
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical stereoselective synthesis of polysubstituted 1,4-dicarbonyl Z-alkenes via three-component coupling of sulfoxonium ylides and alkynes with water†
Hao-Ran
Li
,
Yi-An
Ran
,
Yu-Yi
Zhu
,
Weisi
Guo
 ,
Shao-Fei
Ni
*,
Li-Rong
Wen
,
Ming
Li
and
Lin-Bao
Zhang
*
,
Shao-Fei
Ni
*,
Li-Rong
Wen
,
Ming
Li
and
Lin-Bao
Zhang
*
State Key Laboratory Base of Eco-Chemical Engineering, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: sfni@stu.edu.cn; zhang_linbao@126.com
First published on 25th April 2024
Abstract
The first straightforward strategy for the synthesis of 1,4-dicarbonyl Z-alkenes has been developed via an electrochemical cross-coupling reaction of sulfoxonium ylides and alkynes with water. The metal-free protocol showed an easy-to-handle nature, good functional group tolerance, and high Z-stereoselectivity, which is rare in previous cases. The proposed reaction mechanism was convincingly established by carrying out a series of control experiments, cyclic voltammetry experiments, and density functional theory (DFT) studies.
Introduction
1,4-dicarbonyl Z-alkenes constitute a privileged structural motif of many bioactive natural products and pharmaceuticals, including marine natural products, sesquiterpenes, steroids, antitumor agents, and antifungal agents.1 In addition, due to their distinctive electrophilicity and electron affinity, 1,4-dicarbonyl Z-alkenes can serve as versatile precursors in the construction of various heterocycles such as furans, thiophenes, pyrroles, pyrazines, and indolizines.2 Also, they have been used as highly reactive dienophiles for the construction of complex fused ring systems by Diels–Alder [2 + 4] cycloaddition and as Michael acceptors.3 Moreover, the synthetic methodologies of 1,4-dicarbonyl alkenes have attracted considerable attention, and remarkable progress has been made over the past few decades.4 Conventionally, 1,4-dicarbonyl Z-alkenes were synthesized by oxidative ring opening of furan and thiophene derivatives, breakdown of α-diazo carbonyl compounds, and the Wittig reaction.5,6 Recently, the Maulide group developed Ru-catalyzed cross olefination of diazo compounds with sulfoxonium ylides to give disubstituted 1,4-dicarbonyl Z-alkenes. However, poor stereoselectivity, with 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 13:1 mixtures of Z/E olefin products, was observed.6a Similarly, Wang and coworkers described a method for the synthesis of alkenes through three-component Z-selective olefinic coupling of alkynes, α-diazo sulfonium triflates, and water-based on photoredox catalysis (Scheme 1b).6b In 2023, the group of Gurubrahamam established the reactivity of alkynyl hydrazone with halonium ions that provides tetrasubstituted 4-oxo-2,3-dihaloenoates with complete Z-stereoselectivity (Scheme 1c).6c However, these approaches often suffer from several drawbacks, such as the utilization of expensive reagents/catalysts, harsh conditions, and well-tailored substrates. In addition to these shortfalls, formidable challenges remain in the synthesis of unsymmetric 1,4-diaryl-substituted 1,4-dicarbonyl Z-alkenes due to their relative instability.
:1 to 13:1 mixtures of Z/E olefin products, was observed.6a Similarly, Wang and coworkers described a method for the synthesis of alkenes through three-component Z-selective olefinic coupling of alkynes, α-diazo sulfonium triflates, and water-based on photoredox catalysis (Scheme 1b).6b In 2023, the group of Gurubrahamam established the reactivity of alkynyl hydrazone with halonium ions that provides tetrasubstituted 4-oxo-2,3-dihaloenoates with complete Z-stereoselectivity (Scheme 1c).6c However, these approaches often suffer from several drawbacks, such as the utilization of expensive reagents/catalysts, harsh conditions, and well-tailored substrates. In addition to these shortfalls, formidable challenges remain in the synthesis of unsymmetric 1,4-diaryl-substituted 1,4-dicarbonyl Z-alkenes due to their relative instability.
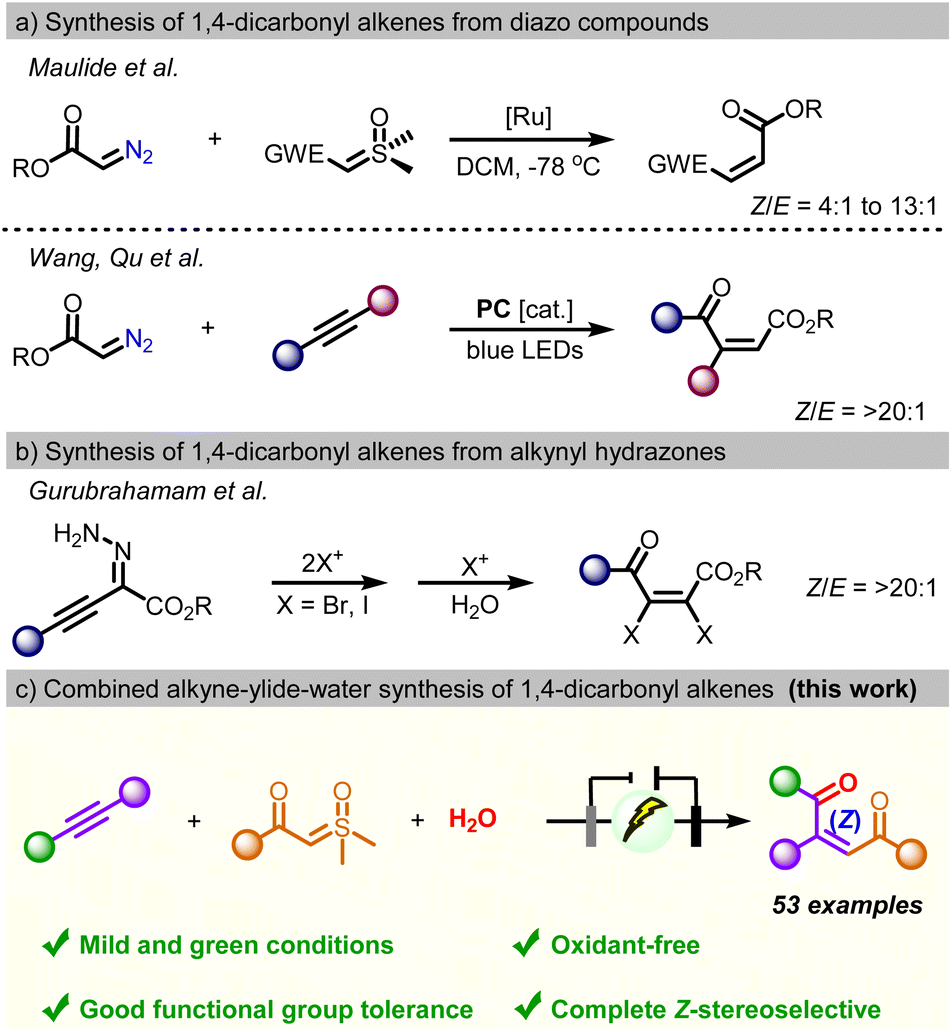 | ||
| Scheme 1 Strategies for the synthesis of 1,4-dicarbonyl alkenes; (a) synthesis from α-halo ketones. (b) Synthesis from diazo compounds. (c) Synthesis from alkynyl hydrazones. (d) Electrochemical three-component coupling. | ||
Over the last few years, organic electrosynthesis has re-emerged as a major synthesis platform.7 Electric current is a superior option to harmful external redox reagents and expensive metal catalysts, and offers higher efficiency and selectivity.8 On the other hand, multi-component cross-coupling has been explored by experts in electrochemistry to fabricate a broad spectrum of attractive and functional reactions.9 In 2023, the group of Lin proposed three-component cross-electrophile coupling to realize regioselective electrochemical dialkylation of alkenes.9a Cheng's group reported a straightforward approach for the electrochemical synthesis of tertiary α-substituted amino acid derivatives via three-component reductive coupling.9b In particular, Baran and coworkers proposed a diverse range of cross-coupling tactics using electrochemistry.9c,d
Electrochemical participation in multi-component reactions has been a hot topic - due to the irreconcilable incompatibility in the redox potential of the individual components under given electrochemical conditions. Here, our interest in electrochemical transformation and green chemistry prompts us to develop a mild electro-oxidative methodology for the fabrication of 1,4-dicarbonyl Z-alkenes via three-component coupling of sulfoxonium ylides and alkynes with water (Scheme 1d).10 The process applies to various alkynes and ylides, and it generates the corresponding 1,4-dicarbonyl Z-alkenes with simple operation and without the requirement of metal catalysts or oxidants. Based on the results from the control experiment and DFT calculations, the observed Z-selectivity could be attributed to the presence of a crucial stereochemically rigid furan intermediate in the process.
Results and discussion
We started our study with the reaction of asymmetric internal alkynes with sulfoxonium ylides in the presence of various solvents and electrodes under electrochemical conditions (Table 1). After a series of preliminary investigations, the best result was obtained when 1a, 2a, and H2O were allowed to react under a constant current flow of 5 mA for 2.25 h at 0 °C by using n-Bu4NBF4 as a supporting electrolyte with graphite felt as the electrode in DCM and HFIP as the cosolvent (Table 1, entry 1). Solvent screening experiments revealed that HFIP and DCM used as the solo solvent in this transformation led to a rapid decrease in the yield (Table 1, entries 2 and 3). Inexpensive graphite felt with excellent performance was vital to the cathode as well as the anode, whereas a medium collection rate product was obtained when the C plate was used as the anode or Pt electrode was used as the cathode instead of graphite felt (Table 1, entries 4 and 5). Different supporting electrolytes were also investigated. When n-Bu4NPF6 and Et4NBF4 were employed in the system, the desired product 3a was obtained with reduced yields (Table 1, entries 6 and 7). Decreasing or increasing the amount of electrolytes did not give better results (Table 1, entries 8 and 9). The effect of temperature on the reaction was also investigated, and it was observed that increasing or lowering the temperature did not benefit the reaction (Table 1, entry 10). Performing the reaction at a higher current or reducing the current provided inferior results (Table 1, entries 11 and 12). In addition, the reaction failed in the absence of an electric current, suggesting the key role of electric energy in the reaction (Table 1, entry 13).|
|
||
|---|---|---|
| Entrya | Variation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), H2O (0.1 mmol), n-Bu4NBF4 (0.4 mmol), DCM (4.0 mL), HFIP (1.0 mL), graphite felt anode (1.0 cm × 1.0 cm × 0.5 cm), graphite felt cathode (1.0 cm × 1.0 cm × 0.5 cm), undivided cell, 5 mA, air, 0 °C, 2.25 h (4.20 F mol−1), undivided cell. b Isolated yields. c NR = no reaction. | ||
| 1 | None | 84 |
| 2 | HFIP as solvent | 40 |
| 3 | DCM as solvent | Trace |
| 4 | C (+)/GF (−) | 60 |
| 5 | GF (+)/Pt (−) | 55 |
| 6 | n-Bu4NPF6 instead of n-Bu4NBF4 | 63 |
| 7 | Et4NBF4 instead of n-Bu4NBF4 | 52 |
| 8 | 0.3 mmol n-Bu4NBF4 | 72 |
| 9 | 0.5 mmol n-Bu4NBF4 | 41 |
| 10 | −20 °C or RT instead of 0 °C | 68/50 |
| 11 | 3 mA instead of 5 mA, 3.75 h | 53 |
| 12 | 7 mA instead of 5 mA, 1.60 h | 56 |
| 13 | No electricity | NRc |
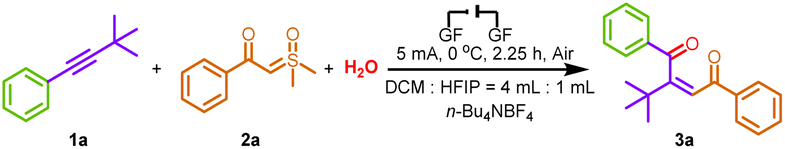
With an optimized set of conditions in hand, the scope of this transformation was explored, as illustrated in Scheme 2. The substrate scope of alkynes was investigated first. The alkynes 1a–1e prepared from alkyl- and phenyl-substituted iodobenzene and 3,3-dimethyl-1-butyne were found to be suitable substrates for this reaction, and the corresponding 1,4-dicarbonyl Z-alkenes were formed in moderate to good yields (3a–3e). Moreover, the structure of 3a was further confirmed by X-ray crystallography analysis (see the ESI† for details; CCDC: 2340616). For substrates containing methyl (1f), ethyl (1g), n-heptyl (1h), and cyclopropyl (1i) groups, the corresponding products 3f–3i were successfully delivered with yields of 49–68%. To further broaden the range of substrates for the reaction, the adoption of symmetrical internal alkynes (1j–1l) gave the coupling products we foresee, demonstrating the universal applicability in terms of symmetrical or asymmetric alkynes. The products 3a–3l indicated that the steric hindrance did not affect the efficiency of electrochemical transformations. Remarkably, to illustrate the suitability of this approach in drug molecules, we smoothly transformed alkynes embedded in the ibuprofen, fenofibric acid, loxoprofen, and probenecid frameworks into the trisubstituted alkenes 3m–3p in moderate yields. However, the E-isomer was obtained in these cases since the Z-type alkenes are unstable after isolation and rapidly transform into E-type.
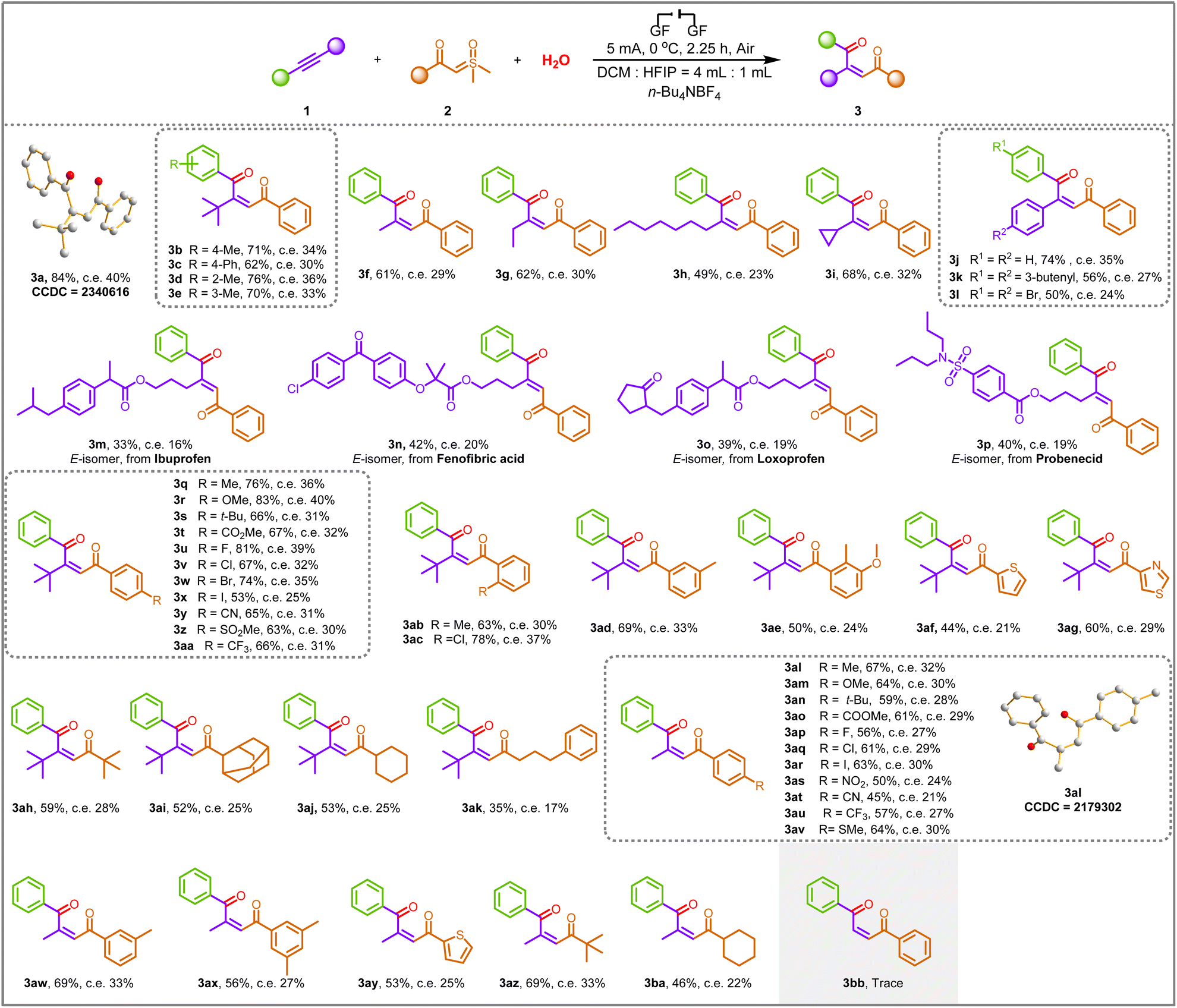 | ||
| Scheme 2 Substrate scope of alkynes and sulfoxonium ylides. Reaction condition: 1a (0.1 mmol), 2a (0.2 mmol), H2O (0.1 mmol), n-Bu4NBF4 (0.4 mmol), DCM (4.0 mL), HFIP (1.0 mL), graphite felt anode (1.0 cm × 1.0 cm × 0.5 cm), graphite felt cathode (1.0 cm × 1.0 cm × 0.5 cm), undivided cell, 5 mA, under air, 0 °C, 2.25 h (4.20 F mol−1), undivided cell. c. e. = current efficiency. | ||
Next, a range of sulfoxonium ylides 2 with structural diversity was investigated under the electrochemical conditions. Ylides 2 with distinct electronic properties (i.e., electron-rich and -deficient) at various positions in the benzene ring proved to be suitable substrates (3q–3ad). The desired transformation was achieved in good to excellent yields (53–83% yields). Moreover, the transformation was further extendable to a substrate with double substitution patterns, and the 1,4-dicarbonyl Z-alkene product 3ae was obtained in 50% yield. In addition, heterocyclic functional groups, such as thiophene and thiazole, were compatible with the newly developed reaction conditions, enhancing the scope of the synthetic protocol (3af–3ag). Interestingly, fatty ylides containing tert-butyl (2v), adamantine (2w), cyclohexyl (2x), and 4-phenyl-n-butyl (2y) groups were also found to be compatible under optimal conditions, and the corresponding products 3ah–3ak were obtained in moderate to good yields, which are inaccessible by other means. The coupling of low-resistance methylphenylethyne to ylides with different substituents proceeded successfully, providing the desired products 3al–3ba in good yields, demonstrating that steric effects have less impact on stereoselectivity. Moreover, the structure of 3al was further confirmed by X-ray crystallography analysis (see the ESI† for details; CCDC: 2179302). Unfortunately, terminal alkyne was proved to be an unavailable substrate since only a trace amount of 3bb was generated under standard conditions. It is worth mentioning that all product configurations were determined by analogy with the NMR data of 3a and 3al.
To explore the practicality of the electrochemical protocol, a gram-scale experiment was conducted (Scheme 3A) and a 75% yield was obtained under the standard conditions.
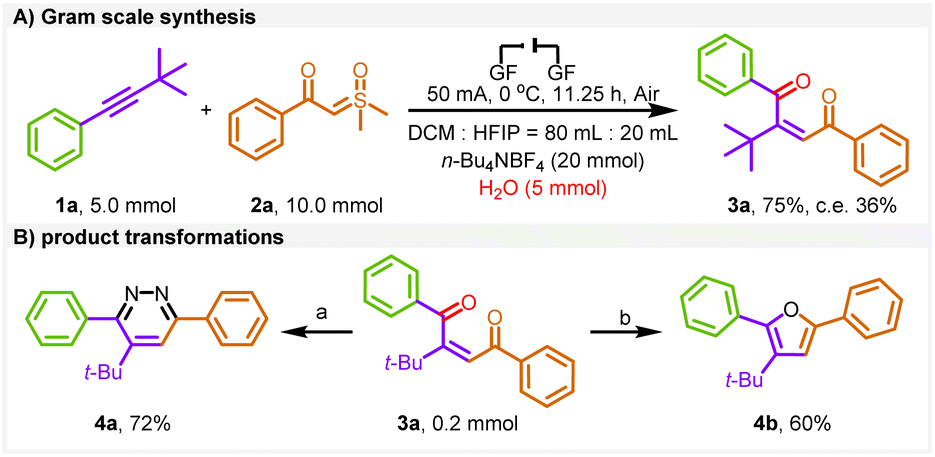 | ||
| Scheme 3 Gram scale synthesis and product transformations. Reaction conditions: (A) NH2NH2·H2O, MeOH, 50 °C. (B) NaBH4, 90% C2H5OH, 20 h. c. e. = current efficiency. | ||
To enhance the applicability of the product (Scheme 3B), we performed the conversion of pyridazine (4a) and furan (4b) by utilizing one-step follow-up transformations of 3a, providing effective access to multisubstituted heterocycles.
To give a better understanding of the reaction mechanism, the density functional theory (DFT) calculations were applied in this work. As shown in Scheme 4a, the enol intermediate Int1 converts to 1,4-dicarbonyl Z-alkenes P–Z, the favored product, through the black pathway. From Int1, the C–S bond cleavage takes place via transition state TS1 to afford a stable five-membered ring intermediate Int2, followed by a ring-opening process viaTS2 with water and the deprotonation process to form the product P–Z. The calculated reaction barriers for these two transition states are 8.7 and 9.3 kcal mol−1, respectively. The whole process is exothermic at about 66.0 kcal mol−1. As for the unfavored 1,4-dicarbonyl E-alkene P–E pathway (in red color), the reaction starts with the rotation of the C–C bond in Int1 to form Int2′, followed by a rapid C–S bond cleavage and deprotonation process to P–E. Specifically, Int1 takes 14.2 kcal mol−1 of energy for the C–C bond rotation in the transition state TS1′, which is higher than that for the route to the main product. In addition, Int2 is more stable than Int2′. Thus, the route leading to Int2 is more favorable than that for Int2′ both kinetically and thermodynamically. Moreover, no transition states in these proton transfer processes were located, and the O–H bond scan confirmed that these deprotonation processes are barrier-less (Scheme 4b).
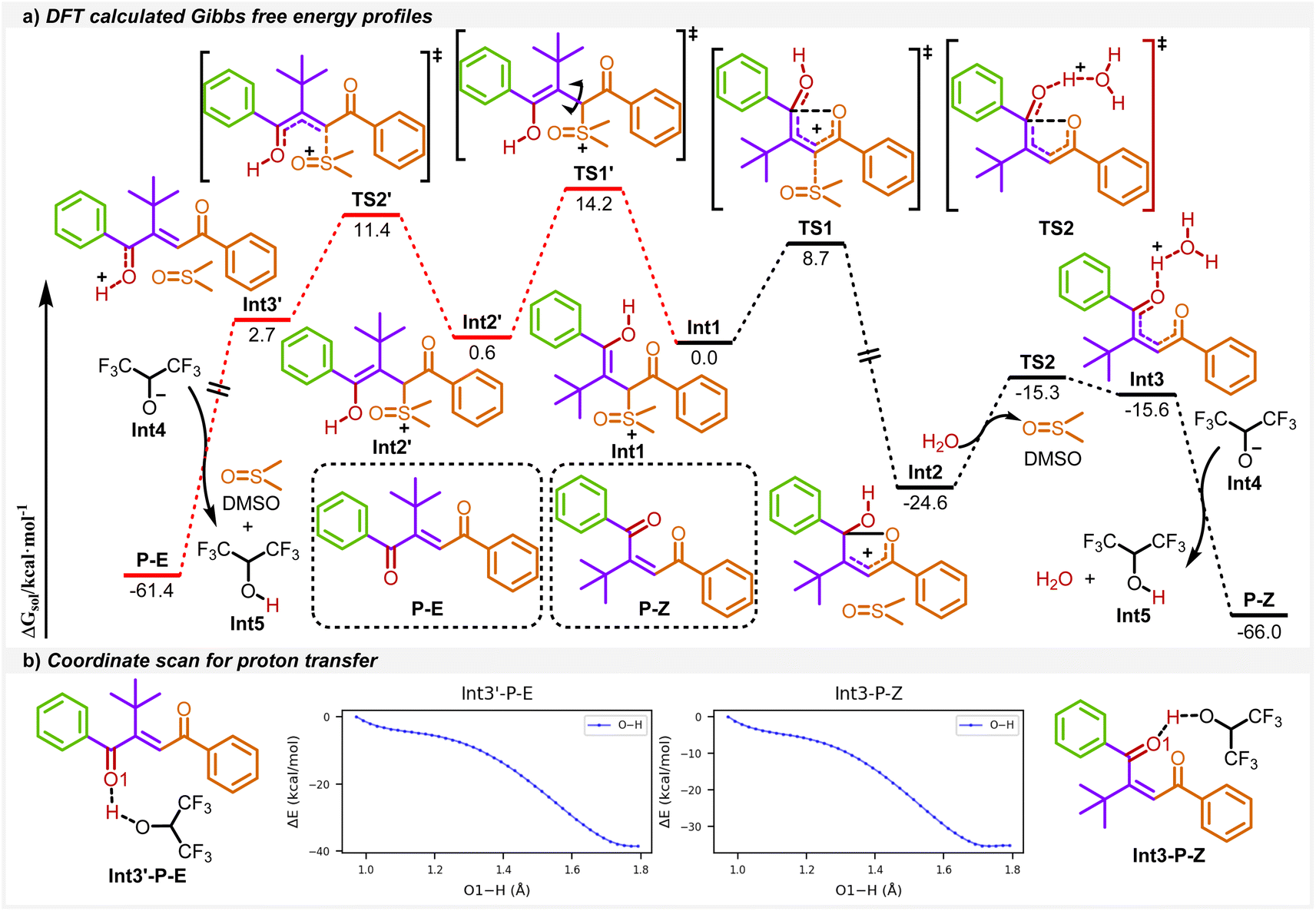 | ||
| Scheme 4 DFT calculations. | ||
To gain some insights into the reaction mechanism, control experiments were performed. First of all, cyclic voltammetry (CV) experiments were performed on 1a and 2a and the results revealed that the onset potential of 1a was 1.79 V. However, the onset potential of 2a was 1.02 V, indicating that 2a was oxidized preferentially at the anode. Moreover, in the presence of alkyne 1a, the anodic current increased significantly, indicating that the ylide radical had a rapid interaction with alkyne 1a (Fig. 1). In addition, the oxidation peak of 2a emerged at 2.73 V for the dilution system, indicating that the concentration may influence the efficiency of the reaction. (see the ESI for details, Fig. S13†)
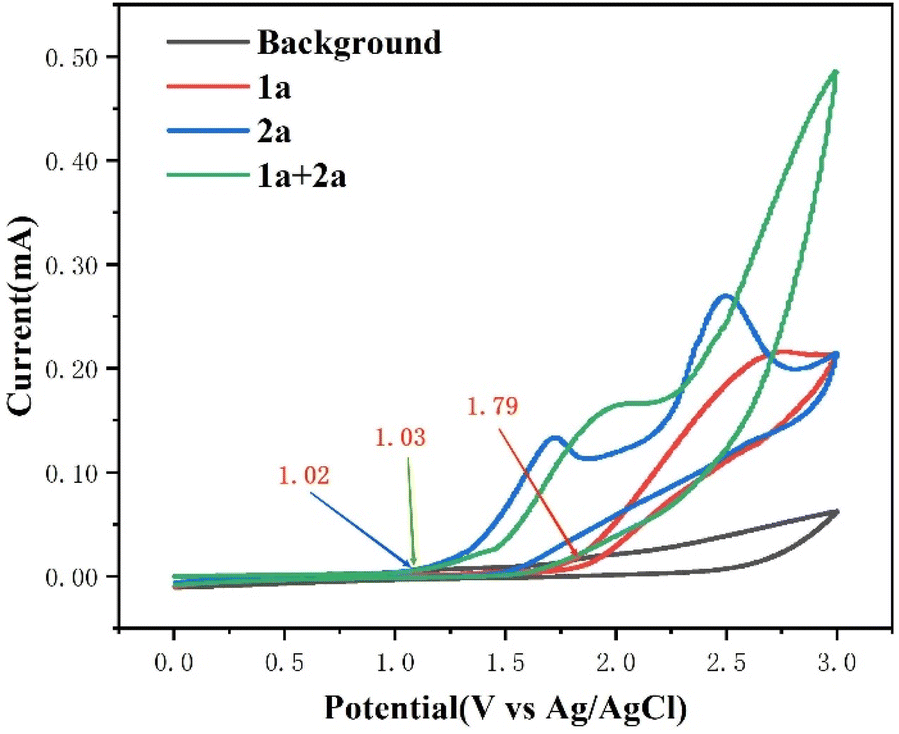 | ||
| Fig. 1 Cyclic voltammetry experiments of 1a and 2a in a solvent (4.0 mL DCM, 1.0 mL HFIP) containing H2O (0.1 mmol) and n-Bu4NBF4 (0.1 mmol). The scan rate was 100 mV s−1. Glassy carbon as the working electrode, Pt wire as the counter electrode, and Ag/AgCl as the reference electrode. | ||
The mechanistic insights into the coupling reaction were further investigated through a range of control experiments (Scheme 5). The reaction was carried out in the presence of free-radical-trapping reagents (FRT = BHT, and DPE), while no desired product was yielded under these conditions (Scheme 5a), indicating that the radical pathway might be involved in the process. In addition, compounds 3bc and 3bd were detected by HPLC-MS analysis (see the ESI for details, Fig. S18–S19†), further demonstrating the presence of the sulfoxonium ylide radical. To trace the source of oxygen in alkene products, the coupling reaction of 1f, 2a, and H218O was performed, giving 18O labeled alkene 3f′′ detected by HPLC-MS analysis (see the ESI for details, Fig. S20†). The 18O-labeling experiment supported the fact that H2O provides the source of the oxygen atom in the 1,4-dicarbonyl Z-alkene (Scheme 5b). The reaction was also conducted under an inert atmosphere (N2 and Ar), and 3a was isolated with a comparable yield. Notably, the conversion of 3a was hindered without water added to the reaction system. Trace amounts of the product could be attained, which could be attributed to residual water present in the solvent (Scheme 5c). Under standard conditions, the reaction time was reduced to 1 h, and Int2 was detected by HRMS analysis (see the ESI for details, Fig. S21†), which could be utilized as evidence of the existence of an intermediate state Int2 for the coupling process (Scheme 5d).
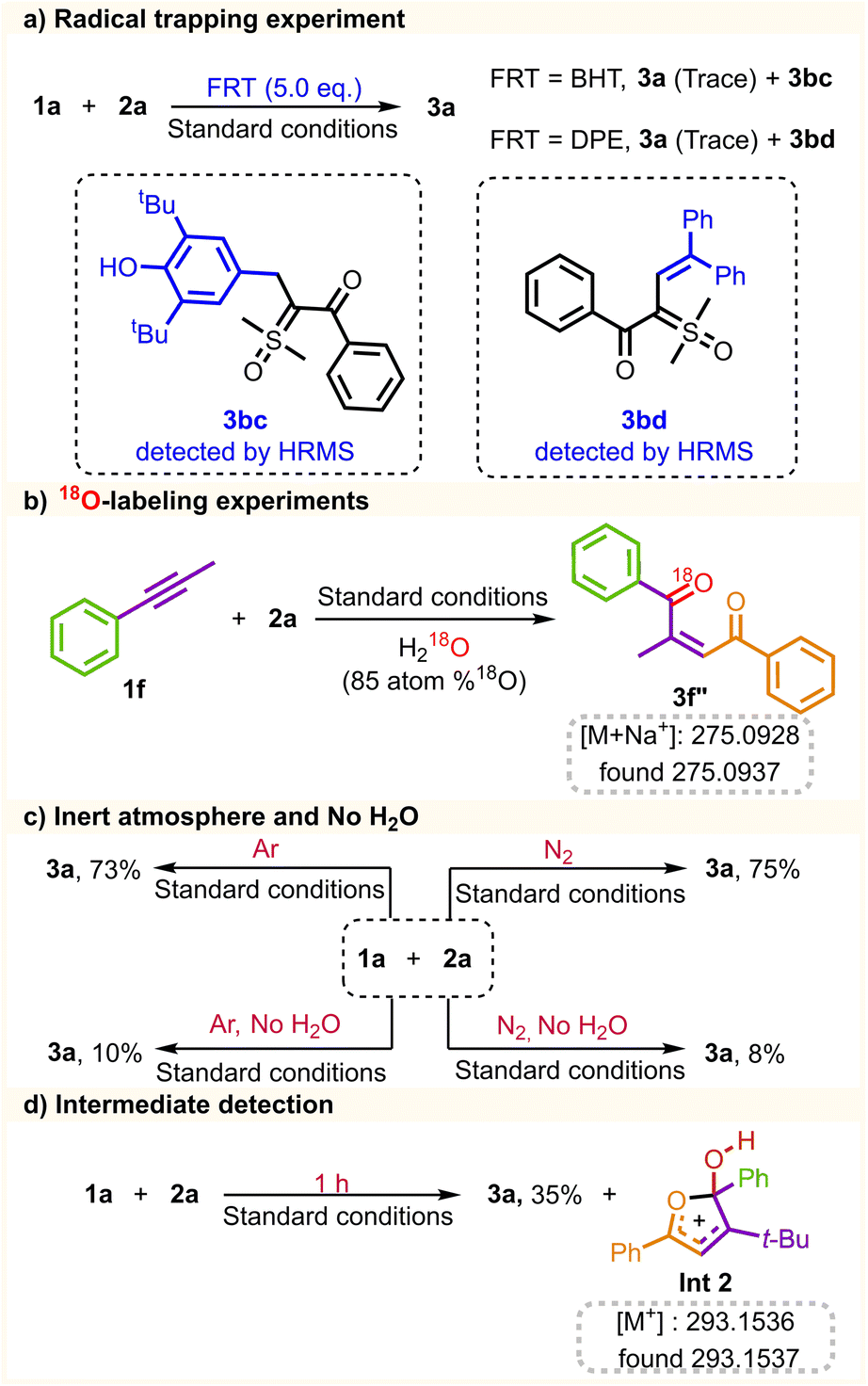 | ||
| Scheme 5 Mechanistic studies: (a) radical trapping experiment. (b) 18O-labeling experiments. (c) The inert atmosphere and no H2O. (d) Intermediate detection. | ||
Based on our mechanistic studies, a plausible route is presented in Scheme 6. Initially, sulfoxonium ylide was oxidized by a single-electron transfer process at the anode to generate the sulfoxonium ylide radical I, which is then coupled to alkyne to form alkenyl radical II. Secondly, it is further oxidized at the anode to form the alkenyl cation III, which is attacked by H2O to form Int1. Subsequently, a cyclization reaction occurred to generate a crucial five-membered ring intermediate Int2 by removing DMSO. Then, Int2 underwent a rapid, open-ring reaction to form Int3 due to the attack of water. Finally, a deprotonation process with the hexafluoroisopropanol anion occurred to form product 3a. Meanwhile, during this electrochemical reaction, protons were simultaneously reduced, releasing H2 as a byproduct at the surface of the cathode.
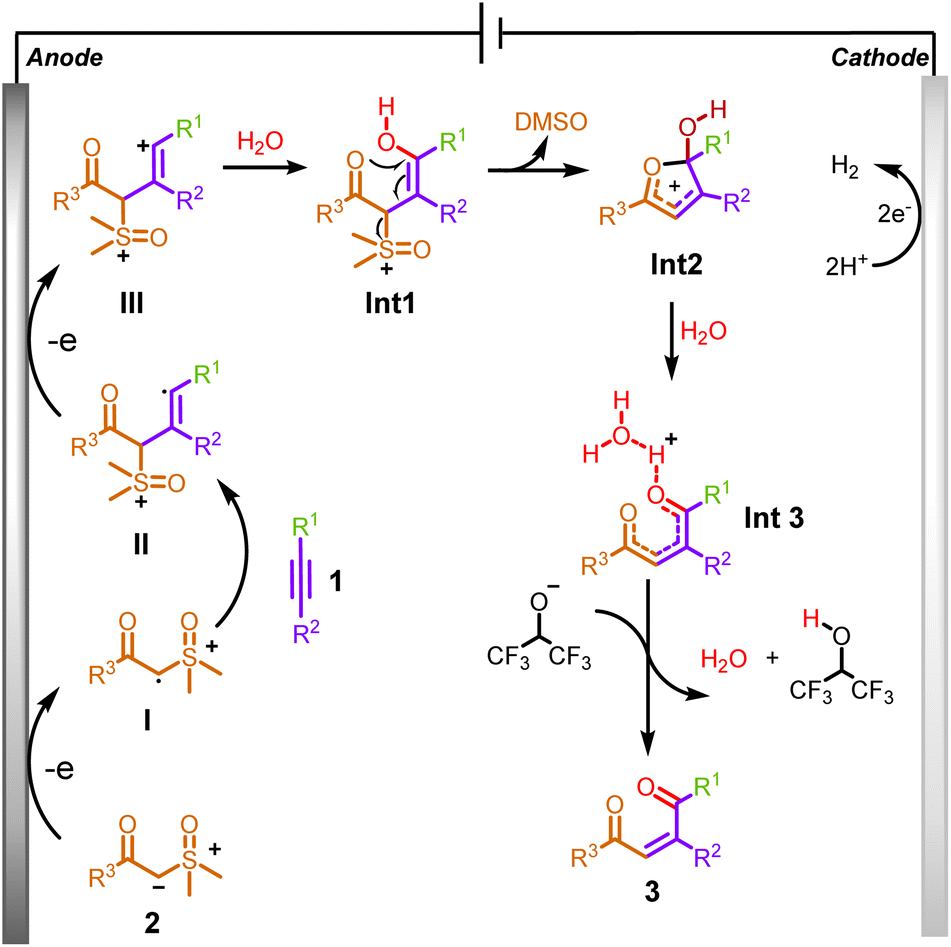 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed the first electrochemical method for the direct synthesis of symmetric and asymmetric 1,4-dicarbonyl Z-type alkenes via the three-component coupling of readily available alkynes and ylides with water. The reason for the high Z-stereoselectivity is attributed to the generation of a crucial furan intermediate in the process. Moreover, the source of carbonyl oxygen was water and was demonstrated through a series of controlled experiments. The protocol possesses several notable features: metal-free and oxidant-free conditions, broad substrate scope, easy-to-handle nature, and scaled-up operation, which makes the method attractive and full of synthetic potential.Data availability
All experimental, computational and crystallographic data associated with this study can be found in the article or in the ESI.†Author contributions
L.-B. Zhang conceived and designed the experiments. H.-R. Li and Y.-A. Ran performed the experiments. S.-F. Ni and Y.-Y. Zhu performed the DFT calculations. M. Li, W. Guo, and L.-R. Wen analyzed the experiments. H.-R. Li and L.-B. Zhang revised the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21801152) and the Natural Science Foundation of Shandong Province (ZR2019BB005) for financial support. We thank the Youth Innovation Science and Technology Plan of Colleges and Universities in Shandong Province (2021KJ076).Notes and references
- (a) J. Salva and D. J. Faulkner, J. Org. Chem., 1990, 55, 1941–1943 CrossRef CAS; (b) J. S. Webb, D. B. Cosulich, J. H. Mowat, J. B. Patrick, R. W. Broschard, W. E. Meyer, R. P. Williams, C. F. Wolf, W. Fulmor, C. Pidacks and J. E. Lancaster, J. Am. Chem. Soc., 1962, 84, 3185–3187 CrossRef CAS; (c) P. Bollinger and T. Zardin-Tartaglia, Helv. Chim. Acta, 1976, 59, 1809–1820 CrossRef CAS; (d) K. K. Chexal, C. Tamm, J. Clardy and K. Hirotsu, Helv. Chim. Acta, 1979, 62, 1129–1142 CrossRef CAS.
- (a) C. R. Bauer and R. E. Lutz, J. Am. Chem. Soc., 1953, 75, 5997–6002 CrossRef CAS; (b) H. Surya Prakash Rao and S. Jothilingam, Tetrahedron Lett., 2001, 42, 6595–6597 CrossRef CAS; (c) H. Surya Prakash Rao and S. Jothilingam, J. Org. Chem., 2003, 68, 5392–5394 CrossRef PubMed; (d) J. Lu, D. M. Ho, N. J. Vogelaar, C. M. Kraml, S. Bernhard, N. Byrne, L. R. Kim and R. A. Pascal, J. Am. Chem. Soc., 2006, 128, 17043–17050 CrossRef CAS PubMed; (e) G. Yin, Z. Wang, A. Chen, M. Gao, A. Wu and Y. Pan, J. Org. Chem., 2008, 73, 3377–3383 CrossRef CAS PubMed; (f) V. Rajeshkumar, C. Neelamegam, L. John and S. Anandan, ChemistrySelect, 2018, 3, 11606–11609 CrossRef CAS.
- (a) Y. Wang, H. Li, Y.-Q. Wang, Y. Liu, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2007, 129, 6364–6365 CrossRef CAS PubMed; (b) W.-M. Shu, Y. Yang, D.-X. Zhang, L.-M. Wu, Y.-P. Zhu, G.-D. Yin and A.-X. Wu, Org. Lett., 2013, 15, 456–459 CrossRef CAS; (c) W. Zhou, X. Su, M. Tao, C. Zhu, Q. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 14853–14857 CrossRef CAS PubMed; (d) X. Dou, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 6739–6743 CrossRef CAS PubMed.
- (a) X.-Y. Lu, G. Zhu and S. Ma, Chin. J. Chem., 1993, 11, 267–271 CrossRef CAS; (b) J. Q. Yu and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 3232–3233 CrossRef CAS PubMed; (c) B. Crone and S. F. Kirsch, Chem. Commun., 2006, 764–766 RSC; (d) D.-J. Dong, H.-H. Li and S.-K. Tian, J. Am. Chem. Soc., 2010, 132, 5018–5020 CrossRef CAS PubMed; (e) K. Xu, Y. Fang, Z. Yan, Z. Zha and Z. Wang, Org. Lett., 2013, 15, 2148–2151 CrossRef CAS PubMed; (f) C. Sämann, M. A. Schade, S. Yamada and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 9495–9499 CrossRef PubMed; (g) Y. He, Z. Zheng, Q. Liu, G. Song, N. Sun and X. Chai, J. Org. Chem., 2018, 83, 12514–12526 CrossRef CAS PubMed.
- (a) W. Baratta, A. D. Zotto and P. Rigo, Chem. Commun., 1997, 2163–2164 RSC; (b) A. Del Zotto, W. Baratta, G. Verardo and P. Rigo, Eur. J. Org Chem., 2000, 2000, 2795–2801 CrossRef; (c) K. A. Runcie and R. J. K. Taylor, Chem. Commun., 2002, 974–975 RSC; (d) C. Asta, J. Conrad, S. Mika and U. Beifuss, Green Chem., 2011, 13, 3066–3069 RSC; (e) M. Nandakumar, R. Sivasakthikumaran and A. K. Mohanakrishnan, Eur. J. Org Chem., 2012, 2012, 3647–3657 CrossRef CAS; (f) M. Duy Vu, W.-L. Leng, H.-C. Hsu and X.-W. Liu, Asian J. Org. Chem., 2019, 8, 93–96 CrossRef CAS; (g) C. Ye, B.-G. Cai, J. Lu, X. Cheng, L. Li, Z.-W. Pan and J. Xuan, J. Org. Chem., 2021, 86, 1012–1022 CrossRef CAS PubMed.
- (a) J. D. Neuhaus, A. Bauer, A. Pinto and N. Maulide, Angew. Chem., Int. Ed., 2018, 57, 16215–16218 CrossRef CAS PubMed; (b) X. Y. Wang, W.-Y. Tong, B. Huang, S. Cao, Y. L. Li, J. C. Jiao, H. Huang, Q. Yi, S. L. Qu and X. Wang, J. Am. Chem. Soc., 2022, 144, 4952–4965 CrossRef CAS PubMed; (c) A. Sharma, P. Jamwal and R. Gurubrahamam, Org. Lett., 2023, 25, 7236–7241 CrossRef CAS.
- (a) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (b) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (c) C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS PubMed; (d) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC; (e) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS; (f) S. J. Harwood, M. D. Palkowitz, C. N. Gannett, P. Perez, Z. Yao, L. J. Sun, H. D. Abruña, S. L. Anderson and P. S. Baran, Science, 2022, 375, 745–752 CrossRef CAS; (g) Y. F. Li, L. R. Wen and W. S. Guo, Chem. Soc. Rev., 2023, 52, 1168–1188 RSC.
- (a) S. Gnaim, A. Bauer, H.-J. Zhang, L. Chen, C. Gannett, C. A. Malapit, D. E. Hill, D. Vogt, T. Tang, R. A. Daley, W. Hao, R. Zeng, M. Quertenmont, W. D. Beck, E. Kandahari, J. C. Vantourout, P.-G. Echeverria, H. D. Abruna, D. G. Blackmond, S. D. Minteer, S. E. Reisman, M. S. Sigman and P. S. Baran, Nature, 2022, 605, 687–695 CrossRef CAS PubMed; (b) L.-H. Jie, B. Guo, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2022, 144, 2343–2350 CrossRef CAS PubMed; (c) D. F. Yang, Z. P. Guan, Y. N. Peng, S. X. Zhu, P. J. Wang, Z. L. Huang, H. Alhumade, D. Gu, H. Yi and A. Lei, Nat. Commun., 2023, 14, 1476–1484 CrossRef CAS PubMed; (d) Y.-F. Tan, D. Yang, Y.-H. Yang, J.-F. Lv, L.-X. Zong, Z. Guan and Y.-H. He, Green Chem., 2023, 25, 9388–9393 RSC; (e) T. von Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, Science, 2023, 379, 1036–1042 CrossRef PubMed; (f) Y. W. Liu, P. F. Li, Y. W. Wang and Y. Qiu, Angew. Chem., Int. Ed., 2023, e202306679 CAS.
- (a) L. X. Lu, Y. Wang, W. Zhang, W. Zhang, K. A. See and S. Lin, J. Am. Chem. Soc., 2023, 145, 22298–22304 CrossRef CAS PubMed; (b) F. Liu, W. J. Ding, J. C. Lin and X. Cheng, Org. Lett., 2023, 25, 7617–7621 CrossRef CAS PubMed; (c) Y. Gao, B. Zhang, J. He and P. S. Baran, J. Am. Chem. Soc., 2023, 145, 11518–11523 CrossRef CAS PubMed; (d) Y. Hioki, M. Costantini, J. Griffin, K. C. Harper, M. P. Merini, B. Nissl, Y. Kawamata and P. S. Baran, Science, 2023, 380, 81–87 CrossRef CAS PubMed.
- (a) Z.-C. Wang, R.-T. Li, Q. Ma, J.-Y. Chen, S.-F. Ni, M. Li, L.-R. Wen and L.-B. Zhang, Green Chem., 2021, 23, 9515–9522 RSC; (b) Z.-H. Fu, H.-D. Tian, S.-F. Ni, J. S. Wright, M. Li, L.-R. Wen and L.-B. Zhang, Green Chem., 2022, 24, 4772–4777 RSC; (c) H.-D. Tian, Z.-H. Fu, C. Li, H.-C. Lin, M. Li, S.-F. Ni, L.-R. Wen and L.-B. Zhang, Org. Lett., 2022, 24, 9322–9326 CrossRef CAS PubMed; (d) M.-Q. Ping, M.-Z. Guo, R.-T. Li, Z.-C. Wang, C. Ma, L.-R. Wen, S.-F. Ni, W.-S. Guo, M. Li and L.-B. Zhang, Org. Lett., 2022, 24, 7410–7415 CrossRef CAS; (e) R.-T. Li, D.-F. Yuan, M.-Q. Ping, Y.-Y. Zhu, S.-F. Ni, M. Li, L.-R. Wen and L.-B. Zhang, Chem. Sci., 2022, 13, 9940–9946 RSC; (f) C. Li, Z. Chen, X.-Y. Guo, L.-R. Wen, M. Li and L.-B. Zhang, Chem. Commun., 2023, 59, 12164–12167 RSC; (g) Z. Chen, C. Li, K. Liu, L.-R. Wen, M. Li and L.-B. Zhang, Org. Chem. Front., 2024, 11, 477–483 RSC; (h) M.-Z. Guo, M.-J. Mou, Z. Chen, S.-F. Ni, M. Li, L.-R. Wen and L.-B. Zhang, Chin. J. Chem., 2024, 42, 585–591 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2179302 and 2340616. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01141d |
| This journal is © The Royal Society of Chemistry 2024 |