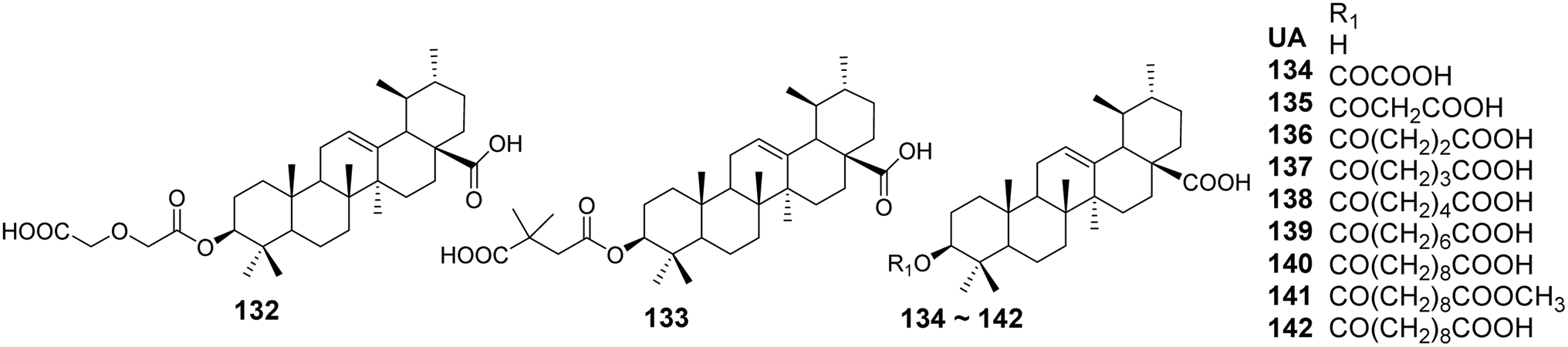
The structural modifications of UA have been investigated to enhance its antiviral activity. Various derivatives have been synthesized to explore their potential effects against different viral strains. Min et al.85 reported that the 3-O-acyl derivatives of ursolic acid (compounds 132–142, Fig. 40) exhibited significant inhibitory effects on HIV replication in human T lymphocytes (H9 cells), with IC50 values of 0.31 and 2.1 μmol L−1, respectively. Compared to the lead compound ursolic acid (IC50 value of 4.4 μmol L−1), these two compounds demonstrated improved anti-HIV activity and safety profiles. They further reported that a series of anhydrides were introduced at the C-3 position of UA to form corresponding dicarboxylic acid monoesters, evaluating the impact of the length of the anhydride chain on anti-HIV activity. Analysis of the IC50 values revealed that, when introducing anhydride chains containing up to six carbons, the anti-HIV activity of the compounds increased with the number of carbon atoms. However, when two methylene units were added, a slight decrease in activity was observed. Additionally, when the carboxyl groups of the anhydride formed carboxylic acid methyl esters, the anti-HIV activity of the compounds decreased, and in some cases, was completely lost.
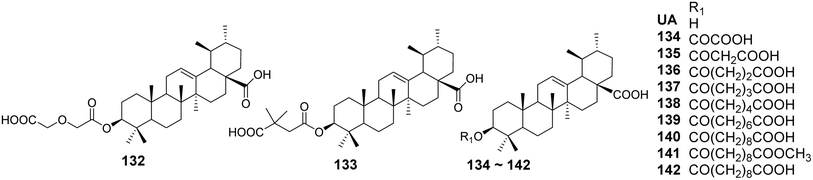 |
| | Fig. 40 Structure of UA derivatives 132 to 142. | |
6.3 Ursolic acid 3-β ester derivatives and corresponding activity evaluation
UA has garnered attention for its versatile pharmacological properties, and the development of 3-β ester derivatives is a promising approach to enhance its biological activity. Various studies have explored the synthesis of these ester derivatives and evaluated their potential in different therapeutic areas.
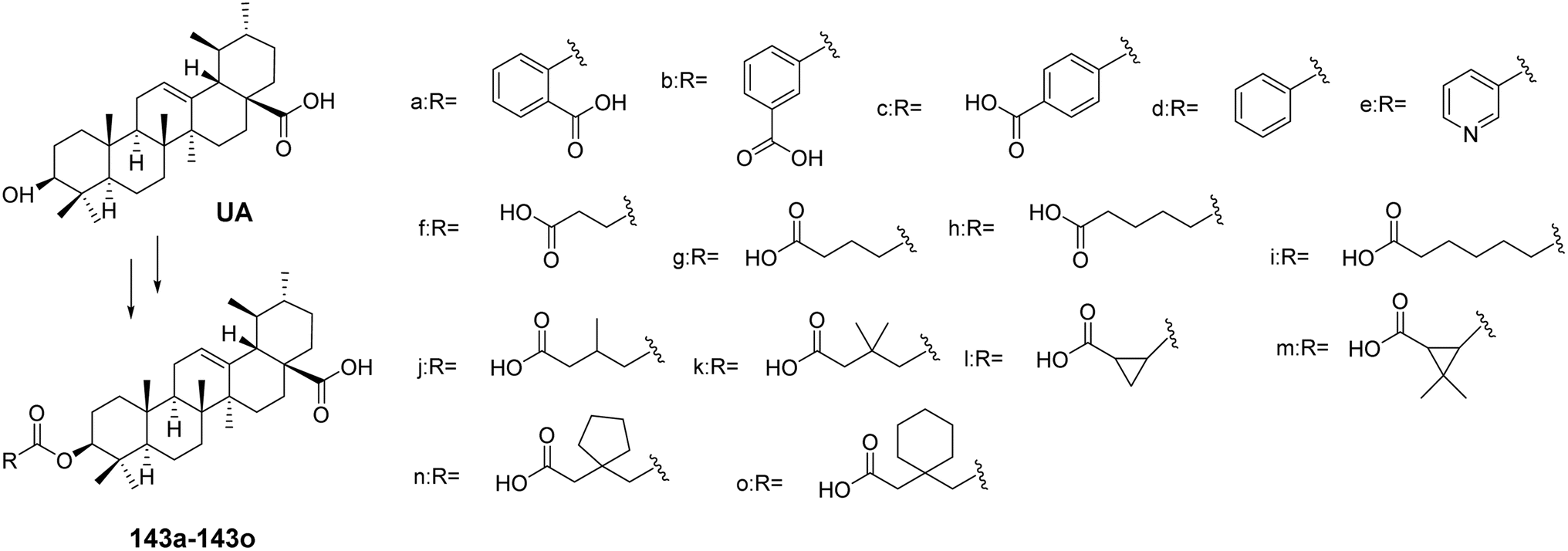
For instance, Chen et al.86 synthesized a series of UA 3-β ester derivatives (143a–143o, Fig. 41). Activity assays indicated that compounds 143a and 143b, which contain phthalate esters or isophthalate esters, exhibited moderate inhibitory effects on cholesterol ester transfer protein (CETP), while the inhibitory effect of derivative 143c, containing terephthalate esters, was completely lost. Further comparison of derivatives 143b, 143d, and 143e revealed that the carboxyl group on the backbone chain is essential for enhancing inhibitory activity. In contrast to derivative 143g, the derivatives containing succinate esters (143j and 143k) lost their inhibitory effects on CETP, possibly due to steric hindrance from the methyl group. Derivative 143l, which contains 1,2-cyclopropanedicarboxylate esters, exhibited the strongest CETP inhibitory activity, with an IC50 of 2.4 μmol L−1. The presence of a carboxyl group at the end of the backbone chain and the length of the backbone chain are critical factors for maintaining or enhancing CETP inhibitory activity. These types of 3-β ester triterpenoid derivatives (e.g., derivative 143l) hold promise as lead compounds with potent CETP enzyme inhibitory activity. Xu et al.87 found that glycosylated corosolic acid derivatives possess certain α-glucosidase inhibitory activity and improved water solubility, which is associated with the presence of hydroxyl groups at the C-2 and C-3 positions. This activity is also closely related to the substituents at the C-28 position.88,89
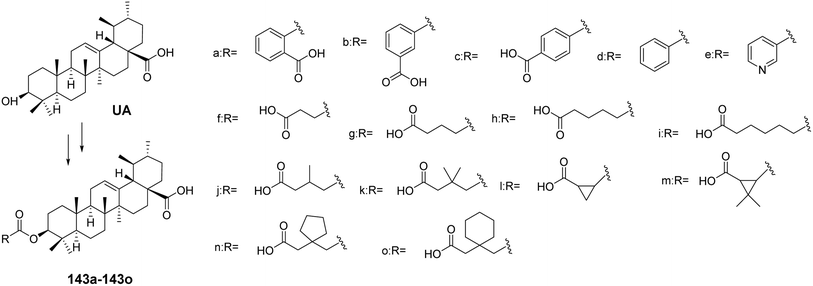 |
| | Fig. 41 Synthesis of ursolic acid 3-β ester derivatives (143a–143o). | |
7 Structural modifications and pharmacological activities of celastrol
Celastrol, also known as tripterine, is a triterpenoid compound belonging to the oleanane-type pentacyclic triterpenoids. It was first extracted from the traditional Chinese medicinal herb Tripterygium wilfordii by the renowned organic chemist Zhao Chenggu in 1936.90 As the first active compound isolated from the root bark of Tripterygium wilfordii, celastrol is the most abundant bioactive component in this plant and has been demonstrated to exhibit potent anticancer activity.91 Compared to some conventional chemotherapeutic agents, celastrol shows similar or even superior anticancer effects, particularly against multidrug-resistant cancer cells. In 2007, it was listed by Cell magazine as one of the five natural products most likely to be developed into modern pharmaceuticals.92 Despite extensive research into its potential anticancer properties, its further application remains limited due to challenges such as poor water solubility, low bioavailability, and high toxicity.93
7.1 Structural modifications of celastrol at the C-29 position and its anticancer activity
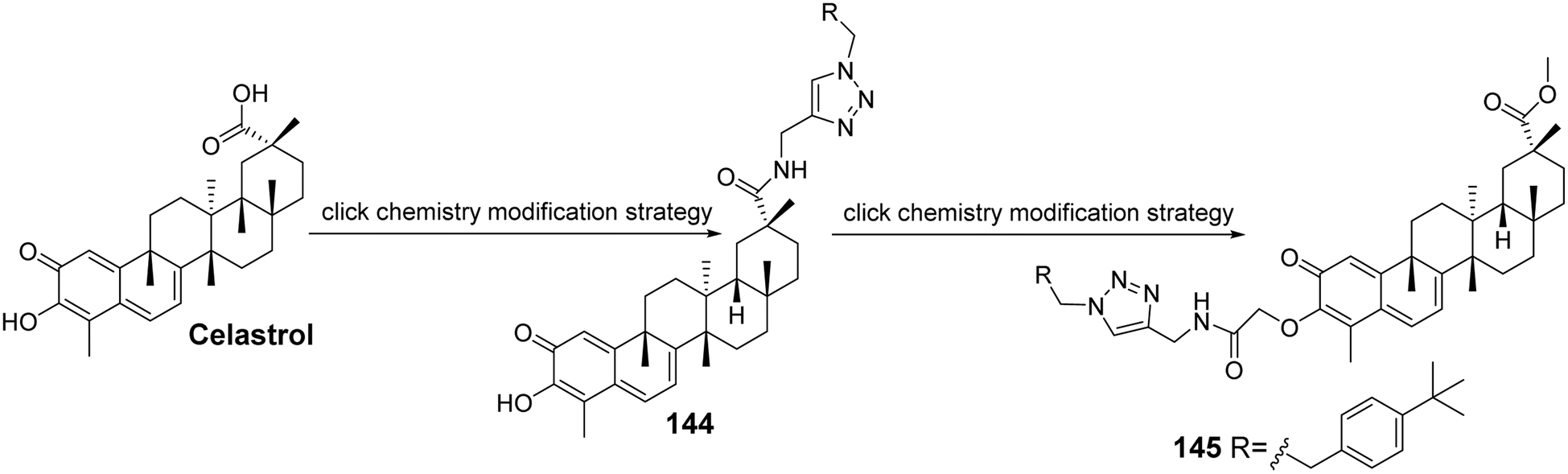
Feng et al.94 modified the hydroxyl group at C-3 and the carboxyl group at C-29 of celastrol to synthesize a series of 1,2,3-triazole derivatives of celastrol (compound 145, Fig. 42). They evaluated the anti-glioma activity of these derivatives in vitro against four human glioma cell lines (A172, LN229, U87, and U251). Structure–activity relationship studies indicated that the modifications at C-3 and C-29 significantly enhanced the anti-proliferative activity compared to celastrol, with the C-29 carboxyl group derivatives exhibiting stronger anti-proliferative activity than the C-3 hydroxyl group derivatives across the four human glioma cell lines. Among these, compound 145 demonstrated the highest anti-proliferative activity in the U251 cell line, with an IC50 of 0.94 μmol L−1. Further studies revealed that its effective anti-glioma activity is associated with the activation of the receptor–interacting protein 1/receptor–interacting protein 3/mixed lineage kinase domain-like protein (RIPI/RIP3/MLKL) pathway.
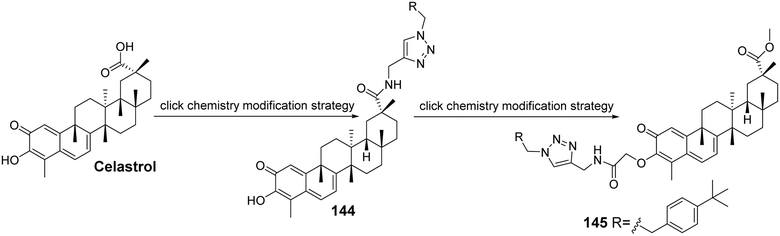 |
| | Fig. 42 Synthesis of celastrol derivative 145. | |
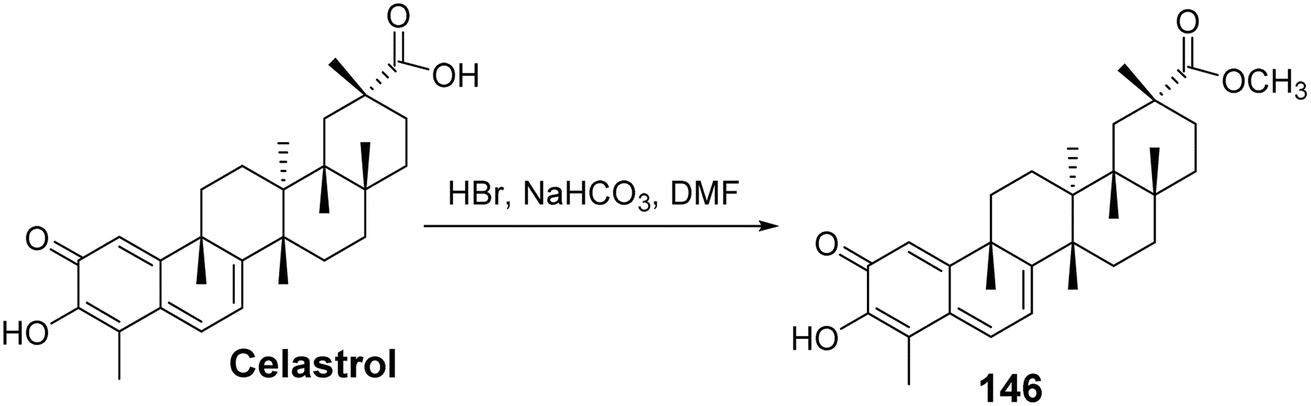
Ferulic acid (FA) is a type of phenolic acid that has been demonstrated to possess various pharmacological activities, including antioxidant, anti-diabetic, anti-inflammatory, and neuroprotective effects. FA and its derivatives have also shown significant anticancer efficacy both in vitro and in vivo.95 Coghi et al.96 designed and synthesized a series of novel hybrids of celastrol and ferulic acid, evaluating their antitumor activity. Structure–activity relationship (SAR) studies indicated that different linkers influenced the pharmacological effects, with a two-carbon linker exhibiting stronger anticancer capabilities compared to a three-carbon linker. Moreover, when a methoxy group was substituted on the FA moiety, the hybrids displayed improved efficacy. Among these, the most active derivative (compound 146, Fig. 43) exhibited the strongest inhibitory capacity. Mechanistic studies revealed that compound 146 could disrupt the heat shock protein Hsp90–Cdc37 complex while also inducing abnormal regulation of heat shock protein (Hsp90) receptors (p-Akt and Cdk4), leading to concentration-dependent cell cycle arrest in the G0/G1 phase.
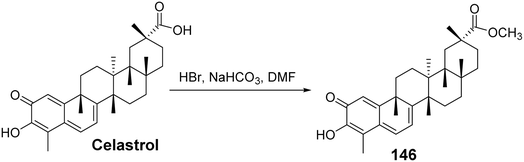 |
| | Fig. 43 Synthesis of celastrol derivative 146. | |
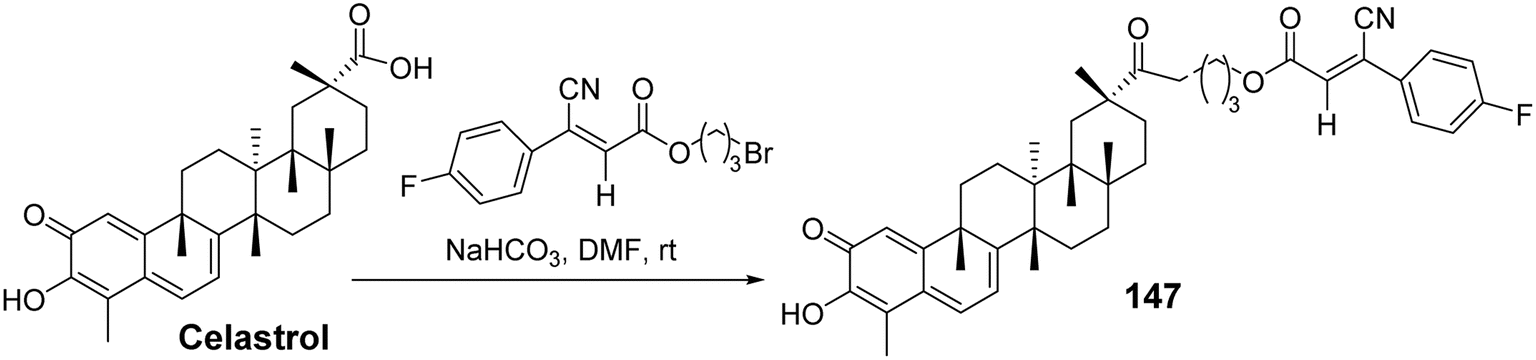
Disrupting the interaction between Hsp90 and Cdc37 has emerged as a promising strategy for investigating the antitumor activity of celastrol.97 To further explore the effects of celastrol C-29 derivatives on the Hsp90–Cdc37 complex, Li et al.98 introduced several lipophilic fragments into celastrol C-29, synthesizing 48 new celastrol derivatives and testing their effects on the Hsp90–Cdc37 complex and antitumor cell proliferation activity. SAR studies revealed that the length of the linker significantly influenced the activity of the compounds. Using two-carbon and three-carbon linkers resulted in much higher inhibition rates on tumor cells compared to four-carbon linkers, with 8-cyanide groups exhibiting better efficacy than α-hydroxyl groups. Among the three-amine substitutions, introducing electron-donating groups onto the phenyl ring enhanced the antiproliferative activity of the compounds. One particularly active derivative (compound 147, Fig. 44) demonstrated excellent antiproliferative activity against relevant cancer cells (IC50: 0.41–0.94 μmol L−1). Further mechanistic studies indicated that compound 147 could reduce the expression of the Hsp90–Cdc37 complex in A549 cells and form a complex with Cdc37 in tumor cells, thereby disrupting the Hsp90–Cdc37 interaction.
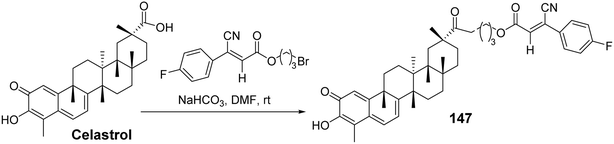 |
| | Fig. 44 Synthesis of celastrol derivative 147. | |
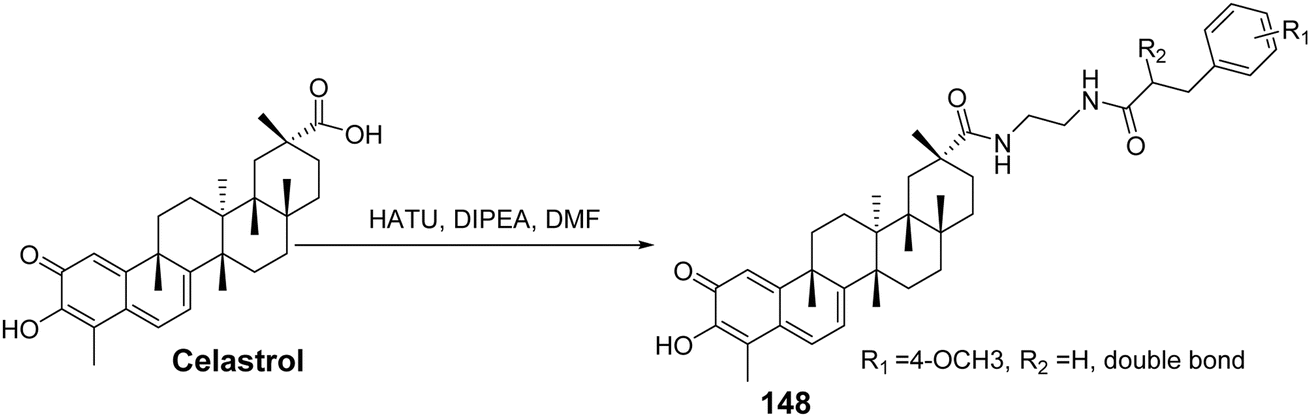
CSCs are associated with cancer progression, tumor recurrence, metastasis, and chemotherapy resistance. To develop effective and selective anti-CSC agents, Li et al.99 synthesized two series of celastrol derivatives with cinnamoylamine linkers, utilizing ethylenediamine and piperazine as connecting chains. Their antitumor activity against ovarian cancer was evaluated. SAR analysis revealed that most compounds exhibited stronger antiproliferative activity compared to celastrol. Additionally, no significant differences in activity were observed between the two series of compounds with different linker chains. However, the celastrol derivative bearing a 3,4,5-trimethoxy cinnamoylamine side chain (compound 148, Fig. 45) demonstrated the strongest antiproliferative effect against ovarian cancer cells (IC50 = 0.6 μmol L−1). Further pharmacological tests indicated that compound 148 significantly inhibited the colony formation ability of tumor cells, reduced the number of tumor spheres, and decreased the percentage of ovarian CSCs.
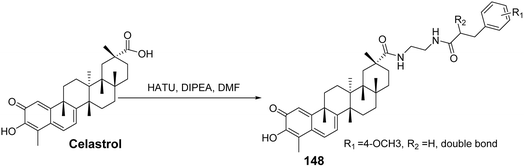 |
| | Fig. 45 Synthesis of celastrol derivative 148. | |
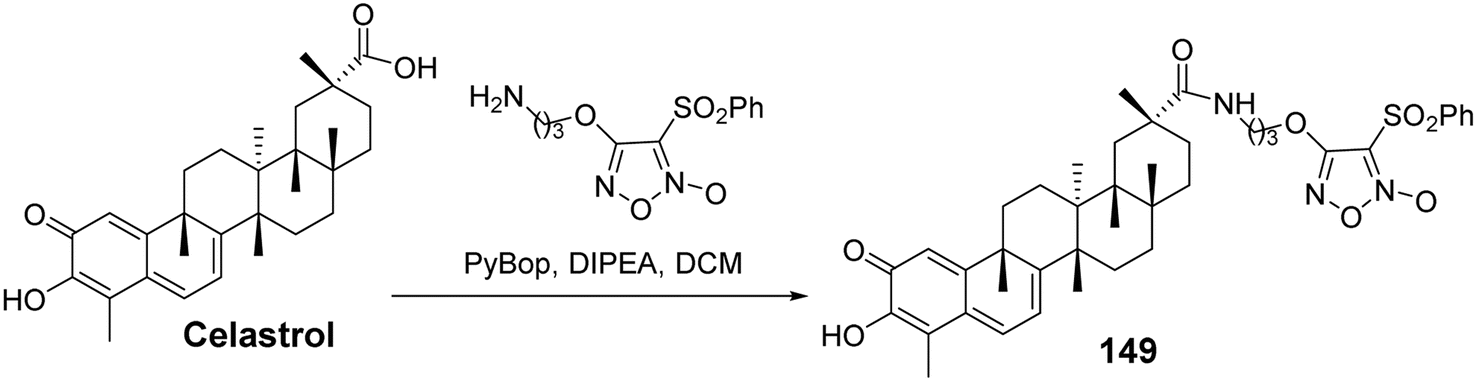
Nitric oxide (NO) is a key signaling and effector molecule in tumor development. Generally, high concentrations of NO can induce apoptosis in tumor cells, while low concentrations typically offer cellular protection. Based on this, Tang et al.100 synthesized a series of celastrol nitric oxide donor drugs and evaluated their NO release capabilities and antiproliferative activities. The results showed that the most active derivative (compound 149, Fig. 46) released the highest levels of NO in vitro and exhibited greater antiproliferative activity compared to other compounds (A549: IC50 = 0.48 μmol L−1). SAR analysis indicated that the C-29 amide bond was beneficial for enhancing antiproliferative activity. Furthermore, furoxan nitrogen oxides demonstrated stronger antiproliferative activity as NO-releasing components than nitrate esters. Additional pharmacological studies revealed that compound 149 could induce dysregulation of the Hsp90 receptor, apoptosis, and cell cycle arrest at the G0/G1 phase. These findings suggest that the inhibition of Hsp90 and NO release have a synergistic effect in tumor cells, providing new directions for the discovery of antitumor drugs.
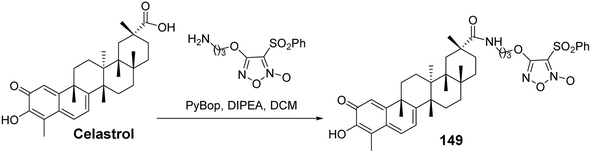 |
| | Fig. 46 Synthesis of celastrol derivative 149. | |
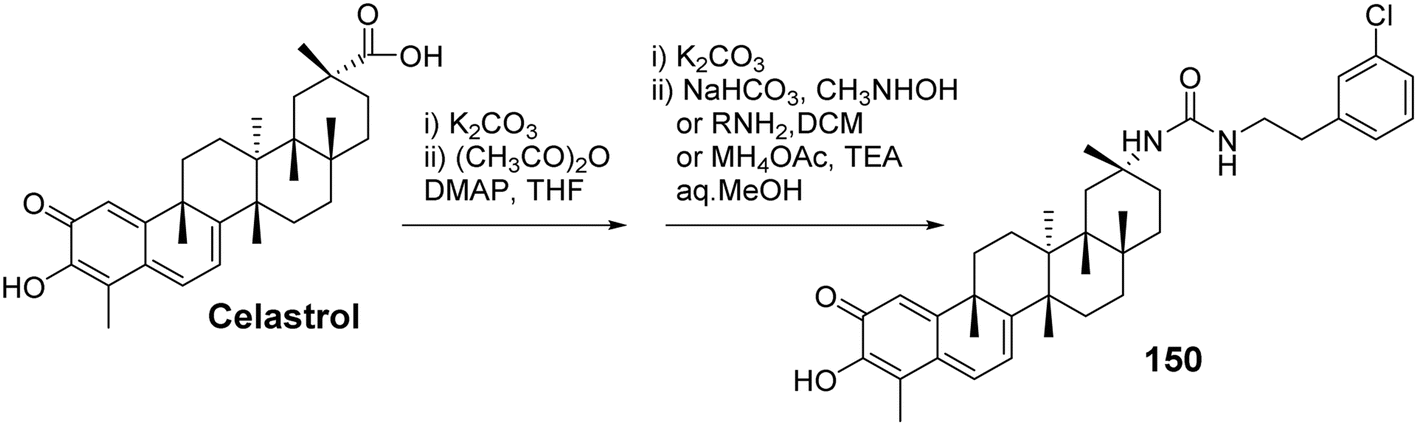
7.2 A/B ring-modified celastrol derivatives and their anticancer activity
In the exploration of the anticancer activity of celastrol, the modification of its A/B rings has become an important strategy. By chemically modifying the A/B rings of celastrol, it is possible to enhance its biological activity and improve its drug properties. Tang et al.101 synthesized a series of derivatives of celastrol with urea moieties as linkers at the C-29 position and modifications at the A/B rings, followed by testing their antitumor cell proliferation activity. SAR analysis revealed that the C-29 carboxy urea derivatives exhibited enhanced cytotoxicity against tumor cell lines compared to celastrol. However, modifications to the A/B rings resulted in diminished antiproliferative activity of the derivatives. This suggests that the intact quinone methide structure of the A/B rings and the reactive Michael addition site at position 6 may be essential groups for the anticancer effects of celastrol. Among these, the most active derivative (compound 150, Fig. 47) demonstrated superior antiproliferative activity with greater selectivity. Preliminary studies on its mechanism of action indicated that compound 150 induces apoptosis through the activation of cysteine protease-8 (caspase-8) and cysteine protease-3 (caspase-3), as well as the cleavage of poly (ADP-ribose) polymerase (PARP) via an extrinsic pathway. Additionally, it induced downregulation of p53 in the SKOV-3 cell line, which is a p53-mutant cell line.
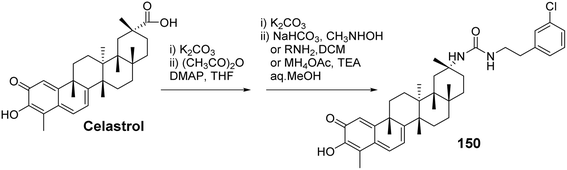 |
| | Fig. 47 Synthesis of celastrol derivative 150. | |
7.3 C-6 modified celastrol derivatives and anticancer activity
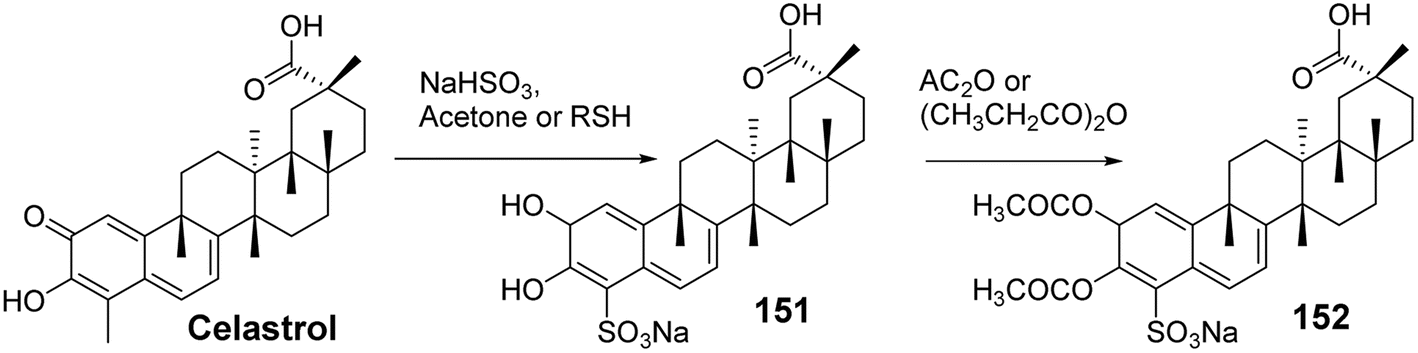
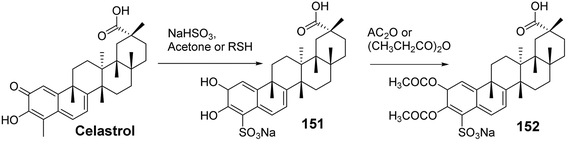
To explore the impact of A-ring and C-6 modifications on the antitumor activity of celastrol, Tang et al.102 synthesized a series of C-6 sulfonated, thiolated, and carbon-modified celastrol derivatives via a Michael addition reaction. The cytotoxicity of these derivatives was evaluated in human cancer cell lines (BGC-823, H4, Bel7402, H522, Colo205, HepG2, and MDA-MB468). SAR analysis showed that C-6 sulfonated and thiolated derivatives exhibited stronger cytotoxicity compared to carbon-modified derivatives at the C-6 position, with 3-hydroxy acetylation proving more effective than propionylation. Among these derivatives, compound 152 (Fig. 48) demonstrated the most potent bioactivity in the Colo205 cell line (IC50 = 0.06 μmol L−1). Further experimental results revealed that, in nude mice bearing human colorectal cancer xenografts, compound 152 effectively inhibited the growth of Colo205 xenografts in vivo and exhibited better safety profiles compared to celastrol.
 |
| | Fig. 48 Synthesis of celastrol derivative 152. | |
8 Conclusion
Natural products, as an important source of lead compounds for drug development, play a crucial role in pharmaceutical research. However, their inherent limitations, such as poor water solubility, low bioavailability, and significant toxicity, have led researchers to focus increasingly on structural modifications to obtain more efficient and less toxic new drugs. Among these, pentacyclic triterpenes have garnered growing attention due to their diverse biological activities, with research on these compounds becoming progressively more in-depth in recent years. Despite the structural diversity of pentacyclic triterpenes, their limited content in plants, complex extraction processes, and our insufficient understanding of their biosynthetic pathways, effective chemical modifications offer promising opportunities for their development. Structural modifications, particularly aimed at their hydrophobic framework, are essential for further exploration.
OA, BA and UA are pentacyclic triterpenes widely distributed in nature, known for their anti-inflammatory, antiviral, and anticancer properties, showcasing significant potential for therapeutic applications. However, due to their poor water solubility and low bioavailability, structural modifications can effectively improve these properties by enhancing water solubility, increasing bioactivity, and reducing toxic side effects. Although numerous bioactive derivatives have been developed, few have advanced to clinical application. Research into the structural modification of OA and BA has yielded significant progress in the field of anticancer therapy, identifying key targets and mechanisms of action. SAR studies have provided insights that will guide the design of more potent pentacyclic triterpene derivatives in the future.103–106 Additionally, as research continues, UA has also become a focal point for anticancer drug development through structural modification. The continued exploration of OA, BA, and UA derivatives for the development of highly effective, low-toxicity anticancer drugs remains a promising area of research.
The structural modification of celastrol has mainly focused on the C-29 position, with relatively little research on the 3-OH group and A/B rings. However, the high reactivity of the A/B rings may be crucial to celastrol's pharmacological effects. Therefore, further studies on the modification of the A/B rings are essential, and even ring-opening reactions could be explored to generate celastrol derivatives with reduced toxicity and enhanced efficacy. This could also lead to a more comprehensive understanding of the SAR of celastrol. Moreover, biotransformation could provide novel approaches for the structural modification of celastrol, potentially yielding derivatives that are difficult to obtain through chemical methods, thus offering new directions for celastrol's modification.
GA is another pentacyclic triterpene with broad biological activities, particularly in the fields of anti-inflammatory and anticancer research, showcasing immense potential for therapeutic applications. Similar to OA, BA, and UA, GA's poor water solubility and high toxicity have limited its clinical application. Recent studies on GA's structural modifications have led to the discovery of derivatives with promising anticancer activities, along with initial insights into their mechanisms of action, laying the groundwork for the development of GA as an anticancer agent. Further optimization of GA's structural modifications to enhance bioactivity and reduce toxicity will facilitate the clinical translation of GA and its derivatives.
In conclusion, structural modifications of natural products hold great potential for the development of novel anticancer drugs. The modification studies on pentacyclic triterpenes, such as OA, BA, UA, and celastrol, not only enrich the theoretical foundation of chemistry and pharmacology but also provide new avenues for drug discovery.
Data availability
No new data were created or analyzed in this study. This article is a review of existing literature, and all data supporting the findings are derived from previously published sources, which are appropriately cited in the manuscript. Should any further clarification on data sources be needed, the corresponding author can provide additional information upon request.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
This research was funded by Guiyang City Science and Technology Plan Project (No. [2024]2-37), the Guizhou Provincial Basic Research Program (Natural Science) (No. ZK[2023] general 404 and ZK [2024] general 370), the Research Project on Traditional Chinese Medicine and Ethnic Medicine Science and Technology of Guizhou Provincial Administration of Traditional Chinese Medicine (QZYY-2024-130), High-level Innovative Talents of Guizhou Province (QianKeHe platform talents-GCC [2023] 047), Guizhou Province Innovation Talent Team Construction for the Efficient and Comprehensive Utilization of Unique Food and Medicinal Materials (Platform Talent-CXTD[2023]020), Natural Science Research Project of Guizhou Department of Education (Qianjiaoji [2023]069) and the Projects of Guizhou Province (Qian Jiao He KY Zi [2022]253, gzwkj2022-232 and gzwkj2022-467).
References
- D. Wang, B. Wang and Y. Liu, et al., Synthetic pathway and fermentation process optimization of oleanolic acid in yeast cell factory, China J. Chin. Mater. Med., 2014, 39(14), 2640–2645 CAS.
- J. Pollier and A. Goossens, Oleanolic acid, Phytochemistry, 2012, 77, 10–15 CrossRef CAS PubMed.
- J. Wei, H. Liu and M. Liu, et al., Oleanolic acid potentiates the antitumor activity of 5-fluorouracil in pancreatic cancer cells, Oncol. Rep., 2012, 28, 1339–1345 CrossRef CAS.
- M. Zhu, C. Wang and W. Sun, et al., Boosting 11-oxo-beta-amyrin and glycyrrhetinic acid synthesis in Saccharomyces cerevisiae via pairing novel oxidation and reduction system from legume plants, Metab. Eng., 2018, 45, 43–50 CrossRef CAS PubMed.
- M. A. Fernández, B. I. Heras and M. D. García, et al., New insights into the mechanism of action of the anti-inflammatory triterpene lupeol, J. Pharm. Pharmacol., 2010, 53, 1533–1539 CrossRef.
- W. N. Setzer and M. C. Setzer, Plant-derived triterpenoids as potential antineoplastic agents, Mini-Rev. Med. Chem., 2003, 3(6), 540–556 CrossRef CAS.
- K. Qian, R.-Y. Kuo and C.-H. Chen, et al., Anti-AIDS agents 81. design, synthesis, and structure-activity relationship study of betulinic acid and moronic acid derivatives as potent HIV maturation inhibitors, J. Med. Chem., 2010, 53(8), 3133–3141 CrossRef CAS PubMed.
- E. Pisha, H. Chai and I.-S. Lee, et al., Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis, Nat. Med., 1995, 1(10), 1046–1051 CrossRef CAS.
- E. Urano, S. D. Ablan and R. Mandt, et al., Alkyl amine bevirimat derivatives are potent and broadly active HIV-1 maturation inhibitors, Antimicrob. Agents Chemother., 2016, 60(1), 190–197 CrossRef CAS.
- T. Lin, D. Wang and Z. Dai, et al., Creation of a Saccharomyces cerevisiae cell factory for the fermentation production of lupeol, China J. Chin. Mater. Med., 2016, 41(6), 1008–1015 Search PubMed.
- C. Z Lu, C. Zhang and F. Zhao, et al., Biosynthesis of ursolic acid and oleanolic acid in Saccharomyces cerevisiae, AIChE J., 2018, 64, 3794–3802 CrossRef.
- X. He and R. H. Liu, Triterpenoids isolated from apple peels have potent antiproliferative activity and may be partially responsible for apple's anticancer activity, J. Agric. Food Chem., 2007, 55, 4366–4370 CrossRef CAS PubMed.
- B. D. Abrosca, A. Fiorentino and P. Monaco, et al., Annurcoic acid : A new antioxidant ursane triterpene from fruits of cv. Annurca apple, Food Chem., 2006, 98, 285–290 CrossRef.
- E. Vranová, D. Coman and W. Gruissem, Network analysis of the MVA and MEP pathways for isoprenoid synthesis, Annu. Rev. Plant Biol., 2013, 64, 665–700 CrossRef PubMed.
- Z. Luo, K. Zhang and X. Ma, et al., Research Progress in Synthetic Biology of Triterpenoid Saponins, Chin. Tradit. Herb. Drugs, 2016, 47(10), 1806–1814 CAS.
- Z. Zhao, L. Xing and Y. Zhou, et al., Preparation of Oleanolic Acid Sodium Salt and Its Solubility, Chin. Tradit. Pat. Med., 199l, 13(8), 32 Search PubMed.
- G. Zhai, H. Lou and L. Zou, et al., Research progress on drug-phospholipid complexes, Chin. J. Pharm., 2001, 36(12), 800–806 CAS.
- Z. Li, Z. Sun and H. Liu, The protective effect of sodium oleanolate on chemical liver injury in rats, Guizhou Med. J., 2004, 28(7), 582–583 Search PubMed.
- Y. Zhang, J. X. Li and J. Zhao, et a1., Synthesis and Activity of Oleanolic Acid Derivatives, A Novel Class of Inhibitors of Osteoclast Formation, Bioorg. Med. Chem. Lett., 2005, 15, 1629–1632 CrossRef CAS.
- Y. Meng, H. Nie and X. Wang, et al., Synthesis and Anti-Tumor Activity of Oleanolic Acid Derivatives, Guizhou Med. J., 2011, 28(7), 582–583 Search PubMed.
- Y. Zhang, L. Gu and L. Su, et al., Synthesis of Oleanolic Acid Derivatives
Containing Amino Phosphonate and Their Anti-Tumor Activity, J. Anhui Agric. Sci., 2012, 40(5), 2737–2738 CAS.
- W. Huang, J. Huang and D. Zhang, et al., Study on the Anti-Invasion and Apoptosis-Inducing Effects of Pentacyclic Triterpenoids on Human Lung Cancer Cells, Chin. J. Lung Cancer, 2003, 6(4), 254–257 CAS.
- Z. Huang, H. Li and L. Sun, et al., Study on the Anti-Metastatic Effect of Oleanolic Acid on Rat Transplantable Liver Cancer, J. Anhui Agric. Sci., 2011, 39(18), 10807–10808 CAS.
- H. Sun, C. Hu and W. Fang, et al., Synthesis of 3-Oxo-Oleanolic Acid Amino Acid Conjugates, Determination of Aqueous Solubility, and Research on Anticancer Activity, Chin. J. Med. Chem., 2008, 18(1), 11–15 CAS.
- Q. Li, J. Gu and L. Li, et al., Synthesis of Oleanolic Acid Derivatives, Synth. Chem., 2010, 18(3), 290–296 CAS.
- J. Xi, Q. Chang and C. K. Chan, et al., Formulation Development and Bioavailability Evaluation of a Self-nanoemulsified Drug Delivery System of Oleanolic Acid, AAPS PharmSciTech, 2009, 10, 172–182 CrossRef CAS.
- Y. Zhang, J. X. Li and J. Zhao, et al., Synthesis and activity of oleanolic acid derivatives, a novel class of inhibitors of osteoclast formation, Bioorg. Med. Chem. Lett., 2005, 15(6), 1629–1632 CrossRef CAS.
- T. Akihisa, S. Kamo and T. Uchiyama, et al., Cytotoxic activity of Perilla frutescens var. japonica leaf extract is due to high concentrations of oleanolic and ursolic acids, J. Nat. Med., 2006, 60, 331–333 CrossRef CAS.
- W. Li, S. das and K. Ng, et al., Formulation, biological and pharmacokinetic studies of sucrose ester-stabilized nanosuspensions of oleanolic acid, Pharm. Res., 2011, 28, 2020–2033 CrossRef CAS.
- L. Wang, G. Han and W. Liu, et al., Chemical and Pharmacological Research on Oleanolic Acid, J. Pharm. Pract., 2001, 19(2), 104–107 Search PubMed.
- H. Yang, M. Deng and H. Jia, et al., A review of structural modification and biological activities of oleanolic acid, Chin. J. Nat. Med., 2024, 22(1), 15–30 CAS.
- F. Jannus, J. Sainz and F. J. Reyes-Zurita, et al., Principal Bioactive Properties of Oleanolic Acid, Its Derivatives and Analogues, Molecules, 2024, 29, 3291–3312 CrossRef CAS PubMed.
- A. Spivak, R. Khalitova and D. Nedopekina, et al., Synthesis and evaluation of anticancer activities of novel c-28 guanidine-functionalized triterpene acid derivatives, Molecules, 2018, 23, 3000–3022 CrossRef PubMed.
- K. Chouaïb, A. Romdhane and S. Delemasure, et al., Regiospecific synthesis by copper-and ruthenium-catalyzed azide–alkyne 1, 3-dipolar cycloaddition, anticancer and anti-inflammatory activities of oleanolic acid triazole derivatives, Arabian J. Chem., 2019, 12(8), 3732–3742 CrossRef.
- E. F. Khusnutdinova, G. N. Apryshko and A. V. Petrova, The Synthesis and Selective Cytotoxicity of New Mannich Bases, Derivatives of 19-and 28-Alkynyltriterpenoids, Russ. J. Bioorg. Chem., 2018, 44, 123–127 CrossRef CAS.
- X. Liu, B. Li and Z. Zhang, et al., Synthesis and discovery novel anti-cancer stem cells compounds derived from the natural triterpenoic acids, J. Med. Chem., 2018, 61(23), 10814–10833 CrossRef CAS PubMed.
- Y.-Q. Meng, Z.-Y. Kuai and S.-W. Zhan, et al., Synthesis and anti-tumor activity of oleanolic acid derivatives, Acta Pharm. Sin., 2011, 46(10), 1215–1220 CAS.
- Z. Chen, K.-Y. Huang and Y. Ling, et al., Discovery of an oleanolic acid/hederagenin–nitric oxide donor hybrid as an EGFR tyrosine kinase inhibitor for non-small-cell lung cancer, J. Nat. Prod., 2019, 82(11), 3065–3073 CrossRef CAS.
- D. S. Raghuvanshi, N. Verma and S. Singh, et al., Design and synthesis of novel oleanolic acid based chromenes as anti-proliferative and anti-inflammatory agents, New J. Chem., 2018, 42(20), 16782–16794 RSC.
- A. Froelich, B. Bednarczyk-Cwynar and L. Zaprutko, et al., Beckmann rearrangement within the ring C of oleanolic acid lactone: Synthesis, structural study and reaction mechanism analysis, J. Mol. Struct., 2017, 1136, 173–181 CrossRef CAS.
- L. Zaprutko, D. Partyka and B. Bednarczyk-Cwynar, Triterpenoids. Part 21: Oleanolic acid azaderivatives as percutaneous transport promoters, Bioorg. Med. Chem. Lett., 2004, 14(18), 4723–4726 CrossRef CAS.
- J. Xu, X. Wang and H. Zhang, et al., Synthesis of triterpenoid derivatives and their anti-tumor and anti-hepatic fibrosis activities, Nat. Prod. Res., 2020, 34(6), 766–772 CrossRef CAS PubMed.
- J. F. Li, R. Z. Huang and G. Y. Yao, et al., Synthesis and biological evaluation of novel aniline-derived asiatic acid derivatives as potential anticancer agents, Eur. J. Med. Chem., 2014, 86, 175–188 CrossRef CAS.
- N. Heisi, B. Siewert and D. Strohl, et al., A simple but unusual rearrangement of an oleanane to a taraxerane28, 14β-olide, Steroids, 2021, 172, 108853–108858 CrossRef.
- T. Honda, B. V. Rounds and G. W. Gribble, et al., Design and synthesis of 2-cyano-3, 12-dioxoolean-1, 9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages, Bioorg. Med. Chem. Lett., 1998, 8(19), 2711–2714 CrossRef CAS PubMed.
- R. Borella, L. Forti and L. Gibellini, et al., Synthesis and anticancer activity of CDDO and CDDO-Me, two derivatives of natural triterpenoids, Molecules, 2019, 24(22), 4097–4117 CrossRef CAS PubMed.
- Y.-Y. Wang, Y.-X. Yang and H. Zhe, et al., Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties, Drug Des., Dev. Ther., 2014, 2075–2088 Search PubMed.
- M. H. L. Wong, I. K. Bryan and I. M. Copple, et al., Design and synthesis of irreversible analogues of bardoxolone methyl for the identification of pharmacologically relevant targets and interaction sites, J. Med. Chem., 2016, 59(6), 2396–2409 CrossRef CAS PubMed.
- F. Gao, Q. Zuo and T. Jiang, et al., A newly synthesized oleanolic acid derivative inhibits the growth of osteosarcoma cells in vitro and in vivo by decreasing c-MYC-dependent glycolysis, J. Cell. Biochem., 2019, 120(6), 9264–9276 CrossRef CAS PubMed.
- F. Kang, Y. Ai and Y. Zhang, et al., Design and synthesis of new hybrids from 2-cyano-3, 12-dioxooleana-9-dien-28-oic acid and O2-(2, 4-dinitrophenyl) diazeniumdiolate for intervention of drug-resistant lung cancer, Eur. J. Med. Chem., 2018, 149, 269–306 CrossRef CAS.
- W. Yan, C. Zhang and B. Li, et al., A Series of oleanolic acid derivatives as anti-hepatitis B virus agents: Design, synthesis, and in vitro and in vivo biological evaluation, Molecules, 2016, 21(4), 402–413 CrossRef PubMed.
- W. Li, F. Yang and L. Meng, et al., Synthesis, structure activity relationship and anti-influenza A virus evaluation of oleanolic acid-linear amino derivatives, Chem. Pharm. Bull., 2019, 67(11), 1201–1207 CrossRef CAS PubMed.
- L. Meng, Y. Su and F. Yang, et al., Design, synthesis and biological evaluation of amino acids-oleanolic acid conjugates as influenza virus inhibitors, Bioorg. Med. Chem., 2019, 27(23), 115147–115156 CrossRef CAS PubMed.
- M. Medina-O'Donnell, F. Rivas and F. J. Reyes-Zurita, et al., Oleanolic acid derivatives as potential inhibitors of HIV-1 protease, J. Nat. Prod., 2019, 82(10), 2886–2896 CrossRef PubMed.
- J. W. Wang, W. R. Xu, P. Yong, et al., Glycyrrhetinic acid-30 amide derivatives and their use, US2008214636(A1), 2008.
- J. Yong, Research Progress on the Synthesis and Bioactivity of Glycyrrhizic Acid and Its Triterpenoid Saponin Derivatives, Shizhen Guoyi Guoyao, 2012, 23(6), 1495–1498 CAS.
- J. Tatsuzaki, M. Taniguchi and K. F. Bastow, et al., Antitumor agents glycyrrhetinic acid dehydrozingerone conjugates as cytotoxic agents, Bioorg. Med. Chem., 2007, 15, 6193–6199 CrossRef CAS PubMed.
- Y. Zhang, Research on Chiral Ketones of Galactose and Triterpenoid Saponins and Isoxazole and Benzimidazole Ring Isoxazolidinones, PhD Dissertation, Shandong University, 2007.
- N. Zhang, X. Cui and X. Zhao, et al., Synthesis and Anti-Liver Cancer Activity of Glycyrrhetinic Acid Derivatives, Chin. J. Exp. Tradit. Med. Formulae, 2015, 10, 37–41 Search PubMed.
- R. Csuk, S. Schwarz and R. Kluge, et al., structure-Activity Relationships of Glycyrhetinic Acid Derivatives Modified at
PositionC-11, Arch. Pharm., 2012, 345(1), 28–32 CrossRef CAS PubMed.
- S. Schwarz and R. Csuk, et al., synthesis and antitumour activity of glycyrhetinic acid derivativesy, Bioorg. Med. Chem., 2010, 18(21), 7458–7474 CrossRef CAS.
- Y. Gao, X. Guo and L. Zhao, et al., The synthesis of Glycyrhetinic Acid Derivatives Containing A Nitrogen Heterocycle and Their Antiproliferative Effects in Human Leukemia Cells, Molecules, 2010, 15, 4439–4449 CrossRef CAS PubMed.
- Y. Meng, Z. Yang and M. Zhang, et al., Synthesis of Glycyrrhetinic Acid C3, C11, and C30 Derivatives and Their Anti-Tumor Activity In Vitro, Drugs Clin., 2014, 29(1), 6–10 CAS.
- X. Su, N. Vicker and H. Lawrence, et al., Inhibition of human and rat 11β-hydroxysteroid dehydrogenase type 1 by 18β-glycyrrhetinic acid derivatives, J. Steroid Biochem. Mol. Biol., 2007, 104(3–5), 312–320 CrossRef CAS PubMed.
- R. C. Santos, J. A. R. Salvador and S. Marín, et al., Novel semisynthetic derivatives of betulin and betulinic acid with cytotoxic activity, Bioorg. Med. Chem., 2009, 17(17), 6241–6250 CrossRef CAS PubMed.
- E. R. Trumbull, E. Bianchi and D. J. Eckert, et al., inhibitory agents from Vauquelinia corymbosa (Rosaceae), J. Pharm. Sci., 1976, 65(9), 1407–1408 CrossRef CAS.
- P. Chatterjee, S. A. Kouzi and J. M. Pezzuto, et al., Biotransformation of the antimelanoma agent betulinic acid by Bacillus megaterium ATCC 13368, Appl. Environ. Microbiol., 2000, 66(9), 3850–3855 CrossRef CAS.
- R. Mukherjee, M. Jaggi and P. Rajendran, et al., Betulinic acid and its derivatives as anti-angiogenic agents, Bioorg. Med. Chem. Lett., 2004, 14(9), 2181–2184 CrossRef CAS.
- J. Li, M. Goto and X. Yang, et al., Fluorinated betulinic acid derivatives and evaluation of their anti-HIV activity, Bioorg. Med. Chem. Lett., 2016, 26(1), 68–71 CrossRef CAS PubMed.
- O. V. Tsepaeva, A. V. Nemtarev and L. R. Grigor'eva, et al., Synthesis of C (28)-linker derivatives of betulinic acid bearing phosphonate group, Russ. Chem. Bull., 2021, 70, 179–182 CrossRef CAS.
- A. Y. Spivak, D. A. Nedopekina and E. R. Shakurova, et al., Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity, Russ. Chem. Bull., 2013, 62, 188–198 CrossRef CAS.
- Y. Chen, S. Y. Sit and J. Chen, et al., The design, synthesis and structure-activity relationships associated with C28 amine-based betulinic acid derivatives as inhibitors of HIV-1 maturation, Bioorg. Med. Chem. Lett., 2018, 28, 1550–1557 CrossRef CAS.
- V. Reutrakul, N. Anantachoke and M. Pohmakotr, et al., Anti-HIV-1 and anti-inflammatory lupanes from the leaves, twigs, and resin of Garcinia hanburyi, Planta Med., 2010, 76(4), 68–71 CrossRef.
- J. Li, W. Guo and Q. Yang, Effects of ursolic acid and oleanolic acid on human colon carcinoma cell line HCT15, World J. Gastroenterol., 2002, 8(3), 493–495 CrossRef CAS PubMed.
- H. S. Young, H. Y. Chung and C. K. Lee, et al., Ursolic acid inhibits aflatoxin B1-induced mutagenicity in a Salmonella assay system, Biol. Pharm. Bull., 1994, 17(7), 990–992 CrossRef CAS.
- E. Kassi, Z. Papoutsi and H. Pratsinis, et al., Ursolic Acid, a Naturally Occurring Triterpenoid, Demonstrates Anticancer Activity on Human Prostate Cancer Cells, J. Cancer Res. Clin. Oncol., 2007, 133, 493–500 CrossRef CAS.
- D. Zhang and L. L. Zhao, Synthesis and Anti-hepatocellular Carcinoma In Vitro of Ursolic Acid Derivatives Bearing Benzylphosphonic Acid Diethyl Ester Acetamide Moieties, Chin. J. Mod. Appl. Pharm., 2022, 39(06), 738–744 Search PubMed.
- T. Tian, Synthesis Characterization and Anticancer Activity Research of Three Pentacyclic Triterpenoid Nitrogen-Containing Derivatives, PhD Dissertation, Liaoning Normal University, 2016.
- K. Bai, F. Chen and Y. Guo, Synthesis and In Vitro Anticancer Activity of Polyethylene Glycol Conjugated Ursolic Acid Derivatives, Chin. J. New Drugs, 2012, 13(21), 1536–1540 Search PubMed.
- K. Bai, F. Chen and Y. Guo, Synthesis and In Vitro Anticancer Activity of
Ursolic Acid Derivatives on Prostate Cancer PC-3 Cells, Chin. J. New Drugs, 2012, 22(21), 2667–2673 Search PubMed.
- K. Bai, F. Chen and Y. Zheng, et al., Synthesis and In Vitro Anti-Gastric Cancer Activity of Amino Acid Modified Ursolic Acid Derivatives, Chin. J. Pharm., 2012, 4(47), 265–269 Search PubMed.
- Y. Zhao, A-ring multi-oxidized derivatives of pentacyclic triterpenes, their preparation methods, and applications, J. Chin. Med. Mater., 2006,(05), 427–428 Search PubMed.
- J. Wu, Z. H. Zhang and L. H. Zhang, et al., Design, synthesis, and screening of novel ursolic acid derivatives as potential anti-cancer agents that target the HIF-1α pathway, Bioorg. Med. Chem. Lett., 2019, 29(6), 853–858 CrossRef CAS PubMed.
- W. Wang, L. Lei and Z. Liu, et al., Design, synthesis, and biological evaluation of novel nitrogen heterocycle-containing ursolic acid analogs as antitumor agents, Molecules, 2019, 24(5), 877–891 CrossRef PubMed.
- Q. Min, Synthesis of 3,4-Fissured Pentacyclic Triterpenoid-3,4-Lactone Derivatives and Their Anti-Hepatitis B Virus Activity, PhD Dissertation, Southern Medical University, 2015.
- C. Chen, R. Sun and Y. Sun, et al., Synthesis,biological evaluation and SAR studies of ursolic acid 3β-ester derivatives as novel CETP inhibitors, Bioorg. Med. Chem. Lett., 2020, 30(2), 126–824 CrossRef.
- J. Xu, X. Nie and Y. Hong, et al., Synthesis of water soluble glycosides of pentacyclic dihydroxytriterpene carboxylic acids as inhibitors of α-glucosidase, Carbohydr. Res., 2016, 424, 42–53 CrossRef CAS PubMed.
- Z. Zeng, X. Yin and X. Wang, et al., Synthesis of water soluble pentacyclic dihydroxyterpene carboxylic acid derivatives coupled amino acids and their inhibition activities on α-glucosidase, Bioorg. Chem., 2019, 86, 277–287 CrossRef CAS.
- X. Liu, X. Zang and X. Yin, et al., Semi-synthesis of C28- modified triterpene acid derivatives from maslinic acid or corosolic acid as potential α-glucosidase inhibitors, Bioorg. Chem., 2020, 97, 103694–103740 CrossRef CAS PubMed.
- A. R. Setty and L. H. Sigal, Herbal medications commonly used in the practice of rheumatology: mechanisms of action, efficacy, and side effects, Semin. Arthritis Rheum., 2005, 34(6), 773–784 CrossRef PubMed.
- M. Jing, J. Yang and L. Zhang, et al., Celastrol inhibits rheumatoid arthritis through the ROS-NF-κB-NLRP3 inflammasome axis, Int. Immunopharmacol., 2021, 98, 107–879 CrossRef.
- Z. Li, R. Liu and Z. Guo, et al., Celastrol-based nanomedicine promotes corneal allograft survival, J. Nanobiotechnol., 2021, 19(1), 1–19 CrossRef.
- J. Zhang, K. Zhou and X. Zhang, et al., Celastrol ameliorates inflammation in human retinal pigment epithelial cells by suppressing NF-κB signaling, J. Ocul. Pharmacol. Ther., 2019, 35(2), 116–123 CrossRef CAS.
- Y. Feng, W. Wang and Y. Zhang, et al., Synthesis and biological evaluation of celastrol derivatives as potential anti-glioma agents by activating RIP1/RIP3/MLKL pathway to induce necroptosis, Eur. J. Med. Chem., 2022, 229, 114070–114086 CrossRef CAS PubMed.
- M. Xu, N. Li and Z. Zhao, et al., Design, synthesis and antitumor evaluation of novel celastrol derivatives, Eur. J. Med. Chem., 2019, 174, 265–276 CrossRef CAS.
- P. Coghi, J. P. L. Ng and O. Kadioglu, et al., Synthesis, computational docking and biological evaluation of celastrol derivatives as dual inhibitors of SERCA and P-glycoprotein in cancer therapy, Eur. J. Med. Chem., 2021, 224, 113676–113690 CrossRef CAS PubMed.
- N. Li, M. Xu and B. Wang, et al., Discovery of Novel Celastrol Derivatives as Hsp90–Cdc37 Interaction Disruptors with Antitumor Activity, J. Med. Chem., 2019, 62(23), 10798–10815 CrossRef CAS.
- X. Li, J. Ding and N. Li, et al., Synthesis and biological evaluation of celastrol derivatives as anti-ovarian cancer stem cell agents, Eur. J. Med. Chem., 2019, 179, 667–679 CrossRef CAS PubMed.
- N. Li, M. Xu and N. Bao, et al., Discovery of novel NO-releasing celastrol derivatives with Hsp90 inhibition and cytotoxic activities, Eur. J. Med. Chem., 2018, 160, 1–8 CrossRef CAS PubMed.
- W. J. Tang, J. Wang and X. Tong, et al., Design and synthesis
of celastrol derivatives as anticancer agents, Eur. J. Med. Chem., 2015, 95, 166–173 CrossRef CAS PubMed.
- Y. Jing, G. Wang and Y. Ge, et al., Synthesis, anti-tumor and anti-angiogenic activity evaluations of asiatic acid amino acid derivatives, Molecules, 2015, 20(4), 7309–7324 CrossRef CAS PubMed.
- K. Tang, J. Huang and J. Pan, et al., Design, synthesis and biological evaluation of C (6)-indole celastrol derivatives as potential antitumor agents, RSC Adv., 2015, 5(25), 19620–19623 RSC.
- R. G. Brahmachari, N. C. Mandal and R. Roy, et al., A new pentacyclic triterpene with potent antibacterial activity from Limnophila indica Linn.(Druce), Fitoterapia, 2013, 90, 104–111 CrossRef.
- A. A. Lasisi, B. W. Ayinde and A. O. Adeleye, et al., New triterpene isovanniloyl and antibacterial activity of constituents from the roots of Paullinia pinnata Linn (Sapindaceae), J. Saudi Chem. Soc., 2015, 19(2), 117–122 CrossRef.
- L. J. Shai, L. J. McGaw and M. A. Aderogba, et al., Four pentacyclic triterpenoids with antifungal and antibacterial activity from Curtisia dentata (Burm.f) CA Sm. Leaves, J. Ethnopharmacol., 2008, 119(2), 238–244 CrossRef CAS.
- A. Parra, S. Martin-Fonseca and F. Rivas, et al., Semi-synthesis of acylated triterpenes from olive-oil industry wastes for the development of anticancer and anti-HIV agents, Eur. J. Med. Chem., 2014, 74, 278–301 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Juan Cai†,
Li Juan Liu,
Zhang Chao Dong,
Ying Zhou*,
Ting Ting Feng* and
Yang Shi*
,
Juan Cai†,
Li Juan Liu,
Zhang Chao Dong,
Ying Zhou*,
Ting Ting Feng* and
Yang Shi*