
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConsecutive π-Lewis acidic metal-catalysed cyclisation/photochemical radical addition promoted by in situ generated 2-benzopyrylium as the photoredox catalyst†
Masahiro
Terada
 *,
Ryohei
Yazaki
,
Ren
Obayashi
,
Zen
Iwasaki
,
Shigenobu
Umemiya
and
Jun
Kikuchi‡
*,
Ryohei
Yazaki
,
Ren
Obayashi
,
Zen
Iwasaki
,
Shigenobu
Umemiya
and
Jun
Kikuchi‡
Department of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai, Miyagi 980-8578, Japan. E-mail: mterada@tohoku.ac.jp
First published on 19th March 2024
Abstract
A π-Lewis acidic metal-catalysed cyclisation/photochemical radical addition sequence was developed, which utilises in situ generated 2-benzopyrylium cation intermediates as photoredox catalysts and electrophilic substrates to form 1H-isochromene derivatives in good yields in most cases. The key 2-benzopyrylium intermediates were generated through the activation of the alkyne moiety of ortho-carbonyl alkynylbenzene derivatives by such π-Lewis acidic metal catalysts as AgNTf2 and Cu(NTf2)2, and the subsequent intramolecular cyclisation and proto-demetalation using trifluoroacetic acid. Further photo-excitation of the 2-benzopyrylium intermediates facilitated single-electron transfer from a benzyltrimethylsilane derivative as a donor molecule to promote the radical addition of arylmethyl radicals to the 2-benzopyrylium intermediates.
Introduction
Photo-excited compounds exhibit redox properties and high reactivity by transitioning from the ground state to the excited state after absorbing light energy, resulting in a large redox potential. Light-absorbing molecules for the photo-oxidation reaction are primarily divided into metal cations and organic cations. Organic cations are good electron acceptors and thus the excited ones have a larger oxidation potential and a shorter lifetime than excited metal cations, making them potentially applicable to substrates that cannot be oxidized by metal cations.1 Among organic cations, iminium salts have long been used in photoreactions.2,3 Meanwhile photochemical radical reactions initiated by organic cations as catalysts have been actively developed during the past decade and are based on two methods: one is the use of organic cations as photocatalysts, and the other is the catalytic generation of cationic species in situ for use as photo-oxidation agents. Several candidates for organic cations1c,4 are used as photocatalysts, with acridinium5 and pyrylium6 salts as representative examples.On the other hand, examples of methods for catalytically generating organic cations and using them as photo-excitation species include secondary amine catalysis for the formation of iminium cations7 and acid catalysis for the formation of benzopyrylium cations8 (Scheme 1). In 2017, Melchiorre and co-workers reported an enantioselective benzylation that used toluene derivatives, which involved the photo-excitation of an iminium cation intermediate generated in situ from a chiral secondary amine catalyst and α,β-unsaturated aldehydes (Scheme 1a).7a In 2021, we reported a method for activating the benzylic C–H bond of toluene derivatives by photo-excitation of a 1-benzopyrylium cation intermediate generated in situ from a chromenol derivative using a Brønsted acid catalyst (Scheme 1b).8 In this work, we sought to develop an enantioselective variant using a chiral phosphoric acid (CPA) catalyst. However, the catalytic generation of organic cations used as oxidants by photo-excitation is mainly limited to the above two methods. The scope of organic cations generated under catalytic conditions is still narrow even though cationic species are readily available through various methods.9 Therefore, it would be a more attractive approach if the catalytically generated organic cations could be used to broaden the range of cationic species applicable to photochemical reactions.
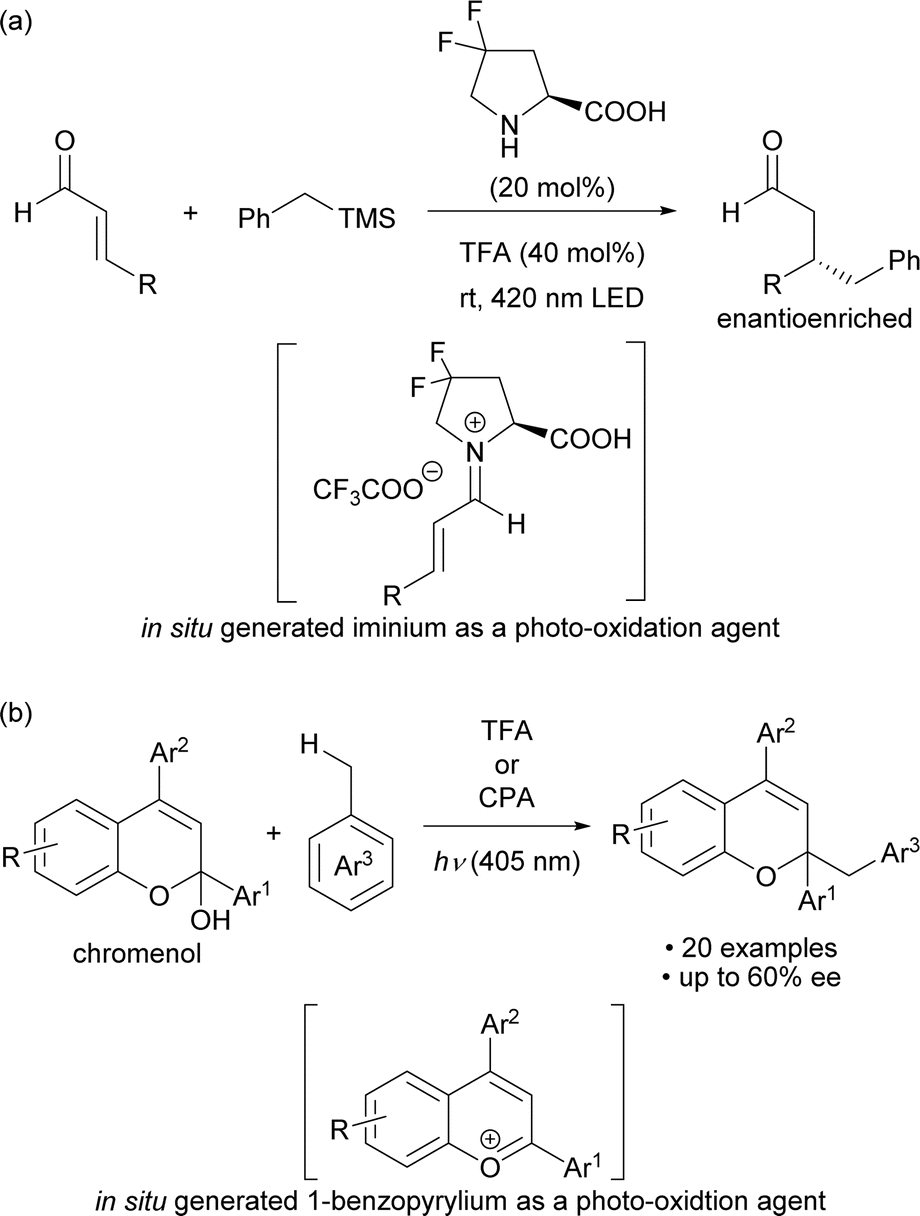 | ||
| Scheme 1 (a) Photochemical enantioselective benzylation of an iminium cation intermediate generated in situ from a chiral secondary amine catalyst and an α,β-unsaturated aldehyde. (b) Photochemical reaction of 1-benzopyrylium cations generated in situ from a chromenol derivative using a Brønsted acid catalyst. | ||
The intramolecular cyclisation reaction of alkynylbenzene derivatives having a carbonyl or related functional group at the ortho-position, which is initiated by the activation of the alkyne moiety using a π-Lewis acidic metal catalyst ([M]X), is a typical method for constructing heterocyclic skeletons (Scheme 2a).10 The cationic intermediates produced in this cyclisation reaction are trapped by nucleophiles, enabling the construction of functionalised heterocycles in a single step. Based on this background, we envisioned the use of 2-benzopyrylium cation intermediates A as new photo-oxidation agents, which are generated in situ through the activation of the alkyne moiety of ortho-carbonyl alkynylbenzene derivatives 1 by [M]X and subsequent intramolecular cyclisation and proto-demetalation by HA (proton source) (Scheme 2b). Herein we report a π-Lewis acidic metal-catalysed cyclisation/photochemical radical addition sequence using 1 as the cation precursor. The use of benzyltrimethylsilane derivatives 2 as the donor molecules11 for the single-electron transfer (SET) to catalytically generated 2-benzopyrylium intermediates A under light irradiation results in the formation of radical cations B for further transformations. The nucleophilic nature of radicals C, which are generated by desilylation from corresponding radical cations B, facilitates radical addition to 2-benzopyrylium intermediates A, giving rise to corresponding 1H-isochromene derivatives 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 through further reduction by SET from radical intermediates D. Meanwhile the SET oxidises D to regenerate 2-benzopyrylium intermediates A that function not only as electrophilic substrates but also as photoredox catalysts.
12 through further reduction by SET from radical intermediates D. Meanwhile the SET oxidises D to regenerate 2-benzopyrylium intermediates A that function not only as electrophilic substrates but also as photoredox catalysts.
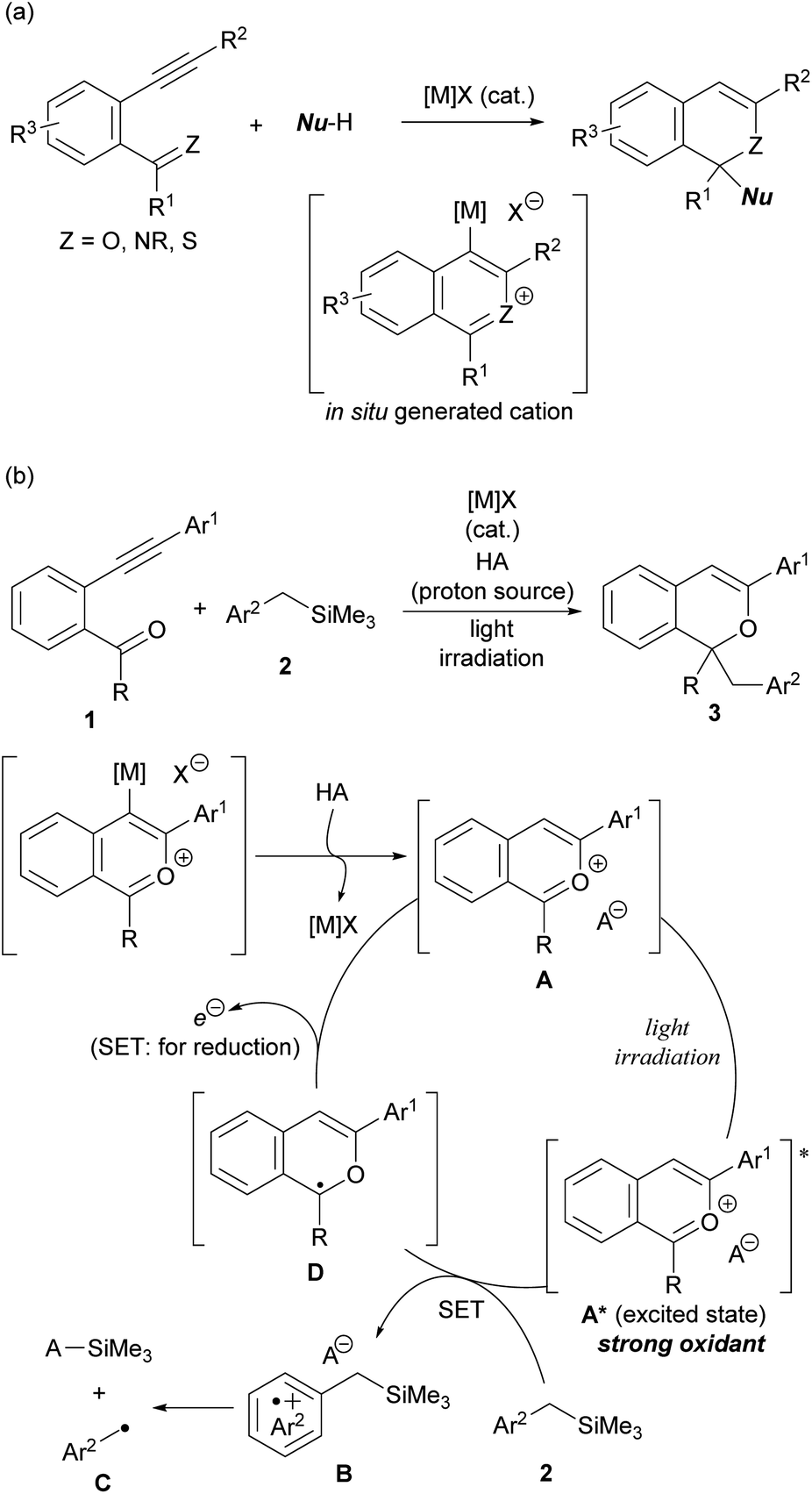 | ||
| Scheme 2 (a) π-Lewis acidic metal-catalysed cyclisation of alkynylbenzene derivatives having a C = Z functional group at the ortho-position. (b) Consecutive π-Lewis acidic metal-catalysed cyclisation/photochemical radical addition promoted by in situ generated 2-benzopyrylium intermediates A as photoredox catalysts. | ||
Results and discussion
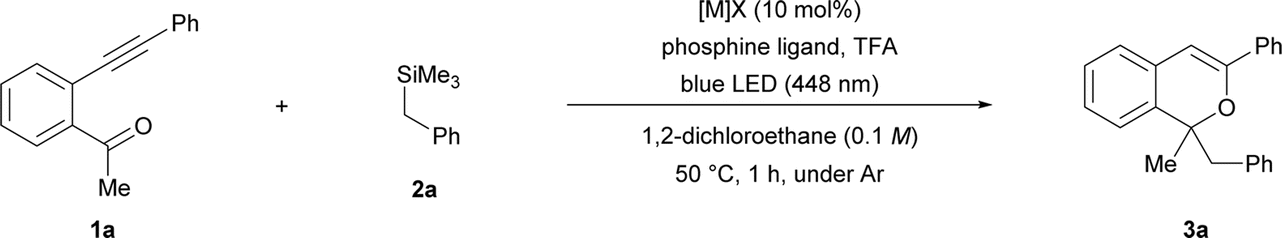
At the outset of our studies to verify the intended consecutive transformation, AgNTf2 was used as the π-Lewis acidic metal catalyst for promoting the cyclisation of ortho-alkynyl acetophenone 1a, and subsequent proto-demetalation was performed using trifluoroacetic acid (TFA) to generate the 2-benzopyrylium intermediate. The initial screening was conducted using 10 μmol (10 mol%) of AgNTf2, 0.1 mmol of 1a, 0.25 mmol (2.5 equiv.) of benzyltrimethylsilane (2a) as the donor molecule, and 0.25 mmol (2.5 equiv.) of TFA under light irradiation (blue LED: λmax = 448 nm) at 0 °C for 1 h in 1 mL of dichloromethane. As shown in Table 1, entry 1, the reaction conducted in the absence of a phosphine ligand afforded corresponding product 3a in low yield, and significant deposition of silver was observed during the reaction. In order to suppress the deposition, we added a phosphine ligand to stabilize the silver salt (entries 2–4), and the yield of 3a was slightly improved even though it was dependent on the electronic properties of the phosphine ligand. Further screening of reaction conditions using 20 mol% of P(4-CF3C6H4)3 as the ligand was conducted by elevating the reaction temperature to 50 °C (see ESI† for screening details) and increasing the amounts of TFA or 2a (entries 5–8). Increasing the amounts of TFA and 2a significantly improved the chemical yield of 3a (entry 7),§ although a further increase in the amount of TFA lowered the yield (entry 8). When AgNTf2 was replaced by other π-Lewis acidic metal catalysts such as copper and gold complexes (entries 9–13), Cu(NTf2)2 exhibited a comparable result to AgNTf2 (entry 7 vs. 10), affording corresponding product 3a in good yield.§ When the light source was changed from λmax = 448 nm to 405 nm, a marked reduction in chemical yield was observed (entry 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | [M]X | Phosphine ligand (mol%) | Equiv. of TFA | Equiv. of 2a | Yieldb (%) |
| a Unless otherwise specified, all reactions were carried out using 0.1 mmol of 1a, the indicated amount of 2a, 10 μmol (10 mol%) of metal catalyst [M]X, 20 μmol (20 mol%) of the phosphine ligand, and TFA in 1,2-dichloroethane (1 mL: 0.1 M of 1a) at 50 °C for 1 h. b Yield was calculated by NMR analysis using 1,1,2,2-tetrabromoethane as the internal standard. Isolated yield is indicated in parentheses. c In dichloromethane at 0 °C. d Purple LED (λmax = 405 nm) was used. | |||||
| 1c | AgNTf2 | — | 2.5 | 2.5 | 28 |
| 2c | AgNTf2 | PPh3 (20) | 2.5 | 2.5 | 37 |
| 3c | AgNTf2 | P(C6F5)3 (20) | 2.5 | 2.5 | 23 |
| 4c | AgNTf2 | P(4-CF3C6H4)3 (20) | 2.5 | 2.5 | 43 |
| 5 | AgNTf2 | P(4-CF3C6H4)3 (20) | 2.5 | 2.5 | 50 |
| 6 | AgNTf2 | P(4-CF3C6H4)3 (20) | 2.5 | 10 | 65 |
| 7 | AgNTf 2 | P(4-CF 3 C 6 H 4 ) 3 (20) | 5 | 10 | 76 (81) |
| 8 | AgNTf2 | P(4-CF3C6H4)3 (20) | 10 | 10 | 64 |
| 9 | AgOTf | P(4-CF3C6H4)3 (20) | 5 | 10 | 71 |
| 10 | Cu(NTf 2 ) 2 | P(4-CF 3 C 6 H 4 ) 3 (20) | 5 | 10 | 78 |
| 11 | Cu(OTf)2 | P(4-CF3C6H4)3 (20) | 5 | 10 | 31 |
| 12 | CuOTf·C6H6 | P(4-CF3C6H4)3 (20) | 5 | 10 | 34 |
| 13 | AuNTf2 | P(4-CF3C6H4)3 (10) | 2.5 | 10 | 38 |
| 14d | AgNTf2 | P(4-CF3C6H4)3 (20) | 5 | 10 | 17 |
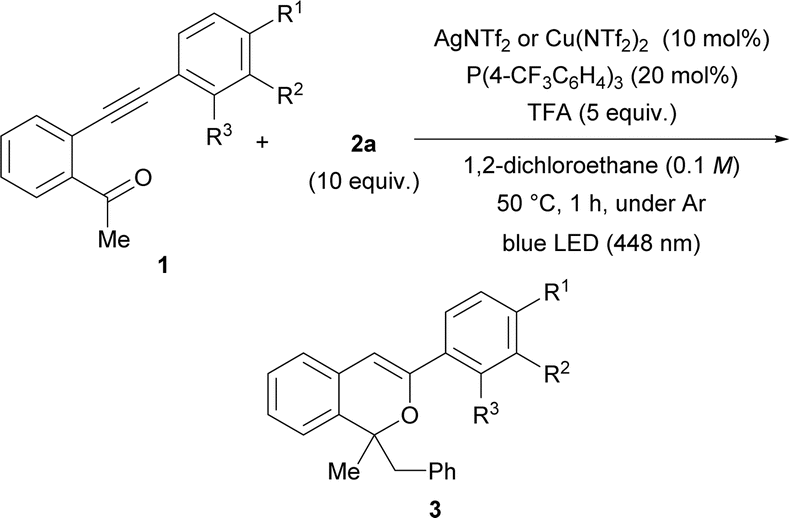
As shown in Table 1 entries 7 and 10, AgNTf2 and Cu(NTf2)2 catalysts were effective for the present consecutive transformation. Next, the scope of substrates in which various substituents were introduced to the terminal phenyl group was investigated in the presence of the AgNTf2 or Cu(NTf2)2 catalyst under the optimal reaction conditions shown in Table 1 (entries 7 and 10). As shown in Table 2, the use of the AgNTf2 or Cu(NTf2)2 catalyst resulted in the formation of product 3 in low to fairly good yields (see ESI† for details). When substrate 1b, which has an electron-donating methoxy group at the para-position (entry 1), was used, the reaction temperature had to be lowered to 0 °C and the reaction time had to be extended to 6 h to obtain product 3b in moderate yield (44%). In the case of substrates 1c having a methyl substituent (a weak electron-donating group) and 1d having a bromo substituent (a weak electron-withdrawing group) at the para-position, the Cu(NTf2)2 catalyst accelerated the reaction efficiently, affording 3c and 3d, respectively, in moderate to good yields (entries 2 and 3). In contrast, for substrate 1e having a strong electron-withdrawing CF3 group, the AgNTf2 catalyst promoted the formation of 3e in good yield (entry 4). Substrates having a methyl, bromo, or trifluoromethyl group at the meta-position gave products 3 in acceptable yields using the suitable metal catalyst for each reaction (entries 6–8), although, in the reaction of 1f having a methoxy group at the meta-position, 3f was formed in moderate yield using the AgNTf2 catalyst (entry 5). The reaction of 1j having a methyl group at the ortho-position afforded 3j in good yield (entry 9).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | R1 | R2 | R3 | 3 | [M]X | Yield of 3b (%) |
| a Unless otherwise specified, all reactions were carried out using 0.1 mmol of 1, 1.0 mmol (10 equiv.) of 2a, 10 μmol (10 mol%) of AgNTf2 or Cu(NTf2)2, 20 μmol (20 mol%) of P(4-CF3C6H4)3, and 5 equiv. of TFA in 1,2-dichloroethane (1 mL: 0.1 M of 1) at 50 °C for 1 h. b Isolated yield. c At 0 °C for 6 h. | |||||||
| 1c | 1b | MeO | H | H | 3b | Cu(NTf2)2 | 44 |
| 2 | 1c | Me | H | H | 3c | Cu(NTf2)2 | 47 |
| 3 | 1d | Br | H | H | 3d | Cu(NTf2)2 | 77 |
| 4 | 1e | CF3 | H | H | 3e | AgNTf2 | 72 |
| 5 | 1f | H | MeO | H | 3f | AgNTf2 | 39 |
| 6 | 1g | H | Me | H | 3g | Cu(NTf2)2 | 58 |
| 7 | 1h | H | Br | H | 3h | AgNTf2 | 59 |
| 8 | 1i | H | CF3 | H | 3i | Cu(NTf2)2 | 63 |
| 9 | 1j | H | H | Me | 3j | AgNTf2 | 74 |
Subsequently, the substituent at the carbonyl moiety was investigated (Scheme 3). Phenyl ketone 1k was not fully consumed when the Cu(NTf2)2 catalyst was used. The use of AgNTf2 resulted in a slight increase in yield, but the yield remained low. The optimal reaction conditions were not suitable for aldehyde 1l. Nevertheless, the yield of product 3l was improved to 65% by decreasing the amount of TFA to 2.5 equiv. and the concentration of 1l to 0.05 M and prolonging the reaction to 2 h when the AgNTf2 catalyst was used. The modified reaction conditions were applied to aldehyde 1m having a 4-bromophenyl group at the alkynyl terminus, however 3m was obtained in low yield even by using either AgNTf2 or Cu(NTf2)2.
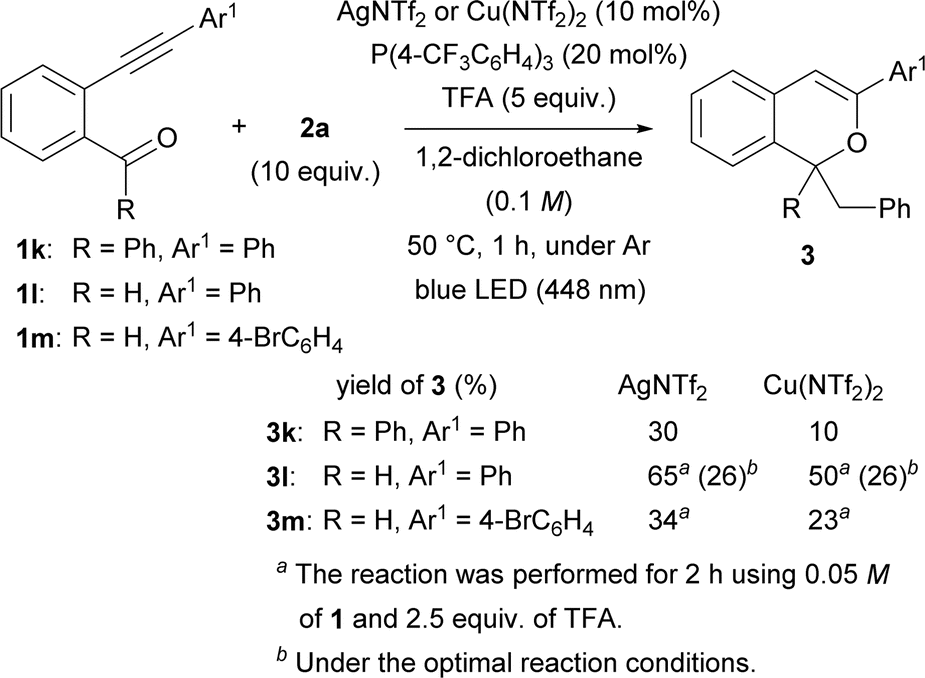 | ||
| Scheme 3 Consecutive transformation of phenyl ketone 1k and aldehydes 1l and m. | ||
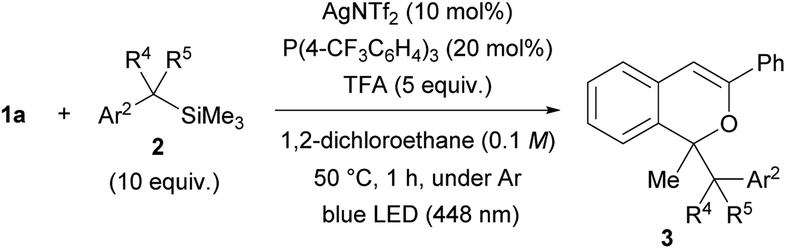
The scope of donor molecules 2b–e was further investigated (Table 3). The reactions of 2b and 2c, in which an electron-withdrawing CF3 group and an electron-donating MeO group13 were introduced to the para-position of benzyltrimethylsilane, respectively, were conducted using the AgNTf2 catalyst (entries 1 and 2). Product 3n and 3o were formed in low (18%) and moderate (54%) yields, respectively, under the optimal reaction conditions. In each case, extending the reaction time from 1 hour to 2 hours did little improve the yield. Donor molecule 2d having a methyl substituent at the benzylic position underwent the reaction smoothly, affording a diastereomeric mixture (65:35) of 3p in 62% yield (entry 3). However, introduction of a dimethyl substituent at the benzylic position, i.e., sterically congested 2e, completely suppressed the formation of 3q (entry 4).
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 2 | Ar2 | R4 | R5 | 3 | Yield of 3b (%) |
| a Unless otherwise specified, all reactions were carried out using 0.1 mmol of 1, 1.0 mmol (10 equiv.) of 2, 10 μmol (10 mol%) of AgNTf2, 20 μmol (20 mol%) of P(4-CF3C6H4)3, and 5 equiv. of TFA in 1,2-dichloroethane (1 mL: 0.1 M of 1) at 50 °C for 1 h. b Isolated yield. c The reaction was conducted for 2 h. d NR: no reaction. | ||||||
| 1c | 2b | 4-CF3C6H4 | H | H | 3n | 18 (21)c |
| 2 | 2c | 4-BrC6H4 | H | H | 3o | 54 (50)c |
| 3 | 2d | Ph | Me | H | 3p | 62 |
| 4 | 2e | Ph | Me | Me | 3q | NRd |
Finally, in order to investigate whether cationic intermediates A function as photo-oxidation agents, we conducted several control experiments, UV-Vis measurements, and Stern–Volmer luminescence quenching experiments. First, in the control experiments (see ESI† for details), no product was formed when the reaction was performed without the metal catalyst or light irradiation. In the absence of TFA, the product was obtained, although the yield remained quite low (13% with Cu(NTf2)2 catalyst). These results indicate that the present consecutive transformation is a photochemical reaction and 2-benzopyrylium intermediate A is the key component. The formation of 2-benzopyrylium intermediate A was further confirmed by UV-Vis measurement.14 As shown in Fig. 1, no absorption was observed in the visible region for substrate 1a alone (dark blue line) and substrate 1a/TFA mixture (gray line). On the other hand, when the substrate and AgNTf2 were mixed, a small absorption peak appeared in the visible region (orange line). When TFA was added to this mixture, a large absorption peak was observed around 430 nm (light blue line). In addition, we performed Stern–Volmer luminescence quenching experiments using a 2-benzopyrylium cation and donor molecule 2 to confirm quenching of the photo-excited 2-benzopyrylium cation by 2 (see ESI† for details). As a result, the emission intensity of the 2-benzopyrylium cation derived from 1b decreased as the amount of 2a increased. From this observation, SET from 2 to the photo-excited 2-benzopyrylium cation was verified. These results indicate that cationic intermediate A excited by irradiation at λmax = 448 nm, which is close to the maximum absorption wavelength, functioned as a photo-oxidation agent. However, as shown in Table 1, entry 14, when the light source was changed to purple LED (λmax = 405 nm) which is also close to the maximum absorption wavelength, product 3a was obtained in low yield presumably because of product degradation under the reaction conditions.
 | ||
| Fig. 1 UV-Vis spectra of related species in dichloromethane (5.0 × 10−4 M of 1a). | ||
The plausible catalytic cycles are depicted in Fig. 2. The whole process is composed of three catalytic cycles, namely, catalytic cycles I and II and a photoredox cycle of the photocatalyst. Catalytic cycle I generates key 2-benzopyrylium intermediates A from substrates 1 using π-Lewis acidic metal ([M]X) as the catalyst for the intramolecular cyclisation and TFA as the proto-demetalation agent. Subsequently, in catalytic cycle II, generated cationic intermediates A function not only as photocatalysts but also as electrophiles for the radical addition reaction with nucleophilic radicals C, which are generated by SET from donor molecules 2 to photo-excited cationic intermediates A* followed by the desilylation of radical cations B (see Scheme 2b). The radical addition of C to A produces radical cations E, which, in turn, are reduced by SET from radical intermediates D, a reduced form of photoredox catalyst A, affording corresponding 1H-isochromene derivatives 3. The photoredox cycle is also completed by regenerating cation intermediates A from radical intermediates D by SET. In the carbon–carbon bond forming event, the radical coupling of radicals C with radical intermediates D is also assumed. However, on the basis of our previous study on the reaction of 1-benzopyrylium derivatives with the same benzyl radical (see Scheme 1b),8 it is considered that the radical addition of C to A is more energetically favorable than the radical coupling.
 | ||
| Fig. 2 Plausible catalytic cycles. | ||
Conclusions
We have developed a π-Lewis acidic metal-catalysed cyclisation/photochemical radical addition sequence that affords 1H-isochromene derivatives in good yields in most cases. In the present consecutive transformation, ortho-carbonyl alkynylbenzene derivatives were used as precursors for the formation of 2-benzopyrylium intermediates generated in situ through the activation of the alkyne moiety of the precursor by a π-Lewis acidic metal catalyst ([M]X) and the subsequent intramolecular cyclisation followed by proto-demetalation by TFA. Further photo-excitation of 2-benzopyrylium intermediates facilitated SET from benzyltrimethylsilane derivatives as donor molecules to initiate the radical addition of arylmethyl radicals to 2-benzopyrylium intermediates. The most distinctive feature of this consecutive transformation is that the in situ generated 2-benzopyrylium intermediates were used not only as electrophilic substrates but also as new photo-oxidation agents. On the basis of the present method, it can be considered that in situ generated organic cations other than 2-benzopyrylium will be used as photoredox catalysts, and hence other methodologies that can expand the possibilities of photoredox catalysis are expected. In addition, a flow photoreaction system is also applicable to enhance the reaction efficiency and scale-up the present consecutive transformation. Further studies of the development of other photoredox reactions, particularly those utilizing a variety of organic cations generated in situ, and these applications to flow photoreactions are in progress in our laboratory.Data availability
The exploratory investigation, experimental procedures, and characterization data are available.Author contributions
M. T. contributed conceptualization, project administration, writing – original draft & editing, supervision, and funding acquisition. R. Y., R. O., and Z. I. contributed experimental studies, data curation, and formal analysis. S. U. contributed formal analysis and writing – review & editing. J. K. contributed conceptualization, design of the work, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (JP17H06447) and a Grant-in-Aid for Transformative Research Areas (A) “Green Catalysis Science for Renovating Transformation of Carbon-Based Resources” (JP23H04908) from MEXT, Japan and a Grant-in-Aid for Young Scientists (JP19K15552) from JSPS. We would like to express our gratitude to Prof. T. Iwamoto and Prof. S. Ishida (Tohoku University) who supported the Stern–Volmer luminescence quenching experiments.Notes and references
- (a) M. L. Mari, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- For selected examples or reviews using iminium cations, see: (a) P. S. Mariano, Tetrahedron, 1983, 39, 3845–3879 CrossRef CAS; (b) P. S. Mariano, Acc. Chem. Res., 1983, 16, 130–137 CrossRef CAS; (c) C. Chen, V. Chang, X. Cai, E. Duesler and P. S. Mariano, J. Am. Chem. Soc., 2001, 123, 6433–6434 CrossRef CAS PubMed.
- R. M. Borg, R. O. Heuckeroth, A. J. Y. Lan, S. L. Quillen and P. S. Mariano, J. Am. Chem. Soc., 1987, 109, 2728–2737 CrossRef CAS.
- K. Tanaka, Y. Iwama, M. Kishimoto, N. Ohtsuka, Y. Hoshino and K. Honda, Org. Lett., 2020, 22, 5207–5211 CrossRef CAS PubMed.
- For selected examples and reviews using acridinium ions, see (a) A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L.-C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed; (b) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 CrossRef CAS PubMed; (c) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 16219–16223 CrossRef CAS PubMed; (d) A. R. White, L. Wang and D. A. Nicewicz, Synlett, 2019, 30, 827–832 CrossRef CAS PubMed; (e) F. Tan, X. He, W. Tian and Y. Li, Nat. Commun., 2020, 11, 6126 CrossRef CAS PubMed; (f) C. Fischer, C. Kerzig, B. Zilate, O. S. Wenger and C. Sparr, ACS Catal., 2020, 10, 210–215 CrossRef CAS; (g) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS PubMed; (h) Y.-X. Cao, G. Zhu, Y. Li, N. L. Breton, C. Gourlaouen, S. Choua, J. Boixel, H.-P. J. de Rouville and J.-F. Soulé, J. Am. Chem. Soc., 2022, 144, 5902–5909 CrossRef CAS PubMed; (i) M. Mishraa, P. P. Singha, P. Nainwalc, S. Tivarid and V. Srivastavad, Tetrahedron Lett., 2023, 129, 154749 CrossRef.
- For a review and selected paper using pyrylium ions, see: (a) M. A. Miranda and H. Garcia, Chem. Rev., 1994, 94, 1063–1089 CrossRef CAS; (b) N. J. Gesmundo and D. A. Nicewicz, Beilstein J. Org. Chem., 2014, 10, 1272–1281 CrossRef PubMed; (c) E. Alfonzo, F. S. Alfonso and A. B. Beeler, Org. Lett., 2017, 19, 2989–2992 CrossRef CAS; (d) P. Chandu, K. G. Ghosh and D. Sureshkumar, J. Org. Chem., 2019, 84, 8771 CrossRef CAS; (e) E. Holaa and J. Ortyl, Eur. Polym. J., 2021, 150, 110365 CrossRef; (f) L. Bao, J.-T. Cheng, Z.-X. Wang and X.-Y. Chen, Org. Chem. Front., 2022, 9, 973–978 RSC; (g) M. M. Nielsen, T. Holmstrøm and C. M. Pedersen, Angew. Chem., Int. Ed., 2022, 61, e202115394 CrossRef CAS PubMed.
- For selected examples using iminium cations, see: (a) M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed; (b) M. Berger, D. Carboni and P. Melchiorre, Angew. Chem., Int. Ed., 2021, 60, 26373–26377 CrossRef CAS PubMed; (c) T. Uchikura, N. Kamiyama, T. Mouri and T. Akiyama, ACS Catal., 2022, 12, 5209–5216 CrossRef CAS; (d) M. Berger, D. Ma, Y. Baumgartner, T. H.-F. Wong and P. Melchiorre, Nat. Catal., 2023, 6, 332–338 CrossRef CAS.
- J. Kikuchi, S. Kodama and M. Terada, Org. Chem. Front., 2021, 8, 4153–4159 RSC.
- For a review and selected examples for the generation of organic cation species, see: (a) M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700–6704 CrossRef CAS PubMed; (b) J. Yoshida, Y. Ashikari, K. Matsumoto and T. Nokami, J. Synth. Org. Chem., Jpn., 2013, 1136–1144 CrossRef CAS; (c) Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969–9973 CrossRef CAS; (d) C. Kuang, X. Zhou, Q. Xie, C. Ni, Y. Gu and J. Hu, Org. Lett., 2020, 22, 8670–8675 CrossRef CAS PubMed; (e) A. C. Keuper, K. Fengler, F. Ostler, T. Danelzik, D. G. Piekarski and O. G. Mancheño, Angew. Chem., Int. Ed., 2023, 62, e2023047 CrossRef PubMed.
- For selected examples and representative reviews of cyclisation reaction using π-acidic metal catalysts, see: (a) N. Asao, T. Nogami, K. Takahashi and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 764–765 CrossRef CAS PubMed; (b) T. Yao, X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 11164–11165 CrossRef CAS PubMed; (c) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS PubMed; (d) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536–6544 RSC; (e) R. K. Shiroodi and V. Gevorgyan, Chem. Soc. Rev., 2013, 42, 4991–5001 RSC; (f) K. Saito, Y. Kajiwara and T. Akiyama, Angew. Chem., Int. Ed., 2013, 52, 13284–13288 CrossRef CAS PubMed; (g) C. Obradors and A. M. Echavarren, Chem. Commun., 2014, 50, 16–28 RSC; (h) M. Terada, F. Li and Y. Toda, Angew. Chem., Int. Ed., 2014, 53, 235–239 CrossRef CAS PubMed; (i) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (j) W. Debrouwer, T. S. A. Heuge-baert, B. I. Roman and C. V. Stevens, Adv. Synth. Catal., 2015, 357, 2975–3006 CrossRef CAS; (k) A. M. Asiri and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471–4503 RSC; (l) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091–14200 CrossRef CAS PubMed; (m) Z. Zhang, V. Smal, P. Retailleau, A. Voituriez, G. Frison, A. Marinetti and X. Guinchard, J. Am. Chem. Soc., 2020, 142, 3797–3805 CrossRef CAS PubMed.
- For recent examples using benzyltrimethylsilane derivatives as donors under photoredox conditions, see: (a) S. Dutta, J. E. Erchinger, F. Schäfers, A. Das, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202212136 CrossRef CAS PubMed; (b) L. Hou, Y. Zhou, H. Yu, T. Zhan, W. Cao and X. Feng, J. Am. Chem. Soc., 2022, 144, 22140–22149 CrossRef CAS PubMed; (c) L. Feng, X. Chen, N. Guo, Y. Zhou, L. Lin, W. Cao and X. Feng, Chem. Sci., 2023, 14, 4516–4522 RSC.
- For selected examples for the synthesis of 1H-isochromene derivatives, see: (a) S. Mondal, T. Nogami, N. Asao and Y. Yoshinori, J. Org. Chem., 2003, 68, 9496–9498 CrossRef CAS PubMed; (b) E. Tomás-Mendivil, J. Starck, J.-C. Ortuno and V. Michelet, Org. Lett., 2015, 17, 6126–6129 CrossRef PubMed; (c) F.-H. Li, J. Li, S.-Y. Wang and S.-J. Ji, Tetrahedron, 2017, 73, 5731–5737 CrossRef CAS; (d) W. Ieawsuwan, S. Limjirawatthana, P. Ploypradith and S. Ruchirawat, Asian J. Org. Chem., 2023, e202300578 Search PubMed.
- For the formal redox potental of donor 2c, see: (a) T. Maruyama, Y. Mizuno, I. Shimizu, S. Suga and J. Yoshida, J. Am. Chem. Soc., 2007, 129, 1902–1903 CrossRef CAS PubMed; (b) S. Montanaro, D. Ravelli, D. Merli, M. Fagnoni and A. Albini, Org. Lett., 2012, 14, 4218–4221 CrossRef CAS PubMed.
- UV-Vis spectra measurement of 2-benzopyrlyrium cations, see: B. Guo, Y. Zhou, L. Zhang and R. Hua, J. Org. Chem., 2015, 80, 7635–7641 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00808a |
| ‡ Current address: Graduate School of Pharmaceutical Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan. |
| § Benzyltrimethylsilane (2a), which was not consumed in the corresponding radical reaction, was almost completely recovered in either catalytic reaction. |
| This journal is © The Royal Society of Chemistry 2024 |