
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA scalable approach using a gC3N4-covalent organic framework hybrid catalyst towards sustainable hydrogen production from seawater and wastewater†
Kiran
Asokan
ab,
T. M.
Bhagyasree
ab,
George
Devasia
bc,
Sailaja
Krishnamurty
bc,
Sabah
Solim
d,
Lina
Rueda
d,
Dhabia M.
Al-Mohannadi
e,
Mohammed
Al-Hashimi
 e,
Konstantinos
Kakosimos
e and
Sukumaran
Santhosh Babu
*ab
e,
Konstantinos
Kakosimos
e and
Sukumaran
Santhosh Babu
*ab
aOrganic Chemistry Division, National Chemical Laboratory (CSIR-NCL), Dr Homi Bhabha Road, Pune 411008, India. E-mail: sb.sukumaran@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201 002, India
cPhysical and Materials Chemistry Division, National Chemical Laboratory (CSIR-NCL), Dr Homi Bhabha Road, Pune 411008, India
dQatar Shell Research & Technology Centre, Qatar Science & Technology Park, Education City, Doha, Qatar
eChemical Engineering Department, Texas A&M University at Qatar, Doha, Qatar
First published on 18th July 2024
Abstract
The photocatalytic generation of H2 using covalent organic frameworks (COFs) is gaining more interest. While numerous reports have focused on the production of H2 from deionized water using COFs, the inability to produce H2 from industrial wastewater or seawater is a common limitation in many reported catalysts. Additionally, many of these reports lack a clear path to scale up the catalyst synthesis. In this study, we explore the prospect of hybridizing a COF with gC3N4 to create a robust photocatalyst for efficient H2 generation. This hybrid exhibits outstanding performance not only in deionized water, but also in wastewater, and simulated seawater. Furthermore, we explore the feasibility of the bulk-scale synthesis and successfully produce a 20 g hybrid catalyst in a single batch, and the synthesis method is scalable to achieve the commercial target. Remarkably, a maximum HER rate of 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 873 μmol g−1 h−1 and 109125 μmol g−1 h−1 was obtained for the hybrid catalyst from industrial wastewater and simulated seawater, respectively. The performance of bulk-scale batches closely matches that of the small-scale ones. This research paves the way for the utilization of organic photocatalysts on a commercial scale, offering a promising solution for sustainable large-scale H2 production.
873 μmol g−1 h−1 and 109125 μmol g−1 h−1 was obtained for the hybrid catalyst from industrial wastewater and simulated seawater, respectively. The performance of bulk-scale batches closely matches that of the small-scale ones. This research paves the way for the utilization of organic photocatalysts on a commercial scale, offering a promising solution for sustainable large-scale H2 production.
Introduction
The incorporation of renewable resources in energy generation is an essential pillar for a sustainable future. Among the potential alternatives, hydrogen (H2), the most prevalent molecule in the universe, stands out as a clean fuel with the potential to supplant conventional fossil fuels. This transition to H2 is driven by advantages including abundance, emission-free combustion, high energy efficiency, and inherent renewability.1 Nevertheless, the practical adoption of H2 encounters certain constraints, particularly concerning long-distance transport and extended-term storage. Issues such as flammability pose challenges in the entire supply and demand chain, and the production of green H2 at a large scale may entail elevated costs.1 The conventional methods for H2 production involve sources such as natural gases, naphtha, heavy oil, coal, and electrolysis, and industrial-scale production primarily relies on the Bosch process.2 However, when exploring sustainable avenues for producing green H2, one cannot overlook the fact that 71% of our planet is covered with water, making it an abundant source of H2. By harnessing solar energy and employing catalytic processes to extract H2 from water, we have the opportunity to create a bountiful, clean energy source that aligns with the goals of decarbonization.Over the past decade, organic photocatalysts have gained increasing prominence in the field of water splitting.3–5 In 2014, Lotsch and co-workers reported, for the first time, photocatalytic generation of H2 (1970 μmol g−1 h−1) from water using a hydrazone-based covalent organic framework (COF) as the photosensitizer.6 Subsequently, numerous research groups have explored the use of various COFs for the photocatalytic generation of H2 from water.7,8 Over time, several strategies have been employed to enhance the stability of photocatalysts and improve H2 evolution efficiency. They include molecular engineering,9,10 multivariate synthesis,11 defect engineering,12 dye sensitization,13 electron transfer mediators,14 hybridizations,15–23etc. These efforts have led to significant advancements in the field, demonstrating the versatility and potential of COFs in sustainable H2 production. Hybridization of COFs using metallic conductors, MOFs, inorganic semiconductors, and polymers to form heterojunctions within COFs has recently emerged as a strategy for photocatalytic H2 evolution (PHE).24 Even though COF hybridization with gC3N4 was reported earlier, the synthesis procedure follows the solvothermal method which limits the opportunity for bulk-scale synthesis.25,26 Moreover, the photocatalytic performance was tested in deionized water and the H2 evolution efficiency was not up to the mark. Ming et al. reported a metal–insulator–semiconductor-based photosystem comprising Tp-COF with polyvinylpyrrolidone (PVP) coated Pt nanoparticles (NPs) for the photocatalytic production of H2.27 Unlike bare Pt NPs, here the photoexcited π-electrons in the n-type Tp-COF semiconductor were effectively extracted and tunnelled towards Pt NPs through an ultrathin PVP insulating layer which further enhanced the activity. While significant progress has been made in PHE, most high-performing photocatalysts have been tested using deionized water for H2 production.28–45 It's important to note that only a small fraction of Earth's water resources, about 1%, is fresh water. Therefore, exploring PHE from alternative sources such as seawater, industrial wastewater, or non-potable water is of great importance. However, there are limited reports on H2 generation from seawater using COFs due to the challenges posed by side reactions occurring on the catalyst surface and thereby reducing the efficiency of photogenerated charge carriers in photocatalytic processes.46–48
However, in 2023, Yue et al. reported a significant breakthrough with a reasonably high-performance H2 production rate of 41300 μmol g−1 h−1 using Tp-Pa, outperforming many other organic, inorganic, and hybrid materials employed for PHE from seawater under visible light irradiation.49 However, the absence of reports addressing sustainable H2 production using scalable photocatalysts, despite numerous studies demonstrating high rates of H2 evolution using various synthetic methodologies, is a significant gap in the research field. Herein, we introduce a hybrid catalyst comprising Tp-Pa and gC3N4 capable of reasonably high H2 evolution from both seawater and industrial wastewater. In this work, we aim to tackle two major challenges: (1) H2 production from industrial wastewater and seawater, and (2) bulk-scale synthesis of the photocatalyst (Scheme 1). These efforts not only expand the application of COFs in environmentally significant areas but also make the technology more accessible for practical, large-scale use.
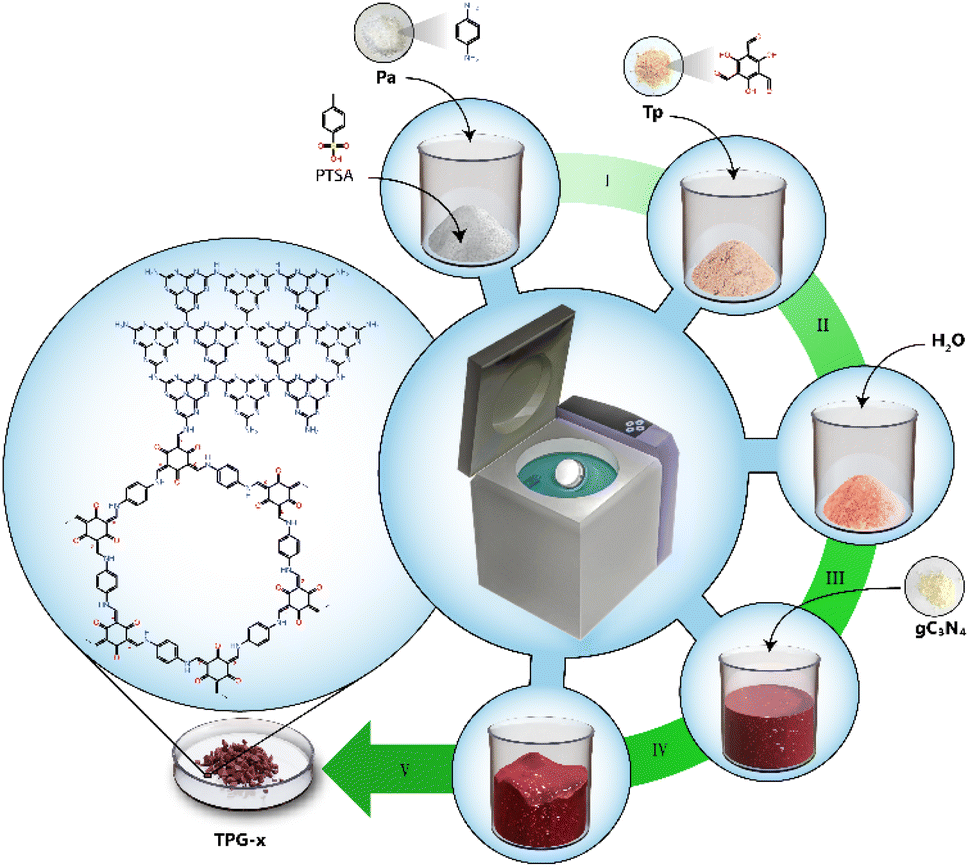 | ||
| Scheme 1 Bulk scale synthesis strategy adopted for the preparation of TPG-x photocatalysts. Step I: mix PTSA and Pa in a planetary mixer. Step II: add Tp and mix again. Step III: add an adequate amount of H2O to the reaction mixture and mix. Step IV: add gC3N4 to the reaction mixture and mix. Step V: heat the reaction mixture in an oven at 90 °C, followed by washing and drying to yield the final catalyst in powder form. | ||
Results and discussion
All the COFs including hybrid COFs Tp-Pa–gC3N4-xs (TPG-x, x is the percentage of gC3N4 to the total quantity of Tp and Pa and it varies as 10, 25, 50, 75, and 100) were prepared using mechanochemical synthesis on a 2 g scale (Schemes S1† and 1, and Table S1†).50 A representative chemical structure of TPG-x is shown in Scheme 1. All the COFs were characterized by FT-IR and intense peaks at around ∼1226 cm−1 (–C–N) and ∼1551 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) revealed the formation of a β-ketoenamine linked framework for both Tp-Pa and TPG-xs (Fig. 1 and S1†). Also, the stretching frequencies of nitrogen-containing heterocycles were evident on both gC3N4 and TPG-xs. A broad peak at around ∼3250 cm−1 was obtained for gC3N4, which corresponds to the stretching frequency of N–H bonds. Furthermore, the intensity of N–H stretching frequency was reduced for TPG-xs, thereby confirming the bond formation between the free aldehyde group of Tp-Pa and NH2 of gC3N4, which is in line with the previous literature report.27,51 Solid-state 13C cross-polarization magic angle spinning (CP-MAS) NMR spectroscopy was utilized to provide evidence of hybrid COF formation. The peaks at around 104.6 and 182 ppm for Tp-Pa and TPG-x correspond to –CO and –CC– bond formation, respectively; this provided critical insights into the composition and structural characteristics of the synthesized materials, further validating hybrid COF formation and enhancing our understanding of these novel materials. At the same time, pristine gC3N4 shows peaks at around 156 and 165 ppm, corresponding to the C–N and C–C bonds of heptazine units. As evident from the graph, hybrid COFs show a gradual increase in the intensity of peaks (heptazine unit) from 0 to 100% of gC3N4 in TPG-x (Fig. 1 and S2†). The crystallinity of the samples was analyzed using powder X-ray diffraction (PXRD). All TPG-xs show a sharp crystalline peak around 2θ of 4.8, corresponding to reflection from the (100) plane (Fig. 1 and S3†). Similarly, small intense peaks at around 8.07 and 12.9 were also observed, indicating reflection from the (200), and (210) planes, respectively. Apart from this, another peak at 27.5 corresponding to reflection from the (001) plane is also visible, which confirms the π–π stacking arising in the COF. gC3N4 shows one sharp intense peak at around 2θ ≈ 27.6 arising from the stacking of the conjugated heptazine ring.52 Notably, the intensity of the peak from the (001) plane increases gradually as the ratio of gC3N4 increases because of its overlap with the peak from the (001) plane of Tp-Pa.53 Thermogravimetric analysis (TGA) of COFs shows thermal stability up to 400 °C corresponding to a robust framework. All samples of TPG-x showed two major degradation peaks, at around 400 °C corresponding to Tp-Pa and at around 600 °C corresponding to gC3N4 (Fig. 1 and S4†). The decomposition of all the samples in air points to the composition of the samples.
C) revealed the formation of a β-ketoenamine linked framework for both Tp-Pa and TPG-xs (Fig. 1 and S1†). Also, the stretching frequencies of nitrogen-containing heterocycles were evident on both gC3N4 and TPG-xs. A broad peak at around ∼3250 cm−1 was obtained for gC3N4, which corresponds to the stretching frequency of N–H bonds. Furthermore, the intensity of N–H stretching frequency was reduced for TPG-xs, thereby confirming the bond formation between the free aldehyde group of Tp-Pa and NH2 of gC3N4, which is in line with the previous literature report.27,51 Solid-state 13C cross-polarization magic angle spinning (CP-MAS) NMR spectroscopy was utilized to provide evidence of hybrid COF formation. The peaks at around 104.6 and 182 ppm for Tp-Pa and TPG-x correspond to –CO and –CC– bond formation, respectively; this provided critical insights into the composition and structural characteristics of the synthesized materials, further validating hybrid COF formation and enhancing our understanding of these novel materials. At the same time, pristine gC3N4 shows peaks at around 156 and 165 ppm, corresponding to the C–N and C–C bonds of heptazine units. As evident from the graph, hybrid COFs show a gradual increase in the intensity of peaks (heptazine unit) from 0 to 100% of gC3N4 in TPG-x (Fig. 1 and S2†). The crystallinity of the samples was analyzed using powder X-ray diffraction (PXRD). All TPG-xs show a sharp crystalline peak around 2θ of 4.8, corresponding to reflection from the (100) plane (Fig. 1 and S3†). Similarly, small intense peaks at around 8.07 and 12.9 were also observed, indicating reflection from the (200), and (210) planes, respectively. Apart from this, another peak at 27.5 corresponding to reflection from the (001) plane is also visible, which confirms the π–π stacking arising in the COF. gC3N4 shows one sharp intense peak at around 2θ ≈ 27.6 arising from the stacking of the conjugated heptazine ring.52 Notably, the intensity of the peak from the (001) plane increases gradually as the ratio of gC3N4 increases because of its overlap with the peak from the (001) plane of Tp-Pa.53 Thermogravimetric analysis (TGA) of COFs shows thermal stability up to 400 °C corresponding to a robust framework. All samples of TPG-x showed two major degradation peaks, at around 400 °C corresponding to Tp-Pa and at around 600 °C corresponding to gC3N4 (Fig. 1 and S4†). The decomposition of all the samples in air points to the composition of the samples.
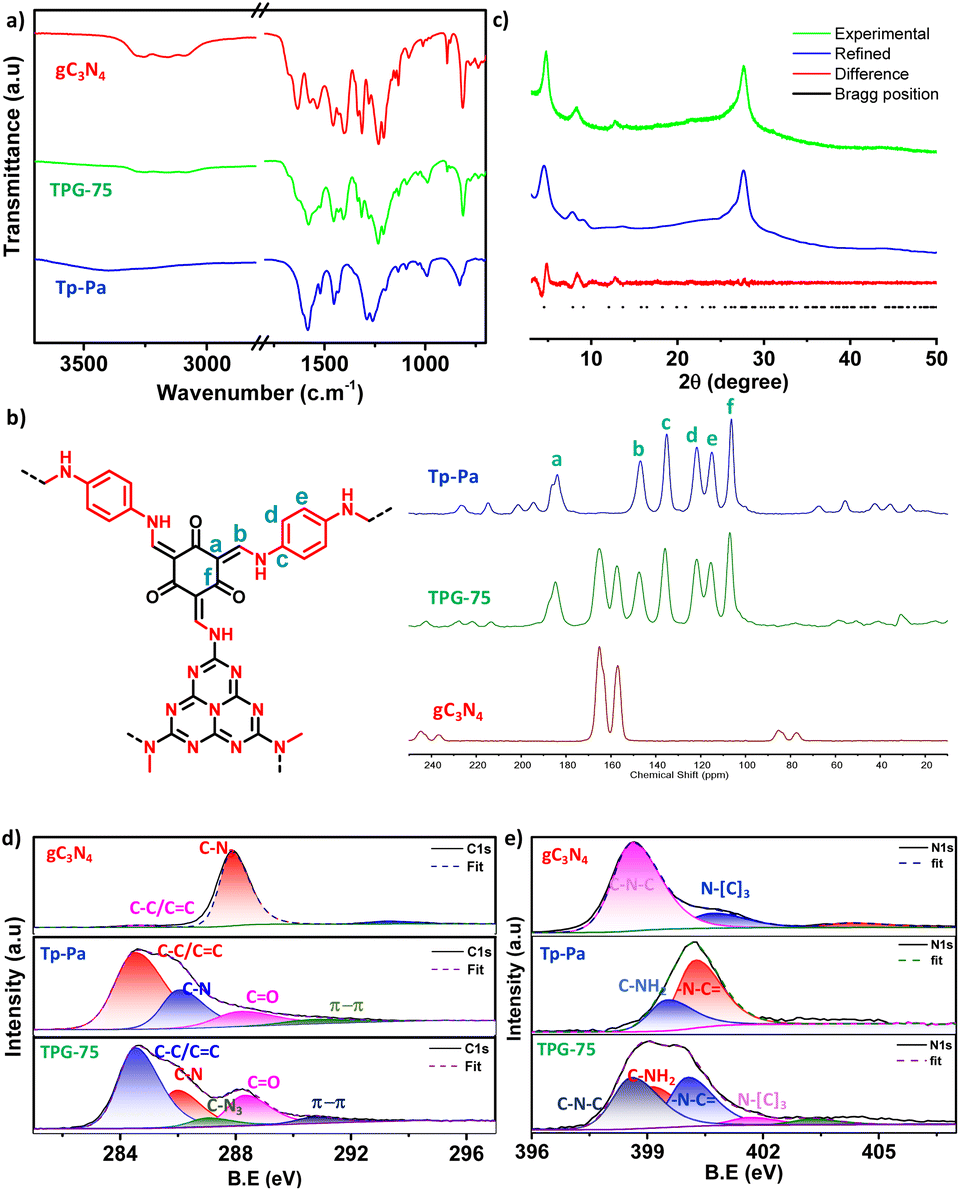 | ||
| Fig. 1 Comparison of (a) FT-IR, (b) 13C CP MAS NMR, (c) PXRD and XPS for (d) C 1s, and (e) N 1s of Tp-Pa, gC3N4 and TPG-75. | ||
As a representative example, the detailed characterization of TPG-75 is provided in Fig. 1 and 2. X-ray photoelectron spectroscopy (XPS) revealed the presence of C 1s, N 1s, and O 1s in both pristine and hybrid COFs (Fig. 1 and S5†). Furthermore, the C 1s spectrum of gC3N4, Tp-Pa, and TPG-75 was deconvoluted into different peaks.27,51 As shown in Fig. 1d, the C 1s spectrum of pristine gC3N4 can be deconvoluted into one major individual peak at 287.9 eV corresponding to the C–N bond along with an adventitious carbon peak at 284.6 eV. At the same time, the spectrum of the hybrid COF can be deconvoluted into 4 individual peaks at 284.6, 285.9, 288.3, and 290.7 corresponding to C–C/CC, C–N, CO, and π–π interaction, respectively. Similarly, a full survey of the N 1s spectrum reveals the individual N1s peaks of both gC3N4 and Tp-Pa in TPG-75 (Fig. 1e). Hence XPS of TPG-75 shows a combination of gC3N4 and Tp-Pa. The morphology of the samples was analyzed using Field Emission Scanning Electron Microscopy (FE-SEM) and High-Resolution Transmission Electron Microscopy (HR-TEM) (Fig. 2a, b and S6–S12†). gC3N4 showed a sheet-like morphology with a thickness of around 20–30 nm and stacking of sheets to form a layered structure (Fig. S7†). HR-TEM images also confirmed the sheet-like morphology of TPG-75 having crystalline fringes with a d-spacing of 0.2 nm (Fig. 2b). The selected area electron diffraction (SAED) pattern also confirmed the crystallinity of the samples at a lower amount of gC3N4. As the percentage increased, the fringes were not visible, especially for TPG-100 (Fig. S12†). At the same time, the characteristic morphology of gC3N4 was more evident in TPG-75 and TPG-100 (Fig. S11 and S12†). Elemental mapping using Scanning Tunnelling Electron Microscopy (STEM) was also carried out and it confirmed the presence of C (81%), N (15%), and O (4%) in TPG-75 (Fig. S13†).
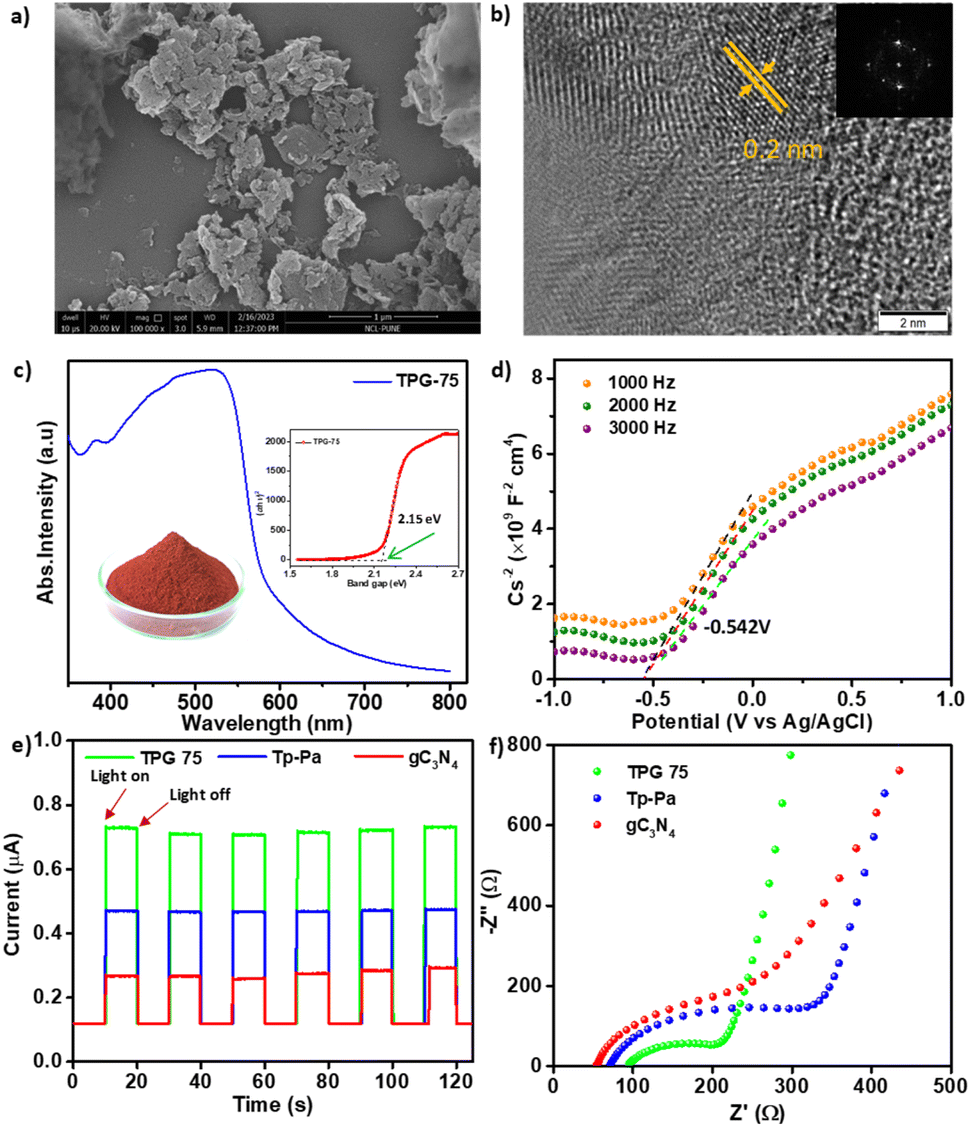 | ||
| Fig. 2 (a) FE-SEM image of TPG-75. (b) HR-TEM image of TPG-75 showing lattice fringes and d-spacing; SAED pattern is shown as an inset. (c) UV-vis DRS pattern of TPG-75 (inset shows the corresponding Tauc plot). (d) Mott–Schottky measurement of TPG-75. (e) Photocurrent measurements of TPG-75, Tp-Pa, and gC3N4 under visible light irradiation on and off conditions with a time interval of 20 seconds. (f) EIS of TPG-75, Tp-Pa, and gC3N4. | ||
The permanent porosity and surface area of the samples were analyzed using Brunauer–Emmett–Teller (BET) and Density Functional Theory (DFT) pore size distribution methods. TPG-xs showed a lower surface area compared to pristine Tp-Pa due to the incorporation of gC3N4. A BET surface area of 652 m2 g−1 was obtained for TPG-75 which is less compared to the surface area of Tp-Pa (1171 m2 g−1). It is to be noted that gC3N4 exhibited a very low surface area of 87 m2 g−1 and increasing gC3N4 content gradually decreased the surface area of TPG-xs (Fig. S14–S20†). Ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) was used to measure the absorption features and all COFs exhibited a broad absorption ranging from 300 to 750 nm whereas the absorption of gC3N4 is limited to 300 to 400 nm (Fig. S21†). The optical energy band gaps were calculated from the Tauc plot for all catalysts and it was found that gC3N4 has a broader band gap of 3.1 eV whereas Tp-Pa exhibits a narrow band gap of 2.14 eV (Fig. 2c). The obtained band gap values are comparable with previously reported values.27,51 The band gap of TPG-75 was 2.15 eV, which is similar to the band gap of Tp-Pa (Fig. S22†). At the same time, the Mott–Schottky measurements exhibit a positive slope denoting n-type semiconductor characteristics for the hybrid catalyst (Fig. 2d), pristine gC3N4, and Tp-Pa (Fig. S23†). In order to understand the light responses, the photocurrent measurement of TPG-75 was conducted and compared with that of Tp-Pa and gC3N4 (Fig. 2e). TPG-75 shows a higher photocurrent response compared to Tp-Pa and gC3N4 during several on–off photoirradiation cycles under identical experimental conditions. A sequential increment in photoresponse was noticed from Tp-Pa to gC3N4 and further to TPG-75. Furthermore, the results from the electrochemical impedance spectra (EIS) reveal a notable reduction in charge transfer resistance for charge separation in TPG-75 compared to that of Tp-Pa and gC3N4 (Fig. 2f). These findings collectively affirm that the hybridization process has brought about a substantial enhancement in charge separation within the COF platform.
PHE of all hybrid COFs, Tp-Pa, and gC3N4 was carried out in a quartz round bottom flask of 50 mL capacity under visible light irradiation in the presence of SED and a cocatalyst. In a typical experiment, 5 mg of catalyst was dispersed in an aqueous solution of 0.056 M (200 mg) ascorbic acid (AA) in 20 mL H2O. PVP-coated Pt NPs were synthesized according to the reported literature54 and used as such as the cocatalyst. The HER rate of Tp-Pa was found to be 26126 μmol g−1 h−1, which is comparable with that in the previous report (Fig. 3a).49 It has to be noted that gC3N4 showed no activity with AA as SED under identical experimental conditions (Fig. 3a). Surprisingly, hybrid catalysts with different percentages of gC3N4 performed much better than Tp-Pa, delivering a nearly three-fold increase in activity. For instance, TPG-75 showed a remarkably high catalytic activity of 85617 and 179064 μmol g−1 h−1 under visible light and simulated solar light illumination, respectively (Fig. 3b). An increase in activity under simulated solar light can be attributed to the high light absorption and generation of more electron–hole pairs by the hybrid catalyst. An increase in gC3N4 content resulted in high photocatalytic activity up to 75 wt% gC3N4; however, TPG-100 showed lower performance than TPG-75 (Fig. 3a).
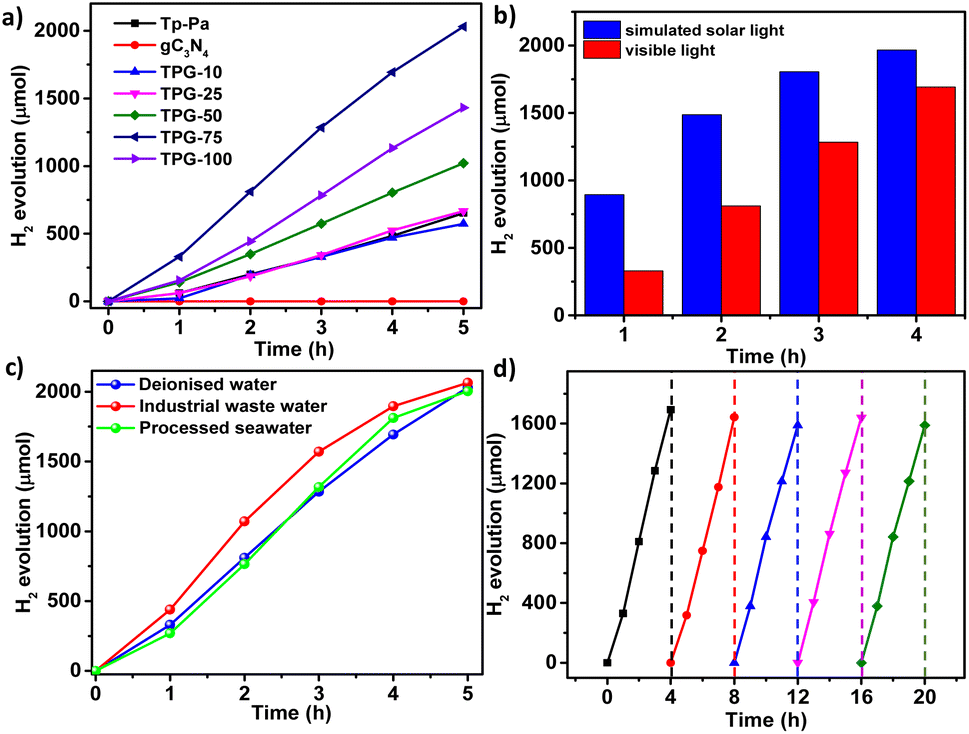 | ||
| Fig. 3 (a) Comparison of the HER rate of TPG-xs with Tp-Pa and gC3N4. (b) Comparison of the HER rate of TPG-75 with visible light and simulated solar light. (c) Comparison of the HER rate of TPG-75 with different water sources. (d) Cycling stability of the PHE of TPG-75 with deionized water up to 5 cycles. | ||
Optimizations in PHE was performed by varying quantities of the catalyst, SED, and cocatalyst. The effect of the photocatalyst amount was evaluated with TPG-75 under standard conditions. With 2 mg and 10 mg catalyst loading under the same optimized conditions, a maximum of 129416 μmol g−1 h−1 and 38717 μmol g−1 h−1 ofHER rate was obtained, respectively (Fig. S24†). Although the rate is high for 2 mg of TPG-75, the cumulative production of H2 was only 1294 μmol compared to 2030 μmol with a 5 mg catalyst and 1770 μmol with a 10 mg catalyst. At the same time, a reduction in production was observed with 10 mg of the TPG-75 catalyst because more catalyst concentration can hinder light absorption. After confirming the optimum catalyst amount, the quantity of SED used was optimized. Initially, 100 mg SED was used by keeping other conditions the same and tested for PHE. It was noted that by increasing the AA quantity from 100 to 200 mg, a drastic increase in the HER rate was observed. Furthermore, an increase in AA from 200 to 300 and 400 mg yielded nearly the same HER rate (Fig. S25†). Later, the quantity of PVP-Pt NPs was optimized with 5 mg of TPG-75, and upon increasing the loading of PVP-Pt NPs from 100 to 400 μL, a decrease in H2 evolution was observed (Fig. S26†). The optimization experiments concluded that a combination of 5 mg of TPG-75, 20 mL H2O, 200 mg of AA, and 100 μL of PVP-Pt NPs is the optimum condition for the HER. Different sacrificial agents were screened for the PHE studies of TPG-75. Besides AA, triethanolamine, sodium ascorbate (SA), lactic acid, and citric acid were used for PHE. Interestingly, TPG-75 showed a steady performance of 26433 μmol g−1 h−1 with SA as SED. At the same time, the hybrid catalyst did not show any H2 evolution with other SEDs (Fig. S27†). TPG-75 exhibits a more remarkable performance than many of the reported COF catalysts in terms of PHE from deionized water (Table S2†). The enhanced HER activity has arisen from the synergistic effect of Tp-Pa and gC3N4 parts in the TPGs.
As an extensive study, we employed our catalyst in the photocatalytic HER from water sources other than deionized water (Table S3†). First, the performance of TPG-75 was studied in industrial wastewater containing 0.1% formic acid, propionic acid, and butyric acid. TPG-75 could give a 109432 μmol g−1 h−1 HER rate under visible light irradiation and optimized conditions (Fig. 3c). This is the highest value reported for any organic photocatalyst from industrial wastewater.55 Also, the performance of the same catalyst with processed seawater was 90602 μmol g−1 h−1 which is also the best performance, higher than that from deionized water under similar conditions of PHE. From these studies, we found that the performance of the catalyst is not degrading in wastewater and processed seawater and is stable in both cases to deliver a good HER. The reusability and cycling studies of the hybrid photocatalyst under the optimized conditions under visible light irradiation were conducted, and it was found that the photocatalyst is stable with similar performance for more than 5 cycles (Fig. 3d). The recycled sample was filtered again, recovered fully, and tested for chemical and morphological changes, if any. It is worth noting that even after repeated cycles, the catalyst remained the same without any considerable physical or chemical degradation. HR-TEM images show layered sheet-like morphology with Pt NPs of size 2–4 nm embedded in the sheet (Fig. S28†). STEM analysis and elemental mapping of the sample were also conducted which proved the incorporation of Pt NPs all over the surface and between layers of TPG-75 (Fig. S29†). In addition, Pt NPs were distributed in a segregated way all over the catalyst surface owing to the affinity of NPs to the heteroatoms of the hybrid catalyst. The FT-IR spectrum of the recycled catalyst shows no extra peak confirming the stability of the catalyst (Fig. S30†). To understand this further, the HER was performed by physically mixing Tp-Pa and gC3N4 in the same ratio as that adopted for TPG-75 before irradiation. Interestingly, we could observe a decline in the HER compared to TPG-75 (Fig. S31†). This indicates the need to physically connect the two components to boost HER performance. The apparent quantum yield (AQY) of the best-performing sample TPG-75 was investigated at different wavelengths. For this, monochromatic wavelength filters ranging from 450, 500, and 550 nm were chosen for irradiation. Interestingly, an AQY of 10.4% was obtained for TPG-75 in deionized water at 450 nm whereas 8.34% and 4% AQY were obtained at 500 nm and 550 nm, respectively. The performance of TPG-75 was also studied under direct sunlight. In a typical experiment with the same optimized conditions, the performance of TPG-75 under direct sunlight was nearly comparable with the H2 evolution using a solar simulator (Fig. S32†). However, the intensity of light irradiation controls the H2 evolution in direct sunlight experiments (Fig. S32†). It is clearly understood that a similar performance of TPG-75 under a solar simulator was retained with half the light intensity under direct sunlight. The intensity of the light used from the solar simulator was 300 mW cm−2 whereas the average intensity from sunlight was nearly 152 mW cm−2.
To understand the synergistic effect between pristine COF and gC3N4 in hybrid TPG-x, different experiments were conducted. The steady-state emission spectrum of the hybrid photocatalyst was measured and compared with that of Tp-Pa and gC3N4 (Fig. S33†). Accordingly, gC3N4 shows an emission peak at around 445 nm; however, Tp-Pa and TPG-x were not emissive. In fact, the emission of the gC3N4 counterpart in TPG-x was quenched due to the charge transfer (CT) between Tp-Pa and gC3N4 and this was supported by a decrease in emission lifetime of gC3N4 in the presence of Tp-Pa (Fig. S34 and Table S4†). To elucidate this assumption, we performed additional experiments by preparing control samples. Pristine gC3N4 was ground with different ratios of Tp-Pa (100:0.01, 100:0.05, 100:1, and 100:5), and the corresponding emission spectra were recorded (Fig. S35†). We could observe a gradual quenching of the emission intensity of gC3N4 upon mixing with Tp-Pa (Fig. S36†). The intensity of emission maxima decreased as the amount of Tp-Pa increased, along with a slight blue shift in emission maxima. This could point out the CT between Tp-Pa and gC3N4.44,56 Correspondingly, the emission lifetime of control samples was also measured (Fig. S37 and Table S5†). The band structure of Tp-Pa and gC3N4 was confirmed by ultraviolet photoelectron spectroscopy (UPS) experiments.57 From UPS studies, the energy of the valence band maximum (EVBM) was calculated by subtracting the width of the UPS spectrum from the excitation energy of the He I source (21.22 eV). EVBM of Tp-Pa and gC3N4 was found to be −7.45 and −6.73 eV, respectively (Fig. S38†). Similarly, the energy of the conduction band minimum (ECBM) was calculated from the Tauc plot and EVBM and was found to be −5.31 and −3.63 eV, respectively.
DFT calculations were used to understand the charge distribution between gC3N4 (001) and Tp-Pa (001) surfaces. First, the slab structures of gC3N4 and Tp-Pa were optimized for further calculations (Fig. 4a and b). Fig. 4c and d show the electrostatic potential of gC3N4 and Tp-Pa where it is observed that the work functions of gC3N4 and Tp-Pa are 4.58 eV and 4.71 eV, respectively. Work function differences between the heterojunction integrant give us an idea about charge distribution and hence induce electric field formation. This in turn affects the CT process and separates photogenerated electrons and holes.58 As the work function of gC3N4 is smaller than that of Tp-Pa, charges will move from gC3N4 to the COF until the Fermi level equilibrium is reached. Also, the Fermi level position of Tp-Pa (−4.23 eV) being lower than that of gC3N4 (−3.37 eV) will accelerate the CT from gC3N4 to Tp-Pa. Furthermore, to understand the CT process, the charge density difference between Tp-Pa and gC3N4 was calculated. For this calculation, a heterostructure model consisting of a 3 × 3 cell of gC3N4 (001) and one unit cell of Tp-Pa (001) was taken. We could see that after optimizing, there is a pronounced distortion in the surfaces resulting from the interaction between gC3N4 and Tp-Pa (Fig. S39†). Also, a three-dimensional (3D) charge density differential for the Tp-Pa/gC3N4 heterostructure was obtained to understand the CT process (Fig. 4e and f). The yellow and cyan regions show positive and negative charges on the heterostructure. It is evident from the calculation that Tp-Pa is mostly dominated by the cyan region (negative) and gC3N4 is mostly dominated by the yellow region (positive) indicating a good amount of charge accumulation and reduction on Tp-Pa and gC3N4, respectively. When in contact, the charge accumulation and reduction occur and ultimately cause a band edge bending phenomenon on gC3N4 and Tp-Pa, respectively. The band bending phenomenon causes the band edge of Tp-Pa to move downwards whereas the band edge of gC3N4 moves upwards and the PHE of TPG-xs can be explained based on these conclusions (Fig. 5). Upon irradiating with light, the electrons of gC3N4 and Tp-Pa will get excited to higher energy levels. Due to the band bending up/downward phenomenon, the excited electrons of Tp-Pa will recombine with the holes of gC3N4, hence decreasing the charge recombination process within gC3N4. Furthermore, the excited state electrons of gC3N4 will be transferred to Pt NPs. At the same time, the holes generated in Tp-Pa will be carried forward by AA and oxidized to dehydroascorbic acid. The above observation was further confirmed by PHE studies and emission studies of control samples (Fig. S36†). It must be remembered that gC3N4 is not performing well with AA suggesting that whatever holes were generated in gC3N4 could not be transferred to AA and oxidized. Also, from emission, it was evident that due to CT between gC3N4 and Tp-Pa, the emission of gC3N4 was quenched by adding Tp-Pa.
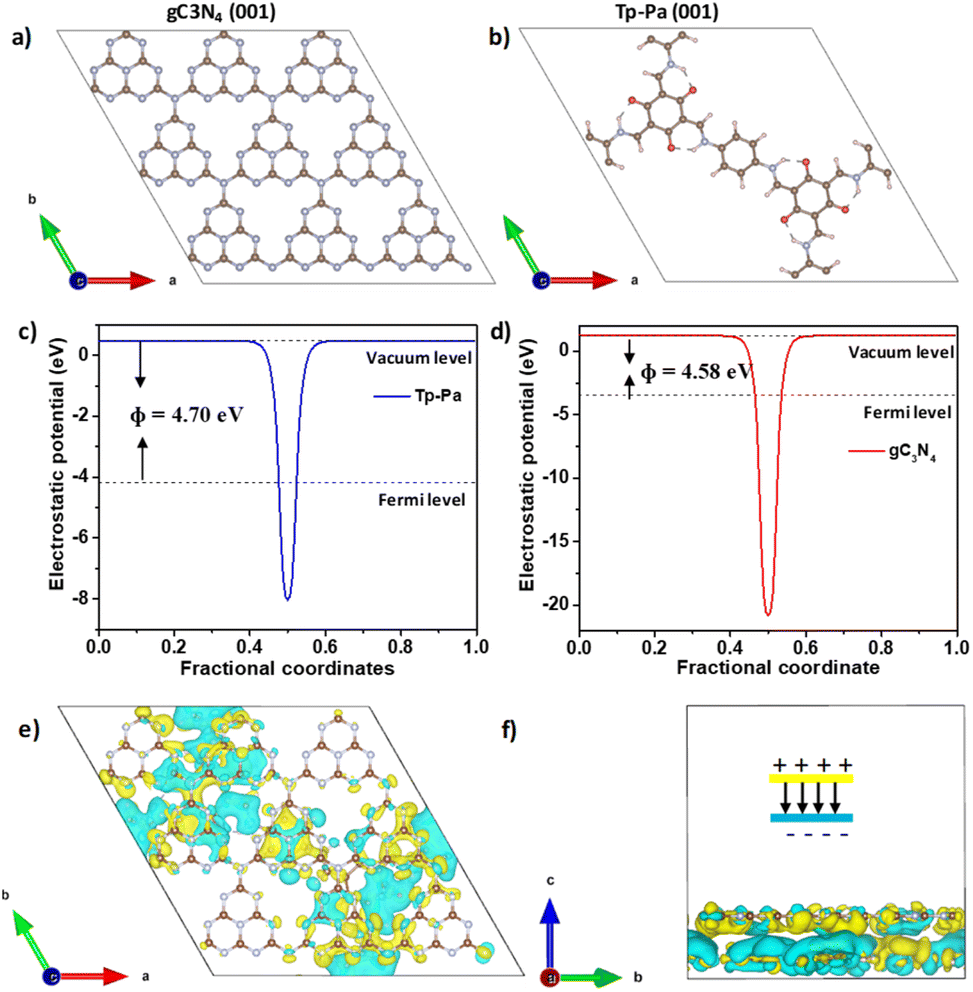 | ||
| Fig. 4 Optimized slab structure of (a) gC3N4 and (b) Tp-Pa COF. Theoretical electrostatic potential of (c) Tp-Pa (001) and (d) gC3N4 (001). Three-dimensional calculated charge density difference of the Tp-Pa/gC3N4 heterostructure from the (e) top view and (f) side view. | ||
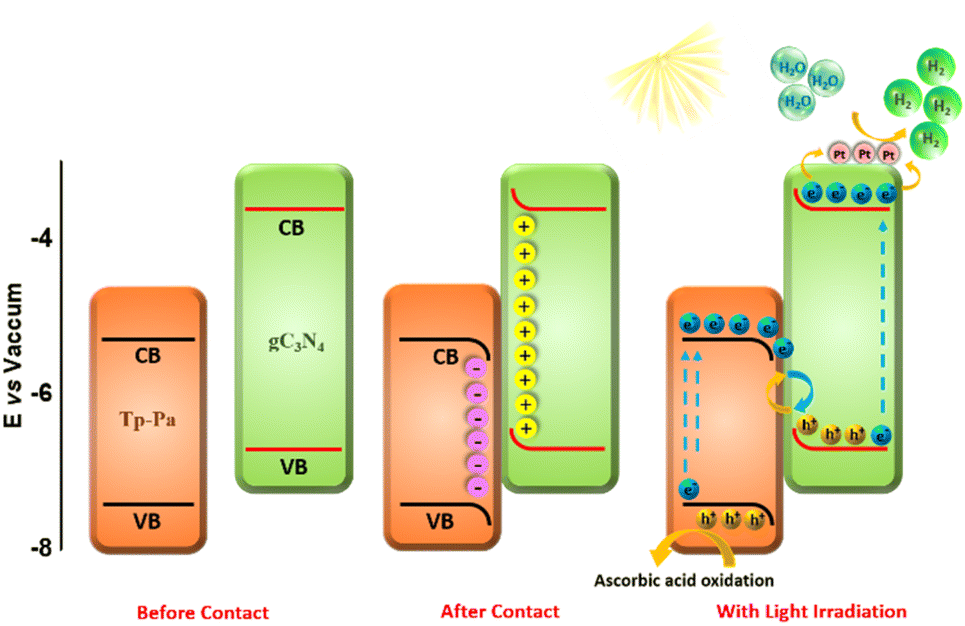 | ||
| Fig. 5 Band structure of Tp-Pa and gC3N4 with a plausible PHE mechanism upon irradiating with light. | ||
We prepared TPG-75 on a 20 g scale using a planetary mixer and the detailed procedure is provided in the experimental section. After extensive washing and purification, TPG-75-20G was characterized by 13C NMR, FT-IR, XPS, BET, HR-TEM, PXRD (Fig. S40†), FE-SEM, and STEM elemental mapping (Fig. S41†). All the experimental results are in line with the small-scale synthesis (2 g) and confirm the suitability of the synthesis protocol for the large-scale production of TPGs. PHE of TPG-75-20G exhibited a comparable performance of 84383 μmol g−1 h−1 with deionized water and no significant reduction in the H2 evolution rate upon scale-up synthesis (Fig. 6a and b, and Video S1†). Similarly, comparable performance to that of TPG-75 was noticed with processed seawater (90371 μmol g−1 h−1) and industrial wastewater (94873 μmol g−1 h−1) (Fig. 6b). To further extend the scope of using the catalyst for the production of H2 from seawater, we prepared simulated seawater using a standard protocol59 and examined it for PHE studies. Using optimized PHE conditions, the performance of TPG-75-20G from simulated seawater was found to be 109125 μmol g−1 h−1 which is one of the highest reported values among different photocatalysts studied to date (Fig. 6a). The exceptional increment in the HER rate from simulated seawater can be attributed to the in situ polarization of the framework arising from the adsorption of metal salts. The polarization effect plays a crucial role in amplifying the dielectric constant of the organic semiconductor. This, in turn, reduces the exciton dissociation energy, enhancing the charge separation and transfer. Consequently, it facilitates the promotion of H2 production from seawater.49 We compared these results with reported literature on the photocatalytic HER from seawater and found that our sample is one of the best-performing catalysts among organic, inorganic, and hybrid materials (Fig. 6c and Table S6†). The cycling stability of TPG-75-20G was also investigated, revealing stability for more than 5 cycles with minimal degradation observed in hydrogen evolution using deionized water (Fig. S42†).
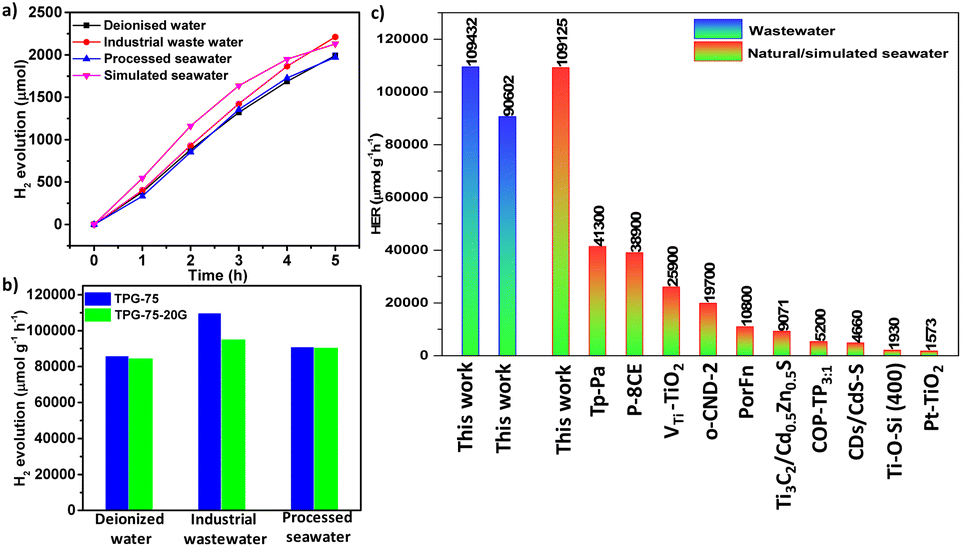 | ||
| Fig. 6 (a) HER performance of TPG-75-20G with different water sources and (b) comparison of HER performance of TPG-75 and TPG-75-20G with different water sources. (c) Comparison of the HER rate of TPG-75 with that of different hybrid materials reported so far from wastewater and simulated or natural seawater. | ||
To further support the results, we have obtained photocurrent responses and conducted EIS studies of TPG-75-20G in different water sources. From photocurrent responses, we could see that the light responses of TPG-75-20G were more prominent in industrial wastewater and simulated seawater compared to deionized water (Fig. S43†) proving the higher H2 evolution from industrial wastewater and simulated seawater. The EIS studies also revealed small charge transfer resistance for TPG-75-20G with different water sources (Fig. S43†). In short, all the supporting experiments point to the merit of our work to achieve scalable synthesis and remarkable H2 production from seawater and industrial wastewater.
Conclusions
We present, for the first time, the large-scale synthesis of an organic photocatalyst with outstanding HER efficiency. We successfully synthesized a hybrid photocatalyst based on Tp-Pa and gC3N4 through a scalable mechanochemical method and comprehensively characterized it using various experimental techniques. The hybrid photocatalyst exhibited high crystallinity and surface area. Our comprehensive research, incorporating electrochemical methods and theoretical calculation, suggests that the presence of both Tp-Pa and gC3N4 components results in synergistic effects. Consequently, the semiconducting properties of the hybrid catalyst can be finely tuned, making it a promising photocatalyst for H2 evolution from water. The improved photocatalytic HER rate can be attributed to the enhanced charge dissociation and exciton formation within the hybrid catalyst. Notably, the hybrid catalyst achieves a remarkable photocatalytic HER rate of 85617 and 179064 μmol g−1 h−1 under visible light and simulated solar light, respectively. It also performs exceptionally well with different water sources, demonstrating rates of 109432 μmol g−1 h−1 for industrial wastewater and 90602 μmol g−1 h−1 for processed seawater. This represents the highest reported rate for a photocatalyst that is effective in deionized water, wastewater, and seawater.
Furthermore, we have explored the bulk synthesis of these photocatalysts, a critical step toward potential industrial applications. A facile mechanochemical synthesis procedure using a planetary mixer enabled us to successfully synthesize 20 g of photocatalyst in a single batch while retaining crystallinity, surface area, and nearly the same H2 evolution. Remarkable HER rates of 84383, 90371, 94873, and 109125 μmol g−1 h−1 were obtained for deionized water, processed seawater, industrial wastewater, and simulated seawater, respectively. Our work contributes valuable insights into the development of organic semiconductors for industrial applications, emphasizing scalability and mass production targeting potential commercialization.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. S. B. conceived the research idea. K. A. synthesized and characterized COFs and conducted all photocatalytic HER experiments. B. T. M. designed and performed all the electrochemistry experiments. G. D. and S. K. carried out the DFT calculation and provided the corresponding write-up. The results were regularly discussed and all the contributing authors commented and provided significant suggestions. K. A. and S. S. B. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of this work from the Qatar National Research Fund (QNRF), project number NPRP12S-0228-190188, led and co-funded by the Qatar Shell Research and Technology Center (QSRTC). We thank Flavia Cassiola, Product Quality Lead, Renewable Feedstocks, Shell, for the inputs and feedback on the manuscript.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 15729–15735 CrossRef CAS PubMed
.
- K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2017, 9, 1523 CrossRef CAS
- Y. Li, X. Song, G. Zhang, L. Wang, Y. Liu, W. Chen and L. Chen, ChemSusChem, 2022, 15, e202200901 CrossRef CAS PubMed
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814 CrossRef CAS PubMed
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789 RSC
- T. He and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed
- C.-C. Gu, F.-H. Xu, W.-K. Zhu, R.-J. Wu, L. Deng, J. Zou, B.-C. Weng and R.-L. Zhu, Chem. Commun., 2023, 59, 7302 RSC
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902 CrossRef CAS PubMed
- J. Yang, A. Acharjya, M. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. Van De Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., Int. Ed., 2021, 60, 19797 CrossRef CAS PubMed
- T. Zhou, X. Huang, Z. Mi, Y. Zhu, R. Wang, C. Wang and J. Guo, Polym. Chem., 2021, 12, 3250 RSC
- J. Wang, X.-X. Tian, L. Yu, D. J. Young, W.-B. Wang, H.-Y. Li and H.-X. Li, J. Mater. Chem. A, 2021, 9, 25474 RSC
- Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, e202305355 CAS
- Z. Mi, T. Zhou, W. Weng, J. Unruangsri, K. Hu, W. Yang, C. Wang, K. A. I. Zhang and J. Guo, Angew. Chem., Int. Ed., 2021, 60, 9642 CrossRef CAS PubMed
- Y.-H. Yao, J. Li, H. Zhang, H.-L. Tang, L. Fang, G.-D. Niu, X.-J. Sun and F.-M. Zhang, J. Mater. Chem. A, 2020, 8, 8949 RSC
- Y.-J. Cheng, R. Wang, S. Wang, X.-J. Xi, L.-F. Ma and S.-Q. Zang, Chem. Commun., 2018, 54, 13563 RSC
- Y.-H. Yao, Y. Yang, Y. Wang, H. Zhang, H.-L. Tang, H.-Y. Zhang, G. Zhang, Y. Wang, F.-M. Zhang and H. Yan, J. Colloid Interface Sci., 2022, 608, 2613 CrossRef CAS PubMed
- F.-M. Zhang, J.-L. Sheng, Z.-D. Yang, X.-J. Sun, H.-L. Tang, M. Lu, H. Dong, F.-C. Shen, J. Liu and Y.-Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 12106 CrossRef CAS PubMed
- H.-Y. Zhang, Y. Yang, C.-C. Li, H.-L. Tang, F.-M. Zhang, G.-L. Zhang and H. Yan, J. Mater. Chem. A, 2021, 9, 16743 RSC
- C.-C. Li, M.-Y. Gao, X.-J. Sun, H.-L. Tang, H. Dong and F.-M. Zhang, Appl. Catal., B, 2020, 266, 118586 CrossRef CAS
- H. Yan, Y.-H. Liu, Y. Yang, H.-Y. Zhang, X.-R. Liu, J.-Z. Wei, L.-L. Bai, Y. Wang and F.-M. Zhang, Chem. Eng. J., 2022, 431, 133404 CrossRef CAS
- Y. Chen, D. Yang, Y. Gao, R. Li, K. An, W. Wang, Z. Zhao, X. Xin, H. Ren and Z. Jiang, Research, 2021, 2021, 9798564 CAS
- M. Luo, Q. Yang, W. Yang, J. Wang, F. He, K. Liu, H. Cao and H. Yan, Small, 2020, 16, 2001100 CrossRef CAS PubMed
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445 CrossRef PubMed
- P. Dong, A. Zhang, T. Cheng, J. Pan, J. Song, L. Zhang, R. Guan, X. Xi and J. Zhang, Chin. J. Catal., 2022, 43, 2592 CrossRef CAS
- M. Luo, Q. Yang, K. Liu, H. Cao and H. Yan, Chem. Commun., 2019, 55, 5829–5832 RSC
- J. Ming, A. Liu, J. Zhao, P. Zhang, H. Huang, H. Lin, Z. Xu, X. Zhang, X. Wang, J. Hofkens, M. B. J. Roeffaers and J. Long, Angew. Chem., Int. Ed., 2019, 58, 18290 CrossRef CAS PubMed
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180 CrossRef CAS PubMed
- T. Banerjee, F. Haase, G. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228 CrossRef CAS PubMed
- J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845 CrossRef CAS PubMed
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862 CrossRef CAS PubMed
- K. Gottschling, G. Savasci, H. Vignolo-González, S. Schmidt, P. Mauker, T. Banerjee, P. Rovó, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2020, 142, 12146 CrossRef CAS PubMed
- Y.-P. Zhang, H.-L. Tang, H. Dong, M.-Y. Gao, C.-C. Li, X.-J. Sun, J.-Z. Wei, Y. Qu, Z.-J. Li and F.-M. Zhang, J. Mater. Chem. A, 2020, 8, 4334 RSC
- Y. Bai, Y. Liu, M. Liu, X. Wang, S. Shang, W. Gao, C. Du, Y. Qiao, J. Chen, J. Dong and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202113067 CrossRef CAS PubMed
- Y. Yang, N. Luo, S. Lin, H. Yao and Y. Cai, ACS Catal., 2022, 12, 10718 CrossRef CAS
- W. Dong, Z. Qin, K. Wang, Y. Xiao, X. Liu, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202216073 CrossRef CAS PubMed
- S. Ma, T. Deng, Z. Li, Z. Zhang, J. Jia, Q. Li, G. Wu, H. Xia, S. Yang and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208919 CrossRef CAS PubMed
- W. Zhou, Q. Deng, H. He, L. Yang, T. Liu, X. Wang, D. Zheng, Z. Dai, L. Sun, C. Liu, H. Wu, Z. Li and W. Deng, Angew. Chem., Int. Ed., 2023, 62, e202214143 CrossRef CAS PubMed
- L. Sun, W. Wang, T. Kong, H. Jiang, H. Tang and Q. Liu, J. Mater. Chem. A, 2022, 10, 22531 RSC
- G. Yan, X. Sun, K. Zhang, Y. Zhang, H. Li, Y. Dou, D. Yuan, H. Huang, B. Jia, H. Li and T. Ma, Small, 2022, 18, 2201340 CrossRef CAS PubMed
- B. Cai, L. Cao, R. Zhang, N. Xu, J. Tang, K. Wang, Q. Li, B. Xu, Y. Liu and Y. Fan, ACS Appl. Energy Mater., 2023, 6, 930 CrossRef CAS
- L. Dai, A. Dong, X. Meng, H. Liu, Y. Li, P. Li and B. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300224 CrossRef CAS PubMed
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S.-W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145(15), 8364–8374 CrossRef CAS PubMed
- H. Zhang, Z. Lin, P. Kidkhunthod and J. Guo, Angew. Chem., Int. Ed., 2023, 62, e202217527 CrossRef CAS PubMed
- R. Shen, G. Liang, L. Hao, P. Zhang and X. Li, Adv. Mater., 2023, 2303649 CrossRef CAS PubMed
- S. Fukuzumi, Y.-M. Lee and W. Nam, ChemSusChem, 2017, 10, 4264 CrossRef CAS PubMed
- K. C. Ranjeesh, L. George, A. Maibam, S. Krishnamurty and S. S. Babu, ChemCatChem, 2021, 13, 1717 CrossRef CAS
-
Y. Lu, Y.-X. Liu, S. Cao and X.-Y. Yang, Photo-Driven Seawater Splitting
for Hydrogen Production, ed. X.-Y. Yang, Springer Nature Singapore, Singapore, 2023, pp. 99–164 Search PubMed
- Q. Yue, G. Li, P. Fu, B. Meng, F. Ma, Y. Zhou and J. Wang, Nano Res., 2023, 16, 6251 CrossRef CAS
- K. Asokan, M. K. Patil, S. P. Mukherjee, S. B. Sukumaran and T. Nandakumar, Chem.–Asian J., 2022, 17, e202201012 CrossRef CAS PubMed
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240 CrossRef CAS PubMed
- H. Ou, L. Lin, Y. Zheng, P. Yang, Y. Fang and X. Wang, Adv. Mater., 2017, 29, 1700008 CrossRef PubMed
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed
- Z. Tang, D. Geng and G. Lu, J. Colloid Interface Sci., 2005, 287, 159–166 CrossRef CAS PubMed
- S. S. Tak, O. Shetye, O. Muley, H. Jaiswal and S. N. Malik, Int. J. Hydrogen Energy, 2022, 47, 37282 CrossRef CAS
- K. Wang, Y. Zhong, W. Dong, Y. Xiao, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202304611 CrossRef CAS PubMed
- J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845–23853 CrossRef CAS PubMed
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042 CrossRef CAS
- J. Lyman and R. H. Fleming, J. Mar. Res., 2018, 3, 134–146 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, characterization of the photocatalysts and HER experiments. See DOI: https://doi.org/10.1039/d4sc01387e |
| This journal is © The Royal Society of Chemistry 2024 |