
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlleviating the competitive adsorption of hydrogen and hydroxyl intermediates on Ru by d–p orbital hybridization for hydrogen electrooxidation†
Youkai
Feng
a,
Siguang
Lu
a,
Luhong
Fu
b,
Fulin
Yang
*a and
Ligang
Feng
 *a
*a
aSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225002, China. E-mail: yangfl@yzu.edu.cn; ligang.feng@yzu.edu.cn
bCollege of Materials Science and Engineering, Huaqiao University, Xiamen, Fujian 361021, China
First published on 29th December 2023
Abstract
Strengthening the hydroxyl binding energy (OHBE) on Ru surfaces for efficient hydrogen oxidation reaction (HOR) in alkaline electrolytes at the expense of narrowing the effective potential window (EPW) increases the risk of passivation under transient conditions for the alkaline exchange membrane fuel cell technique. Herein, an effective Ru/NiSe2 catalyst was reported which exhibits a gradually enhanced intrinsic activity and slightly enlarged EPW with the increased degree of coupling between Ru and NiSe2. This promotion could be attributed to the optimized electron distribution and d-band structures of Ru surfaces weakening the hydrogen binding energy and especially the OHBE through the strong d–p orbital hybridization between Ru and NiSe2. Unlike the conventional way of strengthened OHBE enhancing the oxidative desorption of hydrogen intermediates (Had) via the bi-functional mechanism, the weakened OHBE on this Ru/NiSe2 model catalyst alleviates the competitive adsorption between Had and the hydroxyl intermediates (OHad), thereby accelerating the HOR kinetics at low overpotentials and hindering the full poisoning of the catalytic surfaces by strongly adsorbed OHad spectators at high overpotentials. The work reveals a missed but important approach for Ru-based catalyst development for the fuel cell technique.
Introduction
Alkaline exchange membrane fuel cells (AEMFCs) are regarded as next-generation technology for hydrogen-to-electric energy conversion.1–3 One of the most important advantages is that the electrocatalysts for the cathodic oxygen reduction reaction could get rid of the dependence on platinum group metals (PGMs) compared to the proton exchange membrane-based counterpart operated under harsh acidic conditions.4–6 Nevertheless, the anodic hydrogen oxidation reaction (HOR) in high pH electrolytes suffers from more complex catalytic mechanisms and thus sluggish kinetics.7,8 Especially for benchmarked commercial Pt/C, two orders of magnitude lowered catalytic activity results in the incremental usage of PGMs, inevitably raising the system cost.9 Therefore, developing highly effective and low-cost alkaline HOR catalysts is critically desirable for the further commercialization of AEMFCs.Ru is a promising alternative catalyst in alkaline HOR because of its moderate activity but significantly lower price.10–12 Compared to Pt, elementary Ru possesses slightly stronger hydrogen binding energy (HBE) and greatly enhanced oxophilicity (hydroxyl binding energy, OHBE), which are two crucial thermodynamic descriptors related to hydrogen electrocatalysis.10,13 Meanwhile, the relationship between the alkaline HOR catalytic behaviors and the OHBE of the Ru surfaces is still under debate.14–19 Conventionally, Ru due to its exceedingly strong oxophilicity is susceptible to being occupied by oxygenated species like the adsorbed hydroxyl (OHad) in a high coverage.11,14,20 In this case, the OHad species would act as the spectators and inhibit the dissociative adsorption of H2. Hence, Ru-based catalysts usually exhibit a narrowed effective potential window (EPW) for the HOR, falling short of the needed 0.3 V vs. a reversible hydrogen electrode (RHE) to greatly reduce the risk of passivation under transient conditions for practical AEMFC devices.21,22 Another opinion is that the OHBE of Ru is still insufficient especially when near the hydrogen equilibrium potential (∼0 V vs. RHE).18,19,23 Appropriate enhancement of the OHBE could also promote the desorption of Had through the bi-functional pathways, that is, the enriched oxygenated species at the electrode/electrolyte interfaces could participate in the reaction directly to accelerate the formation of H2O. For example, strengthening the OHBE of the Ru surface via either material design (like alloying and doping)17–19 or electrolyte engineering (like the alkali metal cation effect)14,24 can indeed improve the HOR activity. These two contradictory viewpoints make it difficult to provide a general strategy to optimize Ru-based catalysts. Moreover, the enhanced OHBE would also result in the ultimately narrowed EPW due to the fully adsorbed OHad poisoning the active sites for H2 dissociative adsorption, which seems closely related to the OHBE but has been less discussed.24 It could be argued that the OHBE enhancement approaches are at the expense of the EPW to accelerate the alkaline HOR kinetics at low overpotentials. Hence, revealing the exact functions of OHad species on Ru surfaces towards alkaline HOR is of great importance to understanding the reaction mechanism and guiding the design of high-performance catalysts without narrowing the EPW.
In this work, a support-optimization strategy is conducted to investigate the alkaline HOR catalytic behaviors of Ru-based catalysts as well as the roles of OHad species. NiSe2, a typical transition metal selenide with good electrical conductivity,25 chemical stability, and relatively weak oxophilicity,26,27 has been adopted to gradually replace the commercial carbon black supports to anchor Ru nanocrystals, denoted as Ru/NiSe2/C-X (where X = 10, 20, 30, or 80, representing the mass percent of NiSe2 in the catalysts). Among them, Ru/NiSe2/C-20 exhibits a 2–3 times enhanced mass activity compared to Ru/C. It is found that the strong interactions between Ru and NiSe2 lead to significant electron transfer from NiSe2 to Ru, which is in contrast to what is observed for carbon-supported Ru catalysts. Both experiments and density functional theory (DFT) calculations confirm the weakened HBE and especially OHBE, resulting in improved HOR activity near the hydrogen equilibrium potential and a broadened EPW. Two catalytic modes have been proposed to describe the roles of weakly/strongly adsorbed OHad on Ru-based catalysts, elucidating the OHBE-related EPW and the reason that either weakening or strengthening the OHBE can accelerate the alkaline HOR kinetics.
Results and discussion
To obtain the well-defined structures of the support surfaces, NiSe2 octahedrons were synthesized via a solvothermal method with the exposure of (111) facets.28 The cubic phase and morphology could be confirmed by the X-ray diffraction (XRD) pattern (PDF#65-1843) and scanning electron microscope (SEM) image shown in Fig. S1.† Then, a series of Ru/NiSe2/C-X catalysts were constructed through microwave reduction29 in the presence of RuCl3 as the precursor and NiSe2 octahedrons or carbon blacks as the supports (see the Experimental section in the ESI† for details). Fig. 1a shows the XRD patterns of the Ru/NiSe2/C-X catalysts. Among them, Ru/NiSe2/C-80 is simply denoted as Ru/NiSe2 due to no extra addition of carbon supports. Apart from Ru/C which was prepared without adding any selenide during the synthesis, all the samples exhibit distinct diffraction peaks of cubic NiSe2. However, there is no obvious pattern of Ru species possibly due to the small size and weak crystallinity, consistent with that of Ru/C. X-ray photoelectron spectroscopy (XPS) was further performed to explore the surface components and chemical states, which confirmed the existence of Ru, Ni, Se, C, and O species (Fig. S2†). To avoid the interferences from the signal of C 1s, Fig. 1b exhibits the high-resolution XPS spectra of Ru 3p for the five samples.30,31 The two groups of characteristic peaks located at around 462 and 485 eV could be attributed to Ru 3p3/2 and 3p1/2 for metallic Ru0, respectively.31 Interestingly, these peaks gradually shift to negative binding energy with the increased amount of NiSe2, from 462.2 eV of Ru 3p3/2 for Ru/C to 461.1 eV for Ru/NiSe2. This suggests the formation of electron-rich Ru surfaces owing to the interactions between the metals and the supports that cause partial electron transfer from NiSe2 to Ru.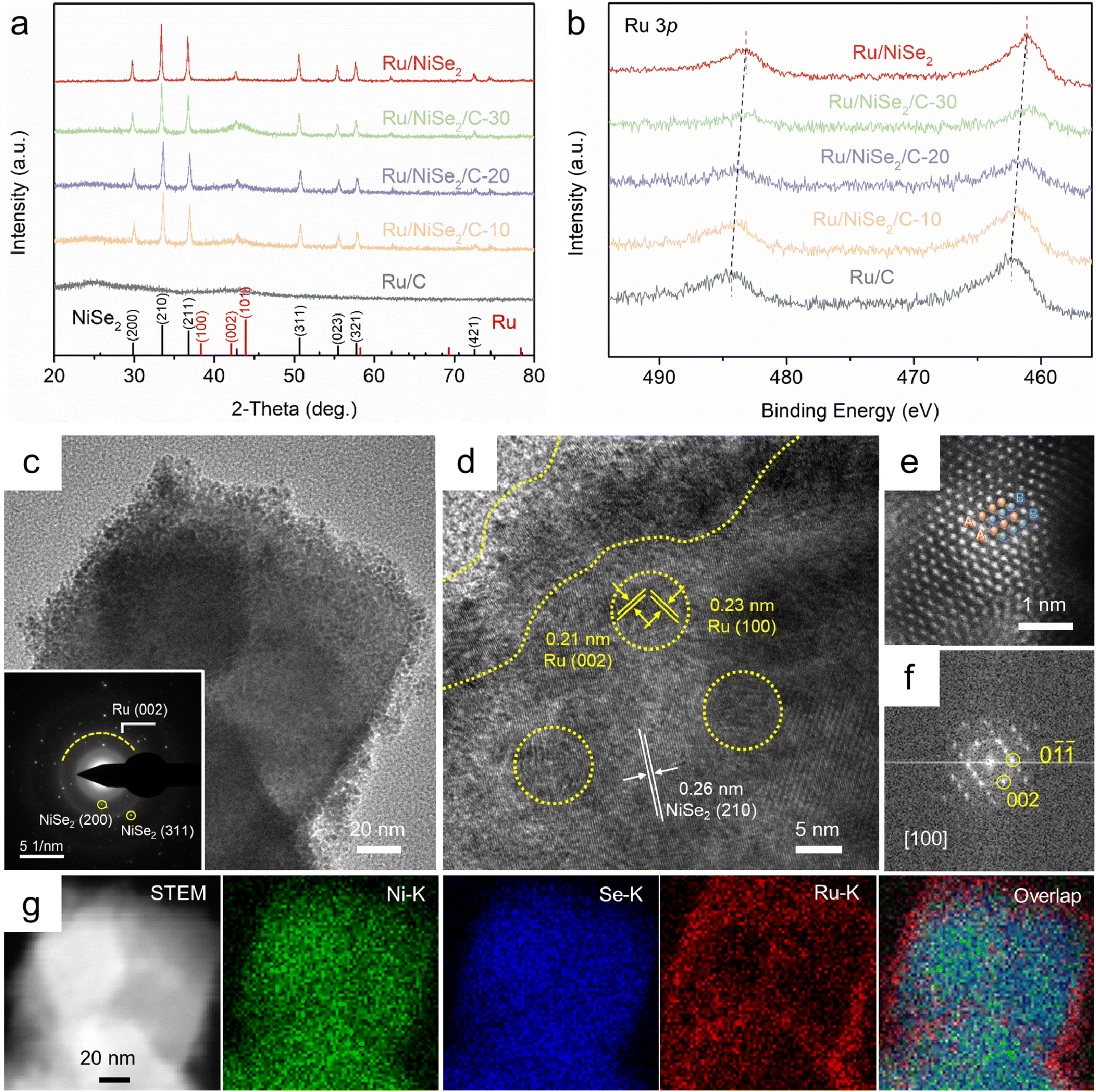 | ||
| Fig. 1 (a) XRD patterns and (b) high-resolution XPS spectra of Ru 3p for Ru/C, Ru/NiSe2/C-10, Ru/NiSe2/C-20, Ru/NiSe2/C-30, and Ru/NiSe2. (c) TEM (inset: SAED pattern) and (d) HRTEM images of Ru/NiSe2/C-20, where Ru nanocrystals are marked by dotted yellow lines and circles. (e) HAADF-STEM image. (f) Corresponding FFT pattern of a single Ru nanocrystal. (g) EDS mapping images of Ru/NiSe2/C-20. | ||
Transmission electron microscopy (TEM) was conducted to further confirm the structures of Ru/NiSe2/C-20. Fig. 1c and S3† show that small Ru nanoparticles with an average size of 2.7 nm are well dispersed on the surfaces of NiSe2 octahedrons. Similar structures could also be observed according to Fig. S4† for Ru/NiSe2, suggesting the anchoring effect of NiSe2 to obtain the ultra-fine Ru nanoparticles. The selected area electron diffraction (SAED) pattern of Ru/NiSe2/C-20 shows the polycrystalline diffraction character where the ring corresponds to the (002) plane of Ru, and the two spots marked by yellow circles correspond to the (200) and (311) planes of NiSe2 (inset of Fig. 1c). For comparison, the TEM image of Ru/C is exhibited in Fig. S5,† showing the Ru nanoparticles with an average size of 3.2 nm dispersed on carbon supports. The high-resolution TEM (HRTEM) image of Ru/NiSe2/C-20 shown in Fig. 1d presents the distinctly exposed interplanar spacing of 0.26 nm inside the polyhedron, corresponding to the (210) facet of NiSe2. Small Ru nanocrystals exist at the surface of NiSe2 as marked by dotted yellow lines, showing the interplanar spacings of 0.21 and 0.23 nm, corresponding to the (002) and (100) facets of hexagonal close-packed (hcp) Ru. The high-angle annular dark field scanning TEM (HAADF-STEM) image as shown in Fig. 1e clearly displays the atomic stacking of “ABABAB…”, indicating the formation of the thermodynamically stable hcp structure, which could be further confirmed by the corresponding fast Fourier transform (FFT) pattern along the [100] zone axis (Fig. 1f).32 The energy dispersive X-ray spectroscopy (EDS) elemental mapping images depict the quasi-core@shell structure, demonstrating the supported Ru nanocrystals on the surfaces of NiSe2 octahedrons (Fig. 1g). The atomic ratios of Ru/Ni for the four samples of Ru/NiSe2/C-X (X = 10, 20, and 30) and Ru/NiSe2 were determined by EDS to be 2.73, 1.89, 1.30, and 0.47, respectively (Table S1†).
To evaluate the alkaline HOR performance of the as-prepared catalysts, rotating disk electrode (RDE) measurements in a standard three-electrode system were carried out in 0.1 M KOH solution. The exact mass ratios of Ru were quantified by inductively coupled plasma-atomic emission spectroscopy (ICP-AES), in which the loadings of Ru on the electrodes for all these samples are presented in Table S2.†Fig. 2a presents the negatively scanned HOR polarization curves at the rotating speed of 1600 rpm in an H2-saturated solution. Meanwhile, the current density would almost disappear in the same potential range in an N2-saturated electrolyte (Fig. S6†), verifying the faradaic process of hydrogen oxidation. The apparent current density gradually rises with the addition of NiSe2 supports, leading to Ru/NiSe2/C-20 exhibiting the highest apparent activity and an enlarged EPW compared to Ru/C. Further increasing the content of NiSe2 would degrade the catalytic performance. Fig. S7† shows the cyclic voltammetric (CV) curves of Ru/NiSe2/C-20 and NiSe2/C, in which no characteristic redox peaks of Ni or Se species could be seen during the HOR. This means that NiSe2 would not contribute to the CV of Ru/NiSe2/C-20. Typically, the kinetic current density (jk) for Ru/NiSe2/C-20 was analyzed to circumvent the effect of mass (H2)-transport by performing the electrochemical tests at different rotating rates from 2500 to 625 rpm (Fig. S8†). The corresponding Koutecky–Levich plot shows a slope of 14.6 cm2 mA−1 rpm−1/2, similar to the theoretical value of 14.8 cm2 mA−1 rpm−1/2,33 indicating a two-electron process of the HOR. Therefore, the jk value after being normalized by the mass of Ru (jk,m) for Ru/NiSe2/C-20 could be extracted according to the Koutecky–Levich equation,34 which reaches 2.51 mA μgRu−1 at the overpotential of 50 mV and is higher than that of Ru/C (0.97 mA μgRu−1), Ru/NiSe2/C-10 (1.90 mA μgRu−1), Ru/NiSe2/C-30 (1.80 mA μgRu−1), Ru/NiSe2 (0.32 mA μgRu−1), and most of the reported Ru-based catalysts (Fig. 2b, and Tables S2 and S3†).
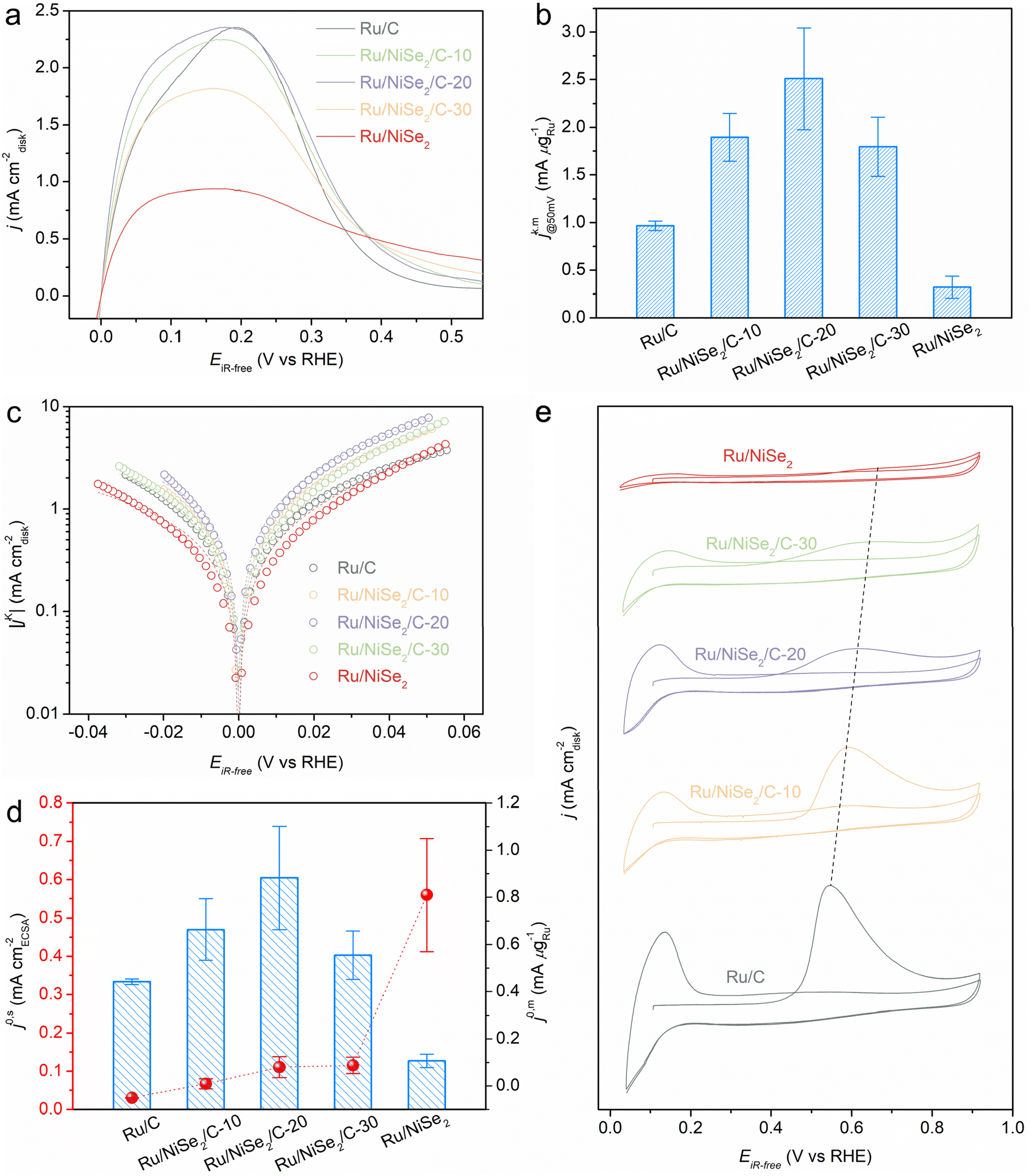 | ||
| Fig. 2 (a) HOR polarization curves and (b) corresponding jk,m values at 0.05 V vs. RHE of Ru/C, Ru/NiSe2/C-10, Ru/NiSe2/C-20, Ru/NiSe2/C-30, and Ru/NiSe2. (c) Tafel plots with Butler–Volmer fitting lines and (d) extracted exchange current densities normalized by the ECSA and the mass of Ru, respectively. (e) CO-stripping curves of the five samples. | ||
Exchange current density (j0) is an important parameter to evaluate the performance of the HOR, which could be extracted from the Tafel plot (Fig. 2c) according to the Butler–Volmer equation.35,36 A similar trend is obtained for the j0 values normalized by the mass of Ru (j0,m) for the above catalysts, in which Ru/NiSe2/C-20 possesses the highest j0,m of 0.88 mA μgRu−1 and surpasses most of the reported Ru-based catalysts (Fig. 2d, Tables S2 and S3†). To reveal the reason for the formation of this volcanic relationship between the mass activities and the contents of NiSe2 supports, electrochemically active surface areas (ECSAs) were further measured via the CO-stripping experiments, showing that the ECSA value decreased gradually from Ru/C to Ru/NiSe2 (Fig. 2e). This is because the specific surface area of the large-sized NiSe2 octahedrons is smaller than that of the carbon blacks, leading to the reduced active sites on the supported Ru nanoparticles. To determine the intrinsic performances of these catalysts, the j0 values are normalized to the corresponding ECSAs to get the specific activities (j0,s). As shown in Fig. 2d, j0,s monotonically increases with the growing content of NiSe2. Ru/NiSe2 exhibits the highest j0,s of 0.56 mA cmECSA−2, which is 18.7 times that of Ru/C, indicating the improvement from NiSe2 octahedrons through the metal–support interactions. Exchange current densities could also be obtained from the linear fitting at the micro-polarization range (−5 to 5 mV vs. RHE),37,38 resulting in similar data as shown in Fig. S9 and Table S2.† Therefore, it is suggested that the monotonically decreased ECSA and monotonically increased j0,s with the addition of incremental content of NiSe2 (that is, the increased coupling degree between Ru and NiSe2) is conducive to the volcanic curve of the mass activities. Specifically, Ru/NiSe2/C-20 shows the highest jk,m and j0,m values, which are 2.6 and 2.0 times those of Ru/C, respectively. In addition, the stability of Ru/NiSe2/C-20 was evaluated by an accelerated durability test (ADT). The HOR polarization curve after 1000 cycles shows negligible change compared to the initial one, indicating excellent stability (Fig. S10†). XRD and TEM characterizations further prove the unchanged structures of the sample after ADT (Fig. S11 and S12†). The chemical state of Ru/NiSe2/C-20 was further characterized by XPS spectra. After 1000 CV cycles, an extra peak appears at 292.1 eV (Fig. S13a†), which could be attributed to the C–F bond from the Nafion used as the binder (see the Experimental section in the ESI†). To avoid interference from C 1s, Ru 3p spectra of the sample before and after the stability test were analyzed (Fig. S13b and Tables S4, S5, S6, and S7†). While there are no obvious changes in the peak positions about Ru 3p3/2 and Ru 3p3/2, the content of Ru0 is increased but it is reduced for the Ru3+, respectively, suggesting a surface reduction process occurred. Therefore, the ADT tests would not lead to the irreversible formation of Ru oxides. The spectral signals of Ni and Se seem too weak to be deconvoluted because of the quasi core (NiSe2 octahedrons)–shell (Ru nanoparticles) structure as well as the insufficient detection depth of the XPS technique. The peaks of Ni 2p at around 856.3 eV and 873.8 eV are still perceptible,39 and the extra peak at ∼862.5 eV is the F KLL due to the existence of perfluorinated sulfonic acid resin from the Nafion binder (Fig. S13c†). Similar to Ru 3p, the Se oxides in the Se 3d spectrum (∼58.8 eV) nearly disappear after ADT tests. Meanwhile, the more pronounced peak of Se–Ni located at ∼55.2 eV can be visible (Fig. S13d†). Therefore, it could be concluded that Ru/NiSe2/C-20 possessed good stability during the ADT tests.
Based on the above analysis of physical characterizations and electrochemical tests, it is speculated that the intrinsic activity of Ru-based catalysts towards alkaline HOR could be facilitated by NiSe2 supports. For the sake of corroborating this viewpoint, DFT calculations and further experiments were executed. Ru clusters with 13 atoms supported on NiSe2(111) or graphene(001) slabs were constructed as the models for simulating Ru/NiSe2 and Ru/C, respectively (Fig. 3a). The geometry-optimized structures show that the Ru13 cluster would be partially embedded into NiSe2(111), which has not been observed on Ru/C. This indicates stronger interactions between Ru and NiSe2 with a calculated “formation energy” (FE) of −12.37 eV compared to those between Ru and the carbon layer (FE = −2.19 eV) as shown in Fig. 3b. Differential charge density distributions confirm that more electrons are transferred between Ru and NiSe2 compared to Ru and the carbon layer (Fig. S14†). Table S8 and Fig. S15† present the Mulliken charge of each Ru atom in the two models. The average Mulliken charges (Fig. 3c) testify to the opposite directions of charge transfer where partial electrons transfer from NiSe2 to Ru but from Ru to the carbon layer, in agreement with the XPS analysis (Fig. 1b). In consideration of Se as a p-block element that has a similar 3p atomic orbital level to the 4d of Ru, a d–p interaction might exist between Ru clusters and NiSe2 supports.40,41 To verify this, the partial density of states (PDOS) of Ru/NiSe2 and Ru/C models has been analyzed as depicted in Fig. 3d. It is found that the energies of Ru 4d and Se 3p bands match better than those of Ru 4d and C 2p bands near the Fermi level (Ef), denoting the strong d–p orbital hybridization between Ru and NiSe2. Moreover, the d-band centers of Ru clusters were also calculated (Fig. 3e), which are usually used as a descriptor to predict the catalytic performance according to the d-band theory proposed by Nørskov et al.42,43 Compared to Ru/C, Ru/NiSe2 shows a slightly downshifted d-band center. Therefore, the dramatically changed electron distribution and d-band structure induced by the d–p orbital coupling between Ru and NiSe2 might optimize the binding energy to the key intermediates such as Had and OHad.
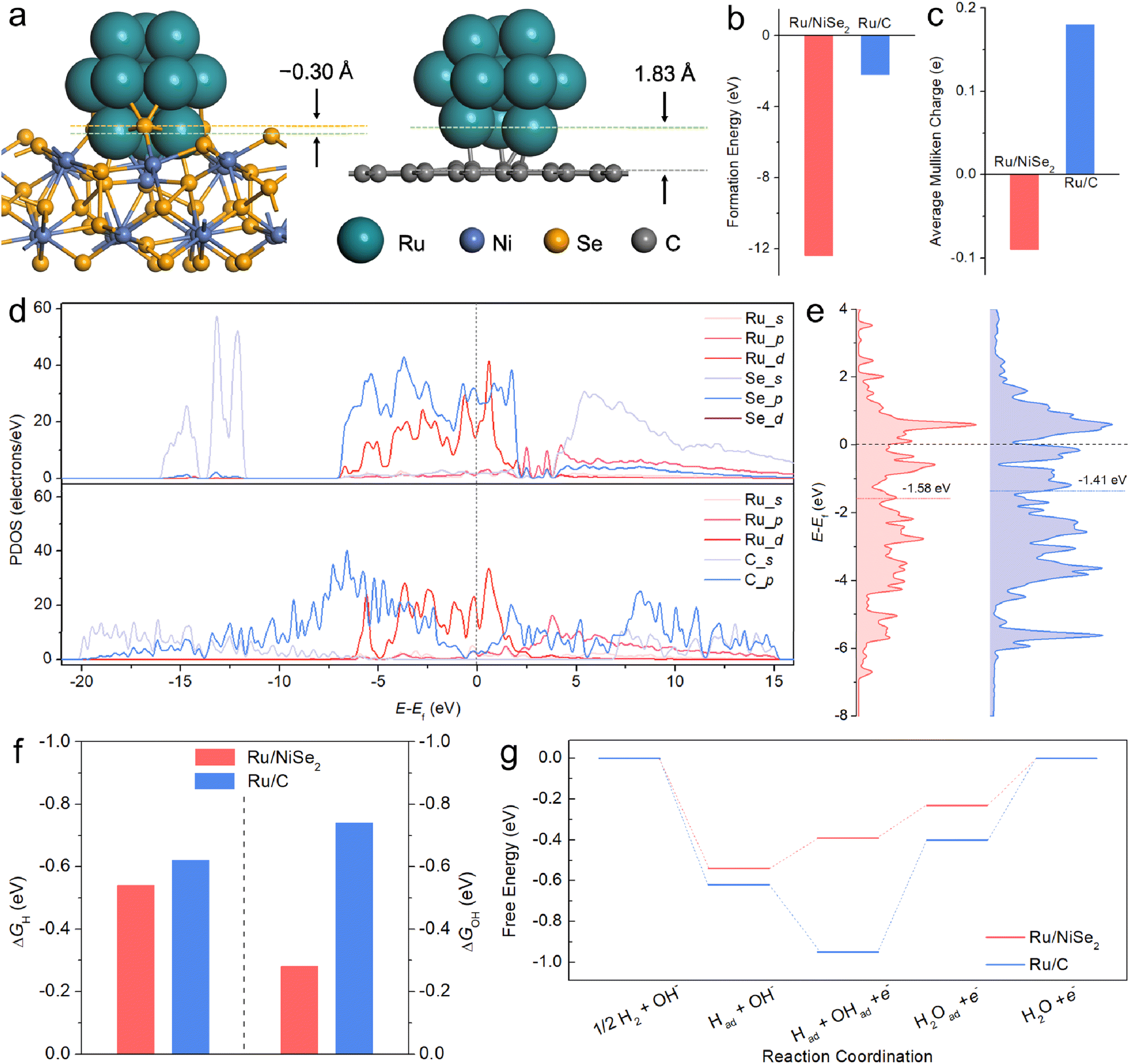 | ||
| Fig. 3 (a) Models of the hcp-Ru13 clusters on NiSe2(111) and graphitized C(001) representing Ru/NiSe2 and Ru/C, respectively. (b) Formation energy of the metal–support coupling process and (c) average Mulliken charge of Ru/NiSe2 and Ru/C. (d) PDOS and (e) corresponding d-band structures of Ru/NiSe2 and Ru/C. (f) Calculated ΔGH and ΔGOH of the most stable Ru sites on Ru/NiSe2 and Ru/C, and (g) corresponding reaction pathways for alkaline HOR. | ||
As depicted in Fig. 3f, the adsorption Gibbs free energies of Had (ΔGH) and OHad (ΔGOH) for the most stable Ru-sites on Ru/NiSe2 are −0.54 and −0.28 eV, respectively, which are weaker than those on Ru/C (−0.62 and −0.74 eV) and are closer to the thermoneutral values. It is also noted that the difference in the HBEs between Ru/NiSe2 and Ru/C is smaller than that in the OHBEs. Other possible active centers were also considered as shown in Fig. S16.† It can be seen that there are roughly two types of sites with weak HBE (ΔGH > −0.4 eV) or strong HBE (ΔGH < −0.5 eV). However, the weak HBE sites are more likely occupied by OHad due to the more negative values of ΔGOH. Additionally, the calculated ΔGOH on NiSe2 showed non-spontaneous OH adsorption (Fig. S16†), indicating that the weak OHBE was unable to activate neighboring H species via the bi-functional path. Hence, we suggest that the weakened OHBE might be the primary reason for the NiSe2-induced enhancement, which is consistent with the result of CO-stripping that the addition of NiSe2 would cause the positive shift of the peak of the CO-oxidation potential (Fig. 2e). Fig. 3g exhibits the corresponding energy profiles for the typical alkaline HOR process. The energy barrier for Ru/NiSe2 is 0.23 eV, much lower than that of Ru/C (0.55 eV), theoretically confirming the enhanced alkaline HOR performance of Ru-sites from the NiSe2 supports.
Therefore, a conclusion could be drawn based on the reported literature and this work that either the rational strengthened or weakened OHBE could accelerate the alkaline HOR catalytic kinetics for Ru-based catalysts and oppositely change their EPWs. The reasons why contradictory approaches lead to the same activity trend should be thoroughly explored. Fig. 4a presents the HOR polarizations normalized to their corresponding maximum current density, which better reflects the changes in the relative current density versus the increased content of NiSe2 supports. It is clearly observed that the increased NiSe2 content causes the gradually raised current density at potentials lower than ∼0.15 V vs. RHE as well as the gradually enlarged potential at the current density dropping to half of its maximum value (denoted as Edec@1/2jmax) as summarized in Fig. 4b. Considering the structural sensitivity of the catalytic surfaces for alkaline HOR, the active sites with different coordination environments, such as the terrace or the edge sites and the supports surrounded or removed sites, usually possess different binding energies to the crucial intermediates and thus different performances on an actual catalyst.32,44 Hence, we divide the HOR region into three parts, the weak Had adsorption area (Hw) at low overpotentials, the strong Had adsorption area (Hs) at high overpotentials, and the OHad adsorption area (poisoned surface) at the potential more positive than ∼0.3 V vs. RHE. On one hand, while Ru/C shows the lowest ratio of Hw, increasing the content of NiSe2 supports can effectively improve the Hw proportion, finally leading to the monotonically enhanced intrinsic activity at low overpotentials. This could be attributed to the weakened adsorption of OHad species originating from the strong interactions between Ru and NiSe2 alleviating the competitive adsorption of Had and OHad. Thereby more active sites for the dissociative adsorption of H2 could be provided. On the other hand, the weakened OHBE would result in more difficult oxidative desorption (Volmer step) of the over-strongly adsorbed Had through the bi-functional mechanism.13,45,46 Thus, it would happen at higher overpotentials, positively shifting the Hs area as well as the Edec@1/2jmax that is defined as a parameter to evaluate the completely poisoned Ru surfaces by OHad spectators. In contrast, the performance enhancement from the strengthened OHBE based on previous research might derive from another mode. We speculate that the stronger OHBE would boost the oxidative desorption of the strongly adsorbed Had and hence accelerate the alkaline HOR kinetics. However, the competitive adsorption between Had and OHad would be more significant, leading to the negatively shifted Edec@1/2jmax. That means the promotion strategy of strengthening the OHBE of Ru-based catalysts is at the expense of the narrowed EPW.
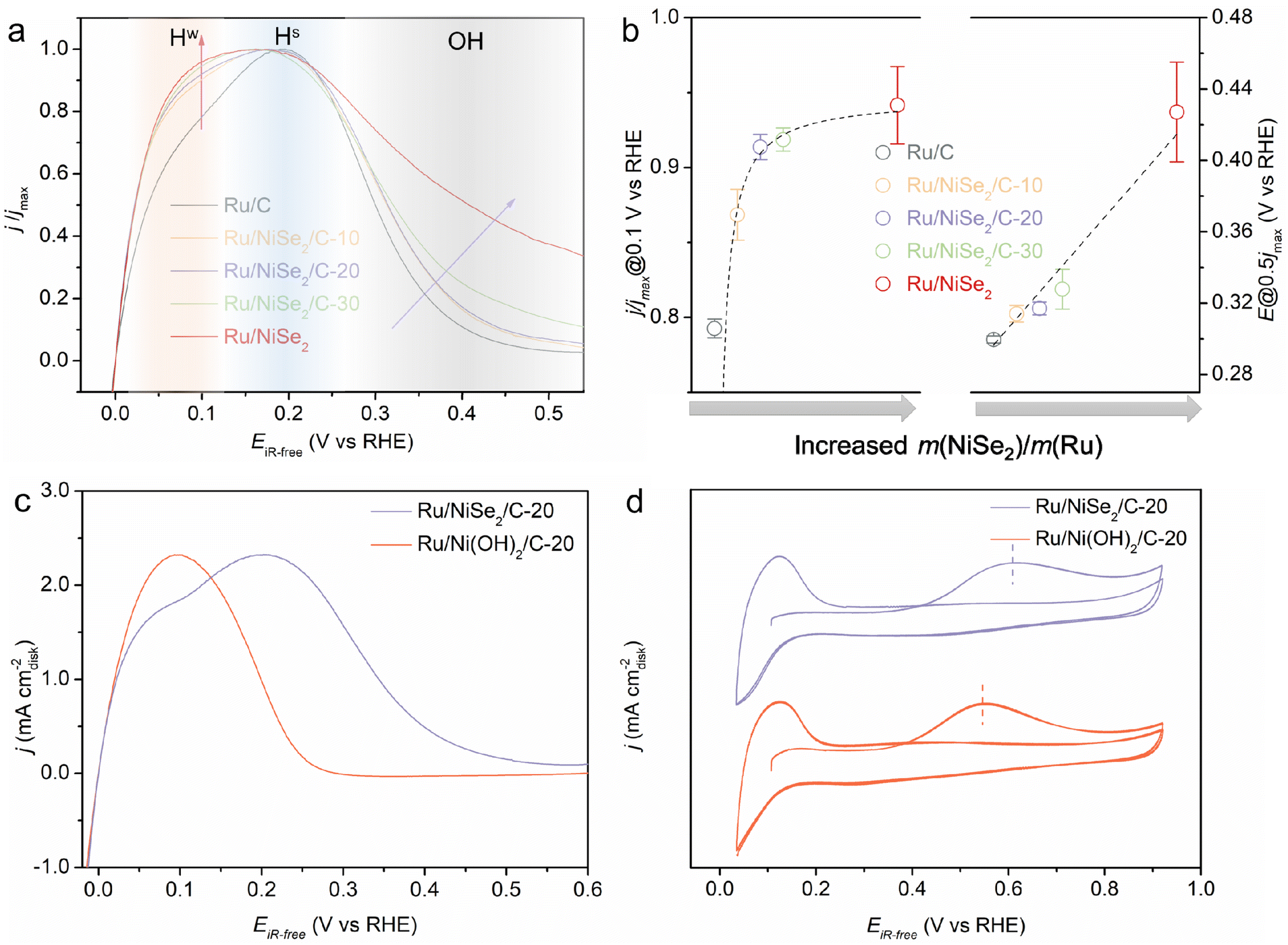 | ||
| Fig. 4 (a) Normalized HOR polarizations and (b) the relationships of the relative current densities at 0.1 V vs. RHE or the potential at the declined current densities dropping to half of their maximum values (Edec@1/2jmax) with the increased content of NiSe2. (c) HOR polarizations and (d) CO-stripping curves of Ru/NiSe2/C-20 and the in situ formed Ru/Ni(OH)2/C-20 after electrooxidation treatment. | ||
For the further verification of this viewpoint, Ru/NiSe2/C-20 was electrochemically treated by 100 cycles of cyclic voltammetry in the potential range between 0.0 and 1.2 V vs. RHE, denoted as Ru/Ni(OH)2/C-20. After that, NiSe2 supports would be irreversibly oxidized into Ni(OH)2 due to the high potential condition,47,48 which could be further confirmed by Raman spectroscopy where a new Raman peak appeared after electrochemical high-potential reconstruction at 1075 cm−1 assigned to the second-order acoustic mode of β-Ni(OH)2 (Fig. S17†).49 It is known that the in situ formed Ni(OH)2 would enhance the OHBE of the supported Ru.50 As shown in Fig. 4c, Ru/Ni(OH)2/C-20 exhibits obviously improved HOR activity at low overpotentials, similar to the reported Ru-based catalysts.14,24,32 However, the HOR current density begins to decline at about 0.1 V with the negatively shifted Edec@1/2jmax. Fig. 4d depicts the negatively shifted CO-stripping peak of the in situ formed Ru/Ni(OH)2/C-20 at 0.54 V vs. RHE compared to that of Ru/NiSe2/C-20 (0.60 V vs. RHE) and even Ru/C (0.55 V vs. RHE). The OHBE of the Ru site on Ru/Ni(OH)2 has also been calculated, which theoretically proves the enhanced oxophilicity of the Ru surface originated from the in situ phase transformation from NiSe2 to Ni(OH)2 (Fig. S18†). These results are in line with the above prediction that the strengthened OHBE could accelerate the alkaline HOR performance at low overpotentials but would make the EPW narrow for Ru-based catalysts. That is, the strengthened OHad is beneficial for the oxidative desorption of the strongly adsorbed Had but would hinder the adsorption of the highly active Had with weak HBE and would result in the easier poisoning of the catalytic surface, as the over-strongly adsorbed OHad can only act as the spectator. In order to further demonstrate that the weakly adsorbed Had would be occupied by OHad, the HOR behaviors in higher pH electrolytes were also investigated. By increasing the OH− concentration from 0.1 M to 1.0 M, the current density (or normalized current density) at the Hw region significantly reduces (Fig. S19†), corresponding to less Had species with relatively weak HBE. This means the higher pH would result in more competitiveness of the oxygenated species to adsorb on Ru sites and make the more weakly adsorbed Had desorb. Therefore, we propose two different promotion modes to qualitatively illustrate that the opposite regulations of the OHBE on the surface of Ru can both facilitate the alkaline HOR kinetics but with the different variation trends of the EPW, as schemed in Fig. 5.
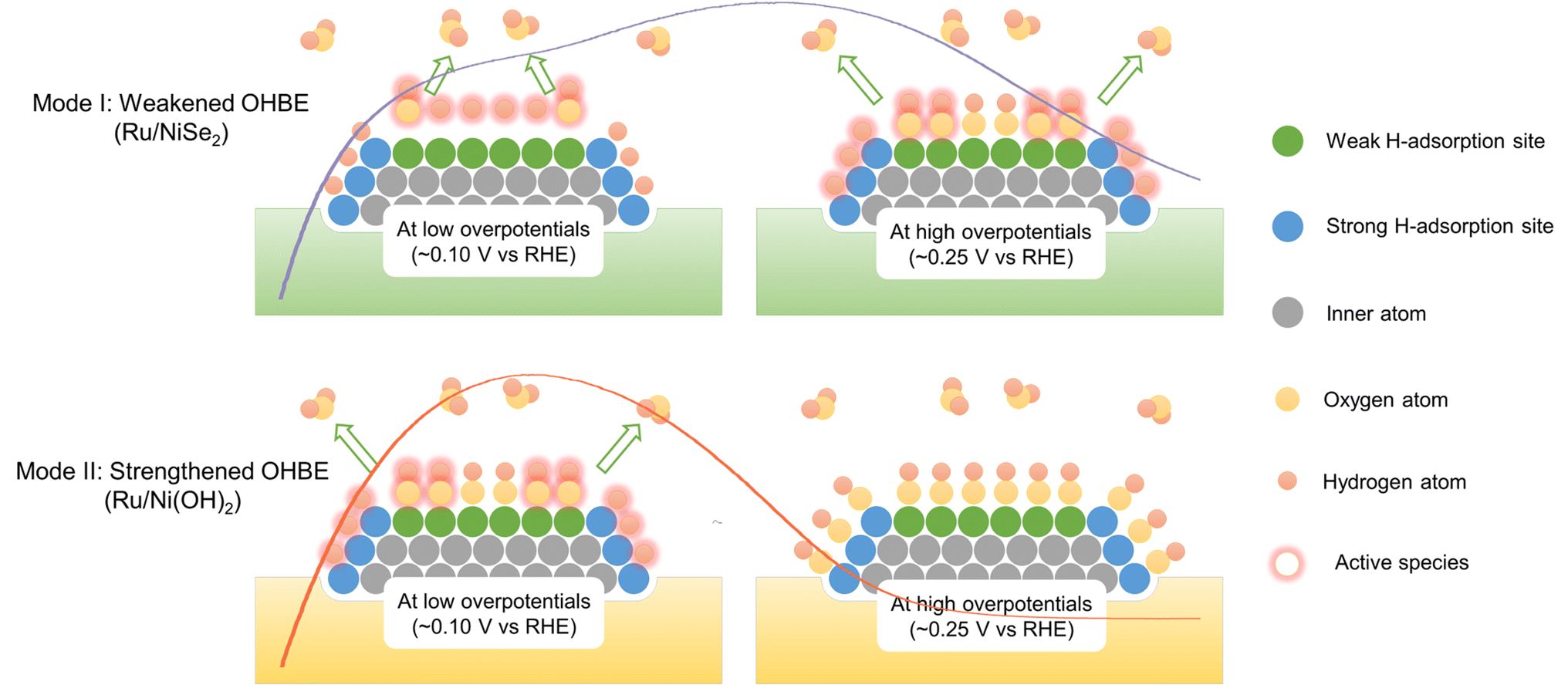 | ||
| Fig. 5 Schematic diagram of the two modes to illustrate the promotion mechanisms towards alkaline HOR on Ru-based catalysts with weakened or strengthened OHBEs, corresponding to the polarization curves of Ru/NiSe2 or Ru/Ni(OH)2, respectively, at both low overpotentials and high overpotentials. In Mode I, weakly adsorbed Had would react with hydroxyl species at low overpotentials and strongly adsorbed Had at high overpotentials, resulting in the relatively wide EPW. In Mode II, strongly adsorbed Had would react with hydroxyl species at low overpotentials and OHad would completely make the surface poison at high overpotentials, resulting in the relatively narrow EPW. The curves in two modes represent the HOR polarization curves of Ru/NiSe2 and Ru/Ni(OH)2 from Fig. 4c as a guide for the eye. | ||
Conclusion
In conclusion, we reported a d–p coupling strategy to improve the alkaline HOR performance of Ru catalysts by using NiSe2 octahedrons as supports. Specifically, Ru/NiSe2/C-20 exhibits the mass-normalized jk (at the overpotential of 50 mV) and j0 values of 2.51 and 0.88 mA μgRu−1, respectively, which is 2.6 and 2.0 times compared to that of Ru/C. Moreover, the intrinsic activity and the EPW monotonically increase with the increased content of NiSe2. The coupling of Ru with NiSe2 can significantly regulate the electron structures and weaken the OHBE, which alleviates the competitive adsorption of Had and OHad and provides more active sites for the formation of weakly adsorbed Had with high activity. Different from the previous results, we propose two possible promotion modes to qualitatively illustrate that the opposite regulations of the OHBE on the surface of Ru yield a similar consequence of the accelerated alkaline HOR kinetics at low overpotentials but a different trend about the EPW. This is finally attributed to the discrepant roles of OHad species with different OHBEs and at different overpotentials. This work provides another approach to design novel Ru-based catalysts with high activity towards alkaline HOR by rationally weakening the OHBE, without the expense of the narrowed EPW.Data availability
Data are available upon request from the authors.Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (22202172, 22272148, 22201085) and the Natural Science Foundation of Jiangsu Province (Grant No. BK20220560).References
- N. Ramaswamy and S. Mukerjee, Chem. Rev., 2019, 119, 11945–11979 CrossRef CAS PubMed.
- J. Cai, X. Zhang, Z. Lyu, H. Huang, S. Wang, L. Fu, Q. Wang, X. Yu, Z. Xie and S. Xie, ACS Catal., 2023, 13, 6974–6982 CrossRef CAS.
- W. Qiao, L. Yu, J. Chang, F. Yang and L. Feng, Chin. J. Catal., 2023, 51, 113–123 CrossRef CAS.
- L. Osmieri, L. Pezzolato and S. Specchia, Curr. Opin. Electrochem., 2018, 9, 240–256 CrossRef CAS.
- K. Veeramani, G. Janani, J. Kim, S. Surendran, J. Lim, S. C. Jesudass, S. Mahadik, H. lee, T.-H. Kim, J. K. Kim and U. Sim, Renewable Sustainable Energy Rev., 2023, 177, 113227 CrossRef CAS.
- C. Wang, A. Schechter and L. Feng, Nano Res. Energy, 2023, 2, e9120056 CrossRef.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- X. Qin, L. Zhang, G. Xu, S. Zhu, Q. Wang, M. Gu, X. Zhang, C. Sun, P. B. Balbuena, K. Amine and M. Shao, ACS Catal., 2019, 9, 9614–9621 CrossRef CAS.
- Y. Zhou, Q. Wang, X. Tian, J. Chang and L. Feng, J. Energy Chem., 2022, 75, 46–54 CrossRef CAS.
- X. Zhang, X. Xiao, J. Chen, Y. Liu, H. Pan, W. Sun and M. Gao, Energy Environ. Sci., 2022, 15, 4511–4526 RSC.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef CAS PubMed.
- Y. Zhao, X. Wang, G. Cheng and W. Luo, ACS Catal., 2020, 10, 11751–11757 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, D. Strmcnik, A. P. Paulikas, D. Myers, V. R. Stamenkovic and N. M. Markovic, Electrocatalysis, 2012, 3, 221–229 CrossRef CAS.
- J. Jiang, S. Tao, Q. He, J. Wang, Y. Zhou, Z. Xie, W. Ding and Z. Wei, J. Mater. Chem. A, 2020, 8, 10168–10174 RSC.
- L. Su, Y. Jin, D. Gong, X. Ge, W. Zhang, X. Fan and W. Luo, Angew. Chem., Int. Ed., 2023, 62, e202215585 CrossRef CAS PubMed.
- Y. Li, C. Yang, C. Ge, N. Yao, J. Yin, W. Jiang, H. Cong, G. Cheng, W. Luo and L. Zhuang, Small, 2022, 18, 2202404 CrossRef CAS PubMed.
- Y. Zhao, D. Wu and W. Luo, ACS Sustainable Chem. Eng., 2022, 10, 1616–1623 CrossRef CAS.
- L. Su, X. Fan, Y. Jin, H. Cong and W. Luo, Small, 2023, 19, 2207603 CrossRef CAS PubMed.
- Y. Kuang, W. Qiao, F. Yang and L. Feng, J. Energy Chem., 2023, 85, 447–454 CrossRef CAS.
- B. P. Setzler, Z. Zhuang, J. A. Wittkopf and Y. Yan, Nat. Nanotechnol., 2016, 11, 1020–1025 CrossRef CAS PubMed.
- S. Qin, Y. Duan, X.-L. Zhang, L.-R. Zheng, F.-Y. Gao, P.-P. Yang, Z.-Z. Niu, R. Liu, Y. Yang, X.-S. Zheng, J.-F. Zhu and M.-R. Gao, Nat. Commun., 2021, 12, 2686 CrossRef CAS PubMed.
- H. Choi, S. Surendran, D. Kim, Y. Lim, J. Lim, J. Park, J. K. Kim, M.-K. Han and U. Sim, Environ. Sci.: Nano, 2021, 8, 3110–3121 RSC.
- Y. Zhao, F. Yang, W. Zhang, Q. Li, X. Wang, L. Su, X. Hu, Y. Wang, Z. Wang, L. Zhuang, S. Chen and W. Luo, CCS Chem., 2022, 4, 1732–1744 CrossRef CAS.
- Y. Lim, S. Surendran, W. So, S. Shanmugapriya, C. Jo, G. Janani, H. Choi, H. S. Han, H. Choi, Y.-H. Yun, T.-H. Kim, M.-J. Kim, K. Jin, J. K. Kim and U. Sim, Mater. Chem. Front., 2023, 7, 5843–5857 RSC.
- Q. Yuan, J. Zhao, D. H. Mok, Z. Zheng, Y. Ye, C. Liang, L. Zhou, S. Back and K. Jiang, Nano Lett., 2022, 22, 1257–1264 CrossRef CAS PubMed.
- V. I. Zaikovskii, K. S. Nagabhushana, V. V. Kriventsov, K. N. Loponov, S. V. Cherepanova, R. I. Kvon, H. Bönnemann, D. I. Kochubey and E. R. Savinova, J. Phys. Chem. B, 2006, 110, 6881–6890 CrossRef CAS PubMed.
- S. Zhu, Q. Li, Q. Wei, R. Sun, X. Liu, Q. An and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 311–316 CrossRef CAS PubMed.
- R. Harpeness, Z. Peng, X. Liu, V. G. Pol, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2005, 287, 678–684 CrossRef CAS PubMed.
- C. L. Bianchi, V. Ragaini and M. G. Cattania, Mater. Chem. Phys., 1991, 29, 297–306 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- T. Zhao, D. Xiao, Y. Chen, X. Tang, M. Gong, S. Deng, X. Liu, J. Ma, X. Zhao and D. Wang, J. Energy Chem., 2021, 61, 15–22 CrossRef CAS.
- Y. Men, D. Wu, Y. Hu, L. Li, P. Li, S. Jia, J. Wang, G. Cheng, S. Chen and W. Luo, Angew. Chem., Int. Ed., 2023, 62, e202217976 CrossRef CAS PubMed.
- J. Zheng, S. Zhou, S. Gu, B. Xu and Y. Yan, J. Electrochem. Soc., 2016, 163, F499–F506 CrossRef CAS.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066 CrossRef PubMed.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- J. Zheng, Y. Yan and B. Xu, J. Electrochem. Soc., 2015, 162, F1470–F1481 CrossRef CAS.
- S. Lu and Z. Zhuang, J. Am. Chem. Soc., 2017, 139, 5156–5163 CrossRef CAS PubMed.
- C. Jo, S. Surendran, M.-C. Kim, T.-Y. An, Y. Lim, H. Choi, G. Janani, S. Cyril Jesudass, D. Jun Moon, J. Kim, J. Young Kim, C. Hyuck Choi, M. Kim, J. Kyu Kim and U. Sim, Chem. Eng. J., 2023, 463, 142314 CrossRef CAS.
- Y. Wang, M. Zheng, Y. Li, C. Ye, J. Chen, J. Ye, Q. Zhang, J. Li, Z. Zhou, X.-Z. Fu, J. Wang, S.-G. Sun and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202115735 CrossRef CAS PubMed.
- L. Wu, L. Su, Q. Liang, W. Zhang, Y. Men and W. Luo, ACS Catal., 2023, 13, 4127–4133 CrossRef CAS.
- B. Hammer and J. K. Nørskov, in Advances in Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- Z. Li, L. An, M. Song, T. Zhao, J. Zhang, C. Zhang, Z. Li and D. Wang, Chin. Chem. Lett., 2023, 34, 107622 CrossRef CAS.
- I. T. McCrum and M. T. M. Koper, Nat. Energy, 2020, 5, 891–899 CrossRef CAS.
- Y.-H. Wang, X.-T. Wang, H. Ze, X.-G. Zhang, P. M. Radjenovic, Y.-J. Zhang, J.-C. Dong, Z.-Q. Tian and J.-F. Li, Angew. Chem., Int. Ed., 2021, 60, 5708–5711 CrossRef CAS PubMed.
- L. Wang, X. Wang, S. Xi, Y. Du and J. Xue, Small, 2019, 15, 1902222 CrossRef PubMed.
- Y. Shi, W. Du, W. Zhou, C. Wang, S. Lu, S. Lu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 22470–22474 CrossRef CAS PubMed.
- M. Aghazadeh and H. Rad, J. Mater. Sci.: Mater. Electron., 2022, 33, 1–17 CrossRef.
- T. Jiang, K. Li, S. Park, K. Zheng, Y. Meng, Y. Yuan, Z. Liu, Z. Zhu, X. Zheng, S. Liu and W. Chen, Nano Lett., 2022, 22, 1741–1749 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05387c |
| This journal is © The Royal Society of Chemistry 2024 |