
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePackage delivered: folate receptor-mediated transporters in cancer therapy and diagnosis
Mohsen
Ahmadi
 *a,
Christoph A.
Ritter
b,
Thomas
von Woedtke
ac,
Sander
Bekeschus
ad and
Kristian
Wende
*a
*a,
Christoph A.
Ritter
b,
Thomas
von Woedtke
ac,
Sander
Bekeschus
ad and
Kristian
Wende
*a
aLeibniz Institute for Plasma Science and Technology (INP), Center for Innovation Competence (ZIK) Plasmatis, Felix Hausdorff-Str. 2, 17489 Greifswald, Germany. E-mail: mohsen.ahmadi@inp-greifswald.de; kristian.wende@inp-greifswald.de
bInstitute of Pharmacy, Section Clinical Pharmacy, University of Greifswald, Greifswald, Germany
cInstitute for Hygiene and Environmental Medicine, Greifswald University Medical Center, Ferdinand-Sauerbruch-Straße, 17475 Greifswald, Germany
dClinic and Policlinic for Dermatology and Venereology, Rostock University Medical Center, Strempelstr. 13, 18057 Rostock, Germany
First published on 17th January 2024
Abstract
Neoplasias pose a significant threat to aging society, underscoring the urgent need to overcome the limitations of traditional chemotherapy through pioneering strategies. Targeted drug delivery is an evolving frontier in cancer therapy, aiming to enhance treatment efficacy while mitigating undesirable side effects. One promising avenue utilizes cell membrane receptors like the folate receptor to guide drug transporters precisely to malignant cells. Based on the cellular folate receptor as a cancer cell hallmark, targeted nanocarriers and small molecule–drug conjugates have been developed that comprise different (bio) chemistries and/or mechanical properties with individual advantages and challenges. Such modern folic acid-conjugated stimuli-responsive drug transporters provide systemic drug delivery and controlled release, enabling reduced dosages, circumvention of drug resistance, and diminished adverse effects. Since the drug transporters' structure-based de novo design is increasingly relevant for precision cancer remediation and diagnosis, this review seeks to collect and debate the recent approaches to deliver therapeutics or diagnostics based on folic acid conjugated Trojan Horses and to facilitate the understanding of the relevant chemistry and biochemical pathways. Focusing exemplarily on brain and breast cancer, recent advances spanning 2017 to 2023 in conjugated nanocarriers and small molecule drug conjugates were considered, evaluating the chemical and biological aspects in order to improve accessibility to the field and to bridge chemical and biomedical points of view ultimately guiding future research in FR-targeted cancer therapy and diagnosis.
 Mohsen Ahmadi | Dr Mohsen Ahmadi earned his PhD in 2019 from the University of Greifswald (Germany). His research revolves around the development and replication of the chemical synthesis of active sites found in molybdoenzymes, with a focus on addressing molybdenum cofactor deficiency. Following this, he joined the Center for Innovation Competence (ZIK) plasmatis at the Leibniz Institute for Plasma Science and Technology (INP) in Germany, where his work centered on the development and activation of drugs/prodrugs for the treatment of cancer and inflammatory diseases. Currently, his primary research interests lie in understanding the mechanisms of drug degradation and prodrug activation through tumor-induced reactive species simulated by cold physical plasma. He also explores the chemistry of mimetic systems to create therapeutic agents. |
 Christoph A. Ritter | Prof. Dr Christoph A. Ritter studied pharmacy from 1991 to 1996 at the Friedrich Alexander University in Erlangen-Nuremberg. In 2000, he received his PhD at the Faculty of Mathematics and Natural Sciences of the Friedrich-Alexander University Erlangen-Nuremberg (Germany). After his postdoc in Hematology–Oncology in 2003 from Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, TN, USA, he joined the Institute of Pharmacology at the University of Greifswald as a junior professorship. In 2007/2008, he obtained his habilitation in the subjects of pharmacology and clinical pharmacy. Since 2009 he has held a W2 professorship for a clinical pharmacy at the University of Greifswald. |
 Thomas von Woedtke | Prof. Dr Thomas von Woedtke studied Pharmacy at the University of Greifswald, Germany. He received the Doctoral Degree in 1995 in Pharmaceutical Technology, followed by a Habilitation degree in 2005. 2008–2023 he was Research Program Manager Plasma Medicine at the Leibniz Institute for Plasma Science and Technology (INP Greifswald), Germany, and since 2020 he is Member of the Board of INP, responsible for the Research Division Health & Hygiene. He holds a professorship for Plasma Medicine at Greifswald University Medicine since 2011. |
 Sander Bekeschus | Prof. Dr Sander Bekeschus is a human biologist trained in immunology, focusing on plasma and redox biology. After short-term scientific missions in New Zealand and the USA, he obtained his PhD from the University of Greifswald (Germany) before starting a third-party-funded research group at the Leibniz Institute for Plasma Science and Technology (INP) in 2016. Since 2023, he is Professor for Translational Plasma Research at the Clinic for Dermatology of Rostock University Medical Center. His main interests are cellular and translational research models and enabling the investigation of immune-related processes using medical plasmas in dermatology, oncology, and redox medicine. |
 Kristian Wende | Dr Kristian Wende received his MSc degree (natural compound chemistry) in 1998 and his PhD degree (natural compound analytics and toxicology) in 2003 from the University of Greifswald (Germany). Since 2010, he joined the Leibniz Institute for Plasma Science and Technology (INP) and after various short term scientific missions to Minneapolis/MN/USA, York/UK, and Eindhoven/NL he became a third-party-funded research group leader in 2017. In 2023 he became a senior scientist at the same institution. His current research comprises analytical and biophysical methods to determine biomolecule oxidation in the context of redox biology. |
1 Introduction
1.1 Cancer therapy – state of the art
Global cancer statistics estimated the incidence and mortality for 36 cancers in 185 countries with 19.3 million new cancer cases and almost 10 million cancer deaths in 2020.1 Breast cancer was diagnosed in 2.3 million patients (11.7%), while the share of brain cancer was only 0.3 million cases (1.6%) due to treatment-associated complications of glioblastoma brain tumours. Europe, with 9.7% of the global population, accounts for 22.8% of all cancer cases and 19.6% of cancer's death toll. Despite the massive effort put into cancer prevention and the advanced approaches developed to tackle cancer in the past decade,2 new methodologies and seminal breakthroughs in cancer therapeutics are desired to cut these numbers. Hope is put in implementing nanotechnology tools, combined with artificial intelligence, to boost structural-based drug transporter design to pave the way for effective and selective cancer therapy.3 Among these approaches, nanocarriers (NCs) have gained a major role. These are nano-transporter systems of one to 500 nm in size utilized as transport modules for drugs. NCs were designed not only to modulate the drug's pharmacokinetics and pharmacodynamics compared to the administration of free drugs but also to increase safety and efficiency by limiting undesired side effects.4 Accordingly, NCs have been designed with high encapsulation capacities, tailored surface chemistry, and clever concepts to conjugate the therapeutic/diagnostic agents.5 Size, shape, and surface characteristics determine the drug delivery efficiency, drug's half-life, and drug cytotoxicity (Fig. 1). In parallel, small molecule–drug conjugates (SMDCs), releasing a potent cytotoxic agent when reaching a destination – e.g., the tumour microenvironment, decreasing the off-target toxicity – have been developed. Here, a small molecule acts as a targeting structure to direct the conjugate, replacing the antibody in the elsewise similar concept of antibody–drug conjugate but without its immunogenic nature.6 NCs and SMDCs are applied to develop passive or active targeting systems to deliver therapeutics to cancer cells.2g,4a The concept of drug delivery via passive targeting was initially utilized, e.g., by taking advantage of the more leaky vasculature of some tumours rendering it more permeable for macromolecules than in healthy tissues. This universal pathophysiological phenomenon allows macromolecular compounds or particles such as albumin or polymer-conjugated drugs beyond certain sizes (above 40 kDa) to accumulate and be retained in the tumour tissue. It was coined as the enhanced permeability and retention effect (EPR, see Fig. 1). However, the EPR effect is not universal due to differences in the tumour microenvironment such as degree of vascularization, lymphatic vasculature, immune systems activity, and angiogenesis patterns.7 As a result, not all tumours may exhibit a substantial EPR effect, limiting the applicability of drug delivery systems relying on this effect. Besides, the lack of cellular specificity of drug transporters in cancer cells impedes drug accumulation and efficiency, consequently leading to drug resistance.8 Meta-analysis studies by Chan et al.9 and Lin et al.10 have indeed shown that the median delivery efficiencies were only 0.7% of administrated drug transporters dose accumulated in high EPR xenografted tumours, which is due to endothelial barriers, endosomal escape, and clearance from the blood via the kidney and liver.4a,11 This highlights the challenges associated with narrow drug accumulation in tumours and confirm the need for more innovative drug delivery strategies to enhance drug delivery to tumours. Hence, active targeting strategies have been developed based on medical, chemical, and structural considerations, revolutionizing medicinal chemistry and grossly enhancing selectivity (Fig. 1).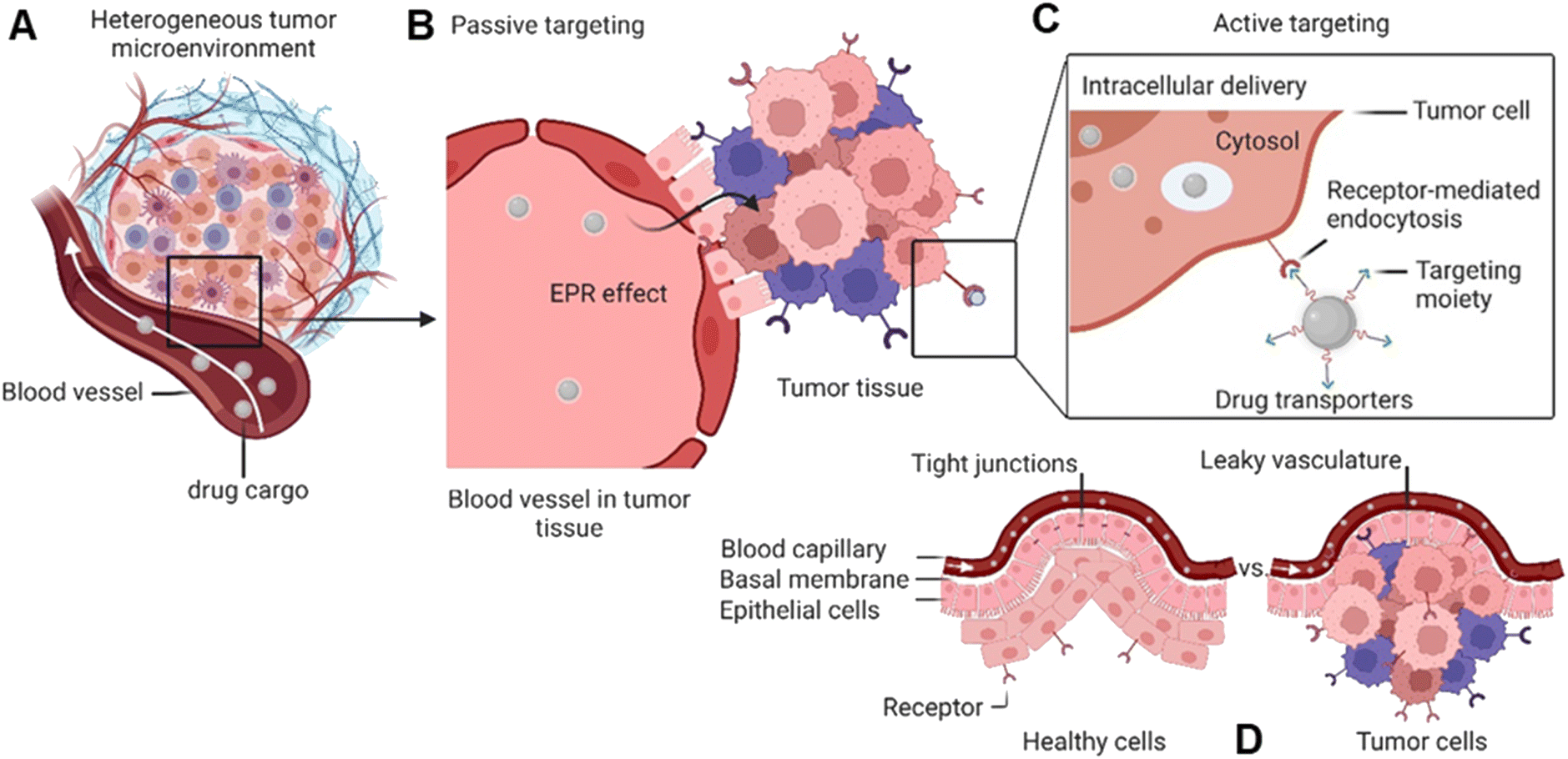 | ||
| Fig. 1 Passive and active targeting systems for delivery of therapeutics into cancer cells. (A) Heterogenous tumour microenvironment. (B) Passive targeting through the EPR effect for accumulating NCs inside the tumour. (C) Drug transporter internalization into the cytosol via receptor-mediated endocytosis. (D) Blood capillary system of healthy cells vs. cancer cells. | ||
Targeted drug transporters facilitate selective delivery to primary cancer sites and metastasis lesions, particularly in cases involving tumours with poor EPR effect.4a Targeting drug delivery utilizing dedicated plasma membrane receptors (Fig. 1) is considered to increase cellular uptake and enhance the cytotoxicity of its cargo.12 Several targeted-based strategies, i.e., receptor-mediated transporters, monoclonal antibodies, carbohydrate-binding proteins (lectins) for cell-surface recognition, and targeting vaccine delivery, have been utilized to modulate targeted drug delivery.13 The most effective targeted delivery systems to accumulate cytotoxic agents rely upon cell surface proteins that tend to be overexpressed in malignant tissues, such as folate receptors,14 glucose transporters,15 epidermal and hepatocyte growth factor receptors,16 transferrin,17 prostate-specific membrane antigen,18 angiopep-2,19 and asialoglycoprotein receptors.20 The FRα expression in metastatic triple-negative breast cancer (TNBC) patients is significantly higher than in early-stage patients.21
On the other hand, the blood–brain barrier and brain–tumour barrier restrict drug delivery into the brain, resulting in poor diagnosis and treatment.22 Transportation of NCs and SMDCs via folate receptor-mediated strategy improves the drug accumulation on tumour site. Apart from that, drug transporters can deliver specific drugs to inhibit the efflux transposers like P-glycoprotein and mediate multidrug resistance in brain tumour treatment.23
Accordingly, promising to overcome the passive targeting limitations, innovative folic acid-conjugated drug transporter systems have been given significant attention in recent years. Most of our understanding of FR-targeted drug transporters is based on in vitro and in vivo models using carcinoma cell lines and mouse xenografts (Fig. 2). Hence, the translation into clinical models is needed to explore the full potential of SMDCs and NCs in human or humanized model systems. Hence, the intrinsic relationship between the drug transporter's chemistry and biology might regulate the boundary that needs further justifications to address these knowledge gaps.
 | ||
| Fig. 2 Reported pertinent carcinoma cell lines corresponding to various FA-conjugated NCs and SMDCs for in vitro evaluation of distinct tumours. | ||
To this end, the present review attempts to collect, sort, and consider the available evidence of drug transporter chemistry and related physical properties, as well as its delivery and release mechanisms over the past five years. A wealth of original contributions has been published in this considered time frame. In order to keep the review and the number of citations in a manageable scale, we selected based on the comprehensiveness of the material characterization, data reliability as far as it could be judged from the publication, and on originality and chemical aspects of the approach. We will focus on brain and breast cancers since both malignancies have different biological backgrounds and physiological barriers impeding access (e.g., blood–brain barrier). A further major aspect is shedding light on the relation the chemical modification of drug transporters into their biological aspect to the outlook of forthcoming directions in targeted cancer therapy and diagnosis. Apart from chemical interpretation, we discuss pathophysiological and pre-clinical challenges and barriers toward an effective and safe translation into clinical application.
1.2 Drug transporters
The concept of targeted drug delivery has been around for two centuries, and active targeting remains a fascinating approach for scientists to design multi-functionalized therapeutics.24 Despite the rapidly growing domain of small molecule–drug conjugates (SMDCs), only Lutathera (177Lu-DOTATATE) targeting peptide receptor is approved for gastroenteropancreatic neuroendocrine tumours.6a,25 In addition, the folate receptor targeted SMDCs, such as vintafolide (folatedesacetylvinblastine hydrazide), OTL-38 (Pte-Tyr-NIR-dye), EC17 (folate–fluorescein isothiocyanate), etarfolatide (folate-99mTc), etc. are in the clinical trial.6a,26 On the other hand, various types of folic acid (FA)-conjugated NCs utilized for targeted drug delivery have been developed and are schematically illustrated in Fig. 3A. To this end, the percentage of reported FA-conjugated NCs and SMDCs constructed for cancer diagnosis and therapy over the past years underlines their importance (Fig. 3B). The current landscape of Food and Drug Administration (FDA)-approved and currently in clinical phases tested drug transporters have been reviewed (Corrie et al.,27 and Anselmo and Mitragotri et al.28). Liposomes, PEGylated liposomes, protein-based NCs, and polymeric NCs in general are the main NCs that have been approved as nano vehicles for drug delivery (Table 1). | ||
| Fig. 3 (A) Various types of nanocarriers (NCs) utilized for targeted drug delivery. (B) The approximate percentage of reported FA-conjugated NCs for cancer management for all types of cancer (a) and specifically in brain and breast cancer (b). (C) Schematic visualization of NCs regarding the physical properties. | ||
| Name | Vehicle (loaded drug) | Cancer type | Ref. |
|---|---|---|---|
| a FDA-approved nanocarrier (Cyelax in European union (EU)) composed of hydrogenated soy phosphatidylcholine (HSPC), cholesterol, and DSPE-PEG2k. b Known as MM-398. c European Medicines Agency (EMA)-approved nanocarrier composed of egg phosphatidylcholine (EPC) and cholesterol. d FDA-approved nanocarrier. e Approved in China. f Approved in South Korea (composed of the polylactide-block-PEGs copolymer). g Developed by MediGene (composed of cationic dioleoyltrimethylammoniumpropane (DOTAP) and neutral dioleoylphosphatidylcholine (DOPC)). | |||
| Doxila | PEGylated liposome (doxorubicin) | Breast and ovarian cancer | 37 |
| Onivydeb | PEGylated liposome (irinotecan) | Solid tumour entities: metastatic pancreatic cancer and breast cancer (phase I) | 38 |
| Myocetc | Liposome (doxorubicin) | Metastatic breast cancer | 39 |
| Abraxaned | Albumin-bounded NC (paclitaxel) | Metastatic breast cancer | 40 |
| Lipusue | Liposome (paclitaxel) | Breast cancer and non-small cell lung carcinoma (NSCLC) | 41 |
| Genexol-PMf | Copolymeric micelle (paclitaxel) | Breast cancer and NSCLC | 42 |
| EndoTAG-Ig | Liposome (paclitaxel) | Triple-negative breast cancer | 43 |
Nanocarriers represent an excellent promise for efficient drug delivery due to their high surface area and volume ratio for drug encapsulation, enhancing drug pharmacokinetics and biodistribution, and cytotoxicity via active targeting strategies.29 The physicochemical properties of NCs can be tuned as desired depending on the target cancer via altering their composition, morphology, size, shape, surface, and conjugation chemistry, ultimately significantly impacting their biological activity along the way and after reaching the tumour site.30 Surface charge is a distinct property of NPs and refers to the net electric charge present on the surface of the particles due to charged functional groups or ions. The amphiphilic characteristics of NPs dictated by their hydrophobic and hydrophilic properties, which are fundamental determinants controlling their interactions within complex biological matrices. However, the surface charge and hydrophobicity/hydrophilicity can influence each other to some extent. For instance, charged functional groups on the NP's surface can contribute to its hydrophilicity, making it more likely to interact with water molecules. A neutrally charged surface may be hydrophilic (using, e.g., zwitterions or poly(ethylene glycol)). In contrast, a charged surface may be hydrophobic if the (negative or positive) charge density is low because of, for example, hydrophobic linkers.
In parallel, the zeta (ζ)-potential needs to be considered as a parameter that depends on the surface charge directly related to the colloidal stability of NCs in suspension over time and influences their early adsorption (or adhesion) onto the cell membrane circulation time, metabolism, clearance, and recognition by cells of the immune system. Thus, various aspects of interfacial phenomena regarding the ζ-potential in chemistry that satisfyingly interplayed with biology evaluations have been studied.31 The schematic visualization of NCs regarding the physical properties is depicted in Fig. 3C. The ζ-potential should not be considered an absolute criterion on its own. The ζ-potential, which is the electrical potential at the plane of shear or the hydrodynamic slip plane near a solid surface, serves as an indicator of the electrostatic repulsion forces acting between particles. The repulsion force helps to prevent the aggregation or flocculation of NPs. Particles have high ζ-potentials (either positive or negative), the electrostatic repulsion between them promoting dispersion and stability. A range of ±25 mV is often considered a guideline for sufficient repulsion force to maintain colloidal stability. The ζ-potential is not static and can shift depending on the environment. For example, in a physiological medium, the high concentration of counter ions (such as salts) screens the electrostatic repulsion, reduces the effective ζ-potential and weakens the repulsion forces, which may cause NC agglomeration, even if their potential is beyond ±25 mV in deionized water. Moreover, highly charged NCs will interact strongly with proteins (protein corona) and other macromolecules, making them less stable in serum than neutrally charged but hydrophilic NCs. Therefore, only ζ-potential values may not fully capture the NP's stability in complex biological environments.
Apart from the surface charge, particle size mainly affects the drug pharmacokinetics via the biodistribution of drug-loaded cargo to the cancer tissue by the EPR effect. Indeed, the optimal particle size is between 20–200 nm to prevent particle clearance in the kidney and liver. Larger particles are recognized and phagocytosed by Kupffer cells in the liver from the bloodstream. In comparison, smaller particles below the renal filtration threshold (typically around 5–6 nm) can be excreted through kidney filtration and eliminated via urination.9 It is worth noting that particle size alone is not the only factor determining NP's clearance. Other factors, such as surface charge, surface modifications, and surface coatings, can also influence the interaction with the immune system and clearance pathways.32 For example, the choice of spacers and linkers in the chemical modification of NCs and SMDCs holds the potential to influence crucial factors such as size, shape, and charge.33 In parallel, these selections can also exert a significant impact on loading capacity, circulation time within the bloodstream, and the subsequent release dynamics upon accumulation at the tumour site.34 (refer to Section 1.3). Zhang et al. recently reported the chemical structure of charge-reversal NCs to enhance their cellular uptake to achieve prolonged blood circulation and decreased systemic toxicity.35 These factors were interpreted by Patra et al. in detail to control renal clearance and improve the success rate of clinical translation of NCs in cancer diagnosis and therapy36 (refer to Section 4 for more details).
1.3 Structural design, loading, and release chemistry
1.3.2.1 Linkers. Linkers carrying modifiable functional groups such as thioether (sulphide, sulfoxide, thioketal),48 acetal (ketal),49 carbamate,50 amine and hydrazine,51 hydroxyl,52 borate ester,51,53 disulfide,54 acetyl-hydrazone,55 and carbodiimide,56 (in particular via EDC-NHS cross-linked method)57 are necessary for a facile conjugation with or release of the cargo drug from NC and SMDCs44d (Fig. 4A). EDC-NHS cross-linking method is commonly employed to conjugate carboxylic acid (–COOH) moieties with primary amine (–NH2) groups, resulting in the formation of an amide bond. For example, amino acids such as glycine, serine, and lysine contain both amino and carboxyl groups, and can therefore serve as linkers. Disulphide linkers are responsive to the reducing environment found in intracellular compartments that can be selectively fractured, for instance, by intracellular glutathione, enabling intracellular drug release.58 Clickable linkers such as azides or alkynes allow for specific and rapid conjugation reactions with complementary functional groups.59 Light-responsive linkers such as photocaged C40-oxidized abasic site (PC4AP) incorporated into peptide– and protein–drug conjugates that undergo photo-decaging in response to light irradiation.60
 | ||
| Fig. 4 (A) Schematic view of FA-conjugated drug transporter including folic acid, linker, spacer, and drug payload. (B) Stimuli-responsive drug release triggered via internal or external stimuli at the site of action. | ||
The incorporation of stimuli-cleavable linkers into drug delivery systems provides a powerful strategy for on-demand drug release. Structural modifications of the heterobifunctional linker may control the physicochemical properties of NCs,2c SMDCs,44d and antibody–drug conjugates,6b resulting in more effective cancer therapy and diagnosis. For example, disulphide-containing linkers displayed superior activity against folate receptor-positive FR(+) cells54,61 and could lead to the payload release upon reduction by glutathione.54 According to Song, Ding, and Yang et al., the utilization of amide, diselenide, and ester linkers has significantly promoted on-demand drug release.62 Notably, pH-responsive linkers such as hydrazine and acetal linkers can be disintegrated from acid-liable functional counterparts due to a lower endosomal and lysosomal pH than cytosol pH.63 Drugs such as mitomycin C64 and camptothecin65 are masked using benzyl carbamate disulphide and disulphide carbonate, respectively. In a different example, a thioether propargyl carbamate linker can be conjugated to a cysteine residue through site-specific protein modification.66
1.3.2.2 Spacers. Spacers are flexible molecules with different lengths or polarity that have been extensively utilized in bioconjugate chemistry and need to be biodegradable, non-toxic, and biocompatible, having functional groups to correlate linkers with other bioconjugates, such as folic acid and therapeutic agents (or vice versa) (Fig. 4A). Although spacers and linkers are often equivalently categorized in the literature, they must be classified according to discreet chemical properties and activation (degradation) mechanisms. Hence, spacers could respond to stimuli for degradation after accumulating in tumour tissue (which could be different from linkers) to release the payload. Thus, spacers could have similar structural functionalization to bond with NCs and SMDCs, but not necessarily. However, spacers are generally applied to reduce steric bulkiness for two main reasons: (i) to accelerate the release process (drug release triggered by stimuli like enzyme, redox potential, and reactive species), (ii) to increase the distance between the triggered cleavable bonds conjugated between the folic acid and drug transporter. Spacers are not only used for stimuli-responsive payload NCs,67 but also utilized for SMDCs,60 and prodrugs concepts68 for on-demand drug release.
1.4 Folate receptors – distinct cellular markers
Folate receptors (FRs) are single-chain glycoprotein-based receptors (35–40 kDa) that are expressed in four isoforms (FRα, FRβ, FRγ, and FRδ).61 Those isoforms display almost 70% amino acid sequence identity. FRα, FRβ, and FRδ are glycosyl-phosphatidylinositol-anchored proteins, whereas FRγ lacks the GPI-anchor region.75 Cellular uptakes of folic acid (FA) occur via FRα and FRβ, which are located on the cell surface by a c-terminal GPI-anchor. Despite the sequence divergence of FRα and FRβ on their carboxy-terminal, the binding affinities to FA and its reduced folate forms (i.e., methyltetrahydrofolate and tetrahydrofolate) are relatively similar. In this process, FA and reduced folate bind to the FRs (binding affinity (Kd) ∼10−10 M) in the extracellular milieu and are then internalized into the cell, followed by the subsequent release of FA into the cytosol. Dann et al. reported structural models of the endocytic trafficking of FRs and their pH-dependent conformational changes.76 Changes in FR conformation at pH 7.4 before the association of folate in an open state (Fig. 5A). In contrast, the FR interacted with folate via amino acid residues aspartic acid (Asp)97, tryptophan (Trp)154, histidine (His)151, and serine (Ser)150 (Fig. 5B). The close form in acidic pH (pH range ∼5.6 to 7.2), the conformation of FR was changed after folate release (Fig. 5C).76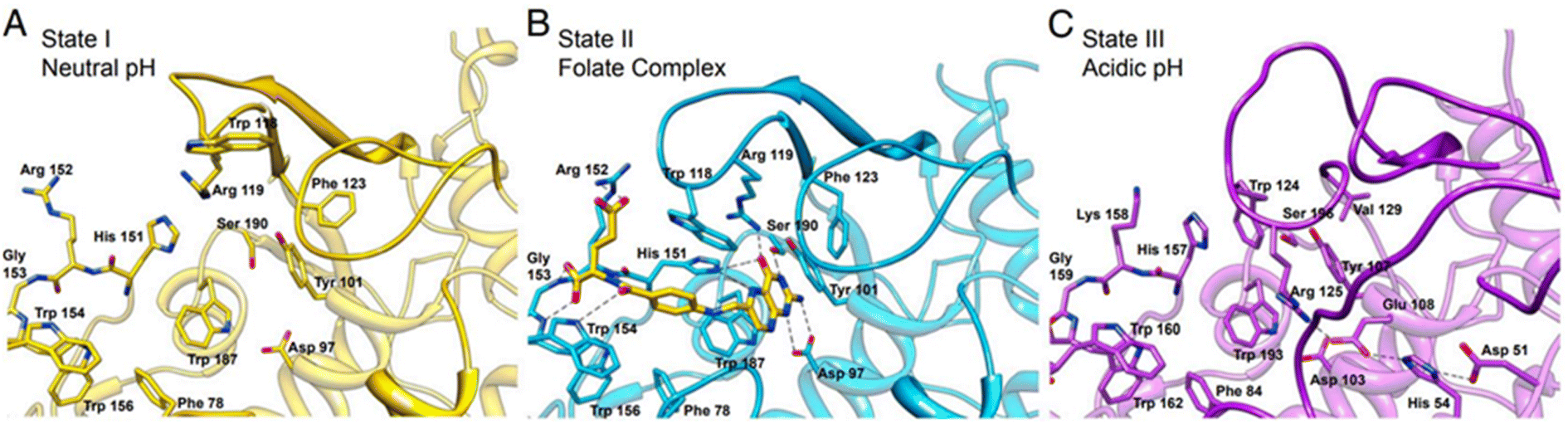 | ||
| Fig. 5 The active site cavity of the folate receptor. (A) Conformational changes in the residues that interact with folate in the open form at neutral pH. (B) The folate complex. (C) The closed form at acidic pH. Reproduced from ref. 76 (CC BY 4.0). | ||
The pterin ring of the folate molecule is located at the end of the active site cavity. At the same time, the 4-aminobenozyl moiety interacts via hydrophobic interactions in the central region of the cavity. In contrast, the γ-carboxylate of the glutamyl tail is partially exposed to solvent.76 This group is more accessible to solvents than the pterin amine (which is poorly reactive), which makes it a preferred site for modification and conjugation while maintaining the affinity of FA to the FR. A very classical route that should be mentioned is the activation of the carboxylic acid to form the folate N-hydroxysuccinimide (NHS) ester, which is then reacted with a primary amine on the bioconjugation partner, forming stable amide bonds. Of note, the pterin amine can potentially participate in chemical reactions. However, the pterin ring system leads to electron delocalization and stabilization of the overall structure, reducing its reactivity and making it less prone to undergo nucleophilic reactions. In the context of drug conjugation, the limited reactivity of this amine requires additional activation or modification steps to enhance its reactivity and enable efficient conjugation with molecules or carriers. However, the conjugation on the pterin amine site of FA decreases the affinity to the FR.
FRα is predominantly overexpressed in brain, colon, kidney, ovarian, breast, and lung cancers.77 In contrast, the expression of FRβ is detected mainly in activated macrophages due to stimulation by mediators of inflammation.78 The expression of FRs in carcinomas is approximately 300-fold higher than in healthy cells, estimated to be 1–10 million copies per cancer cell,44d,79 and the receptor-recycling rate is higher in malignant than in non-malignant cells.80 Of note, FA is a non-immunogenic water-soluble B vitamin that can be converted to tetrahydrofolate via dihydrofolate reductase. Besides, the FA is an essential cofactor in single-carbon methylation reactions and two steps of de novo purine biosynthesis, which is required for amino acid metabolism, DNA synthesis, and repair.81 In principle, FA endocytosis is crucial for tumour tissues to sustain their chronic proliferation.82 FRs have the most potential for prognostic biomarkers for a selective internalization of FA-conjugated drug transporters via FR-targeting by the cancer cells, known as the – Trojan Horse – for the delivery of therapeutics. Accordingly, the FA molecule can be decorated by glutamic acid (at the α- or γ-positions) to drug transporter, with minimal change of their binding affinity to the FRs (Fig. 6A). Therefore, drug transporter with small nucleotide size to large polymeric or protein constructs have been considered for targeted delivery of drugs and multidrug to the tumour tissue by FR-mediated endocytosis to enter the cytosol.83Fig. 6B provides a schematic illustration demonstrating an FA-conjugated drug transporter and the process of its internalization via FR-mediated pathways. Cellular drug uptake reveals that FA-conjugated drug transporter is internalized into endosomes by FR-mediated endocytosis and detached from FR encountered with a slight drop of pH to about five within the endosome through the action of proton pumps.84 FRs ideally return to the cell surface for further FA-conjugated drug transporter internalization, and the functionally active drug cleaved in the lysosome enables drug accumulation in cancer cells.
 | ||
| Fig. 6 (A) The chemical structure of folic acid (or folate) consists of pterin, aminobenzoic acid, and glutamic acid units. (B) Schematic illustration of FA-conjugated transporters internalization entered the cytosol. | ||
2 Folic acid (FA)-conjugated nanocarriers
2.1 Breast cancer
Breast cancer predominantly arises from mutations affecting steroid receptors, specifically estrogen (ER) and progesterone (PR) receptors.85 This malignancy manifests primarily through several molecular subtypes, with a notable emphasis on hormone receptor-positive variations. These subtypes encompass the ER- and PR-positive Luminal A and ER-positive Luminal B categories. Conversely, the human epidermal growth factor receptor 2 (HER2)-enriched subtype of breast cancer, constituting a distinct category, is characterized by the absence of ER and PR receptor expression, thus leading to a notably more unfavourable prognosis.86 Conclusively, basal-type breast cancer, often referred to as triple-negative cancer, exhibits an absence of ER, PR, and HER2 expression, leading to an even graver prognosis and markedly reduced survival rates. Current treatment options depend on the type, stage, and individual conditions, usually a combination of surgery, chemotherapy, and radiotherapy, and are associated with substantial adverse effects with severe personal and societal impact.87 To ameliorate these challenges, FR-targeted strategies by utilizing the FA-conjugated nanocarriers (NCs) hold considerable promise in facilitating the specific delivery of chemotherapeutics to cancer cells.88 The following section will review the advances in the field of FA-conjugated NCs for treating – or diagnosing – breast cancer in vitro and in vivo.Chitosan is widely utilized to build drug transporters due to its unique properties, such as nontoxicity, hydrophilicity, and water solubility. Chitosan is a linear cationic polysaccharide composed of randomly distributed β-(1 → 4)-linked D-glucosamine and N-acetyl-D-glucosamine that has been considered to fabricate PNPs. Chitosan's properties can be improved and tailored to introduce new functional groups on its skeleton through chemical modifications. Sohail et al.89 grafted thiol and folic acid (FA) onto chitosan to formulate PNPs for delivery of docetaxel (DTX), resulting in an enhanced internalization into MDA-MB-231 cells and improving the oral absorption level of DTX (Fig. 7A). In this method, drug is encapsulated into PNPs using the ionotropic gelation technique with tripolyphosphate (TPP) as the crosslinking agent.90 The positively charged amine groups on chitosan can interact with the negatively charged phosphate groups on TPP to form a nanoparticle structure via ionotropic gelation. In this context, Shao et al.91 and Li et al.92 utilized TPP to formulate cross-linked FA-conjugated chitosan-based NPs to deliver ligustrazine and catechin to breast cancer cells. When NPs are introduced into the body, they may interact with various cell types, including immune cells, endothelial cells, and other healthy cells. The reported formulations91,92 had no significant cytotoxicity in vitro as high as ∼0.5 mg mL−1 of unloaded PNPs. However, Sohail et al.89 first found that PNPs show improved antitumour cytotoxicity (IC50 ∼ 0.58 μg mL−1) against MDA-MB-231 cells, which is significantly lower than free DTX. Additionally, ex vivo analysis demonstrated that in the presence of verapamil (100 μg mL−1), DTX absorption of DTX-loaded thiolated-chitosan-based NPs was enhanced, which is related to the P-glycoprotein (P-gp) efflux pump inhibition. The apparent permeability coefficient enhancement ratio from the apical to the basolateral surface of rat intestine was reported to be about 11-fold higher for the thiolate-modified PNPs due to the inhibitory effect of their thiolated bonds to conjugate with cysteine of the protein tyrosine phosphatase, indicating a promising avenue in FA-conjugated NC research. The impact of thiolation on the chemical, physical, and biological properties of chitosan is extensively reviewed by Bernkop-Schnürch.93 As shown in Fig. 7B, Rafienia et al.94 fabricated MBZ-loaded FA-conjugated chitosan-based NPs cross-linked with TPP to increase their mechanical strength, stability, and drug release properties. The cylindrical subcutaneous implants containing the chitosan-based NPs are implanted in BALB/c mice xenografted with triple-negative 4T1 cells, which are known to be designed for under-skin implantation for sustained release of the drug.95 The implanted NPs in the tumour-bearing mice's flank were degraded after 18 days, released the NPs on 4T1 cells, internalized with FR-mediated endocytosis, and inhibited tumour volume growth.
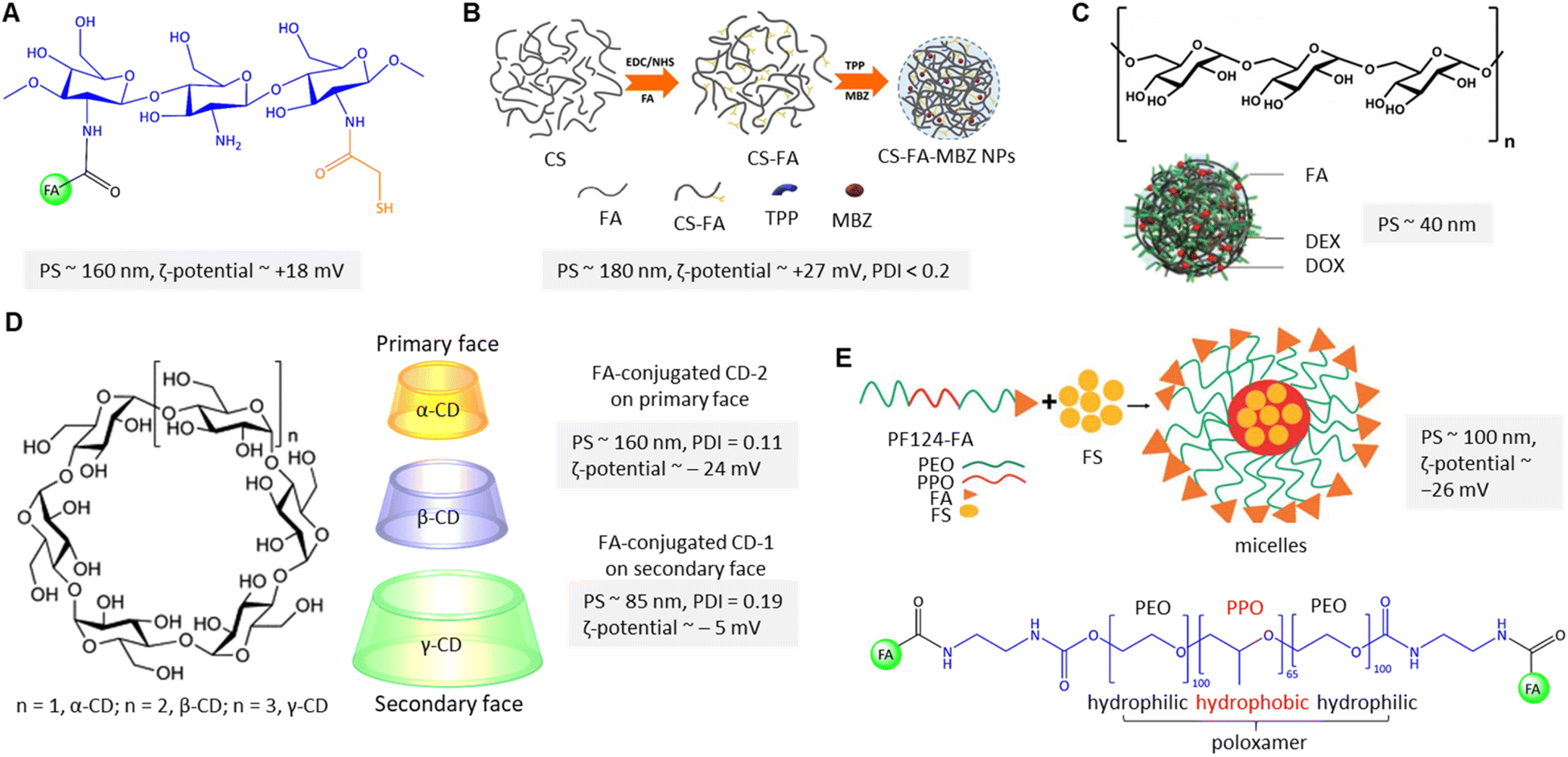 | ||
| Fig. 7 (A) The chemical structure of polymeric FA-conjugated thio-chitosan. (B) The preparation of CS-FA-MBZ NPs. Adapted from ref. 94 (CC BY 4.0). (C) Structure of dextran along with DOX@DEX-FA NPs. Adapted with permission from ref. 97. Copyright 2018, Royal Society of Chemistry. (D) Structure of cyclodextrins along with folate-conjugated CD-1 and CD-2. (E) Schematic view of FS-PF-FA micelle preparation along with the chemical structure of FA-PLGA-FA. Adapted with permission from ref. 103. Copyright 2018, Taylor & Francis. | ||
The degree of folic acid (FA) substitution refers to the number of FA conjugated to each chitosan molecule that significantly affects NPs properties such as size, morphology, release profile, loading efficiency, and loading capacity. Curcumin (CUR)-loaded chitosan-based NPs reported by Bagheri-Khoulenjani et al.96 showed the highest degree of substitution when the 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of FA:H-chitosan (400 kDa) was utilized. However, the 1:1 ratio of FA with L-chitosan (40 kDa) showed better loading efficiency (∼90%), and faster CUR release kinetics by decreasing the pH from 7.4 to 5. However, the choice between H-chitosan and L-chitosan for FA conjugation depends on the specific application and desired properties of the resulting NPs.
:1 ratio of FA:H-chitosan (400 kDa) was utilized. However, the 1:1 ratio of FA with L-chitosan (40 kDa) showed better loading efficiency (∼90%), and faster CUR release kinetics by decreasing the pH from 7.4 to 5. However, the choice between H-chitosan and L-chitosan for FA conjugation depends on the specific application and desired properties of the resulting NPs.
In contrast to chitosan, dextran is a branched polysaccharide consisting of α-1,6 linked glucose monomers with α-1,3 branches that have been used to encapsulate hydrophobic and hydrophilic drugs (Fig. 7C). However, the drug-loaded dextran-based NPs stability and release profile can be affected by the physiological environment, such as pH and ionic strength. Yang and Li et al. explored pH-dependent self-assembled doxorubicin (DOX)-loaded FA-conjugated dextran NPs that can be degraded in an acidic tumour microenvironment.97 The esterification of the accessible γ-COOH of FA and –OH of dextran was reported as the central polymeric core to encapsulate the DOX (Fig. 7C). The DOX release was about 76% at pH 5.5, significantly higher than at pH 7.4 (∼42%). The authors claimed that the high degree of substitution (79 FA molecules/per dextran) is due to protonation/dissociation of the free α-COOH at pKa ∼5.8 not only stabilized dextran NPs but also enhanced in vitro FR-mediated cellular uptake of FA-decorated NPs in FR(+) 4T1 cells. They reported that FA-conjugated PNPs show the highest tumour inhibition, about 75%, compared to non-targeted NPs.
Cyclodextrins (CDs) are amphiphilic cyclic oligosaccharides with 6 to 8 glucopyranose units that can encapsulate poorly water-soluble drugs in the inner hydrophilic cavity and release the content under physiological conditions of tumour tissue (Fig. 7D). Bilensoy et al. reported active targeting delivery of paclitaxel (PTX) via FA-conjugated CD-NPs for reducing toxicity and increasing the PCX antitumour efficacy for metastatic breast cancer.98 In their system, the FA was conjugated through the C6 linker chain onto the CD's derivatives on the secondary face (FCD-1 with neutral surface charge) and primary face (FCD-2 with negative surface charge) to render active targeting (Fig. 7D). The reported PNP formulation has caused cytotoxicity and cellular uptake of FCD-1 NPs into the 4T1 cells. The large number of aliphatic chains of FCD-1 compared to FCD-2 provided stronger interactions with PTX and more sustained drug release. The in vitro PTX release was about 96% after 24 h. Due to the low aqueous solubility of PTX, a mixture of Cremophor EL (CrEL), and dehydrated ethanol (1:1 ratio v/v), a compatible anticancer activity was reported in so-called CrEL formulations.99 Along the same lines, Bilensoy and colleagues state that CrEL-free PTX-loaded FCD-1 and FCD-2 NPs significantly reduced tumour burden.98 It was shown that FCD-1 NPs significantly improved the survival rate of mice by reducing in vivo toxicity to healthy tissues. An enhanced antitumour efficacy was achieved by administrations of 1.25 mg kg−1 of FCD-1 NPs per day for 20 days compared to unloaded FCD NPs.
Sarrafzadeh and Khorramizadeh investigated β-CD with seven glucopyranose units to incorporate zinc oxide (ZnO).100 ZnO with a high surface area and low toxicity has the ability not only to encapsulate the drugs but also to conjugate with CUR, as described by the authors. In addition, ZnO mediates anti-cancer effects on its own. Therefore, ZnO β-CD nanostructures functionalized with 3-mercaptopropionic acid (MPA) and FA in order to target the delivery of CUR to MDA-MB-231 cells. The MPA can be coordinated by substituting the S atom at the ZnO site, while β-CD can bind to the ZnO surface.101 The hydrodynamic particle size was reported at about 120 nm with a ζ-potential of −22 mV.100 The authors claimed that the CUR was mainly placed into β-CD cavities on the surface of ZnO. However, CUR loaded in the outer layer of β-CD is not excluded. The authors reported that FA-conjugated PNPs displayed superior toxicity activity against MDA-MB-231 cells, with no effect on healthy HEK 293 cells.
Poloxamers, also called pluronic, belong to amphiphilic triblock copolymers that have been used to fabricate PNPs suitable as water-insoluble drug carriers due to their core–shell structures, critical micelle concentration value (CMC), and a higher ratio of hydrophilic-lipophilic balance (HLB) in aqueous media.102 Following this rationale, Bothiraja et al. fabricated FA-conjugated triblock pluronic F127 micelles in which festin (FS) is encapsulated in hydrophobic poly(propylene oxide) (PPO) cores (Fig. 7E).103 Rupture of the micelles and full cumulative release of FS were reported within 12 h, while the initial burst release was about 30–40%. Notably, about 80% of FS was released from the micellar cores at pH 5, which was higher than at pH 7.4 (∼50%). In addition, the authors found that FS's cellular uptake from FA-conjugated micelles increased about 6-fold compared to non-targeted micelles. In another study, a mixed pluronic PF127/F68 micelle was utilized by Patil and co-workers.104 In this design, the micelle was conjugated with FA for targeted delivery of chrysin to MCF-7 cells and enhanced the drug's oral bioavailability. Pluronic F68 is composed of a shorter hydrophobic polypropylene core resulting in low loading capacity due to its high CMC value. To address this problem, the proportional contribution of F127 and F68 must be considered to balance the HLB and improve the drug encapsulation efficiency and release.105 The proportion affected micelle size from 152 to 420 nm (ζ-potential ∼ −21 mV), which is attributed to the hydrating of polymer chains.104 The authors found that about 75% of chrysin was released after 24 h from the micelles at pH 6.8. The CMC of the FA-conjugated mixed micelle was 1.52 mg mL−1, which was lower than the FA-conjugated PF127 micelle due to its higher lipophilicity. The GI50 value of the conjugated micelle was reported at about 16.5 mM, higher than free chrysin and non-conjugated micelles.
Several polyesters such as PGA, PBL, PVL, PCL, PLA, PLGA, and PDO have been used for the fabrication of amphiphilic block copolymers. In this context, Vu-Quang and Tran et al. reported a self-assembled pluronic P123-grafted chitosan nanogel conjugated with FA for the co-delivery of PTX/CUR to MCF-7 cells.106 Pluronic P123 was activated by p-nitrophenyl chloroformate (NPC) and substituted with a poly-3-amino-1-propanol sidechain. The resulting NPC-P123-OH is conjugated with –NH2 of chitosan at pH 5 via carbamate formation. The size of the nanogels was distributed about 51 nm utilizing a micelle admixture of chitosan:P123 with a weight ratio of 1:20 and a CMC value of 0.08 mg mL−1. Both PTX and CUR were encapsulated in the hydrophobic PPO core. The cumulative release rate was reported as about 23% of PTX/CUR at pH 5.6 after 48 h. The CMC indicates the polymeric network's micellar stability, size, and viscosity that influence drug loading efficiency and release from the micelles. The authors reported more sustainable stability at a lower concentration of P123 (ζ-potential ∼ +39 mV) and a lower CMC profile (∼0.036 mg mL−1). In addition, the synergistic effect of PTX/CUR was confirmed via observation of a pronounced anticancer activity for dual-loaded micelles (IC50 ∼ 5.7 nM) compared to PTX-loaded micelles (IC50 ∼ 8 nM). In line with the above investigation, the approach was studied in multi-drug resistant MCF-7/ADR cells by Hong et al. utilizing pH-sensitive pluronic L61 unimers for the co-delivery of CUR and DOX.107 Unimers refer to individual polymer chains (micelles) formed in solution with unassembled structures. Micellar copolymer poly-histidine (Phis)-PLA-PEG-PLA-Phis and pluronic 127 (F-pHSM-L61/CUR/DOX) was partially conjugated with FA for two reasons: first, the hydrophilic poly(ethylene oxide) structures of F127 ensure the prolonged circulation of the micelles and could also promote gelation.108 Second, L61/CUR facilitates endosomal escape to overcome the MDR of breast cancer.109 The authors found that the pluronic L61/CUR micelles downregulated the expression of P-gp in response to drug efflux from the cancer cells.107In vivo DiR fluorescence imaging after administration of FA-conjugated DOX/CUR/DiR micelles onto the tumour-bearing mouse model exhibited the accumulation of DiR in the tumour site, cell proliferation inhibition, and mitochondria-mediated cell death. Poly(ADP-ribose) polymerase protein (PARP) cleavage corroborated that the antitumour effect is associated with pro-apoptotic effects. Very recently, Yang and Liu et al. designed dual-targeted pH-sensitive polymeric micelles constructed using the hyaluronic acid-modified poly-histidine (HA-PHis) and FA-conjugated F127.110 Interestingly, the effect of FA-conjugated DTX-loaded micelles on the cell survival rate (IC50) in HepG2 and MCF-7 cells was reported about 2.5 and 10 μg mL−1, respectively.
The α-tocopheryl polyethylene glycol succinate (TPGS) is a water-soluble synthetic derivative of α-tocopherol combining hydrophilic PEG and hydrophobic alkyl chain (Fig. 8A). In this context, Su and Ping et al. utilized TPGS2k, a polymeric carrier, to conjugate the FA and mitoxantrone (MTO) (Fig. 8B).111 This system was designed to deliver MTO via FR-targeting to MCF-7 cells. The optimized CMC of TPGS2k, MCT, and FCT were found to be about 0.0251, 0.072, and 0.0338 mg mL−1, respectively, lower than that of TPGS1k (0.2 mg mL−1). A lower CMC can contribute to improved stability of micelles and resistance to dissociation in certain contexts, such as the bloodstream. The authors found that the initial drug release at pH 5 was 35% for MTO-MCT and 40% for MTO-FMCT. In contrast, the cumulative drug release reached 76%, and 86% after 40 h, remarkably higher than that observed at pH 7.4.
 | ||
| Fig. 8 (A) The chemical structure of TGPS. (B) Schematic diagram of MTO-FMCT NPs. Reproduced with permission from ref. 111. Copyright 2017, American Chemical Society. (C) ALN/FA-decorated PTX-loaded NPs utilized for bone metastatic breast cancer (left) and ex vivo NIR fluorescence images of the isolated tibias of 4T1 tumour-bearing mice at 8 h post-injection with PBS and different DiI-labeled NPs (right). Reproduced with permission from ref. 115. Copyright 2020, Royal Society of Chemistry. | ||
Advanced breast cancers tend to metastasize in bones, lungs, liver, and brain;112 therefore, several studies have been performed utilizing biomarkers for diagnosis and chemotherapy.113 The bones are the first site of action (60–80%) often detected in those with stage IV breast cancer.114 Recently, Chiang and Chiu et al. reported dual bone- and tumour-targeted chemotherapy utilizing a polymeric-based vehicle comprising PLGA core coated with alendronate-modified FA-conjugated TPGS to deliver PTX to 4T1 cells and bone matrix (Fig. 8C).115 Alendronate, a member of the N-containing bisphosphonate, can be conjugated to TPGS, providing additional functionalities such as targeting bone tissue115 or inhibiting osteoclast activity.116 The results demonstrated a superior alendronate-mediated binding affinity for hydroxyapatite in the bone matrix using Rho-labelled NPs. An elevated level of cellular uptake of drug payload via FR-targeting to FR(+) 4T1 cells was reported compared to FR(−) A549 cells. Meanwhile, in vivo PTX accumulation in bone metastases was monitored via enhanced fluorescence signals of the tumour-bearing right tibia compared to the left tibia after intravenous injection of various DiI-loaded PNPs (Fig. 8C).
PLGA enhances the bioavailability of encapsulated drugs from degradation and premature release. Hence, an FA-conjugated PLGA-based NC reported by Debnath et al. for co-delivery of gemcitabine (GEM) and CUR to MDA-MB-231 and MCF-7 cells,117 to address an issue for TNBC that has become increasingly resistant to GEM due to overexpression of hypoxia-inducible factors. The authors reported a biphasic release pattern with an initial burst that was followed by a sustained release of GEM/CUR. The FA-conjugated drug-loaded PNPs led to a strong apoptotic cell death attributed to significantly upregulated p53 and Bax proteins. At the same time, B-cell lymphoma 2, cyclooxygenase-2, NF-κB, and p65 were downregulated in PNP-treated cancer cells. PLGA-based NPs can also be radiolabelled by attaching a chelator to the surface of the NPs that can be complex with the radioisotope. In another study, the authors fabricated technetium-99m (99mTc)-radiolabelled PLGA-based NPs for non-invasive diagnostic imaging and FR-targeted delivery of epigallocatechin-3-gallate against MDA-MB-231 and MCF-7 cells.118 NCs were radiolabelled with 99mTc using stannous chloride dihydrate (SnCl2·2H2O) as a reducing agent, enabling the tracking and non-invasive imaging of the NCs in vivo. The reported scintigraphy images by authors showed higher tumour accumulation of 99mTc-labeled FA-conjugated PNPs than non-targeted PNPs.
In general, the chemical modification of polymers on the surface or core via linkers and lipophilic agents is a promising strategy to improve nanomaterial's performances, solubility, and multi-functionalization ability to conjugate with other molecules. For example, a unique PNP was constructed by Zhang et al. through the conjugation of two units of hydrophobic PCL via S–S bonds to the hydrophilic PEG7.5k segment using mercaptoethanol (Fig. 9A).119 This copolymer was utilized for the co-delivery of DOX and indocyanine green (ICG) as an imaging and hyperthermia agent to EMT-6 cells. DSPE-PEG2k-FA was utilized for FR-targeted delivery; thereby, hydrophobic tails of DSPE interacted with the hydrophobic block and PEG-FA located on PNP's surface. The film hydration method was used in their system to admix PCL-SS-PEG-SS-PCL and DSPE-PEG2k-FA. In the following, DOX/ICG were trapped into polymer after sonication. In line with this polymeric design, Danafar et al. served lysine as a linker to conjugate FA and PEG to form a multifunctional drug delivery system.120 FA can be conjugated to one end of lysine via the –NH2 group, while PEG can be conjugated to the other end of lysine via the –COOH group. The obtained FA-lysine-PEG-PCL micelles were utilized to deliver tamoxifen (TMX) to MCF-7 cells. The TMX-loaded FA-conjugated micelles had a diameter of 97 nm with a ζ-potential of about −23 mV. Cumulative TMX release at pH 5.5 was about 60% within 72 h, twice than that observed at pH 7.4. The authors found that the MCF-7 cell viability was decreased by about 53% using TMX-loaded FA-conjugated micelles instead of non-targeted micelles.120 In another study, they utilized the same micellar system to co-deliver TMX and quercetin to 4T1 cells.121 The authors found that by applying FA-conjugated micelles containing the highest dose of TMX/quercetin (∼20 μg mL−1), the cell viability decreased to about 29%. In another work, an FA-conjugated PEG2k-DSPE nanoemulsion was constructed by Hu et al. using high-pressure homogenization.122 The PTX was loaded into a PEGylated nanoemulsion to achieve in vivo delivery to 4T1 cell-based tumours in mice. Surface modifications via PEGylation are utilized not only to extend their plasma half-life circulation but also to abrogate their systemic elimination via the reticuloendothelial system.123 An in vitro cumulative release of 47% was reported after 12 h, along with a higher uptake into 4T1 cells of the FA-conjugated PNPs compared to the non-targeted NPs. The authors reported in vivo studies focusing on tumour growth inhibition, reduced drug side effects, and prolonged survival, resulting in enhanced antitumour effect and interference of passive and active targeting using PEGylated PNPs.122
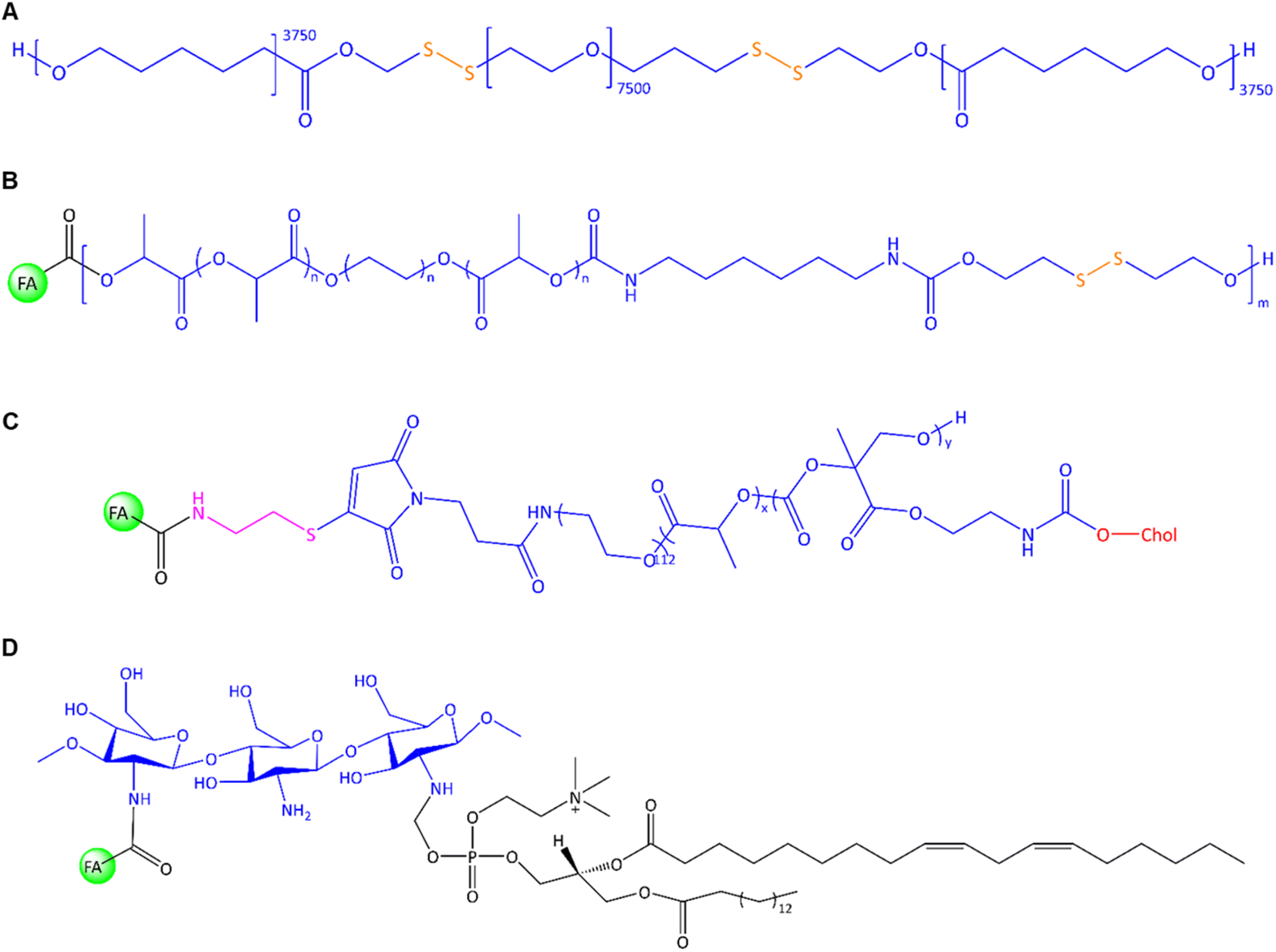 | ||
| Fig. 9 The chemical structure of (A) PCL-ss-PEG-ss-PCL, (B) FA-conjugated chitosan-lipid NPs, (C) FA-PEG-b-p-(MTC-Chol-co-LA) lipopolymer, (D) FA-conjugated chitosan/phospholipids (lipoid S75). | ||
Further, Koul et al. utilized redox-responsive PNPs via ring-opening polymerization of lactide with PEG that was followed by isomerization polymerization of this copolymer and 2-hydroxyethyl disulphide (Fig. 9B).124 The random multiblock FA-PLA-PEG-PLA-urethan-S-S was used to deliver DOX to MCF-7, BT474, and L929 cells. Urethane (carbamate) contributes to the stability and mechanical properties of the NCs, while disulphide linkages can be selectively cleaved in the presence of reducing agents such as glutathione (GSH). The reaction of OH-PLA-PEG-PLA-OH with 2-hydroxyethyl disulphide and hexamethylene diisocyanate under an N2 atmosphere led to the formation of multiblock copolymer that later conjugated with FA in the presence of NHS/DCC. Drug release studies showed different outcomes in neutral and acidic pH in the presence and absence of GSH as a reducing agent. The authors found that about 72% of DOX was released at pH 5.5, higher than at pH 7.4 (∼18%). The drug release profile upon GSH showed accelerated drug release at pH 7.4 and 10 mM GSH (∼55% drug release after 96 h). Enhanced in vitro uptake into MCF-7 cells of up to 22% was reported for FA-conjugated PNPs compared to non-targeted PNPs. In vivo studies of Ehrlich ascites tumours in mice showed that about 91% of the tumour regressed by using FA-conjugated PNPs compared to free DOX-treated mice with only 35% antitumour activity.
In a recent approach, self-assembled lipopolymeric NPs with higher stability than liposomes were utilized by Chitkara et al. to deliver DTX via FR-targeting for the treatment of TNBC using MDA-MB-231 cells.125 The authors grafted an amphiphilic lipopolymer with cholesterol and DL-lactide by microwave-assisted ring-opening polymerization. The microwave energy promotes the opening of cyclic monomers (lactide) and their subsequent polymerization into linear chains enhances reaction rates, and yields uniform polymerization compared to traditional methods. The structure of FA-PEG-b-p-(MTC-Chol-co-LA) lipopolymer is shown in Fig. 9C. Two major advantages of PEG chain biopolymers are: first, the self-assembly of PEG chain co-polymers and the form of disc-like micelles with stacked-like morphology that enable a higher drug payload, and second, linear or branched aliphatic polycarbonates are susceptible to stimuli-responsive degradation.126 However, the authors reported that about 13% of DTX was released in the first 12 h, while the cumulative release reached around 77% after 7 days.125 The fabricated FA-conjugated lipopolymeric NPs offered a desirable property profile and showed significant in vitro and in vivo stability, prolonged DTX release on the tumour site, a significant intracellular uptake, improved pharmacokinetic profile, enhanced EPR effect, improved cytotoxicity, apoptosis, and change in expression levels of Bcl-2, BAX, and Ki-67.
Following these findings, Li and Zhu et al. reported that the Bax, Bcl-2, caspase-3, and caspase-9 were activated in apoptotic cells by extrinsic and intrinsic pathways utilizing FA-conjugated chitosan-based NPs via co-delivery of DOX and oleanolic acid.127 The highest mRNA expression levels were exhibited for those genes and induced apoptosis in MDA-MB-231 cells. This concept was further exploited in an exciting study by Khan and Madni et al., utilizing FA-conjugated chitosan-phosphatidylcholine-based NPs to enhance the antitumour efficiency of cisplatin toward SK-OV-3, A2780, and MCF-7 cells.128 In this system, the phosphate head group of lipoid S75, consisting of 70% phosphatidylcholine, engages in interactions with the positively charged FA-conjugated chitosan (Fig. 9D). Notably, the ratio of lipid:FA–chitosan in the ionic gelation method impacts NP's size and polydispersity index and the encapsulation efficiency of cisplatin. Gel-like particles can be created by inducing the cross-linking of polymers through electrostatic interactions between oppositely charged ions. They found a sustained release profile of up to 90% within 48 h. Folate receptors mediate higher cellular uptake of FA-conjugated cisplatin-loaded PNPs and enhanced cytotoxicity of cisplatin-loaded PNPs compared to free cisplatin in vitro.
PEGylation has been applied for clinical NC formulation to shield particles from opsonization and reduce the rapid uptake by the reticuloendothelial system of the blood.123b Another study by Arias et al. showed the great potential of FA-conjugated PEGylated PLGA NPs for targeted 5-FU delivery.129 The authors optimized several polyvinyl alcohol (PVA) concentrations (0.5–1.5% w/v) and sonication time (from 0.5 to 5 min) to stabilize uniform size distribution, polydispersity, and optimal formulation of PLGA-PEG-FA NPs. The negative surface charge of FA-PEG-PLGA NPs at about −15 mV exhibited a relatively lower value before FA conjugation. By protonation of –NH2 groups of FA, the negative charge on PLGA is diminished. The authors reported that the initial burst release of 5-FU was only 25% after 1 h, attributed to 5-FU release that is weakly bound on the surface. In contrast, complete polymer degradation after 6 days led to about 80% 5-FU release. Cytotoxicity studies in FR(+) MCF-7 and HT-29 cells demonstrated that the IC50 of FA-conjugated PNPs was 4-fold lower than that of the non-targeted PNPs in vitro.
An interpenetrating polymeric network (IPN) is a hydrogel-based drug carrier comprising at least two polymers cross-linked – simultaneously or sequentially – with each other.130 An IPN refers to a unique type of polymer structure where two or more polymer networks are intertwined or interlocked at a molecular level without covalent bonds. Raj et al. utilized an IPN comprising carboxymethyl cellulose and egg white (EW) that was cross-linked with PEG and PVA to deliver cyclophosphamide (CP) to MCF-7 cells.131 The authors blended the carboxymethyl cellulose with EW via the heat coagulation process to improve the mechanical properties of IPN and CP release efficiency. In principle, hydrogen bonds in cellulose hydrogels enhanced physicochemical properties and pH sensitivity expanding its applications.132 The low drug loading is attributed to the steric barrier of cross-linked PEG, which prevents IPN aggregation and stabilizes its structure. Notably, the hydrodynamic size of FA conjugation on CP-loaded IPN was reported at about 239 nm (DPI ∼0.19) with a ζ-potential of −36 mV, confirming grafting of FA-EW conjugate on the polymer surface. The encapsulation efficiency of CP-loaded FA-conjugated IPN was reported at about 94% higher than carboxymethyl cellulose-EW IPN (∼64%) because of the higher capacity of cross-linked PEG/PVA to entrap the CP. Furthermore, the authors found that the CP release from FA-EW/CP IPN at pH 5 (∼55%) is relatively higher than at pH 7.4 (∼29%) after 48 h.131
Multi-shelled hollow capsules, including organic, polymer, metal oxides, and metallic-based capsules, are mainly utilized in drug carriers due to their layer-by-layer assembly to create a unique internal cavity to carry drugs and the well-controlled release upon stimuli. The choice of materials depends on the desired properties of the capsules, such as biocompatibility, stability, and responsiveness to external stimuli. The distinct compartments within the capsules can be loaded with different drugs, enabling combination therapies or sequential release of multiple therapeutic agents. In a pioneering study, Kim et al. reported FA-conjugated hollow polymeric capsules (HPCs) for delivery of DOX to MCF-7 cells and mouse embryonic fibroblast (NIH/3T3) cells.133 As shown in Fig. 10G, the benzenedimethanol-based HPCs, and naphthalenedimethanol-based HPCs were synthesized via a self-assembly Friedel–Crafts polymerization composed of hydroxyl-branched hollow capsules. The authors assume that the –OH was converted to –COOH in order to conjugate with the FA molecule and stabilize DOX through π–π interactions within the aromatic structure. The authors have developed an acid–base interaction-mediated self-assembly method to generate in situ functionalized HPCs with tuneable wall thickness.134 The particle's porosity provided a maximum DOX encapsulation of up to 86%. An efficient drug release of up to 50% was reported after 30 h in an acidic medium. In comparison, the cumulative release was only 16% after 150 h under neutral and weak basic conditions due to the pH-responsible release performance of PNPs. Furthermore, the in vitro delivery of DOX to MCF-7 cells showed that FA-HPCs had higher cellular uptake than non-targeted HPCs. Noteworthy, naphthalenedimethanol-based capsules had stronger DOX fluorescence inside the nuclei due to higher π–π interactions. Multi-shelled structures possess several desirable properties, including high loading capacity, sequential drug release, and the ability for multifunctional modification, making them versatile and attractive for receptor-mediated targeted therapies.
 | ||
| Fig. 10 Schematic illustration of the FA-conjugated hollow polymeric capsule FA-HPCs for delivery of DOX. (a) Self-assembly of HPCs, (b) FA-conjugated HPC synthesis, (c) illustration of drug delivery to cancer cells. Reproduced with permission from ref. 133. Copyright 2021, American Chemical Society. | ||
:9 ratio of dipalmitoylphosphatidylcholine (DPPC): 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) was utilized. The gold rods stabilize the liposome and prevent premature drug release. In contrast, the drug was trapped during the film hydration and sonication process. The complete NC disintegration and subsequently DOX release and uptake by MDA-MB-231 cells was reported upon near-infrared irradiation (NIR, λ = 750 nm) at pH 2 within 12 h. Au nanorod/liposome system was aggregated after irradiation, while hydrolytic lipase led to full disintegration of liposome at acidic pH of tumour microenvironment, and consequently the DOX release. This NC system also displayed a good contrast after NIR exposure in computer tomography as well as transmission electron microscopy imaging. Similarly, the luteolin (LUT)-loaded liposomal system coated with poly-lysine-FA, as reported by Mudavath et al.,136 is an interesting formulation that delivered the payload upon NIR laser at λ = 808 nm. The size of the FA-conjugated LUT-loaded liposome was about 180 nm with a positive surface charge of +33 mV. LUT was reported to inhibit cell migration and proliferation by regulating vascular endothelial growth factor (VEGF) expression and induced apoptosis via up-regulation of caspase-3.
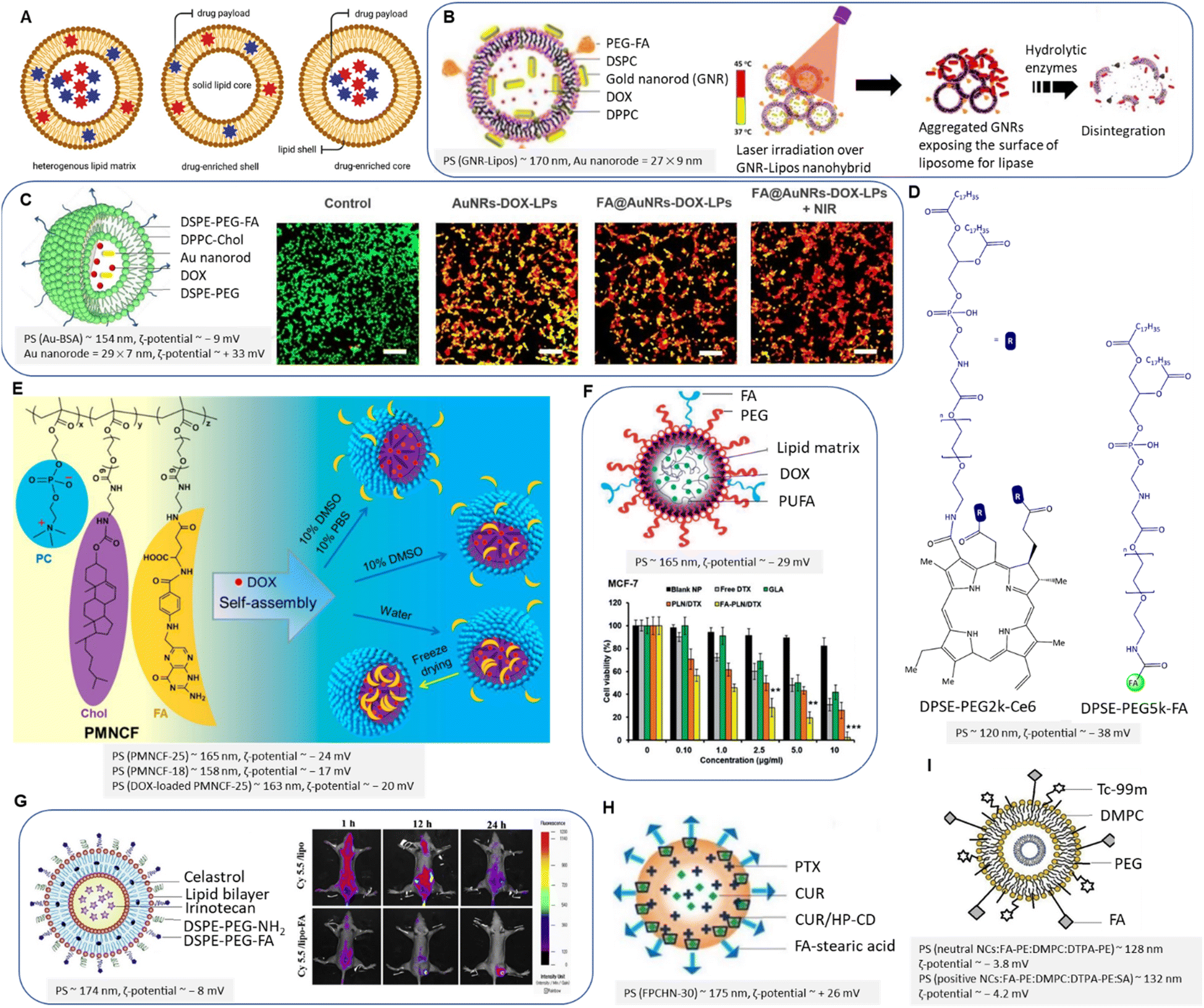 | ||
| Fig. 11 (A) Schematic representation of lipid matrix, drug-enriched shell model and drug-enriched core model. (B) Schematic illustration of gold nanorods-liposome “FA-PEG-GNR-Lipos” (left) and the schematic release of embedded liposomes upon NIR (right). Adapted with permission from ref. 135. Copyright 2018, American Chemical Society. (C) Schematic of FA@AuNRs-DOX-LPs (left) and CLSM images of calcein-AM/EthD-1 stained 4T1 cells treated with NCs upon NIR (right). Rearranged with permission from ref. 137. Copyright 2018, Elsevier. (D) Chemical structure of PTX@FA-NLC-PEG-Ce6. (E) Schematic illustration of PMNCF structure and micelle formation. Reproduced with permission from ref. 140. Copyright 2019, American Chemical Society. (F) FA-conjugated PUFA-based LNPs and antitumour activity of NCs in vitro after 24 h. Reproduced from ref. 143 (CC BY). (G) FA-conjugated liposomes (left) and in vivo biodistribution of NCs on MDA-MB-231 tumour-bearing mice (right). Reproduced with permission from ref. 145. Copyright 2018, Elsevier. (H) PTX/CUR-HP-CD co-loaded LNPs. Adapted from ref. 148 (CC BY 3.0). (I) FA-conjugated radiolabeled liposome. Reproduced with permission from ref. 155. Copyright 2019, Elsevier. | ||
PEGylated artificial phospholipid vesicles were mainly used to stabilize the chemotherapeutics and prolong blood circulation. PEGylation forms a hydrophilic layer on the liposome surface, resulting in reduced affinity to the mononuclear phagocyte system, reduced systemic toxicity, and clearance immunogenicity. Han, Park, and Choi et al. introduced a relevant liposomal platform intending to evaluate in vivo activity of breast tumour regression by the synergistic effect of PT and DOX chemotherapy.137 The liposomes are composed of DPPC/cholesterol/DSPE-PEG2k. The seedlessly synthesized Au nanorods were coated with bovine serum albumin (BSA) to reduce the toxicity caused by cetrimonium bromide as an emulsifier. The co-loaded DOX and Au nanorods were decorated with FA-conjugated liposomes (Fig. 11C). About 46% of encapsulated DOX was released at endosomal environmental pH upon exposure to NIR (λex = 808 nm) for 5 min. FA-conjugated liposomes induced significantly higher toxicity against 4T1 cells (IC50 ∼ 3.1 μg mL−1) than non-targeted carriers. Cell viability decreased upon NIR irradiation, and a higher dose of DOX entered the cell (IC50 ∼ 1.9 μg mL−1), which is attributed to local hyperthermia. Confocal laser scanning microscopy imaging of calcein-AM/EthD-1 stained 4T1 cells before and after treatment indicated that the most efficient anti-tumour effects belong to synergistic therapy using FA-conjugated NPs and NIR (Fig. 11C). In another study, Feng et al. constructed an FA-conjugated PEGylated nanostructured lipid carrier loaded with PTX and photosensitizer chlorin e6 (Ce6) for effective photothermal therapy.138 The amine group of DSPE-PEG2k was conjugated with Ce6 to enhance water solubility, while FA interacted with amphiphilic DSPE-PEG5k-NH2 guided targeted drug delivery (Fig. 11D). This nanocarrier system enhanced the solubility of PTX and Ce6, increased their intracellular uptake, and produced sufficient local ROS, such as singlet oxygen139 that was triggered by laser irradiation via electron intersystem crossing, eventually inducing increased cytotoxicity on MDA-MB-231 cells by moderate synergistic effects. The cell viability of cancer cells was reported at about 95 μg mL−1 of FA-conjugated LNPs irradiated with light of wavelength 660 nm.138 The cumulative release value of PTX was about 55% within 72 h. The in vivo imaging of tumour-bearing nude mice after NPs injection showed increased fluorescence intensity regarding FA-conjugated NPs than non-targeted NPs (Fig. 11D).
Contrary to liposomes, micelles are closed lipid monolayers with a hydrophobic or hydrophilic core with hydrophobic fatty acids on the surface (known as an inverted micelle). Micelles are extensively utilized not only for efficient endosomal escape due to their self-assembly structure having a hydrophobic core and a hydrophilic shell but also related to their higher affinity to accumulate in cancer cells. In this context, Gong et al. reported on FA-conjugated cell membrane mimetic copolymeric micelles (PMNCF) constructed via amidation reaction of the –O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of PMN with the –NH2 of phosphorylcholine zwitterion and cholesterol.140 Of note, free-radical copolymerization was utilized by the authors to develop PMN copolymers.141 The FA molecule conjugated at the end of the polymer side chains bearing the amino group (Fig. 11E). The FA conjugation and equal ionic charges of phosphorylcholine zwitterion affect cancer cell targeting and cellular uptake. By increasing the percentage of dimethyl sulfoxide to 10% of the solution, the authors reported better FA solubility and higher FA connectivity to the micelle surface. Hence, the killing efficacy was enhanced to 160% upon the above optimization. The molecular weight of PMNCF influences the NPs size, ζ-potential, and consequently cell viability. Cell viabilities of DOX-loaded micelles (0.02 mg mL−1) reduced free DOX toxicity to 20% for normal L929 cells. The strong hydrophobicity of the cholesterol core led to the well-controlled release of hydrophobic DOX at pH 7.4 and decreased cytotoxicity. Increasing the hydrophobicity of the micellar core induced a higher loading capacity and sustained DOX release, which follows previous research.142
O of PMN with the –NH2 of phosphorylcholine zwitterion and cholesterol.140 Of note, free-radical copolymerization was utilized by the authors to develop PMN copolymers.141 The FA molecule conjugated at the end of the polymer side chains bearing the amino group (Fig. 11E). The FA conjugation and equal ionic charges of phosphorylcholine zwitterion affect cancer cell targeting and cellular uptake. By increasing the percentage of dimethyl sulfoxide to 10% of the solution, the authors reported better FA solubility and higher FA connectivity to the micelle surface. Hence, the killing efficacy was enhanced to 160% upon the above optimization. The molecular weight of PMNCF influences the NPs size, ζ-potential, and consequently cell viability. Cell viabilities of DOX-loaded micelles (0.02 mg mL−1) reduced free DOX toxicity to 20% for normal L929 cells. The strong hydrophobicity of the cholesterol core led to the well-controlled release of hydrophobic DOX at pH 7.4 and decreased cytotoxicity. Increasing the hydrophobicity of the micellar core induced a higher loading capacity and sustained DOX release, which follows previous research.142
Polyunsaturated fatty acids (PUFAs) are another group of cell membrane-compatible molecules that was utilized by Yong and Kim et al. to design FA-conjugated PUFA-based lipid NPs to increase the efficacy of DTX in multi-resistant metastatic breast cancers (Fig. 11F).143 This compatibility can enhance the effectiveness and bioavailability of these NCs in drug delivery applications. The results corroborated that the PUFA synergistically improved the anticancer efficacy of DTX against MCF-7 and MDA-MB-231 cells by inducing a G2/M phase arrest and cell apoptosis in line with other investigations. A dose-dependent cytotoxic effect reported by exposing cells to 10 μg mL−1 of DTX yielded 50% cell death in MCF-7 cells. One-half of the loaded DTX was released from FA-conjugated NPs after 96 h. The authors also reported that the PUFA/DTX combination not only downregulated the expression of PARP, caspase-3, and caspase-9 but also blocked the phosphorylation of the Akt signalling pathway in tumour models revealed by western blot analysis. This phenomenon is in accordance with the downregulation of the phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt) signalling pathway in breast cancer to regulate cell growth, cell proliferation, and apoptosis.144 In addition, the authors found that the Bcl-xl as a transmembrane protein family was markedly downregulated upon treatment with FA-conjugated lipid NPs.
FR-targeted liposomes loaded with bioactive agents exhibited selective cytotoxicity against FR(+) breast cancer cells. As depicted in Fig. 11G, FA-conjugated celastrol- and irinotecan-loaded liposomes were fabricated and evaluated by Yong and Kim et al. for treating FR(+) MCF-7 and MDA-MB-231 cells.145 Liposomal NPs were prepared by a thin-film hydration technique146 utilizing DPPC, cholesterol, and DSPE-PEG-FA. Irinotecan and celastrol with different solubility rates were safely encapsulated in lipophilic and aqueous environments of the lipid bilayer resulting in a sustained release mechanism. Of note, irinotecan has gastrointestinal toxicity and myelosuppression, limiting its usage and administration.147In vitro uptake of both drugs was reported for FR(+) cells using FA-conjugated PEGylated liposomes, whereas their uptake in A549 as FR(−) lung cancer cells was insignificant. This was demonstrated by Cyanine 5.5 loaded liposomes that yielded higher intensity using FA-conjugated liposomes than non-targeted liposomes (Fig. 11G). In addition, tumour cell volumes, angiogenesis, and cell proliferating markers CD31 and Ki-67 were significantly downregulated, while the PARP and caspase-3 were upregulated by treating with FA-conjugated drug-loaded liposomes. Unlike the above research, for the purpose of overcoming MDR in MCF-7 and ADR cells, a sequential release of encapsulated CUR in the lipophilic cavity of 2-hydroxypropyl-β-cyclodextrin (HP-β-CD) and PTX-trapped in FA-conjugated LNPs reported by Baek and co-workers (Fig. 11H).148 The hydroxypropyl groups introduced into the β-CD molecule improve its solubility and enhance its ability to interact with hydrophobic molecules. This molecule was utilized to improve drug stability and water-solubility for earlier release of CUR compared to PTX. Several clinical trials utilizing CUR have reported its impact on the expression and regulation of growth factors, protein kinases, inflammatory cytokines, and apoptosis-related proteins.149 However, a faster CUR release enables P-gp mediated efflux pump inhibition,150 which allows increased cellular uptake and cytotoxicity of PTX. It is known that P-gp suppression in a dose-dependent manner of CUR can be achieved by downregulating the PI3K, AKt, and NF-kB pathways.151
Solid lipid NCs were designed by admixing glyceryl monostearate and TPGS in the oil phase to the polysorbate 80 in the aqueous phase and blended with stearic acid and FA in the organic lipid phase.148 However, they found that the lipophilicity, location of drugs on lipid NPs, amount of used HP-β-CD, the lipid matrix, surfactant concentration, and solubility of the drugs could affect the release profile of drugs from NPs.148 The same strategy was employed using dual CUR/GEM-loaded PNPs.117 In another work, lipoprotein-based NCs were fabricated by Pandita et al., comprised of phosphatidylcholine, cholesterol, and stearyl amine.152 The natural biocompatibility and targeting capabilities make lipoprotein a promising platform for targeted drug delivery, imaging, and diagnostics, e.g., by incorporating fluorescent dyes or contrast agents into low-density lipoproteins. The authors found that the FA was conjugated to BSA by amino groups and oriented outward lipophilic center of NCs. Resveratrol (RSV) was loaded into LNPs, and about 91% of the drug was released within 72 h. FA-conjugated LNPs inhibited the growth of MCF-7 and A549 cells with an IC50 value of 9.6 and 16.8 μg mL−1, respectively.
Strategies using radiolabeled NCs are one of the major studies on the limitation of endogenous (interstitial) radiotherapy.153 For example, technetium-99m (99mTC) and indium-111 (111In), gallium-67 (67Ga), gadolinium-153 (153Gd), iodine-123 (123I), and copper-67 (67Cu) are known as γ-emitting radionuclides that have been employed for non-invasive monitoring of the biodistribution and accumulation of the drug via single photon emission computed tomography (SPECT), while iodine-131 (131I) has been used as β+ emitter in positron emission tomography (PET).154 To visualize liposome distribution and their accumulation sites, a 99mTc-radiolabeled liposomal platform was employed by Silindir-Gunay et al. for molecular tumour imaging SPECT and CT.155 In principle, NCs such as liposomes can be labeled by indirect labeling that involves attaching a radiolabeled molecule (such as a chelator or a targeting ligand) to the surface of previously prepared NCs using conjugation chemistry156 or by direct labeling via incorporating a radiolabeled ligand or chelator to label metal radionuclides into the NC's surface during the preparation.157 The authors reported neutral and positive charged FA-conjugated and PEGylated DTPA-PE containing liposomes (Fig. 11I). DTPA was applied as a metal chelating agent for direct radiolabeling of liposomes with 99mTc. The authors formulated this liposomal platform according to the film hydration method using DMPC, PEG2k-DSPE, cholesterol, and DTPA-PE.155 In this design, the particle size increased by adding a positive charge inducer, such as stearyl amine, to liposomes. FA-conjugated liposomes (either neutral and positively charged) were effective as tumour-imaging agents and exhibited a significant uptake enhancement and brighter fluorescence than unmodified liposomes in 4T1 breast cancer cells in vitro.
 | ||
| Fig. 12 (A) Schematic preparation of 5-FU-loaded Fe3O4@Bio-MOF-FC. Rearranged with permission from ref. 162. Copyright 2019, Elsevier. (B) Schematic of DOX-loaded Mag-Alg-PEG-FAG (left) and magnetic hyperthermia effect on DOX release from MNPs along with confocal fluorescence microscopy images of the uptake of rhodamine-labelled Mag-Alg-PEG-FA NPs by the MDA-MB 231 cells after 24 h under a static magnetic field (right). Reproduced from ref. 167 (CC BY). (C) 3D illustration of DOX-loaded CMC-ARG-FA MNPs along with the chemical structure of CMC-ARG-FA_DOX. Reproduced with permission from ref. 168. Copyright 2020, Royal Society of Chemistry. (D) Schematic representation of the FA-mPEG-PAMAM G3-CUR@SPIONs. Adapted from ref. 172 (CC BY). (E) The chemical structure of FA-conjugated 64Cu-labeled MNPs. | ||
Alginate (Alg)-PEG copolymer was employed by Angelopoulou et al. to coat the condensed magnetic iron oxide NPs (termed co-MIONs; Fig. 12B) not only to improve the DOX loading efficiency by about 10% via Alg shell but also for a better response than conventional magnetic nanocrystals to a magnetic field in MRI by employing co-MIONs.167 PEG (OH-PEG-NH2) is conjugated to the carboxylic acid end group of Alg, while FA is conjugated to the hydroxyl terminal group of PEG to produce FA-functionalized pegylated co-MIONS. The MNPs exhibited sustained DOX release of about 60% within 48 h, responsive to pH and magnetic hyperthermia (Fig. 12B). In the acidic tumour microenvironment, the –COOH of Alg protonated and facilitated DOX release. The granular distribution of the MNPs in the cytoplasm after 24 h (Fig. 12B). The FA-conjugated MNPs enhanced DOX uptake and increased apoptosis and cytotoxicity against the MDA-MB-231 cells under a 0.5 T magnetic field. Similar MNPs named “all-in-one nanosoldier” were reported by Mansur et al. to treat MDA-MB-231 cells through DOX release, magnetic hyperthermia, and ROS-induced therapy.168 The carboxymethyl cellulose (CMC) was utilized to coat FA-conjugated MNPs (Fig. 12C). The FA was coordinated to L-argenine (Arg) grafted on CMC through amide bonds. The DOX was loaded by electrostatic interactions between negatively charged carboxylate of CMC and Arg, while the FA interacted with protonated –NH2 of the DOX. The initial burst release of DOX within the first 5 h resulted in an accumulation of approximately 50%. The authors reported that the release profile was not significantly affected by the pH of the medium at pH 5.5 and 7.4, which showed overall DOX/CMC interactions balance and DOX solubility. Their findings indicated ferroptosis contributed to the magnetic hyperthermia, while DNA dysfunction was attributed to the intracellular release of DOX. Furthermore, ROS therapy and DOX chemotherapy utilizing FA-conjugated MNPs led to cell death in FR(+) cells than FR(−) cells. Following the above-reported investigations, Zhang and Zhao et al. introduced DOX-loaded SPIONs coated with PEG/PEI polymers and conjugated with FA for MRI-guided targeting chemotherapy.169 SPIONs refer to iron oxide NPs that can be uniformly dispersed in a solution without significant aggregation. They exhibit superparamagnetic properties under an external magnetic field but lose their magnetization when the field is removed. Their uniform size distribution and superparamagnetic properties make them ideal for targeted drug delivery, MRI, magnetic hyperthermia, and in vitro/in vivo cell labeling and tracking. Monodispersed SPIONs can be developed by the polyol method due to good colloidal stability with a predicted small hydrodynamic size.170 However, Zhang and Zhao et al. used PEG/PEI polymers first to improve the stability and dispersion of SPIONs in aqueous solutions. Second, amino groups of PEI can be co-conjugated to the carboxylic group from the FA.169 Indeed, PEI provides dispersion stability by promoting repulsion between NPs, preventing aggregation or precipitation. Conversely, PEG contributes to hydrogen bonding on the SIPON surface and more prolonged circulation time, enhancing the overall performance and biodistribution of the NCs. DOX was loaded into the polymer's network through electrostatic attraction and hydrogen bonding, which allowed the DOX to be released at acidic pH (about 90% of DOX was released at pH 5 within 48 h). Tumour growth was inhibited by in vivo magnetic hyperthermia treatment towards mice bearing MCF-7 xenograft tumour upon intravenous administration of FA-conjugated SPIONs. Monitoring of SPIONs aggregation in cancer cells by MRI using a superconducting quantum interference device (SQUID) exhibited high saturation magnetization with a negative value of T2 contrast agent and r2 relaxivity of about 81 mM−1 S−1.
In another study, Zarrabi and Makvandi et al. utilized FA-conjugated MNPs coated with SiO2 and hyperbranched polyglycerol (hPG), wherein MRI signal intensity using MNPs showed a relation between increasing the NPs uptake in the MCF-7 cells and decreasing the signal related to the T2 relaxation time.171 The hPG belonging to the dendritic polymer is used to coat MNPs in order to improve their dispersibility in aqueous solutions and enhance stability. They found that FA-conjugated MNPs showed a higher relaxivity of about 23 mM−1 S−1 than non-targeted MNPs. Interestingly, a synthesized SPION system by Ghaznavi and Shakeri-Zadeh et al. was coated with two branched polymers, including methoxy-PEGylated poly(amidoamine) and amino-propyl triethoxysilane and trimethoxysilane, for FR-targeted delivery of CUR to treat breast cancer.172 Crosslinking of polymers on the surface of NPs is a process that involves creating covalent or physical cross-linkages between polymer chains to enhance stability, controlled release, and tailored surface properties. As shown in Fig. 12D, the cross-linked polymeric micelles on the surface of SPOINs can be triggered by pH and AMF. The release rate of CUR at pH 5.5 was about 40% compared to that observed at pH 7.4 (∼20%). The authors claimed that cell death from necrosis to apoptosis triggered by thermo-chemotherapy strategy upon AMF treated with FA-conjugated MNPs was significantly higher than non-targeted MNPs towards KB and MCF-7 cells. In the following, genipin cross-linked aminated starch and zinc oxide were utilized by Maji et al. to coat FA-conjugated iron oxide MNPs to deliver CUR to human lymphocytes, HepG2, and MCF-7 cancer cells.173 The average size of MNPs has been reported to be about 88 nm with a positive ζ-potential of +43 mV. The ZnO network could reduce aminated starch's toxicity and increase the CUR loading capacity. The authors applied a variation of ZnO concentration to find that the 0.5% of ZnO with 3% genipin cross-linked aminated starch led to the highest encapsulation efficiency (∼76.8%) and up to 58% of cell viability decreased after MNPs optimization. Notably, the oxidative stress via ROS production in HepG2 cells was enhanced with increasing concentration of ZnO and reduced tumour growth.
In a pioneering study and as discussed in Section 2.1.2 regarding radiolabelled NCs, Yu et al. synthesized dual-mode MNPs for PET- and MRI-based diagnosis of cancer cells.174 Citrate-stabilized Fe3O4 NPs were modified with hydrazine to allow conjugation with FA and 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) (Fig. 12E). In the last step, 64Cu was chelated with NOTA. Besides, other N-functionalized polyazacycloalkane chelators, e.g., DOTA,175 TETA,176 AAZTA/DATA,177 and DOTA/PCTA178 are frequently used to capture the 64Cu radiolabel. The nitrogen atoms within the polyazacycloalkane structure serve as donor sites to coordinate with the radioisotope. At the same time, the functional groups such as carboxylic acids – for example, in NOTA – can provide additional coordination sites and enhance the stability of the metal complex. Interestingly, Yu et al. found a significant radiochemical purity of about 82% in NOTA-64Cu MNPs, which was stable in buffer solution and human serum within 24 h.174 They found a higher uptake in FR(+) KB cells than FR(−) cells such as A549 and SKBR3. Furthermore, numerous radiotracers have been used to image the drug distribution and assess therapeutic response in clinical investigations. In particular, radionucleotide imaging is often used to diagnose and management of breast cancer patients because of the overexpression of HER2.179 By analysing the distribution and intensity of the radioisotope uptake, physicians can obtain valuable information about the extent of HER2-positive tumour lesions, their size, and their metastatic spread. Notably, radionuclide imaging is often used with other imaging modalities, e.g., mammography, ultrasound, or MRI, to evaluate breast cancer patients. As an example, the biodistribution and safety of 64Cu-NOTA-trastuzumab have been studied as a PET tracer for HER2(+) cancer patients.180 Similarly, the first in-patient breast cancer HER-PET studied utilizing [18F]GE-226 radiotracer, for non-invasive HER2 imaging in primary and metastatic tumours.181
Furthermore, non-magnetic metal NCs can be coated with proteins, polymers, and silica via a layer-by-layer assembly, sol–gel, reduction, seed-mediated growth, laser-induced, and photo-induced methods.185 FA-conjugated and silica-coated AuNPs were reported by Salehi and Alizadeh et al. as a way to deliver methotrexate (MTX) toward MDA-MB-231 and MCF-7 cells.186 In their system, the thiol group binds to the silica surface, forming thiol-functionalized silica-coated NPs. Subsequently, AuNPs were immobilized into a sol–gel matrix via thiol linkers. MTX, as well as FA, were loaded into the Au@SiO2 platform. The FA-conjugated AuNPs had a mean size of about 105 nm with a ζ-potential of about +13 mV that was changed to −19 mV after MTX loading.
In principle, MTX can tightly bind to dihydrofolate reductase and inhibit the synthesis of DNA, purines, and thymidylate.187 In addition, 5-FU is a thymidylate synthase inhibitor that limits the thymidine substrate for DNA synthesis and cell division in TNBC tumours.188 Both MTX and 5-FU can have off-site toxic effects on cells, which may contribute to side effects experienced by patients. By addressing the challenges associated with 5-FU resistance, poor bioavailability, and off-site toxic effects, researchers aim to optimize the use of these drugs in the management of TNBC tumours. In this context, Singh et al. fabricated chitosan-coated FA-conjugated 5-FU-loaded AuNPs and tested its impact in MCF-7, HepG2, and HEK293 cells cancer cell lines.189 The aminopolysaccharide chitosan offers several hydroxyl and amino groups as binding sites for 5-FU, FA, and AuNP. In this system, trisodium polyphosphate (TPP)-decorated AuNPs loaded with 5-FU and coated with FA-conjugated chitosan. The hydrodynamic sizes of the FA-conjugated AuNPs were reported to be about 149 nm with a highly positive ζ-potential of about +57 mV, which is related to the high stability of the colloid dispersion. The release efficiency of 5-FU had a partial pH-dependent manner within 72 h (∼90% at pH 5 and ∼86% at pH 7.4).
For delivery of 5-FU, Kim et al. utilized citrate/PEG (CPEG)-stabilized AuNPs decorated with the thiol group of thioglycolic acid (TGA) in order to conjugate to the –COOH of TGA-AuNPs (Fig. 13).190 The carboxyl-terminated PEG (CPEG) acts as a stabilizing agent, yielding higher stability of AuNPs by preventing agglomeration.191 The author claimed that the carboxyl moieties on the surface of AuNPs (Au⋯S–CH2–COOH) can be conjugated with the –NH2 of the FA molecule. The targeted AuNPs exhibited over 50% 5-FU release at pH 5, while the drug release at pH 7.4 was only reported about 24% within 12 h. The Au–S bonds are cleaved at pH ≤ 6 in the tumour microenvironment, led to nanostructure disintegration and 5-FU release to suppress MCF-7 cell growth via a combination of cytotoxic effects of 5-FU and antifolate activity of FA-TGA to prevent FA metabolism and prevent cell proliferation. In vitro cytotoxicity studies displayed no toxicity to healthy cells up to 200 μg mL−1 of 5-FU and inhibited the proliferation of MCF-7 cells at a concentration of 25 μg mL−1.
 | ||
| Fig. 13 The chemical structure of TGA-AuNPs. | ||
A unique polysaccharide-coated fluorescein isothiocyanate (FITC)-labelled AuNPs was designed by Mahesh and Kandasamy et al.192 The carboxyl groups of the polysaccharide (extracted from the gum kondagogu to capped the AuNPs) conjugated with the –OH of FITC and –NH2 of FA molecule. The cellular uptake of FA-conjugated AuNP (NP's size ∼37 nm, ζ-potential ∼ −23 mV) displayed a significant FITC delivery to FR(+) MCF-7 cells than FR(−) A549 cells for cellular imaging.
In principle, disruption of intracellular redox homeostasis by ROS inducers causes DNA mutation and apoptosis of cancer cells.193 ROS-induced cancer therapy is considered to regulate pro- and anti-apoptotic proteins, e.g., caspases and Bcl-2 family proteins.194 In this context, Wang et al. constructed FA-conjugated copper oxide nanoparticles (CuONPs) that could alter the expression of Bcl-2 and upregulation of cytochrome-C, Bax, caspase-3, and caspase-9 expressions via activation of mitochondrial ROS generation.195 CuONPs were coated with aminated starch to deliver Helianthus tuberosus extracts to MDA-MB-231 cells. According to the literature, the aminated starch was used as a capping agent for CuONPs and as a linkage to conjugate with –COOH of FA.196 The authors reported that FA-conjugated CuONPs triggered cancer cell apoptosis via regulating Bcl-2/Bax and caspase cascade activation from ROS-stressed cytochrome C of mitochondria.195 Generally, NPs targeted mitochondria could significantly increase ROS release, membrane integrity loss, EPR effect initiation, and leak of cancer cells' nuclear materials into the cytosol.197
Human serum albumin (HSA) is the most abundant protein in human blood plasma. Of note, a single-chain polypeptide of HSA containing 585 amino acids helps to increase the solubility of lipophilic drugs. It has a three-dimensional structure with multiple α-helices and many disulfide bonds. HSA is an attractive candidate for protein-based NCs due to its biocompatibility, drug-loading capacity, and ability to self-assemble into NPs. Hence, Akbarian et al. employed HSA to construct protein-based NPs to deliver artemether via its encapsulation into protein by desolvation technique.203 The hydrodynamic diameter of NPs has been reported to be about 198 nm with a ζ-potential of −23 mV. The authors claimed that FA conjugations to the HSA NPs significantly enhanced the artemether uptake in the MDA-MB-231 cells than non-targeted NPs.203 In another case, Mi and Fan et al.204 utilized bovine serum albumin (BSA is a protein derived from cow's blood plasma that shares 80% sequence homology with HSA205) to enhance ginsenoside Rg5 solubility and effective delivery to breast cancer cells via folate receptor. The size of FA-conjugated BSA-NPs was about 200 nm (DPI ∼0.08) with a ζ-potential of −22 mV. The authors found that the Rg5's release efficiency at pH 5 was significantly faster than at pH 7.4. The cumulative release reached about 46% at pH 5 within 48 h and then increased to 86% after 96 h. Moreover, in an MCF-7 xenograft mouse model, FA-conjugated Rg5-loaded BSA NPs inhibited tumour growth more efficiently than Rg5-BSA NPs and Rg5 itself. Real-time biodistribution was followed by DiR-labeled NPs revealing that Rg5-BSA NPs could also accumulate in tumours via the EPR effect. A similar formulation method was used by Kunjiappan and Panneerselvam et al. to fabricate FA-conjugated myricetin-loaded BSA-based NPs but with a smaller particle size of about 78 nm (PDI ∼0.54) and a positive ζ-potential of +38 mV.206 In another example, Danafar and Davaran et al. reported another FA-conjugated BSA-based NPs for the delivery of chrysin.207 The NPs had a spherical shape, with a diameter of about 97 nm (DPI ∼0.18) with a negative ζ-potential of −11 mV.206 The loading capacity was reported at about 2%, and only 20% of the myricetin was released at pH 7.4. The subsequent release reached 35% after 96 h, while the subsequent release enhanced to 57% at pH 5.8 due to NPs dissociation and release of the encapsulated myricetin. Besides, the myricetin release from FA-conjugated NPs was higher than non-targeted NPs, with higher efficiency at pH 5.4 compared to pH 7.4. An effective decrease in the viability of MCF-7 cells by the targeted NPs compared to non-targeted NPs was observed.
Despite the several advantages, including their low cytotoxicity, abundant sources, and significant uptake into the targeted tumour cells, they face challenges like less stability to maintain their integrity and functionality and manufacturing complexity and cost compared to other synthetic carriers.
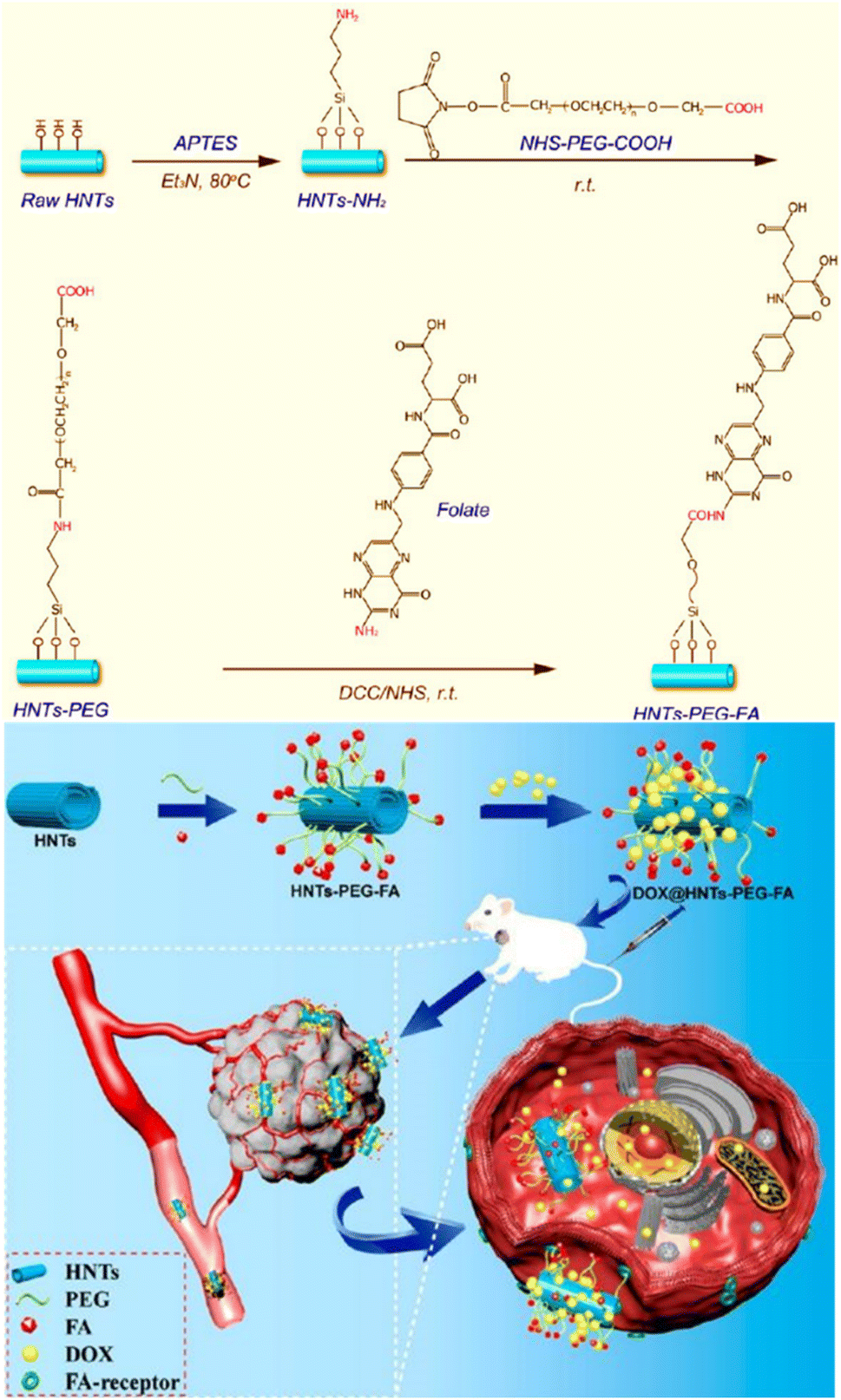 | ||
| Fig. 14 The representation of the chemical structure of HNTs-PEG-FA along with the schematic synthesis procedure for DOX-loaded HNTs-PEG-FA (top) and the targeting release of DOX from NCs in the cancer cells (bottom). Reproduced with permission from ref. 232. Copyright 2018, American Chemical Society. | ||
The current synthetic progress along with drug delivery, bio-imaging, and biomedical applications of silica-209 and carbon-210 based NCs have been so far addressed. The π–π stacking interactions in carbon-based NCs dominate excellent internal interactions with drugs via supramolecular forces and facilitate binding with drugs and biomolecules.211 Silica-based NPs offer high surface area, tunable porosity, and excellent biocompatibility. They can be loaded with therapeutic agents for targeted drug delivery, and their surface can be modified for optical imaging and photothermal therapy (PTT). In this context, FA-conjugated mesoporous silica NPs (MSNs) were developed by Capan et al. to deliver DOX to breast cancer cell lines.212 They functionalized MSNs (∼50 nm, ∼ +27 mV) using 3-aminopropyltriethoxysilane to produce MSNs-NH2 that was conjugated with FA (∼60 nm, ∼ +11 mV) via NHS/EDC protocol. Obtained results show that FA-conjugated NPs have superior anti-cancer effects on ZR-75-1 and T-47D cells, without notable toxicity on L929 cells. Similarly, Mehravi et al. utilized MSNs for gadolinium (Gd3+) delivery to image breast cancer cells.213 In this method, the silica-diethylenetriamine tetraacetic acid-Gd3+ complex was conjugated via siloxane linkage to the surfactant-extracted FA-MSNs and doped with rhodamine B isothiocyanate to fabricate fluorescent-doped nanoprobe. Relaxometry showed that NCs have good T1-weighted MRI contrast agents by delivering a sufficient amount of Gd3+ as contrast agents into cancer cells. This is attributed to their ability to deliver sufficient Gd3+ into cells through FR endocytosis efficiently. Another example of carbon-based NCs having a large surface area is based on fullerenes. Serda et al. recently reported triple-bonded [60]fullerene triazoles that successfully localized in MCF-7 cells.214 Tuning [60]fullerene via functionalization through, for instance, 1,2,3-triazole linker group tailored electronic properties and interactions with (bio)molecules. Nano onions with multi-shelled structures are a unique and fascinating class of NCs that offer a high surface area-to-volume ratio for drug loading. In this context, Wang and He et al. reported that silica-carbon nano onion targets tumour vasculature to specifically release P-gp inhibitor and control DOX delivery into tumour cells.215 They showed the superior light absorption property of nano onion in the NIR, leading to controlled P-gp inhibitor and DOX release at a low NIR power.
Graphene oxide (GO) and the above-mentioned carbon- and silica-based NCs are distinct nanostructures with unique properties. GO possesses a large surface area and excellent optical properties, making it suitable for cellular imaging and targeted drug delivery. Additionally, the photothermal properties of GO can also be exploited for PTT. For the purpose of supramolecular force engagement, Chen et al. fabricated a pH-sensitive FA-conjugated GO NPs for the targeted delivery of DOX to MCF-7 cells.216 In their system, FA-conjugated DOX-loaded GO (GOFA) was further encapsulated in a thermo-sensitive hyaluronic acid-chitosan-g-poly(N-isopropylacrylamide) (HACPN) hydrogel. Of note, hydrogels are stimuli-responsive nanomaterials utilized for drug delivery and wound dressing.217 The release of DOX was 5-fold higher at pH 5.5 than at pH 7.4. An augmented in vitro cytotoxicity of FA-conjugated NCs against MCF-7 was reported with an IC50 value of 7.3 μg mL−1 compared to non-targeted NCs (IC50 ∼ 10 μg mL−1) and free DOX (IC50 ∼ 32 μg mL−1). The administration of NCs to mice xenografted with MCF-7 cells (MCF-7/Luc) yielded a tumour volume decrease (2-fold/21 days). In another example, methyl acrylate (MA), as a pH-sensitive polymer, can be conjugated to the GO surface through amide and ether linkers218 that are utilized to improve the degradability of GO after accumulation in a physiological environment.219 Rajan et al. utilized MA to graft on GO surface (GO-g-MA) that further loaded with PTX and conjugated with FA to FR-targeting of NCs into MDA-MB-231 cells.220 The GO-g-MA was produced by in situ atom transfer radical polymerization.221 The PTX release from such NCs at pH 5.5 was about 65% after 24 h.220 The NCs showed significant cytotoxicity (IC50 ∼ 75 μg mL−1). In addition, increased levels of caspase-8, caspase-3, and cytochrome c activities were reported using FA-conjugated NCs. In vivo assessment showed a significant reduction in tumour growth in rats during the 6 weeks of treatment which is attributed to cell-cycle arrest induction, followed by mitochondria-mediated apoptosis.
Carbon quantum dots (CQDs) are fluorescent NPs with excellent biocompatibility, easy surface functionalization, good aqueous solubility, and outstanding optical properties. In an attempt to improve visualization, Shuang et al. implemented FA-conjugated fluorescent carbon dots (CDs) for photostability fluorescence imaging of FR(+) HepG2 cells compared to FR(−) PC-12 cells.222 The CDs were synthesized utilizing dandelion leaf as the carbon source and ethylenediamine as the nitrogen source in a hydrothermal process. Incorporation of nitrogen atoms into CD structures is an effective way to augment their quantum yield, thereby extending their utility in cellular labelling and bioimaging applications. The reported CDs demonstrated a quantum yield of 13.9%. Additionally, these carbon dots featured amino groups on their surfaces for conjugation with FA moieties. The reported average size was 3.5 nm with a ζ-potential of −15 mV. The fluorescence spots in the cytoplasm attributed to the FA-conjugated CDs in cancer cells, revealing successful FR-targeting. In the following, Dong et al. studied the uptake of fluorescent FA-immobilized CDs by MCF-7 and HepG-2 cells for intracellular bioimaging.223 The average size was reported to be about 3.4 nm with a ζ-potential of about −22 mV. A high quantum yield of 17% was reported when –NH2 groups of the CD surface were conjugated with –COOH groups of FA molecule. In line with the above observation, hyaluronan-conjugated nitrogen-doped carbon quantum dots (nCQDs) were reported by Ravi and co-workers.224 CQDs were conjugated with protoporphyrin IX as a natural ligand of CD44 receptors for bioimaging.224 Notably, fluorescent CDs conjugated with protoporphyrin IX were frequently employed for bioimaging and targeting cancer cells via singlet oxygen (1O2) formation by utilization of the molecule's photosensitizer capabilities.225 Moreover, Li and Qu et al. utilized FA to produce nitrogen-doped FA-derived CDs by hydrothermal-assisted method for HeLa cell imaging.226 The hydrothermal method holds significant appeal due to its inherent advantages, which encompass relatively gentle reaction conditions and the inherent potential for facile functionalization. The average CD size was reported as about 5.4 nm (lattice spacing: 0.21 nm). The authors reported high fluorescence quantum yields up to 94% (Kr ∼6.14 × 107 S−1 > Knr ∼0.36 × 107 S−1) via condensation of FA in water. They also found a direct effect of pH on the fluorescence intensity of the obtained CDs. In a similar study, Shuang et al. reported CDs formed by active dry yeast and then conjugated with FA molecule.227 These CDs were reported spherical and monodispersed with an average size of 3.4 nm with a ζ-potential of about −16 mV. The FA-conjugated CDs provided superior internalization into FR(+) HepG2 cells than FR(−) PC12 cells, resulting in a much stronger green fluorescence in FR(+) cells. Recently, Farhadian et al. reported nitrogen-doped CQDs modified with FA and DOX conjugation on the surface for bioimaging.228 The size of CQDs was reported to be about 7 nm. A higher cytotoxicity effect was observed for CQD-FA-DOX toward 4T1 and MCF7 cells compared to free DOX. Interestingly, about 86% of the loaded DOX was released from CQD-FA-DOX after 72h at pH 5.5. In a unique study, Chatterjee et al. designed graphene quantum dots (GQDs) by a bottom-up approach through pyrolysis of citrate and conjugation with FA molecule by carbodiimide chemistry.229 It is worth mentioning that two versatile methodologies provide adaptable pathways for crafting carbon dots (CDs) with properties tailored for specific applications. In bottom-up approaches, molecular precursors are assembled from smaller carbon units while top-down strategies entail the disintegration of diverse carbon nanomaterials. These approaches collectively offer a range of options to create CDs optimized for various application requirements. The quantum yield for FA-GQD was reported at about 9%. The size range was 3.5–8 nm with a ζ-potential of about −13 mV. The authors claimed that GQDs were non-toxic to healthy cells at a concentration of 1 mg mL−1. However, 2.5 μg mL−1 of FA-GQDs reduced the cancer cell viability up to 75% after 48 h. In a similar approach, Zheng et al. utilized citrate as the carbon source and diethylamine as the nitrogen source to fabricate FA-conjugated nitrogen-doped GQDs used MCF-7 cells.230 Interestingly, increasing the amount of nitrogen doping resulted in more binding sites on nGQDs for FA conjugation and emitting a stronger fluorescence intensity after entry into tumour cells. At an average size of 5 nm (lattice spacing ∼0.24 nm), the cell viability was reported to be 97% after 24 h incubation. At the same time, fluorescence stability of FA-conjugated nGQDs after incorporation by FR(+) MCF-7 cells was observed. Alternatively, the incorporation of sulfur into GQDs is a feasible approach, as evidenced by the work of Kadian and Manik et al.231 The average size was reported about 5 nm with a lattice spacing of 0.35. Irradiation of FA-sGQDs at λex 370 nm exhibited a blue fluorescence with an emission band at 455 nm. Fluorescence microscopy of FR(+) MCF-7 and FR(−) CHO cells utilizing FA-conjugated sGQDs confirmed successful FR-targeting.
Carbon nanotubes (CNTs) exhibit remarkable mechanical, thermal, and electrical attributes, rendering them suitable for diverse applications such as drug delivery, photodynamic therapy (PDT), and imaging. In recent times, intrinsically mesoporous halloysite nanotubes (HNTs) have emerged as promising alternatives to CNTs. This is attributed to their advantages of lower cost, superior water dispersibility, and reduced toxicity. The presence of halloysite nanotubes, featuring a silica outer layer and an alumina inner surface, within NCs significantly modulate drug loading and release characteristics. In a pioneering study, He and Liu et al. fabricated HNTs conjugated with PEG and FA as DOX carriers to MCF-7, 4T1, L02, and HepG2 cells (Fig. 14).232 The HNT length is shortened to about 200 nm by ultrasonic scission. PEGylation of aluminosilicate HNTs-NH2 using NHS-PEG-COOH followed by FA conjugation not only provide an FR-targeting platform but also prolonged HNTs circulation time and controls their dosing interval. HNTs are negatively charged (ζ-potential ∼ −24 mV), so after PEGylation and FA conjugation, the NCs become nearly neutral (ζ-potential ∼ +1 mV). DOX release from FA-conjugated HNTs is reported to be up to 35% at pH 5.3 and induces significant FR(+) MCF-7 cell death and apoptosis compared to FR(−) L02 cells in vitro. In addition, the authors reported that the level of caspase-3 activity utilizing 4T1 cells treated with FA-conjugated HNTs is increased, which is higher than non-targeted NCs and free DOX. In contrast, Bcl-2 activity is decreased when treated with the FA-conjugated HNTs.
An emerging and interesting approach is smart lipid-polymer nanohybrids. Due to their core–shell nanostructures that combine biodegradable PNPs with biomimetic lipid-based NPs, ensuring adequate drug encapsulation and release upon stimulation. One example is the FA-conjugated chitosan-coated solid lipid NPs designed to deliver the steroid-mimetic letrozole (LTZ) to MCF-7 and PC-12 cells (Kashanian et al.).243 Tripalmitin glyceride: stearic acid in a 2:3 ratio with 5 mg of LTZ and 20 mg chitosan was mixed and homogenized based on an oil-in-water homogenization protocol. The obtained particle had a size of 148 nm (PDI ∼0.301) with a positive ζ-potential of about +6 mV. Of note, the electrostatic repulsion between NPs with low ζ-potential – closer to zero, is reduced. This reduction in repulsive forces could potentially lead to an NPs aggregation. However, the aggregation tendency is influenced by other factors like particle charge (highly charged NPs interact with proteins and macromolecules), as well as the physiological medium. The cytotoxicity study using FA-conjugated LTZ-loaded nanohybrid for MCF-7 cells with an IC50 value of 79 nM proved the efficiency of FR targeting compared to free LTZ that did not reach the IC50 value in the investigated concentrations after 24 h. In another study, a PEGylated phytosomal phospholipid bilayer enveloping casein-loaded micelles decorated with FA was reported by Elzoghby et al. targeted delivery of fungal-derived Monascus yellow pigments (MYPs) and resveratrol (RSV) to MCF-7 cells.244 A high colloidal stability of NCs with a size of 137 nm (ζ-potential ∼ −21 mV, PDI ∼0.27), and 272 nm (ζ-potential ∼ −36 mV, PDI ∼0.21) was reported for FA-casein micelles and PEGylated PC-casein micelles, respectively. Both FA and PEGylated micelles significantly reduced vascular endothelial growth factor (VEGF), aromatase, CD1, and NF-κB activities compared to the free drugs. In addition, the caspase-3 activity was found at an elevated level compared to the control groups.
Ding and Wang et al. developed a PTX-loaded mesoporous silica NPs decorated with FA and arginine-glycine-aspartate (Arg-Gly-Asp) RGD tripeptide sequence with a high affinity for FR and integrin αvβ3 expressed on the surface of human breast cancer MCF-7 cells.245 This approach exploits the finding that integrin expression in metastatic breast cancer cells is higher than in healthy cells like non-malignant MCF-10A cells or HeLa cells.246 Accordingly, NHS-PEG-FA and NHS-PEG-RGD conjugation onto the NP's surface provides an active tumour-targeting therapy via FR and integrin αvβ3.245 The long PEG chains enhance the stability of the NPs in vivo. The positive ζ-potential value is attributed to the interaction between –NH2 groups on the surface with tripeptide sequence opposite to the negative ζ-potential of MSNs-NPs (ζ-potential ∼ −18 mV). The calculated IC50 value of free PTX and PTX-loaded NCs on MCF-7 cells after 48 h was 35 and 22 ng mL−1, respectively, indicating a 1.6-fold greater inhibitory efficacy (antitumour activity) of PTX-loaded NPs than that of free PTX.
2.2 Brain cancer
Metastatic brain tumours represent about one-third of all primary brain tumours. The heterogeneous microenvironment of glioblastoma and the blood–brain barrier (BBB) restricts the transport of therapeutic or diagnostic agents, impeding effective intervention significantly. Since the discovery of the BBB concept by Paul Ehrlich in 1885, drug transportation via the microvascular unit using specific transcellular transporters has been intensely studied, and a number of options to foster drug accumulation have been identified.247 Especially the leaky BBB of tumours might contribute to overcome the above limitations.248 Nanocarriers (NCs) became promising approaches for brain cancer treatment, and among them, lipid-based NCs and liposomes, micelles, and polymeric NCs are in clinical trial investigations. A number of plasma membrane proteins may be utilized to target the brain cancer cells, e.g., drug efflux transporters, including folate receptor (FR), organic anion-transporting polypeptides (OATPs), and P-gp similar to the breast cancer cells. The FR-targeting and dual-receptor targeting NCs could increase the BBB cross rate to reach tumour cells. Many advances and challenges are reported in NC design, blood circulation journey, and uptake via FR-targeting that needs to be updated.4a,5 As shown in Fig. 15A, Liao et al. reported pH-sensitive FA-conjugated chitosan-coated magnetic nanoparticles (MNPs) to deliver DOX and TPP to U87 cells.249 The cationic structure of chitosan provides substitutions via nitrogen and oxygen atoms to cross-link with DOX and TPP. The release profile of DOX from uncoated NPs is less stable than from chitosan-coated NPs, and chitosan-coated MNPs are more potent in DOX release at pH 5.7. The authors reported a successful cellular uptake of DOX and tumour growth suppression of human glioblastoma U87 cells in vitro that were boosted by the application of magnetic fields. The in vivo evaluations utilizing immune-incompetent BALB/c nude mice revealed a decreased tumour growth upon magnetic guidance of MNPs by the enhanced local DOX release. In another study, Khoei et al. utilized SPION coated with triblock copolymer PEG-PBA-PEG to deliver temozolomide (TMZ) (Fig. 15B).250 In this design, the FA molecule is conjugated on both sides of the triblock copolymer for dual-targeting. A self-assembled spherical nanostructure was formed to provide a hydrophobic core to load the lipophilic TMZ, along with a hydrophilic shell that stabilizes NCs in aqueous media without the need for an additional stabilizer. Of note, the alkyl group of TMZ at the oxygen-6 and nitrogen-7 positions of guanine causes DNA mismatch repair to double-strand breaks and leads to cancer cell apoptosis. Higher FBS serum concentration led to a higher initial release rate of TMZ.250 MRI images of MNPs administered to glioma-bearing rats exhibited high-intensity signals in the T2-weighted imaging in pre-injection independent of an external local magnetic field (Fig. 15B). In contrast, post-injection images reflected a negative contrast (black dots) enhancement utilizing FA-conjugated MNPs by passing through BBB and accumulating in the rat tumour area in the presence of an external magnetic field.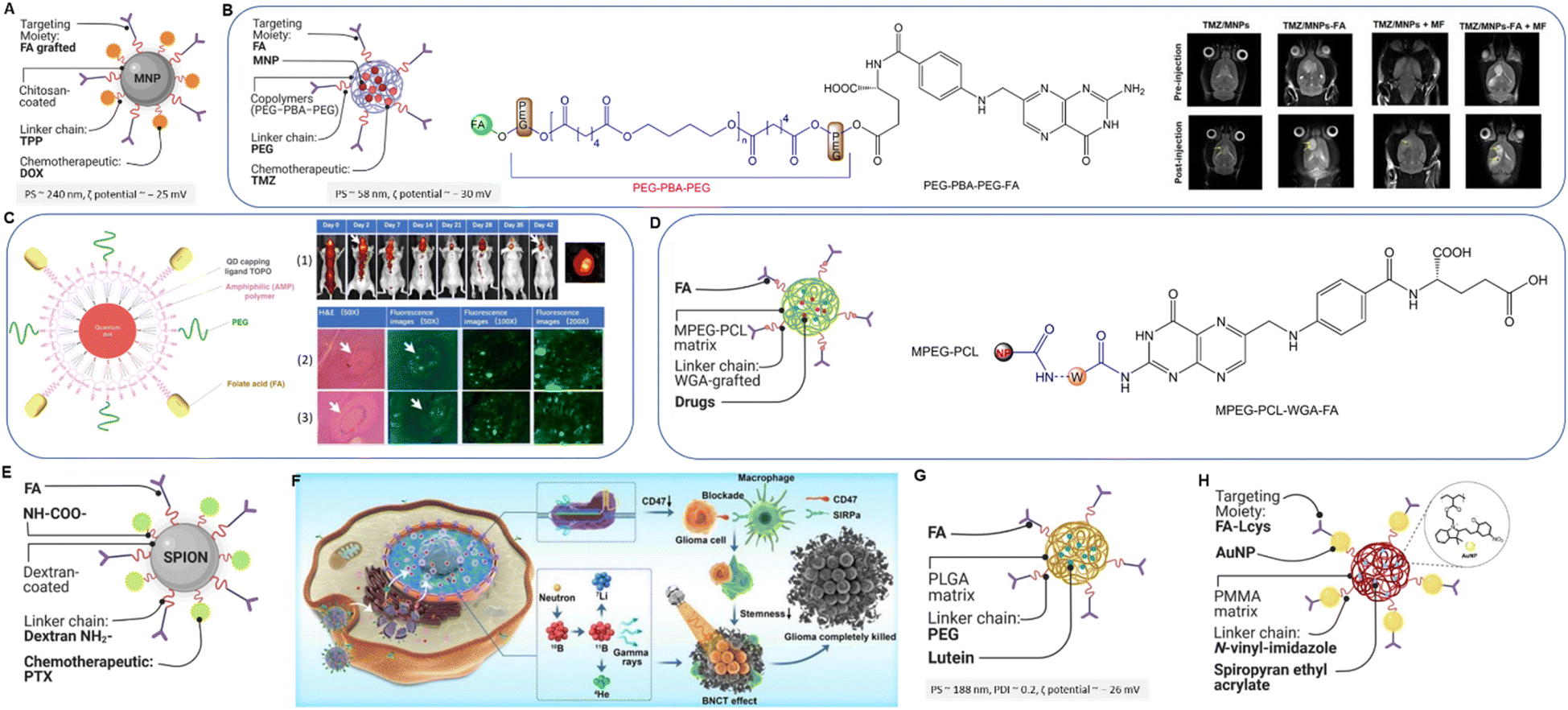 | ||
| Fig. 15 (A) Schematic illustration of chitosan-coated NCs. (B) Schematic illustration of SPION-based NCs (left) and it's copolymeric structure (middle) along with MRI images of glioma-bearing rats administered with different types of NCs upon external magnetic field (right). Reproduced with permission from ref. 250. Copyright 2019, American Chemical Society. (C) Schematic illustration of QD-FA NCs, (1) in vivo fluorescence imaging of tumour in mice brain after administration of QD800-FA over time, (2) ex vivo NIR imaging of brain after 15 days of intrathecal injection of FA-conjugated NCs, (3) H&E staining and fluorescence microscopy of U87MG tumour tissue slices from mice brains after intrathecal injection of QD800-FA after 2 days (c) and 14 days. Reproduced with permission from ref. 253. Copyright 2019, American Chemical Society. (D) Schematic illustration of polymeric NCs (left) and copolymerization structure of MPEG along with PCL conjugated with WGA and FA (right). (E) Schematic illustration of PTX-loaded FA-conjugated MNPs. (F) Therapeutic delivery via boron-containing liposome to glioma cell. Reproduced from ref. 261 (CC BY 4.0). (G) Schematic illustration of lutein-loaded PLGA-PEG-FA NPs. (H) Schematic illustration of FA-conjugated PGPNPs immobilized Au3+ ions. | ||
A similar copolymeric MNP strategy was utilized by Khoei et al. for delivering TMZ to glioblastoma C6 cells.251 The hydrodynamic particle size was reported to be about 48 nm with a ζ-potential of −28 mV. The authors found that more than 90% of TMZ was released within the first 2 h, while the sustained release was decreased due to the poor encapsulation of the drug in the inner hydrophobic core. The uptake of targeted MNPs into C6 cells is about 2.5-fold higher than that of non-targeted MNPs. The same MNPs were employed in another study as a carrier of TMZ for targeted chemotherapy and radiofrequency hyperthermia toward C6 cells.252 About 55% of TMZ were quickly released after 10 min in the presence of an alternating magnetic field (AMF), driving local hyperthermia (∼43 °C) due to the magnetic properties of SPIONs. At body temperature (37 °C), TMZ release remained low. Lin and Li et al. reported a fascinating approach for in vivo imaging of mice brains using FA-conjugated NIR quantum dots (QDs, Fig. 15C).253 FA-conjugated QD800-PEG (CdSeTe/ZnS) NPs were delivered via intrathecal injection in a mouse model with orthotopic transplanted FR (+) U87MG glioma cells, which can be activated at λ = 800 nm (Fig. 15C (1)). By replicating the physiological and biomechanical conditions of tumours in their native tissues by transplanting tumour cell lines into animal models, orthotopic models provide valuable insights into the real-world behaviour of tumours, e.g., primary tumour growth, invasiveness, and metastatic activity and the effectiveness of FR-targeted treatment. The study was evaluated for six weeks after injection in vivo. A high fluorescence signal appeared in the spinal cord and brain after 1 hour post-injection of FA-conjugated QDs, and a contrast enhancement was reported within two days (Fig. 15C (2,3)). The tumour region of interest exhibited a higher uptake of targeted QDs (∼90% percentage of injected dose delivered (ID) g−1) compared to the non-targeted QD800 (∼20% ID g−1). Although in vivo fluorescence imaging can provide valuable information about the distribution and localization of fluorescently labelled compounds, the reported values are only semi-quantitative. In a similar study, Jayasree and Ajayaghosh et al. developed an FA-conjugated gold quantum cluster localized on C6 rat glial cells for fluorescent imaging and real-time tracking of PDT, which can be activated by NIR (λ = 1270 nm) via local generation of 1O2 during the relaxation protoporphyrin IX exciting state to the ground state.254
To deliver etoposide, nitrogen mustard carmustine, and DOX across BBB to target human glioblastoma U87MG cells, Kuo et al. constructed MPEG-PCL NPs grafted with wheat germ agglutinin (WGA) and FA (Fig. 15D).255 The copolymerization of MPEG and PCL was performed using a microemulsion-solvent evaporation method and finally conjugated with WGA and FA. The authors found that a shorter PCL chain in multidrug-loaded PNPs resulted in smaller NPs. In comparison, the longer PCL chain led to stronger hydrophobicity, enhancing drug entrapment efficiency. Notably, the incorporated WGA in nanostructured polymers has a high affinity to N-acetylglucosamine and sialic acid residues to bind cell surface receptors, which enhances cellular internalization and increases bioavailability.255 In an innovative study by the same authors, tamoxifen- and lactoferrin-conjugated solid lipid NPs were utilized to deliver carmustine across the BBB to glioblastoma multiforme cells.256 The presence of tamoxifen and lactoferrin improved the sustained release of carmustine and enhanced the transendothelial electrical resistance, permeability coefficient, and relative fluorescence of intracellular calcein-AM. Of note, tamoxifen could reverse efflux transporters like p-glycoprotein, while lactoferrin is utilized to modulate the receptor-mediated transcytosis across the BBB. Targeted drug delivery to glioblastoma U-87MG cells was assessed in vitro by Farhadi and co-workers, utilizing FA-conjugated ZnO NPs.257 The viability for U87MG cells decreased significantly at concentrations of 1.25 and 2.5 mg mL−1 of the NPs, showing a dose-dependent effect.
Kang et al. reported that targeted chemo-proton therapy (TCPT) on C6 cells utilizing PTX-loaded FA-conjugated dextran-coated SPIONs as a means to improve the PTX efficacy in brain cancer treatments (Fig. 15E).258 The authors found that PTX disrupted cell replication, while the non-toxic concentration of PTX (200 ng mL−1) did not affect the cell viability. In addition, PTX was employed as a radiosensitizer to enhance the efficiency of photon beams in TCPT. In a similar manner, boron neutron capture therapy (BNCT) is utilized as a non-invasive approach via the accumulation of isotope 10B for non-operable tumours with a high ability to absorb neutrons upon irradiation to generate an epithermal neutron beam.259
| 10B + 1n → [11B]* → 4He ↔ 7Li + γ |
Isolectin phospholipid-based liposomes contain hydrophilic boron NPs and cyanine dye 5 (Cy5) NIR fluorescent dye developed by Krishnan and Prasad et al.260 The BNCT platform was utilized for the selective destruction of C6 cells. The surface of the liposome was coated using PMAO and PEG to improve stability and bioavailability. Polymer coating minimized opsonization and phagocytosis in blood circulations, and FA conjugation increased liposome uptake via FR-targeting. The authors found a significant in vitro cellular uptake of boron (2.06 × 1011 atoms per cell) using the targeted liposomes in rat C6 glioblastoma cells and a better in vitro BBB model crossing compared to non-targeted liposomes. This level is well above the required level of ∼109 boron atoms per cell to facilitate BNTC. Inductively coupled plasma mass spectrometry analysis revealed that BBB transmissivity for the liposomes was higher than for the dye Cy5 itself. Alongside, endocytosis of targeted liposomes carrying boron was higher than that of the non-targeted counterpart. In the same direction, non-targeted liposomal NCs were recently developed by Chen et al. to deliver DOX and carborane to the nucleus of GL261 cells (Fig. 15F).261 Although the constructed liposomes lack receptor-mediated endocytosis, combining boron agents with chemotherapeutics led to tumour stemness reduction and improved prognosis compared to borocaptate sodium as a clinical drug. Neuroblastoma SK-N-BE(2) cells were utilized by Sambalingam and Renukuntla et al. to investigate the role of FA conjugation in the targeted delivery of lutein-loaded PLGA-PEG NPs (Fig. 15G).262 The lutein uptake was enhanced about 2-fold after FA conjugation to the PNPs. In addition, a significant lutein accumulation was observed (6.5 μg per 106 cells).
In a unique study, Mahdavian et al. introduced a spiropyran (SP) to merocyanine (MC) by photoisomerization by UV light as a probe for enhanced photodynamic therapy.263 Photoresponsive FA-conjugated Au-decorated polymeric NPs were developed for this purpose (Fig. 15H). In this protocol, the acrylic NPs functionalized with SP, and imidazole groups were immobilized with Au3+ ions to obtain photoresponsive Au-decorated PNPs (named PGPNPs). FA conjugation via an L-cysteine linker improves intracellular uptake by FR-targeting and provides a high local photothermal efficiency. In contrast, AuNPs immobilization enhanced plasmon-enhanced fluorescence and consequently higher ROS photogeneration. Of note, the author utilized non-polar SPs known as photoswitchable materials converting zwitterionic MC isomers under UV irradiation. In contrast, the coloured MC isomer is susceptible to an efficient triplet-singlet intersystem crossing.
3 Folic acid (FA)-conjugated small molecule–drug conjugates
Compared to nanocarriers, in this case, the cytotoxic drugs are directly conjugated to folic acid, partly with the aid of a linker molecule. The resulting chimeric molecules are referred to as cytotoxic FA-conjugated small molecule drug conjugates (SMDCs). Like the nanocarriers (NCs) reviewed above, the FA moiety allows a folate receptor (FR)-based targeting that is exploited for cancer diagnosis and therapy.44d The non-immunogenic nature and low molecular weight of SMDCs enable an effective penetration in solid tumours compared to the much larger antibody–drug conjugates.6a In FR-targeting, FA molecule conjugates by glutamic acid group (at the α- or γ-positions) to multifunctional self-immolative linkers and spacers.61 Spacers minimize the steric hindrance between the structure of the drug transporter and the FA molecule. At the same time, linkers were utilized due to their higher release kinetics upon stimuli and to improve the connectivity and stability of SMDCs.Along this line, conjugated chemotherapeutic to albumin-binding moieties can be used to increase drug delivery and reduce the side effects. Hence, Gao and Chen et al. reported a FA/PTX-conjugated prodrug conjugated with Evans blue (EB) that binds to albumin with strong affinity, resulting in prolonged blood circulation and enhanced accumulation in the tumour tissue.264 Notably, EB was frequently used as a marker for plasma volume determination in animal models.265 As shown in Fig. 16A, the FA-PTX-EB ester prodrug was constructed by coupling a Fmoc-Cys(Trt)-OH linker with PTX via maleimide bond, while the linker's –NH2 bonded via an amide bond to FA-PTX.264 The aqueous solubility of final prodrugs was reported at about 7 mg mL−1. In vitro PTX release (t½) from FA-PTX-EB was 9.15 h, which was a more sustainable release than FA-PTX (<4 h). Notably, the FA-PTX, PTX-EB, and FA-PTX-EB prodrugs showed an increased circulation half-life in mice of 3.82, 4.41, and 7.51 h, respectively, compared to free PTX (2.19 h). The uptake of FA-PTX-EB in MDA-MB-231 cells was about 66%, which is twice that of PTX-EB (∼35%). In mice bearing MDA-MB-231 tumour xenografts, stronger EB fluorescence signals were observed for FA-PTX-EB than other prodrugs without FA. Meanwhile, in vivo therapeutic experiments of FA-conjugated PTX-EB resulted in improved tumour growth inhibition (∼74%) compared to only PTX-EB (∼50%). The authors reported that the expression level of CD46 in the presence of FA-PTX-EB was significantly decreased, which is relatively high in breast cancer cells to protect them from immune response, indicating an effective targeted cancer therapy.
 | ||
| Fig. 16 (A) Schematic illustration of FA/PTX-conjugated prodrugs: PTX-EB, FA-PTX-EB, and FA-PTX. Reproduced from ref. 264 (CC BY-NC 4.0). (B) PTX-lytic peptides conjugate structures. Rearranged with permission from ref. 268. Copyright 2019, Elsevier; (C) DOTA-folate (6R- and 6S-RedFol-1) prodrug. Adapted from ref. 269 (CC BY 4.0). | ||
Lytic peptides have a cationic amphipathic character that could arrange into the amphipathic structure of the lipid membrane and display potent cell penetration.266 In this context, peptides are widely utilized for drug delivery into cells via cell-penetrating peptides.267 Consistent with this, Qian and co-workers synthesized PTX-lytic peptides that were substituted on 16-site cysteine-substituted named “P3–P7” targeting FR and showed enhanced cytotoxicity to MCF-7 and A2780 cells.268 As shown in Fig. 16B, the thiol group of cysteine-containing peptides conjugated with PTX maleimide via Michael additions, while the N-terminal of peptide coordinated to FA molecule. The authors reported that FA-P7-PTX possessed a more substantial effect on cell toxicity (IC50 ∼ 2.9 μM) than FA-P3-PTX, attributed to the more robust membrane-disrupting activity in MCF-7 cells. The authors found that drug conjugates induced cell death by apoptosis via a mitochondria-dependent pathway. Thus, a significant increase in cleaved caspase-3 and cytochrome-C release indicated mitochondrial dysfunction and caspase-3-dependent apoptotic cell death. Furthermore, FA-P7-PTX reduced the growth of solid tumours by about 69% in an in vivo tumour model in mice (H22 cells), better than free PTX (∼49%).
Despite high efforts for cancer therapy utilizing SMDCs, a combination of targeted radionuclides is highly desirable. In an ingenious work, Müller et al. studied a preclinical evaluation of lutetium-177 (177Lu)-radiolabelled albumin-binding DOTA conjugates with 5-methyltetrahydrofolate (6R- and 6S-RedFol-1), as depicted in Fig. 16C, to examine the effect of 177Lu-DOTA-RedFol-1 isomers on FR(+) KB cells.269In vitro cellular uptake was reported at about 42–53%, higher than in 177Lu-OxFol-1 conjugates reported in a previous study by Müller and co-workers.270 (Fig. 16C). In vivo, uptake of RedFol-1 in tumour cells was increased 3-fold compared to OxFol-1.270 Therefore, the authors concluded that the methylation of position 6 of FA could increase the affinity of the RedFol-1 (ref. 269) to mouse and human plasma proteins and increase blood retention compared to the OxFol-1 analog.270 Accordingly, the effect of 177Lu-DOTA-FA conjugates as a preclinical therapy was explored over 70 days on NF9006 tumours in mice in another work by Müller and co-workers.271 Meanwhile, they found that the radiolabelled conjugates enhanced immune response to anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) immunotherapy. Similar cellular uptake and internalization were reported for NF9006 cells as found for FR(+) KB cells with lower FR expression after 4 h incubation. In contrast, in vitro signal intensity of images on NF9006 cells was about 5-fold lower (∼21%) than the signal in KB cells. SPECT and CT imaging along with biodistribution studies, revealed a significant accumulation of FA-conjugated 177Lu-DOTA in NF9006. The authors found that the tumour growth was delayed after administration of FA-conjugated [177Lu]Lu-DOTA in mice prior to anti-CTLA-4 therapy.271 They also claimed that the radioactive isotope therapy enhanced the response to immune checkpoint (cytotoxic CTLA-4) inhibitors via killer CD8+ T cell infiltration of innate immune cells that have been recently reported.272
In line with peptide-based SMDCs, Costi et al. designed an FA conjugate with anticancer peptides that are able to bind human thymidylate synthase (hTS) to enter cancer cells through highly expressed FRα by decreasing the DHFR expression.273 As shown in Fig. 17A, the FA was conjugated with the γ-position of the glutamic moiety of LSCQLYQR peptide from the amide bond, while N10-(trifluoroacetyl)pteroic acid condensed with [γGlu0]-LSCQLYQR peptide to obtain FA-[DGln4]LSCQLYQR with free –OH moiety that was considered to inhibit the hTS activity. The orientation of the pteridine ring provided several H-bonds and π–π interactions, while the peptidic tail interacted with glutamine (Gln)-100, tryptophane (Trp)-102, and asparagine (Asn)-133 residues (Fig. 17B). The authors investigated the binding effect of FA–peptide conjugates with pemetrexed (PMX) and 5-FU as classical anticancer compounds directed to the TS active site. They found a reduced expression level of the hTS by about 20% using FA-[DGln4]LSCQLYQR, which were 2.5-fold upregulated by PMX and slightly increased in the presence of 5-FU. In short, they concluded that hTS, DHFR, heat shock protein HSP 90-α (HSP90AA1), heat shock protein 75 kDa, and mitochondrial precursor (TRAP1) expression were modulated that represent binding of the FA peptide at the monomer–monomer interface of hTS (Fig. 17C). Furthermore, the FA peptides can be combined with cisplatin, raltitrexed, and 5-FU to overcome drug resistance.
 | ||
| Fig. 17 (A) Synthesis procedure of the FA-[DGln4]-LR conjugate. (B) FA-[DGln4]LR/FRα complexes (PDB: 4LRH). (C) Immunoblot analysis of hTS, DHFR, HSP90AA1, and TRAP1 in IGROV-1 cells, and pemetrexed (PMX) after 48 h in the presence of synthesized prodrugs. Reproduced from ref. 273 (CC BY 4.0). | ||
To date, a few non-invasive molecular imaging using small-molecule conjugates have been reported.274 A study conducted by Guo, Zhang, Khong, and Chen et al. further confirmed the use of 177Lu-DOTA-PEG prodrugs conjugated with albumin truncated EB and fibroblast activation protein (FAP) for SPECT imaging.275 The authors reported significant tumour growth suppression and high uptake in U87MG tumour cells after 96 h post-injection of 177Lu-EB-FAP even without PEG linkers. In an ingenious work, Gois et al. reported a modular platform for constructing drug conjugates comprising tripodal boronate complexes featuring reversible covalent bonds with PEG and FA to deliver bortezomib to MDA-MB-231 cells.276 The linear construction of drug conjugates, along with the boron assembly and FA conjugation chemistry (Fig. 18A).276 In this system, bortezomib (Btz), as a potent proteasome inhibitor, is conjugated through a boron atom to the product of the 4-hydroxy acetophenone, while the aminophenol components modified with a small PEG chain on one side and an azide in another side (Fig. 18B). The strain-promoted alkyne–azide cycloaddition was utilized by authors to post-functionalize with FA and cyclooctyne units. Without FA conjugation, the B-complex was inactive at a concentration of 100 nM, whereas after FA conjugation exhibited improved potency (IC50 ∼ 67 nM) against MDA-MB-231 cells. Interestingly, the bivalent FA-conjugated molecule exhibited similar activity (IC50 ∼ 62 nM). Both reported drug conjugates were only cytotoxic at higher concentrations (1–100 μM) against 4T1 cells. However, as shown in Fig. 18C, boronic acids in this B-complex underwent oxidative cleavage, 1,6-rearrangement, and quinone methide/fluorescent coumarin release triggered by ROS in MDA-MB-231 and 4T1 cells incubated for 10 and 30 min that also refers to confocal fluorescence microscopy images.
 | ||
| Fig. 18 (A) Linear construction of TDCs along with their boron-promoted assembly. (B) FA conjugation via SPAAC called “B-complexes”. (C) Confocal fluorescence microscopy analysis of MDA-MB-231 cells incubated with B-complex. Reproduced with permission from ref. 276. Copyright 2017, Wiley-VCH. | ||
4 Challenges and barriers
Despite the advantages of active targeting via the folate receptor and the significant progress made in recent years, the freshly designed NCs and SMDCs face physiological barriers in the body – not to forget the financial and legal barriers that need to be taken before an application in humans is possible. Significant efforts are needed to overcome these challenges, and only a few of the proposed constructs will have the chance to advance closer to the market in the coming years.4.1 Drug delivery challenges
Between their site of administration and the target site, multiple biochemical and physiological barriers of the body impede the desired accumulation of cancer cells. On the one hand, the stability of the construct in the (bio)chemical reality of the application site and, if relevant, the blood or other liquids of the body, predetermines its successful uptake into the cancer cells. On the other hand, adsorption to bulk proteins, recognition by the immune system, and the release kinetics at the target site modulate the maximum available amount of drug. Thirdly, the decay kinetics of the released drug, either due to clearance by drug transporters or metabolic processes are major driving forces that must be considered during NCs and SMDCs development strategies to achieve a successful formulation.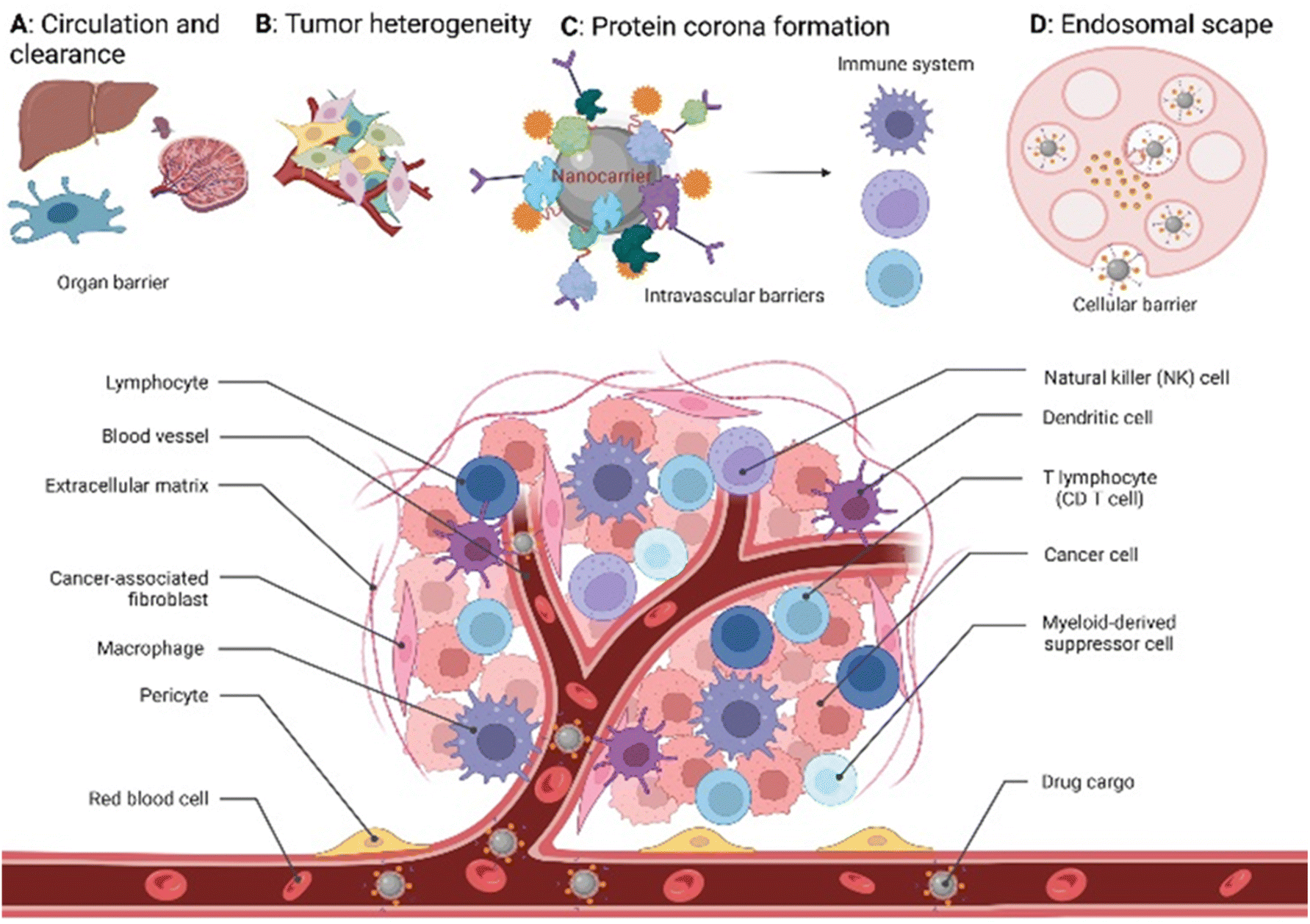 | ||
| Fig. 19 Schematic illustration of tumour microenvironment along with main challenges that NCs face from their route of administration to the site of action. | ||
4.2 Translation into the clinic
Nanocarrier-induced toxicity is a major and increasingly recognized challenge. Several modes of action have been proposed for NC's toxicity. The predominant mechanism is supposed to be an increased production of intracellular ROS. This, in turn, may introduce damage to several cellular structures, e.g., the cell membrane, components of the cytoskeleton, DNA, mitochondria, and lysosomes, which results in cellular dysfunctions of intracellular transport, cell energy imbalance, autophagy, and degradation of macromolecules, mutagenesis, and finally the release of inflammatory mediators and apoptosis. These acute toxicity effects have to be systematically investigated in appropriate in vitro model systems.288 In addition to acute toxic effects, chronic exposure toxicity is expected and therefore needs to be assessed as well. Hence, a number of ex vivo nanosafety assays and animal model studies (i.e., organ toxicity – liver and kidney – , metabolic toxicity, pathological/haematological toxicity, and immune system toxicity) along with pharmacokinetics analysis (i.e., evidence-based ADME) to assess in vivo biocompatibility of NCs have been studied,289 still the step into the clinics was not yet taken due to the aforementioned risks. However, residence times of NCs are not characterized well and may differ between persistent and degradable NCs, which requires different assessment strategies.290 This also includes investigations of biodistribution and pharmacokinetics in appropriate animal models. It has been observed that the distribution of NCs might quite differ between different species.291 Thus, it is crucial to develop standardized models and methods to assess the pharmacokinetics and chronic toxicity after long-term NC exposure.Evidence levels for the safety and efficacy of drug transporters need to be rigorously established, ideally through clinical trials. Preclinical studies provide essential insights, but clinical testing in humans remains a prerequisite. To pave the way for clinical trials, several preconditions must be met. These include details on how they are absorbed, distributed, metabolized, and excreted (pharmacokinetics), where they go in the body (biodistribution), and whether are there any long-term safety concerns, especially after extended exposure. Standardized models and methods for assessing chronic toxicity after extended NC exposure are essential. The costs and legal barriers associated with drug transporters are certainly pertinent, but equally critical are the scientific and safety preconditions that must be satisfied before these innovative therapies can be tested and utilized in human patients.
5 Conclusion and outlook
Targeted compound delivery made a significant progress in the recent years, enhancing therapeutic or diagnostic agent transportation to tumour sites. Despite this, several meta-analysis studies demand further improvements in the median delivery efficiency. Thus, different innovative strategies have been adopted to overcome limitations such as non-specific distribution or poor targeted precision. We here reviewed work that centres the folate receptor – folic acid conjugate axis, aiming to tackle the lingering question: to what extent does folate receptor targeting genuinely bolster delivery efficiency across various in vivo studies? Among the reported in vivo studies, notable improvements were shown in drug delivery efficiency, tumour size reduction, survival rates, and toxicity levels in FR(+) models compared to FR(−) counterparts. While there is variability in the results (due to a lack of standardized experimental conditions), particularly regarding the extent of improvement, FR targeting generally demonstrates enhanced delivery efficiency. Variations may also be linked to tumour types, FR expression levels, or other factors. Reduced side effects and improved therapeutic effects align with this trend. To evaluate FA-conjugated nanocarriers and small molecule drug conjugates based on the challenges and barriers outlined in Section 4, it is imperative to intensify the exploration of compatible carriers such as quantum dots, PEGylated, magnetic, and radiolabelled nanocarriers. These FA-conjugated nanocarriers have demonstrated remarkable potential, significantly enhancing mouse survival rates by decreasing in vivo toxicity to healthy tissues when compared to non-targeted nanocarriers. These platforms have consistently exhibited improved cell proliferation inhibition, induced mitochondria-mediated cell death, minimized drug side effects, and extended survival, ultimately enhancing antitumour effects. Progress in vivo diagnostics, achieved through the tracking and non-invasive imaging of nanocarriers via radiolabelled and magnetic nanocarriers for various imaging modalities like MRI, CT, PET, and SPECT, is noteworthy.As discussed in this review, structure-based de novo design strategies must meet the requirements of the FR-targeted drug transporters to cancer cells, harnessing their structural potential for precise drug delivery. Release mechanisms, triggered by physicochemical stimuli, drive the liberation of therapeutic or diagnostic agents within the tumour microenvironment. This emphasizes the major importance of modified and biodegradable linkers including thioether, disulphide, amide, and ester linkers, as well as adaptable spacers like polypeptides, amino acid residues, and PEG chains for the carrier design. Integration of versatile, self-immolative spacers and linkers, known for their biodegradability, non-toxicity, and biocompatibility, such as PEG, PE, and thiol linkers, further augments these strategies. Utilizing lipid-based and polymeric nanocarriers, primarily focused on ensuring optimal drug encapsulation efficiency, low toxicity, high biocompatibility, and finely tuned release dynamics at tumour sites, deserve special mentioning. In the last half-decade, significant progress has been made in overcoming the obstacles, which had traditional drug delivery reliant on the EPR effect. The meticulous tailoring of FA-conjugated carrier attributes, encompassing chemistry, morphology, delivery modalities, and pharmacodynamics, has certainty-guided precision in drug transporter design, concurrently enhancing efficiency while reducing elimination through the immune system, by improving the compatibility of functionalized carriers decorated with suitable stimuli-responsive linkers and spacers. Based on the substantial folate receptor over-expression in many cancer cells/cancer types, folate receptor-based strategies facilitated the drug delivery notably, but not throughout. Remarkable enhancements were observed for enveloped carriers with biocompatible polymers such as PEGylation and encapsulating drugs within the inner carrier layer, as opposed to surface loading, mitigating opsonization and phagocytosis in the bloodstream, thus reducing off-target toxic effects.
Further approaches have been explored, including liposomes, polymeric, and metal-based NCs, emphasizing the synergy between intricate (bio-) chemical design and insight into (patho-) physiological processes. Fabrication methodologies commonly involve the application of non-toxic stimuli-responsive linkers and spacers, aiming to augment the accumulation of drug-loaded transporters within tumour tissues and regulating or even controlling in vivo pharmacokinetics. Notable chemical examples encompass carbodiimide chemistry, employing EDC/NHS for folic acid conjugation.
A vast number of in vitro studies assessing the interaction between specific FA-conjugated transporters and FRs overexpressed on cancer cell surfaces, paving the ground for future developments. Complementary evaluations, such as carrier cytotoxicity, cellular uptake (IC50) in FR(+) vs. FR(−) cells, drug release kinetics (cumulative vs. sustained), drug release upon stimuli, cell labelling and tracking via fluorescent dyes, and pharmacokinetic profile provide a precast on the drug delivery efficacy, cytotoxicity, biocompatibility, and mechanism of action in vivo. A critical factor is the desired increase in toxicity of FA-conjugated transporters/drugs when compared to their non-targeted counterparts across various FR(+) cell lines in contrast to the FR(−) counterparts and non-malignant cells. This was observed consistently in the reviewed in vitro studies. The downstream effects on the targeted cells depends on the type of drug, but often comprises apoptosis or cell cycle inhibition, followed by expression level changes of pro- and anti-apoptotic proteins (i.e., Bcl-2, caspase-3, -9, PARP, and cytochrome-C), apoptotic-oncoproteins (i.e., p53, p65, Bax) and cell proliferation markers (i.e., Ki-67, VEGF), or membrane proteins (CD1, CD31, CD46).
It's essential to acknowledge that despite significant forward steps in FR-targeted drug delivery, the potential of improvement in delivery efficiency remains substantial. While most studies have delved into in vitro experiments employing cell lines, only a limited number have ventured into animal models. Prolonged blood circulation, carrier–protein interaction, and endosomal escape of FA-conjugated transporters in vivo remains extremely challenging. However, to advance the development of these drug transporters to a clinical level, it is crucial to consider the complex interplay between transport and elimination processes, accumulation in the target region, immune system interactions, cellular uptake, and impact on tumour cells.
While comprehensively addressing all parameters in drug transporter design remains a difficult task, emphasizing their functional attributes during development is pivotal. It's worth recognizing that not all introduced drug transporters possess the inherent potential or necessary financial backing to advance to practical application. Only the most promising strategies that cover these key chemical factors can contribute to bolstering the stability, mobility, and responsiveness of FA-conjugated nanocarriers, thereby enhancing their potential for improved biodistribution and pharmacokinetics may evolve into therapeutic drug/diagnostic systems:
• surface functionalization: covalent attachment of folic acid (FA) to the nanocarrier's surface through stable chemical linkers like amide bonds ensures that the targeting ligand remains firmly attached, preventing premature detachment during circulation.
• Biodegradable linkers/spacers: using biodegradable linkers, such as disulphide or ester linkages, or spacers such as polypeptides and PEG chains can enhance carrier responsiveness. Linkers can be designed to break in response to specific stimuli like reducing environments (e.g., glutathione) or pH changes, facilitating drug release at the target site. Conjugating multiple chemical moieties to the nanocarrier, including both folic acid (FA) and stimuli-responsive components can provide versatility and responsiveness, improving the carrier's ability to navigate complex in vivo environments.
• Stimuli-responsive carriers: smart polymers that respond to the cancers environmental cues can be incorporated into the nanocarrier's structure. These polymers can enable controlled drug release in response to specific conditions within the tumour microenvironment. Incorporating PEGylation on the nanocarrier surface can also enhance stability and circulation time by reducing opsonization, immune recognition, and clearance by the reticuloendothelial system. PEGylation also contributes to improved biodistribution.
• Drug encapsulation and release chemistry: effective drug encapsulation within the carrier matrix, through hydrophobic or electrostatic interactions, ensures drug stability during circulation and controlled release chemistry at the target site upon stimuli. This encapsulation can be fine-tuned chemically to optimize drug loading and release kinetics.
• Chemical stability and non-toxicity: ensuring that the nanocarrier itself is chemically non-toxic and stable under physiological conditions is critical. Biocompatible materials that do not degrade rapidly in the bloodstream are essential to maintain carrier integrity during circulation.
• Size, morphology, and surface charge control: fine-tuning the size, morphology, and surface charge of the nanocarrier through chemical methods not only impacts its mobility and circulation properties but also influences interactions with biological components. Well-dispersed carriers often exhibit improved biodistribution.
Continued research efforts, in collaborative synergy among chemists, material scientists, life sciences researchers, and clinicians will prove pivotal in fully realizing the clinical viability of FR-mediated drug delivery.
Abbreviations
| NC | Nanocarrier |
| SMDC | Small molecule–drugs conjugate |
| NP | Nanoparticle |
| FR | Folate receptor |
| FR(+) | Folate receptor-positive |
| FR(−) | Folate receptor-negative |
| FA | Folic acid |
| TNBC | Triple-negative breast cancer |
| NIR | Near-infrared |
| MRI | Magnetic resonance imaging |
| AMF | Alternating magnetic field |
| BBB | Blood–brain barrier |
| LNP | Lipid-based nanoparticle |
| PNP | Polymeric nanoparticle |
| MNP | Magnetic nanoparticle |
| AuNP | Gold nanoparticle |
| nm | Nanometer |
| nM | Nanomolar |
| μM | Micromolar |
| mM | Millimolar |
| mV | Millivolt |
| mL | Milliliter |
| ζ | Zeta |
| PS | Particle size |
| IC50 | Half maximal inhibitory concentration |
| PDI | Polydispersity index: the square of the standard deviation divided by the mean particle diameter |
Author contributions
M. A. conceptualized and reviewed the articles, analysed data, and drafted the manuscript. M. A. and K. W. designed the review procedure. M. A. and C. A. R. reviewed and drafted the challenges and barriers in targeted drug delivery. K. W. and S. B. revised and edited the manuscript and provided general oversight. T. v. W. undertook overall management of the manuscript. All the authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is funded by the German Federal Ministry of Education and Research (GN: 03Z22DN12 to K.W.). All original figures were created with https://biorender.com/.Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, C. A.: Cancer J. Clin., 2021, 71, 209–249 Search PubMed.
- (a) B. Yang and J. Shi, Angew. Chem., Int. Ed., 2020, 59, 21829–21838 CrossRef CAS PubMed; (b) K. Yang, S. Qi, X. Yu, B. Bai, X. Zhang, Z. Mao, F. Huang and G. Yu, Angew. Chem., Int. Ed., 2022, 61, e202203786 CrossRef CAS; (c) T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CrossRef CAS; (d) T. J. Anchordoquy, Y. Barenholz, D. Boraschi, M. Chorny, P. Decuzzi, M. A. Dobrovolskaia, Z. S. Farhangrazi, D. Farrell, A. Gabizon, H. Ghandehari, B. Godin, N. M. La-Beck, J. Ljubimova, S. M. Moghimi, L. Pagliaro, J.-H. Park, D. Peer, E. Ruoslahti, N. J. Serkova and D. Simberg, ACS Nano, 2017, 11, 12–18 CrossRef CAS; (e) D. Sun, S. Zhou and W. Gao, ACS Nano, 2020, 14, 12281–12290 CrossRef CAS PubMed; (f) J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed; (g) X. Fu, Y. Shi, T. Qi, S. Qiu, Y. Huang, X. Zhao, Q. Sun and G. Lin, Signal Transduction Targeted Ther., 2020, 5, 262 CrossRef.
- J. A. Kemp and Y. J. Kwon, Nano Convergence, 2021, 8, 34 CrossRef CAS PubMed.
- (a) D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed; (b) S. T. Stern, M. N. Martinez and D. M. Stevens, Drug Metab. Dispos., 2016, 44, 1934–1939 CrossRef CAS.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS.
- (a) T. K. Patel, N. Adhikari, S. A. Amin, S. Biswas, T. Jha and B. Ghosh, New J. Chem., 2021, 45, 5291–5321 RSC; (b) S. Cazzamalli, A. Dal Corso, F. Widmayer and D. Neri, J. Am. Chem. Soc., 2018, 140, 1617–1621 CrossRef CAS PubMed.
- (a) P. Boix-Montesinos, P. M. Soriano-Teruel, A. Armiñán, M. Orzáez and M. J. Vicent, Adv. Drug Delivery Rev., 2021, 173, 306–330 CrossRef CAS; (b) H. W. Song, K. L. Foreman, B. D. Gastfriend, J. S. Kuo, S. P. Palecek and E. V. Shusta, Sci. Rep., 2020, 10, 12358 CrossRef CAS PubMed; (c) K. Krüger, L. Silwal-Pandit, E. Wik, O. Straume, I. M. Stefansson, E. Borgen, Ø. Garred, B. Naume, O. Engebraaten and L. A. Akslen, Sci Rep, 2021, 11, 3388 CrossRef; (d) Y. H. Bae, J. Controlled Release, 2009, 133, 2–3 CrossRef CAS PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- Y.-H. Cheng, C. He, J. E. Riviere, N. A. Monteiro-Riviere and Z. Lin, ACS Nano, 2020, 14, 3075–3095 CrossRef CAS.
- M. M. T. van Leent, B. Priem, D. P. Schrijver, A. de Dreu, S. R. J. Hofstraat, R. Zwolsman, T. J. Beldman, M. G. Netea and W. J. M. Mulder, Nat. Rev. Mater., 2022, 7, 465–481 CrossRef.
- Z. Zhao, A. Ukidve, J. Kim and S. Mitragotri, Cell, 2020, 181, 151–167 CrossRef CAS PubMed.
- (a) N. Rodrigues Mantuano, M. Natoli, A. Zippelius and H. Läubli, J. Immunother. Cancer, 2020, 8, e001222 CrossRef PubMed; (b) S. Jin, Y. Sun, X. Liang, X. Gu, J. Ning, Y. Xu, S. Chen and L. Pan, Signal Transduction Targeted Ther., 2022, 7, 39 CrossRef CAS PubMed; (c) S. Wang, Y. Meng, C. Li, M. Qian and R. Huang, Nanomaterials, 2015, 6, 3 CrossRef.
- E. McCord, S. Pawar, T. Koneru, K. Tatiparti, S. Sau and A. K. Iyer, ACS Omega, 2021, 6, 4111–4118 CrossRef CAS.
- (a) C. Scafoglio, B. A. Hirayama, V. Kepe, J. Liu, C. Ghezzi, N. Satyamurthy, N. A. Moatamed, J. Huang, H. Koepsell, J. R. Barrio and E. M. Wright, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4111–E4119 CrossRef CAS PubMed; (b) M. Pliszka and L. Szablewski, Cancers, 2021, 13, 4184 CrossRef CAS.
- (a) C. Du, Y. Qi, Y. Zhang, Y. Wang, X. Zhao, H. Min, X. Han, J. Lang, H. Qin, Q. Shi, Z. Zhang, X. Tian, G. J. Anderson, Y. Zhao, G. Nie and Y. Yang, ACS Nano, 2018, 12, 10785–10796 CrossRef CAS; (b) M. Akbarzadeh Khiavi, A. Safary, J. Barar, A. Ajoolabady, M. H. Somi and Y. Omidi, Cell. Mol. Life Sci., 2020, 77, 997–1019 CrossRef CAS PubMed.
- T. Koneru, E. McCord, S. Pawar, K. Tatiparti, S. Sau and A. K. Iyer, ACS Omega, 2021, 6, 8727–8733 CrossRef CAS PubMed.
- L. Rosenfeld, A. Sananes, Y. Zur, S. Cohen, K. Dhara, S. Gelkop, E. Ben Zeev, A. Shahar, L. Lobel, B. Akabayov, E. Arbely and N. Papo, J. Med. Chem., 2020, 63, 7601–7615 CrossRef CAS PubMed.
- (a) H. Lu, T. Chen, Y. Wang, Y. He, Z. Pang and Y. Wang, Sci. Rep., 2022, 12, 2610 CrossRef CAS; (b) S. Habib and M. Singh, Polymers, 2022, 14, 712 CrossRef CAS PubMed.
- (a) L. Gu, F. Zhang, J. Wu and Y. Zhuge, Front. Mol. Biosci., 2022, 8, 804396 CrossRef; (b) S. A. Igdoura, Curr. Opin. Lipidol., 2017, 28, 209–212 CrossRef CAS PubMed.
- (a) N. Norton, B. Youssef, D. W. Hillman, A. Nassar, X. J. Geiger, B. M. Necela, H. Liu, K. J. Ruddy, M.-Y. C. Polley, J. N. Ingle, F. J. Couch, E. A. Perez, M. C. Liu, J. M. Carter, R. A. Leon-Ferre, J. C. Boughey, E. B. Somers, K. R. Kalari, D. W. Visscher, M. P. Goetz and K. L. Knutson, npj Breast Cancer, 2020, 6, 4 CrossRef CAS PubMed; (b) D.-G. Song, Q. Ye, M. Poussin, J. A. Chacon, M. Figini and D. J. Powell, J. Hematol. Oncol., 2016, 9, 56 CrossRef.
- C. D. Arvanitis, G. B. Ferraro and R. K. Jain, Nat. Rev. Cancer, 2020, 20, 26–41 CrossRef CAS.
- R. W. Robey, K. M. Pluchino, M. D. Hall, A. T. Fojo, S. E. Bates and M. M. Gottesman, Nat. Rev. Cancer, 2018, 18, 452–464 CrossRef CAS.
- K. Strebhardt and A. Ullrich, Nat. Rev. Cancer, 2008, 8, 473–480 CrossRef CAS PubMed.
- (a) https://www.ema.europa.eu/en/medicines/human/EPAR/lutathera#authorisation-details-section, 2017; (b) https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/208700Orig1s000ltr.pdf, 2018.
- (a) K. N. Moore, L. P. Martin, D. M. O'Malley, U. A. Matulonis, J. A. Konner, R. P. Perez, T. M. Bauer, R. Ruiz-Soto and M. J. Birrer, J. Clin. Oncol., 2017, 35, 1112–1118 CrossRef CAS; (b) R. J. Lutz, Transl. Cancer Res., 2015, 4, 118–126 CAS; (c) L. Teng, J. Xie, L. Teng and R. J. Lee, Expert Opin. Drug Delivery, 2012, 9, 901–908 CrossRef CAS.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 CrossRef.
- P. N. Navya, A. Kaphle, S. P. Srinivas, S. K. Bhargava, V. M. Rotello and H. K. Daima, Nano Convergence, 2019, 6, 23 CrossRef CAS PubMed.
- (a) C. Martinelli, C. Pucci and G. Ciofani, APL Bioeng., 2019, 3, 011502 CrossRef PubMed; (b) R. Ridolfo, S. Tavakoli, V. Junnuthula, D. S. Williams, A. Urtti and J. C. M. van Hest, Biomacromolecules, 2021, 22, 126–133 CrossRef CAS PubMed.
- S. Kamble, S. Agrawal, S. Cherumukkil, V. Sharma, R. V. Jasra and P. Munshi, ChemistrySelect, 2022, 7, e202103084 CrossRef CAS.
- D. Jiang, Z. T. Rosenkrans, D. Ni, J. Lin, P. Huang and W. Cai, Acc. Chem. Res., 2020, 53, 1869–1880 CrossRef CAS.
- A. Prokop and J. M. Davidson, J. Pharm. Sci., 2008, 97, 3518–3590 CrossRef CAS PubMed.
- (a) A. Dal Corso, L. Pignataro, L. Belvisi and C. Gennari, Chem.–Eur. J., 2019, 25, 14740–14757 CrossRef CAS; (b) A. Oake, P. Bhatt and Y. V. Pathak, in Surface Modification of Nanoparticles for Targeted Drug Delivery, ed. Y. V. Pathak, Springer International Publishing, Cham, 2019, pp. 1–17, DOI:10.1007/978-3-030-06115-9_1; (c) K. Parmar and J. K. Patel, in Surface Modification of Nanoparticles for Targeted Drug Delivery, ed. Y. V. Pathak, Springer International Publishing, Cham, 2019, pp. 221–236, DOI:10.1007/978-3-030-06115-9_12; (d) V. Wiwanitkit, in Surface Modification of Nanoparticles for Targeted Drug Delivery, ed. Y. V. Pathak, Springer International Publishing, Cham, 2019, pp. 167–181, DOI:10.1007/978-3-030-06115-9_9.
- P. Zhang, D. Chen, L. Li and K. Sun, J. Nanobiotechnol., 2022, 20, 31 CrossRef CAS.
- G. H. Zhu, A. B. C. Gray and H. K. Patra, Trends Pharmacol. Sci., 2022, 43, 709–711 CrossRef CAS.
- J. W. Park, Breast Cancer Res., 2002, 4, 95 CrossRef CAS.
- H. Zhang, OncoTargets Ther., 2016, 9, 3001–3007 CrossRef CAS.
- E. Beltrán-Gracia, A. López-Camacho, I. Higuera-Ciapara, J. B. Velázquez-Fernández and A. A. Vallejo-Cardona, Cancer Nanotechnol., 2019, 10, 11 CrossRef.
- V. Roy, B. R. LaPlant, G. G. Gross, C. L. Bane and F. M. Palmieri, Ann. Oncol., 2009, 20, 449–453 CrossRef CAS.
- R. Haddad, N. Alrabadi, B. Altaani and T. Li, Polymers, 2022, 14, 658 CrossRef CAS PubMed.
- K. S. Lee, H. C. Chung, S. A. Im, Y. H. Park, C. S. Kim, S.-B. Kim, S. Y. Rha, M. Y. Lee and J. Ro, Breast Cancer Res. Treat., 2008, 108, 241–250 CrossRef CAS.
- P. Chowdhury, U. Ghosh, K. Samanta, M. Jaggi, S. C. Chauhan and M. M. Yallapu, Bioact. Mater., 2021, 6, 3269–3287 CAS.
- (a) Y. Huang, X. Li, S. Xu, H. Zheng, L. Zhang, J. Chen, H. Hong, R. Kusko and R. Li, Environ. Health Perspect., 2020, 128, 067010 CrossRef CAS PubMed; (b) J. Li, C. Wang, L. Yue, F. Chen, X. Cao and Z. Wang, Ecotoxicol. Environ. Saf., 2022, 243, 113955 CrossRef CAS PubMed; (c) A. Rana and S. Bhatnagar, Bioorg. Chem., 2021, 112, 104946 CrossRef CAS; (d) I. R. Vlahov and C. P. Leamon, Bioconjugate Chem., 2012, 23, 1357–1369 CrossRef CAS PubMed; (e) F. Salahpour Anarjan, Nano-Struct. Nano-Objects, 2019, 19, 100370 CrossRef CAS; (f) G. Onzi, S. S. Guterres, A. R. Pohlmann and L. A. Frank, in The ADME Encyclopedia: A Comprehensive Guide on Biopharmacy and Pharmacokinetics, Springer International Publishing, Cham, 2021, pp. 1–13, DOI:10.1007/978-3-030-51519-5_109-1; (g) P. Tagde, G. T. Kulkarni, D. K. Mishra and P. Kesharwani, J. Drug Delivery Sci. Technol., 2020, 56, 101613 CrossRef CAS; (h) B. Frigerio, C. Bizzoni, G. Jansen, C. P. Leamon, G. J. Peters, P. S. Low, L. H. Matherly and M. Figini, J. Exp. Clin. Cancer Res., 2019, 38, 125 CrossRef; (i) A. Narmani, M. Rezvani, B. Farhood, P. Darkhor, J. Mohammadnejad, B. Amini, S. Refahi and N. Abdi Goushbolagh, Drug Dev. Res., 2019, 80, 404–424 CrossRef CAS PubMed.
- S. Chen, Y. Wu, F. Lortie, J. Bernard, W. H. Binder and J. Zhu, Macromol. Rapid Commun., 2022, 43, 2200168 CrossRef CAS PubMed.
- (a) B. K. Wilson, P. J. Sinko and R. K. Prud'homme, Mol. Pharm., 2021, 18, 1093–1101 CrossRef CAS PubMed; (b) Q. Li, X. Li and C. Zhao, Front. Bioeng. Biotechnol., 2020, 8, 437 CrossRef PubMed.
- S. Waheed, Z. Li, F. Zhang, A. Chiarini, U. Armato and J. Wu, J. Nanobiotechnol., 2022, 20, 395 CrossRef PubMed.
- M. Geven, R. d'Arcy, Z. Y. Turhan, F. El-Mohtadi, A. Alshamsan and N. Tirelli, Eur. Polym. J., 2021, 149, 110387 CrossRef CAS.
- (a) N. Yu, Y. Xu, T. Liu, H. Zhong, Z. Xu, T. Ji, H. Zou, J. Mu, Z. Chen, X.-J. Liang, L. Shi, D. S. Kohane and S. Guo, Nat. Commun., 2021, 12, 5532 CrossRef CAS; (b) B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306–2317 CrossRef CAS PubMed.
- D. Aydin, M. Arslan, A. Sanyal and R. Sanyal, Bioconjugate Chem., 2017, 28, 1443–1451 CrossRef CAS PubMed.
- Q. Wang, C. Wang, S. Li, Y. Xiong, H. Wang, Z. Li, J. Wan, X. Yang and Z. Li, Chem. Mater., 2022, 34, 2085–2097 CrossRef CAS.
- (a) D. Wang, X. Mu, X. Chen, H. Huang, L. Zhou and S. Wei, Carbohydr. Polym., 2021, 273, 118608 CrossRef CAS PubMed; (b) G. Wang, F. Chen, N. K. Banda, V. M. Holers, L. Wu, S. M. Moghimi and D. Simberg, Front. Immunol., 2016, 7, 418 Search PubMed.
- F. M. F. Santos, A. I. Matos, A. E. Ventura, J. Goncalves, L. F. Veiros, H. F. Florindo and P. M. P. Gois, Angew Chem. Int. Ed. Engl., 2017, 56, 9346–9350 CrossRef CAS.
- Q. Wang, J. Guan, J. Wan and Z. Li, RSC Adv., 2020, 10, 24397–24409 RSC.
- (a) S. J. Sonawane, R. S. Kalhapure and T. Govender, Eur. J. Pharm. Sci., 2017, 99, 45–65 CrossRef CAS; (b) H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- S. Akhshabi, E. Biazar, V. Singh, S. H. Keshel and N. Geetha, Int. J. Nanomed., 2018, 13, 4405–4416 CrossRef CAS.
- C. R. Cammarata, M. E. Hughes and C. M. Ofner, Mol. Pharm., 2015, 12, 783–793 CrossRef CAS PubMed.
- Z. Donglu, F.-O. D. Aimee, S. D. Peter, H. P. Thomas, D. S. Jack, R. K. Katherine, T. C. Robert, L. Liling, D. Yuzhong, L. Yichin, E. C. A. H. Cornelis and S. C. Khojasteh, Drug Metab. Dispos., 2019, 47, 1156 CrossRef.
- (a) W. H. Binder, L. Petraru, R. Sachenshofer and R. Zirbs, Monatsh. Chem., 2006, 137, 835–841 CrossRef CAS; (b) N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717–16734 RSC.
- C. Zang, H. Wang, T. Li, Y. Zhang, J. Li, M. Shang, J. Du, Z. Xi and C. Zhou, Chem. Sci., 2019, 10, 8973–8980 RSC.
- M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790–810 RSC.
- J. Zhang, Y. Lin, Z. Lin, Q. Wei, J. Qian, R. Ruan, X. Jiang, L. Hou, J. Song, J. Ding and H. Yang, Adv. Sci., 2022, 9, 2103444 CrossRef CAS PubMed.
- C. J. Choy, J. J. Geruntho, A. L. Davis and C. E. Berkman, Bioconjugate Chem., 2016, 27, 824–830 CrossRef CAS PubMed.
- P. D. Senter, W. E. Pearce and R. S. Greenfield, Drug Discovery Today, 1990, 55, 2975–2978 CAS.
- W. A. Henne, D. D. Doorneweerd, A. R. Hilgenbrink, S. A. Kularatne and P. S. Low, Bioorg. Med. Chem. Lett., 2006, 16, 5350–5355 CrossRef CAS PubMed.
- B. J. Stenton, B. L. Oliveira, M. J. Matos, L. Sinatra and G. J. L. Bernardes, Chem. Sci., 2018, 9, 4185–4189 RSC.
- A. D. Wong, M. A. DeWit and E. R. Gillies, Adv. Drug Delivery Rev., 2012, 64, 1031–1045 CrossRef CAS PubMed.
- (a) A. Alouane, R. Labruère, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed; (b) M. Ximenis, A. Sampedro, L. Martínez-Crespo, G. Ramis, F. Orvay, A. Costa and C. Rotger, Chem. Commun., 2021, 57, 2736–2739 RSC.
- M. Gisbert-Garzarán, M. Manzano and M. Vallet-Regí, Chem. Eng. J., 2018, 340, 24–31 CrossRef.
- (a) W. S. Saw, T. Anasamy, T. T. A. Do, H. B. Lee, C. F. Chee, U. Isci, M. Misran, F. Dumoulin, W. Y. Chong, L. V. Kiew, T. Imae and L. Y. Chung, Macromol. Biosci., 2022, 22, 2200130 CrossRef CAS PubMed; (b) O. Shelef, S. Gnaim and D. Shabat, J. Am. Chem. Soc., 2021, 143, 21177–21188 CrossRef CAS.
- T. Tedeschini, B. Campara, A. Grigoletto, M. Bellini, M. Salvalaio, Y. Matsuno, A. Suzuki, H. Yoshioka and G. Pasut, J. Controlled Release, 2021, 337, 431–447 CrossRef CAS.
- K. Bozovičar and T. Bratkovič, Int. J. Mol. Sci., 2021, 22, 1611 CrossRef.
- (a) M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC; (b) F. Nazir, T. A. Tabish, F. Tariq, S. Iftikhar, R. Wasim and G. Shahnaz, Drug Discovery Today, 2022, 27, 1698–1705 CrossRef CAS PubMed; (c) R. Salve, P. Kumar, K. R. Gajbhiye, R. J. Babu and V. Gajbhiye, in Stimuli-Responsive Nanocarriers, ed. V. Gajbhiye, K. R. Gajbhiye and S. Hong, Academic Press, 2022, pp. 29–60, DOI:10.1016/B978-0-12-824456-2.00013-8.
- (a) X. Dong, R. K. Brahma, C. Fang and S. Q. Yao, Chem. Sci., 2022, 13, 4239–4269 RSC; (b) H. Alimoradi, S. S. Matikonda, A. B. Gamble, G. I. Giles and K. Griesh, in Nanostructures for Drug Delivery, ed. E. Andronescu and A. M. Grumezescu, Elsevier, 2017, pp. 327–354, DOI:10.1016/B978-0-323-46143-6.00010-5.
- M. Scaranti, E. Cojocaru, S. Banerjee and U. Banerji, Nat. Rev. Clin. Oncol., 2020, 17, 349–359 CrossRef PubMed.
- A. S. Wibowo, M. Singh, K. M. Reeder, J. J. Carter, A. R. Kovach, W. Meng, M. Ratnam, F. Zhang and C. E. Dann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15180–15188 CrossRef CAS PubMed.
- (a) J. B. Schnoell J, L. Kadletz-Wanke, S. Stoiber, E. Gurnhofer, M. Schlederer, G. Heiduschka and L. Kenner, OncoTargets Ther., 2022, 15, 531–538 CrossRef; (b) M. Bartouskova, B. Melichar and B. Mohelnikova-Duchonova, Pteridines, 2015, 26, 1–12 CrossRef CAS; (c) H. Shi, J. Guo, C. Li and Z. Wang, Drug Des., Dev. Ther., 2015, 9, 4989–4996 CAS; (d) L. S. F. Boogerd, M. C. Boonstra, A.-J. Beck, A. Charehbili, C. E. S. Hoogstins, H. A. J. M. Prevoo, S. Singhal, P. S. Low, C. J. H. van de Velde and A. L. Vahrmeijer, Oncotarget, 2016, 7, 17442–17454 CrossRef PubMed.
- (a) W. Han, R. Zaynagetdinov, F. E. Yull, V. V. Polosukhin, L. A. Gleaves, H. Tanjore, L. R. Young, T. E. Peterson, H. C. Manning, L. S. Prince and T. S. Blackwell, Am. J. Respir. Cell Mol. Biol., 2015, 53, 50–59 CrossRef CAS PubMed; (b) D. Chandrupatla, C. F. M. Molthoff, A. A. Lammertsma, C. J. van der Laken and G. Jansen, Drug Delivery Transl. Res., 2019, 9, 366–378 CrossRef CAS PubMed.
- N. Parker, M. J. Turk, E. Westrick, J. D. Lewis, P. S. Low and C. P. Leamon, Anal. Biochem., 2005, 338, 284–293 CrossRef CAS PubMed.
- I. Mellman and Y. Yarden, Cold Spring Harbor Perspect. Biol., 2013, 5, a016949 CrossRef PubMed.
- (a) A. Annibal, R. G. Tharyan, M. F. Schonewolff, H. Tam, C. Latza, M. M. K. Auler, S. Grönke, L. Partridge and A. Antebi, Nat. Commun., 2021, 12, 3486 CrossRef CAS PubMed; (b) V. Pareek, A. M. Pedley and S. J. Benkovic, Crit. Rev. Biochem. Mol. Biol., 2021, 56, 1–16 CrossRef CAS PubMed.
- M. R. Sullivan, A. M. Darnell, M. F. Reilly, C. A. Lewis and M. G. Vander Heiden, bioRxiv, 2020, preprint, DOI:10.1101/2020.06.12.149005.
- (a) S. Su and P. M. Kang, Pharmaceutics, 2020, 12, 837 CrossRef CAS PubMed; (b) A. DeCarlo, C. Malardier-Jugroot and M. R. Szewczuk, Bioconjugate Chem., 2021, 32, 512–522 CrossRef CAS PubMed.
- R. J. Lee, S. Wang and P. S. Low, Biochim. Biophys. Acta, Mol. Cell Res., 1996, 1312, 237–242 CrossRef PubMed.
- P. G. Alluri, C. Speers and A. M. Chinnaiyan, Breast Cancer Res., 2014, 16, 494 CrossRef PubMed.
- A. Bahreyni, Y. Mohamud and H. Luo, J. Nanobiotechnol., 2020, 18, 180 CrossRef PubMed.
- J. Haussmann, S. Corradini, C. Nestle-Kraemling, E. Bölke, F. J. D. Njanang, B. Tamaskovics, K. Orth, E. Ruckhaeberle, T. Fehm, S. Mohrmann, I. Simiantonakis, W. Budach and C. Matuschek, Radiat. Oncol., 2020, 15, 71 CrossRef PubMed.
- L. Yin, J.-J. Duan, X.-W. Bian and S.-c. Yu, Breast Cancer Res., 2020, 22, 61 CrossRef PubMed.
- M. Sajjad, M. I. Khan, S. Naveed, S. Ijaz, O. S. Qureshi, S. A. Raza, G. Shahnaz and M. F. Sohail, AAPS PharmSciTech, 2019, 20, 81 CrossRef CAS PubMed.
- K. G. Desai, Crit. Rev. Ther. Drug Carrier Syst., 2016, 33, 107–158 CrossRef PubMed.
- L. Cheng, H. Ma, M. Shao, Q. Fan, H. Lv, J. Peng, T. Hao, D. Li, C. Zhao and X. Zong, Mol. Med. Rep., 2017, 16, 1101–1108 CrossRef CAS.
- B. Y. Liu, Y. L. Wang, Q. J. Yu, D. P. Li and F. Li, CyTA--J. Food, 2018, 16, 868–876 CrossRef CAS.
- N. Hock, G. F. Racaniello, S. Aspinall, N. Denora, V. V. Khutoryanskiy and A. Bernkop-Schnürch, Adv. Sci., 2022, 9, 2102451 CrossRef CAS PubMed.
- A. Kefayat, M. Hosseini, F. Ghahremani, N. A. Jolfaie and M. Rafienia, J. Nanobiotechnol., 2022, 20, 169 CrossRef CAS.
- (a) E. O. Bakhrushina and N. B. Demina, Pharm. Chem. J., 2022, 56, 396–402 CrossRef CAS; (b) S. A. Stewart, J. Domínguez-Robles, R. F. Donnelly and E. Larrañeta, Polymers, 2018, 10, 1379 CrossRef PubMed.
- S. Esfandiarpour-Boroujeni, S. Bagheri-Khoulenjani, H. Mirzadeh and S. Amanpour, Carbohydr. Polym., 2017, 168, 14–21 CrossRef CAS PubMed.
- Y. Tang, Y. Li, R. Xu, S. Li, H. Hu, C. Xiao, H. Wu, L. Zhu, J. Ming, Z. Chu, H. Xu, X. Yang and Z. Li, Nanoscale, 2018, 10, 17265–17274 RSC.
- N. Erdoğar, G. Esendağlı, T. T. Nielsen, G. Esendağlı-Yılmaz, D. Yöyen-Ermiş, B. Erdoğdu, M. F. Sargon, H. Eroğlu and E. Bilensoy, J. Drug Targeting, 2018, 26, 66–74 CrossRef PubMed.
- F. Raza, H. Zafar, M. W. Khan, A. Ullah, A. U. Khan, A. Baseer, R. Fareed and M. Sohail, Mater. Adv., 2022, 3, 2268–2290 RSC.
- S.-B. Ghaffari, M.-H. Sarrafzadeh, Z. Fakhroueian and M. R. Khorramizadeh, Mater. Sci. Eng. C, 2019, 103, 109827 CrossRef CAS PubMed.
- S.-B. Ghaffari, M.-H. Sarrafzadeh, Z. Fakhroueian, S. Shahriari and M. R. Khorramizadeh, Mater. Sci. Eng. C, 2017, 79, 465–472 CrossRef CAS.
- L. H. Dang, M. T. Vu, J. Chen, C. K. Nguyen, L. G. Bach, N. Q. Tran and V. T. Le, ACS Omega, 2019, 4, 4540–4552 CrossRef CAS.
- A. Pawar, S. Singh, S. Rajalakshmi, K. Shaikh and C. Bothiraja, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 347–361 CrossRef CAS PubMed.
- D. Baidya, J. Kushwaha, K. Mahadik and S. Patil, Drug Dev. Ind. Pharm., 2019, 45, 852–860 CrossRef CAS PubMed.
- (a) P. Singla, O. Singh, S. Sharma, K. Betlem, V. K. Aswal, M. Peeters and R. K. Mahajan, ACS Omega, 2019, 4, 11251–11262 CrossRef CAS PubMed; (b) S. Salwa, H. Shahrul Sahul and K. Noor Haida Mohd, Pharmacogn. Res., 2017, 9, 12–20 CrossRef; (c) K. Al Khateb, E. K. Ozhmukhametova, M. N. Mussin, S. K. Seilkhanov, T. K. Rakhypbekov, W. M. Lau and V. V. Khutoryanskiy, Int. J. Pharm., 2016, 502, 70–79 CrossRef CAS.
- V. T. Nguyen, T. H. Nguyen, L. H. Dang, H. Vu-Quang and N. Q. Tran, J. Nanomater., 2019, 2019, 1067821 Search PubMed.
- W. Hong, H. Shi, M. Qiao, Z. Zhang, W. Yang, L. Dong, F. Xie, C. Zhao and L. Kang, Sci. Rep., 2017, 7, 42465 CrossRef CAS PubMed.
- A. M. Pragatheeswaran and S. B. Chen, Langmuir, 2013, 29, 9694–9701 CrossRef CAS PubMed.
- Y. Gao, L. Jia, Q. Wang, H. Hu, X. Zhao, D. Chen and M. Qiao, ACS Appl. Mater. Interfaces, 2019, 11, 16296–16310 CrossRef CAS.
- D. Yang, Z. Li, Y. Zhang, X. Chen, M. Liu and C. Yang, Pharmaceutics, 2023, 15, 1580 CrossRef CAS PubMed.
- N. E. Guissi, H. Li, Y. Xu, F. Semcheddine, M. Chen, Z. Su and Q. Ping, Mol. Pharm., 2017, 14, 1082–1094 CrossRef CAS PubMed.
- R. K. Tahara, T. M. Brewer, R. L. Theriault and N. T. Ueno, in Breast Cancer Metastasis and Drug Resistance: Challenges and Progress, ed. A. Ahmad, Springer International Publishing, Cham, 2019, pp. 105–129, DOI:10.1007/978-3-030-20301-6_7.
- (a) N. Mohammadi Ghahhari, M. K. Sznurkowska, N. Hulo, L. Bernasconi, N. Aceto and D. Picard, Nat. Commun., 2022, 13, 2104 CrossRef CAS PubMed; (b) Z. Tian, C. Yu, W. Zhang, K.-L. Wu, C. Wang, R. Gupta, Z. Xu, L. Wu, Y. Chen, X. H. F. Zhang and H. Xiao, ACS Cent. Sci., 2022, 8, 312–321 CrossRef CAS PubMed; (c) W. Jiang, Y. Rixiati, H. Huang, Y. Shi, C. Huang and B. Jiao, Cancer Med., 2020, 9, 8173–8185 CrossRef CAS PubMed; (d) K.-H. Lee, K. J. Lee, T.-Y. Kim, F. Hutomo, H. J. Sun, G. J. Cheon, S. I. Park, S. W. Cho and S.-A. Im, J. Bone Miner. Res., 2020, 35, 1838–1849 CrossRef CAS PubMed.
- A. Parkes, K. Clifton, A. Al-Awadhi, O. Oke, C. L. Warneke, J. K. Litton and G. N. Hortobagyi, npj Breast Cancer, 2018, 4, 2 CrossRef PubMed.
- S.-H. Chen, T.-I. Liu, C.-L. Chuang, H.-H. Chen, W.-H. Chiang and H.-C. Chiu, J. Mater. Chem. B, 2020, 8, 3789–3800 RSC.
- A. Larrañaga-Vera, K. S. Toti, J. S. Flatow, A. J. Haraczy, E. Warnick, H. Rao, Z.-G. Gao, S. M. Sussman, A. Mediero, P. Leucht, K. A. Jacobson and B. N. Cronstein, Arthritis Res. Ther., 2022, 24, 265 CrossRef PubMed.
- R. Mukhopadhyay, R. Sen, B. Paul, J. Kazi, S. Ganguly and M. C. Debnath, Pharm. Res., 2020, 37, 56 CrossRef CAS.
- J. Kazi, R. Sen, S. Ganguly, T. Jha, S. Ganguly and M. Chatterjee Debnath, Int. J. Pharm., 2020, 585, 119449 CrossRef CAS PubMed.
- C. Hu, F. Fan, Y. Qin, C. Huang, Z. Zhang, Q. Guo, L. Zhang, X. Pang, W. Ou-Yang, K. Zhao, D. Zhu and L. Zhang, J. Biomed. Nanotechnol., 2018, 14, 2018–2030 CrossRef CAS PubMed.
- M. Zamani, K. Rostamizadeh, H. K. Manjili and H. Danafar, Eur. Polym. J., 2018, 103, 260–270 CrossRef CAS.
- M. Zamani, M. Aghajanzadeh, K. Rostamizadeh, H. K. Manjili, M. Fridoni and H. Danafar, J. Drug Delivery Sci. Technol., 2019, 54, 101283 CrossRef CAS.
- B. Song, S. Wu, W. Li, D. Chen and H. Hu, Pharm. Res., 2020, 37, 242 CrossRef CAS PubMed.
- (a) M. Schulz and W. H. Binder, Macromol. Rapid Commun., 2015, 36, 2031–2041 CrossRef CAS PubMed; (b) V. V. Sheffey, E. B. Siew, E. E. L. Tanner and O. Eniola-Adefeso, Adv. Healthcare Mater., 2022, 11, 2101536 CrossRef CAS PubMed.
- A. Kumar, S. V. Lale, M. R. Aji Alex, V. Choudhary and V. Koul, Colloids Surf., B, 2017, 149, 369–378 CrossRef CAS PubMed.
- S. Sharma, S. S. Pukale, D. K. Sahel, D. S. Agarwal, M. Dalela, S. Mohanty, R. Sakhuja, A. Mittal and D. Chitkara, AAPS PharmSciTech, 2020, 21, 280 CrossRef CAS PubMed.
- (a) T. Şucu and M. P. Shaver, Polym. Chem., 2020, 11, 6397–6412 RSC; (b) A. Domiński, T. Konieczny, K. Duale, M. Krawczyk, G. Pastuch-Gawołek and P. Kurcok, Polymers, 2020, 12, 2890 CrossRef.
- S. Niu, G. R. Williams, J. Wu, J. Wu, X. Zhang, H. Zheng, S. Li and L.-M. Zhu, Chem. Eng. J., 2019, 369, 134–149 CrossRef CAS.
- M. M. Khan, A. Madni, N. Filipczak, J. Pan, M. Rehman, N. Rai, S. A. Attia and V. P. Torchilin, Nanomed. Nanotechnol. Biol. Med., 2020, 28, 102228 CrossRef CAS.
- M. M. El-Hammadi, V. Delgado Á, C. Melguizo, J. C. Prados and J. L. Arias, Int. J. Pharm., 2017, 516, 61–70 CrossRef CAS PubMed.
- N. Raina, R. Rani, A. Khan, K. Nagpal and M. Gupta, Polym. Bull., 2020, 77, 5027–5050 CrossRef CAS.
- V. Raj, P. Priya, R. Renji, M. Suryamathi and S. Kalaivani, Iran. Polym. J., 2018, 27, 721–731 CrossRef CAS.
- (a) F. Lin, X. Lu, Z. Wang, Q. Lu, G. Lin, B. Huang and B. Lu, Cellulose, 2019, 26, 1825–1839 CrossRef CAS; (b) C. O. Crosby, B. Stern, N. Kalkunte, S. Pedahzur, S. Ramesh and J. Zoldan, Rev. Chem. Eng., 2022, 38, 347–361 CrossRef CAS PubMed; (c) N. Naseri, B. Deepa, A. P. Mathew, K. Oksman and L. Girandon, Biomacromolecules, 2016, 17, 3714–3723 CrossRef CAS PubMed.
- W. Song, Y. Zhang, D. G. Yu, C. H. Tran, M. Wang, A. Varyambath, J. Kim and I. Kim, Biomacromolecules, 2021, 22, 732–742 CrossRef CAS PubMed.
- (a) W. Song, Y. Zhang, A. Varyambath and I. Kim, ACS Nano, 2019, 13, 11753–11769 CrossRef CAS PubMed; (b) W. Song, Y. Zhang, A. Varyambath, J. S. Kim and I. Kim, Green Chem., 2020, 22, 3572–3583 RSC.
- D. S. Chauhan, R. Prasad, J. Devrukhkar, K. Selvaraj and R. Srivastava, Bioconjugate Chem., 2018, 29, 1510–1518 CrossRef CAS PubMed.
- Y. H. Dinakar, A. Karole, S. Parvez, V. Jain and S. L. Mudavath, Biochim. Biophys. Acta, Gen. Subj., 2023, 1867, 130396 CrossRef CAS PubMed.
- V. D. Nguyen, H.-K. Min, C.-S. Kim, J. Han, J.-O. Park and E. Choi, Colloids Surf., B, 2019, 173, 539–548 CrossRef CAS PubMed.
- Q. Zhang, J. Zhao, H. Hu, Y. Yan, X. Hu, K. Zhou, S. Xiao, Y. Zhang and N. Feng, Int. J. Pharm., 2019, 569, 118595 CrossRef CAS PubMed.
- N. Kwon, H. Kim, X. Li and J. Yoon, Chem. Sci., 2021, 12, 7248–7268 RSC.
- K. Ding, R. Li, Y. Ma, N. Li, T. Zhang, X. Cheng-Mei, H.-T. Jiang and Y.-K. Gong, Langmuir, 2019, 35, 1257–1265 CrossRef CAS PubMed.
- Y.-K. Gong, L.-P. Liu and P. B. Messersmith, Macromol. Biosci., 2012, 12, 979–985 CrossRef CAS PubMed.
- K. Liang, J. E. Chung, S. J. Gao, N. Yongvongsoontorn and M. Kurisawa, Adv. Mater., 2018, 30, 1706963 CrossRef PubMed.
- T. Ramasamy, P. Sundaramoorthy, H. B. Ruttala, Y. Choi, W. H. Shin, J. H. Jeong, S. K. Ku, H. G. Choi, H. M. Kim, C. S. Yong and J. O. Kim, Drug Delivery, 2017, 24, 1262–1272 CrossRef CAS PubMed.
- N. Hinz and M. Jücker, Cell Commun. Signaling, 2019, 17, 154 CrossRef CAS PubMed.
- Z. C. Soe, R. K. Thapa, W. Ou, M. Gautam, H. T. Nguyen, S. G. Jin, S. K. Ku, K. T. Oh, H. G. Choi, C. S. Yong and J. O. Kim, Colloids Surf., B, 2018, 170, 718–728 CrossRef CAS PubMed.
- H. Zhang, in Liposomes: Methods and Protocols, ed. G. G. M. D'Souza, Springer New York, New York, NY, 2017, pp. 17–22, DOI:10.1007/978-1-4939-6591-5_2.
- Y. Gao, W. Li, J. Chen, X. Wang, Y. Lv, Y. Huang, Z. Zhang and F. Xu, Acta Pharm. Sin. B, 2019, 9, 157–166 CrossRef PubMed.
- J. S. Baek and C. W. Cho, Oncotarget, 2017, 8, 30369–30382 CrossRef PubMed.
- (a) A. Kumar, C. Harsha, D. Parama, S. Girisa, U. D. Daimary, X. Mao and A. B. Kunnumakkara, Phytother. Res., 2021, 35, 6768–6801 CrossRef PubMed; (b) H. Bashang and S. Tamma, Biotechnol. Appl. Biochem., 2020, 67, 171–179 CrossRef CAS PubMed; (c) H. Wang, K. Zhang, J. Liu, J. Yang, Y. Tian, C. Yang, Y. Li, M. Shao, W. Su and N. Song, Front. Oncol., 2021, 11, 660712 CrossRef CAS PubMed.
- (a) D. Waghray and Q. Zhang, J. Med. Chem., 2018, 61, 5108–5121 CrossRef CAS PubMed; (b) P. Famta, S. Shah, E. Chatterjee, H. Singh, B. Dey, S. K. Guru, S. B. Singh and S. Srivastava, Curr. Res. Pharmacol. Drug Discov., 2021, 2, 100054 CrossRef PubMed.
- P. Neerati, Y. A. Sudhakar and J. R. Kanwar, J. Cancer Sci. Ther., 2013, 5, 313–319 Search PubMed.
- N. Poonia, V. Lather, J. K. Narang, S. Beg and D. Pandita, Mater. Sci. Eng. C, 2020, 114, 111016 CrossRef CAS PubMed.
- P. Desai, R. Rimal, S. E. M. Sahnoun, F. M. Mottaghy, M. Möller, A. Morgenroth and S. Singh, Small, 2022, 18, 2200673 CrossRef CAS PubMed.
- J. Pellico, P. J. Gawne and R. T. M. de Rosales, Chem. Soc. Rev., 2021, 50, 3355–3423 RSC.
- M. Silindir-Gunay, M. Karpuz, N. Ozturk, A. Y. Ozer, S. Erdogan and M. Tuncel, J. Drug Delivery Sci. Technol., 2019, 50, 321–328 CrossRef CAS.
- M. R. Edelmann, RSC Adv., 2022, 12, 32383–32400 RSC.
- (a) V. Kozlovskaya, M. Ducharme, M. Dolmat, J. M. Omweri, V. Tekin, S. E. Lapi and E. Kharlampieva, Biomacromolecules, 2023, 24, 1784–1797 CrossRef CAS PubMed; (b) D. Śmiłowicz, S. Eisenberg, S. H. Ahn, A. J. Koller, P. P. Lampkin and E. Boros, Chem. Sci., 2023, 14, 5038–5050 RSC.
- D. Mertz, O. Sandre and S. Bégin-Colin, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1617–1641 CrossRef CAS PubMed.
- P. Kush, P. Kumar, R. Singh and A. Kaushik, Asian J. Pharm. Sci., 2021, 16, 704–737 CrossRef.
- A. Mashhadi Malekzadeh, A. Ramazani, S. J. Tabatabaei Rezaei and H. Niknejad, J. Colloid Interface Sci., 2017, 490, 64–73 CrossRef CAS PubMed.
- M. I. Anik, M. K. Hossain, I. Hossain, A. M. U. B. Mahfuz, M. T. Rahman and I. Ahmed, Nano Sel., 2021, 2, 1146–1186 CrossRef CAS.
- V. Nejadshafiee, H. Naeimi, B. Goliaei, B. Bigdeli, A. Sadighi, S. Dehghani, A. Lotfabadi, M. Hosseini, M. S. Nezamtaheri, M. Amanlou, M. Sharifzadeh and M. Khoobi, Mater. Sci. Eng., C, 2019, 99, 805–815 CrossRef CAS PubMed.
- Z. Zhou, R. Bai, J. Munasinghe, Z. Shen, L. Nie and X. Chen, ACS Nano, 2017, 11, 5227–5232 CrossRef CAS.
- D. Laha, K. Pal, A. R. Chowdhuri, P. K. Parida, S. K. Sahu, K. Jana and P. Karmakar, New J. Chem., 2019, 43, 217–229 RSC.
- K. J. Campbell and S. W. G. Tait, Open Biol., 2018, 8, 180002 CrossRef.
- B. J. Aubrey, G. L. Kelly, A. Janic, M. J. Herold and A. Strasser, Cell Death Differ., 2018, 25, 104–113 CrossRef CAS PubMed.
- A. Angelopoulou, A. Kolokithas-Ntoukas, C. Fytas and K. Avgoustakis, ACS Omega, 2019, 4, 22214–22227 CrossRef CAS PubMed.
- A. A. P. Mansur, H. S. Mansur, A. G. Leonel, I. C. Carvalho, M. C. G. Lage, S. M. Carvalho, K. Krambrock and Z. I. P. Lobato, J. Mater. Chem. B, 2020, 8, 7166–7188 RSC.
- Y. Huang, K. Mao, B. Zhang and Y. Zhao, Mater. Sci. Eng., C, 2017, 70, 763–771 CrossRef CAS PubMed.
- (a) L. Shang, Q.-Y. Wang, K.-L. Chen, J. Qu, Q.-H. Zhou, J.-B. Luo and J. Lin, RSC Adv., 2017, 7, 47715–47725 RSC; (b) F. Fiévet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J. Y. Piquemal, L. Sicard and G. Viau, Chem. Soc. Rev., 2018, 47, 5187–5233 RSC.
- H. Heydari Sheikh Hossein, I. Jabbari, A. Zarepour, A. Zarrabi, M. Ashrafizadeh, A. Taherian and P. Makvandi, Molecules, 2020, 25, 4053 CrossRef.
- A. Montazerabadi, J. Beik, R. Irajirad, N. Attaran, S. Khaledi, H. Ghaznavi and A. Shakeri-Zadeh, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 330–340 CrossRef CAS PubMed.
- C. Saikia, M. K. Das, A. Ramteke and T. K. Maji, Carbohydr. Polym., 2017, 157, 391–399 CrossRef CAS PubMed.
- S. Park, B. B. Cho, J. R. Anusha, S. Jung, C. Justin Raj, B. C. Kim and K. H. Yu, J. Nanosci. Nanotechnol., 2020, 20, 2040–2044 CrossRef CAS PubMed.
- H. Yang, F. Gao, B. McNeil, C. Zhang, Z. Yuan, S. Zeisler, J. Kumlin, J. Zeisler, F. Bénard, C. Ramogida and P. Schaffer, EJNMMI Radiopharm. Chem., 2021, 6, 3 CrossRef.
- T. Le Bihan, C. H. S. Driver, T. Ebenhan, N. Le Bris, J. R. Zeevaart and R. Tripier, ChemMedChem, 2021, 16, 809–821 CrossRef CAS.
- L. Greifenstein, D. Späth, J. P. Sinnes, T. Grus and F. Rösch, Radiochim. Acta, 2020, 108, 555–563 CrossRef CAS.
- T. T. Huynh, S. Sreekumar, C. Mpoy and B. E. Rogers, Oncotarget, 2022, 13, 360–372 CrossRef.
- A. V. F. Massicano, B. V. Marquez-Nostra and S. E. Lapi, Mol. Imaging, 2018, 17, 1536012117745386 CrossRef.
- I. Lee, I. Lim, B. H. Byun, B. I. Kim, C. W. Choi, S.-K. Woo, K. I. Kim, K. C. Lee, J. H. Kang, M.-K. Seong, H.-A. Kim, W. C. Noh and S. M. Lim, EJNMMI Res., 2021, 11, 8 CrossRef CAS PubMed.
- L. M. Kenny, F. J. Gilbert, G. Gopalakrishnan, P. Aravind, T. Barwick, N. Patel, D. R. Hiscock, I. Boros, S. Kealey, F. I. Aigbirhio, J. Lozano-kuehne, S. J. Cleator, B. Fleming, P. Riddle, R. Ahmad, S. Chua, S. R. D. Johnston, J. Mansi, G. J. Cook and E. O. Aboagye, J. Clin. Oncol., 2022, 40, 3069 CrossRef.
- P. Gao, X. Chang, D. Zhang, Y. Cai, G. Chen, H. Wang and T. Wang, Acta Pharm. Sin. B, 2021, 11, 1175–1199 CrossRef CAS PubMed.
- (a) E. Domínguez-Álvarez, B. Rácz, M. A. Marć, M. J. Nasim, N. Szemerédi, J. Viktorová, C. Jacob and G. Spengler, Drug Resistance Updates, 2022, 63, 100844 CrossRef; (b) V. Chandrakala, V. Aruna and G. Angajala, Emergent Mater., 2022, 5, 1593–1615 CrossRef CAS.
- W. Li, Z. Cao, R. Liu, L. Liu, H. Li, X. Li, Y. Chen, C. Lu and Y. Liu, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 4222–4233 CrossRef CAS.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- R. Agabeigi, S. H. Rasta, M. Rahmati-Yamchi, R. Salehi and E. Alizadeh, Nanoscale Res. Lett., 2020, 15, 62 CrossRef CAS PubMed.
- H. Shamshad, R. Bakri and A. Z. Mirza, Mol. Biol. Rep., 2022, 49, 6659–6691 CrossRef CAS PubMed.
- S. Song, B. Tian, M. Zhang, X. Gao, L. Jie, P. Liu and J. Li, Clin. Exp. Pharmacol. Physiol., 2021, 48, 279–287 CrossRef CAS PubMed.
- J. Akinyelu and M. Singh, Appl. Nanosci., 2019, 9, 7–17 CrossRef CAS.
- G. Mani, S. Kim and K. Kim, Biomacromolecules, 2018, 19, 3257–3267 CrossRef CAS PubMed.
- R. Javed, M. Zia, S. Naz, S. O. Aisida, N. u. Ain and Q. Ao, J. Nanobiotechnol., 2020, 18, 172 CrossRef PubMed.
- S. S. D. Kumar, A. Mahesh, M. G. Antoniraj, H. S. Rathore, N. N. Houreld and R. Kandasamy, Int. J. Biol. Macromol., 2018, 109, 220–230 CrossRef CAS PubMed.
- M. Azmanova and A. Pitto-Barry, ChemBioChem, 2022, 23, e202100641 CrossRef CAS PubMed.
- D. Tang, R. Kang, T. V. Berghe, P. Vandenabeele and G. Kroemer, Cell Research, 2019, 29, 347–364 CrossRef CAS PubMed.
- A. V. A. Mariadoss, K. Saravanakumar, A. Sathiyaseelan, K. Venkatachalam and M. H. Wang, Int. J. Biol. Macromol., 2020, 164, 2073–2084 CrossRef CAS PubMed.
- K. S. Siddiqi and A. Husen, Biomater. Res., 2020, 24, 11 CrossRef CAS PubMed.
- M. Izci, C. Maksoudian, B. B. Manshian and S. J. Soenen, Chem. Rev., 2021, 121, 1746–1803 CrossRef CAS PubMed.
- (a) A. Lath, A. R. Santal, N. Kaur, P. Kumari and N. P. Singh, Biotechnol. Genet. Eng. Rev., 2022, 1–40, DOI:10.1080/02648725.2022.2082157; (b) E. Kluza, D. W. J. van der Schaft, P. A. I. Hautvast, W. J. M. Mulder, K. H. Mayo, A. W. Griffioen, G. J. Strijkers and K. Nicolay, Nano Lett., 2010, 10, 52–58 CrossRef CAS PubMed.
- A. Pudlarz and J. Szemraj, Open Life Sci., 2018, 13, 285–298 CAS.
- G. Wei, Y. Wang, X. Huang, H. Hou and S. Zhou, Small Methods, 2018, 2, 1700358 CrossRef.
- (a) S. Hong, D. W. Choi, H. N. Kim, C. G. Park, W. Lee and H. H. Park, Pharmaceutics, 2020, 12, 604 CrossRef CAS PubMed; (b) A. Varanko, S. Saha and A. Chilkoti, Adv. Drug Delivery Rev., 2020, 156, 133–187 CrossRef CAS PubMed; (c) Y. Miao, T. Yang, S. Yang, M. Yang and C. Mao, Nano Convergence, 2022, 9, 2 CrossRef CAS PubMed; (d) A. Jain, S. K. Singh, S. K. Arya, S. C. Kundu and S. Kapoor, ACS Biomater. Sci. Eng., 2018, 4, 3939–3961 CrossRef CAS PubMed.
- Z. Kayani, A.-K. Bordbar and O. Firuzi, Biomed. Pharmacother., 2018, 107, 945–956 CrossRef CAS PubMed.
- A. Akbarian, M. Ebtekar, N. Pakravan and Z. M. Hassan, Int. J. Biol. Macromol., 2020, 152, 90–101 CrossRef CAS PubMed.
- Y. Dong, R. Fu, J. Yang, P. Ma, L. Liang, Y. Mi and D. Fan, Int. J. Nanomed., 2019, 14, 6971–6988 CrossRef CAS PubMed.
- D. C. Carter and J. X. Ho, in Advances in Protein Chemistry, ed. C. B. Anfinsen, J. T. Edsall, F. M. Richards and D. S. Eisenberg, Academic Press, 1994, vol. 45, pp. 153–203 Search PubMed.
- S. Kunjiappan, S. Govindaraj, P. Parasuraman, M. Sankaranarayanan, S. Arunachalam, P. Palanisamy, U. P. Mohan, E. Babkiewicz, P. Maszczyk, S. Vellaisamy and T. Panneerselvam, Nanotechnology, 2020, 31, 155102 CrossRef CAS PubMed.
- H. Nosrati, R. Abbasi, J. Charmi, A. Rakhshbahar, F. Aliakbarzadeh, H. Danafar and S. Davaran, Int. J. Biol. Macromol., 2018, 117, 1125–1132 CrossRef CAS PubMed.
- (a) J. Ahlawat, S. Masoudi Asil, G. Guillama Barroso, M. Nurunnabi and M. Narayan, Biomater. Sci., 2021, 9, 626–644 RSC; (b) A. Mokhtari-Farsani, M. Hasany, I. Lynch and M. Mehrali, Adv. Funct. Mater., 2022, 32, 2105649 CrossRef CAS; (c) K. D. Patel, R. K. Singh and H.-W. Kim, Mater. Horiz., 2019, 6, 434–469 RSC.
- A. A. Nayl, A. I. Abd-Elhamid, A. A. Aly and S. Bräse, RSC Adv., 2022, 12, 13706–13726 RSC.
- N. Kumar, P. Chamoli, M. Misra, M. K. Manoj and A. Sharma, Nanoscale, 2022, 14, 3987–4017 RSC.
- (a) A. Jafari, K. Khanmohammadi Chenab, H. Malektaj, F. Farshchi, S. Ghorbani, A. Ghasemiamineh, M. Khoshakhlagh, B. Ashtari and M.-R. Zamani-Meymian, FlatChem, 2022, 100381, DOI:10.1016/j.flatc.2022.100381; (b) E. M. Pérez and N. Martín, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC.
- H. Tonbul, A. Sahin, E. Tavukcuoglu, G. Ultav, S. Akbas, Y. Aktas, G. Esendaglı and Y. Capan, J. Drug Delivery Sci. Technol., 2021, 63, 102535 CrossRef CAS.
- S. Z. Hosseinabadi, S. Safari, M. Mirzaei, E. Mohammadi, S. M. Amini and B. Mehravi, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2020, 11, 045010 CAS.
- M. Serda, K. Malarz, J. Korzuch, M. Szubka, M. Zubko and R. Musioł, ACS Biomater. Sci. Eng., 2022, 8, 3450–3462 CrossRef CAS PubMed.
- H. Wang, Y. Liang, Y. Yin, J. Zhang, W. Su, A. M. White, J. Bin, J. Xu, Y. Zhang, S. Stewart, X. Lu and X. He, Nat. Commun., 2021, 12, 312 CrossRef CAS PubMed.
- Y. T. Fong, C. H. Chen and J. P. Chen, Nanomaterials, 2017, 7 CrossRef CAS PubMed.
- X. Xu, Y. Liu, W. Fu, M. Yao, Z. Ding, J. Xuan, D. Li, S. Wang, Y. Xia and M. Cao, Polymers, 2020, 12, 580 CrossRef CAS PubMed.
- P. Bhawal, S. Ganguly, T. K. Chaki and N. C. Das, RSC Adv., 2016, 6, 20781–20790 RSC.
- F. Ofridam, M. Tarhini, N. Lebaz, É. Gagnière, D. Mangin and A. Elaissari, Polym. Adv. Technol., 2021, 32, 1455–1484 CrossRef CAS.
- K. Vinothini, N. K. Rajendran, A. Ramu, N. Elumalai and M. Rajan, Biomed. Pharmacother., 2019, 110, 906–917 CrossRef PubMed.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497–7505 CrossRef CAS PubMed.
- X. Zhao, J. Zhang, L. Shi, M. Xian, C. Dong and S. Shuang, RSC Adv., 2017, 7, 42159–42167 RSC.
- Y. Jiao, H. Sun, Y. Jia, Y. Liu, Y. Gao, M. Xian, S. Shuang and C. Dong, Microchem. J., 2019, 146, 464–470 CrossRef CAS.
- B. B. Karakoçak, A. Laradji, T. Primeau, M. Y. Berezin, S. Li and N. Ravi, ACS Appl. Mater. Interfaces, 2021, 13, 277–286 CrossRef PubMed.
- J. R. Aguilar Cosme, H. E. Bryant and F. Claeyssens, PLoS One, 2019, 14, e0220210 CrossRef CAS PubMed.
- H. Liu, Z. Li, Y. Sun, X. Geng, Y. Hu, H. Meng, J. Ge and L. Qu, Sci. Rep., 2018, 8, 1086 CrossRef PubMed.
- J. Zhang, X. Zhao, M. Xian, C. Dong and S. Shuang, Talanta, 2018, 183, 39–47 CrossRef CAS PubMed.
- A. Khoshnood, N. Farhadian, K. Abnous, M. M. Matin, N. Ziaee and E. Yaghoobi, J. Photochem. Photobiol., A, 2023, 444, 114972 CrossRef CAS.
- S. J. Bansal S, U. Kumari, I. P. Kaur, R. P. Barnwal, R. Kumar, S. Singh, G. Singh and M. Chatterjee, Int. J. Nanomed., 2019, 4, 809–818 CrossRef PubMed.
- S. Feng, J. Pan, C. Li and Y. Zheng, Nanotechnology, 2020, 31, 135701 CrossRef CAS PubMed.
- S. Kadian, G. Manik, N. Das and P. Roy, Microchim. Acta, 2020, 187, 458 CrossRef CAS PubMed.
- Y. P. Wu, J. Yang, H. Y. Gao, Y. Shen, L. X. Jiang, C. R. Zhou, Y. F. Li, R. R. He and M. X. Liu, ACS Appl. Nano Mater., 2018, 1, 595–608 CrossRef CAS.
- (a) G. Choi, N. S. Rejinold, H. Piao and J.-H. Choy, Chem. Sci., 2021, 12, 5044–5063 RSC; (b) H. Tian, T. Zhang, S. Qin, Z. Huang, L. Zhou, J. Shi, E. C. Nice, N. Xie, C. Huang and Z. Shen, J. Hematol. Oncol., 2022, 15, 132 CrossRef PubMed.
- A. Jahangiri-Manesh, M. Mousazadeh, S. Taji, A. Bahmani, A. Zarepour, A. Zarrabi, E. Sharifi and M. Azimzadeh, Pharmaceutics, 2022, 14, 664 CrossRef CAS PubMed.
- (a) D.-K. Lim, A. Barhoumi, R. G. Wylie, G. Reznor, R. S. Langer and D. S. Kohane, Nano Lett., 2013, 13, 4075–4079 CrossRef CAS PubMed; (b) W. Yang, B. Xia, L. Wang, S. Ma, H. Liang, D. Wang and J. Huang, Mater. Today Sustain., 2021, 13, 100078 CrossRef.
- G. Chauhan, V. Chopra, A. Tyagi, G. Rath, R. K. Sharma and A. K. Goyal, Eur. J. Pharm. Sci., 2017, 96, 351–361 CrossRef CAS PubMed.
- M. Ma, H. Li, Y. Xiong and F. Dong, Mater. Des., 2021, 198, 109367 CrossRef CAS.
- S. Malekmohammadi, H. Hadadzadeh, H. Farrokhpour and Z. Amirghofran, Soft Matter, 2018, 14, 2400–2410 RSC.
- S. K. Maji, S. Sreejith, A. K. Mandal, X. Ma and Y. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 13648–13656 CrossRef CAS PubMed.
- X. Liu, X. Wu, Y. Xing, Y. Zhang, X. Zhang, Q. Pu, M. Wu and J. X. Zhao, ACS Appl. Bio Mater., 2020, 3, 2577–2587 CrossRef CAS PubMed.
- H. Ghaznavi, S. Hosseini-Nami, S. K. Kamrava, R. Irajirad, S. Maleki, A. Shakeri-Zadeh and A. Montazerabadi, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1594–1604 CAS.
- R. Prasad, S. B. Agawane, D. S. Chauhan, R. Srivastava and K. Selvaraj, Bioconjugate Chem., 2018, 29, 4012–4019 CrossRef CAS PubMed.
- A. Hemati Azandaryani, S. Kashanian and K. Derakhshandeh, Pharm. Res., 2017, 34, 2798–2808 CrossRef CAS PubMed.
- S. W. El-Far, M. W. Helmy, S. N. Khattab, A. A. Bekhit, A. A. Hussein and A. O. Elzoghby, Nanomedicine, 2018, 13, 1463–1480 CrossRef CAS PubMed.
- H. Yan, Y. You, X. Li, L. Liu, F. Guo, Q. Zhang, D. Liu, Y. Tong, S. Ding and J. Wang, Front. Pharmacol, 2020, 11, 898 CrossRef CAS PubMed.
- Z. Shen, Y. Li, K. Kohama, B. Oneill and J. Bi, Pharmacol. Res., 2011, 63, 51–58 CrossRef CAS PubMed.
- M. W. Dewhirst and T. W. Secomb, Nat. Rev. Cancer, 2017, 17, 738–750 CrossRef CAS PubMed.
- R. Villaseñor, J. Lampe, M. Schwaninger and L. Collin, Cell. Mol. Life Sci., 2019, 76, 1081–1092 CrossRef PubMed.
- C. L. Yang, J. P. Chen, K. C. Wei, J. Y. Chen, C. W. Huang and Z. X. Liao, Nanomaterials, 2017, 7, 85 CrossRef CAS PubMed.
- R. Afzalipour, S. Khoei, S. Khoee, S. Shirvalilou, N. Jamali Raoufi, M. Motevalian and M. R. Karimi, ACS Biomater. Sci. Eng., 2019, 5, 6000–6011 CrossRef CAS PubMed.
- S. Emamgholizadeh Minaei, S. Khoei, S. Khoee and M. R. Karimi, Int. J. Biochem. Cell Biol., 2019, 108, 72–83 CrossRef CAS PubMed.
- S. E. Minaei, S. Khoei, S. Khoee, F. Vafashoar and V. P. Mahabadi, Mater. Sci. Eng., C, 2019, 101, 575–587 CrossRef CAS PubMed.
- Z. Liang, Y. Yang, F. Jia, K. Sai, S. Ullah, C. Fidelis, Z. Lin and F. Li, ACS Appl. Bio Mater., 2019, 2, 1432–1439 CrossRef CAS PubMed.
- L. V. Nair, S. S. Nazeer, R. S. Jayasree and A. Ajayaghosh, ACS Nano, 2015, 9, 5825–5832 CrossRef CAS PubMed.
- Y. C. Kuo, Y. H. Chang and R. Rajesh, Mater. Sci. Eng., C, 2019, 96, 114–128 CrossRef CAS PubMed.
- Y.-C. Kuo and S.-J. Cheng, Int. J. Pharm., 2016, 499, 10–19 CrossRef CAS PubMed.
- Z. H. Marfavi, M. Farhadi, S. B. Jameie, M. Zahmatkeshan, V. Pirhajati and M. Jameie, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 2783–2790 CrossRef CAS PubMed.
- S. H. Kang, S. P. Hong and B. S. Kang, Int. J. Radiat. Biol., 2018, 94, 1006–1016 CrossRef CAS PubMed.
- M. A. Dymova, S. Y. Taskaev, V. A. Richter and E. V. Kuligina, Cancer Commun., 2020, 40, 406–421 CrossRef PubMed.
- A. Singh, B. K. Kim, Y. Mackeyev, P. Rohani, S. D. Mahajan, M. T. Swihart, S. Krishnan and P. N. Prasad, J. Biomed. Nanotechnol., 2019, 15, 1714–1723 CrossRef CAS PubMed.
- J. Chen, Q. Dai, Q. Yang, X. Bao, Y. Zhou, H. Zhong, L. Wu, T. Wang, Z. Zhang, Y. Lu, Z. Zhang, M. Lin, M. Han and Q. Wei, J. Nanobiotechnol., 2022, 20, 102 CrossRef CAS PubMed.
- P. K. Bolla, V. Gote, M. Singh, V. K. Yellepeddi, M. Patel, D. Pal, X. Gong, D. Sambalingam and J. Renukuntla, J. Microencapsulation, 2020, 37, 502–516 CrossRef CAS PubMed.
- J. Keyvan Rad, A. R. Mahdavian, S. Khoei and S. Shirvalilou, ACS Appl. Mater. Interfaces, 2018, 10, 19483–19493 CrossRef CAS PubMed.
- L. Shan, X. Zhuo, F. Zhang, Y. Dai, G. Zhu, B. C. Yung, W. Fan, K. Zhai, O. Jacobson, D. O. Kiesewetter, Y. Ma, G. Gao and X. Chen, Theranostics, 2018, 8, 2018–2030 CrossRef CAS PubMed.
- (a) O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2016, 27, 2239–2247 CrossRef CAS PubMed; (b) J.-P. Richalet, D. Marchant, J.-L. Macarlupu and N. Voituron, Ann. Biomed. Eng., 2018, 46, 2189–2195 CrossRef PubMed; (c) R. Tian, O. Jacobson, G. Niu, D. O. Kiesewetter, Z. Wang, G. Zhu, Y. Ma, G. Liu and X. Chen, Theranostics, 2018, 8, 735–745 CrossRef CAS PubMed.
- X. Chen, H. Liu, A. Li, S. Ji and H. Fei, J. Biol. Chem., 2021, 297, 101364 CrossRef CAS PubMed.
- L. Gui, X.-H. Zhang, Z.-Y. Qiao and H. Wang, ChemNanoMat, 2020, 6, 1138–1148 CrossRef CAS.
- Y. Dai, X. Cai, X. Bi, C. Liu, N. Yue, Y. Zhu, J. Zhou, M. Fu, W. Huang and H. Qian, Eur. J. Med. Chem., 2019, 171, 104–115 CrossRef CAS PubMed.
- P. Guzik, M. Benesova, M. Ratz, J. M. Monne Rodriguez, L. M. Deberle, R. Schibli and C. Muller, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 972–983 CrossRef CAS PubMed.
- K. Siwowska, S. Haller, F. Bortoli, M. Benešová, V. Groehn, P. Bernhardt, R. Schibli and C. Müller, Mol. Pharm., 2017, 14, 523–532 CrossRef CAS PubMed.
- P. Guzik, K. Siwowska, H. Y. Fang, S. Cohrs, P. Bernhardt, R. Schibli and C. Müller, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 984–994 CrossRef CAS PubMed.
- H. Raskov, A. Orhan, J. P. Christensen and I. Gögenur, Br. J. Cancer, 2021, 124, 359–367 CrossRef CAS PubMed.
- G. Marverti, C. Marraccini, A. Martello, D. D'Arca, S. Pacifico, R. Guerrini, F. Spyrakis, G. Gozzi, A. Lauriola, M. Santucci, G. Cannazza, L. Tagliazucchi, A. S. Cazzato, L. Losi, S. Ferrari, G. Ponterini and M. P. Costi, J. Med. Chem., 2021, 64, 3204–3221 CrossRef CAS PubMed.
- (a) F. Krutzek, C. K. Donat, M. Ullrich, K. Zarschler, M.-C. Ludik, A. Feldmann, L. R. Loureiro, K. Kopka and S. Stadlbauer, Cancers, 2023, 15, 2638 CrossRef CAS PubMed; (b) Y. Miao, G. Lv, Y. Chen, L. Qiu, M. Xie and J. Lin, Bioorg. Med. Chem. Lett., 2020, 30, 127572 CrossRef CAS PubMed.
- X. Wen, P. Xu, M. Shi, J. Liu, X. Zeng, Y. Zhang, C. Shi, J. Li, Z. Guo, X. Zhang, P.-L. Khong and X. Chen, Theranostics, 2022, 12, 422–433 CrossRef CAS PubMed.
- F. M. F. Santos, A. I. Matos, A. E. Ventura, J. Gonçalves, L. F. Veiros, H. F. Florindo and P. M. P. Gois, Angew. Chem., Int. Ed., 2017, 56, 9346–9350 CrossRef CAS PubMed.
- Y. Wei, L. Quan, C. Zhou and Q. Zhan, Nanomed.: Nanotechnol. Biol. Med., 2018, 13, 1495–1512 CrossRef CAS PubMed.
- M. J. Ernsting, M. Murakami, A. Roy and S.-D. Li, J. Controlled Release, 2013, 172, 782–794 CrossRef CAS PubMed.
- R. Ge, Z. Wang and L. Cheng, npj Precis. Oncol., 2022, 6, 31 CrossRef CAS PubMed.
- J. Ren, N. Andrikopoulos, K. Velonia, H. Tang, R. Cai, F. Ding, P. C. Ke and C. Chen, J. Am. Chem. Soc., 2022, 144, 9184–9205 CrossRef CAS PubMed.
- (a) A. Abdelkhaliq, M. van der Zande, A. Punt, R. Helsdingen, S. Boeren, J. J. M. Vervoort, I. Rietjens and H. Bouwmeester, J. Nanobiotechnol., 2018, 16, 70 CrossRef PubMed; (b) Y. Arezki, F. Delalande, C. Schaeffer-Reiss, S. Cianférani, M. Rapp, L. Lebeau, F. Pons and C. Ronzani, Nanoscale, 2022, 14, 14695–14710 RSC.
- A. Tomak, S. Cesmeli, B. D. Hanoglu, D. Winkler and C. Oksel Karakus, Nanotoxicology, 2021, 15, 1331–1357 CrossRef CAS PubMed.
- Z. Wang and J. S. Brenner, AAPS J., 2021, 23, 105 CrossRef CAS PubMed.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 CrossRef CAS PubMed.
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. Shi and O. C. Farokhzad, J. Controlled Release, 2014, 190, 485–499 CrossRef CAS PubMed.
- (a) W. Zhen, S. An, S. Wang, W. Hu, Y. Li, X. Jiang and J. Li, Adv. Mater., 2021, 33, 2101572 CrossRef CAS PubMed; (b) M. M. Hegde, S. Prabhu, S. Mutalik, A. Chatterjee, J. S. Goda and B. S. Satish Rao, J. Pharm. Invest., 2022, 52, 49–74 CrossRef CAS.
- A. Saminathan, M. Zajac, P. Anees and Y. Krishnan, Nat. Rev. Mater., 2022, 7, 355–371 CrossRef.
- M. Ajdary, M. A. Moosavi, M. Rahmati, M. Falahati, M. Mahboubi, A. Mandegary, S. Jangjoo, R. Mohammadinejad and R. S. Varma, Nanomaterials, 2018, 8, 634 CrossRef PubMed.
- (a) S. Sharma, R. Parveen and B. P. Chatterji, Curr. Pathobiol. Rep., 2021, 9, 133–144 CrossRef CAS PubMed; (b) S. Hua, M. B. C. de Matos, J. M. Metselaar and G. Storm, Front. Pharmacol, 2018, 9, 790 CrossRef PubMed.
- W. Najahi-Missaoui, R. D. Arnold and B. S. Cummings, Int. J. Mol. Sci., 2021, 22, 385 CrossRef CAS PubMed.
- P. A. Chiarelli, R. A. Revia, Z. R. Stephen, K. Wang, M. Jeon, V. Nelson, F. M. Kievit, J. Sham, R. G. Ellenbogen, H.-P. Kiem and M. Zhang, ACS Nano, 2017, 11, 9514–9524 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |