
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence4-Fluorobenzyl cyanide, a sterically-hindered solvent expediting interfacial kinetics in lithium-ion batteries†
Mingsheng
Qin
ab,
Ziqi
Zeng
*a,
Qiang
Wu
a,
Xiaowei
Liu
c,
Qijun
Liu
ab,
Shijie
Cheng
a and
Jia
Xie
 *a
*a
aState Key Laboratory of Advanced Electromagnetic Technology, School of Electrical and Electronic Engineering, Huazhong University of Science and Technology, Wuhan 430074, Hubei, China. E-mail: xiejia@hust.edu.cn; ziqizeng@hust.edu.cn
bState Key Laboratory of Materials Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan 430074, Hubei, China
cState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430074, Hubei, China
First published on 12th March 2024
Abstract
The electrochemical performance of lithium-ion batteries (LIBs) is plagued by sluggish interfacial kinetics. Fortunately, the Li+ solvation structure bridges the bulk electrolyte and interfacial chemistry, providing a pathway for promoting electrochemical kinetics in LIBs. Herein, we improve the interfacial kinetics by tuning the Li+ coordination chemistry based on solvent molecular engineering. Specifically, 4-fluorobenzyl cyanide (FBCN), featuring steric hindrance and a weak Lewis basic center, is designed to construct a bulky coordination structure with Li+, weakening ion–dipole interaction (Li+–solvents) but promoting coulombic attraction (Li+–anions) at a normal Li salt concentration. This sterically-controlled solvation chemistry reduces the interfacial barrier and thus contributes to improved rate performance, as demonstrated practically in LiFePO4//graphite pouch cells. This study provides fresh insights into solvent steric control and coordination chemistry engineering, opening a new avenue for enhancing electrochemical kinetics in LIBs.
1. Introduction
The advancement of electrified transportation calls for superior lithium-ion batteries (LIBs) featuring fast-charging capability, long-term stability and high safety levels.1–3 However, current LIBs based on carbonate electrolytes experience sluggish kinetics, exacerbated by interfacial effects, leading to performance decay and safety issues at high rates.4–6 Specifically, common carbonate electrolyte (CCE, 1 M LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC)) exhibits strong affinity with Li+ because of its high dielectric constant and donor number, enabling a conductivity over 10 mS cm−1 at ambient temperature.7,8 Nevertheless, LIBs containing CCE still exhibit an unsatisfactory rate performance, which highlights the importance of interfacial kinetics and reliable passivation during the electrochemical process.9,10 Furthermore, typical carbonates are highly flammable, exacerbating the safety risks at a high rate. Therefore, it is imperative to explore alternative solvents with high safety levels while preserving favourable electrochemical performance.Highly concentrated electrolytes (HCEs) exhibit facilitated kinetics at a graphite surface, because of the participation of anions in the solvation shell, which actually weakens the ion–dipole interaction (Li+–carbonates) and results in an inorganic-rich SEI.11–13 However, high cost and viscosity hinder their applications. Fortunately, weakly coordinating solvents provide another opportunity to tune the Li+–solvent affinity. For example, 1,3-dimethoxypropane (DMP) and 1,2-DMP featuring additional methyl groups exhibit decreased solvating power and better Li metal compatibility than DME.14,15 Moreover, bis(2-fluoroethyl)ether enables decent fast-charging capability in lithium–metal batteries with tridentate coordination chemistry.16 Trifluoropropylene carbonate, as expected, also exhibits better graphite compatibility than PC after molecular fluorination.17 These results indicate that solvent molecular engineering can affect the micro-solvating Li+ affinity and dictate the coordination chemistry, which in turn provides an novel avenue for pursuing superior electrochemical performance. However, the deliberate tuning of the micro-solvation chemistry (e.g. binding energy and coordination number) and interfacial process (e.g. SEI and desolvation) through solvent molecular control is still challenging. Moreover, some small solvents (e.g. acetonitrile) featuring strong Li+–solvent binding require rigid and bulky substituents to reliably weaken the binding, and this is not often reported.
In this contribution, we design 4-fluorobenzyl cyanide (FBCN) as a new electrolyte solvent for dynamically favourable LIBs with sterically-controlled coordination chemistry. The bulky FBCN coordinates with Li+ through partially deactivated Lewis basic centers and the fluorobenzene substituent incurs additional steric hindrance in a space-confined solvation shell, leading to a loose coordination configuration and enhanced electrochemical kinetics on the graphite surface. As a result, an improved power capability is realized in the LiFePO4//graphite pouch cell using this EC-free electrolyte despite its inferior ionic conductivity, establishing the significance of interfacial kinetics and passivation for fast charging. Moreover, this electrolyte enables good graphite compatibility, wide-temperature adaptability and high safety levels. The solvent molecular control and sterically confined coordination chemistry open a feasible pathway toward formulating low-energy-barrier electrolytes in LIBs.
2. Results and discussion
2.1 Molecular design and physicochemical properties of the electrolyte
An “ideal” solvent for an electrolyte must be capable of dissociating Li salts, which relies on Lewis basic sites in the molecule.18,19 Acetonitrile (ACN) contains Lewis basic nitrogen and its basicity is increased by an electron-donating alkyl group, leading to strong Li+–ACN binding and tight coordination, as validated by the uneven charge distribution in molecular electrostatic potential (ESP) results (Fig. 1a). Unfortunately, the adoption of ACN as an electrolyte in LIBs is always accompanied by interfacial degradation and gas inflation, even after modifications by film-forming additives (Fig. S1 and S2†).20 Fluorobenzene (FB) is inert towards Li+ but shows preferential adsorption on the graphite surface via π–π stacking, typically serving as an inert cosolvent for better electrochemical performance (Fig. 1a).21,22 Herein, we graft a fluorobenzene substituent on active acetonitrile to construct 4-fluorobenzyl cyanide (FBCN), which shows finely tuned affinity to Li+, because of the additional steric hindrance (a bulky fluorobenzene group) in the micro-solvating structure compared to pristine ACN (Fig. 1a). Note that the designed FBCN is different from FB or ACN and exhibits different properties and superior electrochemical performance. Moreover, the high flash point (108 and −15 °C for FBCN and FB, respectively), high boiling point (238 and 85 °C for FBCN and FB, respectively) and good Li salt dissociation capability make FBCN worth studying as a promising solvent (Table S1†). Briefly, the rigid benzene ring increases the intermolecular repulsion in the space-confined solvation shell (steric hindrance), leading to relatively weak Li+–FBCN binding (ion–dipole interaction) and thus a loose solvation shell.22 As a result, this FBCN-dominated electrolyte might exhibit enhanced interfacial kinetics and enable improved rate capability. | ||
Fig. 1 (a) Design scheme and ESP results for ACN, FBCN and FB. (b) C1 and H1 designations in molecular structures. (c) 13C-NMR spectra and (d) 1H-NMR spectra for FBCN and ACN. (e) Binding energies for Li+ and various solvents. (f) Molecular structures, optical images and (g) conductivities for various Li salts in FBCN (Li salt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :FBCN MRs of 1:9). :FBCN MRs of 1:9). | ||
The weakened solvation of FBCN is validated by ESP calculations, showing an ESPmin of −41.26 kcal mol−1, which falls right in the middle of ACN and FB (Fig. 1a). The higher ESPmin of FBCN indicates a lower thermodynamic stability of the interaction with Li+ compared to ACN, favoring a loose coordination structure.23,24 NMR results for various solutions were recorded to validate this weakened affinity between Li+ and FBCN (Fig. S3†). The C1 in the cyano group shows a downshift for FBCN and ACN after Li salt dissociation, indicating deshielding effects once coordinated with Li+ (Fig. 1b and c).25 Note that the downshift value for FBCN (ΔC1 = 0.3) is smaller than that of ACN (ΔC1 = 0.5) at the same LiFSI/solvent molar ratio (MR of 1:9), indicating the partially reduced Li+–FBCN coordination. This reduced deshielding effect as a result of weak Li+–FBCN binding is also validated by a smaller downshift for H1 near the cyano group (Fig. 1d). First-principle calculations were performed to probe the ion–dipole interaction between various solvents and Li+, as expressed by the binding energy (Eb) (Fig. S4†). EC exhibits the lowest Eb, because of its high dielectric constant (89.8) and donor number, indicating the strongest Li+–EC binding (Fig. 1e).26 This tight coordination in the EC-based electrolyte results in sluggish desolvation and hampered electrochemical kinetics. In contrast, FBCN exhibits an Eb of −0.73 eV, which is higher than those of ACN (−0.89 eV) and EC (−0.98 eV), indicating the weak Li+–FBCN binding.
FBCN shows selectivity in dissolving Li salts because of the partially neutralized Lewis basic center. Different Li salts are added into FBCN with fixed MR (Li salts/FBCN with a MR of 1:9) (Fig. 1f). Conventional lithium salts including LiPF6 and LiBF4 exhibit inadequate dissociation in FBCN and form colloid electrolytes, as per the Tyndall effect. Note that LiDFOB exhibits negligible solubility in FBCN and forms a suspension because of the high lattice energy of LiDFOB.27 In contrast, LiFSI and LiTFSI are dissolved completely and provide sufficient ionic carriers within FBCN, because of the large size of the anions and delocalization of the negative charge,8 forming a transparent and light yellow solution (FBCN itself is light yellow). The conductivities for different Li salt-enabled electrolytes are compared (Fig. 1g). The LiFSI-enabled electrolyte exhibits a moderate conductivity of 3.5 mS cm−1 at ambient temperature. Note that LiTFSI increases the viscosity and contributes to a relatively low conductivity (2.45 mS cm−1), which leads to an inferior rate capability at high rates compared to that of the LiFSI-based electrolyte (Fig. S5†).28 Additionally, the conductivities of LiFSI in various solvents (LiFSI/solvents MR of 1:9) are compared (Fig. S6†), showing the lowest conductivity in FBCN solvent compared to cyclic (e.g. EC) and linear carbonates (e.g. DMC), which is caused by the weak Li+–FBCN interaction and relatively poor Li salt dissociation.
2.2 Electrochemical characterizations and high-rate capability
The interfacial compatibility towards graphite is investigated. LiFSI–9FBCN (LiFSI:FBCN MR of 1:9) goes through reductive decomposition on graphite as demonstrated by the endless discharge at 0.5 V and limited initial coulombic efficiency (ICE) below 4% (Fig. 2a). For this reason, we added 2 wt% fluoroethylene carbonate (FEC) and 2 wt% LiDFOB to synergistically passivate the graphite surface, and this modified electrolyte is defined as the FBCN-based electrolyte. Note that just FEC (FBCN + FEC) or LiDFOB (FBCN + LiDFOB) results in limited improvements of the interfacial compatibility. For example, graphite (Fig. S7–S10†) and LiFePO4 (Fig. S11–S14†) in FBCN + FEC suffer from limited capacity (ICE of 76.6% and capacity of 318 mA h g−1 at 1C) and unstable cycling, because of the unreliable interfacial passivation. Moreover, FBCN + LiDFOB contributes to cell failure in graphite//Li cells (no reversible capacity) and fast capacity fading in LiFePO4//Li cells (less than 8 cycles) due to thick interfacial passivation. In contrast, the FBCN-based electrolyte (modified by FEC and LiDFOB) exhibits good graphite compatibility, which is comparable to that of common carbonate electrolyte (CCE) and EC-based electrolyte (CCE with 2 wt% FEC and 2 wt% LiDFOB) (Fig. 2a). Note that same additives are added in EC-based electrolyte to better understand the superiority of FBCN itself. Moreover, the lattice and morphologies of cycled graphite in different electrolytes were studied by XRD patterns (Fig. S15†), SEM images (Fig. S16†) and EDS mappings (Fig. S17†), showing substantially improved interfacial stability in the FBCN-based electrolyte. The following electrochemical characterizations are based on the optimized FBCN-based electrolyte. Note that the additional steric hindrance (fluorobenzene substituent) cannot enable reversible graphite in FBCN, but it does show superiority compared to ACN (failure even with additives, Fig. S1 and S2†) with the help of additives.
 | ||
| Fig. 2 (a) Initial charge–discharge curves, (b) CV results at 0.1 mV s−1 and (c) rate performance of graphite//Li cells in various electrolytes. (d) Rate capabilities of LiFePO4//Li cells and (e) LiFePO4//graphite pouch cells in various electrolytes. (f) Charge–discharge curves and (g) long-term stability of 1.6 A h LiFePO4//graphite pouch cell in FBCN-based electrolyte. (h) DSC results for various electrolytes. (i) Cycling stability of Li4Ti5O12//Li cells under cold conditions. | ||
The graphite//Li cell in the FBCN-based electrolyte shows lower electrochemical polarization, as exhibited in Fig. 2b. Moreover, overlapping CV curves demonstrate its good reliability towards the graphite anode (Fig. S18†), which is consistent with its long-term stability (Fig. S19†).29 The rate performance of graphite in the FBCN-based electrolyte is assessed (Fig. 2c), showing capacities of 255 and 188 mA h g−1 at 2C and 3C, respectively, despite its unsatisfactory conductivity (Fig. S20†) and moderate interfacial wettability (Fig. S21†). In contrast, the graphite//Li cell in CCE exhibits a limited capacity of 50 mA h g−1 at 3C, regardless of the decent conductivity over 10 mS cm−1. Moreover, the cell in the EC-based electrolyte exhibits failure at a high rate, even with film-forming additives (e.g. FEC and LiDFOB). The feasibility and compatibility of the FBCN-based electrolyte towards LiFePO4 are validated, showing lower polarization (Fig. S22†), superior cycling stability (Fig. S23†) and better rate capability (Fig. 2d and S24†) compared to the EC-based electrolyte. Benefiting from the decent oxidative tolerance of nitriles,20 the FBCN-based electrolyte exhibits better high-voltage stability than the EC-based electrolyte (Fig. S25†). As a result, the FBCN-based electrolyte enables the reversible cycling of LiCoO2//Li (2.8–4.2 V) (Fig. S26†) and NCM811//Li cells (2.8–4.3 V) (Fig. S27†). Note that the FBCN-based electrolyte exhibits poor compatibility towards Li metal (Fig. S28 and S29†), yet the superior electrochemical performance in the half cell configuration in turn highlights interfacial improvements from graphite, LiFePO4, LiCoO2 and NCM811 sides. These results indicate that FBCN is more suitable for LIBs than LMBs.22
The improved rate performance is demonstrated in LiFePO4//graphite pouch cells (Fig. 2e). A similar initial capacity (150 mA h g−1) is obtained at 0.2C for cells in the FBCN-based and EC-based electrolytes. However, the discharge capacity exhibits a large reduction with an increase in the current density for the cell in the EC-based electrolyte. In contrast, the cell in the FBCN-based electrolyte sustains a higher capacity retention even at 3C, indicating the superiority of the FBCN-based electrolyte despite its low conductivity (3.5 mS cm−1). Note that all electrode materials are microscale without any modifications, and a superior performance can be obtained once the electrode is optimized.30 As a model, the FBCN-based electrolyte is validated in a 1.6 A h LiFePO4//graphite pouch cell (Fig. 2f), enabling 96.5% capacity retention over 600 cycles at 0.33C (Fig. 2g). Furthermore, the FBCN-based electrolyte shows a wide liquid range (−40–156 °C) and inferior flammability (Fig. S30†), while the EC-based electrolyte exhibits solidification below −26 °C and can be ignited immediately (Fig. 2h). A higher discharge capacity of 120 mA h g−1 is obtained for the cell in the FBCN-based electrolyte at −20 °C, indicating the facile interfacial kinetics and good low-temperature adaptability (Fig. 2i). As a result, the FBCN-based electrolyte exhibits inferior ionic conductivity in the bulk electrolyte, but enables promoted interfacial kinetics in LIBs, which is in contrast to the common understanding of positive correlations between ionic conductivity and rate capability.8,31 This mechanism will be discussed later.
2.3 Steric hindrance and solvation structure
The abnormal rate capability hints at an innovative solvation structure in the FBCN-based electrolyte. As shown in Fig. 3a, the 13C NMR spectrum for FBCN exhibits a downshift upon Li salt dissociation, indicating coordination between FBCN and Li+, which is consistent with the downshift in 19F spectra (Fig. 3b). The C and H signals for different groups on FBCN are also monitored and show coordination between FBCN and Li+ (Fig. S31 and S32†).32,33 The local coordination configuration is identified by AIMD simulations (Fig. 3c and S33†). All solvating species are listed and most Li+ are surrounded by three FBCN in the FBCN-based electrolyte (Fig. S34†). In contrast, the main solvate contains two EC and one DMC in the EC-based electrolyte (Fig. S35†). The average coordination number for FBCN is 2.61 in the FBCN-based electrolyte (Fig. 3d), while the average coordination numbers for EC and DMC are 1.92 and 1.19, respectively, in the EC-based electrolyte (Fig. S33†). The large FBCN dominates this space-confined Li+ solvation and contributes to less coordinated solvents for the FBCN-based electrolyte. Moreover, the average distance for coordinated FBCN (2.17 Å) is larger than for the anions (2.03 Å) (Fig. 3e), which is because of the intermolecular repulsion on bulky FBCN and thus an increased Li+–FBCN binding distance. In addition, the distances for coordinated EC (2.07 Å) and DMC (2.07 Å) are smaller than that for FBCN (2.17 Å) (Fig. S33†), indicating a loose solvation shell for the latter since the bulky FBCN cannot bind with Li+ as closely as small solvents (e.g. EC and DMC).34 | ||
| Fig. 3 (a) 13C-NMR and (b) 19F-NMR spectra for various solutions. (c) Snapshot of the AIMD simulation and main species in the electrolyte. (d) Coordination numbers and (e) radial distributions calculated from the AIMD simulation. Illustrations of solvation structures and interfacial behaviors in (f and h) EC-based and (g and i) FBCN-based electrolytes. Adsorption energies of (j) FBCN, (k) EC, (l) DMC and (m) FEC on the graphite (010) surface. | ||
As illustrated in Fig. 3f, there is an absence of steric hindrance in the EC-based electrolyte because of the combination of linear and cyclic carbonates, and a tight solvation shell is formed because of the strong ion–dipole attraction between Li+ and carbonate (e.g. EC and DMC).7,21 This solvation configuration exhibits sluggish interfacial kinetics and leads to lithium dendritic growth at a high rate (Fig. 3h). The bulky substituent on FBCN, nevertheless, contributes to intermolecular repulsion in this space-confined solvation structure.14,35 The steric hindrance leads to loose coordination by weakening the Li+–FBCN strength and increasing the Li+–FBCN distance (Fig. 3g), which accounts for easier desolvation and improved rate capability (Fig. 3i). Different from HCEs or localized HCEs, this strategy relies on the molecular steric effect and dispenses with a high Li salt concentration, resulting in adequate solvation in the bulk electrolyte and efficient desolvation at the graphite interphase.
DFT calculations are performed to study the interfacial behaviors of different solvents under ideal conditions.36 The optimized geometrical configurations on the graphite (010) plane for FBCN, EC, DMC and FEC are illustrated (Fig. 3j–m). The preferential engagement of FBCN on the edge plane of the graphite is revealed by its shortest distance (d = 2.43 Å). Moreover, calculated adsorption energies for FBCN, EC, DMC and FEC are −0.40, −0.26, −0.36, and −0.28 eV, respectively, indicating the energetic favorability of FBCN on the graphite surface. Given the polycrystalline nature of the graphite and possible exposure of every lattice plane at the interphase, the adsorption behavior on the graphite (001) plane is calculated, showing similar results (Fig. S36 and Table S2†). First, the adsorption of FBCN alters the local environment (e.g. preferential species, concentration gradient and interfacial shielding) and contributes to a FBCN-rich environment at the graphite surface, affecting the interfacial chemistry.22,37 Second, the interaction (e.g. van der Waals force) between FBCN and graphite might weaken the Li+–FBCN binding at the interface because of the competitive force on Li+–FBCN–graphite complexes, helping with the breakup of Li+–FBCN solvates (desolvation process).38,39 All of these effects might influence the interfacial behavior and lead to improved electrochemical performance.
2.4 Interfacial chemistry and electrode characterization
The interfacial chemistry was studied. FBCN exhibits the lowest unoccupied molecular orbital (LUMO) energy of −0.51 eV and is inclined to obtain electrons on the graphite surface (Fig. 4a). The reductive instability of FBCN is revealed by LSV results (Fig. 4b), showing a dominant cathodic current at 0.9 V (decomposition of FSI−) and 0.4 V (decomposition of FBCN) in LiFSI–9FBCN. LiDFOB and FEC are helpful for kinetically stabilizing FBCN through the construction of LiF-rich and B-containing SEI, which is revealed by additional cathodic peaks for LiDFOB (1.7 V) and FEC (1.3 V) in LSV curves in the FBCN-based electrolyte.40,41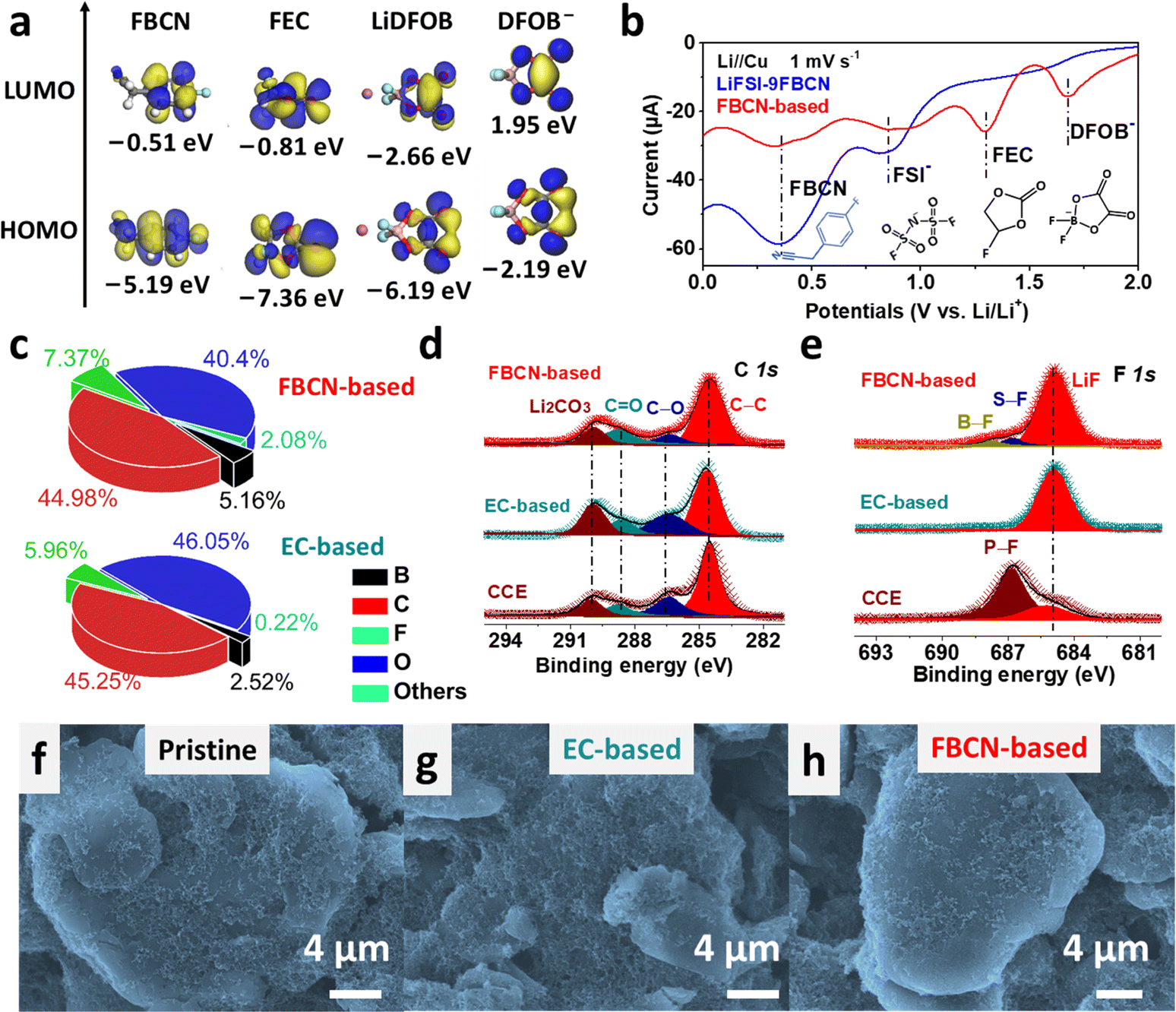 | ||
| Fig. 4 (a) LUMO and HOMO energy levels for different components. (b) LSV results at 1 mV s−1. (c) Elemental contents for graphite cycled in various electrolytes. (d) C and (e) F spectra recorded at the graphite surface. SEM images of (f) pristine graphite and graphite cycled in (g) EC-based electrolyte and (h) FBCN-based electrolyte. | ||
The SEI formed in the FBCN-based electrolyte exhibits higher contents of F (7.37%) and B (5.16%) than those of the EC-based electrolyte (Fig. 4c), indicating its reliance on the decomposition of FEC and LiDFOB for kinetically stabilizing the graphite (Fig. S37†). Similar interfacial components are detected for graphite in various electrolytes, containing Li2CO3, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O species (Fig. 4d and S38†).42,43 Note that a dominant LiF peak is observed in the FBCN-based electrolyte with subtle signals from the decomposition of LiDFOB (B–F) and LiFSI (S–F) (Fig. 4e).40 Similarly, the EC-based electrolyte also exhibits a predominant peak of LiF, indicating inorganic-rich SEI derived from film-forming additives (e.g. LiDFOB and FEC). Nevertheless, CCE exhibits a relatively weak intensity from LiF but a dominant peak from the incomplete decomposition of LiPF6 (P–F), which indicates poor interfacial passivation (Fig. S39†).44 The SEI is monitored by TEM images (Fig. S40†), and a thick and uneven coverage (>40 nm) is observed in CCE. In contrast, a thin but uneven coverage (>20 nm) is exhibited in the EC-based electrolyte. Interestingly, a thin and even SEI is formed in the FBCN-based electrolyte, indicating superior passivation after additive modification. Benefiting from the good interfacial protection in the FBCN-based and EC-based electrolytes, the graphite surface is uniformly covered and well maintained after cycling as revealed by SEM images (Fig. 4f–h and S41†).
O and C–O species (Fig. 4d and S38†).42,43 Note that a dominant LiF peak is observed in the FBCN-based electrolyte with subtle signals from the decomposition of LiDFOB (B–F) and LiFSI (S–F) (Fig. 4e).40 Similarly, the EC-based electrolyte also exhibits a predominant peak of LiF, indicating inorganic-rich SEI derived from film-forming additives (e.g. LiDFOB and FEC). Nevertheless, CCE exhibits a relatively weak intensity from LiF but a dominant peak from the incomplete decomposition of LiPF6 (P–F), which indicates poor interfacial passivation (Fig. S39†).44 The SEI is monitored by TEM images (Fig. S40†), and a thick and uneven coverage (>40 nm) is observed in CCE. In contrast, a thin but uneven coverage (>20 nm) is exhibited in the EC-based electrolyte. Interestingly, a thin and even SEI is formed in the FBCN-based electrolyte, indicating superior passivation after additive modification. Benefiting from the good interfacial protection in the FBCN-based and EC-based electrolytes, the graphite surface is uniformly covered and well maintained after cycling as revealed by SEM images (Fig. 4f–h and S41†).
2.5 Interfacial kinetics and mechanism
Theoretical calculations were performed to understand the kinetic barrier for FBCN-involved and EC-involved solvates. A four-coordination number of Li+–3[FBCN]–[FSI−] is considered in the FBCN-based electrolyte because of its high content.45,46 For comparison, the coordination chemistry of Li+–3[EC]–[FSI−] is considered to exclude the disturbance from anions, and a simple theoretical model is used (Fig. S42,† more details are discussed in ESI Experimental parts†).47,48 Note that this model and calculation are used for an ideal situation to help with only the theoretical understanding of the interfacial kinetics. The energies required for isolating bare Li+ from the various solvates are compared to study the stabilities of the Li+ solvates (Fig. 5a and b). The Li+–3[EC]–[FSI−] shows a higher energy barrier for segregating Li+ (6.86 eV) than Li+–3[FBCN]–[FSI−] (6.37 eV), indicating an energy-consuming step for isolating Li+ in tight EC-involved solvates, which hints at strong Li+–EC binding and a sluggish interfacial process. In addition, the energy required for removing individual species from Li+ solvates is studied. As shown in Fig. S43,† EC is involved in a more energy-consuming step than FBCN, indicating the strong Li+–EC binding. Moreover, the anions exhibit a higher energy barrier (ΔE = 3.35 eV) than the solvents (e.g. EC and FBCN), indicating a stronger coulombic attraction than ion–dipole interaction (Tables S3 and S4†).39 Note that these calculations cannot reflect the true interfacial process, and obtained values do not equal the desolvation energy, but the always lower energy for FBCN-involved solvates theoretically reflects the relatively facile kinetics compared to EC-involved solvates. These results further demonstrate a weaker Li+–FBCN binding than that of EC.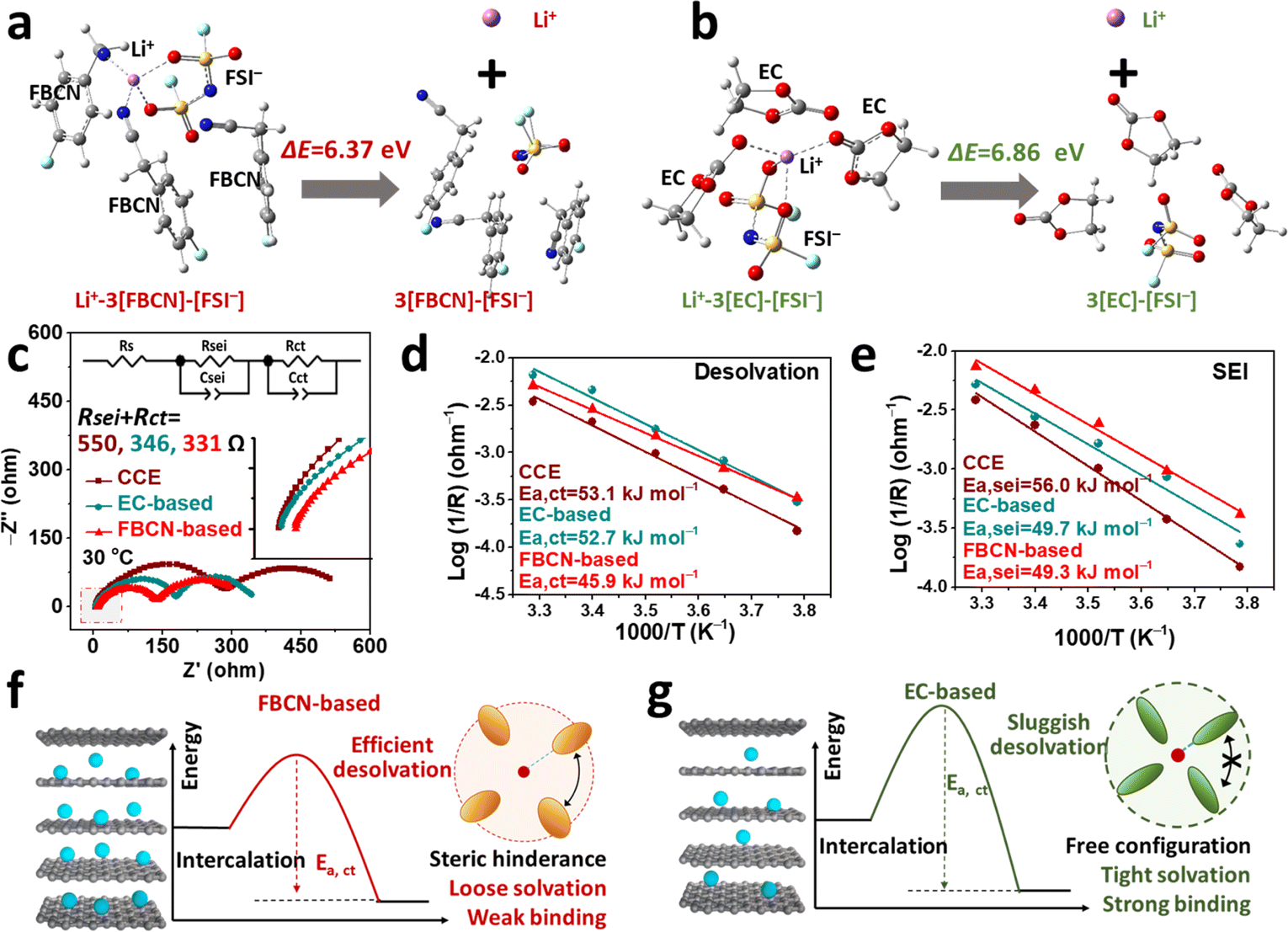 | ||
| Fig. 5 Energies for removing Li+ from (a) FBCN-dominated solvates and (b) EC-dominated solvates. (c) EIS results and fitting curves for graphite//graphite cells in various electrolytes. Calculated activation energies for (d) desolvation and (e) Li+ transport across the SEI in various electrolytes. Illustrations of regulated solvation structures and desolvation in (f) FBCN-based and (g) EC-based electrolytes. | ||
Based on the Nyquist plots of symmetric cells, the interfacial resistance for the EC-based electrolyte (346 Ω) is smaller than that of CCE (550 Ω) (Fig. 5c), which is a result of the additive modification (e.g. FEC and LiDFOB). Encouragingly, the resistance experiences a further decrease in the FBCN-based electrolyte (331 Ω), indicating facilitated interfacial kinetics in this loose Li+ solvation (Fig. S44†).49 The activation energies for Li+ desolvation (Ea,ct) and its transport across the SEI (Ea,sei) are calculated based on temperature-dependent EIS from 263 to 303 K (Fig. S45†).5,50 The Rct (charge-transfer resistance) and Rsei (Li+ transport across the SEI) are deconvoluted from the equivalent circuit in Fig. 5c. As a result, Ea,ct values for the EC-based electrolyte and CCE are 52.7 and 53.1 kJ mol−1, respectively, as a result of the tight binding between Li+ and EC/DMC.51 On switching to the FBCN-based electrolyte, Ea,ct plummets to 45.9 kJ mol−1, indicating a facile desolvation process. Simultaneously, Ea,sei decreases to 49.7 and 49.3 kJ mol−1 for EC-based and FBCN-based electrolytes (Fig. 5e), which is caused by the additive-modified SEI. In contrast, CCE goes through a higher Ea,sei (56 kJ mol−1), because of sluggish Li+ diffusion in the organic-rich SEI.9 The DRT results show a weak Rct evolution for the FBCN-based electrolyte over a wide-temperature range, indicating the kinetically favorable process for the Li+–FBCN solvates (Fig. S46†). As a result, CCE suffers from sluggish interfacial kinetics, which is a result of the organic-rich SEI and sluggish desolvation (strong Li+–EC binding).5,51 Moreover, the EC-based electrolyte benefits from the additive-modified SEI (lower Ea,sei) but still exhibits strong Li+–EC binding (higher Ea,ct). In contrast, the FBCN-based electrolyte shows fast interfacial kinetics, as demonstrated by the lower Ea,sei (additive modification) and Ea,ct (weakened Li+–FBCN binding).
Based on above results, two scenarios are illustrated to comprehend the origin of the high rate capability in the FBCN-based electrolyte. As shown in Fig. 5f, the bulky substituent on FBCN leads to intermolecular repulsion within the space-confined solvation structure, forming a loose coordination structure with a weakened ion–dipole interaction (Li+–FBCN). This solvate exhibits a lower interfacial barrier (e.g. efficient desolvation and stable interphase) at the inorganic-rich graphite surface, exhibiting superior rate performance despite its inferior ionic transfer within the bulk electrolyte. In contrast, tight coordination and strong Li+–EC binding are obtained in the EC-based electrolyte, which suffers from a higher interfacial barrier and unsatisfactory interfacial passivation (Fig. 5g), leading to a relatively poor rate capability, even with high ionic conductivity. Despite the superior effect of steric hindrance on the solvent, quantifying this effect is challenging, because of complicated relationships between the steric hindrance, molecular structure, coordination chemistry and interfacial behavior. This work provides a good model for understanding the steric hindrance and an alternative solvent for advanced electrolytes.
3. Conclusions
In summary, we herein demonstrate a new FBCN solvent for use in the design of a low-barrier electrolyte. The bulky fluorobenzene group on the FBCN leads to intermolecular repulsion within the space-confined Li+ solvation shell, forming a loose solvation environment featuring weakened Li+–FBCN binding and increased binding distance. This sterically modified solvate results in a lower interfacial barrier on the graphite surface, accounting for a substantially improved rate capability despite the inferior ionic conductivity within the bulk electrolyte. This work provides a new route for solvation engineering by molecular structure design of solvents.Data availability
Data are available upon reasonable request from the corresponding authors.Author contributions
Z. Zeng and J. Xie proposed the concept. M. Qin prepared the materials, collected relevant data, and wrote the manuscript. All authors participated in data analysis and manuscript discussion.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2022YFB2404800). The authors gratefully acknowledge the Analytical and Testing Center of HUST for allowing us to use its facilities. The authors thank Shiyanjia Lab (https://www.shiyanjia.com) and eceshi (https://www.eceshi.com) for helping with materials characterization.References
- Y. Gao, T. Rojas, K. Wang, S. Liu, D. W. Wang, T. H. Chen, H. Y. Wang, A. T. Ngo and D. H. Wang, Nat. Energy, 2020, 5, 534–542 CrossRef CAS
.
- M. Qin, Z. Zeng, S. Cheng and J. Xie, Interdiscip. Mater., 2023, 2, 308–336 CrossRef CAS
- T. Zhang, W. He, W. Zhang, T. Wang, P. Li, Z. Sun and X. Yu, Chem. Sci., 2020, 11, 8686–8707 RSC
- E. R. Logan and J. R. Dahn, Trends Chem., 2020, 2, 354–366 CrossRef CAS
- K. Xu, A. von Cresce and U. Lee, Langmuir, 2010, 26, 11538–11543 CrossRef CAS PubMed
- F. Cheng, X. Zhang, Y. Qiu, J. Zhang, Y. Liu, P. Wei, M. Ou, S. Sun, Y. Xu, Q. Li, C. Fang, J. Han and Y. Huang, Nano Energy, 2021, 88, 106301 CrossRef CAS
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS
- k. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed
- Y. X. Yao, X. Chen, N. Yao, J. H. Gao, G. Xu, J. F. Ding, C. L. Song, W. L. Cai, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2022, 62, e202214828 CrossRef PubMed
- Q. Y. Li, D. P. Lu, J. M. Zheng, S. H. Jiao, L. L. Luo, C. M. Wang, K. Xu, J. G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 42761–42768 CrossRef CAS PubMed
- X. H. Zhang, L. F. Zou, Y. B. Xu, X. Cao, M. H. Engelhard, B. E. Matthews, L. R. Zhong, H. P. Wu, H. Jia, X. D. Ren, P. Y. Gao, Z. H. Chen, Y. Qin, C. Kompella, B. W. Arey, J. Li, D. Y. Wang, C. M. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2020, 10, 2000368 CrossRef CAS
- P. Iamprasertkun, A. Ejigu and R. A. W. Dryfe, Chem. Sci., 2020, 11, 6978–6989 RSC
- Z. Wang, L.-P. Hou, Q.-K. Zhang, N. Yao, A. Chen, J.-Q. Huang and X.-Q. Zhang, Chin. Chem. Lett., 2024, 35, 108570 CrossRef CAS
- E. Park, J. Park, K. Lee, Y. Zhao, T. Zhou, G. Park, M.-G. Jeong, M. Choi, D.-J. Yoo, H.-G. Jung, A. Coskun and J. W. Choi, ACS Energy Lett., 2022, 8, 179–188 CrossRef
- J.-L. Liang, S.-Y. Sun, N. Yao, Z. Zheng, Q.-K. Zhang, B.-Q. Li, X.-Q. Zhang and J.-Q. Huang, Sci. China: Chem., 2023, 66, 3620–3627 CrossRef CAS
- G. Zhang, J. Chang, L. Wang, J. Li, C. Wang, R. Wang, G. Shi, K. Yu, W. Huang, H. Zheng, T. Wu, Y. Deng and J. Lu, Nat. Commun., 2023, 14, 1081 CrossRef CAS PubMed
- J. Yang, Q. Liu, K. Z. Pupek, T. L. Dzwiniel, N. L. D. Rago, J. Cao, N. Dandu, L. Curtiss, K. Liu, C. Liao and Z. Zhang, ACS Energy Lett., 2021, 6, 371–378 CrossRef CAS
- A. Shyamsunder, W. Beichel, P. Klose, Q. Pang, H. Scherer, A. Hoffmann, G. K. Murphy, I. Krossing and L. F. Nazar, Angew. Chem., Int. Ed., 2017, 56, 6192–6197 CrossRef CAS PubMed
- X. Peng, Y. Lin, Y. Wang, Y. Li and T. Zhao, Nano Energy, 2022, 96, 107102 CrossRef CAS
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegash, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed
- M. Qin, Z. Zeng, Q. Wu, H. Yan, M. Liu, Y. Wu, H. Zhang, S. Lei, S. Cheng and J. Xie, Energy Environ. Sci., 2023, 16, 546–556 RSC
- M. Qin, Z. Zeng, X. i. Liu, Y. Wu, R. He, W. Zhong, S. Cheng and J. Xie, Adv. Sci., 2023, 10, 2206648 CrossRef CAS PubMed
- C. Zhu, C. Sun, R. Li, S. Weng, L. Fan, X. Wang, L. Chen, M. Noked and X. Fan, ACS Energy Lett., 2022, 7, 1338–1347 CrossRef CAS
- S. Chen, J. Fan, Z. Cui, L. Tan, D. Ruan, X. Zhao, J. Jiang, S. Jiao and X. Ren, Angew. Chem., Int. Ed., 2023, 62, e202219310 CrossRef CAS PubMed
- M. Qin, M. Liu, Z. Zeng, Q. Wu, Y. Wu, H. Zhang, S. Lei, S. Cheng and J. Xie, Adv. Energy Mater., 2022, 12, 2201801 CrossRef CAS
- Y. X. Yao, X. Chen, C. Yan, X. Q. Zhang, W. L. Cai, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef CAS PubMed
- J. L. Allen, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Power Sources, 2011, 196, 9737–9742 CrossRef CAS
- D. Yaakov, Y. Gofer, D. Aurbach and I. C. Halalay, J. Electrochem. Soc., 2010, 157, A1383–A1391 CrossRef CAS
- J. Shi, N. Ehteshami, J. Ma, H. Zhang, H. Liu, X. Zhang, J. Li and E. Paillard, J. Power Sources, 2019, 429, 67–74 CrossRef CAS
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv.
Energy Mater., 2021, 11, 2101126 CrossRef CAS
- X. Ma, J. Li, S. L. Glazier, L. Ma, K. L. Gering and J. R. Dahn, Electrochim. Acta, 2018, 270, 215–223 CrossRef CAS
- X. Liu, X. Shen, H. Li, P. Li, L. Luo, H. Fan, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Adv. Energy Mater., 2021, 11, 2003905 CrossRef CAS
- T. Hou, G. Yang, N. N. Rajput, J. Self, S.-W. Park, J. Nanda and K. A. Persson, Nano Energy, 2019, 64, 103881 CrossRef CAS
- H. Zhang, Z. Zeng, M. Liu, F. Ma, M. Qin, X. Wang, Y. Wu, S. Lei, S. Cheng and J. Xie, Chem. Sci., 2023, 14, 2745–2754 RSC
- Y. Lu, W. Zhang, S. Liu, Q. Cao, S. Yan, H. Liu, W. Hou, P. Zhou, X. Song, Y. Ou, Y. Li and K. Liu, ACS Nano, 2023, 17, 9586–9599 CrossRef CAS PubMed
- E. S. Kryachko and E. V. Ludeña, Phys. Rep., 2014, 544, 123–239 CrossRef CAS
- L. Chen, W. Sun, K. Xu, Q. Dong, L. Zheng, J. Wang, D. Lu, Y. Shen, J. Zhang, F. Fu, H. Kong, J. Qin and H. Chen, ACS Energy Lett., 2022, 7, 1672–1678 CrossRef CAS
- Y. Wang, Z. Ma, Z. Cao, T. Cai, G. Liu, H. Cheng, F. Zhao, L. Cavallo, Q. Li and J. Ming, Adv. Funct. Mater., 2023, 33, 2305974 CrossRef CAS
- A. Wang, L. Wang, Y. Wu, Y. He, D. Ren, Y. Song, B. Zhang, H. Xu and X. He, Adv. Energy Mater., 2023, 13, 2300626 CrossRef CAS
- B. S. Parimalam and B. L. Lucht, J. Electrochem. Soc., 2018, 165, A251–A255 CrossRef CAS
- X. Shen, P. Li, X. Liu, S. Chen, X. Ai, H. Yang and Y. Cao, Chem. Sci., 2021, 12, 9037–9041 RSC
- M. Qin, Z. Zeng, Q. Wu, X. Liu, Q. Liu, S. Cheng and J. Xie, J. Energy Chem., 2023, 85, 49–57 CrossRef CAS
- W. N. Xu, X. B. Liao, W. W. Xu, C. L. Sun, K. N. Zhao, Y. Zhao and C. G. Hu, Nano Energy, 2021, 88, 106237 CrossRef CAS
- Y.-G. Cho, M. Li, J. Holoubek, W. Li, Y. Yin, Y. S. Meng and Z. Chen, ACS Energy Lett., 2021, 6, 2016–2023 CrossRef CAS
- Y. Wu, A. Wang, Q. Hu, H. Liang, H. Xu, L. Wang and X. He, ACS Cent. Sci., 2022, 8, 1290–1298 CrossRef CAS PubMed
- A. Wang, Z. Zou, D. Wang, Y. Liu, Y. Li, J. Wu, M. Avdeev and S. Shi, Energy Storage Mater., 2021, 35, 595–601 CrossRef
- K. Leung and C. M. Tenney, J. Phys. Chem. C, 2013, 117, 24224–24235 CrossRef CAS
- T. Hou, K. D. Fong, J. Wang and K. A. Persson, Chem. Sci., 2021, 12, 14740–14751 RSC
- X. Zheng, Z. Gu, J. Fu, H. Wang, X. Ye, L. Huang, X. Liu, X. Wu, W. Luo and Y. Huang, Energy Environ. Sci., 2021, 14, 4936–4947 RSC
- K. Xu, J. Electrochem. Soc., 2007, 154, A162 CrossRef CAS
- J. Xu, X. Wang, N. Yuan, J. Ding, S. Qin, J. M. Razal, X. Wang, S. Ge and Y. Gogotsi, Energy Storage Mater., 2019, 23, 383–389 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00013g |
| This journal is © The Royal Society of Chemistry 2024 |