
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA practical method for the synthesis of small peptides using DCC and HOBt as activators in H2O–THF while avoiding the use of protecting groups†
Karina Herrera-Guzmána,
Miguel Ángel Jaime-Vasconcelosa,
Eréndira Toralesa,
Itzel Chacóna,
Rubén Gaviñoa,
Eréndira García-Ríosa,
Jorge Cárdenas *a and
José A. Morales-Serna*b
*a and
José A. Morales-Serna*b
aInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Ciudad de Mexico, 04510, Mexico. E-mail: rjcp@unam.mx
bCentro de Investigaciones Científicas, Instituto de Química Aplicada, Universidad del Papaloapan, Tuxtepec, Oaxaca, 68301, Mexico. E-mail: joseantonio.moralesserna@gmail.com
First published on 19th December 2024
Abstract
The synthesis of peptides in solution proceeds through successive steps involving the removal of a protecting group and the formation of the peptide bond. While most methodological efforts have focused on the development of new protecting groups and coupling agents, methodologies based on minimal protecting groups have been less explored. In this research, a peptide synthesis methodology was developed using DCC and HOBt in THF–H2O, avoiding the use of protecting groups, reducing reaction times, and reusing HOBt during successive couplings. The reaction conditions allow the production of peptides that can directly serve as the starting material for the next coupling, leading to the creation of small peptide sequences, which in turn are precursors to biologically important molecules. Here we explore the example of Sansalvamide as a template for other active peptides. Unlike SPPS, our methodology constructs the sequence from the N-terminus to C-terminus. This unique approach could streamline peptide synthesis and facilitate the development of complex peptides efficiently.
Introduction
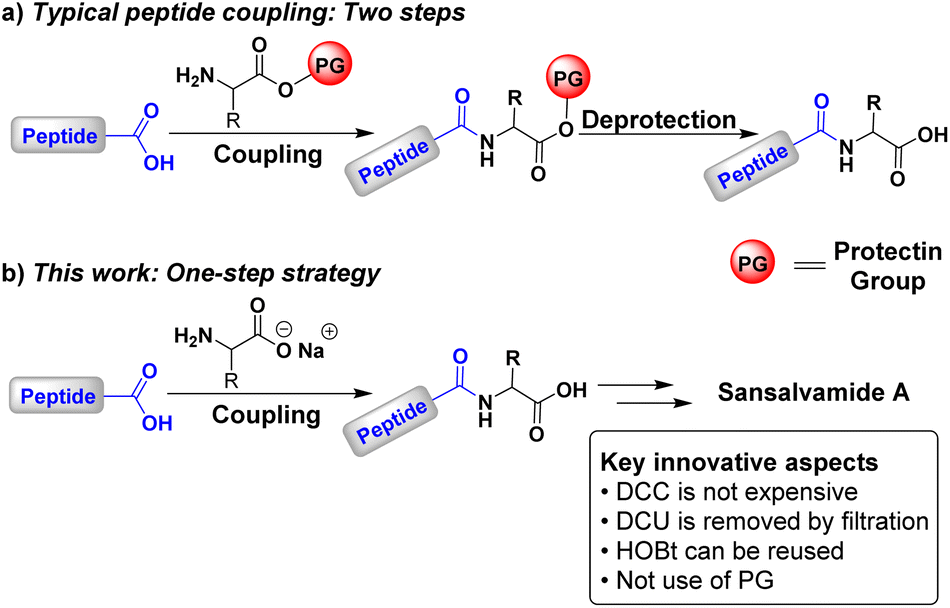
Peptide synthesis is at the cutting edge of contemporary research, as peptides play an important role in many fields. Peptide-based materials with diverse functionalities address biomaterials with significant interest because they complement small molecule therapeutics.1 However, peptide synthesis remains a challenging task due to the inherent thermodynamic limitations of amide bond formation. From an energetic standpoint, the formation of an amide from a carboxylic acid and an amine is thermodynamically unfavourable.2 To facilitate this process, it is essential to exploit carboxylic acid derivatives, such as acyl halides, anhydrides, and esters.3–5 For peptide bond formation, the most common approach involves activating the carboxylic acid using coupling agents, which enables bond formation under mild reaction conditions.6–9The synthesis of peptides with a defined sequence of amino acids is a demanding task, despite it only involves forming a series of amide bonds to build the desired peptide sequence.10,11 This complexity arises from the need to protect functional groups not involved in the amide bond formation12,13 and in order to prevent undesired side reactions, such as the formation of N-acyl ureas, oxazolones,14 and peptide epimers.15–17 Peptide bond formation typically proceeds through successive coupling and deprotection steps,18–21 requiring two reactions for each new C–N bond (Scheme 1a). While the correct choice of protecting groups ensures the desired sequence, the appropriate selection of coupling agents and reaction conditions is crucial to avoid racemisation or epimerisation of the final product.12–17
 | ||
| Scheme 1 Synthetic strategy. | ||
Although established methodologies exist,11,21 recent efforts have focused on developing new coupling strategies,22–30 coupling agents,31–41 and protecting groups,42–44 as well as on establishing synthetic protocols where amino acids are mostly free of protecting groups45–49 and employing environmentally friendly solvents.50–56 Here, we propose a strategy for synthesising small peptides to contribute to this kind of methodologies. Our approach is based on the use of N,N′-dicyclohexylcarbodiimide (DCC) and 1-hydroxybenzotriazole (HOBt) in a THF–H2O mixture. This approach eliminates the need for protecting groups and, consequently, extra deprotection steps (Scheme 1b). Furthermore, the peptide obtained from the reaction can be transferred directly to the next coupling without purification. This straightforward approach resembles solid-phase peptide synthesis (SPPS) since intermediates are not isolated.57–60 Nonetheless, it is important to highlight the absence of intermediate deprotection steps.61–65
Another significant contribution from our strategy is the use of DCC as a coupling agent. Despite being cost-effective compared to other water-soluble carbodiimides, DCC is often avoided due to the challenges associated with removing its byproduct, 1,3-dicyclohexyl urea (DCU).66 To facilitate the removal, we took advantage of the environmentally friendly solvent mixture THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O as precipitation and elimination of DCU is very easy by filtration, avoiding the need for chromatographic techniques. Additionally, our approach allows for the reuse of HOBt during peptide chain growth, as previously reported.67
:H2O as precipitation and elimination of DCU is very easy by filtration, avoiding the need for chromatographic techniques. Additionally, our approach allows for the reuse of HOBt during peptide chain growth, as previously reported.67
Given the current interest in developing efficient protocols for synthesising cyclic peptides68,69 and the need for synthetic samples in biological assays,70–74 we aim to apply our methodology to the synthesis of different depsipeptides, focusing first on Sansalvamide A as target.75 This application will help to demonstrate the potential scope and impact of our work.
Results and discussion
To establish optimal reaction conditions, we first evaluated various conditions for synthesising the dipeptide N-Boc-Pro-Phe-OH 6 from N-Boc-Pro 1 and the zwitterion of L-phenylalanine 4. The process begins with the formation of benzotriazole esters 2 and 3 through the reaction of DCC and HOBt in CH2Cl2 (Scheme 2). After 30 minutes, thin-layer chromatography (TLC) confirmed that the conversion to activated esters was complete. Dichloromethane was chosen for this step due to its effectiveness in quantitatively forming these intermediates.76–79 Following solvent removal, the mixture of esters 2 and 3 was redissolved in THF exploring its water miscibility, a key feature in peptide bond formation under our reaction conditions.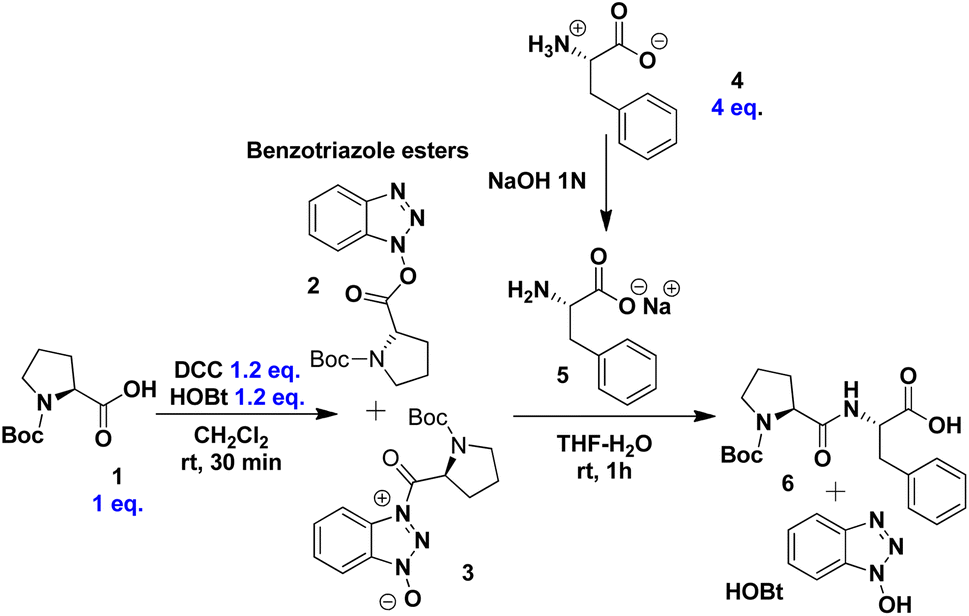 | ||
| Scheme 2 Synthesis of dipeptide 6. | ||
Meanwhile, the zwitterion of L-phenylalanine 4 was dissolved in THF. To obtain the predominant carboxylate form 5, an equimolar amount of a 1 N aqueous solution of NaOH was added. Consequently, the amino group acts as the most nucleophilic species for peptide bond formation. To form dipeptide 6, the solution containing 5 was slowly added to the mixture of benzotriazol esters 2 and 3, with constant stirring for 5 minutes. After stirring for an additional 55 minutes, the reaction mixture was filtered to remove DCU, yielding a product consisting of dipeptide 6 and HOBt (Scheme 2).
Next, we established a kinetic profile varying the equivalents of 4, which helped determine the highest yield of reaction when an excess of 4 equivalents of zwitterion were added (Fig. 1a). Additionally, the reaction yield for 6 reached a maximum of 80% with no further increase despite extended reaction time (Fig. 1b). Yields were quantified by 1H NMR of the crude reaction mixture.
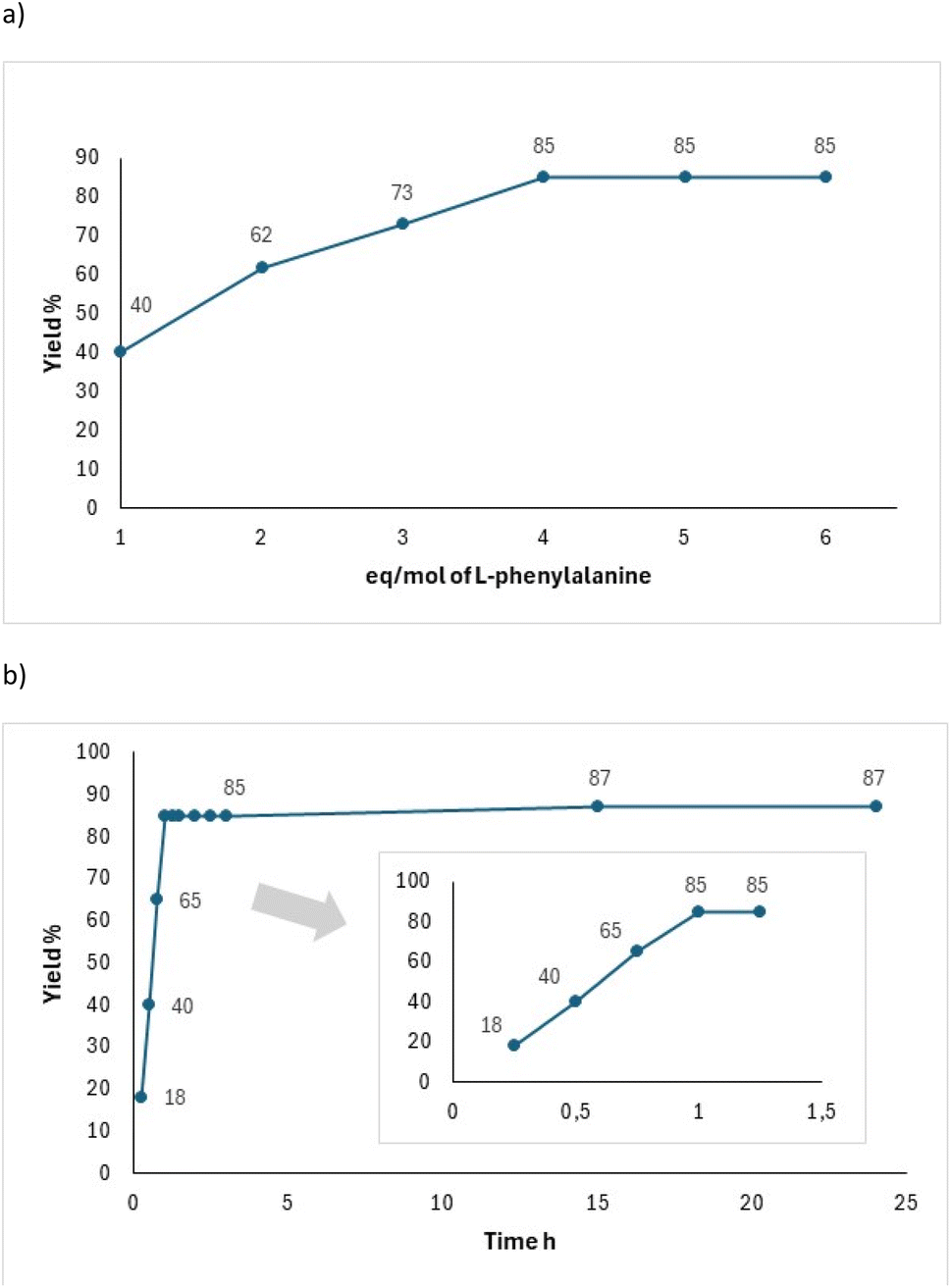 | ||
| Fig. 1 Reaction kinetic to obtain 6: (a) varying equivalents of L-phenylalanine 4 and (b) varying the reaction time. | ||
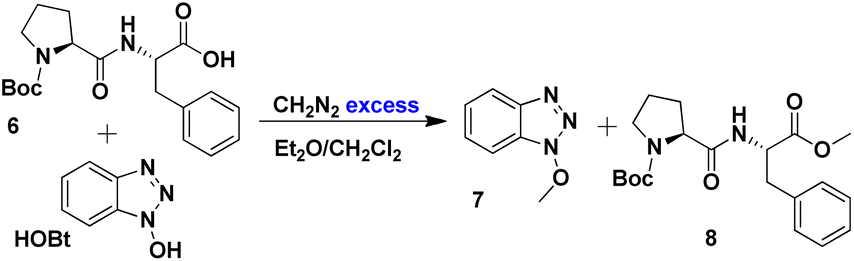
The mixture of HOBt and dipeptide 6 underwent a methylation reaction with diazomethane, resulting in the quantitative formation of the methylated derivatives of benzotriazole 7 and dipeptide 8 (Scheme 3). The reaction products were separated by column chromatography, and 8 was characterised by NMR, with no epimer formation observed. The optical rotation reported for compound 8 in methanol was −53.4° (λ = 589 nm),80 while our value at 25 °C was −53.8° (λ = 589 nm), establishing that no epimerisation occurred during peptide bond formation.
 | ||
| Scheme 3 Methylation reaction with diazomethane. | ||
To further explore our methodology, we synthesised peptides 9–15 from Boc-L-Pro 1, with yields determined by 1H NMR (Table 1). The lowest yield (50%) was observed with non-chiral glycine, while the highest (80%) was seen with leucine.
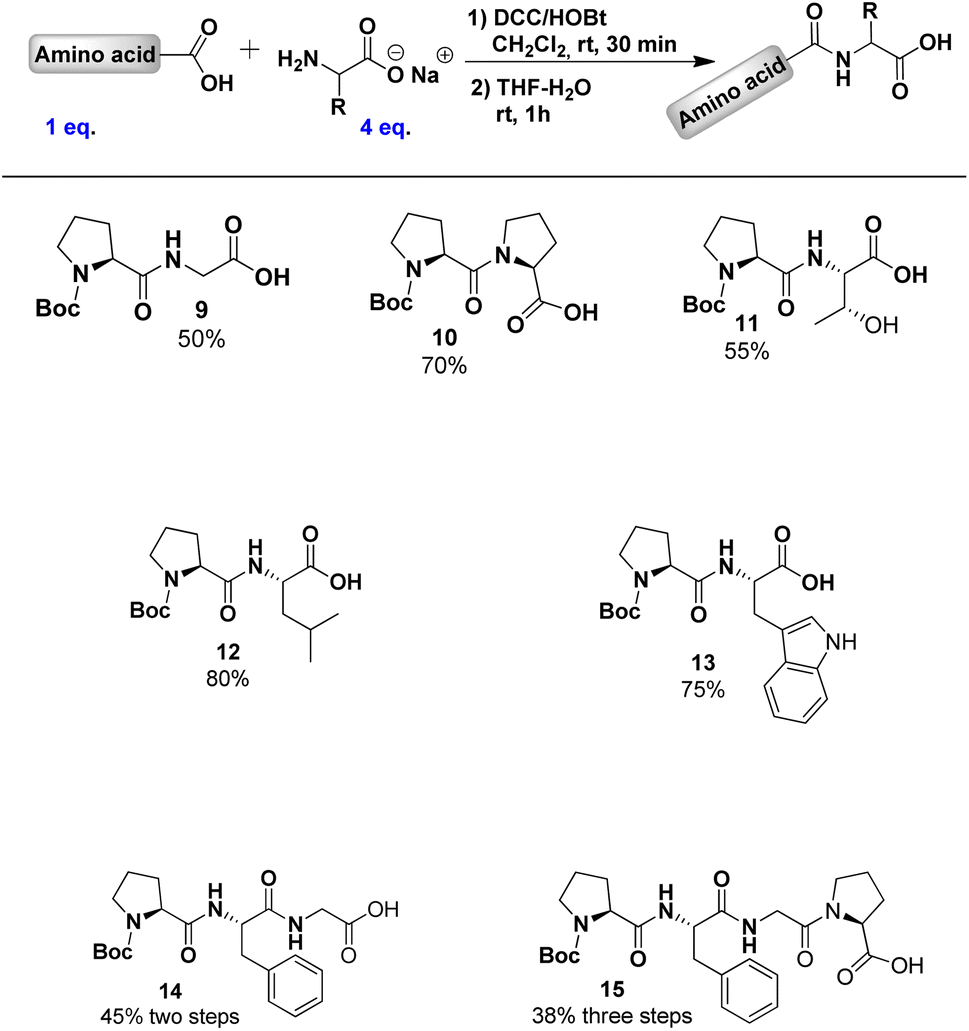 |
Next, we focused on synthesising Sansalvamide A 23 to demonstrate the applicability of our peptide chain elongation strategy to biologically relevant molecules. Sansalvamide A is a cyclopenta depsipeptide isolated from the fungus Fusarium sp.,75 noted for its inhibitory activity against the topoisomerase of the Molluscum contagiosum81 virus and its cytotoxicity against various colon cancer cell lines (COLO 205, SKMEL-2, and HCT 116, HT-29).82–84
The initial step in synthesising Sansalvamide A involved coupling α-hydroxy acid 16 with the zwitterion of L-valine 17 (Scheme 4). The corresponding benzotriazole esters of 16 were first formed in the presence of DCC and HOBt in CH2Cl2. The formation of activated esters was verified by TLC, and after 30 minutes of reaction, the conversion was found to be quantitative. After solvent removal, the mixture of benzotriazole esters was dissolved in THF. As established with 4, L-valine 17 was meanwhile being dissolved in a THF/aqueous NaOH solution. Following the addition of 16 to 17, dipeptide 18 in a mixture with HOBt was used without further purification for the next step. Subsequent coupling steps lead to completion of the primary structure of Sansalvamide A until peptide 22 without intermediate purification. Peptide 22 is the direct precursor of the cyclodepsipeptide Sansalvamide A (Scheme 4). The formation of each peptide bond was monitored via 1H NMR analysis of the crude reaction mixture.
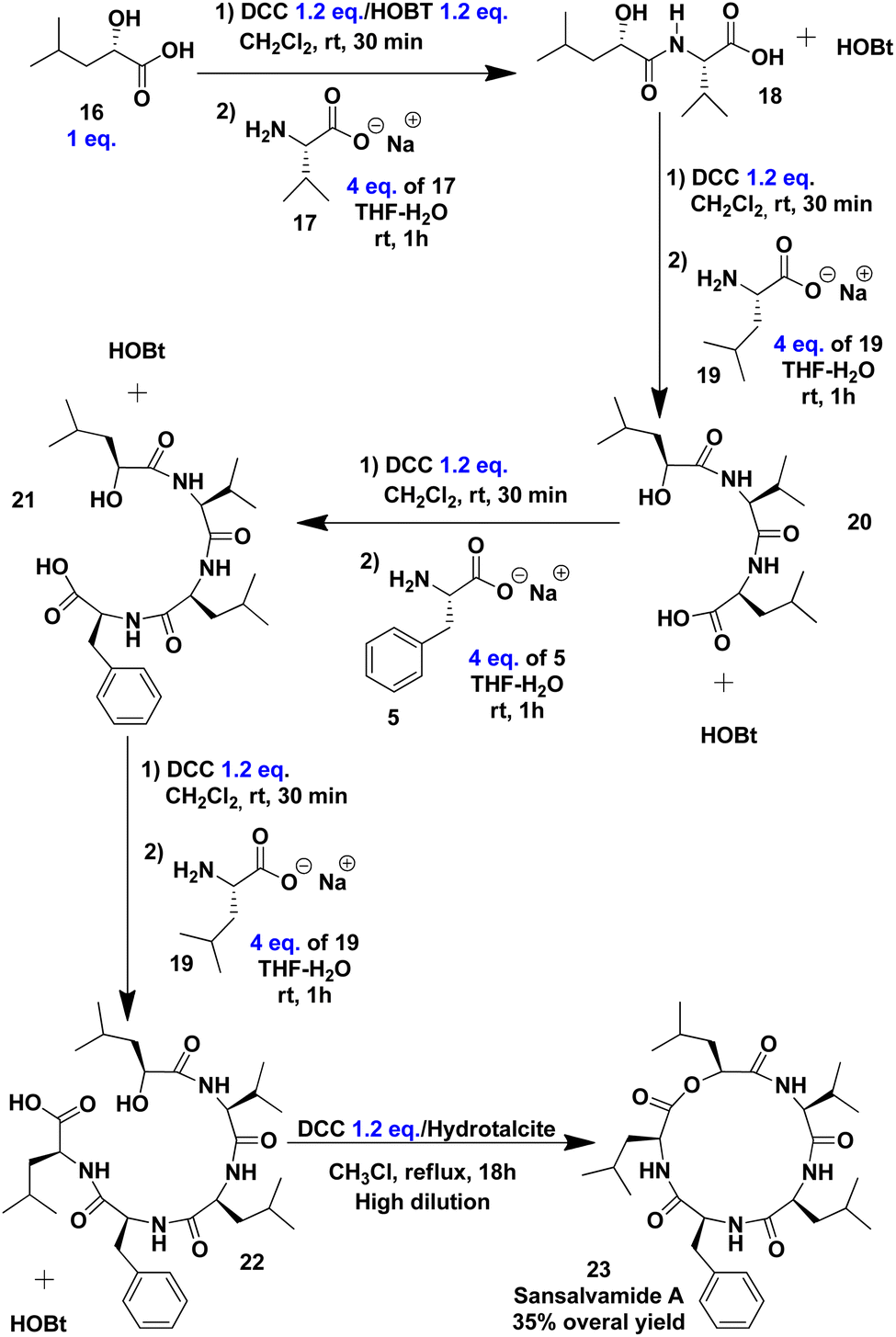 | ||
| Scheme 4 Synthesis of Sansalvamide A. | ||
The macrolactonization as the final step took place under high-dilution conditions in the presence of DCC and hydrotalcite, a solid catalytic material serving as a solid base.85 The resulting mixture was purified by column chromatography, yielding cyclodepsipeptide 23 as a white solid, which was characterised by NMR and electrospray ionisation ion trap (ESI-IT) mass spectrometry (see ESI†). The NMR data matched those reported for the natural product (Table 2), and the purity of 23 was confirmed by HPLC, with no epimers detected.
| 1H NMR (CD3OD) | 13C NMR (CD3OD) | |||
|---|---|---|---|---|
| Ref. 74 | This work | Ref. 74 | This work | |
| OLeu | ||||
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
172.9 | 172.8 | ||
| α-CH | 5.06 (dd, J = 8.8, 4.9 Hz) | 5.06 (dd, J = 9, 4.8 Hz) | 76.2 | 76.3 |
| β-CH2 | 1.76–1.88 (m) | 1.62–1.88 (m) | 42.1 | 41.8 |
| 1.58–1.64 (m) | ||||
| γ-CH | 1.66–1.78 (m) | 1.62–1.67 (m) | 26.1 | 26.1 |
| δ-CH3 | 0.99 (d, J = 6.4 Hz) | 0.99 (d, J = 6.4 Hz) | 23.4 | 23.3 |
| 0.96 (d, J = 6.4 Hz) | 0.96 (d, J = 6.4 Hz) | 22.2 | 22.1 | |
|
||||
| Val | ||||
| CO |
173.8 | 174.0 | ||
| α-CH | 4.09 (d, J = 8.8 Hz) | 4.09 (d, J = 8.5 Hz) | 60.9 | 60.7 |
| β-CH | 2.10 (m) | 2.07 (m) | 32.2 | 32.1 |
| γ-CH3 | 0.96 (d, J = 6.4 Hz) | 0.92 (d, J = 6.6 Hz) | 20.0 | 19.9 |
| 0.92 (d, J = 6.8 Hz) | 0.86 (d, J = 6.6 Hz) | 19.0 | 18.8 | |
|
||||
| Leu-1 | ||||
| CO |
174.1 | 174.09 | ||
| α-CH | 3.83 (br dd, J = 8.8, 5.4 Hz) | 3.74 (dd, J = 9, 5.2 Hz) | 56.2 | 56.4 |
| β-CH2 | 1.72 (m) | 1.72 (m) | 40.1 | 39.7 |
| 1.38 (m) | 1.38 (m) | |||
| γ-CH | 1.41 (m) | 1.41 (m) | 26.2 | 26.2 |
| δ-CH3 | 0.85 (d, J = 6.4 Hz) | 0.85 (d, J = 6.5 Hz) | 23.2 | 23.1 |
| 0.81 (d, J = 5.9 Hz) | 0.81 (d, J = 6.2 Hz) | 22.2 | 22.1 | |
|
||||
| Phe | ||||
| CO |
173.7 | 173.6 | ||
| α-CH | 4.60 (dd, J = 10.8, 4.4 Hz) | 4.57 (dd, J = 10.8, 4.8 Hz) | 58.1 | 58.1 |
| β-CH2 | 3.23 (dd, J = 13.9, 4.6 Hz) | 3.23 (dd, J = 13.9, 4.8 Hz) | 38.4 | 38.1 |
| 3.06 (dd, J = 13.7, 10.7 Hz) | 3.07 (dd, J = 13.9, 10.8 Hz) | |||
| 1 Ar | 138.8 | 138.7 | ||
| 2 and 6 Ar | 7.25–7.32 (m) | 7.22–7.29 (m) | 130.4 | 130.2 |
| 3 and 5 Ar | 7.25–7.32 (m) | 7.22–7.29 (m) | 129.7 | 129.7 |
| 4 Ar | 7.22 (m) | 7.22–7.29 (m) | 128.0 | 128.0 |
|
||||
| Leu-2 | ||||
| CO |
171.3 | 171.4 | ||
| α-CH | 4.73 (dd, J = 9.8, 5.9 Hz) | 4.72 (dd, J = 9.7, 5.8 Hz) | 52.6 | 52.5 |
| β-CH2 | 1.64–1.86 (m) | 1.64–1.86 (m) | 41.6 | 41.4 |
| γ-CH | 1.62 (m) | 1.62 (m) | 26.0 | 25.9 |
| δ-CH3 | 0.99 (d, J = 6.4 Hz) | 0.99 (d, J = 6.4 Hz) | 23.5 | 23.5 |
| 0.96 (d, J = 6.4 Hz) | 0.96 (d, J = 6.4 Hz) | 22.4 | 22.4 | |
After purifying the final product, the overall yield of the process was 35%. To enhance this yield, we tested THF instead of CH2Cl2 for benzotriazole ester formation, but the overall yield dropped to 20%. A comparison of our methodology with those previously reported for Sansalvamide A showed our yield is not different from those works using LPPS86 or SPPS.87,88 Moreover, the time required for peptide bond formation is strikingly reduced, as detailed in Table 3.
| Strategy | ||||
|---|---|---|---|---|
| LPPSa | LPPSa | SPPSb | SPPSb | |
| a LPPS: Liquid Phase Peptide Synthesis.b SPPS: Solid Phase Peptide Synthesis. | ||||
| Coupling reagent | DCC/HOBt | EDC/HOBt | HBTU | PyBOP |
| Reuse of coupling reagent | No/Yes | No/No | No | No |
| Solvent | THF/H2O | CH2Cl2 | NMP | CH3CN |
| Time for coupling (h) | 1.5 | 16 | 6 | 18 |
| Steps | 5 | 9 | 13 | 12 |
| Overall yield (%) | 35 | 45 | 40 | 13 |
| Reference | This work | 86 | 87 | 88 |
Importantly, while DCC is typically avoided due to challenges in removing its byproduct, DCU, our use of a THF–H2O solvent system facilitates its precipitation and removal by filtration. Furthermore, our methodology minimises waste generation, as HOBt is reused throughout the process. These achievements of our approach undoubtedly represent significant environmental and economic benefits.89–93
Conclusions
This work focused on the development of an efficient methodology for the coupling of peptides without protective groups in a THF–H2O system. Our approach not only allows the coupling of amino acids in a shorter time than other methodologies, but it is also cost-effective due to the use of DCC and recycling of HOBt. This results in a significant reduction in the waste produced during the reaction.Finally, we tested this methodology with the total synthesis of Sansalvamide A, a cyclodepsipeptide with anticancer activity. We strongly believe this approach will facilitate the development of libraries for the study of cyclopeptides, thus contributing to the advancement of research on biologically active compounds.
Experimental
General information
All the chemicals were purchased from Sigma-Aldrich and used without further purification. Yields refer to materials that are chromatographically and spectroscopically homogeneous (1H and 13C) unless otherwise stated. Reactions were monitored by TLC on 0.25 mm Macherey Nagel silica gel plates. Developed TLC plates were visualised under a short-wave UV lamp and by heating plates dipped in Ce(SO4)2 or ninhydrin reagent 2% ethanol solution. Flash column chromatography (FCC) was performed using flash silica gel (230–400) with solvent polarity correlated to TLC mobility. Optical rotations were measured at 598 nm on a Jasco DIP-370 digital polarimeter using a 100 mm cell. NMR experiments were conducted on aVarian-Gemini 200 MHz, Varian Unity 300 MHz and, Bruker-Avance 300 MHz instruments, using CDCl3 (99.9% D) as the solvent. Chemical shifts (δ) were referenced to internal standards CDCl3 (7.26 ppm 1H, 77.0 ppm 13C) or Me4Si as an internal reference (0.00 ppm). Chemical shifts are reported in parts per million (ppm). HPLC/MS were collected using a Waters 1525 chromatograph coupled with a Bruker Esquire 600 ESI-IT using a ZORBAX Eclipse Plus C18 3.5 μm 2.1 × 100 mm (Agilent), the mobile phase flow-rate was 0.2 mL min−1 and the detection range was 200–600 nm. Elution solvents were A (water/formic acid 99.9/0.1, v/v) and B (methanol) and the elution program was from 40 to 100% of B during 15 min followed by isocratic elution with 100% of B during 7 min.General procedure for peptide bond formation
Preparation of diazomethane
N-Nitroso-N-methylurea (250 mg) was added to ethyl ether (20 mL), and suspended over a 50% aqueous solution of KOH (6 mL) at 0 °C. The reaction mixture was stirred for 20 minutes, then transferred to a separatory funnel where the phases were separated. The ether phase was dried over KOH pellets.:1), and the ethereal solution of diazomethane was added at 0 °C. The reaction mixture was stirred for 15 minutes and then left at room temperature for 20 hours. The solvents were eliminated under reduced pressure, and the crude reaction mixture crude was separated by column chromatography. The dipeptide 8 was obtained from the elution mixture of EtOAc–hexane (20:80). The product was recrystallised from an Et2O–hexane mixture, yielding a white product (99% yield) with a melting point of 67–69 °C.Synthesis of Sansalvamide A 24
Synthesis of 18. Flask A: a solution of α-hydroxy acid 16 (1 g, 7.575 mmol), DCC (1.872 g, 9.090 mmol) and, HOBt (1.227 g, 9.090 mmol) in CH2Cl2 (30 mL) was stirred at room temperature for 30 minutes. CH2Cl2 was evaporated under reduced pressure, and the residue was dissolved in THF (50 mL). Flask B: to a solution of zwitterion of L-valine 17 (3.545 g, 30.300 mmol) in THF (60 mL), 30 mL of 1 N NaOH was added, and the mixture was stirred for 5 minutes at room temperature. The solution from flask B was added to flask A over 5 minutes at room temperature. After 55 minutes, the reaction mixture was filtered, and the filtrate was placed in a separatory funnel to be extracted with EtOAc (2 × 50 mL). The aqueous phase was adjusted to pH 3 and subsequently extracted with EtOAc (3 × 50 mL). The organic phases were combined and washed with brine (3 × 50 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product contains peptide 18 and HOBt. The yield (90%) was quantified by 1H NMR of the crude reaction mixture, and the mixture was used without purification in the next step.
Synthesis of 20. Following the general procedure, the reaction was carried out with zwitterion of L-leucine 19 (3.572 g, 27.268 mmol) and DCC (1.685 g, 8.180 mmol). Yield (85%) was determined by 1H NMR of the crude reaction mixture, and the mixture was used without purification in the next step.
Synthesis of 21. Following the general procedure, the reaction was carried out with zwitterion of L-phenylalanine 5 (3.824 g, 23.176 mmol) and DCC (1.432 g, 6.952 mmol). Yield (80%) was quantified by 1H NMR of the crude reaction mixture, and peptide 21 was used in the next step without purification.
Synthesis of 22. Following the general procedure, the reaction was carried out with zwitterion of L-leucine 19 (2.428 g, 18.540 mmol) and DCC (1.145 g, 5.562 mmol). Peptide 22 (77%) and HOBt as a white solid were obtained. Peptide 22 was quantified by 1H NMR from the crude reaction mixture and used in the next step without purification.
Synthesis of depsipeptide 24 via a macrolactonization reaction. A solution of DCC (881 mg, 4.281 mmol) and hydrotalcite (200 mg) in ethanol-free chloroform (100 mL) was brought to reflux. A solution of peptide 22 and HOBt in 20 mL of THF was infused via a syringe pump over 18 h, the reaction mixture was then filtered and evaporated. It was then diluted with EtOAc (75 mL), washed with 10% citric acid solution (2 × 50 mL), 10% NaHCO3 solution (2 × 50 mL), brine (2 × 50 mL), dried over Na2SO4 and concentrated under vacuum. The crude product was purified through a silica gel column chromatography, first eluting with hexane and then with 70% EtOAc–hexane. Finally, Sansalvamide A 23 was obtained as white solid (1.553 g, 75%, m.p. 144–146 °C).
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. C. and J. A. M. S. conceived the project and acquired the funds. M. A. J. V., R. G., J. C., and J. A. M. S. designed the experiments. M. A. J. V., K. H. G., E. T., I. C., R. G., E. G. R., J. C., and J. A. M. S. conducted the experimental work. J. C. and J. A. M. S. coordinated the whole project. M. A. J. V., K. H. G., R. G., E. G. R., J. C., and J. A. M. S. wrote the manuscript. All the authors contributed to the discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT-UNAM, Project No. IN209818). E. T. is grateful to Consejo Nacional de Ciencia y Tecnología (CONACyT-355820) for the PhD fellowship.Notes and references
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Trends in peptide drug discovery, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- P. Gao, M. M. Rahman, A. Zamalloa, J. Feliciano and M. Szostak, Classes of amides that undergo selective N–C amide bond activation: the emergence of ground-state destabilization, J. Org. Chem., 2022, 88, 13371–13391 CrossRef PubMed.
- P. Acosta-Guzmán, A. Ojeda-Porras and D. Gamba-Sánchez, Contemporary approaches for amide bond formation, Adv. Synth. Catal., 2023, 365, 4359–4391 CrossRef.
- E. Massolo, M. Pirola and M. Benaglia, Amide bond formation strategies: latest advances on a dateless transformation, Eur. J. Org Chem., 2020, 4641–4651 CrossRef CAS.
- J. Singh and A. Sharma, Green and sustainable visible light-mediated formation of amide bonds: an emerging niche in organic chemistry, New J. Chem., 2022, 46, 16220–16242 RSC.
- J. Yang, H. Huang and J. Zhao, Active ester-based peptide bond formation and its application in peptide synthesis, Org. Chem. Front., 2023, 10, 1817–1846 RSC.
- T. I. Al-Warhi, H. M. Al-Hazimi and A. El-Faham, Recent development in peptide coupling reagents, J. Saudi Chem. Soc., 2012, 16, 97–116 CrossRef CAS.
- S. Y. Han and Y. A. Kim, Recent development of peptide coupling reagents in organic synthesis, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS.
- E. Valeur and M. Bradley, Amide bond formation: beyond the myth of coupling reagents, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- A. Sharma, A. Kumar, B. G. De La Torre and F. Albericio, Liquid-phase peptide synthesis (LPPS): a third wave for the preparation of peptides, Chem. Rev., 2022, 122, 13516–13546 CrossRef CAS PubMed.
- P. Lloyd-Williams, F. Albericio and E. Giralt, in Chemical Approaches to the Synthesis of Peptides and Proteins, CRC Press, Boca Raton, 1997 Search PubMed.
- I. Ramakrishna, T. Hattori, T. Ishiguro and H. Yamamoto, Triisobutylaluminium-mediated regioselective protection of sterically hindered amide NH of cyclo-(AA-Gly): key building block for next-generation peptide synthesis, Synlett, 2024, 35, 1113–1120 CrossRef CAS.
- H. Itoh and M. Inoue, Full solid-phase total synthesis of macrocyclic natural peptides using four-dimensionally orthogonal protective groups, Org. Biomol. Chem., 2019, 17, 6519–6527 RSC.
- S. R. Manne, D. C. Akintayo, O. Luna, A. El-Faham, B. G. De La Torre and F. Albericio, Tert-butylethylcarbodiimide as an efficient substitute for diisopropylcarbodiimide in solid-phase peptide synthesis: understanding the side reaction of carbodiimides with OxymaPure, Org. Process Res. Dev., 2022, 26, 2894–2899 CrossRef CAS.
- V. Di Matteo, G. Esposito, V. Costantino, G. Della Sala, R. Teta and A. Mangoni, When synthesis gets it wrong: unexpected epimerization using PyBOP in the synthesis of the cyclic peptide Thermoactinoamide A, J. Nat. Prod., 2024, 87, 948–953 CrossRef CAS PubMed.
- M. Kobayashi, K. Fujita, K. Matsuda and T. Wakimoto, Streamlined chemoenzymatic synthesis of cyclic peptides by non-ribosomal peptide cyclases, J. Am. Chem. Soc., 2023, 145, 3270–3275 CrossRef CAS PubMed.
- G. Liu, Y. Guo, M. Wang and Y. Gao, Recent advances in asymmetric synthesis of chiral amides and peptides: racemization-free coupling reagents, Org. Biomol. Chem., 2024, 24, 4420–4435 Search PubMed.
- M. Anand, M. Alagar, J. Ranjitha and V. Selvaraj, Total synthesis and anticancer activity of a cyclic heptapeptide from marine sponge using water soluble peptide coupling agent EDC, Arabian J. Chem., 2019, 12, 2782–2787 CrossRef CAS.
- C. A. Arbour, L. G. Mendoza and J. L. Stockdill, Recent advances in the synthesis of C-terminally modified peptides, Org. Biomol. Chem., 2020, 18, 7253–7272 RSC.
- I. F. Eggen, F. T. Bakelaar, A. Petersen, P. B. W. Ten Kortenaar, N. H. S. Ankone, H. E. J. M. Bijsterveld, G. H. L. Bours, F. E. Bellaj, M. J. Hartsuiker, G. J. Kuiper and E. J. M. Ter Voert, A novel method for repetitive peptide synthesis in solution without isolation of intermediates, J. Pept. Sci., 2005, 11, 633–641 CrossRef CAS PubMed.
- G. K. Rathod, R. Misra and R. Jain, Advances in Peptide Synthesis, in Recent Advances in Pharmaceutical Innovation and Research, ed. P. P. Singh, Springer, Singapore, 2023, pp. 193–213 Search PubMed.
- A. Yamada, T. Takei, T. Kawakami, Y. Taniguchi, K. Sekiguchi and H. Hojo, Application of cysteinyl prolyl ester for the synthesis of cyclic peptides containing an RGD sequence and their biological activity measurement, Front. Chem., 2024, 12, 1391678 CrossRef CAS PubMed.
- X. Y. Liu, W. Cai, N. Ronceray, A. Radenovic, B. Fierz and J. Waser, Synthesis of fluorescent cyclic peptides via gold (I)-catalyzed macrocyclization, J. Am. Chem. Soc., 2023, 145, 26525–26531 CrossRef CAS PubMed.
- A. J. Mijalis, D. A. Thomas III, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, A fully automated flow-based approach for accelerated peptide synthesis, Nat. Chem. Biol., 2017, 13, 464–466 CrossRef CAS PubMed.
- Y. Mifune, H. Nakamura and S. Fuse, A rapid and clean synthetic approach to cyclic peptides via micro-flow peptide chain elongation and photochemical cyclization: synthesis of a cyclic RGD peptide, Org. Biomol. Chem., 2016, 14, 11244–11249 RSC.
- H. Li, J. Li, J. Chao, Z. Zhang and C. Qin, Head-to-tail cyclization for the synthesis of naturally occurring cyclic peptides on organophosphorus small-molecular supports, Org. Chem. Front., 2022, 9, 946–952 RSC.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Ligation technologies for the synthesis of cyclic peptides, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed.
- A. Wu, I. Ramakrishna, T. Hattori and H. Yamamoto, Silicon-based hydrophobic tags applied in liquid-phase peptide synthesis: protected DRGN-1 and poly alanine chain synthesis, Org. Biomol. Chem., 2022, 20, 8685–8692 RSC.
- A. Wróblewska, I. I. Bak-Sypien, P. Paluch, E. Wielgus, J. Zając, A. Jeziorna, S. Kaźmierski and M. J. Potrzebowski, Solvent-free mechanosynthesis of oligopeptides by coupling peptide segments of different lengths–elucidating the role of cesium carbonate in ball mill processes, Chem.–Eur. J., 2024, 30, e202400177 CrossRef PubMed.
- A. K. Mishra, G. Parvari, S. K. Santra, A. Bazylevich, O. Dorfman, J. Rahamim and A. M. Szpilman, Solar and visible light assisted peptide coupling, Angew. Chem., Int. Ed., 2021, 60, 12406–12412 CrossRef CAS PubMed.
- A. Sakurada, M. Sato, K. Higashida and M. Sawamura, Gold–zinc co–catalyzed alkynoate hydrocarboxylation with N-protected amino acids for preparation of storable acylating reagents and racemization–free peptide synthesis, Adv. Synth. Catal., 2024, 366, 2507–2513 CrossRef CAS.
- J. Yang, D. Zhang, Y. Chang, B. Zhang, P. Shen, C. Han and J. Zhao, TFPN–mediated racemization/epimerization–free amide and peptide bond formation, Org. Chem. Front., 2024, 11, 5422–5428 RSC.
- J. Xu, S. Yuan, M. Miao and Z. Chen, 1-Hydroxybenzotriazole-assisted, N-heterocyclic carbene catalyzed β-functionalization of saturated carboxylic esters: access to spirooxindole lactones, J. Org. Chem., 2016, 81, 11454–11460 CrossRef CAS PubMed.
- T. Nobuta, N. Tsuchiya, Y. Suto and N. Yamagiwa, Hexylsilane-mediated direct amidation of amino acids with a catalytic amount of 1,2,4-triazole, Org. Biomol. Chem., 2024, 22, 703–707 RSC.
- S. Xu, D. Jiang, Z. Peng, L. Hu, T. Liu, L. Zhao and J. Zhao, Ynamide-mediated peptide bond formation: mechanistic study and synthetic applications, Angew. Chem., Int. Ed., 2022, 61, e202212247 CrossRef CAS PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Large–scale applications of amide coupling reagents for the synthesis of pharmaceuticals, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- J. García-Gros, Y. Cajal, A. M. Marqués and F. Rabanal, Synthesis of the antimicrobial peptide Murepavadin using novel coupling agents, Biomolecules, 2024, 14, 526 CrossRef PubMed.
- D. Uehara, S. Adachi, A. Tsubouchi, Y. Okada, V. V. Zhdankin, A. Yoshimura and A. Saito, Peptide coupling using recyclable bicyclic benziodazolone, Chem. Commun., 2024, 60, 956–959 RSC.
- K. M. Freiberg, R. D. Kavthe, R. M. Thomas, D. M. Fialho, P. Dee, M. Scurria and B. H. Lipshutz, Direct formation of amide/peptide bonds from carboxylic acids: no traditional coupling reagents, 1–Pot, and green, Chem. Sci., 2023, 14, 3462–3469 RSC.
- O. Al Musaimi, R. Wisdom, P. Talbiersky, B. G. De La Torre and F. Albericio, Propylphosphonic anhydride (T3P®) as coupling reagent for solid–phase peptide synthesis, ChemistrySelect, 2021, 6, 2649–2657 CrossRef CAS.
- D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. Watson, Evaluation of alternative solvents in common amide coupling reactions: replacement of dichloromethane and N,N-dimethylformamide, Green Chem., 2013, 15, 596–600 RSC.
- W. Li, N. M. O'Brien-Simpson, M. A. Hossain and J. D. Wade, The 9–fluorenylmethoxycarbonyl (Fmoc) group in chemical peptide synthesis–its past, present, and future, Aust. J. Chem., 2019, 73, 271–276 CrossRef.
- D. Takahashi, T. Inomata and T. Fukui, AJIPHASE®: a highly efficient synthetic method for one–pot peptide elongation in the solution phase by an Fmoc strategy, Angew. Chem., 2017, 129, 7911–7915 CrossRef.
- T. Yamamoto, T. C. Chang and K. Tanaka, Epoc group: transformable protecting group with Gold (III)–catalyzed fluorene formation, Chem. Sci., 2021, 12, 10703–10709 RSC.
- T. Hattori and H. Yamamoto, Peptide bond formation between unprotected amino acids: convergent synthesis of oligopeptides, J. Am. Chem. Soc., 2024, 146, 25738–25744 CrossRef CAS PubMed.
- C. Palomo, A. L. Palomo, F. Palomo and A. Mielgo, Soluble α–amino acid salts in acetonitrile: practical technology for the production of some dipeptides, Org. Lett., 2002, 4, 4005–4008 CrossRef CAS PubMed.
- A. Nagaya, S. Murase, Y. Mimori, K. Wakui, M. Yoshino, A. Matsuda and N. Nishizawa, Extended solution–phase peptide synthesis strategy using isostearyl–mixed anhydride coupling and a new C–terminal silyl ester–protecting group for N-methylated cyclic peptide production, Org. Process Res. Dev., 2021, 25, 2029–2038 CrossRef CAS.
- M. Diamandas, R. Moreira and S. D. Taylor, Solid–phase total synthesis of dehydrotryptophan–bearing cyclic peptides Tunicyclin B, Sclerotide A, CDA3a, and CDA4a using a protected β–hydroxytryptophan building block, Org. Lett., 2021, 23, 3048–3052 CrossRef CAS PubMed.
- K. Hojo, Y. Manabe, T. Uda and Y. Tsuda, Water–based solid–phase peptide synthesis without hydroxy side chain protection, J. Org. Chem., 2022, 87, 11362–11368 CrossRef CAS PubMed.
- A. S. Galanis, F. Albericio and M. Grøtli, Solid–phase peptide synthesis in water using microwave–assisted heating, Org. Lett., 2009, 11, 4488–4491 CrossRef CAS PubMed.
- A. S. Galanis, F. Albericio and M. Grøtli, Solid–phase peptide synthesis in water using microwave–assisted heating, Org. Lett., 2009, 11, 4488–4491 CrossRef CAS PubMed.
- T. M. Postma and F. Albericio, N-Chlorosuccinimide, an efficient peptide disulfide bond–forming reagent in aqueous solution, RSC Adv., 2013, 3, 14277–14280 RSC.
- R. S. Dhiman, L. G. Opinska and R. Kluger, Biomimetic peptide bond formation in water with aminoacyl phosphate esters, Org. Biomol. Chem., 2011, 9, 5645–5647 RSC.
- M. Cortes-Clerget, J. Y. Berthon, I. Krolikiewicz-Renimel, L. Chaisemartin and B. H. Lipshutz, Tandem deprotection/coupling for peptide synthesis in water at room temperature, Green Chem., 2017, 19, 4263–4267 RSC.
- S. B. Lawrenson, R. Arav and M. North, The greening of peptide synthesis, Green Chem., 2017, 19, 1685–1691 RSC.
- S. Hazra, F. Gallou and S. Handa, Water: An underestimated solvent for amide bond–forming reactions, ACS Sustainable Chem. Eng., 2022, 10, 5299–5306 CrossRef CAS.
- W. Hou, X. Zhang and C. F. Liu, Progress in chemical synthesis of peptides and proteins, Trans. Tianjin Univ., 2017, 23, 401–419 CrossRef.
- L. Posada, L. Rey, J. Villalba, S. Colombo, L. Aubriot, N. Badagian and G. Serra, Cyclopeptides natural products as herbicides and inhibitors of cyanobacteria: Synthesis of Versicotides E and F, ChemistrySelect, 2022, 7, e202201956 CrossRef CAS.
- M. I. Muhajir, A. Hardianto, J. Al-Anshori, D. Sumiarsa, T. Mayanti, N. Nurlelasari and R. Maharani, Total synthesis of Nocardiotide A by using a combination of solid– and solution–phase methods, ChemistrySelect, 2021, 6, 12941–12946 CrossRef CAS.
- H. Luo, H. Yin, C. Tang, P. Wang and F. Liang, Synthesis of cyclic peptide Reniochalistatin E and conformational isomers, Chin. Chem. Lett., 2018, 29, 1143–1146 CrossRef CAS.
- S. R. Tivari, S. V. Kokate, E. M. Sobhia, S. G. Kumar, U. B. Shelar and Y. S. Jadeja, A series of novel bioactive cyclic peptides: synthesis by head-to-tail cyclization approach, antimicrobial activity and molecular docking studies, ChemistrySelect, 2022, 7, e202201481 CrossRef CAS.
- N. Jain and S. H. Friedman, A tetra-orthogonal strategy for the efficient synthesis of scaffolds based on cyclic peptides, Int. J. Pept. Res. Ther., 2018, 24, 535–542 CrossRef CAS PubMed.
- C. Fagundez, D. Sellanes and G. Serra, Synthesis of cyclic peptides as potential anti-malarials, ACS Comb. Sci., 2018, 20, 212–219 CrossRef CAS PubMed.
- H. Masui and S. Fuse, Recent advances in the solid-and solution-phase synthesis of peptides and proteins using microflow technology, Org. Process Res. Dev., 2022, 26, 1751–1765 CrossRef CAS.
- S. Noki, B. G. de la Torre and F. Albericio, Safety-catch linkers for solid-phase peptide synthesis, Molecules, 2024, 29, 1429 CrossRef CAS PubMed.
- H. Nzama, S. R. Manne, O. Marder, G. Orosz, B. G. de la Torre and F. Albericio, Unveiling the quaternary carbodiimide symphony: harmonizing green chemistry in peptide synthesis, Green Chem. Lett. Rev., 2024, 17, 2392826 CrossRef.
- Y. J. Pu, R. K. Vaid, S. K. Boini, R. W. Towsley, C. W. Doecke and D. Mitchell, A Practical method for functionalized peptide or amide bond formation in aqueous–ethanol media with EDC as activator, Org. Process Res. Dev., 2009, 13, 310–314 CrossRef CAS.
- A. Méndez-Ardoy, I. Insua, J. R. Granja and J. Montenegro, Cyclization and self-assembly of cyclic peptides, in Peptide Macrocycles. Methods and Protocols, ed. M. B. Coppock and A. J. Winton, Humana, New York, 2022, pp. 449–466 Search PubMed.
- S. C. Stolze and M. Kaiser, Case studies of the synthesis of bioactive cyclodepsipeptide natural products, Molecules, 2013, 18, 1337–1367 CrossRef CAS PubMed.
- N. Lalani, S. Tivari, V. Jain and Y. Jadeja, Review on therapeutic potential of peptides: advancements in synthesis methods, linear and cyclic peptides, and strategies for overcoming challenges, Pept. Sci., 2024, e24343 CrossRef CAS.
- D. S. Nielsen, N. E. Shepherd, W. Xu, A. J. Lucke, M. J. Stoermer and D. P. Fairlie, Orally absorbed cyclic peptides, Chem. Rev., 2017, 117, 8094–8128 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Cyclic peptide therapeutics: past, present and future, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- M. R. Naylor, A. T. Bockus, M. J. Blanco and R. S. Lokey, Cyclic peptide natural products chart the frontier of oral bioavailability in the pursuit of undruggable targets, Curr. Opin. Chem. Biol., 2017, 38, 141–147 CrossRef CAS PubMed.
- Y. Lee, C. Phat and S. C. Hong, Structural diversity of marine cyclic peptides and their molecular mechanisms for anticancer, antibacterial, antifungal, and other clinical applications, Peptides, 2017, 95, 94–105 CrossRef CAS PubMed.
- G. N. Belofsky, P. R. Jensen and W. Fenical, Sansalvamide: a new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium, Tetrahedron Lett., 1999, 40, 2913–2916 CrossRef CAS.
- M. C. Sheikh, S. Takagi, T. Yoshimura and H. Morita, Mechanistic studies of DCC/HOBt-mediated reaction of 3-phenylpropionic acid with benzyl alcohol and studies on the reactivities of ‘active ester’ and the related derivatives with nucleophiles, Tetrahedron, 2010, 66, 7272–7278 CrossRef CAS.
- J. A. Morales-Serna, A. Sauza, G. P. de Jesús, R. Gaviño, G. G. de la Mora and J. Cardenas, Facile and efficient addition of terminal alkynes to benzotriazole esters: synthesis of d-erythro-sphingosine using ynones as the key intermediate, Tetrahedron Lett., 2013, 54, 7111–7114 CrossRef CAS.
- J. A. Morales-Serna, E. García-Ríos, J. Bernal, E. Paleo, R. Gaviño and J. Cardenas, Reduction of carboxylic acids using esters of benzotriazole as high-reactivity intermediates, Synthesis, 2011, 1375–1382 CAS.
- J. A. Morales-Serna, A. Vera, E. Paleo, E. García-Ríos, R. Gaviño, G. G. de la Mora and J. Cardenas, Using benzotriazole esters as a strategy in the esterification of tertiary alcohols, Synthesis, 2010, 4261–4267 CAS.
- L. A. Carpino, E. S. M. Mansour and D. Sadat-Aalaee, tert-Butyloxycarbonyl and benzyloxycarbonyl amino acid fluorides. New, stable rapid-acting acylating agents for peptide synthesis, J. Org. Chem., 1991, 56, 2611–2614 CrossRef CAS.
- Y. Hwang, D. Rowley, D. Rhodes, J. Gertsch, W. Fenical and F. Bushman, Mechanism of inhibition of a poxvirus topoisomerase by the marine natural product Sansalvamide A, Mol. Pharmacol., 1999, 55, 1049–1053 CrossRef CAS PubMed.
- B. K. Chagaleti, K. Baby, S. I. Pena-Corona, G. Leyva-Gomez, S. M. Sindhoor, N. R. Naveen, J. Jose, A. A. Aldahish, J. Sharifi-Rad and D. Calina, Anti-cancer properties of Sansalvamide A, its derivatives, and analogs: an updated review, Naunyn-Schmiedeberg's Arch. Pharmacol., 2024, 397, 7337–7351 CrossRef CAS PubMed.
- C. L. Carroll, J. V. Johnston, A. Kekec, J. D. Brown, E. Parry, J. Cajica and S. R. McAlpine, Synthesis and cytotoxicity of novel Sansalvamide A derivatives, Org. Lett., 2005, 7, 3481–3484 CrossRef CAS PubMed.
- K. Otrubova, T. J. Styers, P. S. Pan, R. Rodriguez, K. L. McGuire and S. R. McAlpine, Synthesis and novel structure–activity relationships of potent Sansalvamide A derivatives, Chem. Commun., 2006, 1033–1034 RSC.
- J. A. Morales-Serna, M. Á. Jaime-Vasconcelos, E. García-Ríos, A. Cruz, D. Angeles-Beltrán, L. Lomas-Romero, G. E. Negrón-Silva and J. Cárdenas, Efficient activity of magnesium–aluminium hydrotalcite in the synthesis of amides, RSC Adv., 2013, 3, 23046–23050 RSC.
- J. A. Morales-Serna, E. Sánchez, R. Velázquez, J. Bernal, E. García-Ríos, R. Gaviño and J. Cárdenas, Highly efficient macrolactonization of ω-hydroxy acids using benzotriazole esters: synthesis of Sansalvamide A, Org. Biomol. Chem., 2010, 8, 4940–4948 RSC.
- Y. Lee and R. B. Silverman, Rapid, high-yield, solid-phase synthesis of the antitumor antibiotic Sansalvamide A using a side-chain-tethered phenylalanine building block, Org. Lett., 2000, 2, 3743–3746 CrossRef CAS PubMed.
- L. Chen, M. Zheng, Y. Zhou, H. Liu and H. Jiang, Ionic-liquid-supported total synthesis of Sansalvamide A peptide, Synth. Commun., 2008, 38, 239–248 CrossRef CAS.
- O. Al Musaimi, B. G. de la Torre and F. Albericio, Greening Fmoc/tBu solid-phase peptide synthesis, Green Chem., 2020, 22, 996–1018 RSC.
- L. Ferrazzano, M. Catani, A. Cavazzini, G. Martelli, D. Corbisiero, P. Cantelmi and A. Tolomelli, Sustainability in peptide chemistry: current synthesis and purification technologies and future challenges, Green Chem., 2022, 24, 975–1020 RSC.
- M. H. A. Somehsaraie, V. F. Vavsari, M. Kamangar and S. Balalaie, Chemical wastes in the peptide synthesis process and ways to reduce them, Iran. J. Pharm. Res., 2022, 21, e123879 CAS.
- K. Venkateswarlu, Ashes from organic waste as reagents in synthetic chemistry: a review, Environ. Chem. Lett., 2021, 19, 3887–3950 CrossRef CAS.
- J. Lopez, F. Gallou, I. Kekessie, K. Wegner, I. Martinez, M. E. Kopach and L. Vandenbos, Process Mass Intensity (PMI): A holistic analysis in current peptide manufacturing processes, informing sustainability in peptide synthesis, J. Org. Chem., 2024, 89, 4261–4282 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and spectra of the synthesised compounds. See DOI: https://doi.org/10.1039/d4ra07847k |
| This journal is © The Royal Society of Chemistry 2024 |