
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect imaging of dynamic heterogeneous lithium–gold interaction at the electrochemical interface during the charging/discharging processes†
Jiaxin
Mao‡
,
Guopeng
Li‡
,
Dongwei
Xu‡
and
Rui
Hao
 *
*
Department of Chemistry, Research Center for Chemical Biology and Omics Analysis, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: haor@sustech.edu.cn
First published on 17th January 2024
Abstract
Lithium can smoothly plate on certain lithium alloys in theory, such as the Li–Au alloy, making the alloy/metal films promising current collectors for high energy density anode-free batteries. However, the actual performance of the batteries with alloy film electrodes often rapidly deteriorates. It remains challenging for current imaging approaches to provide sufficient details for fully understanding the process. Here, a “see-through” operando optical microscopic approach that allows direct imaging of Li–Au interaction with high spatiotemporal and chemical resolution has been developed. Through this approach, a two-step Li–Au alloying process that exhibits interesting complementary spatiotemporal evolution paths has been discovered. The alloying process regulates the nucleation of further Li deposition, while the Li nucleation sites generate pores on the electrode film. After several cycles, film rupture occurs due to the generation of an increased number of pores, thus explaining the previously unclear mechanism of poor cycling stability. We have also elucidated the deterioration mechanism of silver electrodes: the growth of defect pores in size, independent of the alloying process. Overall, this new imaging approach opens up an effective and simple way to monitor the dynamic heterogeneity of metal–metal interaction at the electrochemical interface, which could provide helpful insight for designing high-performance batteries.
Introduction
Li-ion batteries are currently the dominant power source for mobile electronic products. However, the theoretical specific capacity of common commercial anode materials, such as graphite, falls remarkably short of satisfying the need for high-energy-density devices. Nevertheless, anode-free electrodes—which omit anode materials and employ only current collectors—can significantly improve the energy density of batteries and could be key to realizing ideal high-energy-density systems.1 However, anode-free batteries are prone to generating dendrites/dead Li via electrodeposition/dissolution, leading to serious safety concerns and diminished cycling performance.2Lithium could interact with other metals during the electrochemical charging and discharging process in diverse ways, including alloying/dealloying and/or electrochemical deposition/dissolution. Investigations on anode-free batteries have proven that the Li electrodeposition can be effectively regulated using Li alloys, which serve as a buffer layer to eliminate nucleation barriers and overpotential,3,4 resulting in smooth, dendrite-free Li deposition. Recent results indicate that a Au film could enable uniform Li growth/stripping in anode-free solid-state batteries in a dozen or so cycles.5 In particular, Au and Li can be alloyed to form Li-rich intermetallic compounds (up to Au4Li15)6 with a zero Li nucleation barrier potential on the surface of alloys. Thus, in theory, Li deposition in anode-free batteries can be regulated using a Li–Au alloy film. However, the performance of Au thin-film anodes deteriorates rapidly during cycling.3 Consequently, Au is typically deposited onto a substrate in a selective or restrictive manner to improve the electrochemical Li deposition behavior, for example, by loading Au nanoparticles inside hollow carbon spheres7 or carbon nanofibers.8
Although investigations have been conducted on Li–Au alloy systems,5,9–14 the discrepancy between theoretical predictions and actual outcomes of Li deposition on Li–Au alloy films has not been fully addressed. Speculations about electrode fracture mechanisms, such as those targeting the internal strain caused by frequent structural transformation,15 the presence of a low-energy barrier, and/or the occurrence of barrierless spinodal decomposition,16 have not been confirmed; this is due to the lack of methods permitting in situ direct high-resolution observation of the interfacial changes during the alloying/dealloying process. Similarly, studies on Ag films17,18 and nanoparticles19,20 alloyed with Li in solid solution reactions have extensively analyzed Li–Ag alloying, which can also create pores on the surface that can harm battery performance but have not elucidated the causes of pore formation.
The dynamic electrochemical interface evolution in batteries can be characterized using multiple operando imaging techniques including X-ray imaging,21–23 neutron imaging,24–26 and magnetic resonance imaging.27,28 Notably, the spatial resolution of these techniques, which is typically on the micrometer scale, hinders the collection of detailed microscopic/nanoscopic-level information on processes. Moreover, in situ transmission electron microscopy (TEM),29–32 scanning electron microscopy (SEM),33–35 and atomic force microscopy36–38 can provide information with high spatial resolution, but typically with low chemical resolution or deviating from battery working conditions. For instance, the expansion/shrinkage of an Au film during Li electrodeposition/dissolution has been observed by in situ liquid-cell TEM.6 However, factors such as the use of high-energy electron-beam radiation may prevent nanoscopic TEM observations from fully reflecting the details of the process of interest.
Therefore, to investigate the nucleation site evolution on the Au surface during alloying and dealloying and to comprehensively analyze the causes of homogeneity or heterogeneity during Li electrodeposition/dissolution, spatiotemporally resolved in situ imaging techniques with high chemical resolution must be developed. Real-time microscopic observation of the dynamics and details of electrodeposition on alloy electrode surfaces can guide the development of Li–Au and Li–Ag systems as well as other important alloy elements in Li-ion batteries,39 such as Si,40 Zn,41 Bi, Sn,42,43 and Al.44
In situ or operando optical microscopy (OM) is an advantageous noninvasive technique for analyzing nanoscale electrochemical interfaces under non-vacuum ambient conditions.45,46 Electrochemical interfaces with systems such as single particles,47,48 single molecules,49,50 and single nanobubbles51,52 have recently been probed by optical imaging. Moreover, OM—which is suitable for studying dynamic electrodeposition53,54—has been used to investigate the metal electrodeposition behaviour and morphological evolution.55–57 However, direct and detailed observation of the processes occurring on the metal electrode surface is hindered by the obstructive nature of the metal electrodes.58 Nevertheless, a few methods can be employed to study the evolution of compounds by observing the electrode–electrolyte interface with high spatial as well as chemical resolutions.
To extensively assess Li interaction with Au or Ag during the battery charging/discharging process, “see-through” operando observation was achieved with a transparent metal film electrode in simple trans-48 and epi-illumination59,60 imaging modes. The obstruction of light by metal electrodes is thus avoided and the dynamic electrochemical interfacial phenomena can be monitored with ease. Due to the absorbance/reflectance difference of the different alloys, the progress of the alloying/dealloying process can be monitored. Gold-film-based imaging is an established characterization technique for evaluating the nanoparticle dynamics, depending on its low reflectivity61,62 or surface plasmon resonance.63,64 However, different from the previous reports, in our research, the Au layer is the object of observation, rather than the medium for electron transfer.
Complementarity spatiotemporal evolution paths of the two Li–Au alloying/dealloying processes and the consequential Li heterogeneous deposition/dissolution are revealed via the “see-through” imaging approach. The alloying processes show strong regulating effects on the following lithium deposition: Li always randomly nucleates outside the initial sites of both two alloying steps and forms disk morphology at later stages. Lithium deposition also affects the gold film: during the discharging process, the lithium nucleation sites generate pores in the Au film. With multiple charging/discharging cycles, the randomly formed Li nucleation sites create an increasing number of pores, leading to cracking and failure of the electrode. In contrast, pore generation in the Li–Ag system was not affected by the alloying process but the alloying/dealloying process could also corrode the silver film, leading to enlarged pores.
Thus, the dynamic heterogeneous interactions between lithium and gold/silver as the essential cause of film electrode failure were elucidated. These findings provide deeper insights into the mechanism governing electrochemical alloying, thereby helping guide the design of high-performance anode-free materials. The novel and universal “see-through” imaging approach could provide a convenient and effective tool for revealing previously obscured details of key interfacial electrochemical processes occurring in various electrochemical storage systems.
Results and discussion
Imaging of Li–Au alloying processes
First, Li metal coin-type cells were assembled to verify that an Au film could effectively enhance their coulombic efficiency (CE) in short term. Cu foil, which was used as an effective current collector and substrate, and a 20 nm-thick Au film plated on the Cu foil by electron-beam evaporation (denoted as Au/Cu) were employed as the Li metal cell electrodes. LiPF6 (1.0 M) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol% mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) with 5% fluoroethylene carbonate (FEC) was employed as the electrolyte for the Li metal cells. Li‖Au/Cu initially exhibited a persistent, more stable CE (∼92.9%) than that of Li‖Cu (Fig. S1†). Notably, the CE of Au/Cu in the first cycle (91.59%) was lower than that of Cu owing to excessive solid electrolyte interphase (SEI) formation during the alloying and Li metal deposition. However, the CE of Au/Cu increased over the next five cycles, but then quickly dropped to the same level as that of Cu thereafter. Therefore, the dynamic Li alloying–dealloying mechanism had to be clarified to guide the design of high-performance electrodes and the exploration of other electrodeposition behaviors by investigating the electrochemical deposition and dissolution of Li on Au surfaces. Other metal materials including Au/Ni and Au/Ni/Cu (Fig. S1†) were also tested. Au plating on a Ni substrate with good electrical conductivity as an anode-free electrode material can also improve battery performance, proving that the substrate does not play a major role in the influence of Au and Li alloys on the performance of the battery.
:1 vol% mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) with 5% fluoroethylene carbonate (FEC) was employed as the electrolyte for the Li metal cells. Li‖Au/Cu initially exhibited a persistent, more stable CE (∼92.9%) than that of Li‖Cu (Fig. S1†). Notably, the CE of Au/Cu in the first cycle (91.59%) was lower than that of Cu owing to excessive solid electrolyte interphase (SEI) formation during the alloying and Li metal deposition. However, the CE of Au/Cu increased over the next five cycles, but then quickly dropped to the same level as that of Cu thereafter. Therefore, the dynamic Li alloying–dealloying mechanism had to be clarified to guide the design of high-performance electrodes and the exploration of other electrodeposition behaviors by investigating the electrochemical deposition and dissolution of Li on Au surfaces. Other metal materials including Au/Ni and Au/Ni/Cu (Fig. S1†) were also tested. Au plating on a Ni substrate with good electrical conductivity as an anode-free electrode material can also improve battery performance, proving that the substrate does not play a major role in the influence of Au and Li alloys on the performance of the battery.
To observe the morphological dynamics and alloying heterogeneity during Li electrodeposition, a Li-ion electrochemical cell was designed using a transparent electrode (Fig. 1A). The key to the design of this ultrathin electrode, which is commonly used in in situ electron microscopy, is configuring its structure to obtain a clear view of the interior of the interface, thereby helping better correlate microscopic information with macroscopic phenomena. Integrating the electrode with trans- and epi-illumination techniques enabled observation of the inner evolution of the electrode (Fig. 1A). Changes in the absorption and reflection of light during the alloying, as reflected in the in situ UV-vis spectrum (Fig. S2†), indicated that the process could be identified by the two imaging modes. The trans-illumination mode measured the transmission of light through the sample. As shown in the UV-vis spectrum, the alloying reaction resulted in a reduction of the electrode's transmittance. The epi-illumination mode measured the reflectivity of the sample and the reflection increased when Li was alloyed with Au. Transmittance is subsequently normalized by establishing the ratio between the transmitted intensity of the original and the initial frame. The absorbance of the sample can also be calculated accordingly. The variations in the initial alloying process were more pronounced in the trans mode, whereas the dendrite dissolution was clearer in the epi mode. The transmittance and reflectivity of the metal film can be calculated with the intensity variation data obtained from the trans- and epi-recordings.60 For non-metal materials, it is relatively simple to determine the absorbance of the sample qualitatively. However, considering the complicated interaction between light and the metal film, it is more challenging to quantify the process, but qualitative analysis can be achieved. Li was electrodeposited on the surface of a pure Cu film as dendrites (Fig. S3†), proving that the pattern observed on the electrode surface was Li alloyed with Au, independent of Cu. The Li–Au electrochemical alloying and dealloying were investigated at a current of 35.3 μA, which corresponded to a current density of 0.5 mA cm−2. Other currents of 7.1 μA, 177 μA, 0.707 mA, 1.06 mA, and 1.77 mA—corresponding to current densities of 0.1, 2.5, 10, 15, and 25 mA cm−2, respectively—were also tested. In a coin cell with an area capacity of 0.5 mA h cm−2, these current densities corresponded to rates of 0.2, 5, 20, 30, and 50C, respectively.
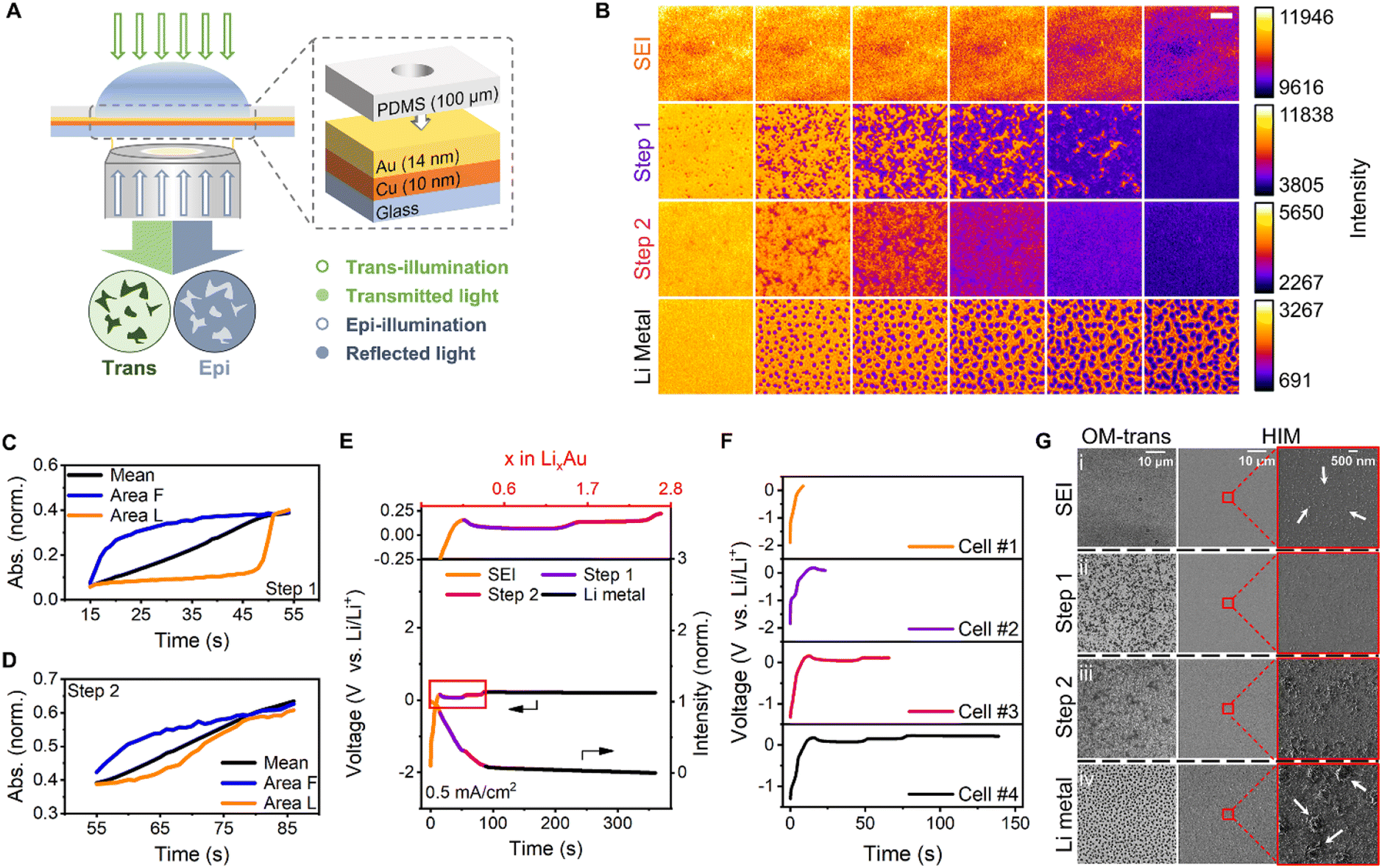 | ||
| Fig. 1 Experimental apparatus, electrochemical measurements and imaging results. (A) Schematic of the in situ experimental setup and electrochemical cell integrated with the trans-illumination (green) and epi-illumination (blue) modes to monitor the alloying. In the trans-illumination mode, the dark and light areas are the alloyed and unalloyed regions, respectively. In the epi-illumination mode, the light and dark areas are the alloyed and unalloyed regions, respectively. (B) Montages of images obtained via trans-mode recording of four Li deposition stages on Au at a current density of 0.5 mA cm−2. The montages from top to bottom are SEI formation (t = 0–15 s, 3 s interval, SEI for short), the first alloying step (t = 17–52 s, 7 s interval, step 1 for short), the second alloying step (t = 55–85 s, 6 s interval, step 2 for short), and Li metal deposition (t = 87–357 s, 54 s interval, Li metal for short), respectively. The montages of each row were normalized according to their intensity range. The reading order of all montages is from left to right. Scale bar, 5 μm. Normalized absorbance versus time curve of different regions in the first alloying step (C) and the second alloying step (D). The normalized absorbance for the whole was denoted as the mean. The initial and the final alloyed regions were denoted as ‘Area Init’ and ‘Area Fnl’, respectively. (E) Galvanostatic curve of Au (left) and the mean intensity–time curve (right) obtained via trans-mode recording during Li deposition at a current density of 0.5 mA cm−2. The different colors represent distinct stages (orange, SEI formation; purple, the first alloying step; magenta, the second alloying step; and black, Li metal deposition). (F) Galvanostatic curves of Au with four Li deposition stages. The curves of four electrochemical cells from top to bottom belong to cell #1 finishing at SEI formation, cell #2 finishing at the first alloying step, cell #3 finishing at the second alloying step, and cell #4 finishing at Li metal deposition, respectively. (G) Images obtained via optical microscopy (OM, left) and helium-ion microscopy (HIM, middle), and magnified versions of the boxed region in the middle of the HIM images (right) at the four Li deposition stages including SEI formation (SEI for short), the first alloying step (step 1 for short), the second alloying step (step 2 for short), and Li metal deposition (Li metal for short). | ||
Various stages of Li electrodeposition on the Au surface at a current density of 0.5 mA cm−2 were observed (Fig. 1B). Initially, the SEI film was formed uniformly and darkened gradually;11 however, no detailed morphology was observed. Moreover, the composition of the electrolyte on the electrode surface was altered, thereby forming a passivation layer. After the SEI film formation, Li and Au were alloyed to form uniform and flat alloy layers in two steps which exhibited heterogeneity in the direction and sites of the alloying. The alloying sites in the first step were analyzed at different current densities (Fig. S4†). After the electrochemical alloying, Li was deposited at uniformly distributed nucleation sites on the alloy surface, possibly because of the low-energy barriers to Li nucleation induced by the alloy layer. Li was also deposited in other regions of the disk-shaped particle, according to its intensity–time curve (Fig. S5†). The intensity–time curves of the epi recording somewhat fluctuated during the Li deposition (Fig. S6†), presumably owing to the migration of Au into Li. The same four-stage behavior can also be observed by an in situ test on the 28 nm-thick Au film (Fig. S7†). The evolution of Li–Au interaction on this film is similar to that on the 14 nm-thick Au film, indicating consistent alloying behaviour in the film thickness range.
Studying the dynamic heterogeneity of batteries65–67 is necessary to understand the evolution of electrode charging and discharging processes. To study the dynamic heterogeneity of two alloying steps, the normalized absorbances of the sample at different regions were calculated from the transmittance in trans-mode. The whole absorbance evolution during the electrochemical alloying and deposition is shown in Fig. S8.† The initial and the final alloyed regions were denoted as ‘Area Init’ and ‘Area Fnl’, respectively.
In the alloying first step, there is a significant starting time difference between the initial and the final alloyed regions (Fig. 1C). The normalized absorbance curve of the first alloying step appears to consist of two distinct stages: rapid growth and flat plateau. During the rapid growth stage, the absorbance increases sharply, and the alloying time is relatively short. In contrast, during the flat plateau stage, the absorbance only changes slightly. In the second alloying step, the difference between the initial and the final alloyed regions at the time of occurrence was not very obvious, as indicated by the slow increase in normalized absorbance (Fig. 1D). The normalized absorbance of other selected regions which were alloyed initially or finally in two alloying steps shows similar results (Fig. S9 and S10†). We speculate that the distinctly different alloying behaviors should be due to the increased lithium diffusion resistance in the second alloying step, which is also reflected as an overpotential in the galvanostatic curve.11
For the whole process, each step is associated with a new voltage plateau and is consistent with the image sequences. The corresponding galvanostatic curves were selected to define the four steps and scrutinized (Fig. 1E; the orange, purple, magenta, and black curves correspond to SEI formation, the first step of the alloying, the second step of the alloying, and Li metal deposition, respectively). Using the respective current and time data, the time axis was converted into the Li content (x) of LixAu. Consequently, two plateaus developed at potentials of 0.07 and 0.14 V, which persisted up to x values of 1.53 and 2.69 respectively, corresponding to two alloying steps. The overpotential should be related to increased lithium diffusion resistance.11 A third plateau was initiated at a potential of 0.23 V, and the Li nucleation overpotential on the surface of the Li–Au alloy was essentially zero.7 In contrast to the expected uniform deposition, selective deposition of Li metal was observed at specific sites. This confirmed the lack of nucleation barriers for Li in the Li–Au alloy, which would have been beneficial for uniform Li metal deposition. The number of Li nucleation sites increased with increasing current density, presumably because of the SEI thickness (Fig. S11†).68 However, the diameter of the Li particle decreased owing to a diffusion-controlled reaction with a high possibility of dendrite growth.69
Helium-ion microscopy (HIM) was employed to examine the nanoscopic details of the four stages and to compare its imaging attributes with those of the OM-based technique reported herein. Ex situ HIM imaging of lithium deposition was performed using four electrochemical cells, which started at the same current density and stopped at different stages. The four stages, namely SEI formation, the first alloying step, the second alloying step, and Li metal deposition, corresponded to galvanostatic curves of four cells in Fig. 1F. Electrochemical cell #1, #2, #3 and #4 finished charging at 0.15 V during SEI formation, at 23.4 s during the first alloying step, at 65.8 s during the second alloying step, and at 139 s during Li metal deposition, respectively. The OM and HIM images of four cells were denoted as (i), (ii), (iii) and (iv), respectively. For a fixed large field of view, the trans-mode images with adequate chemical resolution (Fig. 1G(ii) and (iii)) showed a conspicuous pattern of the alloyed metal with superior contrast. However, the HIM images could hardly distinguish the alloy pattern (Fig. 1G(ii) and (iii)), although they could discern the Li metal deposited in the later period. Magnified HIM images (Fig. 1G(i)) revealed 20–100 nm-diameter particles in the SEI film, which is consistent with previous reports.70–72 Furthermore, ∼700 nm-diameter disk-shaped deposited Li particles were also observed in the images (Fig. 1G(iv)). Notably, OM offers noteworthy advantages for monitoring samples over a large field of view, such as no sample damage, simple operation, and adequate chemical resolution. Furthermore, unlike ion microscopy or TEM, which has extremely stringent sample preparation requirements, OM can permit in situ/operando observation of large samples. The design of the transparent electrode and the combination of trans- and epi-illumination modes enable clear observation and exploration of the inner details of the electrode.
Heterogeneity of electrochemical Li alloying with Au
Sites participating in the two-step alloying and their relationship with those engaged in the later-stage Li metal nucleation were further analyzed. The beginning and end of the acquired image montage (Fig. 1B) were defined by the alloying stages of the 3 mm-diameter electrode system. The morphological evolution of the alloying pattern was uniform for all areas of the working electrode, except for a small time difference relative to the center and edge of the electrode. To better examine the pattern evolution of each alloying step, only the start and end of a random 200 pixel-long 200 pixel-wide area were defined (Fig. 2A).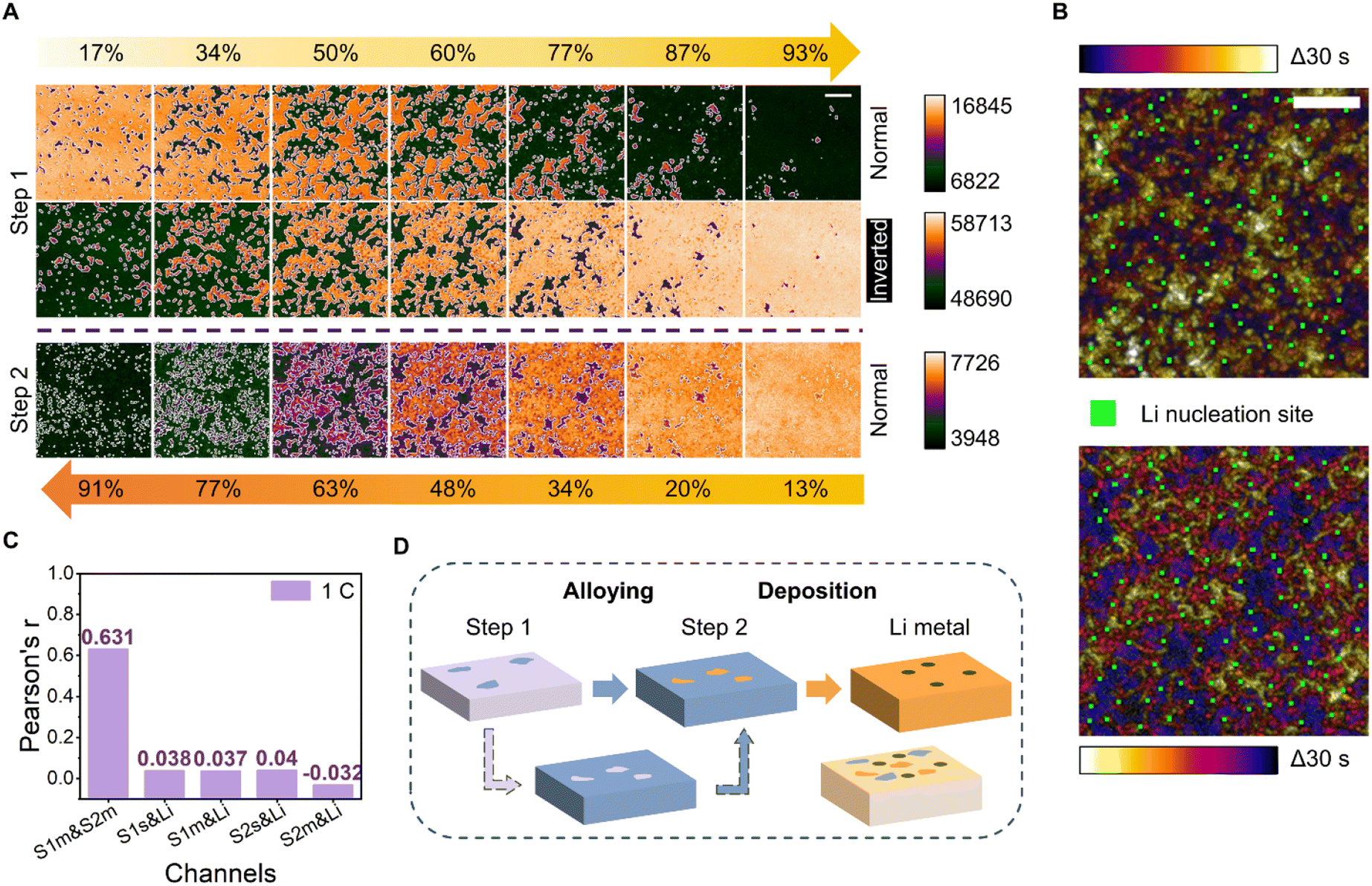 | ||
| Fig. 2 Li–Au alloying and lithium deposition processes. (A) Montages of images acquired via trans-mode recording of the first and second steps of Li deposition on Au (t = 9–38 and 46–73 s, top and bottom rows, respectively) at a current density of 0.5 mA cm−2. The dark and light areas are the alloyed and unalloyed regions, respectively. The direction of the yellow arrows represents the time sequence of alloying evolution. The different labels of the % values represent distinct exact times: t = 13 (17%), 18 (34%), 23 (50%), 26 (60%), 31 (77%), 34 (87%), and 36 s (93%) in the first alloying step; t = 48 (13%), 50 (20%), 53 (34%), 57 (48%), 60 (63%), 65 (77%), and 69 s (91%) in the second alloying step, respectively. The middle set, an intensity-inverted montage of the first step (top), was acquired to relate it to the second step (bottom). The light and dark areas are the alloyed and unalloyed regions, respectively. The montages of each row were normalized according to their intensity range. Notably, the intensity of each image in the second step was an average of 2 frames. Scale bar, 5 μm. (B) Temporal color-coded image (top) of the first alloying step (30 s-long, t = 9–38 s) and its intensity-inverted version (bottom), showing the Li nucleation sites (green). Scale bar, 5 μm. (C) Pearson's r values for the alloyed or unalloyed regions in the initial and middle stages of the two alloying steps and the Li nucleation sites at a deposition rate of 1C, as calculated using a colocalization algorithm. The different x-axis labels—(i) S1m & S2m, (ii) S1s & Li, (iii) S1m & Li, (iv) S2s & Li, and (v) S2m & Li—represent the following channels: (i) unalloyed regions in the middle stages of the first step, and alloyed regions in the middle stages of the second step; (ii) alloying sites in the first step and Li nucleation sites; (iii) alloyed regions in the middle stages of the first step and Li nucleation sites; (iv) alloying sites in the second step and Li nucleation sites; and (v) alloyed regions in the middle stages of the second step and Li nucleation sites. (D) Schematic illustrating the dynamic heterogeneity in electrochemical Li–Au alloying and Li deposition. Specific areas of the electrode surface at each step represent regions that have been alloyed or deposited. The pattern of the image in the lower left corner represents the unalloyed area at the end of the first step and corresponds perfectly to the alloyed pattern of the second step. The image in the lower right corner represents the scenario in which the sites participating in the three steps (two alloying stages and Li metal deposition) are not correlated. | ||
Surprisingly, we find that the alloy paths of the two alloying stages are complementary spatiotemporally. The initial alloying sites of the second step correspond to the last alloying sites of the first step. The “normal” images in Fig. 2A were directly false-color tinted. The emergence of the dark feature in “normal” mode indicates the beginning of the alloying process. The “bright” feature at the end of step 1 (93%) was the region staying unalloyed, the pattern of which is complementary to the initially alloyed region in step 2 (13%). The phenomenon can be further depicted by the inverted images in the second row showing a high degree of similarity to the third-row images.
The complementarity of the two alloying steps was observed even at higher current densities (Fig. S12†). At a low current density of 0.1 mA cm−2, the temporal color-coded image showed a clear dynamic evolution and the heterogeneity of alloying sites for an alloying time of 187 s. At higher current densities, the dynamic changes were more concentrated in the early stages (blue and purple regions) and final stages (yellow region). Temporal color-merged images were also acquired during the second alloying step. With more Li deposition, the contrast of the trans-mode images worsened, and the second alloying step without strong heterogeneity exhibited characteristics inferior to those of the first alloying step.
To further understand the relationship between the alloying sites engaged in the two steps and the nucleation sites of Li metal, an in-depth analysis of the three sites was performed. To that end, a temporal color-coded image of the first alloying step and its intensity-inverted version merged with Li nucleation sites were acquired (Fig. 2B). The position at the end of the first step can be used to represent the start of the second step, meaning that the relationship between the three can be studied using only the pattern of the first alloying and the lithium nucleation sites. Notably, the Li nucleation sites, signified by the green areas in the figure, were absent in the white regions in both the normal and intensity-inverted images. Three samples were tested under the same experimental conditions to prove that this phenomenon was not unique or accidental (Fig. S13†). Moreover, the appearance of Li nucleation sites in the middle region of the alloy persisted at even higher current densities (Fig. S14†).
To quantify the relationship between the alloying locations and Li nucleation sites, Pearson's r values73 were calculated for the alloyed/unalloyed regions of the two steps and the Li nucleation sites using a colocalization algorithm (Fig. 2C). At a current density of 0.5 mA cm−2, which corresponds to a deposition rate of 1C, the Pearson's r value for unalloyed regions in the middle stages of the first step with respect to alloyed regions in the middle stages of the second step, denoted as S1m & S2m, was 0.631, indicating an acceptable correlation; this is a reasonable result given the complementarity and relationship between the two alloying steps. The r values for the alloying sites in the first step with respect to Li nucleation sites (denoted as S1s & Li) and that for the alloying sites in the second step with respect to Li nucleation sites (denoted as S2s & Li) were 0.038 and 0.04, respectively. These values—which were less than 0.5 and close to 0 without colocalization—indicated that Li could nucleate randomly in any region outside the initial alloying sites. Similarly, the values for the alloyed regions in the middle stages of the first or second steps with respect to the Li sites (denoted as S1m & Li and S2m & Li) were 0.037 and −0.032, respectively; these factors remained unrelated. The values corresponding to the samples tested at deposition rates of 0.2C and 5C (Fig. S15†) were consistent, indicating that Li was more likely to nucleate in the middle area, and not in the area of the highest or lowest nucleation barrier (Fig. 2D).
We speculate that the difference in stress level or thickness of the SEI film could modulate the lithium alloying and deposition process. The alloying sites of the second step correspond to the regions less prone to alloying sites of the first step. There might be a tendency for lithium to nucleate under medium stress/thickness conditions. Further investigation of the issue could be a topic for future studies capable of characterizing both the SEI and alloy film in detail simultaneously.
Electrochemical Li dealloying and cycling behavior with Au
To understand the causes of Au film rupture and the association between cracked regions and nucleation sites, Li metal dissolution and dealloying were subsequently examined (Fig. 3A). The corresponding galvanostatic curves in Fig. 3B were selected to define the two steps. Typically, Li dendrites tend to dissolve at their root because of the local current density, leading to a decrease in CE. HIM images (Fig. 3F) indicated that the Li dissolution generated pores in the Li–Au alloy. Obviously, the dissolution of lithium in the first five images of the first montage (Fig. 3A) occurred mainly at the top of the dendrites, with little change in the roots. The formation of holes occurs specifically at the stage of the last two images. Therefore, the black dashed areas of Fig. 3A were comprehensively studied to understand the pore formation. The pore of a single dendrite, which appeared as bright and dark regions in the trans and epi modes, respectively, was visualized (Fig. 3C and D); this feature emerged at the center of the Li metal and expanded.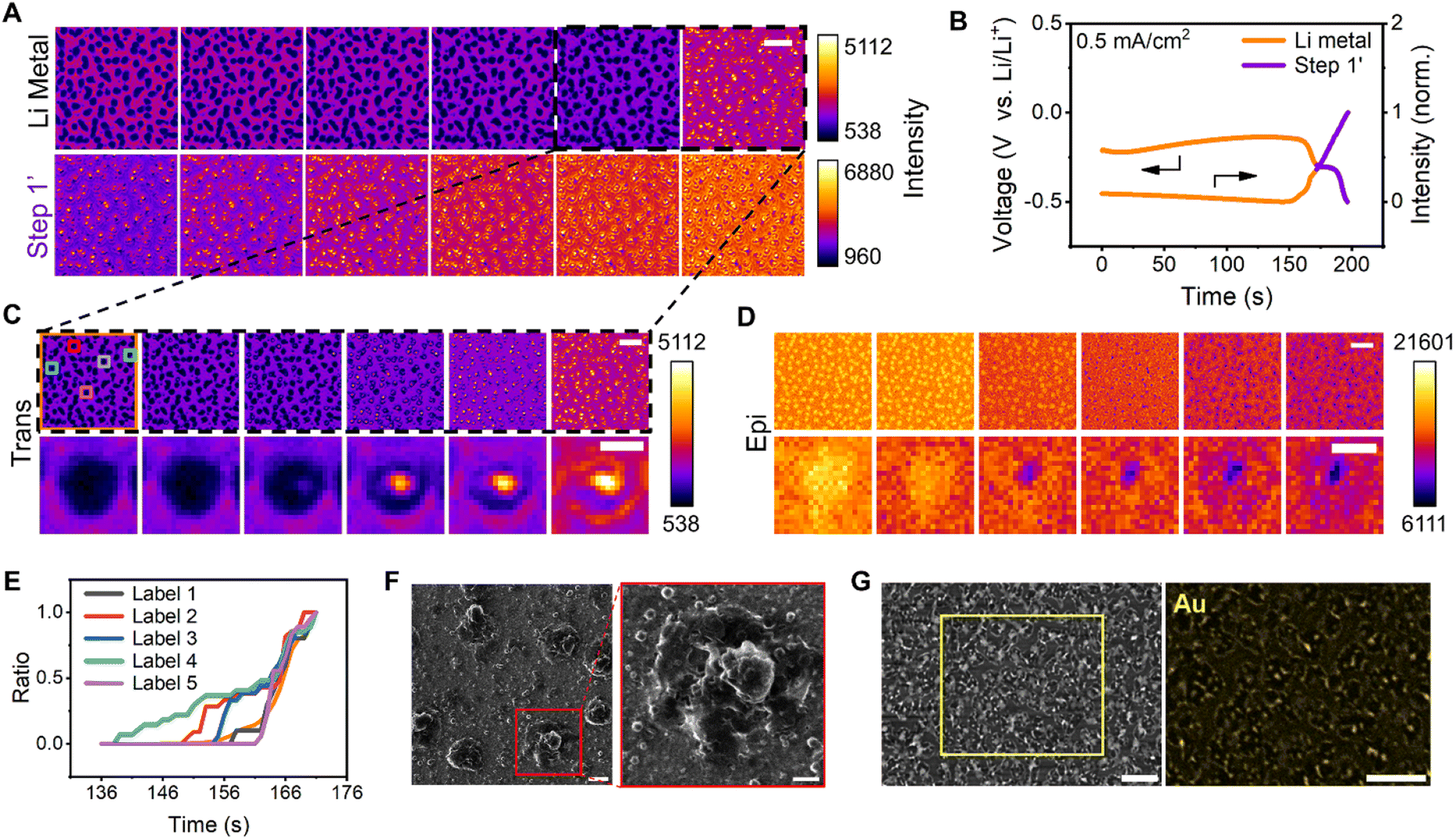 | ||
| Fig. 3 Li–Au dealloying and Li dissolution processes. (A) Montages of images acquired via trans-mode recording of Li metal dissolution (t = 0–170 s, 34 s interval, Li metal for short, top) and Li–Au dealloying (t = 171–196 s, 5 s interval, step 1' for short, bottom). The montages of each row were normalized according to their intensity range. The reading order of all montages is from left to right. Scale bar, 5 μm. (B) Galvanostatic curve (left) of Au and mean intensity–time curve (right) obtained via trans-mode recording of Li dissolution at a current density of 0.5 mA cm−2. The different colors represent distinct stages (orange, Li metal dissolution; purple, the first dealloying step). (C) Montages of images obtained via trans-mode recording (t = 136–171 s, 7 s-interval) of Li dendrites (top) and a representative single dendrite (bottom) in the dashed region shown in (A) during Li metal dissolution. The reading order of all montages is from left to right. Scale bars, 5 μm (top) and 1 μm (bottom). (D) Montages of images obtained via epi-mode recording (t = 110.25–131.5 s, 4.25 s-interval) of Li dendrites (top) and a representative single dendrite during Li metal dissolution. The trans- and epi-recordings were recorded separately from different experiments and different samples at the same deposition and dissolution current density. The reading order of all montages is from left to right. Scale bars, 5 μm (top) and 1 μm (bottom). (E) Area ratio–time curve of different regions in the boxed region shown in (C). (Area ratio refers to the area of a pore at a specific time divided by that of the final pore.) (F) HIM image of the Au surface after Li deposition at a current density of 0.5 mA cm−2 and dissolution until −0.5 V, and the magnified image of the red boxed region. Scale bars, 500 nm (left) and 200 nm (right). (G) Au mapping of the electrode surface by energy-dispersive X-ray spectroscopy (EDX) after Li deposition at a current density of 0.5 mA cm−2 (30 min). Scale bars, 10 μm (left) and 5 μm (right). | ||
Five randomly selected regions and the entire area were normalized by the area of the final pore (Fig. 3E), and the Li metal dissolution and pore formation were found to be heterogeneous over time. The evolutions of the selected five single dendrites during Li metal dissolution and the evolution of the pore size with time are shown in Fig. S16.† The pore emergence time exhibits slight variations, accompanied by differing expansion rates. The distributions of the size of dendrites and pores are shown in Fig. S17.† It indicated that the dissolution of dendrites could induce the formation of a void first, ultimately leading to large pores. The cross-sectional areas of single dendrites were mostly about 0.45 μm2, and Li dissolution and dealloying resulted in more micropores with an area of less than 0.1 μm2.
A cell that was charged for 30 min and discharged to −1 V at a current density of 0.5 mA cm−2 was used to accurately investigate the Au loss. Au mapping by energy-dispersive X-ray spectroscopy (EDX) revealed the near absence of Au in the region corresponding to the pores on the surface of the electrode in the SEM image (Fig. 3G), indicating that the Au film was etched by Li during the Li dissolution and dealloying. Notably, the initial nucleation sites of Li metal destroyed the Au film. It has been reported that the Au particles relocate at a different interface during the charging/discharging process,5 indicating that Au could migrate into Li during Li deposition. Thermodynamic preference was considered as the driving force for Ag migrating into Li.74 We thus speculate that the same principle applies to the Au–Li system.
Trans-mode captures of the Au film in the pristine state and six states generated over three cycles were analyzed (Fig. 4A). The morphology of the deposited Li metal inclined toward dendrites with increased cycling because the alloy surface was gradually covered by the deposited Li layer. Moreover, the deposition was not controlled by the alloy surface. The number of pores on the electrode surface increased with cycling, as did the total area of the pores, thereby impeding subsequent Li deposition. The electrode surface became increasingly rough, and the electrode expanded in volume (Fig. 4B). Furthermore, the Li metal deposited around the preceding pores and its nucleation sites were randomly distributed between the initially alloyed regions during each cycle. The galvanostatic cycling curves acquired over three cycles (Fig. 4C) indicated excessive SEI formation in the first cycle, resulting in a low CE. However, the SEI formation continued in the subsequent cycles owing to the cracking and rupture of the SEI during dissolution.
 | ||
| Fig. 4 Li–Au cycling processes. (A) Trans-mode-derived images of the Au film surface in the pristine state and six other states generated during three cycles. The pristine state of the Au surface before electrochemical deposition is labeled as P. The electrochemical deposition and dissolution are denoted as De and Di, respectively. De1*–De3* and Di1*–Di3* represent the end of electrochemical deposition and dissolution of the three cycles, respectively. Scale bar, 5 μm. (B) Plots of the area ratio and pore count versus charging period for three cycles. (C) Galvanostatic cycling curves at a current density of 0.5 mA cm−2. De1–De3 and Di1–Di3 represent electrochemical deposition and dissolution of the three cycles, respectively. | ||
Electrochemical Li alloying and dealloying behaviour with Ag
The method reported herein was applied to other alloy systems, such as those featuring Ag, to verify its universality and further understand the dynamics of heterogeneous alloying. To that end, the trans-mode recordings were first separated into six stages according to morphological evolution (Fig. 5A). Using the respective galvanostatic current and time data, the time values were converted into the Li content in the Li–Ag alloy (Fig. S18†). The Li–Ag binary phase diagram has indicated the existence of three intermediate phases.75 Like Li–Au, the initial spike in voltage should be attributed to the formation of the SEI layer. Two uniform darkening stages were observed, representing the first and second alloying steps. Two further heterogeneous developments were perceived as the third and fourth alloying steps. Ultimately, no notable stagewise behaviour was detected, and no further alloying steps can be observed in the corresponding electrochemical curve, leading to the assumption that the final stage was associated with the deposition of Li metal. The dissimilarity in Li–Ag alloying75 (4 steps) and Li–Au alloying processes11 (2 steps) should be due to the different phase evolution paths during the lithium alloying process. | ||
| Fig. 5 Li–Ag alloying and cycling processes. (A) Montages of images acquired via trans-mode recording of six stages of Li deposition on Ag at a current density of 0.5 mA cm−2. The montages from top to bottom are SEI formation (t = 0–10 s, 2 s interval, SEI for short), the first, second, third, and fourth alloying steps (t = 12–67, 69–104, 105–150, and 151–216 s, respectively; 11, 7, 9, and 13 s interval, respectively; S1, S2, S3, and S4 for short, respectively), and Li metal deposition (t = 218–718 s, 100 s interval, Li for short), respectively. The montages of each row were normalized according to their intensity range. The reading order of all montages is from left to right. Scale bar, 5 μm. (B) Galvanostatic curve (left) of Ag and the mean intensity–time curve (right) of the trans-mode recording during Li deposition at a current density of 0.5 mA cm−2. The different colors represent the following distinct stages: SEI formation (orange); the first, second, third, and fourth alloying steps (purple, magenta, blue, and green, respectively); and Li metal deposition (black). See also Fig. S18.† (C) Heatmap visualization to enable comparisons between different regions during the Li–Ag alloying. Each cell is colored by the corresponding Pearson's r value, and the four alloying steps are denoted as S1, S2, S3, and S4. The upper left corner presents the coefficient of the unalloyed area in the different alloying steps and pore regions, whereas the lower right corner shows the coefficient of the alloyed area in the different alloying steps and pore regions. (D) Trans-mode images of the Ag film surface exhibiting three states during three cycles. The electrochemical deposition and dissolution are denoted as De and Di, respectively. Di1*–Di3* represent the end of electrochemical dissolution of the three cycles, respectively. Scale bar, 5 μm. (E) Plots of the area ratio and pore count versus the charging period during three cycles. | ||
In our experiment, we observed that after the fourth alloying step, no obvious stratification was observed, which suggests that Li and Ag might not further dissolve with each other. This could be attributed to the limited dynamics of lithium diffusion into the alloys, resulting in direct Li deposition on the surface rather than a uniform Li–Ag alloy film. Moreover, the morphologies of the silver layer exhibit a large degree of similarity after each dealloying process, which, we believe, can hardly be maintained if silver continuously dissolves into lithium (Fig. S19†). Notably, Li–Ag did not exhibit a stable plateau in its galvanostatic charging curve (Fig. 5B), unlike that of Li–Au, presumably owing to differences in the Li–Au and Li–Ag alloying mechanisms, which are the reconstitution reaction and solid-solution reaction,76 respectively.
The surface of the electrode produced a significant number of pores during Li dissolution (Fig. 5D). To understand the relationship between the pores and alloying, Pearson's r values were determined for the regions in the middle stages of the alloying steps and the porous areas (Fig. 5C). The coefficients for the pores with respect to the alloyed or unalloyed regions in the middle stages of the four steps were close to zero, implying the lack of correlation between the alloying and pores. In terms of the calculated colocalization between the different alloying steps, the values for the unalloyed region were higher than those for the alloyed zone; however, they did not exceed 0.5, indicating the absence of a colocalization relationship between these steps. In other words, the alloying steps of Li on the Ag film surface were unrelated, unlike the complementary phenomenon observed on the surface of Au. Additionally, the pore area expanded with cycling, but the pore number did not increase, as evidenced by the trans-mode images acquired at the end of three cycles. The total area of the pores increased and the number of pores decreased with cycling, owing to the expansion and subsequent aggregation of multiple holes (Fig. 5E). In the epi-mode-derived images, the contrast worsened with Li deposition, and the effect was not as evident as that in their trans-mode counterparts (Fig. S19†). The dissolution-induced pores corresponded to areas with higher Li content from the end of the first cycle. Moreover, the bottom of the electrode became uneven in later cycles owing to Li extraction, resulting in a thicker deposited Li layer with inferior contrast.
The difference between the Ag and Au films was that the Ag film only expanded and extended over the pores formed after the first cycle, whereas the Au film randomly formed new Li nucleation sites and generated pores in each cycle. This was the fundamental reason for the preservation and rupture of the Ag and Au film structures, respectively; this finding is consistent with the macroscopic observation in the literature.15 Li corroded the surface of Li–Au and Li–Ag alloys; however, the final outcomes of the Au and Ag films were different owing to the inconsistency of the deposition sites and mechanism. Overall, macroscopic phenomena and the microscopic nature of alloying and dealloying were closely associated using the method reported herein. Additionally, Li alloying with an Ag film verified the universality of this method.
Conclusions
Spatiotemporally and chemically resolved “see-through” operando imaging studies on the electrochemical alloying/dealloying and deposition/dissolution of lithium on gold and silver film electrodes were performed. The dynamic heterogeneity of Li–Au alloying and lithium deposition process was observed, unveiling the unique phenomenon of a complementary two-step alloying evolution pathway and the consequent regulation of Li nucleation sites. The number of Li nucleation sites keeps increasing during the cycling process which could lead to the electrode film fracture eventually. On the other side, the Li–Ag reaction involved solid solution formation, resulting in huge pore generation after multiple charging/discharging processes. Clarifying the mechanisms governing Li alloying/deposition on the surface of metal electrodes will be very beneficial to resolve issues concerning battery performance. Our method can be further utilized as a powerful tool for elucidating the key parameters driving the dynamic morphological and chemical evolution in studies of various battery interfaces.Data availability
The experimental procedures and data are provided in the ESI.†Author contributions
R. H. conceived and supervised the project. D. X. and J. M. designed the experiments. J. M. and D. X. conducted the optical imaging experiments and electrochemical measurements. G. L. performed HIM/SEM characterization. J. M. and G. L. analyzed the data. J. M. and R. H. wrote the paper. J. M., G. L., and D. X. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professor Rui Wen of Institute of Chemistry, Chinese Academy of Sciences for helpful discussions. This work was supported by the National Natural Science Foundation of China (22074059 and 22374064) and start-up fund from SUSTech (Y01216142 and Y01216242).References
- J. F. Qian, B. D. Adams, J. M. Zheng, W. Xu, W. A. Henderson, J. Wang, M. E. Bowden, S. C. Xu, J. Z. Hu and J. G. Zhang, Adv. Funct. Mater., 2016, 26, 7094–7102 CrossRef CAS.
- Y. Tian, Y. L. An, C. L. Wei, H. Y. Jiang, S. L. Xiong, J. K. Feng and Y. T. Qian, Nano Energy, 2020, 78, 105344 CrossRef CAS.
- J. Chen, J. W. Xiang, X. Chen, L. X. Yuan, Z. Li and Y. H. Huang, Energy Storage Mater., 2020, 30, 179–186 CrossRef.
- X. C. Wang, Y. F. He, S. B. Tu, L. Fu, Z. H. Chen, S. Y. Liu, Z. Cai, L. Wang, X. M. He and Y. M. Sun, Energy Storage Mater., 2022, 49, 135–143 CrossRef.
- S. E. Sandoval, J. A. Lewis, B. S. Vishnugopi, D. L. Nelson, M. M. Schneider, F. J. Q. Cortes, C. M. Matthews, J. Watt, M. Tian, P. Shevchenko, P. P. Mukherjee and M. T. McDowell, Joule, 2023, 7, 2054–2073 CrossRef CAS.
- Z. Zeng, W. I. Liang, Y. H. Chu and H. Zheng, Faraday Discuss., 2014, 176, 95–107 RSC.
- K. Yan, Z. D. Lu, H. W. Lee, F. Xiong, P. C. Hsu, Y. Z. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 CrossRef CAS.
- J. W. Xiang, L. X. Yuan, Y. Shen, Z. X. Cheng, K. Yuan, Z. Z. Guo, Y. Zhang, X. Chen and Y. H. Huang, Adv. Energy Mater., 2018, 8, 1802352 CrossRef.
- Y. B. Mo, Y. Gofer, E. J. Hwang, Z. H. Wang and D. A. Scherson, J. Electroanal. Chem., 1996, 409, 87–93 CrossRef.
- H. Dykes, Rosy, D. Sharon, M. Noked and Ö. Çapraz, J. Electrochem. Soc., 2021, 168, 110551 CrossRef CAS.
- P. Bach, M. Stratmann, I. Valencia-Jaime, A. H. Romero and F. U. Renner, Electrochim. Acta, 2015, 164, 81–89 CrossRef CAS.
- P. Bach, I. Valencia-Jaime, U. Rutt, O. Gutowski, A. H. Romero and F. U. Renner, Chem. Mater., 2016, 28, 2941–2948 CrossRef CAS.
- T. L. Kulova, A. M. Skundin, V. M. Kozhevin, D. A. Yavsin and S. A. Gurevich, Russ. J. Electrochem., 2010, 46, 877–881 CrossRef CAS.
- H. Tavassol, M. K. Y. Chan, M. G. Catarello, J. Greeley, D. G. Cahill and A. A. Gewirth, J. Electrochem. Soc., 2013, 160, A888–A896 CrossRef CAS.
- S. M. Zhang, G. J. Yang, Z. P. Liu, S. T. Weng, X. Y. Li, X. F. Wang, Y. R. Gao, Z. X. Wang and L. Q. Chen, ACS Energy Lett., 2021, 6, 4118–4126 CrossRef CAS.
- A. J. Leenheer, K. L. Jungjohann, K. R. Zavadil and C. T. Harris, ACS Nano, 2016, 10, 5670–5678 CrossRef CAS PubMed.
- S. Jin, Y. Ye, Y. Niu, Y. Xu, H. Jin, J. Wang, Z. Sun, A. Cao, X. Wu, Y. Luo, H. Ji and L. J. Wan, J. Am. Chem. Soc., 2020, 142, 8818–8826 CrossRef PubMed.
- S. E. Sandoval, F. J. Q. Cortes, E. J. Klein, J. A. Lewis, P. P. Shetty, D. V. Yeh and M. T. McDowell, J. Electrochem. Soc., 2021, 168, 100517 CrossRef CAS.
- C. Yang, Y. Yao, S. He, H. Xie, E. Hitz and L. Hu, Adv. Mater., 2017, 29, 1702714 CrossRef PubMed.
- R. Zhang, X. Chen, X. Shen, X. Q. Zhang, X. R. Chen, X. B. Cheng, C. Yan, C. Z. Zhao and Q. Zhang, Joule, 2018, 2, 764–777 CrossRef CAS.
- D. P. Finegan, M. Scheel, J. B. Robinson, B. Tjaden, I. Hunt, T. J. Mason, J. Millichamp, M. Di Michiel, G. J. Offer, G. Hinds, D. J. L. Brett and P. R. Shearing, Nat. Commun., 2015, 6, 6924 CrossRef CAS PubMed.
- S. H. Yu, X. Huang, J. D. Brock and H. D. Abruna, J. Am. Chem. Soc., 2019, 141, 8441–8449 CrossRef CAS PubMed.
- M. Sadd, S. Xiong, J. R. Bowen, F. Marone and A. Matic, Nat. Commun., 2023, 14, 854 CrossRef CAS PubMed.
- R. F. Ziesche, T. Arlt, D. P. Finegan, T. M. M. Heenan, A. Tengattini, D. Baum, N. Kardjilov, H. Markotter, I. Manke, W. Kockelmann, D. J. L. Brett and P. R. Shearing, Nat. Commun., 2020, 11, 777 CrossRef CAS PubMed.
- R. F. Ziesche, N. Kardjilov, W. Kockelmann, D. J. L. Brett and P. R. Shearing, Joule, 2022, 6, 35–52 CrossRef CAS.
- R. Bradbury, G. F. Dewald, M. A. Kraft, T. Arlt, N. Kardjilov, J. Janek, I. Manke, W. G. Zeier and S. Ohno, Adv. Energy Mater., 2023, 13, 2203426 CrossRef CAS.
- H. J. Chang, A. J. Ilott, N. M. Trease, M. Mohammadi, A. Jerschow and C. P. Grey, J. Am. Chem. Soc., 2015, 137, 15209–15216 CrossRef CAS PubMed.
- K. Romanenko and A. Jerschow, J. Mater. Chem. A, 2021, 9, 21078–21084 RSC.
- M. Gu, L. R. Parent, B. L. Mehdi, R. R. Unocic, M. T. McDowell, R. L. Sacci, W. Xu, J. G. Connell, P. Xu, P. Abellan, X. Chen, Y. Zhang, D. E. Perea, J. E. Evans, L. J. Lauhon, J. G. Zhang, J. Liu, N. D. Browning, Y. Cui, I. Arslan and C. M. Wang, Nano Lett., 2013, 13, 6106–6112 CrossRef CAS PubMed.
- M. E. Holtz, Y. Yu, D. Gunceler, J. Gao, R. Sundararaman, K. A. Schwarz, T. A. Arias, H. D. Abruna and D. A. Muller, Nano Lett., 2014, 14, 1453–1459 CrossRef CAS PubMed.
- C. Gong, S. D. D. Pu, X. W. Gao, S. X. Yang, J. L. Liu, Z. Y. Ning, G. J. Rees, I. Capone, L. Q. Pi, B. Y. Liu, G. O. Hartley, J. Fawdon, J. Luo, M. Pasta, C. R. M. Grovenor, P. G. Bruce and A. W. Robertson, Adv. Energy Mater., 2021, 11, 2003118 Search PubMed.
- S. W. Tao, M. Li, M. Lyu, L. B. Ran, R. Wepf, I. Gentle and R. Knibbe, Nano Energy, 2022, 96, 107083 CrossRef CAS.
- H. Marceau, C. S. Kim, A. Paolella, S. Ladouceur, M. Lagace, M. Chaker, A. Vijh, A. Guerfi, C. M. Julien, A. Mauger, M. Armand, P. Hovington and K. Zaghib, J. Power Sources, 2016, 319, 247–254 CrossRef CAS.
- G. Rong, X. Zhang, W. Zhao, Y. Qiu, M. Liu, F. Ye, Y. Xu, J. Chen, Y. Hou, W. Li, W. Duan and Y. Zhang, Adv. Mater., 2017, 29, 1606187 CrossRef PubMed.
- C. Y. Chen, A. Sawamura, T. Tsuda, S. Uchida, M. Ishikawa and S. Kuwabata, ACS Appl. Mater. Interfaces, 2017, 9, 35511–35515 CrossRef CAS PubMed.
- C. Shen, G. H. Hu, L. Z. Cheong, S. Q. Huang, J. G. Zhang and D. Y. Wang, Small Methods, 2018, 2, 1700298 CrossRef.
- H. Zhu, J. A. Russell, Z. Fang, P. Barnes, L. Li, C. Efaw, A. Muenzer, J. May, K. Hamal, I. F. Cheng, P. H. Davis, E. Dufek and H. Xiong, Small, 2021, 17, e2105292 CrossRef PubMed.
- Z. Y. Zhang, S. Said, K. Smith, R. Jervis, C. A. Howard, P. R. Shearing, D. J. L. Brett and T. S. Miller, Adv. Energy Mater., 2021, 11, 2101518 CrossRef CAS.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS PubMed.
- D. Ma, Z. Cao and A. Hu, Nano-Micro Lett., 2014, 6, 347–358 CrossRef PubMed.
- S. S. Chi, Q. Wang, B. Han, C. Luo, Y. Jiang, J. Wang, C. Wang, Y. Yu and Y. Deng, Nano Lett., 2020, 20, 2724–2732 CrossRef CAS PubMed.
- F. Xin, H. Zhou, Q. Yin, Y. Shi, F. Omenya, G. Zhou and M. S. Whittingham, ACS Omega, 2019, 4, 4888–4895 CrossRef CAS PubMed.
- C. Wu, J. Maier and Y. Yu, Adv. Funct. Mater., 2015, 25, 3488–3496 CrossRef CAS.
- M. Zhang, L. Xiang, M. Galluzzi, C. Jiang, S. Zhang, J. Li and Y. Tang, Adv. Mater., 2019, 31, e1900826 CrossRef PubMed.
- B. B. Chen, H. Zhang, J. Xuan, G. J. Offer and H. Z. Wang, Adv. Mater. Technol., 2020, 5, 2000555 CrossRef CAS.
- M. Saqib, Y. S. Fan, R. Hao and B. Zhang, Nano Energy, 2021, 90, 106539 CrossRef CAS.
- A. J. Merryweather, C. Schnedermann, Q. Jacquet, C. P. Grey and A. Rao, Nature, 2021, 594, 522–528 CrossRef CAS PubMed.
- R. C. Evans, Z. N. Nilsson and J. B. Sambur, Anal. Chem., 2019, 91, 14983–14991 CrossRef CAS PubMed.
- R. Hao, Z. Peng and B. Zhang, ACS Omega, 2020, 5, 89–97 CrossRef CAS PubMed.
- Y. Fan, R. Hao, C. Han and B. Zhang, Anal. Chem., 2018, 90, 13837–13841 CrossRef CAS PubMed.
- M. Suvira and B. Zhang, Anal. Chem., 2021, 93, 5170–5176 CrossRef CAS PubMed.
- R. Hao, Y. Fan, M. D. Howard, J. C. Vaughan and B. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5878–5883 CrossRef CAS PubMed.
- F. Kanoufi, Encycl. Electrochem., 2021, 26, 1–80 Search PubMed.
- L. Godeffroy, P. Ciocci, J. F. Lemineur and F. Kanoufi, Curr. Opin. Electrochem., 2022, 36, 101165 CrossRef CAS.
- Z. Cai, J. Wang, Z. Lu, R. Zhan, Y. Ou, L. Wang, M. Dahbi, J. Alami, J. Lu, K. Amine and Y. Sun, Angew Chem. Int. Ed. Engl., 2022, 61, e202116560 CrossRef CAS PubMed.
- J. Mao, G. Li, M. Saqib, J. Xu and R. Hao, Chem. Sci., 2022, 13, 12782–12790 RSC.
- G. Li, J. Mao, M. Saqib and R. Hao, Chem.–Asian J., 2022, 17, e202200824 CrossRef CAS PubMed.
- A. J. Sanchez, E. Kazyak, Y. X. Chen, K. H. Chen, E. R. Pattison and N. P. Dasgupta, ACS Energy Lett., 2020, 5, 994–1004 CrossRef CAS.
- V. Shkirskiy and F. Kanoufi, Curr. Opin. Electrochem., 2023, 39, 101259 CAS.
- S. Redor, L. Godeffroy, G. Rousse, A. M. Abakumov, B. Li, F. Kanoufi and J.-M. Tarascon, J. Am. Chem. Soc., 2023, 145, 12823–12836 CAS.
- J. F. Lemineur, J. M. Noël, D. Ausserre, C. Combellas and F. Kanoufi, Angew. Chem., Int. Ed., 2018, 57, 11998–12002 CAS.
- S. Campidelli, R. A. Khachfe, K. Jaouen, J. Monteiller, C. Amra, M. Zerrad, R. Cornut, V. Derycke and D. Ausserré, Sci. Adv., 2017, 3, e1601724 Search PubMed.
- S. Wang, X. Shan, U. Patel, X. Huang, J. Lu, J. Li and N. Tao, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16028–16032 CAS.
- D. Jiang, Y. Y. Jiang, Z. M. Li, T. Liu, X. Wo, Y. M. Fang, N. J. Tao, W. Wang and H. Y. Chen, J. Am. Chem. Soc., 2017, 139, 186–192 CAS.
- J. Wang, H. Hyun, S. Seo, K. Jeong, J. Han, S. Jo, H. Kim, B. Koo, D. Eum, J. Kim, J. Chung, H.-H. Cho, H. N. Han, T. J. Shin, M. Ni, K. Kang and J. Lim, ACS Energy Lett., 2023, 8, 2986–2995 CAS.
- J. Lim, Y. Y. Li, D. H. Alsem, H. So, S. C. Lee, P. Bai, D. A. Cogswell, X. Z. Liu, N. Jin, Y. S. Yu, N. J. Salmon, D. A. Shapiro, M. Z. Bazant, T. Tyliszczak and W. C. Chueh, Science, 2016, 353, 566–571 Search PubMed.
- J. Wang, H. Kim, H. Hyun, S. Jo, J. Han, D. Ko, S. Seo, J. Kim, H. Kong and J. Lim, Small Methods, 2020, 4, 2000551 CAS.
- X. R. Chen, Y. X. Yao, C. Yan, R. Zhang, X. B. Cheng and Q. Zhang, Angew Chem. Int. Ed. Engl., 2020, 59, 7743–7747 CAS.
- X. Q. Ye, M. Saqib, J. X. Mao, G. P. Li and R. Hao, Cell Rep. Phys. Sci., 2021, 2, 100420 CAS.
- A. M. Andersson, A. Henningson, H. Siegbahn, U. Jansson and K. Edstrom, J. Power Sources, 2003, 119, 522–527 Search PubMed.
- Y. Li, Y. Li, A. Pei, K. Yan, Y. Sun, C. L. Wu, L. M. Joubert, R. Chin, A. L. Koh, Y. Yu, J. Perrino, B. Butz, S. Chu and Y. Cui, Science, 2017, 358, 506–510 CAS.
- B. Han, X. Y. Li, S. Bai, Y. C. Zou, B. Y. Lu, M. H. Zhang, X. M. Ma, Z. Chang, Y. S. Meng and M. Gu, Matter, 2021, 4, 3741–3752 CAS.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, C723–C742 CAS.
- J.-S. Kim, G. Yoon, S. Kim, S. Sugata, N. Yashiro, S. Suzuki, M.-J. Lee, R. Kim, M. Badding, Z. Song, J. Chang and D. Im, Nat. Commun., 2023, 14, 782 CAS.
- G. Taillades and J. Sarradin, J. Power Sources, 2004, 125, 199–205 CAS.
- W. J. Zhang, J. Power Sources, 2011, 196, 877–885 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05021a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |