
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beryllium carbonate: a model compound for highest capacity carbon sequestration chemistry†
Gad Lichta,
Kyle Hofstetterb and
Stuart Licht *abc
*abc
aC2CNT LLC, A4 188 Triple Diamond Blvd, North Venice, FL 34275, USA. E-mail: slicht@gwu.edu
bCarbon Corp, 1035 26 St NE, Calgary, AB T2A 6K8, Canada
cDept. of Chemistry, George Washington University, Washington DC 20052, USA
First published on 23rd December 2024
Abstract
Beryllium carbonate has the highest capacity to bind and release the greenhouse gas CO2 compared to amines, ionic liquids, CaCO3 or Li2CO3. The thermodynamic equilibrium for CO2 and BeO from BeCO3 is calculated. TGA of BeCO3 is used to determine the stepwise mechanism of its CO2 release, and the low melting point Li/Sr/BeCO3 is demonstrated.
Introduction
Rising CO2 levels are causing catastrophic climate change, and are an existential threat to the planet. Atmospheric CO2 levels, stable at 235 (±50) ppm for several hundred thousand years prior to the industrial revolution, have nearly doubled to 424 ppm and continue to rise rapidly. The effects of climate change include an increasing probability of a mass species extinction event.1 The ongoing toll on humanity and the planet's habitats are increasingly evident; on a personal note, the first and third authors of this study have just experienced ground zero of two “once in a millennium rainfall events” (Hurricanes Helene and Milton) in a period of two weeks, while the middle author is subject to increasingly frequent forest fires.A principal path to mitigate climate change is CO2 sequestration, and CO2 capture capacity is a measure of the extent of a chemical species binding of CO2 for use in carbon sequestration chemistry. CO2 capture capacity is quantified as mass or mole capacity of CO2 per mass or mole of absorbent. In addition to the general need for effective CO2 trapping materials to mitigate CO2-induced global warming and climate change, other examples of the need for lightest weight carbon capture materials for CO2 air scrubbing, include those needed by submersibles, submarines, and spacecraft.2,3
Several recent reviews have focused on amines (and amino acids), calcium oxide (to calcium carbonate), and ionic liquids to bind and release CO2.4–9 More recently, there has been a growing focus on nanomaterials, such as carbon nanomaterials,10–12 and also on and lithium carbonate and mixed lithium/strontium carbonates13–16 to capture CO2 has emerged. Table 1 summarizes common absorbents including amines, ionic liquids, calcium oxide (to calcium carbonate), and more recently, lithium oxide (to lithium carbonate). In this study, beryllium carbonate is introduced as a model compound, establishing a baseline for among the highest capacities of CO2 captured.
| Chemical absorbent/product | Acronym | Formula | Formula weight, g mol−1 | CO2 storage mechanism | Capacity for CO2, mol CO2/mol absorbent | Source, reference | Capacity for CO2, kg CO2 per kg absorbent |
|---|---|---|---|---|---|---|---|
| 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide | [Bmim][Tf2N] | C10H15F6N3O4S2 | 419.37 | Ionic liquid | 0.68 | 7 | 0.07 |
| 1-Aminopropyl-3-methylimidazolium tetraborafluorate/various products | APMim[BF4] | C7H14N3BF4 | 227.01 | Ionic liquid | 0.34 | 7 | 0.07 |
| 1-Butyl-3-methylimidazolium hexafluorophosphate/various products | [Bmim][PF6] | C8H15F6N2P | 284.186 | Ionic liquid | 0.51 | 7 | 0.08 |
| 1-Butyl-3-methylimidazolium tetrafluoroborate/various products | [Bmim][BF4] | C8H15BF4N2 | 226.03 | Ionic liquid | 0.44 | 7 | 0.09 |
| Taurine to protonated acid, HCO3− or carbamate | C2H7NO3S | 125.14 | Amino acid | 0.7 | 7 | 0.25 | |
| Proline to protonated acid, HCO3− or carbamate | C5H9NO2 | 115.132 | Amino acid | 0.7 | 7 | 0.27 | |
| Glycine to protonated acid, HCO3− or carbamate | C2H5NO2 | 75.067 | Amino acid | 0.6 | 7 | 0.35 | |
| Monoethanolamine to RNHCOO− | MEA | C5H13NO2 | 61.08 | Amine | 0.5 | 7 | 0.36 |
| Diethanolamine to RNHCOO− | DEA | C4H11NO2 | 105.14 | Amine | 0.5 | 7 | 0.21 |
| Methyldiethanolamine to ‘’” | MDEA | C5H13NO2 | 119.164 | Amine | 1 | 7 | 0.37 |
| Diethylethanolamine to “” | DEAE | C6H15NO | 117.192 | Amine | 0.5 | 7 | 0.19 |
| Calcium oxide to carbonate | CaO | 56.08 | CaO + CO2 → CaCO3 | 1 | 9 | 0.78 | |
| Lithium oxide to carbonate | Li2O | 29.88 | Li2O + CO2 → Li2CO3 | 1 | 16 | 1.47 | |
| Beryllium oxide to carbonate | BeO | 24.01 | BeO + CO2 → BeCO3 | 1 | This study | 1.83 |
CO2 can be captured and stored by thermal cycling. In this case, dilute CO2 is generally introduced at a lower temperature and released in a concentrated form at a higher temperature. Thermal cycling can comprise adsorption chemistry, as generally occurs with various amine carbon capture chemistries,5,7 or by chemical reactions, as occurs in the reaction of dissolved or solid calcium oxide with CO2 to calcium carbonate, followed by high-temperature decomposition of calcium carbonate back to calcium oxide.8 Thermal cycling is often accompanied by pressurization and also by different subsequent processes to sequester (Carbon Capture and Storage, CCS) or chemically convert (Carbon Capture Utilization and Storage, CCUS) the captured concentrated CO2. As an alternative to thermal cycling, the CO2 capture can be accomplished by electrolysis (such as the electrochemical splitting of CO2 to C and O2) to a product containing the captured CO2, such as the formation of Carbon NanoTubes (CNTs) from CO2 in molten carbonates.17–20 This latter CCUS process often occurs in a single step.
In Table 1, the capacity for CO2 is compiled for common CO2 absorbents (absorbents referring to both adsorbents, absorbents, and reactants). The capacity for CO2 is presented in units of both mole CO2/mole absorbent and also in more typical units of kg CO2 captured per kg absorbent in the last column. Pragmatic capacities for CO2 will be lower than those compiled when a matrix (such as an inert membrane or solvent stabilizer) is required as an additional mass component in the CO2 capture process.
Amines and amino acids have been widely studied both as absorbents, principally in the liquid phase, to absorb and release CO2, and as adsorbents principally affixed on membranes to adsorb and release CO2. As seen in Table 1, amines and amino acids have respective capacities for CO2 of 0.19–0.37 or 0.27–0.35 kg CO2 per kg (amine or amino acid). Ionic liquids have attained capacities of 0.07–0.09 kg CO2 per kg. CaO/CaCO3 has a capacity for CO2 of 0.78 kg CO2 (determined as of CaO + CO2 → CaCO3 from the 44.01 g per mol FW of CO2 to the 56.08 g per mol FW of CaO). The lighter molecular weight Li2O/Li2CO3 has a capacity for CO2 of 1.47 kg CO2. In order of increasing CO2 capacities, the absorbents are ionic liquids < amino acids & amines < CaO (to CaCO3) < Li2O (to Li2CO3).
In this study, beryllium carbonate, with the lowest melting points of inorganic carbonates, is introduced as a model compound establishing a baseline for maintaining the highest capacities for CO2 (1.83 kg CO2 per kg BeO → BeCO3). Beryllium carbonate is also as an example of a melting point decrease facilitator by addition of BeCO3 to binary Li/SrCO3 electrolyte to become the substantially lower melting point ternary Li/Sr/BeCO3. Binary mixtures typically melt at lower temperatures than pure components because the presence of different molecules disrupts the crystal lattice, weakening intermolecular forces and reducing the energy needed to melt, and in this case CO2 release can then be achieved using a lower thermal energy input.
Results and discussion
Phase changes & CO2 equilibria of alkali & alkali earth carbonates
As summarized in Table 2, the melting point of beryllium carbonate (mp 54 °C), Be2CO3, is low in the extreme compared to that of the other alkali earth or alkali carbonates, or compared to their binary or tertiary carbonate mixtures. It should be noted that reported values of carbonate melting points exhibit a considerable range of 10 °C or more. For example, in 7 studies, the melting point of Li2CO3 is reported from 700 °C to 728 °C.22–28 Furthermore, the CO2 concentration in the atmosphere has increased by 35% since the first of these reports appeared (in 1957). The stability of lithium carbonate increases under 1 atm of CO2, and we also note that many properties, including melting points, of species related to CO2 equilibrium will need to be reevaluated due to the rapidly increasing atmospheric concentration of this greenhouse gas.| Carbonate | Melting point (°C) | Decomposition point (°C) | Ref. |
|---|---|---|---|
| a Li2CO3 decomposition is more rapid under argon than under air.15 | |||
| Be2CO3 | 54 | ∼100 | 21 |
| Li2CO3, Na2CO3 or K2CO3 | 723, 851 or 891 | ∼1300a (Li2CO3) | 30 |
| BaCO3 | 810 | ∼1360 | 31 and 32 |
| MgCO3 | — | ∼350 | 32 |
| CaCO3 | — | ∼850 | 31 and 32 |
| SrCO3 | 1494 | 1494 | 13 |
| Li2/BaCO3; 55/45 mol% | 609 | 31 and 34 | |
| K2CO3/MgCO3; 57/43 mol% | 460 | 35 | |
| Li2/K2CO3; 62/38 mol% | 498 | 35 | |
| Na2/K2CO3; 56/44 mol% | 710 | 35 | |
| Li2/Na2/K2CO3; 43.5/31.5/25 mol% | 397 | 35 | |
| Li2/Sr2CO3; 60/40 wt% | 680 | 13 | |
| Be/Sr/Li2CO3; 33/33/33 wt% | 480 | This study | |
Alkali carbonate binary mix eutectics have lower melting points. The lowest melting alkali carbonate is generally considered to be the LixNayKzCO3 with a melting point of 397 °C (at ∼x = 0.47, y = 0.62, z = 0.5), still considerably higher than the melting point of BeCO3, by 343 °C.
The extent to which an alkali or alkali earth carbonates retains CO2 is given by:
| MCO3 ⇌ CO2(gas) + MO (M = Be, Mg, Li2, Na2, etc.) | (1) |
| K = pCO2 aMO/aMCO3; K(T) = e−ΔG(T)/RT | (2) |
An extensive literature search did not reveal phase diagrams or equilibria for beryllium carbonate. We've calculated the BeCO3/BeO + CO2 equilibrium from the available enthalpy and entropy of the constituent species.29
Fig. 1 presents a comparison of the carbonate/oxide equilibrium constant for binding and releasing of CO2 by beryllium carbonate compared to those for alkali, or other alkali earth carbonates as a function of temperature. Below any of the Fig. 1 equilibrium presented curves, that is, in the high CO2 activity domain, the carbonate salt will spontaneously form from CO2 and the salt's oxide. Above any Fig. 1 equilibrium curve, the low CO2 activity domain (aCO2 aoxide/acarbonate < K), the carbonate salt will spontaneously decompose. For example, as noted in Table 2, solid MgCO3 decomposes at 350 °C, releasing bound CO2, and as seen is the second largest (other than BeCO3) of the eqn (2) carbonate/oxide equilibrium constants. The high industrial carbon footprint conversion process of limestone to lime or cement depends on the solid state decomposition of calcium carbonate, such as aragonite, which occurs at ∼850°.
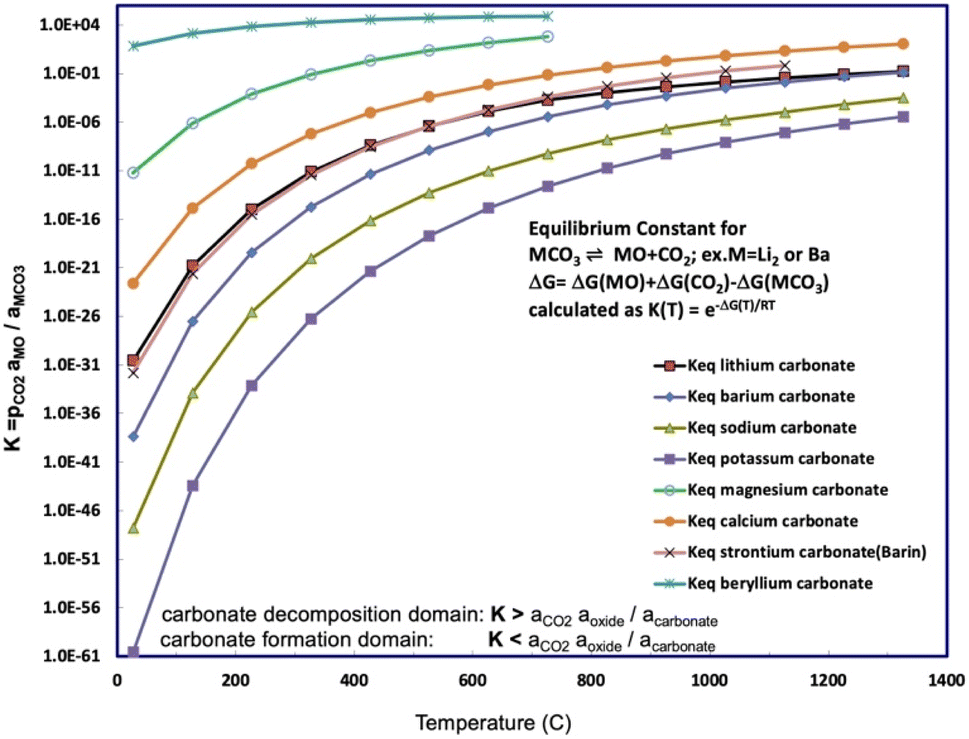 | ||
| Fig. 1 Equilibrium constant for CO2 release calculated for beryllium carbonate and compared to a range of alkali and alkali earth carbonates. The equilibrium constants as a function of temperature for strontium, lithium, sodium, potassium, and barium carbonate. The equilibrium constants are calculated from the free energy according to eqn (5). The free energy is calculated from the metal carbonate, metal oxide, and carbon dioxide enthalpies and entropies.29,36–39 | ||
Li2CO3/Li2O was introduced as among the highest CO2-capture materials, as delineated in the next to last row of Table 1, with a storage capacity of 1.47 kg CO2 per kg Li2O. Under argon, Li2CO3 entirely dissociates to CO2 + Li2O. Specifically, at a TGA rate of 5° min−1 under 1 atm of argon Li2CO3 dissociation starts around the lithium carbonate melting point of 723 °C, and is 98% complete to Li2O by 900 °C, and under 1 atm of pure CO2 also starts at 723 °C, but the dissociation is less than 10% complete by 900 °C.15 However, under even small partial pressures of CO2, such as the 426 ppm (and rising) of atmospheric CO2, Li2CO3 only fractionally dissociates to CO2, attaining 0.3 molal concentration Li2O per kg molten Li2CO3 at 750 °C.36 This high capacity was experimentally realized in the form of the electrolytic splitting of CO2 in molten Li2CO3 to graphene nanocarbons.40–49 The small concentration of dissolved Li2O in molten Li2CO3 under air is sufficient to support high CO2 splitting electrochemical current and continuous renewal of the molten Li2CO3 electrolyte with CO2.50
The relationship between melting and decomposition temperatures for carbonates is complex. Beryllium carbonate has respective melting and decomposition points of BeCO3 (Mp = 54 °C and Dp = 100 °C),21 lithium carbonate Li2CO3 (Mp = 723 °C and Dp = 1300 °C), while as seen in Table 2, solid calcium and barium carbonate do not melt nor sublime, but rather decompose directly to calcium or barium oxide and carbon dioxide, finally, strontium carbonate has equivalent, but very high melting and decomposition points SrCO3 (Mp = Dp = 1494 °C). For binary and ternary mixtures, all the higher melting point carbonates are observed to be highly soluble in lithium carbonate. For example, over 60 wt% SrCO3 is miscible in 750 °C molten Li2CO3.13
The temperature at which individual carbonates do, or do not, melt is observable and reproducible to within a few degrees. However, the decomposition point is much less distinct, occurring over hundreds of degrees. For example, while the formal Li2CO3 decomposition point in Table 2 is ∼1300, substantial decomposition has already occurred at 750 °C with release of CO2 and the resultant Li2O forming as a dissolved salt within the molten Li2CO3.36 Hence, this study focuses on the more precise melting point, rather than the broad range of observable decomposition point temperatures.
Beryllium carbonate as an ultra-high CO2 storage material
When modeled as a specific example of eqn (1), beryllium carbonate is a high-capacity carbon capture storage material. Carbon dioxide is stored in beryllium carbonate and is released in the reaction to CO2 and beryllium oxide:| BeCO3 ⇌ CO2(gas) + BeO | (3) |
Thermodynamically, BeCO3 is the carbonate best suited to initiate storage and release of CO2 at low temperatures. BeCO3 is less prevalent as a salt than calcium, lithium, or strontium carbonate. Be is only the 48th most abundant element in the earth's upper crust,51 and it and its oxide, particularly in powder form, is carcinogenic. However, the storage of CO2 by BeCO3 serves as a model for among the highest carbon storage capacity materials and lowest mass CO2 scrubbers.
The stepwise mechanism of beryllium carbonate CO2 storage
Eqn (1) only provides a thermodynamic overview of a carbonate's capability to release CO2. The individual steps in the process of the binding of CO2 into beryllium carbonate are investigated here by ThermoGravimetric Analysis, TGA.Fig. 2 presents the TGA results of beryllium carbonate conducted from 30 °C, with a 5 °C temperature increase per minute, and in atmospheres of either (1) 80% N2/20% O2 gas mix shown in the orange curve from 30 to 730 °C or (2) 100% N2 shown in the blue curve from 30 °C to 1000 °C. In the figure, the downward trend in the mass is seen to start at approximately, the cited21 54 °C melting of BeCO3. The equivalence of the curves with or without an atmosphere containing O2 provides primary evidence that O2 is neither evolved nor absorbed by beryllium carbonate during the TGA, and that species in equilibrium with O2, including oxides, peroxides, and superoxides, those species are not participants in reactions related to the TGA temperature sweep.
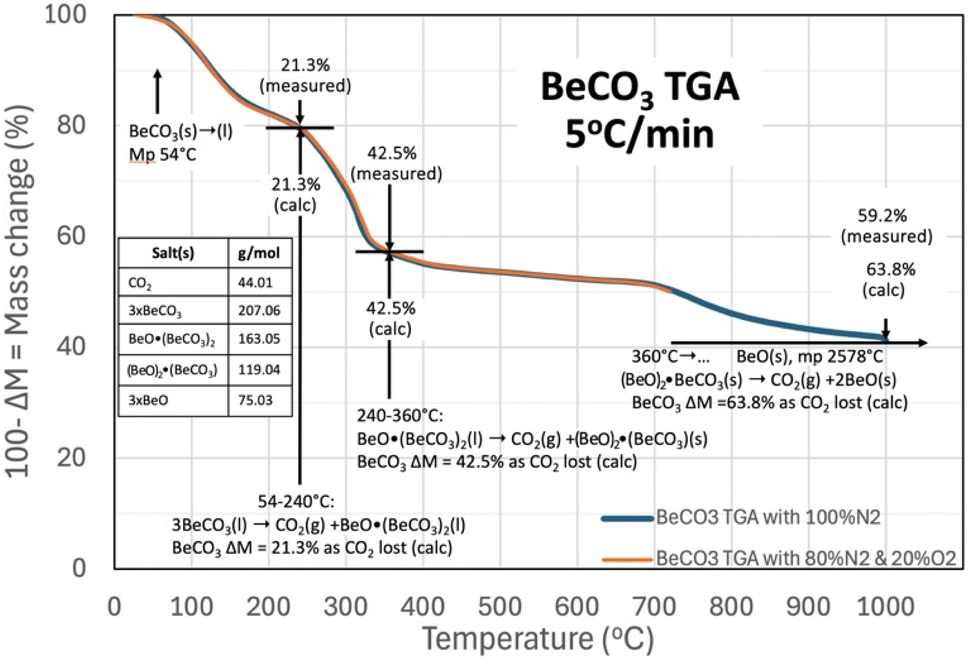 | ||
| Fig. 2 TGA analysis of beryllium carbonate. The TGA is conducted from 30 °C with a 5 °C increase per minute in either (1) 80% N2/20% O2 (from 30 °C to 730 °C) or (2) 100% N2 (from 30 °C to 1000 °C). Note, the observed TGA are identical (from 30 °C to 730 °C) in either the O2 or pure N2 environments. | ||
In Fig. 2 above 54 °C, BeCO3 rapidly evolves CO2 upon melting (at increasing temperature, the release of CO2 from BeCO3 is an exergonic, spontaneous reaction). Released gases diffuse more slowly through a solid than through a liquid.52 Salts evolving CO2 from the liquid, as opposed to from the solid form, facilitate the rapid release of CO2. For example, comparing liquid and solid CO2 amine sorbents, solid gas reactions require much higher minimum work,53 and the concurrent observed increased rate of mass loss acts as an indicator that the salt has melted. In solid salts that can release CO2 by decomposition, CO2 release is constrained by surface depletion and by the slow diffusion of CO2 to the solid surface. Whereas, in the molten state, the liquid surface is continuously replenished, sustaining facile CO2 access to the surface, and to the interior liquid bulk. In the figure, the mass loss and temperature are noted at the start of rapid mass declines with increasing temperature, and a mechanism of CO2 mass loss is then determined by calculating mass consistent changes of the equivalent calcinated beryllium oxide and BeCO3 salts.
As seen in Fig. 2, molten BeCO3 evolves CO2 to become BeO·(BeCO3)2 from ∼54 °C to 240 °C, consistent with the equivalence of both the observed and the calculated mass loss as mass loss (of CO2/mass BeCO3) of 21.3% when one CO2 is evolved from 3 BeCO3 to become BeO·(BeCO3)2, and the rapid mass loss indicative of facile CO2 evolution from a liquid. In the future, several orders of magnitude larger than the TGA mg size samples would be useful to visually corroborate that this is in the liquid (l) phase at these temperatures:
| 3BeCO3(l) → CO2(g) +BeO·(BeCO3)2(l) T = 54–240 °C | (4) |
At increasing temperature, the molten BeO·(BeCO3)2 then evolves CO2 to become (BeO)2·BeCO3 (FW 119.04) from 240 °C to 360 °C; again, as determined by the equivalence of both the observed and the calculated mass loss of 42.5% when 2CO2 are evolved from 3BeCO3 to become BeO·(BeCO3), and once again the rapid mass loss indicative of facile CO2 evolution from a liquid:
| BeO·(BeCO3)2(l) → CO2(g) +(BeO)2·(BeCO3) (s) T = 240–360 °C | (5) |
From 360 °C to ∼700 °C, there is an observed slow, steady rate of CO2 evolution as the mass loss observed in Fig. 2 increases to 50.9% from the original BeCO3. The slow rate of CO2 evolution is evidence that the eqn (5) product may be solid, and the additional (50.9–42.5%) 8.4% mass loss from 3BeCO3 is evidence that the 2BeO·(BeCO3) has evolved an additional 0.4 CO2 over this temperature range with either a lower thermodynamic drive to release CO2, or has reverted to the solid phase.
| (BeO)2·(BeCO3)(s) → 0.4CO2(g) +(BeO)2.4·(BeCO3)0.6(l) T = 360–700 °C | (6a) |
Equivalent to integral molecular values of:
| 5(BeO)2·(BeCO3)(s) → 2CO2(g) +(BeO)12(BeCO3)3(l) T = 360–700 °C | (6b) |
Above 700 °C, the observed rate of mass loss and CO2 evolution again increased, indicative that the product has once again entered a liquid phase as noted on the right side of eqn (6a). In eqn (6b) (BeO)12(BeCO3)3(l) is a generalization of the total equivalence of BeO and BeCO3 in the product, and it is likely that this consists of a solid BeO (mp 2578 °C) in a liquid phase of mixed BeOx/BeCO3. This product then evolves CO2 to become BeO(s) from ∼700 °C onward. As the end product of the beryllium carbonate CO2 loss is a solid, high melting point BeO (mp 2578 °C), and by 1000 °C, the mass observed mass loss has reached 59.2% of the full, calculated 63.8% CO2 mass loss from BeCO3. Holding the TGA temperature at 1000 °C for 4 more hours resulted in a further mass loss of 1.2% to 60.4% of the full, calculated 63.8% CO2 mass loss from BeCO3:
| (BeO)2.4·(BeCO3)0.6(l) → 3BeO(s); T = 700 °C to T > 1000 °C | (7) |
For an overall reaction of:
| 3BeCO3(l) → 3CO2(g) +3BeO; T = 54 °C to T > 1000 °C | (8) |
The thermal release of CO2 from BeCO3 does not result in the formation of powdered BeO, which can be toxic, but rather initially forms BeOy·(BeCO3)y, and then at highest levels of CO2 release temperatures, forms BeO in the TGA as a sintered (solid) mass due to the high temperature of formation, rather than an easily dispersible and potentially toxic powder.
The kinetically and thermodynamic-driven release of CO2 by heating BeCO3 and beryllium oxide/carbonates intermediate compounds has been demonstrated, and will presumably similarly occur by alternatively reducing the pressure over those compounds, or by simultaneously heating and reducing the pressure of those compounds. Thermodynamically, the storage of CO2 by beryllium oxide and beryllium oxide/carbonate intermediates is energetically favored by the reverse process of cooling or pressurizing under CO2 beryllium oxide and beryllium oxide/carbonate intermediates and stores CO2. Beryllium oxide is stable, and this stability to reaction can be overcome by introducing kinetic facilitation to increase the rate of CO2 uptake by cooling and/or with pressurized CO2.
Future studies can probe the likelihood that the reverse beryllium oxide reaction with CO2 to beryllium carbonate can be facilitated by means including: (i) bubbling CO2 through the various molten (liquid phase) stages of beryllium oxide and its beryllium oxide/carbonate intermediates or forming a liquid aerosol combined with CO2, (ii) increasing the surface area of the various solid phases stages of beryllium oxide and its beryllium oxide/carbonate intermediates such as by forming a powder, solid aerosol or fixing it to a high surface membrane or aerogel while combining with CO2, (iii) introducing the CO2 by mixing with a combined solid and liquid phase (slush) of beryllium oxide/carbonate intermediates, (iv) or a multistep reaction to incorporate CO2 into beryllium oxide such as, but not limited to, the (iva) the facile reaction of CO2 with ammonium compounds to form ammonium carbonates and the (ivb) reaction of beryllium oxide with sulfate compounds to form beryllium sulfates, followed by the (ivc) the facile reaction of ammonium carbonates and beryllium sulfates to form BeCO3.
Beryllium-induced carbonate electrolyte melting point decrease
Li2CO3 is expensive due its relative scarcity and due to its increasing demand as a primary resource for EVs, but is useful for CO2 removal and its electrolytic transformation to graphene nanocarbons.16 There is less demand for SrCO3 and its derivative salts, such as SrO, are also much more abundant, and an order of magnitude less expensive than Li2CO3.13,51 We had demonstrated that molten Li2CO3 based electrolytes are effective for CO2 carbon capture by the electrolytic splitting of CO2. Interestingly, we recently found that the replacement of the majority of the Li2CO3 by SrCO3 and SrO is also effective. The low-Li2CO3 electrolytes based on SrCO3 are substantially less expensive than comparable Li2CO3-based electrolytes, and are useful for splitting and transforming CO2 to stable graphene nanocarbons including CNTs and carbon nano-onions (ESI†).13 The use of an electrolyte that is a binary mixture (for example, Sr/Li carbonate or SrO/Li2CO3) that can provide a low melting point electrolyte that facilitates transition metal nucleated growth from CO2 of nanographene carbon allotropes is preferred.13,16,40,42,47 Low-Li2CO3 electrolysis may be performed using a planar, rather than a coiled, and brass, rather than Monel, cathode without substantially affecting low-lithium CNT growth from CO2.13,40,41Pure SrCO3 has a high melting point of 1194 °C, and in accord with Table 2 does not decompose until temperatures ≫1000 °C. As previously noted and as measured by TGA, the rapid decomposition of pure Li2CO3 commences near the 723 °C melting under conditions of no CO2 (argon) up through pure CO2.15 A binary SrCO3/Li2CO3 mix has a melting point of 690–790 °C. The melting point increases as the weight percent of SrCO3 in the binary mix increases from 40 to 65%. The eutectic containing 40 wt% SrCO3 melts at 690 °C, while the binary 50% SrCO3 mix melts at 695 °C (ESI†). Fig. 3 demonstrates that this binary mix melting point is substantially decreased by the inclusion of BeCO3 in a ternary mix. Specifically, Fig. 3 compares the TGA's of a binary mix 50/50 wt% Li2CO3/SrCO3 to that of a ternary mix composed one-third by weight each in Li2CO3, SrCO3, BeCO3. The TGA starts from 30 °C, with a 5 °C min−1 temperature increase, and under an 80% N2/20% O2 gas mix.
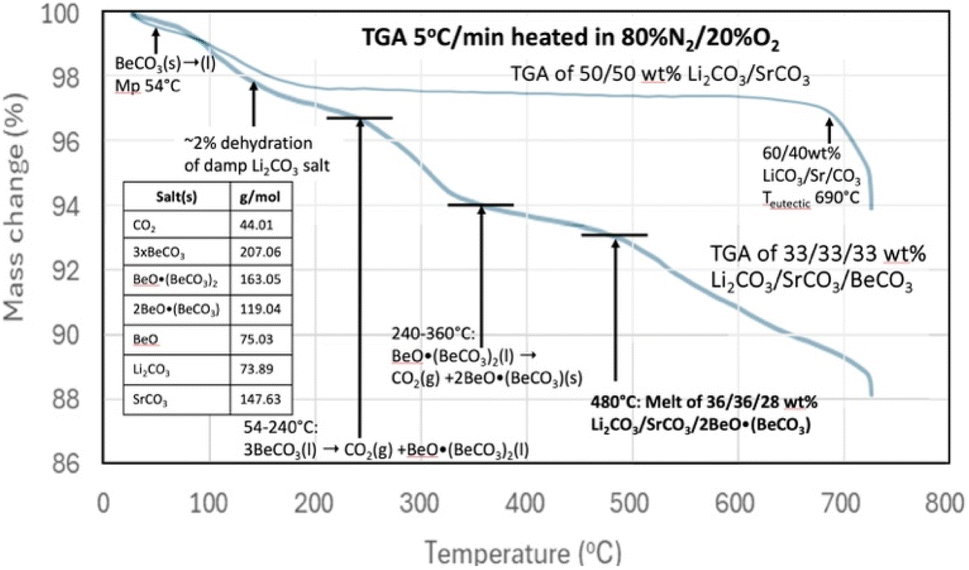 | ||
| Fig. 3 TGA analysis of 50/50 wt% Li2CO3/SrCO3 compared to 33.3/33.3/33.3 wt% Li2CO3/SrCO3/BeCO3. The TGA is conducted from 30 °C to 730 with a 5 °C increase per minute in 80% N2/20% O2. | ||
For the TGA of the binary mix of 50/50 wt% Li2CO3/SrCO3, a few percent weight loss is evident in Fig. 3 at low temperature as the damp material dries. Then the mass is moderately constant, decreasing slowly until a more rapid weight loss occurs around the 690 °C melting point observed for a Li2CO3/SrCO3 mixture. Alternatively, in addition to the low-temperature drying, the 33.3/33.3/33.3 wt% Li2CO3/SrCO3/BeCO3 ternary mix exhibits the hallmarks of pure BeCO3 up to a temperature of 360 °C that were seen in Fig. 2. However, in addition, another sharper decrease in mass loss is observed starting at 480 °C. These are attributed to the melting point of new lower melting ternary mixes of Li2CO3/SrCO3/BeO(BeCO3)2 and specifically of 36/36/28 wt% Li2CO3/SrCO3/BeO when taking into account the loss of CO2 up to 480 °C from the original BeCO3 in the formation of BeO. Note, these wt% masses refer to the measured ratio of masses, as distributed through homogeneous speciation in the oxide dissolved in alkali earth carbonate melt, and not that there is a specific release of CO2 from an isolated alkali earth carbonate within the liquid.
The melting point observed for the Li2CO3/SrCO3/BeCO3 ternary carbonate mix at 480 °C in Fig. 3 is 215 °C lower than the binary Li2CO3/SrCO3mix without the beryllium carbonate addition. Hence, inclusion of BeCO3 can lower the melting point of conventional inorganic carbonates prepared without a mix of BeCO3.
Experimental
Thermodynamic carbonate/CO2 equilibrium calculation
Enthalpies, entropies of species i, i = alkali and alkali earth carbonates, oxides, and CO2 are from standard Barin, NIST (calculated from the available condensed phase thermochemistry data Shomate equations), and NASA data bases.29–39 At any given temperature, the free energy of species “i” was calculated as:| ΔGi(T) = ΔHi(T) − TΔSi(T) | (9) |
The free energy of equilibrium eqn (1) was then calculated as:
| ΔGeqn (1)(T) = ΔGCO2(T) + ΔGMCO(T) − ΔGMCO3(T) | (10) |
The eqn (1) equilibria constants for the various alkali and alkali earth carbonates were then calculated in accord with eqn (2).
TGA measurement, CO2 evolution, and phase change
BeCO3, 99+% purity, was from Chemsavers. Li2CO3 was purchased at a battery grade >99.5%, and used as received. The Li2CO3 had an analyzed composition of 99.8% (Li2CO3, Shanghai Seasongreen Chemical Co). The SrCO3 used was 99.4% pure SrCO3 (Shendong Zhi Chemical Co. Thermal) gravimetric analysis (TGA) was conducted using a PerkinElmer STA 6000 TGA/DSC TGA, under either pure N2 or a mix of 80% N2 and 20% O2. TGAs were conducted with 15 mg of sample using a temperature ramp of 5 °C min−1 over the indicated temperature range. The daily reproducibility of known pure carbonate or graphene samples served as instrumental calibration, and any buildup of residual carbonate was removed by acid wash. In this study, the similarity of the measured mass change with or without the presence of O2 was considered indicative that O2 and related species were not participants in the mass loss sequence. TGA rapid mass loss was considered as gas evolved from the liquid phase, while a low rate of mass loss was considered an indicator of gas evolved from the solid phase as delineated in the text.Conclusions
Rising levels of CO2 in the atmosphere are driving catastrophic climate change, posing an existential threat to the planet. As CO2 concentrations increase, they contribute to global warming, extreme weather events, and ecological disruptions. Addressing this challenge requires innovative solutions, one of which is CO2 capture and sequestration.The capacity for CO2 is a crucial metric in assessing the effectiveness of various chemical species in absorbing, adsorbing or reacting CO2 for sequestration purposes. This capacity is quantified as the amount of CO2 (in kilograms) that can be captured per kilogram of the absorbent material. Common absorbents used in this field include amines, ionic liquids, and calcium oxide, which can transform into calcium carbonate. More recently, lithium oxide (Li2O), which converts to lithium carbonate (Li2CO3), has gained attention as a potential absorbent.
In this study BeCO3 has been introduced as a model compound due to its remarkably high CO2 capture capacity. Although the practical application of beryllium is limited by its scarcity-ranking, as only the 48th most abundant element in the Earth's upper crust, it boasts a CO2 capture capacity of 1.83 kg CO2 per kg of BeO. This is significantly higher than that of other common absorbents: amines range from 0.19–0.37 kg CO2 per kg, ionic liquids capture between 0.07 and 0.09 kg CO2 per kg, calcium carbonate (CaCO3) with a capacity of 0.78 kg CO2 per kg, and lithium carbonate (Li2CO3) that captures 1.47 kg CO2 per kg Li2O.
To better understand the thermodynamics involved, the equilibrium between CO2 and beryllium oxide (BeO) derived from BeCO3 has been calculated and compared to various alkali and alkaline earth carbonates. Thermogravimetric analysis (TGA) of BeCO3 has also been conducted to elucidate the stepwise mechanism of CO2 release, providing insights into how this process in a stepwise release of CO2 at increasing different temperatures.
Additionally, the influence of BeCO3 on the melting point of mixtures has been explored. A comparison of the binary carbonate system consisting of Li2CO3 and strontium carbonate (SrCO3) with a ternary system that includes BeCO3 illustrates how the addition of BeCO3 can substantially depress the melting point. BeCO3 has been presented as a model carbonate to advance the foundation of understanding of the requirements of maximum carbon sequestration. It should be emphasized that beryllium, beryllium carbonate and beryllium oxide are more toxic, less abundant and therefore less available and more expensive than previously studied lower sequestration capacity lithium, magnesium and calcium compounds. This research not only highlights the unique properties of BeCO3, but also contributes to the broader understanding of CO2 capture technologies and their potential role in mitigating climate change.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
G. L. and S. L. designed the research; K.·H., G. L. and S. L. performed the research and analysed the data; G. L. and S. L. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Emissions Reduction Alberta award IT0162473 for partial support of this research.Notes and references
- M. C. Urban, Accelerating extinction risk from climate change, Science, 2015, 348, 571–573 CrossRef.
- H. M. Baek, Possibility of service-regeneration of LiPH for submarines and improvements in CO2 scrubbing performance in LiOH canisters, JAMET, 2022, 46, 115–121 CrossRef.
- S. Satayapal, T. Filburn, J. Trela and J. Strange, Performance and Properties of a Solid Amine Sorbent for Carbon Dioxide Removal in Space Life Support Applications, Energy Fuels, 2001, 15, 250–255 CrossRef.
- C. F. Martín, M. G. Plaza, J. J. Pis, F. Rubiera, C. Pevida and T. A. Centeno, On the limits of CO2 capture capacity of carbons, Sep. Purif. Technol., 2010, 74, 225–229 CrossRef.
- J. Y. Lai, L. H. Ngu and S. S. Hashim, A review of CO2 adsorbents performance for different carbon capture technology processes conditions, Greenhouse Gases:Sci. Technol., 2021, 11, 1076–1117 CrossRef.
- E. Victor, K. Chukwuemeka, O. A. Blessing, I. C. Success, O. Chisom, E. B. Chukwuemeka and N. U. Christian, A Concise Review of Sorbent Materials for Carbon Dioxide Capture and Storage, J. Mat. Sci. Res. Rev., 2022, 10, 72–98 Search PubMed.
- S. Y. W. Chai, L. H. Ngu and B. S. How, Review of carbon capture adsorbents for CO2 utilization, Greenhouse Gases:Sci. Technol., 2022, 12, 394–427 CrossRef.
- Y. Tan, W. Liu, X. Zhang, W. Wei and S. Song, Conventional and optimized testing facilities of calcium looping process for CO2 capture: A systematic review, Fuel, 2024, 358, 130337 CrossRef.
- S. Licht, Efficient solar-driven synthesis, carbon capture and desalinization, STEP, Adv. Mater., 2011, 23, 5592–5612 CrossRef PubMed.
- M. Firdaus, A. Desforges, A. R. Mohamed and B. Vigolo, Progress in adsorption capacity of nanomaterials for carbon dioxide capture: A comparative study, J. Cleaner Prod., 2021, 328, 129553 CrossRef.
- Y. Deng, J. Li, Y. Mia and D. Izikowitz, A comparative review of performance of nanomaterials for Direct Air Capture, Energy Rep., 2021, 7, 3506–3516 CrossRef.
- X. Wang, T. He, J. Hu and M. Liu, The progress of nanomaterials for carbon dioxide capture via the adsorption process, Environ. Sci.:Nano, 2021, 8, 890–912 RSC.
- G. Licht, K. Hofstetter and S. Licht, A new electrolyte for molten carbonate decarbonization, Commun. Chem., 2024, 7, 211 CrossRef CAS.
- Q. Zhu, Y. Zeng and Y. Zheng, Overview of CO2 capture and electrolysis technology in molten salts: operational parameters and their effects, Ind. Chem. Mater., 2023, 1, 59 RSC.
- L. Shi, T. Qu, D. Liu, Y. Deng, B. Yang, and Y. Dai, Process of Thermal Decomposition of Lithium Carbonate, in Materials Processing Fundamentals, J., Lee, S., Wagstaff, G., Lambotte, A., Allanore and F., Tesfaye, The Minerals, Metals & Materials Series, 2020 Search PubMed.
- J. Ren, A. Yu, P. Peng, M. Lefler, F.-F. Li and S. Licht, Recent advances in solar thermal electrochemical process (STEP) for carbon neutral products and high value nanocarbons, Acc. Chem. Res., 2019, 52, 3177–3187 CrossRef PubMed.
- J. Ren, F.-F. Li, J. Lau, L. Gonzalez-Urbina and S. Licht, One-pot synthesis of carbon nanofibers from CO2, Nano Lett., 2015, 15, 6142–6148, DOI:10.1021/jp9044644.
- J. Ren and S. Licht, Tracking airborne CO2 mitigation and low cost transformation into valuable carbon nanotubes, Sci. Rep., 2016, 6, 27760, DOI:10.1038/srep27760.
- G. Licht, K. Hofstetter and S. Licht, Polymer composites with carbon nanotubes made from CO2, RSC Sustainability, 2024, 2, 2496–2504 RSC.
- G. Licht, K. Hofstetter and S. Licht, Buckypaper made with carbon nanotubes derived from CO2, RSC Adv., 2024, 14, 27195–27197 Search PubMed.
- R. C. Ropp, Encyclopedia of the Alkaline Earth Compounds, Elsevier, Netherlands, pp. 359–360, 2013, ISBN 9780444595539 Search PubMed.
- The Differential Thermal Investigations of Clays, ed. T. L. Webb, H. Heystek and R. C. Mackenzie, Mineralogical Society, London, 1957, p.329 Search PubMed.
- A. D. Pelton, C. W. Bale and P. L. Lin, Can. J. Chem., 1984, 62, 457 CrossRef.
- L. Barin, in Thermochemical Data of Pure Substances, VCH, Weinheim, 1989, pp.1–9 Search PubMed.
- N. N. Semenov, T. V. Zablotskii, I. Sib and O. Akad, Nauk SSSR, 1962, 2, 58 Search PubMed.
- E. I. Maslova, I. S. Lileev, I. Sib and O. Akad, Nauk SSSR, 1958, 1, 63 Search PubMed.
- L. S. Itkinaa and nd N. M. Chaplygina, Russ. J. Inorg. Chem., 1962, 7, 1456 Search PubMed.
- Y. Otsubo and K. Yamaguchi, J. Chem.Soc.Japan, 1961, 82, 557 Search PubMed.
- M. Leader, curator, NASA Glenn ThermoBuild thermodynamic database,https://cearun.grc.nasa.gov/ThermoBuild/; last accessed April 17, 2024.
- S. Licht, B. Wang, S. Ghosh, H. Ayub, D. Jiang and J. Ganley, A new solar carbon capture process: STEP carbon capture, J. Phys. Chem. Lett., 2010, 1, 2363–2368 CrossRef CAS.
- S. Licht and B. C. B. Wang, STEP carbon capture: the barium advantage, J. CO2 Util., 2013, 2, 303–312 CrossRef.
- X. Wang, G. Licht, X. Liu and S. Licht, CO2 utilization by electrolytic splitting to carbon nanotubes in non-lithiated, cost effective, molten carbonate electrolytes, Adv. Sustainable Syst., 2022, 2022, 2100481 CrossRef.
- J. Ren, M. Johnson, R. Singhal and S. Licht, Transformation of the greenhouse gas CO2 by molten electrolysis into a wide controlled selection of carbon nanotubes, J. CO2 Util., 2017, 18, 335–344 CrossRef CAS.
- P. Pasierb, R. Gajerski, M. Rokita and M. Rekas, Studies on the binary system Li2CO3-BaCO3, Physica B, 2001, 304, 463–476 CrossRef CAS.
- S. Fangini and A. Masi, Molten carbonates for advanced and sustainable energy applications: Part 1, Int. J. Hydrog. Energy, 2016, 41, 18739–19746 CrossRef.
- J. Ren, J. Lau, M. Lefler and S. Licht, S. The minimum electrolytic energy needed to convert carbon dioxide to carbon by electrolysis in carbonate melts, J. Phys. Chem. C, 2015, 119, 23342–23349 CrossRef CAS.
- P. J. Linstrom and G. Mallard, NIST Chemistry WebBook. NIST Standard Reference Database Number 69, 2001, National Institute of Standards and Technology, J. Chem. Eng. Data, 2001, 46(5), 1059–1063 CrossRef CAS.
- J. Malcom and W. Chase, NIST-JANAF Thermochemical Tables Fourth Ed. Amer Chem Soc, Amer Inst Phys, Nat Inst Stand Tech, 1998, http://webbook.nist.gov, retrieved April 22, 2024 Search PubMed.
- L. Barin, Thermochemical Data of Pure Substances. Part II, VCH, 1989, pp. 1418–1427 Search PubMed.
- X. Liu, G. Licht and S. Licht, Controlled Transition Metal Nucleated Growth of Carbon Nanotubes by Molten Electrolysis of CO2, Catalysts, 2022, 12, 137 CrossRef CAS.
- X. Liu, G. Licht, X. Wang and S. Licht, Controlled Growth of Unusual Nanocarbon Allotropes by Molten Electrolysis of CO2, Catalysts, 2022, 12, 137 CrossRef.
- X. Liu, G. Licht and S. Licht, The green synthesis of exceptional braided, helical carbon nanotubes and nanospiral platelets made directly from CO2, Mater. Today Chem., 2021, 22, 100529 CrossRef.
- X. Wang, G. Licht, X. Liu and S. Licht, One pot facile transformation of CO2 to an unusual 3-D nan-scaffold morphology of carbon, Sci. Rep., 2020, 10, 21518 CrossRef PubMed.
- X. Liu, J. Ren, G. Licht, X. Wang and S. Licht, Carbon nano-onions made directly from CO2 by molten electrolysis for greenhouse gas mitigation, Adv. Sustain. Syst., 2019, 3, 1900056 CrossRef.
- X. Liu, X. Wang, G. Licht and S. Licht, Transformation of the greenhouse gas carbon dioxide to graphene, J. CO2 Util., 2020, 236, 288–294 CrossRef.
- X. Wang, X. Liu, G. Licht and S. Licht, Calcium metaborate induced thin walled carbon nanotube syntheses from CO2 by molten carbonate electrolysis, Sci. Rep., 2020, 10, 15146 CrossRef PubMed.
- X. Wang, F. Sharif, X. Liu, G. Licht, M. Lefler and S. Licht, Magnetic carbon nanotubes: Carbide nucleated electrochemical growth of ferromagnetic CNTs, J. CO2 Util., 2020, 40, 101218 CrossRef.
- M. Johnson, J. Ren, M. Lefler, G. Licht, J. Vicini, X. Liu and S. Licht, Carbon nanotube wools made directly from CO2 by molten electrolysis: Value driven pathways to carbon dioxide greenhouse gas mitigation, Mater. Today Energy, 2017, 5, 230–236 CrossRef.
- J. Ren, M. Johnson, R. Singhal and S. Licht, Transformation of the greenhouse gas CO2 by molten electrolysis into a wide controlled selection of carbon nanotubes, J. CO2 Util., 2017, 18, 335–344 CrossRef.
- G. Licht, K. Hofstetter and S. Licht, Separation of Molten Electrolyte from the Graphene Nanocarbon Product Subsequent to Electrolytic CO2 Capture, Decarbon, 2024, 4, 100044 CrossRef.
- Z. Hu and S. Gao, Upper crustal abundances of trace elements: A revision and update, Chem. Geol., 2008, 253, 205–221 CrossRef.
- A. Kantzas, J. Bryan, S. Taheri, Molecular Diffusion, Chapt. 3 in Fundamentals of Fluid Flow in Porous, Media PERM, 2024, available at: https://perminc.com/resources/fundamentals-of-fluid-flow-in-porous-media/chapter-3-molecular-diffusion/ Search PubMed.
- H. S. Caram, R. Gupta, H. Thomann, F. Ni, S. C. Weston and M. Afeworki, A simple thermodynamic tool for assessing energy requirements for carbon capture using solid or liquid sorbents, Int. J. Greenhouse Gas Control, 2020, 97, 102986 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07753a |
| This journal is © The Royal Society of Chemistry 2024 |