
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper and conjugated carbonyls of metal–organic polymers as dual redox centers for Na storage†
Liubin
Wang
 *a,
Ningbo
Liu
a,
Xiaoying
Zhao
a,
Xiaohan
Wang
a,
Tong
Zhang
c,
Zhiqiang
Luo
*b and
Fujun
Li
*c
*a,
Ningbo
Liu
a,
Xiaoying
Zhao
a,
Xiaohan
Wang
a,
Tong
Zhang
c,
Zhiqiang
Luo
*b and
Fujun
Li
*c
aCollege of Chemistry & Materials Science, Key Laboratory of Analytical Science and Technology of Hebei Province, Hebei University, Baoding 071002, China. E-mail: lbwang@hbu.edu.cn
bTianjin Key Lab for Photoelectric Materials & Devices, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: zhqluo@email.tjut.edu.cn
cFrontiers Science Center for New Organic Matter, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center (RECAST), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: fujunli@nankai.edu.cn
First published on 2nd January 2024
Abstract
Metal–organic polymers (MOPs) are fascinating electrode materials for high-performance sodium-ion batteries due to their multiple redox centers and low cost. Herein, a flower-like π–d conjugated MOP (Cu-TABQ) was synthesized using tetramino-benzoquinone (TABQ) as an organic ligand and Cu2+ as a transition metal node under the slow release of Cu2+ from [Cu(NH3)4]2+ and subsequent dehydrogenation. It possesses dual redox centers of Cu2+/Cu+ and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–O to render a three-electron transfer reaction for each coordination unit with a high reversible capacity of 322.9 mA h g−1 at 50 mA g−1 in the voltage range of 1.0 to 3.0 V. The flower-like structure enhances fast Na+ diffusion and highly reversible organic/inorganic redox centers. This results in excellent cycling performance with almost no degradation within 700 cycles and great rate performance with 198.8 mA h g−1 at 4000 mA g−1. The investigation of the Na-storage mechanism and attractive performance will shed light on the insightful design of MOP cathode materials for further batteries.
O/C–O to render a three-electron transfer reaction for each coordination unit with a high reversible capacity of 322.9 mA h g−1 at 50 mA g−1 in the voltage range of 1.0 to 3.0 V. The flower-like structure enhances fast Na+ diffusion and highly reversible organic/inorganic redox centers. This results in excellent cycling performance with almost no degradation within 700 cycles and great rate performance with 198.8 mA h g−1 at 4000 mA g−1. The investigation of the Na-storage mechanism and attractive performance will shed light on the insightful design of MOP cathode materials for further batteries.
Introduction
Sodium-ion batteries (SIBs) have attracted great attention for large-scale energy storage due to the high abundance and uniform distribution of Na resources.1,2 A wide range of inorganic materials, such as Na3V2(PO4)3, Na0.67MnO2, and Na2FeFe(CN)6, have been attempted as cathodes for SIBs, to deliver capacities below 200 mA h g−1 due to the limited redox of V4+/3+, Mn4+/3+, and Fe3+/2+.3–6 Polymer-based organic electrode materials have been actively studied as host materials for Na+ storage owing to their environmental friendliness and good flexible stability with devisable organic monomers, which could introduce multiple organic/inorganic redox centers to increase theoretical capacity.7–10 However, the electrochemical performances of these polymers are governed by their electrically insulating nature (except for conductive polymers) and disordered molecular chain entanglement, leading to the limited utilization of redox-active sites with low capacity and inferior rate performance.11–13 It is rational to design organic cathode materials with more redox sites and great electrical conductivity for Na+ storage and robust structures for Na+ (de)intercalation for high energy density and reversibility.Redox-active π–d coordination metal–organic polymers (MOPs) exist as the framework structure or linear structure by adjusting the type of inorganic metal nodes and organic ligands, which is favorable for eliminating the dissolution of the hybrid materials in organic electrolytes and avoiding the organic chain agglomeration.14–17 Moreover, the abundantly delocalized electrons and strong metal–ligand interaction bonds in MOPs guarantee high electrical conductivity and good structural stability, promoting the electron transfer to activate redox-active sites with higher capacity.18,19 It is noteworthy that the selection of organic ligands and transition metal centers for MOPs is important to affect the discharge capacity, working potential, and cycling performance. Several typical organic ligands, such as hexaaminobenzene (HAB), 1,2,4,5-tetraaminobenzene (BTA), and 2,3,6,7,10,11-hexaaminotriphenylene (HITP), have been widely used as the anode, while their electrochemical performance is stilled limited by the single active site and low discharge capacity.20–23 Recently, many studies on conjugated carbonyl-derived linkers have indicated to construct a high-capacity MOP-type cathode material because of its low molecular weight, high potential redox sites (carbonyl group, CO), and ortho-diamine groups (–NH2, coordination form of robust metal–nitrogen bonds), like tetramino-benzoquinone (TABQ)16,24–32 This is favorable for increasing the discharge voltage and enhancing the structural stability upon Na+ (de)intercalation. For the transition metal centers, the copper cation (Cu2+) shows stronger competitiveness in terms of cost, toxicity, chemical stability, high redox voltages, and extra capacity compared with other metal ions (e.g., Ni2+, Co2+, Sn2+, etc.).21,33–37 Therefore, it is desirable to combine TABQ and Cu2+ into a MOP skeleton to achieve high energy-density organic cathodes for SIBs and explore the Na+/e− transfer mechanism and structural evolution during the charge/discharge process.
Herein, we synthesized linear Cu-based MOPs using copper (Cu, II) ions as transition metal centers and tetramino-benzoquinone molecules (TABQ, C6H8O2N4) as organic ligands in a mild process. It is formed with a coplanar π–d delocalization skeleton in each unit of Cu-TABQ to enhance Na+ diffusion. Active metal ions (Cu2+) and organic ligands (benzoquinone framework) act as the dual redox centers for Na storage, leading to three Na+ transfers per coordination unit, as revealed in in situ FTIR and ex situ XPS. This provides a high reversible capacity of 322.9 mA h g−1 at 50 mA g−1 within voltages of 1.0–3.0 V for Cu-TABQ. The results show that the Cu-TABQ is an ideal candidate for a high-performance cathode to construct Na-organic batteries and also provides a new direction for the future development of metal-ion batteries.
Results and discussion
The Cu-TABQ MOPs were synthesized via coordination reactions between Cu2+ and TABQ in dimethylsulfoxide (DMSO) with ammonia under ambient air, as shown in Fig. 1a. The coordination chains are gradually developed and eventually form the coplanar structure and polymeric chain by four N atoms and one Cu2+ coordination, which is accompanied by the oxidative dehydrogenation of TABQ ligands by O2 and the release of Cu2+ from [Cu(NH3)4]2+ in Fig. S1a and b.† Cu-TABQ exhibits π–d conjugated coordination due to the hybridization between the π orbitals of TABQ and the d orbitals of Cu2+, which will effectively enhance the structural stability and electronic conductivity of the coordination compound.38 Compared with TABQ, Cu-TABQ presents a lower LUMO energy level and narrow HOMO–LUMO energy gap (ΔEg) in Fig. S1c,† indicating enhanced electronic delocalization and increased electron accepting ability. Besides, the Cu-TABQ powder shows an obvious metallic sheen in the optical photo (Fig. S2†). The molecular structure of the designed Cu-TABQ was investigated by various characterization methods including Fourier transform infrared (FTIR) spectroscopy, Raman spectroscopy, and solid-state 1H nuclear magnetic resonance (1H NMR) spectroscopy. As shown in Fig. 1b, the characteristic triple vibration signal of –NH2 in the region of 3000–3500 cm−1 in the FTIR spectrum of TABQ disappears after coordination, while a new peak (–NH–) appears at around 3276 cm−1 for Cu-TABQ due to the reaction of oxidative dehydrogenation. The coexistence of benzoid amine (C–N) and quinoid imine (CN) peaks at 1264 and 1450 cm−1 in the FTIR spectra of Cu-TABQ indicates the deprotonation of TABQ to form a new coordination bond with Cu2+ and the possession of unpaired electrons around N atoms. This can be confirmed by the red-shifted peak at 1664 cm−1 for the carbonyl bond (CO) in the organic ligand to 1579 cm−1 for TABQ with coordination and attraction between chains.
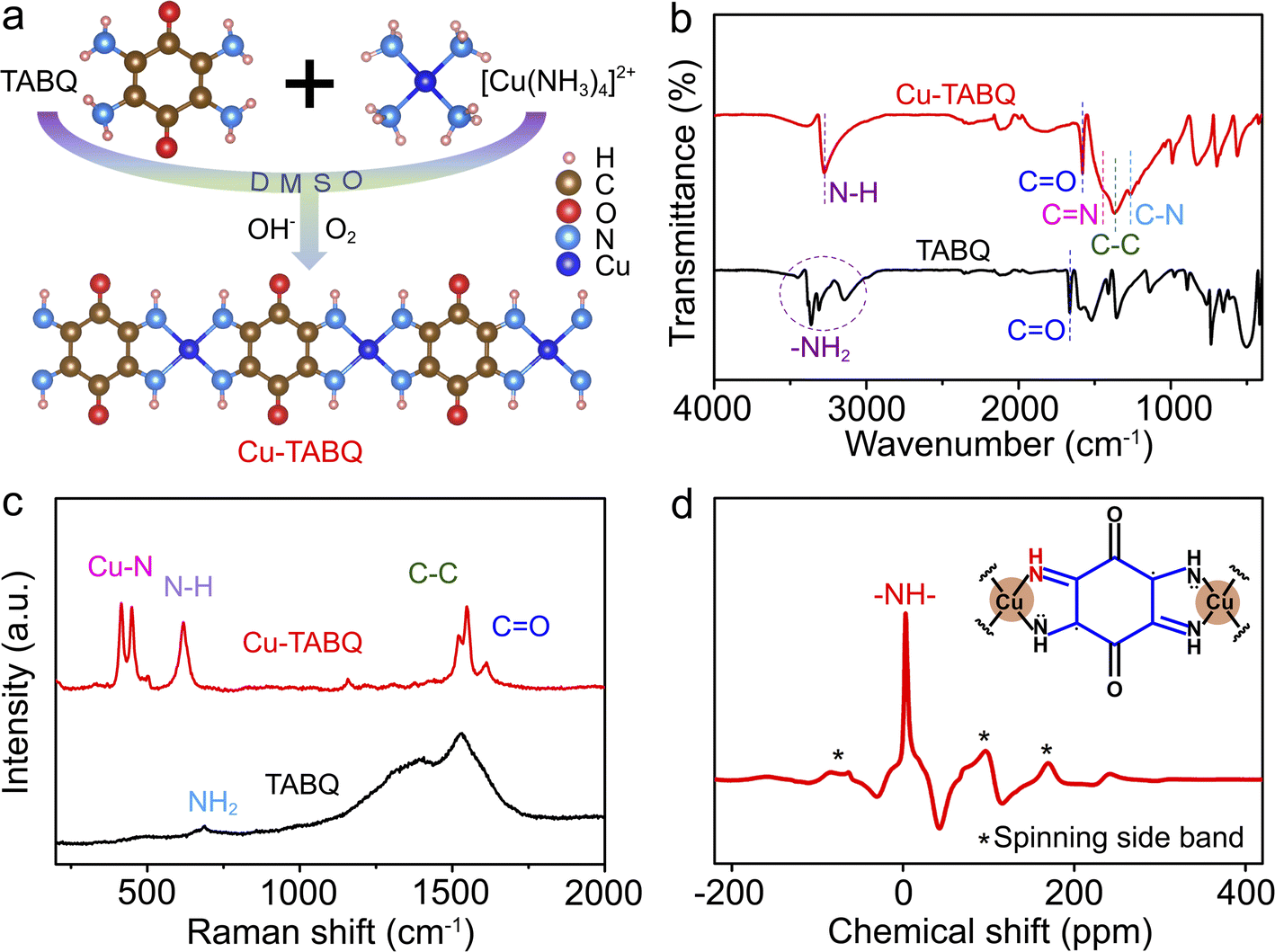 | ||
| Fig. 1 (a) Synthetic scheme of Cu-TABQ; (b) FTIR and (c) Raman spectra of TABQ and Cu-TABQ; (d) solid-state 1H NMR spectrum of Cu-TABQ. | ||
The chemical state of N and O in the Cu-TABQ structure is analyzed by X-ray photoelectron spectroscopy (XPS) in Fig. S3 and S4.† The high-resolution N1s and O1s spectra confirmed the coexistence of C–N and CN bonds and the presence of CO in Cu-TABQ.30,39 The chemical moieties of coordination between Cu2+ and the TABQ ligand can be probed by the emergence of stretching vibrations of Cu–N (414 and 449 cm−1) in the Raman spectrum (Fig. 1c).30,40 The structure information of Cu-TABQ was further verified using the solid-state 1H NMR spectrum. In Fig. 1d, an obvious 1H signal at 3.1 ppm is assigned to hydrogen in the –NH– bond, further confirming the successful synthesis of MOPs with TABQ and Cu2+. The satellite peaks in the Cu 2p spectrum in Fig. S5† reveal the divalent state of Cu. The addition peak at slightly lower binding energy in Cu-TABQ compared to the main Cu 2p3/2 peaks indicates the presence of Cu(I).20 Furthermore, a strong EPR signal with a g-factor of 2.0002 was observed in the sample (Fig. S6†), indicative of the presence of ligand radicals which is consistent with the previously reported MOPs.17,20,32 These suggest that Cu2+ is bonded to TABQ via the coordination bond of Cu–N for Cu-TABQ.
The crystal structure of Cu-TABQ was studied by powder X-ray diffraction (PXRD) and simulated by Rietveld refinement in Fig. 2a. The diffraction peaks located at 22.96°, 28.12°, 35.74°, and 40.20° are assigned to the (210), (111), (40![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (33
) and (33![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) facets, respectively. The refined unit cell parameters for Cu-TABQ are shown in Table S1,† specifically, a = 6.98253 Å, b = 6.98253 Å, c = 3.22703 Å, α = γ = 92.2166°, β = 67.5636°, and V = 145.2689 Å3. Fig. 2b shows the planar and layered packing motif of Cu-TABQ, in which there exists a large planar structure through Cu–N square-planar coordination and hydrogen bonds between chains along the ab plane and the stack cambiums into layered structures along the c-axis. Density functional theory (DFT) calculation was used to investigate the charge configuration and coordination bonds between Cu2+ and TABQ in Cu-TABQ. There is significant overlapping between the d orbitals of Cu atoms and the p orbitals of N atoms in the projected density of states (PDOS) of Cu-TABQ (Fig. 2c), suggesting their strong orbital hybridization. The π–d conjugation of Cu2+ and N atoms with square planar coordination manner guarantees the rigid and coplanar nature of the Cu-TABQ chains, which favors the electronic conductivity of Cu-TABQ and hence enhances the redox activity.
) facets, respectively. The refined unit cell parameters for Cu-TABQ are shown in Table S1,† specifically, a = 6.98253 Å, b = 6.98253 Å, c = 3.22703 Å, α = γ = 92.2166°, β = 67.5636°, and V = 145.2689 Å3. Fig. 2b shows the planar and layered packing motif of Cu-TABQ, in which there exists a large planar structure through Cu–N square-planar coordination and hydrogen bonds between chains along the ab plane and the stack cambiums into layered structures along the c-axis. Density functional theory (DFT) calculation was used to investigate the charge configuration and coordination bonds between Cu2+ and TABQ in Cu-TABQ. There is significant overlapping between the d orbitals of Cu atoms and the p orbitals of N atoms in the projected density of states (PDOS) of Cu-TABQ (Fig. 2c), suggesting their strong orbital hybridization. The π–d conjugation of Cu2+ and N atoms with square planar coordination manner guarantees the rigid and coplanar nature of the Cu-TABQ chains, which favors the electronic conductivity of Cu-TABQ and hence enhances the redox activity.
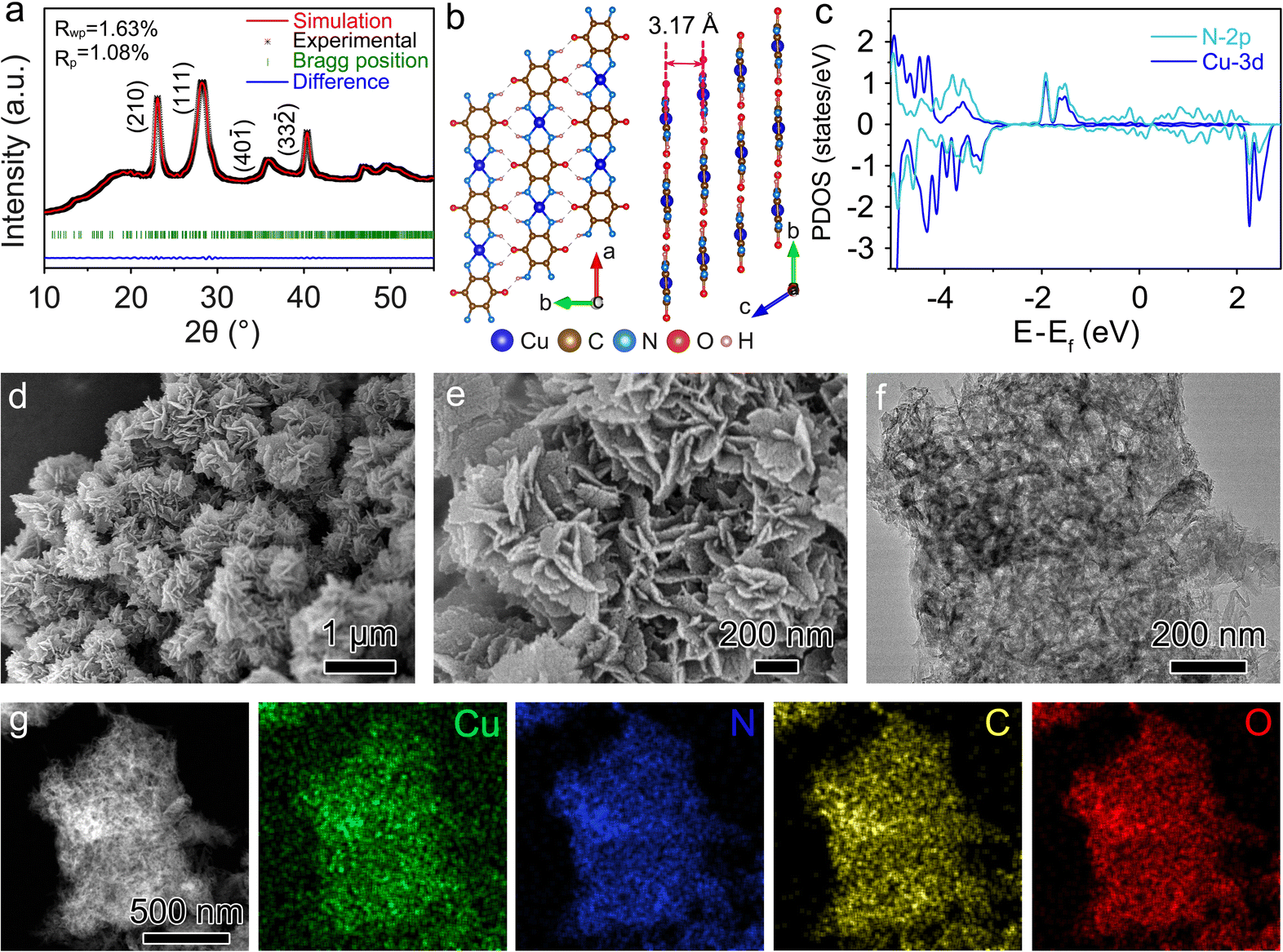 | ||
| Fig. 2 (a) PXRD patterns and Rietveld refinement; (b) diagram of the planar and layered packing motif; (c) PDOS; (d) and (e) SEM images; (f) TEM image; (g) EDS mappings of Cu-TABQ. | ||
The morphology of Cu-TABQ can be controlled by different reaction temperatures. Previous reports have demonstrated that oxygen could increase the possibility of coordination between Cu2+ and TABQ molecules, and finally promote the formation of the cross-linked coordination polymer.21 As shown in Fig. S7–S9,† the coordination temperatures facilely affect the crystallinity and morphology of Cu-TABQ. The integrated SEM and TEM images of Cu-TABQ present 0-dimensional nanoparticles and one-dimensional short rod structures at 30 °C and 60 °C respectively, with a low reaction rate. Whereas the flower-like structure of two-dimensional nanosheets formed at 90 °C for Cu-TABQ because of the fast reaction rate and self-assembly characteristics (Fig. 2d, e, and S10†).41 The TEM image in Fig. 2f shows that Cu-TABQ has higher crystallization at 90 °C with a flower-like shape, which is consistent with XRD results in Fig. S7.† Moreover, the high-resolution TEM (HRTEM) image of Cu-TABQ reveals periodic lattice fringes with an interplanar spacing of 0.32 nm (Fig. S11†), which is assigned to the (001) facet and consistent with the XRD pattern in Fig. 2a. The TEM-EDS-mapping images of Cu-TABQ show the uniform distribution of Cu, C, N, and O elements in Fig. 2g, which is consistent with the elemental analysis results (Table S2†). The Brunauer–Emmett–Teller (BET) curve of Cu-TABQ is obtained from the N2 adsorption isotherm and shown in Fig. S12,† which displays a type II isothermal curve with a large specific surface area of 118.49 m2 g−1. This promises more Na+ storage sites and favors the penetration of electrolytes into cathode materials. The thermal stability of Cu-TABQ was investigated using a thermogravimetric analyzer (TGA) in Fig. S13.† There is ∼3% weight loss at about 180 °C due to the residual DMSO solvent evaporation. Cu-TABQ shows better thermal stability than that of TABQ, which is attributed to the π–d conjugation of Cu2+ and TABQ after coordination. Diffuse reflectance ultraviolet-visible (UV-vis) spectroscopy was used to further explore the electron delocalization and metal–ligand conjugation of Cu-TABQ. As shown in Fig. S14,† Cu-TABQ shows a strong absorption around 439 nm, which could be attributed to the ligand-centered (LC) π–π* transition.42 Its maximum absorption is red-shifted with extended absorption compared with the pristine TABQ revealing electron delocalization between Cu2+ and TABQ and the metal–ligand d–π conjugation.32,42,43 Further, the electronic conductivity of Cu-TABQ was obtained based on I–V curves in Fig. S15† to show a high value of 1.02 × 10−3 S m−1 ensuring good rate capability in batteries.
The Na-storage performances of Cu-TABQ were tested by assembling CR2032 coin cells using Na metal anodes. Cyclic voltammetry (CV) was performed in the voltage range from 1.0 to 3.0 V at 0.1 mV s−1 in Fig. 3a. The irreversible phenomenon in the first cycle is mainly caused by the formation of the SEI film.19 One broad reduction peak located at around 1.55 V in the first cathodic scan originated from the reduction of both Cu2+ and the organic ligand. There are two broad oxidation peaks at 1.7 and 2.4 V attributed to oxidation of C–O bonds and Cu+ in the initial cycle. In the subsequent cycles, two pairs of overlapped redox peaks located at 1.42/1.68 and 1.73/2.31 V are observed, which can be assigned to the redox reaction of the CO/C–O bonds and Cu2+/Cu+, respectively. Fig. 3b shows sloping charge/discharge curves of Cu-TABQ with no clear plateaus at 50 mA g−1 between 1.0 and 3.0 V in selected cycles, which is consistent with the CV curves. It delivers an initial discharge capacity of 426.8 mA h g−1 with a coulombic efficiency of 75.6% in the first cycle. The first 50 cycles show similar overlapping charge/discharge curves, indicating the stable structure of Cu-TABQ during cycling.
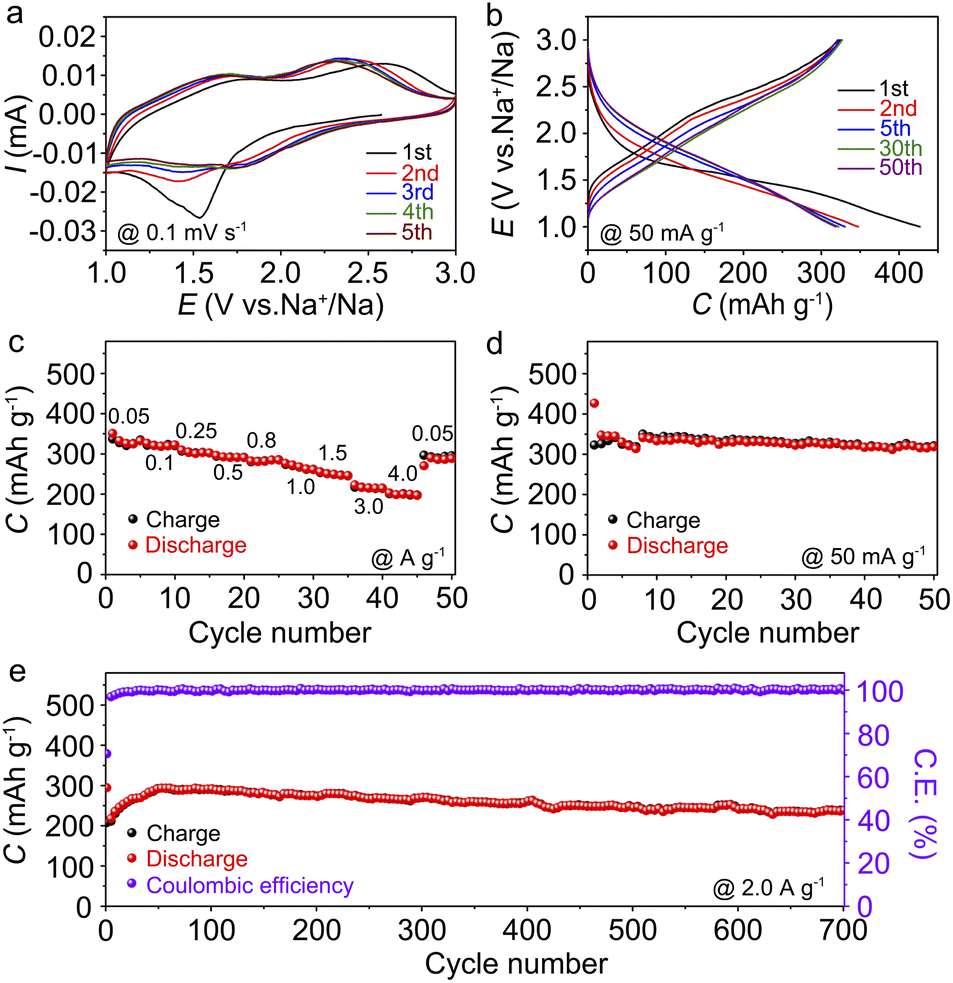 | ||
| Fig. 3 (a) CV curves at 0.1 mV s−1; (b) discharge/charge curves of initial 50 cycles at 50 mA g−1; (c) rate capability from 0.05 to 4.0 A g−1; (d and e) cycling performance of Cu-TABQ at 50 and 2.0 A g−1. | ||
The rate performance of the Cu-TABQ electrode is tested at current densities from 50 to 4000 mA g−1 (Fig. 3c and S16†). It shows the specific capacity of 350.8 mA h g−1 at 50 mA g−1, revealing a three-electron redox process for every coordination unit. Upon cycling at 4000 mA g−1, Cu-TABQ exhibits a capacity of 198.8 mA h g−1, corresponding to 56.7% of its capacity at 50 mA g−1. This excellent rate performance can be attributed to the unique flower-like structure ensuring fast electrolyte infiltration and the inherent π–d conjugated structure providing high conductivity, and thus fast reaction kinetics of Cu-TABQ. The cycling performance was evaluated at 50 mA g−1 to achieve a high capacity of 318.8 mA h g−1 after 50 cycles with a capacity retention of 98.7% in Fig. 3d. After cycling, there is no obvious color change from the disassembled cells (Fig. S17†), which indicates insolubility of the discharged/charged products in the electrolyte. Remarkably, it delivers a capacity of 238.3 mA h g−1 without obvious degradation after 700 cycles when tested at 2000 mA g−1 in Fig. 3e, indicating a high rate stability. The Cu-TABQ electrode shows excellent electrochemical performance in terms of charge/discharge capacity, capacity retention, and rate capability, as compared to the reported metal–organic polymer cathode electrodes (Table S3†), suggesting its promising application in SIBs.
To further understand the chemical states and Na+ storage mechanism of Cu-TABQ, in situ FTIR, ex situ XPS, ex situ Raman, and computational simulations were carried out. In situ FTIR was performed in the voltage range from 1.0 to 3.0 V in the first cycle at 50 mA g−1, as shown in Fig. 4a. During the whole oxidation/reduction process, Cu-TABQ shows a sloping charge/discharge curve, corresponding to a fast reaction kinetics of the Na+ (de)intercalation process due to the dual inorganic (Cu2+) and organic (CO) active sites. In the initial discharge process, the signal peak of CO (1577 cm−1) gradually decreases and eventually disappears, while a new stretching vibration peak is formed around 1199.3 cm−1 that is assigned to C–O bonds. After being fully charged, the intensity of the stretching vibration peak of CO gradually increases and resumes its original positions without any new phases, suggesting the highly reversible redox reactions of CO bonds. The peaks of –N–H at 3276 cm−1 and the C–N bond at 1081 cm−1 exhibit similar behaviour to that of CO bonds, because the insertion of Na+ changes the coordination environment of N and Cu atoms and weakens the N–H⋯Cu(Na+)/N–H⋯N(Na+) hydrogen bonding interaction. During the charging process, the vibrational peak of the amine group appeared again with the extraction of Na+. At the same time, the disappeared peak at 1081 cm−1 and the redshift C–N stretching band after discharging were observed in FTIR spectra, which is related to the π–d conjugation electron rearrangement of the C–N/CN bond after discharge.
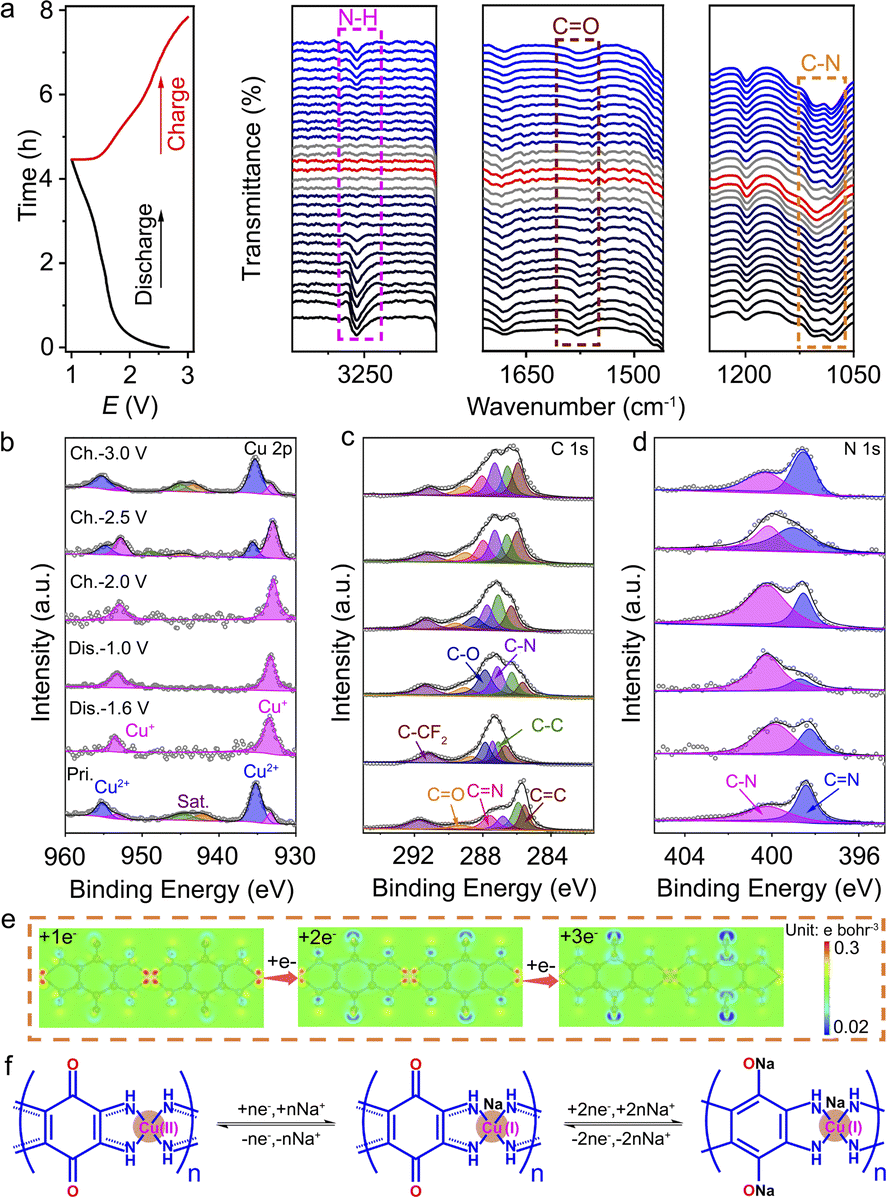 | ||
| Fig. 4 (a) Discharge/charge profile and in situ FTIR spectra of Cu-TABQ; (b–d) ex situ Cu 2p, C 1s, and N 1s XPS spectra of Cu-TABQ; (e) extra charge distribution upon reduction of Cu-TABQ after injection of one, two, and three electrons; and (f) schematic diagram of the Na-storage mechanism. | ||
Ex situ XPS of Cu-TABQ was conducted in Fig. 4b–d, S18, and S19† to investigate the redox reaction and chemical state of the organic ligand. In its original state, a small amount of Cu+ ions at 933.4 eV was detected due to the partially reduced Cu2+ in Cu-TABQ during electrode preparation. When the electrode is discharged to 1.6 V, strong satellite peaks of the Cu 2p spectrum in the pristine electrode disappear, and the peak values of Cu 2p3/2 and Cu 2p1/2 shift to the smaller binding energies of 933.4 and 953.4 eV, indicating the reduction of Cu2+ to Cu+ in Cu-TABQ during the Na+ intercalation process. The Cu 2p spectrum is still dominated by Cu+ further discharged to 1.0 V, which is similar to the redox reaction of Cu2+/Cu+ in other conjugated coordination compounds.31,37 After being fully charged to 3.0 V, the Cu-TABQ can be returned to the initial state. Besides, the C 1s spectra are presented in Fig. 4c, and the peak of the CO bond disappears while the peak of the C–O bond becomes stronger during discharge from 1.6 to 1.0 V, which confirms that CO groups were involved in the redox process. This suggests that the CO bond site acts as one redox centre for Na+ storage. In contrast, the CN bond was strengthened and the C–N bond was weakened after discharge, which is caused by the electron redistribution of C–N/CN bond upon Na+ intercalation.14,44 An Auger electron peak of Na at 536.3 eV can be observed in the O 1s spectrum in Fig. S19† after discharge, corresponding to the sodiation reaction in Cu-TABQ. The N 1s spectrum of Cu-TABQ is shown in Fig. 4c. At the end of discharge, the peak value of the C–N single bond at 400.2 eV increases, while the peak value of the CN double bond at 398.4 eV decreases, which is mainly related to the electron changes in the whole molecular structure, caused by Na+ intercalation in Cu-TABQ.
Fig. S20† shows the Raman spectra of Cu-TABQ in the charging and discharging process. The tensile vibration of the quinone ring remains relatively stable, while the Cu–N bond is weakened and disappears, as in the case of the CO double bond. This indicates that Cu is involved in the redox reaction as the active metal site, and in agreement with the ex situ XPS in Fig. 4b. The ex situ XRD patterns of the Cu-TABQ electrode at different charge/discharge states were recorded to study the structural evolution. As shown in Fig. S21,† the diffraction peaks located at 28.1° (111) shift slightly leftward to 26.8° when discharged to 1.0 V, indicating the increased interlayer (3.3 Å) caused by the Na+ inserted into the space of Cu-TABQ. When it was recharged to 3.0 V, the lattice parameters returned to the initial states, indicating a good reversibility of Cu-TABQ. The molecular electrostatic potential (MESP) of electron-injected Cu-TABQ was further calculated in Fig. 4e to reveal the redox reaction. When one extra electron is injected into the Cu-TABQ unit, the extra electrons tend to locate around N atoms, suggesting the priority of Cu2+ reduction. After another two extra electrons are injected into the Cu-TABQ unit, the extra electrons tend to locate around C and O atoms, indicating the subsequent reduction of CO. Fig. 4f schematically illustrates the Na+ storage pathway of Cu-TABQ via a three-electron redox pathway that involves Cu2+/Cu+ and CO/C–O. Furthermore, Cu-TABQ can undergo a 5 electron transfer reaction and display a high reversible capacity of 776.3 mA h g−1, when the voltage region is extended to 0.1–3.0 V in Fig. S22 and S23,† which is similar to Ni-TABQ.16
The Na+ kinetic behaviour of Cu-TABQ is investigated using CV curves at different scanning rates (Fig. 5a). The shape of the CV curve is almost identical as the scan rate increases. However, the cathodic peaks move towards higher values, and the anodic peaks move negatively, which are caused by polarization. The relationship between the peak current (ip) and scan rate(ν) can be described using the following equation: ip = aνb (lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ip = lga + blgν), where a and b are adjustable parameters. The linear relationship between the peak current and scan rate is depicted in Fig. 5b. The b values of O1, O2, and R1 are 0.86, 0.80, and 0.88, respectively, illustrating that the charge storage process is collaboratively controlled by faradaic intercalation and surface pseudocapacitance.45 The electrochemical reaction kinetics of Cu-TABQ is studied using the galvanostatic intermittent titration technique (GITT), as shown in Fig. 5c. The calculated Na+ diffusion coefficient of the Cu-TABQ cathode is 1.51 × 10−13 to 1.51 × 10−11 cm2 s−1, which is consistent with that based on CV curves at different scan rates in Fig. 5a and EIS in Fig. S24.† DFT calculations were further applied to analyse the diffusion behavior. The Na+ diffusion pathways in crystal structures of Na3Cu-TABQ are shown in Fig. 5d and e. There are two Na+ diffusion pathways in Na3Cu-TABQ, one is across chains (path a) and the other is along the layers (path b). The calculated migration activation energies of the pathways are plotted in Fig. 5f. The diffusion energy barrier along the layers is much smaller than that across chains, resulting in a faster Na+ diffusion and agreeing with the GITT in Fig. 5c. This suggests that the fast diffusion kinetics of Na+ in Cu-TABQ is attributed to the distinctive migration pathways and the large interlayer spacing between MOP chains.
ip = lga + blgν), where a and b are adjustable parameters. The linear relationship between the peak current and scan rate is depicted in Fig. 5b. The b values of O1, O2, and R1 are 0.86, 0.80, and 0.88, respectively, illustrating that the charge storage process is collaboratively controlled by faradaic intercalation and surface pseudocapacitance.45 The electrochemical reaction kinetics of Cu-TABQ is studied using the galvanostatic intermittent titration technique (GITT), as shown in Fig. 5c. The calculated Na+ diffusion coefficient of the Cu-TABQ cathode is 1.51 × 10−13 to 1.51 × 10−11 cm2 s−1, which is consistent with that based on CV curves at different scan rates in Fig. 5a and EIS in Fig. S24.† DFT calculations were further applied to analyse the diffusion behavior. The Na+ diffusion pathways in crystal structures of Na3Cu-TABQ are shown in Fig. 5d and e. There are two Na+ diffusion pathways in Na3Cu-TABQ, one is across chains (path a) and the other is along the layers (path b). The calculated migration activation energies of the pathways are plotted in Fig. 5f. The diffusion energy barrier along the layers is much smaller than that across chains, resulting in a faster Na+ diffusion and agreeing with the GITT in Fig. 5c. This suggests that the fast diffusion kinetics of Na+ in Cu-TABQ is attributed to the distinctive migration pathways and the large interlayer spacing between MOP chains.
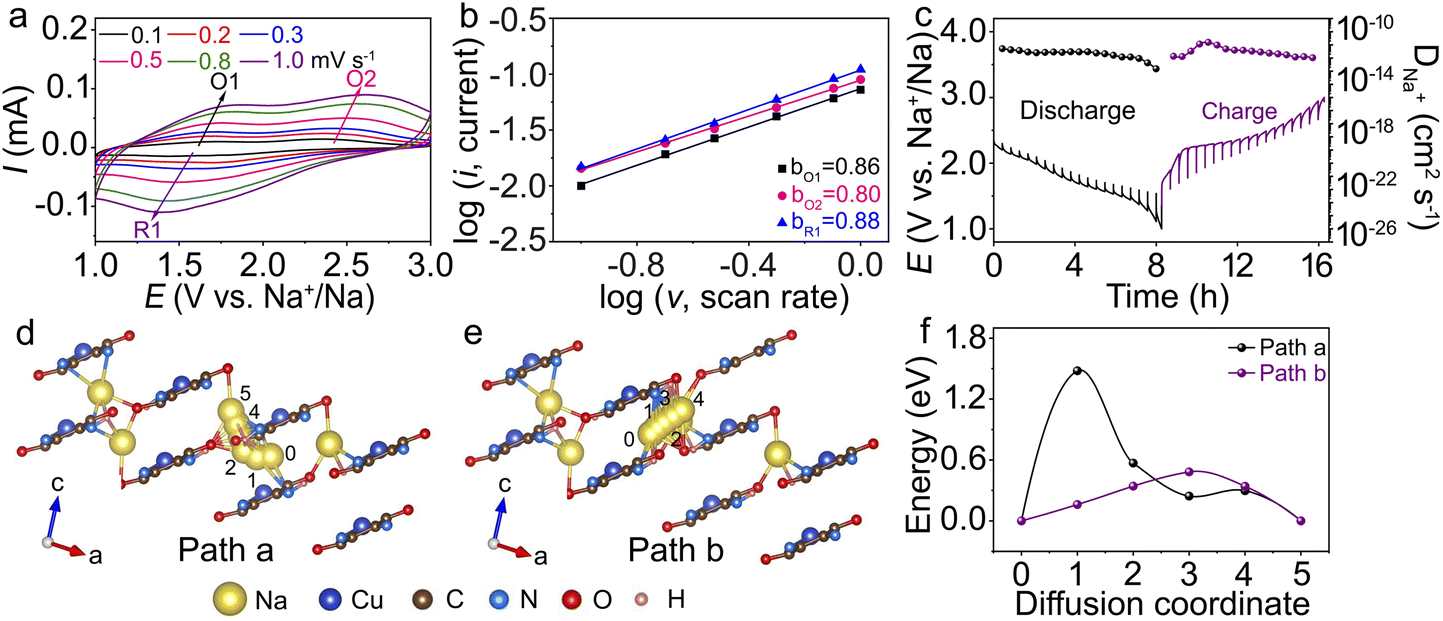 | ||
| Fig. 5 (a) CV curves of Cu-TABQ at scan rates from 0.1 to 1.0 mV s−1; (b) relationship between different peak currents and scan rates of Cu-TABQ; (c) GITT curves and Na+ diffusion coefficient of Cu-TABQ at 50 mA g−1; (d and e) Na+ diffusion pathways in Na3Cu-TABQ (path a across chains, path b along the layers) and (f) diffusion energy barrier. | ||
Conclusions
In summary, a two-dimensional π–d conjugated coordination polymer (Cu-TABQ) is successfully synthesized by a simple wet chemical method and used as a cathode material for Na+ storage. The physicochemical properties, electrochemical properties and sodium storage mechanism of Cu-TABQ are exhaustively studied. It delivers a high reversible capacity of 322.9 mA h g−1 with capacity retention up to 98.7% after 50 cycles. At a high current density of 4000 mA g−1, the capacity can still reach 198.8 mA h g−1. This can be attributed to the stable coordination between Cu2+ and the TABQ ligand with π–d conjugation, which greatly improves the electronic conductivity and hence redox activity for Na+ storage in Cu-TABQ. By means of FTIR, Raman, XPS, and other characterization methods, the sodium storage mechanism is revealed. It is found that Cu-TABQ has dual redox centers of Cu2+/Cu+ (one electron) and CO/C–O (two electrons) for three-electron transfer for each unit and increasing the specific capacity. This shows the great potential of constructing high-performance coordination polymer cathode materials with multiple redox centres and will facilitate the development of advanced cathodes for SIBs.
Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
Liubin Wang: conceptualization, supervision, software, writing – review & editing, project administration. Ningbo Liu: investigation, validation, data curation, formal analysis, writing – original draft. Xiaoying Zhao: software, methodology, formal analysis. Xiaohan Wang: methodology, formal analysis. Tong Zhang: formal analysis. Zhiqiang Luo: supervision, formal analysis, writing – review & editing. Fujun Li: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (22109037, 21905205, and 52171215), Advanced Talents Incubation Program of the Hebei University (521000981408), Chunhui Program of Ministry of Education of China (HZKY20220251), Key Laboratory of Analytical Science and Technology of Hebei Province (22567620H), Post-graduate's Innovation Fund Project of Hebei Province (CXZZSS2023006), and the Laboratory Open Funds of Hebei University (sy202219) is acknowledged.Notes and references
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, A cost and resource analysis of sodium-ion batteries, Nat. Rev. Mater., 2018, 3, 1–11 CrossRef.
- H. S. Hirsh, Y. Li, D. H. Tan, M. Zhang, E. Zhao and Y. S. Meng, Sodium-ion batteries paving the way for grid energy storage, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- J. Xiao, X. Li, K. Tang, D. ang, M. Long, H. Gao, W. Chen, C. Liu, H. Liu and G. Wang, Recent progress of emerging cathode materials for sodium ion batteries, Mater. Chem. Front., 2021, 5, 3735–3764 RSC.
- Y. Liu, J. Li, Q. Shen, J. Zhang, P. He, X. Qu and Y. Liu, Advanced characterizations and measurements for sodium-ion batteries with NASICON-type cathode materials, eScience, 2022, 2, 10–31 CrossRef.
- N. Liu, X. Zhao, B. Qin, D. Zhao, H. Dong, M. Qiu and L. Wang, A high-performance Na-storage cathode enabled by layered P2-type KxMnO2 with enlarged interlayer spacing and fast diffusion channels for sodium-ion batteries, J. Mater. Chem. A, 2022, 10, 25168–25177 RSC.
- J. Peng, W. Zhang, Q. Liu, J. Wang, S. Chou, H. Liu and S. Dou, Prussian blue analogues for sodium-ion batteries: past, present, and future, Adv. Mater., 2022, 34, 2108384 CrossRef CAS PubMed.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Organic batteries for a greener rechargeable world, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef.
- L. Huang, Z. Yu, L. Wang, B. Qin, F. Cai, Z. Yuan and Z. Luo, Spiro-based triphenylamine molecule with steric structure as a cathode material for high-stable all organic lithium dual-ion batteries, J. Energy Chem., 2023, 83, 24–31 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Polymer-based organic batteries, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- J. C. Barbosa, A. Fidalgo-Marijuan, J. C. Dias, R. Gonçalves, M. Salado, C. M. Costa and S. Lanceros-Méndez, Molecular design of functional polymers for organic radical batteries, Energy Storage Mater., 2023, 60, 102841 CrossRef.
- C. Peng, G. H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M. F. Ng, Y. S. Hu, Y. Yang, M. Armand and K. P. Loh, Reversible multi-electron redox chemistry of π-conjugated N-containing heteroaromatic molecule-based organic cathodes, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
- Z. Sang, J. Liu, X. Zhang, L. Yin, F. Hou and J. Liang, One-dimensional π–d conjugated conductive metal–organic framework with dual redox-active sites for high-capacity and durable cathodes for aqueous zinc batteries, ACS Nano, 2023, 17, 3077–3087 CrossRef CAS PubMed.
- S. Lee, J. Hong and K. Kang, Redox-active organic compounds for future sustainable energy storage system, Adv. Energy Mater., 2020, 10, 2001445 CrossRef CAS.
- K. Li, J. Yu, Z. Si, B. Gao, H. G. Wang and Y. Wang, One-dimensional π–d conjugated coordination polymer with double redox-active centers for all-organic symmetric lithium-ion batteries, Chem. Eng. J., 2022, 450, 138052 CrossRef CAS.
- K. Fan, C. Zhang, Y. Chen, G. Zhang, Y. Wu, J. Zou and C. Wang, Regulating the metal nodes of 1D conjugated coordination polymers for enhancing the performance of sodium-ion batteries, J. Mater. Chem. C, 2022, 10, 2592–2599 RSC.
- L. Wang, Y. Ni, X. Hou, L. Chen, F. Li and J. Chen, A two-dimensional metal-organic polymer enabled by robust nickel-nitrogen and hydrogen bonds for exceptional sodium-ion storage, Angew. Chem., Int. Ed., 2020, 59, 22126–22131 CrossRef CAS PubMed.
- Y. Chen, Q. Zhu, K. Fan, Y. Gu, M. Sun, Z. Li, C. Zhang, Y. Wu, Q. Wang, S. Xu, J. Ma, C. Wang and W. Hu, Successive storage of cations and anions by ligands of π–d-conjugated coordination polymers enabling robust sodium-ion batteries, Angew. Chem., Int. Ed., 2021, 133, 18917–18924 CrossRef.
- J. Liu, Y. Chen, X. Feng and R. Dong, Conductive 2D conjugated metalorganic framework thin films: synthesis and functions for (opto-)electronics, Small Struct., 2022, 3, 2100210 CrossRef CAS.
- L. S. Xie, G. Skorupskii and M. Dincă, Electrically conductive metal–organic frameworks, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- J. Park, M. Lee, D. Feng, Z. Huang, A. C. Hinckley, A. Yakovenko, X. Zou, Y. Cui and Z. Bao, Stabilization of hexaaminobenzene in a 2D conductive metal–organic framework for high power sodium storage, J. Am. Chem. Soc., 2018, 140, 10315–10323 CrossRef CAS PubMed.
- Y. Chen, M. Tang, Y. Wu, X. Su, X. Li, S. Xu, S. Zhou, J. Ma, D. Yuan, C. Wang and W. Hu, A one-dimensional π–d conjugated coordination polymer for sodium storage with catalytic activity in negishi coupling, Angew. Chem., Int. Ed., 2019, 131, 14873–14881 CrossRef.
- A. Nazir, H. T. Le, A. G. Nguyen and C. J. Park, Graphene analogue metal organic framework with superior capacity and rate capability as an anode for lithium ion batteries, Electrochim. Acta, 2021, 389, 138750 CrossRef CAS.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dincă, Conductive MOF electrodes for stable supercapacitors with high areal capacitance, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- R. Rajagopalan, Y. Tang, C. Jia, X. Ji and H. Wang, Understanding the sodium storage mechanisms of organic electrodes in sodium ion batteries: issues and solutions, Energy Environ. Sci., 2020, 13, 1568–1592 RSC.
- J. Geng, Y. Ni, Z. Zhu, Q. Wu, S. Gao, W. Hua, S. Indris, J. Chen and F. Li, Reversible metal and ligand redox chemistry in two-dimensional iron-organic framework for sustainable lithium-ion batteries, J. Am. Chem. Soc., 2023, 145, 1564–1571 CrossRef CAS PubMed.
- T. Chen, H. Banda, L. Yang, J. Li, Y. Zhang, R. Parenti and M. Dincă, High-rate, high-capacity electrochemical energy storage in hydrogen-bonded fused aromatics, Joule, 2023, 7, 986–1002 CrossRef CAS.
- J. Yu, X. Chen, H. G. Wang, B. Gao, D. Han and Z. Si, Conjugated ladder-type polymers with multielectron reactions as high-capacity organic anode materials for lithium-ion batteries, Sci. China Mater., 2022, 65, 2354–2362 CrossRef CAS.
- C. Zhang, Z. Li, L. Guan, M. Fu, K. Fan, Y. Chen, G. Zhang, J. Zou and C. Wang, Anti-aggregation triggering molecular transformation and boosting stable sodium storage, Cell Rep. Phys. Sci., 2023, 4, 101290 CrossRef CAS.
- Z. Lin, H. Y. Shi, L. Lin, X. Yang, W. Wu and X. Sun, A high capacity small molecule quinone cathode for rechargeable aqueous zinc-organic batteries, Nat. Commun., 2021, 12, 4424 CrossRef CAS PubMed.
- Q. Lv, Z. Zhu, S. Zhao, L. Wang, Q. Zhao, F. Li, L. A. Archer and J. Chen, Semiconducting metal–organic polymer nanosheets for a photoinvolved Li-O2 battery under visible light, J. Am. Chem, Soc., 2021, 143, 1941–1947 CrossRef CAS PubMed.
- F. Zhang, P. Wang, R. Zhao, Y. Wang, J. Wang, B. Han and Z. Liu, Tuning d-band structure of CuII in coordinated polymer via d–π conjugation for improving CO2 electroreduction selectivity toward C2 products, ChemSusChem, 2022, 15, e202201267 CrossRef CAS PubMed.
- T. Chen, J.-H. Dou, L. Yang, C. Sun, J. J. Oppenheim, J. Li and M. Dincă, Dimensionality modulates electrical conductivity in compositionally constant one-, two-, and three-dimensional frameworks, J. Am. Chem. Soc., 2022, 144, 5583–5593 CrossRef CAS PubMed.
- R. Wei, Y. Dong, Y. Zhang, R. Zhang, M. A. Al-Tahan and J. Zhang, In situ self-assembled hollow urchins F-Co-MOF on rGO as advanced anodes for lithium-ion and sodium-ion batteries, J. Colloid Inter. Sci., 2021, 582, 236–245 CrossRef CAS PubMed.
- J. Liu, D. Xie, X. Xu, L. Jiang, R. Si, W. Shi and P. Cheng, Reversible formation of coordination bonds in Sn-based metal–organic frameworks for high-performance lithium storage, Nat. Commun., 2021, 12, 3131 CrossRef CAS PubMed.
- Z. Liu, F. Zheng, W. Xiong, X. Li, A. Yuan and H. Pang, Strategies to improve electrochemical performances of pristine metal–organic frameworks-based electrodes for lithium/sodium-ion batteries, SmartMat, 2021, 2, 488–518 CrossRef CAS.
- Y. Luo, J. Liu and L. Zhang, A monocrystalline coordination polymer with multiple redox centers as a high-performance cathode for lithium-ion batteries, Angew. Chem., Int. Ed., 2022, 61, e202209458 CrossRef CAS PubMed.
- Q. Chen, O. Adeniran, Z. F. Liu, Z. Zhang and K. Awaga, Graphite-like charge storage mechanism in a 2D π–d conjugated metal-organic framework revealed by stepwise magnetic monitoring, J. Am. Chem. Soc., 2023, 145, 1062–1071 CrossRef CAS PubMed.
- K. Sakaushi and H. Nishihara, Two-dimensional π-conjugated frameworks as a model system to unveil a multielectron-transfer-based energy storage mechanism, Acc. Chem. Res., 2021, 54, 3003–3015 CrossRef CAS PubMed.
- A. Mohtasebi, T. Chowdhury, L. H. Hsu, M. C. Biesinger and P. Kruse, Interfacial charge transfer between phenyl-capped aniline tetramer films and iron oxide surfaces, J. Phys. Chem. C, 2016, 120, 29248–29263 CrossRef CAS.
- D. Pakulski, V. Montes-García, A. Gorczyński, W. Czepa, T. Chudziak, M. Bielejewski, A. Musiał, I. Perez-Juste, P. Samorì and A. Ciesielski, Two-dimensional metal-organic polymers as cathode hybrid materials for high-performance Al-batteries, J. Mater. Chem. A, 2023, 12(1), 440–450 RSC.
- C. Wang, H. Dong, L. Jiang and W. Hu, Organic semiconductor crystals, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- K. Fan, C. Fu, Y. Chen, C. Zhang, G. Zhang, L. Guan, M. Mao, J. Ma, W. Hu and C. Wang, Framework dimensional control boosting charge storage in conjugated coordination polymers, Adv. Sci., 2023, 10, 2205760 CrossRef CAS PubMed.
- X. Wu, Y. Qiu, Z. Chen, B. Guan, X. Hao, A. I. Rykov, Y. Sun, L. Liu, Y. Zou, J. Sun, W. Xu and D. Zhu, Paramagnetic conducting metal-organic frameworks with three-dimensional structure, Angew. Chem., Int. Ed., 2020, 59, 20873–20878 CrossRef CAS PubMed.
- M. Mohammadi Ghaleni, A. Al Balushi, S. Kaviani, E. Tavakoli, M. Bavarian and S. Nejati, Fabrication of Janus membranes for desalination of oil-contaminated saline water, ACS Appl. Mater. Interfaces, 2018, 10, 44871–44879 CrossRef CAS PubMed.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, High-rate electrochemical energy storage through Li+ intercalation pseudocapacitance, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05023h |
| This journal is © The Royal Society of Chemistry 2024 |