
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic structural evolution of MgO-supported palladium catalysts: from metal to metal oxide nanoparticles to surface then subsurface atomically dispersed cations†
Yizhen
Chen‡
 a,
Rachita
Rana‡
a,
Yizhi
Zhang‡
b,
Adam S.
Hoffman
c,
Zhennan
Huang
d,
Bo
Yang
b,
Fernando D.
Vila
e,
Jorge E.
Perez-Aguilar
c,
Jiyun
Hong
c,
Xu
Li
f,
Jie
Zeng
f,
Miaofang
Chi
d,
Coleman X.
Kronawitter
a,
Haiyan
Wang
b,
Simon R.
Bare
*c,
Ambarish R.
Kulkarni
*a and
Bruce C.
Gates
*a
a,
Rachita
Rana‡
a,
Yizhi
Zhang‡
b,
Adam S.
Hoffman
c,
Zhennan
Huang
d,
Bo
Yang
b,
Fernando D.
Vila
e,
Jorge E.
Perez-Aguilar
c,
Jiyun
Hong
c,
Xu
Li
f,
Jie
Zeng
f,
Miaofang
Chi
d,
Coleman X.
Kronawitter
a,
Haiyan
Wang
b,
Simon R.
Bare
*c,
Ambarish R.
Kulkarni
*a and
Bruce C.
Gates
*a
aDepartment of Chemical Engineering, University of California, Davis, California 95616, USA. E-mail: arkulkarni@ucdavis.edu; bcgates@ucdavis.edu
bSchool of Materials Engineering, Purdue University, West Lafayette, Indiana 47907, USA
cStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA. E-mail: srbare@slac.stanford.edu
dOak Ridge National Laboratory, Oak Ridge, Tennessee 37830, USA
eDepartment of Physics, University of Washington, Seattle, Washington 98195, USA
fNational Research Center for Physical Sciences at the Microscale, CAS Key Laboratory of Strongly-Coupled Quantum Matter Physics, Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, Department of Chemical Physics, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China
First published on 4th April 2024
Abstract
Supported noble metal catalysts, ubiquitous in chemical technology, often undergo dynamic transformations between reduced and oxidized states—which influence the metal nuclearities, oxidation states, and catalytic properties. In this investigation, we report the results of in situ X-ray absorption spectroscopy, scanning transmission electron microscopy, and other physical characterization techniques, bolstered by density functional theory, to elucidate the structural transformations of a set of MgO-supported palladium catalysts under oxidative treatment conditions. As the calcination temperature increased, the as-synthesized supported metallic palladium nanoparticles underwent oxidation to form palladium oxides (at approximately 400 °C), which, at approximately 500 °C, were oxidatively fragmented to form mixtures of atomically dispersed palladium cations. The data indicate two distinct types of atomically dispersed species: palladium cations located at MgO steps and those embedded in the first subsurface layer of MgO. The former exhibit significantly higher (>500 times) catalytic activity for ethylene hydrogenation than the latter. The results pave the way for designing highly active and stable supported palladium hydrogenation catalysts with optimized metal utilization.
Introduction
Supported noble metals are among the most widely applied industrial catalysts, ranging in structure from isolated atoms to nanoparticles.1 They are applied in processes ranging from hydrogenation (e.g., of olefins)2 to oxidation (e.g., of CO and hydrocarbons in vehicle exhaust conversion reactors).3 Their structures depend on the reaction environments, which change during operation.4–7 The environments commonly vary from those that are reducing in catalysis to those that are oxidizing in catalyst regeneration. In reducing atmospheres, supported noble metals are typically present as zerovalent nanoparticles,8 and, in oxidizing atmospheres, as cations or metal oxide clusters.9 The transformations of the various structures lead to changes in catalyst performance, and the complexity of the changes, increased by the intrinsic heterogeneity of solid support surfaces, makes it challenging to determine how the various species contribute to catalysis.1,10,11Among the metals used in commercial catalysts, palladium stands out for its wide range of active forms—with the metal having various formal oxidation states, ligand surroundings, and nuclearities.12–14 Our goal was to elucidate the dynamics of structural transformations of supported palladium and compare the catalytic properties of the samples in a range of forms for a simple test reaction, ethylene hydrogenation.15 We chose MgO as the support because it is available as high-surface-area powders consisting of non-reducible crystalline particles that are better-defined structurally than the more common supports such as silica and transition aluminas. Further, MgO consists of low-atomic-number atoms, enabling high Z-contrast with palladium atoms on its surface in high-angle annular dark field (HAADF) scanning transmission electron microscopy (STEM).16,17
Now we report results determined with techniques including X-ray absorption spectroscopy (XAS), HAADF-STEM, and electronic structure calculations at the level of density functional theory (DFT) showing how to oxidatively convert metallic palladium nanoparticles on MgO sequentially into PdO nanoparticles and then into atomically dispersed palladium on the MgO support and further into atomically dispersed palladium in the first subsurface layer of the MgO. The results show how the transformation chemistry proceeds as the metal nanoparticles are oxidized and the resultant PdO nanoparticles are oxidatively fragmented—coincident with the transformation of the support surface from a hydroxide to an oxide. We used ethylene hydrogenation as a probe reaction to differentiate between the atomically dispersed (cationic) palladium on and in the MgO lattice. The data show that surface palladium on MgO is more active catalytically than the subsurface palladium.
Results
Structural transformations of supported palladium species
Pd/MgOwet was synthesized by slowly adding a solution of aqueous Na2PdCl4 to an MgO powder–ethanol slurry by a method similar to that reported for synthesis of Pt/MgO.18 The resultant samples were calcined in flowing air at various temperatures (400, 500, and 700 °C), removing water and giving samples labeled as Pd/MgOT (the subscript is the calcination temperature in °C) (Table S1 in the ESI†). The palladium loading was approximately 0.1 wt% (Table S1†), determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES), a value chosen to minimize the number of different support sites occupied by palladium and thereby simplify the structures.TEM images of Pd/MgO samples (Fig. S1;† “S” refers to the ESI†) show that the average grain size of MgO was approximately 100 nm and that it did not change during calcination. X-ray diffraction (XRD) was used to characterize the Pd/MgO (Fig. S2†). The diffraction pattern of the as-synthesized sample, Pd/MgOwet, is characterized by Bragg peaks that correspond to MgO and Mg(OH)2 phases, with the rock-salt phase of MgO being dominant (Fig. S2†). XRD patterns of each calcined Pd/MgO sample (Fig. S2†) include only peaks associated with MgO, showing that the Mg(OH)2 phase underwent dehydration at temperatures lower than 400 °C.19 The diffraction patterns characterizing the Pd/MgO samples lack discernible peaks characteristic of palladium-containing phases (e.g., palladium metal or PdO), implying that the palladium was highly dispersed.16
Transmission Fourier-transform infrared (IR) spectra characterizing the νOH stretching region of each of the Pd/MgO samples showed the evolution of OH groups as a function of calcination temperature (Fig. S3†). Strong bands representative of OH groups on Mg(OH)2 (at 3700 cm−1) were observed in the spectrum of Pd/MgOwet, suggesting that some surface layers of Mg(OH)2 had formed during synthesis.20–22 This band was not present in the spectra of the calcined samples, implying that the Mg(OH)2 species had been converted to MgO during the calcination, consistent with the XRD results. IR spectra of the calcined Pd/MgO samples include bands in the 3200–4000 cm−1 range associated with hydrogen-bonding of H2O/OH groups and isolated Mg–OH groups (Fig. S3†), and these decreased in intensity with increasing calcination temperature, as dehydration (dehydroxylation) occurred.16,20
Thermogravimetric analysis-mass spectrometry (TGA-MS) was used to track the removal of water from Pd/MgOwet as it was heated from room temperature to 700 °C in flowing air (Fig. S4†). A significant loss of mass from the sample occurred at about 330 °C, concomitant with the detection of water in the mass spectrometer. The observed loss of water and the conversion of the Mg(OH)2 phase detected by XRD and IR spectroscopy indicate the conversion of Mg(OH)2 to MgO, as expected.19 Taken together, the results show that, at temperatures >400 °C, the catalyst support was bulk MgO with surface OH groups and that the MgO did not change in structure or grain size with increasing calcination temperature.
Ex situ HAADF-STEM images of Pd/MgOwet (Fig. 1a and S5†) indicate the presence of palladium-containing nanoparticles with diameters of approximately 4–5 nm. The corresponding energy dispersive X-ray spectroscopy (EDS) maps (Fig. 1b) show the distribution of these palladium-containing nanoparticles on the MgO. HAADF-STEM images of the calcined sample Pd/MgO400 (Fig. 1c and S6†) demonstrate the presence of nanoparticles, now with diameters of 8–12 nm, that is, significantly larger than those in Pd/MgOwet. The corresponding EDS maps (Fig. 1d) show that these were palladium-containing nanoparticles, distributed over the available MgO surface. The EDS data characterizing Pd/MgOwet and Pd/MgO400 (Fig. S5 and S6†) show that water washing and calcination had removed essentially all the chlorine that arose from the precursor.18,23,24
 | ||
| Fig. 1 (a) HAADF-STEM image of Pd/MgOwet. (b) Corresponding EDS elemental map of sample shown in (a). (c) HAADF-STEM image of Pd/MgO400. (d) Corresponding EDS elemental map of sample shown in (c). (e) HAADF-STEM image of Pd/MgO500. (f) Magnified HAADF-STEM image of Pd/MgO500. (g) HAADF-STEM image of Pd/MgO700. (h) Magnified HAADF-STEM image of Pd/MgO700. | ||
Additional changes in structure of the supported palladium resulted when the calcination temperature was increased from 400 to 500 °C, and subsequently to 700 °C. The HAADF-STEM images show that the palladium was then atomically dispersed on the MgO (Fig. 1e–h). We emphasize that it was the calcination at temperatures >500 °C that led to oxidative fragmentation of the larger nanoparticles to give atomically dispersed palladium.
In summary, the HAADF-STEM images and the corresponding EDS maps demonstrate the structural evolution of the palladium during calcination at increasing temperatures, first involving agglomeration to form larger particles and then particle fragmentation.
Ex situ X-ray absorption spectra (XAS) were collected at the palladium K-edge to characterize the various Pd/MgO samples. X-ray absorption near edge structure (XANES) spectra (Fig. S7†) show that the edge position shifted to higher energy with increasing calcination temperature, accompanied by a change in shape and an increase in the white line intensity. The XANES spectrum characterizing Pd/MgOwet is similar to that of palladium metal, and the spectrum of Pd/MgO400 resembles that of PdO. The XAS data characterizing the samples calcined at the higher temperatures, Pd/MgO500 and Pd/MgO700, are characterized by a unique XANES region with an edge energy of 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 355.4–24356.6 eV and a white line shape that does not match that of any of the bulk palladium standards (Fig. S7†).
355.4–24356.6 eV and a white line shape that does not match that of any of the bulk palladium standards (Fig. S7†).
Extended X-ray absorption fine structure (EXAFS) spectra of Pd/MgOwet, Pd/MgO400, and Pd/MgO500 were modeled using a scattering path approach (Fig. 2a, b, S9–S11, and Tables S3–S5†). The best-fit model representing Pd/MgOwet is characterized by a Pd–Pd scattering path with a coordination number of 9.4 ± 0.8 and a distance of 2.74 ± 0.01 Å—a bonding distance, characteristic of metallic palladium (Fig. 2a, S9 and Table S3†). A Pd–O scattering path (at a bonding distance) could not be modeled, consistent with the inference that the sample consisted of metallic palladium nanoparticles on the support. On the basis of the combinations of the EXAFS data with the STEM images, we infer that the palladium precursors had been reduced in the ethanolic solution in the initial synthesis step to form palladium nanoparticles prior to deposition on the MgO (as reported before25), even with the low metal loading that favored site isolation of the palladium on the support.
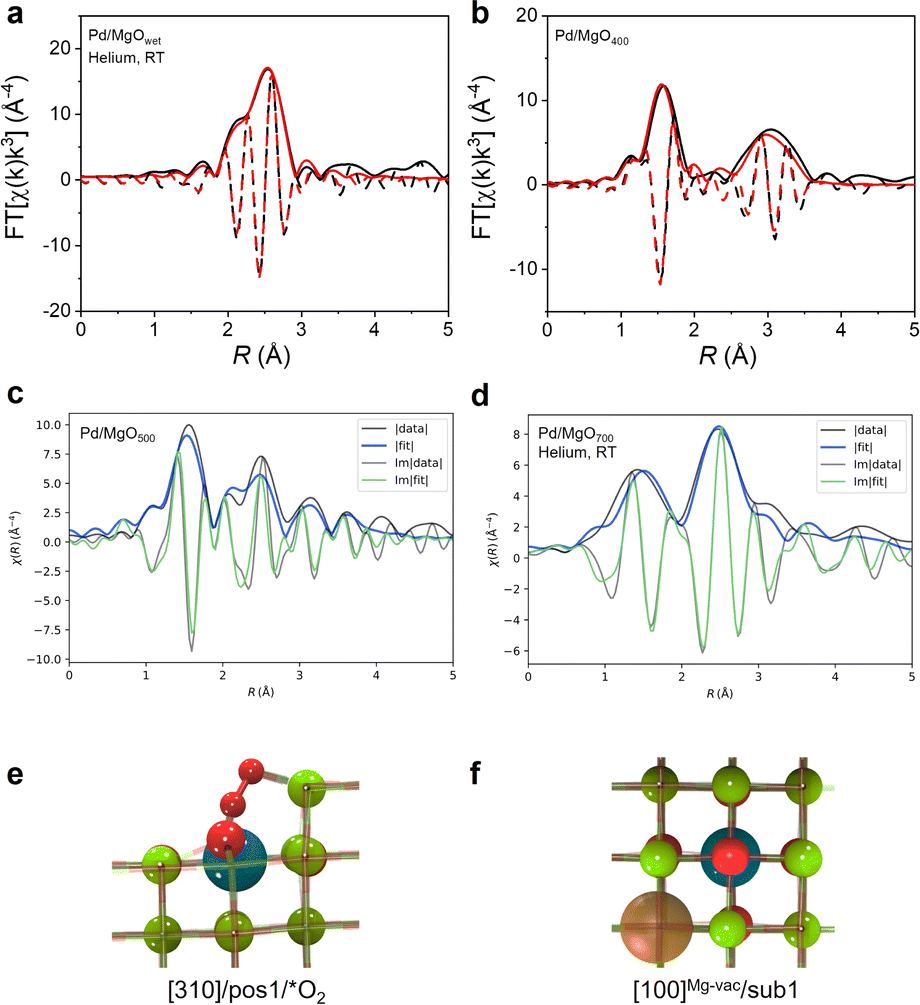 | ||
| Fig. 2 Magnitude (solid lines) and imaginary component (dashed lines) of k3-weighted Fourier transform of EXAFS data and results of conventional EXAFS modeling of (a) Pd/MgOwet in helium flowing at 20 mL (NTP) min−1 at room temperature; (b) Pd/MgO400, and (c) magnitude (solid lines) and imaginary part (dashed lines) of k3-weighted Fourier transform of EXAFS data and results of QuantEXAFS modeling of Pd/MgO500. (d) Magnitude (solid lines) and imaginary part (dashed lines) of k3-weighted Fourier transform of EXAFS data and results of QuantEXAFS modeling of Pd/MgO700 in helium flowing at 20 mL(NTP) min−1 at room temperature. (e and f) DFT-optimized structures corresponding to [310]/pos1/*O2 (used for modeling Pd/MgO500 EXAFS data) and [100]Mg−vac/sub1 (used for modeling Pd/MgO700 EXAFS data), respectively (colors: magnesium, green; oxygen, red; palladium, dark blue; carbon, dark gray; hydrogen, white; magnesium vacancy, orange). | ||
Best-fit (conventional) EXAFS models characterizing the ex situ spectra of Pd/MgO400 and Pd/MgO500 (Fig. 2b, S10, S11, Tables S4 and S5†) include Pd–O scattering paths, with a bonding distance and indicating oxidation of the metallic palladium nanoparticles, consistent with the XANES data. The (conventional) EXAFS fits characterizing Pd/MgO400 were satisfactorily modelled using only scattering paths of PdO (i.e., Pd–O at shorter distance, Pd–O at longer distance (0.36 nm), and Pd–{O}–Pd), indicating that there was a complete transformation of the palladium nanoparticles into PdO nanoparticles. In contrast, it was not possible to model the spectrum of the Pd/MgO500 with a Pd–{O}–Pd scattering path, and instead the data were best represented with a Pd–{O}–Mg scattering path—indicating that fragmentation of the PdO nanoparticles into highly dispersed, single-site or near-single-site palladium species on the MgO surface had occurred at the higher calcination temperature of 500 °C (Fig. S11 and Table S5†). This point is verified qualitatively by the shift to lower distance (R) of the broad second peak in the magnitude of the Fourier transformed data representing Pd/MgO400 (Fig. 2b), from Pd–{O}–Pd to shorter scattering distances (Fig. S11†) that correspond to Pd–{O}–Mg paths.
The lattice fringes in the image of Pd/MgO400 on the support (Fig. S12†) indicate a d-spacing of the particles of 0.27 nm, which matches well with the distance between neighboring PdO(002) planes, 0.267 nm.26 Therefore, the particles were determined to be PdO, consistent with the EXAFS results. This result indicates that, after calcination at 400 °C, the palladium nanoparticles had been transformed into oxidic species, PdO or PdO-like nanoparticles. Particle agglomeration, in combination with the expansion of the lattice as palladium metal was being oxidized to form PdO (associated with the different lattice parameters of the two phases), may account for the change in the observed particle sizes.
When the EXAFS data characterizing the sample calcined at 700 °C, Pd/MgO700, were modelled with a conventional scattering path approach (Fig. S13 and Table S6†), the results showed that it consisted of isolated palladium ions on MgO rather than palladium or PdO nanoparticles. On the basis of our work with Pt/MgO calcined at 700 °C, we anticipated that the palladium would be present in relatively uniform sites,18 and so the EXAFS analysis was performed with a method that is more rigorous than the conventional method mentioned above—with the goal of providing maximum insight into the metal–support bonding and metal–support interfacial structure.
Thus, the DFT-guided QuantEXAFS method18 was used to model EXAFS data characterizing both Pd/MgO500 and Pd/MgO700, whereby the fitting included investigation of all the scattering paths of the plausible structures determined by DFT calculations for well-defined atomically dispersed palladium structures supported on crystalline MgO (Fig. 2d, S14 and Table S7†).
QuantEXAFS workflow and categorization of paths have been elaborately discussed in our previous work.18 The candidate models in the database included 47 DFT structures that were optimized by using the PBESol functional implemented in VASP27—these structures along with our input files are available in the ESI.†
Intending to have a comprehensive database of all the plausible structures, we included three representative MgO facets (terrace sites, [100], [100]Mg-vac, and step sites, [310]); various surface and subsurface locations of palladium atoms in the MgO lattice; adsorbates (*O2, *O); and possible vacancies of Mg and O—all considered among the candidate structures. Because the influence of the choice of the DFT functional in the QuantEXAFS analysis was assessed in our previous work,18 here, we focused only on Pd/MgO structures obtained using the PBEsol functional.27
The best structure representing the Pd/MgO500 sample (Fig. 2c), was found from the DFT-optimized database (Fig. 2e) to be a step site with an isolated palladium atom bound to two oxygen atoms. The coordination numbers CNPd–O and CNPd–Mg are 7 and 9, respectively (Fig. 2e and Table S7†). However, the DFT-optimized results characterizing the Pd/MgO700 data show that the palladium was present predominantly as cations in the first MgO subsurface layer, each next to a magnesium vacancy in the [100] MgO facet (these sites are referred to as [100]Mg-vac/sub1) (Fig. 2f and Table S7†). The Pd–O and Pd–Mg coordination numbers in this dataset were found to be CNPd–O = 6 and CNPd–Mg = 11. Table S7† shows the fit parameters for both Pd/MgO500 and Pd/MgO700 along with the neighboring atoms mapped next to palladium cations, with the respective interatomic distances marked on atoms in units of Å, along with other EXAFS fit parameters, Δσ2 and ΔE, which are defined in ESI.†18
We emphasize that the information arising from the QuantEXAFS fits goes beyond just the coordination numbers and bond distances—the one-to-one mapping of models from DFT structures with the experimental EXAFS enabled us to probe the spatial orientations of neighboring atoms, as shown in Table S7.†
A central point is that the EXAFS data characterizing these calcined supported palladium samples confirm that the metallic palladium nanoparticles present initially (at temperature below 400 °C) had been converted into oxidized nanoparticles (at 400 °C) and then eventually converted—as a result of the harsh oxidation at 700 °C—into atomically dispersed palladium present in the first subsurface MgO layer.
The characterization of the intermediate-temperature sample, that is, Pd/MgO500, is more challenging, and we address it below.
The STEM images of Pd/MgO500 give evidence of isolated palladium atoms (Fig. 1e and f), and the absence of (oxidized) palladium nanoparticles—and the conventional EXAFS modeling results (Fig. S11 and Table S5†) further support the hypothesis that the Pd/MgO500 sample incorporated palladium exclusively as isolated atoms. Hence, we again used the QuantEXAFS method18–now to analyze the EXAFS data characterizing that sample, with a goal of determining the location of palladium on/in the MgO (Fig. 2c, S15 and Table S7†).
Reinforcing the STEM images and conventional EXAFS analysis performed with the scattering path method, the QuantEXAFS results give no evidence of a Pd–Pd scattering path, consistent with the atomic dispersion of the palladium. Moreover, using the database of 47 structures mentioned previously (see ESI† for more details),18 we were unable to identify a single structure that is consistent with the experimental EXAFS.
This result motivated us to explore whether more than one type of isolated palladium site was present in the Pd/MgO500 sample.
To address the challenge of accurately representing the heterogeneity of the Pd/MgO500 sample, we implemented a modified version of the QuantEXAFS approach. This modified approach involves simultaneously fitting the scattering paths obtained from two distinct DFT-optimized structures. In addition to the subsurface site, which describes the Pd/MgO700 sample, we now also consider an alternative palladium site where the O2-bound cation is stabilized at the [310] step (Fig. 2e, denoted as [310]/pos1/*O2). These two sites were chosen because they individually fit the experimental results better than the other possible sites (Fig. 2c, S15 and Table S7†), providing a significant step forward toward capturing the heterogeneity of the Pd/MgO500 sample. The results of this fitting procedure are shown in Fig. S15,† which suggests that the Pd/MgO500 can be effectively described as a 37:63 mixture of [100]Mg-vac/sub1 and [310]/pos1/*O2. Note that the two chosen sites are used as representative models to represent the non-homogenous catalyst; detailed analyses of all possible palladium local environments and their mixtures are beyond the scope of this work.
We next turn to analysis of the DFT-derived structures characterizing the Pd/MgO500 and Pd/MgO700 sites in the context of our XANES data. We calculated Bader charges of the DFT-optimized isolated palladium supported on/in the MgO support. The Bader charge analysis indicates that the subsurface-isolated palladium ([100]Mg-vac/sub1) in Pd/MgO700 (neighboring a magnesium vacancy) is well represented by a value qBader = +1.32e, which is close to that of Pd4+ in the reference compound H2Pd(OH)6, +1.36e.18,28 In contrast, the Bader charge calculated for palladium species in [310]/pos1/*O2 sites in Pd/MgO500 is +1.12e, a value that is intermediate between the value for Pd4+ and that calculated for Pd2+ in Pd(acac)2 (+0.87e) (acac is acetylacetonato), showing that the average oxidation state of palladium in Pd/MgO500 is lower than that of the palladium in Pd/MgO700.
The Bader charge analysis supports the variations in absorption edge position observed in the XANES spectra showing that (1) the palladium in Pd/MgO400 is Pd(II)O-like; (2) that in Pd/MgO700 has the highest absorption edge energy and is Pd(IV)-like; and (3) that in Pd/MgO500 is in between, Pd(II+)-like.
To recap, the ex situ characterization data demonstrate the structural evolution of Pd/MgOwet, consisting of metallic palladium supported on MgO/Mg(OH)2, from nanoparticles into subsurface atomically dispersed palladium supported on MgO via various intermediates determined by the calcination temperature. The foregoing results describe the samples in various states, but they do not yet explain the details of the oxidative fragmentation chemistry.
To address this issue, in situ XAS data were collected at the palladium K-edge to monitor the structural evolution of palladium and its oxidation state and local coordination environment as Pd/MgOwet was calcined during a temperature ramp of 2 °C min−1 from room temperature to 700 °C, with 20% O2 in helium flowing through the sample cell at a rate of 20 mL(NTP) min−1. Fig. 3a shows the evolution of the XANES spectra during the course of the calcination. As the temperature of the sample was increased from room temperature (≈25 °C) to 700 °C, there was a shift in the Pd K-edge position to higher energy that was accompanied by an increase in the white line intensity that confirms the oxidative nature of the process (represented by the XAS analysis summarized above).
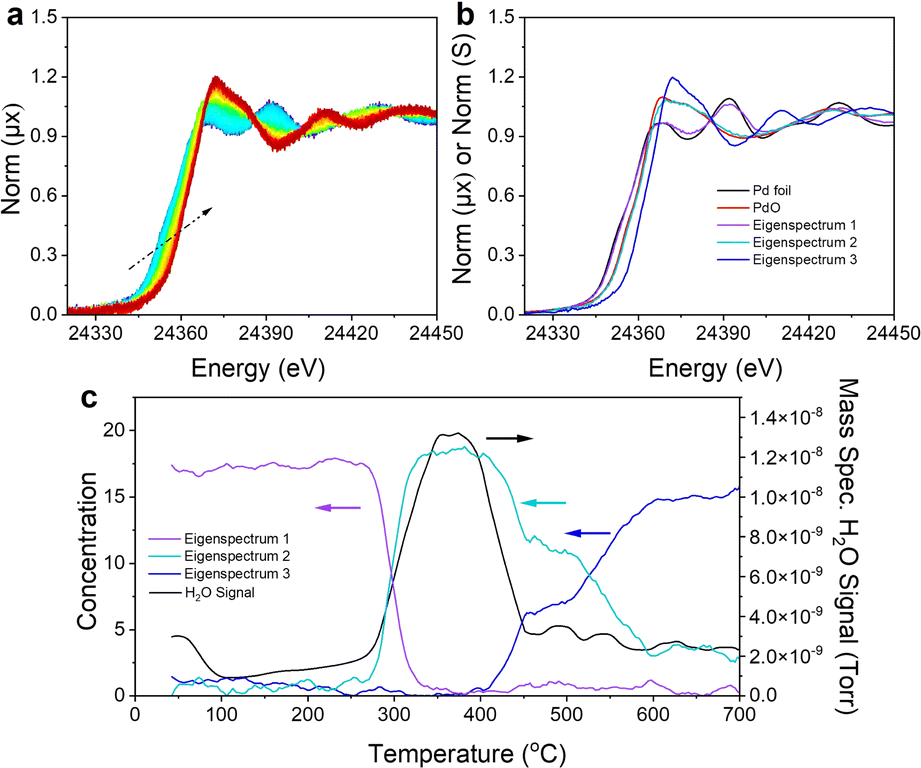 | ||
| Fig. 3 (a) The 9-spectrum rolling average normalized XANES spectra characterizing the Pd/MgO sample during TPO from room temperature to 700 °C in the presence of flowing 20% O2/helium used in the MCR-ALS analysis. The color transition in this figure from blue to dark red corresponds to the time evolution of the sample from the beginning to the end of the TPO. (b) Comparison of the three eigen spectra generated in the MCR-ALS analysis with those of reference compounds. (c) Correlation of the concentration of each eigen spectrum with the measured H2O signal in the mass spectrometer as a function of the TPO temperature. | ||
Principal component analysis (PCA), Fig. S16,† and multivariate curve resolution-alternating least squares (MCR-ALS) analysis (Fig. 3b and c) were used to identify the number and speciation of statistically unique components required to describe the transition between the initial and final palladium K-edge XANES data recorded during the calcination. The PCA scree plot (Fig. S16b†) shows that three components (Fig. S16c†) are sufficient to reconstruct the data with 99.99% accuracy. The eigen spectra calculated in the PCA (Fig. S16c†) also show that the fourth and fifth eigen spectra (components) are dominated by noise rather than spectral signatures matching the scree plot results. MCR-ALS spectral deconvolution was performed by using the assumption that three components describe the system. Fig. 3b shows the three eigen spectra obtained from the MCR-ALS analysis compared with those characterizing bulk reference compounds, palladium metal and PdO. Eigen spectrum 1 is similar to the spectrum of palladium metal; eigen spectrum 2 is similar to the spectrum of PdO; and eigen spectrum 3 is unique, with an edge position at a higher energy (and characterized by a more intense white line) than those of palladium or PdO.
The concentration of each species corresponding to the eigen spectra, together with the H2O signal (m/z = 18) recorded with a mass spectrometer during the in situ calcination, is correlated (Fig. 3c) with the temperature of the sample, showing the evolution of the palladium species and effluent product as a function of the calcination temperature. During the calcination of Pd/MgOwet, the palladium starting in the metallic state was heated to 300–400 °C, and water was removed from the sample, and the metallic palladium component was converted into a component having a XANES spectrum similar to that of PdO. This temperature range coincides with that in which Mg(OH)2 undergoes dehydration (dehydroxylation) to form MgO,29 which explains the substantial loss of water.
This conclusion is corroborated by the ex situ XRD and IR results comparing Pd/MgOwet and Pd/MgO400 (Fig. S2 and S3†), and the temperatures match the temperatures associated with the loss of water in the TGA measurements (Fig. S4†). The formation of PdO in this temperature range is consistent with the ex situ STEM and EXAFS results characterizing Pd/MgO400 (Fig. S12 and Table S4†).
The results therefore suggest that the phase change of the surface Mg(OH)2, the loss of water forming a high local concentration of steam, and the presence of oxygen—or a combination of these—lead to the oxidation of the palladium nanoparticles and the formation of palladium oxide nanoparticles with increasing size.
As the calcination temperature was increased further, to 500 °C, the PdO-like XANES signature was partially converted to that of the final component, corresponding to eigen spectrum 3 (Fig. 3b), as shown by the MCR-ALS analysis. As the temperature was still further increased to 700 °C, all the PdO-like species were converted into the final species.
The QuantEXAFS results characterizing Pd/MgO700 (Fig. 2d, S14 and Table S7†), combined with the Bader charge analysis, bolster our hypothesis that eigen spectrum 3 represents the XANES structure of a sub-surface Pd4+ species. This conclusion is plausible given the additional shift in the edge energy of eigen spectrum 3 compared with the edge positions of the XANES spectra of palladium metal and PdO.
Hence, we performed theoretical XANES calculations using FEFF to compare the experimental XANES with the simulated XANES spectrum characterizing the subsurface palladium site ([100]Mg-vac/sub1) predicted by QuantEXAFS (Fig. S17†).18 Indeed, as shown in Fig. S17,† the simulated spectrum of the subsurface structure is a good candidate to model eigen spectrum 3. Moreover, the fractions corresponding to eigen spectra 2 and 3 that characterize the sample at 500 °C suggest that the palladium was present in surface PdO-like structures as well as subsurface species (Fig. 3b), a result that is consistent with the ex situ EXAFS analysis of the Pd/MgO500 data (Fig. 2c, S11, S15, Tables S5 and S7†). The results thus imply that, after the dehydroxylation of Mg(OH)2 to give MgO, the newly formed palladium oxide nanoparticles began to fragment, with the palladium moving to the MgO subsurface, increasingly at increasing temperatures.
Such fragmentation has been observed in similar materials in which palladium nanoparticles are aged in flowing dilute oxygen at elevated temperatures.9
To provide further insight into the chemistry of the transformations taking place at the higher temperatures, the Pd/MgO400 sample—consisting of supported PdO nanoparticles—was investigated with in situ HAADF-STEM. Images were recorded at various temperatures (Fig. S18†). The Pd/MgO400 was first heated from 25 to 400 °C in the vacuum chamber of the microscope (pressure ≈ 1 × 10−7 mbar). HAADF-STEM images were recorded as the sample was heated from 400 to 700 °C at a rate of 5 °C min−1. The PdO nanoparticles remained essentially unchanged during the heating process, even at 700 °C (Fig. S18†), a result that is contrasted with the changes occurring during heating in the presence of O2 and indicated by the XAS data. Combined, these observations demonstrate that O2 is needed for the fragmentation of the palladium oxide nanoparticles, consistent with the conclusion that the particle break-up to give atomically dispersed palladium was an oxidative fragmentation. We posit that these results imply that the O2 gas-phase chemical potential (even, we suggest, under ambient conditions) has a significant influence on the structural evolution of catalysts such as ours (a conclusion that accords with reported high-pressure scanning tunneling microscopy data30).
Catalytic properties of supported palladium species
Building on the aforementioned structural insights, we examined the catalytic properties of the MgO-supported palladium species using ethylene hydrogenation as a probe reaction. The bare support calcined at various temperatures lacked measurable activity under our conditions (Fig. S19†); therefore, the catalyst performance is attributed to the supported palladium species.31–33The catalytic properties of the Pd/MgOwet sample (which contained metallic palladium nanoparticles) align with those of metallic palladium, as expected and as shown by catalytic testing data that are presented in the ESI† and which corroborate previous findings (Fig. S20†).34–37
Reported results38–42 show that H2 in the feed stream starts to reduce PdO under catalytic hydrogenation conditions at low temperatures, and we infer that this pattern pertains to Pd/MgO400. Thus, we expect that our data are not sufficient to determine steady-state catalyst performance of the supported PdO nanoparticles. Here, we focus only on the catalytic properties of samples consisting of atomically dispersed palladium on MgO supports.
Our experiments were done with a once-through plug-flow reactor operated at atmospheric pressure. The feed partial pressures (mbar) were H2, 100; ethylene, 100; and helium, 800; the total feed flow rate was 40 mL(NTP) min−1, and the catalyst mass was 50 mg. The product stream was analyzed periodically with an on-line gas chromatograph (Table 1, Fig. 4 and S19–S22†).16 The only observed product was ethane.
| Initial form of catalyst | Support | Steady-state rate of ethylene conversion, TOF (molecules·per (Pd atom)·per s) | Reaction conditions |
|---|---|---|---|
| Subsurface isolated palladium species (Pd/MgO700) | MgO | 0.22 | 10% C2H4 + 10% H2 + 80% helium, 40 mL (NTP) min−1, 1 bar, 80 °C, 50 mg of catalyst |
| Mixture of surface and subsurface sites incorporating isolated palladium species (only surface sites are considered to be active sites) (63%; Pd/MgO500) | MgO | 0.60 | 10% C2H4 + 10% H2 + 80% He, 40 mL (NTP) min−1, 1 bar, 45 °C, 50 mg of catalyst |
Ethylene hydrogenation reactions catalyzed by Pd/MgO700 incorporating atomically dispersed palladium in the first subsurface MgO layer were carried out with continuous steady-state flow of reactants at 80 °C for more than 100 h (Fig. S21a†), leading to steady-state conversions. The steady-state TOF ((molecules of ethylene converted) × (Pd atom × s)−1) was found to be 0.22 (Fig. S21a† and Table 1). Similar experiments with this catalyst were carried out at temperatures in the range of about 70–80 °C, with the data (Fig. 4) indicating an apparent activation energy of 120 ± 5 kJ mol−1.
 | ||
| Fig. 4 Arrhenius plot characterizing ethylene hydrogenation on catalysts initially present as Pd/MgO700 and Pd/MgO500. Error bars on TOF values represent standard deviations determined from three independent measurements. | ||
Ethylene hydrogenation reactions catalyzed by Pd/MgO500, which incorporated the aforementioned mixture of isolated surface and subsurface palladium, were performed at a lower reaction temperature (45 °C), chosen because of the higher activity of that sample arising, as discussed below, from the relatively high density of the more accessible isolated surface palladium sites. These flow-reactor experiments were carried out long enough to establish steady-state conversions (Fig. S21b† and Table 1). The steady-state TOF ((molecules of ethylene converted) × (Pd atom × s)−1) was calculated to be 0.60 at 45 °C, a value calculated by normalizing to the above-stated percentage of palladium in surface sites (Table 1). Similar experiments with this catalyst were carried out at temperatures in the range of 35–45 °C, with the results (Fig. 4) indicating an apparent activation energy of 95 ± 5 kJ mol−1. Further, the ratio of the activity of the mononuclear palladium in the first MgO subsurface layer to that of the surface-supported mononuclear palladium was found to be 10−4 to 0.053 at 25 °C, where the TOF values were determined by extrapolation of the Arrhenius plots.
The used catalysts from these experiments were characterized by EXAFS spectroscopy. The data characterizing both Pd/MgO700 and Pd/MgO500 recorded after catalysis were not substantially different from those characterizing the fresh sample (Fig. S23†), implying that the isolated palladium sites ([100]Mg-vac/sub1 and [310]/pos1/*O2) were stable during ethylene hydrogenation catalysis. The results imply that the initial changes in catalyst performance in the flow reactor merely represent adjustments of the local binding environments of the palladium before attainment of steady state in the presence of the reactant stream.
Thus, a central result is that palladium atomically dispersed on the surface of MgO is markedly more active than that present in the first subsurface layer of MgO (Fig. 4 and Table 1) and the catalytic hydrogenation reaction is correspondingly characterized by a lower apparent activation energy. As the Pd/MgO500 sample consists of more accessible palladium cations, we hypothesize that palladium in the [310]/pos1 sites is more active for the hydrogenation reaction than the others. Most supported metal catalysts incorporate nonuniform species, and it is expected that the catalytic activity could be dominated by a minority of species that are highly active, such as metal clusters in the presence of atomically dispersed cationic species.36,43,44 Although the presence of a few highly active minority species cannot be excluded with certainty, the catalysts reported here have been characterized in such depth, including analysis by the QuantEXAFS method,45 that we discount the likelihood that palladium clusters in either Pd/MgO500 or Pd/MgO700 were responsible for a substantial fraction of the observed catalytic activity, which we attribute to isolated palladium sites. These inferences are consistent with observations showing that atomically dispersed iridium on MgO is more active catalytically for ethylene hydrogenation when is has fewer Ir–O support bonds linking it to the support,16 and, similarly, that atomically dispersed platinum on TiO2 is more active for CO oxidation when it is coordinated to fewer support oxygen atoms.46
DFT modelling of catalyst
To explain the experimentally observed trends in catalytic activity of the two samples, we used DFT-based nudged elastic band calculations (using the RPBE/D3-(BJ) functional)47,48 to provide further understanding of the properties of atomically dispersed palladium in the surface and subsurface sites—specifically to investigate the mechanism of ethylene hydrogenation on these sites. The methods are consistent with those reported previously for the calculation of barriers.18Using these methods, we first investigated both the Eley–Rideal and Langmuir–Hinshelwood mechanisms for the subsurface palladium site ([100]Mg-vac/sub1) that represents the Pd/MgO700 sample. Investigating the Eley–Rideal mechanism, we found a relatively high enthalpic barrier (290 kJ mol−1 at 80 °C) that is associated with the reaction of a surface-bound hydrogen with ethylene. Because this calculated value is much higher than the experimentally measured apparent activation energy (120 ± 5 kJ mol−1), we conclude that another mechanism is responsible for the observed catalytic properties.49,50
Our calculations for the Langmuir–Hinshelwood mechanism, shown in Fig. 5a,49–52 agree satisfactorily with experiment, with the enthalpic barrier for hydrogen dissociation (116 kJ mol−1) being close to the experimental apparent activation energy (120 ± 5 kJ mol−1) and indicating that it is the rate-limiting step (the calculated free energy barrier is 118 kJ mol−1). On the basis of these results, we anticipate that the highly coordinated subsurface palladium atom and the neighboring magnesium vacancy activate the oxygen atom to facilitate hydrogen dissociation. The *H bound to the oxygen atom above the subsurface palladium atom stabilizes ethylene adsorption at a neighboring surface magnesium site that draws this *H to form a *C2H5 intermediate, which combines with a second *H to form the stable product ethane gas.
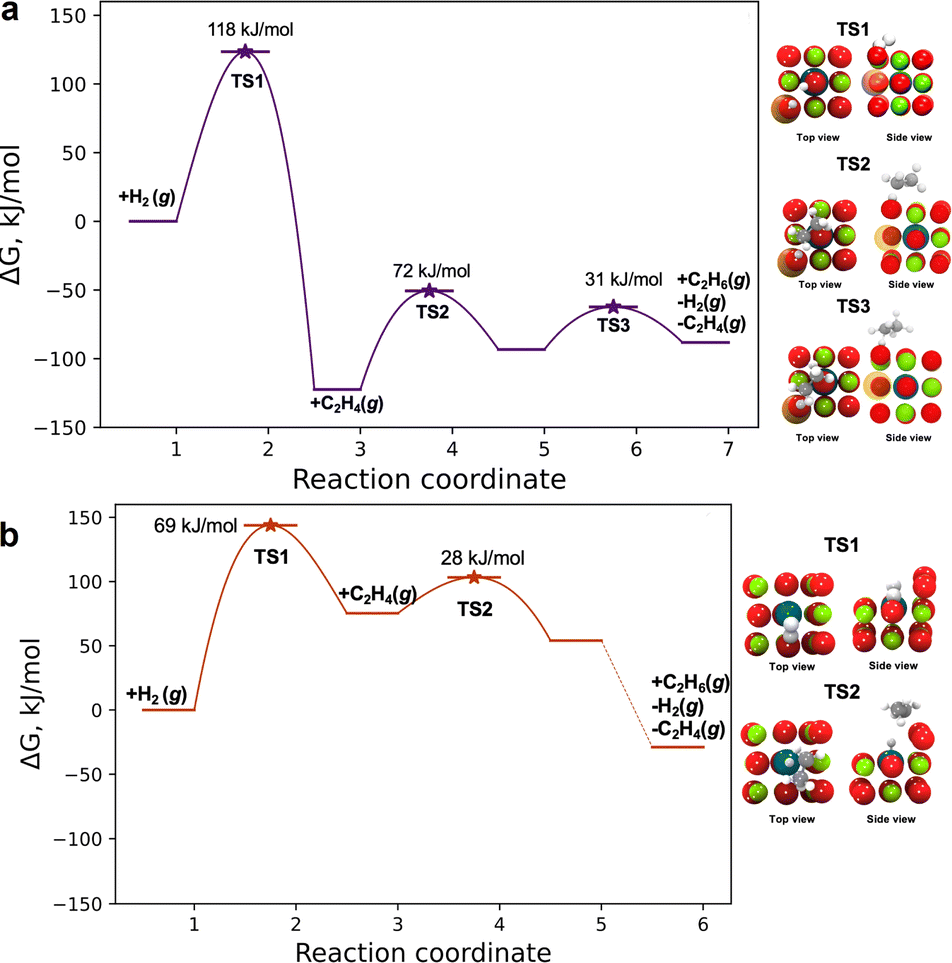 | ||
| Fig. 5 Free energy diagram representing ethylene hydrogenation on (a) [100]Mg-vac/sub1 (Pd/MgO700) at 80 °C and on (b) [310]/pos1/*O2 (Pd/MgO500) at 45 °C (colors: magnesium, green; oxygen, red; palladium, dark blue; carbon, dark gray; hydrogen, white; magnesium vacancy, orange) (the numbers on the horizontal axes are arbitrary). | ||
The above analysis is based on the involvement of the [100] facet of MgO, where the subsurface site corresponds to the Pd/MgO700 site. Because the results of the QuantEXAFS analysis suggest that the surface palladium site may be present in the more active Pd/MgO500 catalyst, we also investigated the catalytic properties of the [310]/pos1/*O2 site invoking an Eley–Rideal mechanism. The first step in the mechanism (Fig. 5b) is the dissociative adsorption of hydrogen to form atomic hydrogen, characterized by a free energy barrier of 69 kJ mol−1 at 45 °C (and an enthalpic barrier of 60 kJ mol−1). This step is inferred to be rate-limiting for ethane formation, and our calculated reaction barriers are close to the values calculated previously for transition metal catalysts.53 Following hydrogen dissociation, the π-bonded ethylene molecule interacts with a hydrogen atom bonded to an oxygen atom of MgO adjacent to the surface palladium atom. This hydrogen atom is transported to form a surface ethyl (*C2H5) intermediate by reacting with a π-bonded ethylene, followed by hydrogenation of *C2H5 to form *C2H6 and desorption of C2H6 in an essentially barrierless step (Fig. 5b). This mechanism is similar to that reported by Stacchiola et al.49
This comparison suggests that the higher activity of the Pd/MgO500 catalyst is likely dominated by the properties of [310] sites that were less prevalent than the less active [100]Mg-vac/sub1 sites. A more detailed analysis of the two-site kinetics is beyond the scope of this investigation, but, consistent with our suggestion, the experimentally observed apparent activation energy (i.e., 95 kJ mol−1) lies between the computationally determined values for the two sites that are likely present in the Pd/MgO500 sample.
Discussion
What distinguishes the work presented here from reported accounts of the evolution of a supported metal catalyst is the variety of the catalytic species and the thoroughness of the structural characterizations and their links to catalyst performance. Palladium appears to be unique among the catalytically important metals in the diversity of its catalytic structures. The involvement of PdO nanoparticles in this chemistry calls to mind the tendency of palladium catalysts to switch between the metallic and metal oxide states, with the switches determined by reaction conditions being essential in controlling the catalytic activity for the industrially important methane oxidation catalysis and providing control of the activity of the catalyst.13The importance of oxidative fragmentation of supported noble metal catalysts has, we infer, quite some generality. Related work has led to similar conclusions about this point: an example, among several, is the work of the group of Zhang,54 who reported that platinum in Fe2O3-supported nanoparticles was volatilized at 800 °C in air to give species that were transported to a reducible Fe2O3 support, where atomically dispersed platinum became bonded, evidently without the need for anion or cation vacancies on the support. Similar chemistry is illustrated by data characterizing platinum on ceria: platinum nanoparticles supported on Al2O3 were mixed with particles of defective CeO2 and calcined in air at temperatures as high as 800 °C,55 leading to the transformation of platinum into isolated atoms on CeO2via gas-phase transport of volatile PtO2. Defect sites on CeO2 played a key role in capturing and stabilizing the atomically dispersed platinum.55,56 Cargnello's group9 showed that palladium nanoparticles on γ-Al2O3 (and on other irreducible metal oxides) that were used to catalyze methane oxidation underwent deactivation as the palladium was converted at high temperatures into atomically dispersed palladium, which was inactive. Also comparable to the chemistry reported here is that reported by the group of Zhang57 showing that air calcination at 900 °C of a mixture of RuO2 powder with particles of iron oxide or iron-substituted MgAl2O4 converted the RuO2 into atomically dispersed ruthenium on the iron-containing supports.
Thus, there are various routes for conversion on metal oxide supports of metallic noble metals into atomically dispersed species, which we posit are generally positively charged. Our work on Pd/MgO provides a contrast to the aforementioned examples, as we not only observed the oxidative fragmentation of the noble metal and the formation of stabilized subsurface metal ions in the support, but also the formation of PdO nanoparticles as intermediates in the process that ultimately led to oxidative fragmentation of metallic nanoparticles. We posit that the fragmentation process was complex, influenced by both kinetics and thermodynamics. We expect that the chemistry of the crystalline MgO support played an important role in the process, but there is little that has been reported that provides mechanistic insight into it. In their insightful review of doped metal oxides,58 McFarland and Metiu noted that doping of metal oxides with metals typically involved cations as dopants and surface oxygen defects likely playing important roles, but, notwithstanding an extensive set of comments about characterization of the samples, these authors did not offer evidence of the mechanisms of the doping. We suggest the relevance of their comments but emphasize that data characterizing the mechanisms of metal doping of metal oxides are lacking. Thus, we suggest that the insights gained from our work may have value for understanding and controlling structural transformations of supported palladium catalysts, but we caution that the results characterizing the MgO support should not be expected to mimic those occurring on other supports.
Conclusions
The data reported here identify the steps in an oxidative pathway for converting palladium nanoparticles via palladium oxide nanoparticles into atomically dispersed species in surface and subsurface sites of MgO. The various palladium structures have been characterized by high-resolution STEM and X-ray absorption spectroscopy, with the data providing evidence of the distributions of the several structures depending on the calcination temperature. The metal nanoparticles underwent fragmentation as the temperature was raised to 700 °C, and oxidation led to a sequence of conversions giving the various forms of the catalyst—which were found to have markedly different catalytic properties for the ethylene hydrogenation reaction. The ratio of values of steady-state TOF values characterizing the mononuclear palladium in the first MgO subsurface layer to that of the surface-supported mononuclear palladium was found to be 10−4 to 0.053 at 25 °C. DFT-calculation results provide insight into the separate mechanisms of the ethylene hydrogenation on the various forms of the catalyst, with the calculated barriers being in satisfactory agreement with the experimental apparent activation energies.Data availability
All the computational results are available on GitHub (https://github.com/kul-group/kul-respository/tree/main/rep_2023_Pd-MgO).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
Y. C. and B. C. G. were supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES) grant DE-FG02-04ER15513. Y. Z. and H. W. acknowledge the support from the U.S. National Science Foundation (DMR-2016453) for microscopy work at Purdue University. B. Y. was supported by the U.S. Office of Naval Research N00014-22-1-2160 and N00014-20-1-2043. The Stanford Synchrotron Radiation Lightsource (SSRL) of SLAC National Accelerator Laboratory is supported by BES under Contract No. DE-AC02-76SF00515. Co-ACCESS, part of the SUNCAT Center for Interface Science and Catalysis, is supported by DOE BES Chemical Sciences, Geosciences, and Biosciences Division. C. X. K. and A. R. K. were supported by DOE BES grant DE-SC0020320. R. R. acknowledges DOE BES DE-SC0020320 for the mechanistic investigation and Co-ACCESS for the QuantEXAFS analysis. Z. H. was supported by DOE BES, Chemical Sciences, Geosciences, and Biosciences Division. Electron microscopy was performed at the Oak Ridge National Laboratory Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility (M. C.). The DFT calculations were performed using the resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231.Notes and references
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- H. Hirata, Catal. Surv. Asia, 2014, 18, 128–133 CrossRef CAS.
- P. Serna and B. C. Gates, J. Am. Chem. Soc., 2011, 133, 4714–4717 CrossRef CAS PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- L. DeRita, S. Dai, K. Lopez-Zepeda, N. Pham, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS PubMed.
- S. H. Krishna, C. B. Jones and R. Gounder, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 115–136 CrossRef CAS PubMed.
- D. Yang, P. Xu, N. D. Browning and B. C. Gates, J. Phys. Chem. Lett., 2016, 7, 2537–2543 CrossRef CAS PubMed.
- E. D. Goodman, A. C. Johnston-Peck, E. M. Dietze, C. J. Wrasman, A. S. Hoffman, F. Abild-Pedersen, S. R. Bare, P. N. Plessow and M. Cargnello, Nat. Catal., 2019, 2, 748–755 CrossRef CAS PubMed.
- B. Zhang, G. Sun, S. Ding, H. Asakura, J. Zhang, P. Sautet and N. Yan, J. Am. Chem. Soc., 2019, 141, 8185–8197 CrossRef CAS PubMed.
- Y. Tang, C. Asokan, M. Xu, G. W. Graham, X. Pan, P. Christopher, J. Li and P. Sautet, Nat. Commun., 2019, 10, 4488 CrossRef PubMed.
- K. Liu, R. Qin and N. Zheng, J. Am. Chem. Soc., 2021, 143, 4483–4499 CrossRef CAS PubMed.
- M. Cargnello, J. J. Delgado Jaén, J. C. Hernández Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gámez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- G. Zhu, J. Han, D. Y. Zemlyanov and F. H. Ribeiro, J. Am. Chem. Soc., 2004, 126, 9896–9897 CrossRef CAS PubMed.
- D. G. Oh, H. A. Aleksandrov, H. Kim, I. Z. Koleva, K. Khivantsev, G. N. Vayssilov and J. H. Kwak, Molecules, 2023, 28, 1–19 Search PubMed.
- A. S. Hoffman, L. M. Debefve, S. Zhang, J. E. Perez-Aguilar, E. T. Conley, K. R. Justl, I. Arslan, D. A. Dixon and B. C. Gates, ACS Catal., 2018, 8, 3489–3498 CrossRef CAS.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- Y. Chen, R. Rana, T. Sours, F. D. Vila, S. Cao, T. Blum, J. Hong, A. S. Hoffman, C. Y. Fang, Z. Huang, C. Shang, C. Wang, J. Zeng, M. Chi, C. X. Kronawitter, S. R. Bare, B. C. Gates and A. R. Kulkarni, J. Am. Chem. Soc., 2021, 143, 20144–20156 CrossRef CAS PubMed.
- L. M. Debefve, A. S. Hoffman, A. J. Yeh, R. C. Runnebaum, S. Shulda, R. M. Richards, I. Arslan and B. C. Gates, J. Phys. Chem. C, 2020, 124, 459–468 CrossRef CAS.
- A. Ansari, A. Ali, M. Asif and Shamsuzzaman, New J. Chem., 2018, 42, 184–197 RSC.
- R. Kurosawa, M. Takeuchi and J. Ryu, RSC Adv., 2021, 11, 24292–24311 RSC.
- S. Coluccia, L. Marchese, S. Lavagnino and M. Anpo, Spectrochim. Acta, Part A, 1987, 43, 1573–1576 CrossRef.
- X. Li, X. I. Pereira-Hernández, Y. Chen, J. Xu, J. Zhao, C. W. Pao, C. Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Nature, 2022, 611, 284–288 CrossRef CAS PubMed.
- Y. Chen, R. Rana, Z. Huang, F. D. Vila, T. Sours, J. E. Perez-Aguilar, X. Zhao, J. Hong, A. S. Ho, X. Li, C. Shang, T. Blum, J. Zeng, M. Chi, M. Salmeron, C. X. Kronawitter, S. R. Bare, A. R. Kulkarni and B. C. Gates, J. Phys. Chem. Lett., 2022, 13, 3896–3903 CrossRef CAS PubMed.
- P. D. Burton, T. J. Boyle and A. K. Datye, J. Catal., 2011, 280, 145–149 CrossRef CAS.
- S. Penner, D. Wang, B. Jenewein, H. Gasbach, B. Klötzer, A. Knop-Gerike, R. Schölgl and K. Hayek, J. Chem. Phys., 2006, 125, 094703 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- S. Chen, S. Li, R. You, Z. Guo, F. Wang, G. Li, W. Yuan, B. Zhu, Y. Gao, Z. Zhang, H. Yang and Y. Wang, ACS Catal., 2021, 11, 5666–5677 CrossRef CAS.
- M. A. Aramendia, V. Borau, C. Jiménez, J. M. Marinas, A. Porras and F. J. Urbano, J. Mater. Chem., 1996, 6, 1943–1949 RSC.
- B. Eren and M. Salmeron, J. Phys. Chem. C, 2019, 123, 8171–8176 CrossRef CAS.
- Y. Guo, H. Qi, Y. Su, Q. Jiang, Y. T. Cui, L. Li and B. Qiao, ChemNanoMat, 2021, 7, 526–529 CrossRef CAS.
- X. Tao, B. Nan, Y. Li, M. Du, L. L. Guo, C. Tian, L. Jiang, L. Shen, N. Sun and L. N. Li, ACS Appl. Energy Mater., 2022, 5, 10385–10390 CrossRef CAS.
- S. Abbet, A. Sanchez, U. Heiz, W. D. Schneider, A. M. Ferrari, G. Pacchioni and N. Rösch, J. Am. Chem. Soc., 2000, 122, 3453–3457 CrossRef CAS.
- A. Binder, M. Seipenbusch, M. Muhler and G. Kasper, J. Catal., 2009, 268, 150–155 CrossRef CAS.
- N. Doudin, S. F. Yuk, M. D. Marcinkowski, M.-T. Nguyen, J.-C. Liu, Y. Wang, Z. Novotny, B. D. Kay, J. Li, V.-A. Glezakou, G. Parkinson, R. Rousseau and Z. Dohnálek, ACS Catal., 2019, 9, 7876–7887 CrossRef CAS.
- J. Resasco, F. Yang, T. Mou, B. Wang, P. Christopher and D. E. Resasco, ACS Catal., 2020, 10, 595–603 CrossRef CAS.
- M. D. Rossell, F. J. Caparrós, I. Angurell, G. Muller, J. Llorca, M. Seco and O. Rossell, Catal. Sci. Technol., 2016, 6, 4081–4085 RSC.
- A. Baylet, P. Marécot, D. Duprez, P. Castellazzi, G. Groppi and P. Forzatti, Phys. Chem. Chem. Phys., 2011, 13, 4607–4613 RSC.
- G. P. Osorio, S. F. Moyado, V. Petranovskii and A. Simakov, Catal. Lett., 2006, 110, 53–60 CrossRef CAS.
- V. Ferrer, A. Moronta, J. Sánchez, R. Solano, S. Bernal and D. Finol, Catal. Today, 2005, 107–108, 487–492 CrossRef CAS.
- H. Zhu, Z. Qin, W. Shan, W. Shen and J. Wang, J. Catal., 2004, 225, 267–277 CrossRef CAS.
- M. Cordoba, F. Coloma-Pascual, M. E. Quiroga and C. R. Lederhos, Ind. Eng. Chem. Res., 2019, 58, 17182–17194 CrossRef CAS.
- L. Liu, D. M. Meira, R. Arenal, P. Concepcion, A. V. Puga and A. Corma, ACS Catal., 2019, 9, 10626–10639 CrossRef CAS PubMed.
- C.-T. Kuo, Y. Lu, L. Kovarik, M. Engelhard and A. M. Karim, ACS Catal., 2019, 9, 11030–11041 CrossRef CAS.
- J. Finzel, K. M. S. Gutierrez, A. S. Hoffman, J. Resasco, P. Christopher and S. R. Bare, ACS Catal., 2023, 13, 6462–6473 CrossRef CAS.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- D. Stacchiola, S. Azad, L. Burkholder and W. T. Tysoe, J. Phys. Chem. B, 2001, 105, 11233–11239 CrossRef CAS.
- M. Yang, B. Han and H. Cheng, J. Phys. Chem. C, 2012, 116, 24630–24638 CrossRef CAS.
- S. Lee, A. Patra, P. Christopher, D. G. Vlachos and S. Caratzoulas, ACS Catal., 2021, 11, 9506–9518 CrossRef CAS.
- M. Neurock and R. A. van Santen, J. Phys. Chem. B, 2000, 104, 11127–11145 CrossRef CAS.
- C. J. Heard, C. Hu, M. Skoglundh, D. Creaser and H. Grönbeck, ACS Catal., 2016, 6, 3277–3286 CrossRef CAS.
- R. Lang, W. Xi, J. C. Liu, Y. T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A. Q. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- R. Alcala, A. DeLaRiva, E. J. Peterson, A. Benavidez, C. E. Garcia-Vargas, D. Jiang, X. I. Pereira-Hernández, H. H. Brongersma, R. ter Veen, J. Staněk, J. T. Miller, Y. Wang and A. Datye, Appl. Catal., B, 2021, 284, 1–9 CrossRef.
- K. Liu, X. Zhao, G. Ren, T. Yang, Y. Ren, A. F. Lee, Y. Su, X. Pan, J. Zhang, Z. Chen, J. Yang, X. Liu, T. Zhou, W. Xi, J. Luo, C. Zeng, H. Matsumoto, W. Liu, Q. Jiang, K. Wilson, A. Wang, B. Qiao, W. Li and T. Zhang, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00035h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |