
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular CH⋯π attraction mediated conformational polymorphism of constrained helical peptides†
Jinming
Sun‡
a,
Zi-You
Tian‡
a,
Jianbo
Liu‡
b,
Chuan
Wan
d,
Chuan
Dai
 b,
Zhihong
Liu
b,
Yun
Xing
a,
Yujie
Wu
e,
Zhanfeng
Hou
a,
Wei
Han
c,
Feng
Yin
*ab,
Yuxin
Ye
*b and
Zigang
Li
*ab
b,
Zhihong
Liu
b,
Yun
Xing
a,
Yujie
Wu
e,
Zhanfeng
Hou
a,
Wei
Han
c,
Feng
Yin
*ab,
Yuxin
Ye
*b and
Zigang
Li
*ab
aState Key Laboratory of Chemical Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: lizg@pkusz.edu.cn
bPingshan Translational Medicine Center, Shenzhen Bay Laboratory, Shenzhen 518118, China
cDepartment of Chemistry, Faculty of Science, Hong Kong Baptist University, Kowloon Town, Hong Kong SAR, China
dCollege of Health Science and Environmental Engineering, Shenzhen Technology University, Shenzhen 518118, China
eOffice of Core Facilities, Shenzhen Bay Laboratory, Shenzhen 518118, China
First published on 9th August 2024
Abstract
In nature, biochemical processes depend on polymorphism, a phenomenon by which discrete biomolecules can adopt specific conformations based on their environment. However, it is often difficult to explore the generation mechanism and achieve polymorphic control in artificial supramolecular assembly systems. In this work, we propose a feasible thought for exploring the transformation mechanism of polymorphism in peptide assembly from the perspective of thermodynamic regulation, which enables polymorphic composition to be limited by switchable intramolecular CH⋯π attraction within a certain temperature range. Combined with the density functional theory calculations, we obtained thermodynamic theoretical data supporting the conformation transition and the underlying polymorphism formation principle. Afterward, we properly designed the peptide to alter the probability of CH⋯π attraction occurring. Then, we selectively obtained a homogeneous morphological form with corresponding molecular conformation, which further demonstrated the important role of molecular conformational manipulation in polymorphism selection. This unique template-based strategy developed in this study may provide scientists with an additional line of thought to guide assembly paths in other polymorphic systems.
Introduction
Natural polymorphism occurs when a discrete macromolecule adopts different conformations and assembles into various structures depending on the external conditions.1 Such a phenomenon is strongly related to biodiversity,2 human diseases,3 genetic variations, and adaptation.4 In biology, various types of supramolecular aggregation can be formed by numerous proteins.5 Of note, most protein polymorphisms originate from changes in packing arrangements, while the structures of monomers remain identical.6 For example, cilia microtubules can form different organizations that respond to ATPase.7 The polymers formed by self-assembling tobacco mosaic virus capsid protein transform between different states in the wake of pH changes.8 The HIV capsid protein can be assembled into multiple shapes, including cones, tubes, and spheres.9 Such polymorphism from the same chemical monomers is important for the high-dimensional structures of proteins to remain adaptable and bioactive. Thus, exploration and control of polymorphism in complex biological systems at the nanometer and micrometer scales can be important steps for chemists in achieving bioinspired architecture.10A great deal of progress has been made in supramolecular assembly in recent decades, particularly in the ability to arrange small molecules into sophisticated architectures.11–16 In nature, self-assembled systems are built using a variety of biomolecules, such as proteins, peptides, and nucleic acids, which usually show morphological and structural–functional diversity.17–22 This has aroused the aspiration of chemists to extend the supramolecular assembly area from specific small-molecular to complicated biomacromolecule systems in the laboratory. For example, mechanistic studies of glucose isomerase suggested that polymorph selection occurs at the earliest stage of the nucleation and exerts control over the system through site-directed mutagenesis.23 Various protein polymorphisms have been constructed by simply altering the chemical structure of the ligand.24 Moreover, the assembly of beta-amyloid peptides, closely related to neurodegenerative diseases, has been extensively explored.25–28
In essence, the polymorphism of biomacromolecules and peptides shares the same principle with self-assembly in nature which is brought about by the precisely located, elaborately balanced, and kT-related weak interaction among the subunits.29–32 Generally, the weak interactions contain hydrogen bonding, π–π stacking, and Coulomb attraction. Induced by these generic interactions, the organization of native proteins or peptides into fibers, micelles, or vesicles is obligatorily subject to analog influences.33 Exploring the driving force behind polymorphism, whether from detailed interactions or thermodynamic and kinetic factors, can provide valuable insights into the allosteric mechanisms of macromolecules.15 However, constrained by the relatively large molecular dimension and disordered chemical heterogeneity, especially the lack of stable secondary conformations, protein-directed generation of polymorphic or monomorphic assemblies remains a great challenge. In addition, the complex interactions are essential for addressing complicated cases, which have been difficult to predict with current theory. Furthermore, polymorphic biomacromolecules are influenced by thermodynamic forces.34 However, the distinct kinetic route and conditions often prefer a local rather than a global minimum, complicating the biomacromolecule as an ideal research subject.35 In contrast, the short peptide offers structural simplicity and robust self-assembly features which prove to be the ideal toolbox and building block to explore the insights of complex protein assemblies in nature.36 Specifically, short peptides with a stable secondary conformation could provide a unitary interaction model to dig out the picture of polymorphism. Benefiting from these advantages, short peptides can facilitate conformational/structural analysis, which has attracted widespread attention for their assembly and medical applications in recent years.37–39
Previously, we developed a chirality-induced helical (CIH) cyclic-pentapeptide system with superior assembly capacity. The types of amino acids in the side chain could manipulate the helicity of the short peptide, resulting in the enhanced self-assembly propensity for the CIH peptides.40,41 Benefiting from the discovery, it was realized that the CIH model could be capable of being finely tuned to explore the diversity of conformation with manipulation of amino acids. In this work, we reported an elaborate conformational polymorphism example based on the CIH peptide system. The peptide molecules could self-assemble into two coexisting and stable peptide crystals under appropriate conditions. The X-ray diffraction data and the DFT calculations revealed that a temperature-controlled intramolecular C–H⋯π interaction plays a pivotal role in switching the thermodynamic conformation of the peptide molecule, thereby adjusting its polymorphism. Moreover, through analysis of the structures and design of the sequence, we achieved the singular stabilization of a specific polymorphic form. In this study, we examined the CIH peptide polymorphic transformation process, affecting variables, and thermodynamically feasible theoretical basis, providing a conformation control model to understand the mechanisms of conformational polymorphism at the atom level.
Results and discussion
The CIH peptides are designed based on an N-terminal acetyl-protected cyclic-pentapeptide scaffold.42 A thioether-containing side chain linker, which had a (R)-configured chiral center at the γ position on the side chain of the C-terminal residue, connects the N-terminal and C-terminal residues (Fig. 1a). Our previous research demonstrated that this (R)-configuration could induce the helical conformation,42 and the phenyl group in the chiral center facilitated self-assembly.40 The synthesis of the CIH peptides was accomplished using a previously developed solid-phase cyclization protocol, which involved a seven-step unnatural amino acid (Fmoc-S5) synthesis.42 The synthesized CIH peptides underwent purification through high-performance liquid chromatography (HPLC) and were subsequently characterized using electrospray ionization mass spectrometry (MS-ESI) (detailed peptide synthesis and characterization results are provided in Section 2 of the ESI†). Circular dichroism (CD) analysis further confirmed that the synthesized CIH peptides adopted a helical conformation in an aqueous solution, aligning with the anticipated one (Fig. S1†).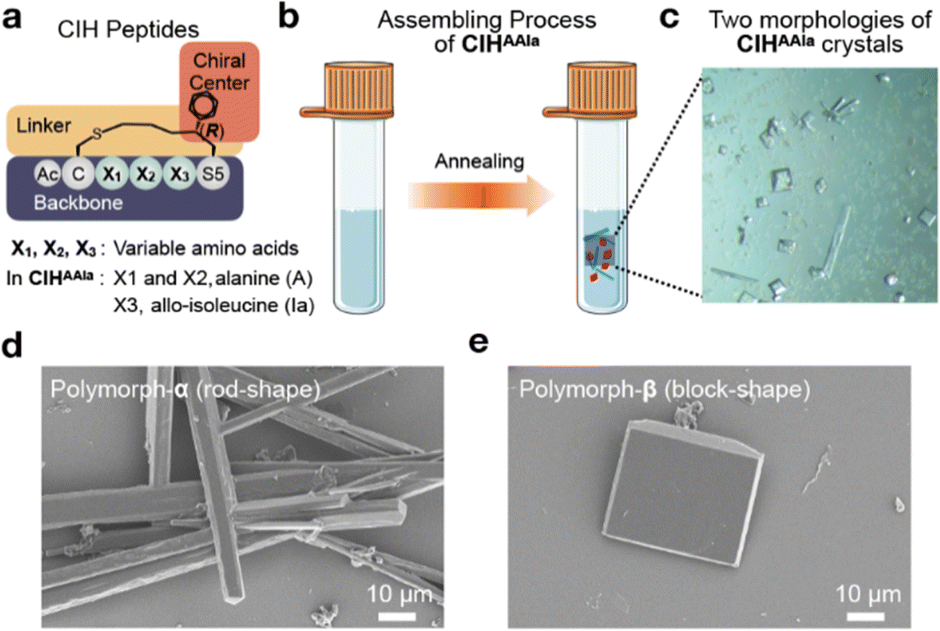 | ||
| Fig. 1 (a) Modular structure model of the CIH peptides. (b) Schematic illustration of the CIHAAIa polymorph-formation process. (c) Optical micrographs of two morphologies of CIHAAIa crystals. (d) SEM morphologies of polymorph-α grown from 5 mM solutions (scale bar: 10 μm). (e) SEM morphologies of polymorph-β grown from 5 mM solutions (scale bar: 10 μm). | ||
Initially, a set of CIH peptides was employed to investigate self-assembling behavior by the ultrasonic method as previously reported (Fig. S2a†). However, peptides containing hydrophobic amino acids exhibited poor solubility and failed to undergo assembly at various concentrations. Subsequently, an alternative heating-annealing method was explored. Specifically, the peptide liquids were subjected to heating at 363 K for varying durations to facilitate the dissolution of the peptide suspension, and the annealing rate was kept constant at 0.1 K min−1. In this way, we obtained a two polymorph self-assembly product after heating for 240 minutes by using CIH peptide Ac-CAAIaS5(Ph)-NH2 (termed CIHAAIa), in which the second, third, and fourth residues are alanine (A), alanine (A), and, allo-isoleucine (Ia), respectively (Fig. 1a, b, and S2b†). Optical microscopy demonstrated that two polymorphs were generated concomitantly but had different morphologies (Fig. 1c). Scanning electron microscopy (SEM) further elucidated that the growth habits of these polymorphs were rods (termed polymorph-α) and blocks (termed polymorph-β), respectively, both exceeding 20 μm in size (Fig. 1d and e).
The crystals of two polymorphs were successfully separated manually for single-crystal X-ray diffraction (SCXRD) experiments according to their different morphologies. The crystal of polymorph-α was found to be orthorhombic with the space group of P212121, and crystals of polymorph-β were tetragonal with the space group of P41212 (Table S1†). Investigations of the crystal structure revealed that CIHAAIa monomers in two polymorphs were both in helical conformation maintained by four sets of hydrogen bonds (Fig. 2b), which were consistent with the CD analysis result (Fig. S1†). Further intermolecular interaction analysis showed that, in conformer-1 (the monomer conformation of CIHAAIa in polymorph-α), a C–H⋯π hydrogen bond was formed inside the benzene and side chain of the fourth residue (the distance from the γ hydrogen atom of the fourth residue to the benzene ring is 2.88 Å) (Fig. 2b, left). However, in conformer-2 (the monomer conformation of CIHAAIa in polymorph-β), the closest distance between the hydrogen atom on the fourth residue and the benzene ring was 3.46 Å (Fig. 2b, right), which exceeds the maximum distance of C–H⋯π contacts.43 Such an intramolecular interaction difference leads to distinct conformations of the side chain of the fourth residue in two conformers (Fig. 2c). Additionally, the C–H⋯π contact within the conformer-1 resulted in a slight rotation as well as a significant displacement of the benzene ring towards the fourth residue, consequently constraining the peptide in a more compact conformation than that in conformer-2 (Fig. 2c).
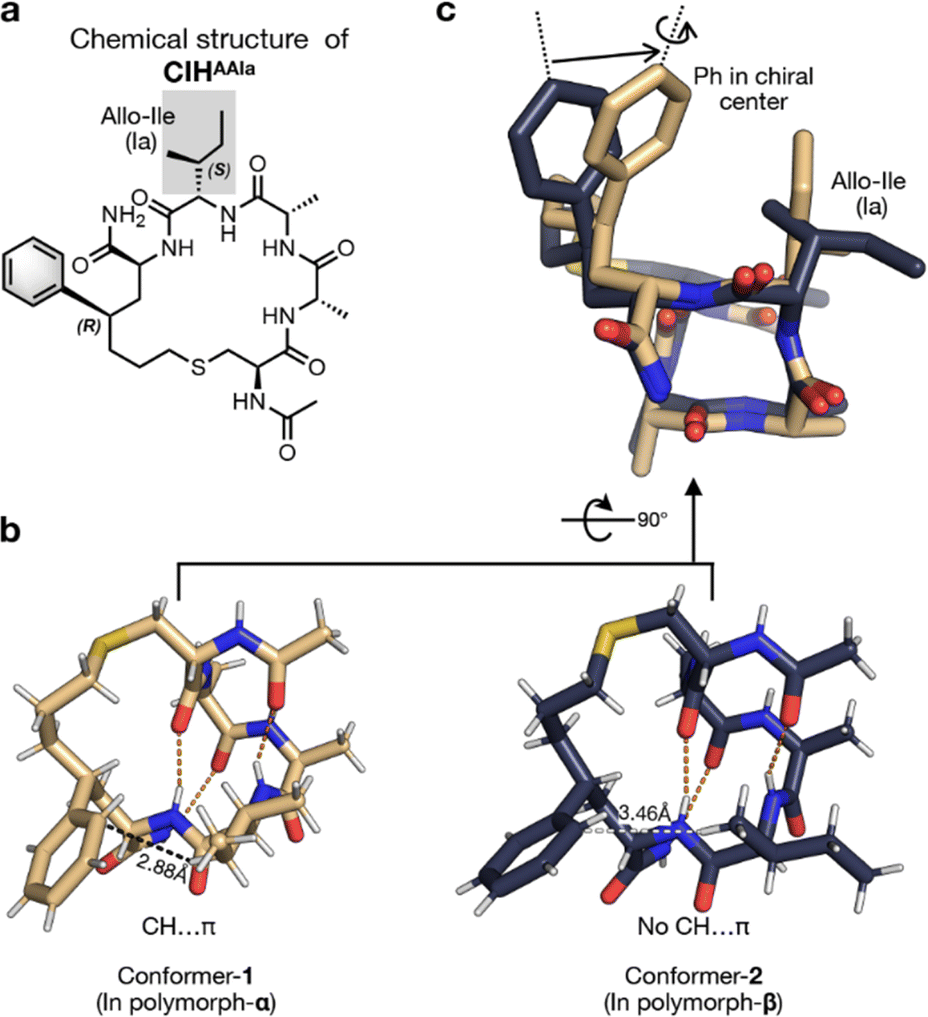 | ||
| Fig. 2 (a) The chemical structure of CIHAAIa. (b) The X-ray structures of a single CIHAAIa molecule in polymorph-α (left, conformer-1, colored in pale yellow) and polymorph-β (right, conformer-2, colored in deep blue). The hydrogen bonds were labeled by orange dashes. The CH⋯π interaction in conformer-1 was labeled by black dashes. The closest distance between the hydrogen atom on the fourth residue allo-Ile and the benzene ring in conformer-2 was labeled by white dashes. (c) Superposition of two conformers of CIHAAIa. | ||
The differential monomer conformations, attributed to the presence or absence of C–H⋯π interaction, give rise to distinct packing arrangements of CIHAAIa molecules in the two polymorphs. In polymorph-α, the molecules assemble into one-dimensional cuboid columns along the a-axis, facilitated by the zipper-like hydrophobic interactions formed by the linker region, benzene group, acetyl group, and the side chains of the second and fourth residues (Fig. 3a). Subsequently, these cuboid columns utilize their amphiphilic surfaces to establish dry- and wet-interfaces with adjacent columns (Fig. 3b and S3†). The dry interface at the column's corners is relatively small and stems from the hydrophobic interactions along the b-axis, while the wet interface constitutes the major portion of the column interface, formed by the solvent-related hydrogen bond network (Fig. S3†). Finally, the dry- and wet-layers stacked along the c-axis to form rod-shaped crystals (Fig. 3c). In polymorph-α, the CIHAAIa molecule diverges from the conventional reliance on hydrogen bonds typically found in contiguous helical structures for its assembly. Instead, it assumes a role as a fundamental building block with a distinctive geometry. The growth of rod-shaped crystals is primarily facilitated by the hydrophobicity effect originating from the linker region and hydrophobic side chains along the a-axis (Fig. 3a and c).
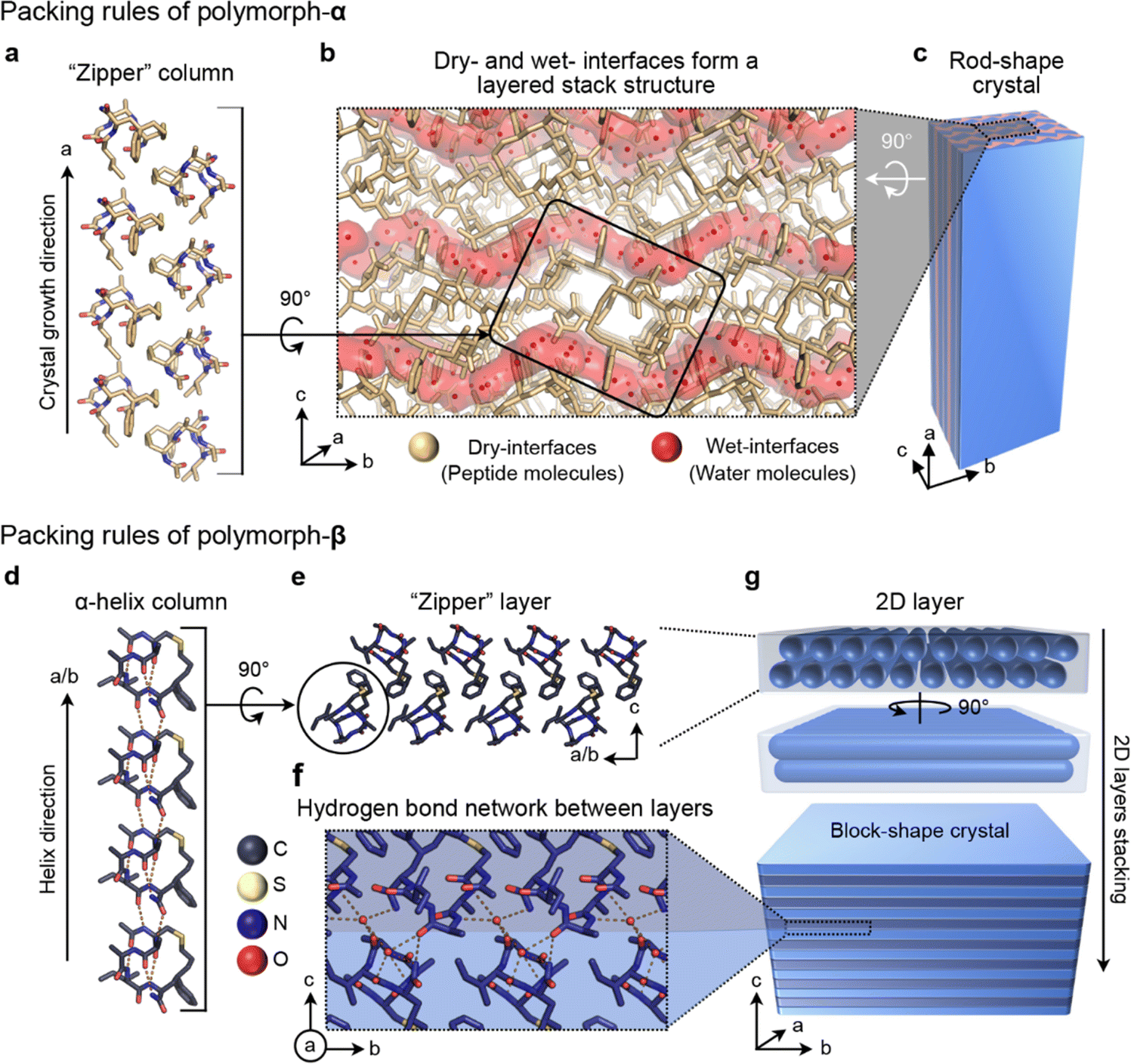 | ||
| Fig. 3 The packing rules of CIHAAIa in polymorph-α (a–c) and polymorph-β (d–g). (a) Side view of the “zipper” columns in polymorph-α. (b) The crystal packing diagrams of polymorph-α. The peptide molecules (colored in pale yellow) formed a dry layer. The wavy water layer (wet-interfaces, colored in red) implies a large number of hydrogen bonds involved in structural stabilization. (c) Schematic illustration of the molecular arrangement inside the rod-shaped crystal. (d) Side view of the helix columns in polymorph-β. (e) The “zipper” layer is maintained by hydrophobic interactions. (f) Hydrogen bond network between layers (C, deep blue; S, yellow; N, blue; O, red). (g) Schematic illustration of the molecular arrangement inside the block-shaped crystal. | ||
In contrast to polymorph-α, the CIHAAIa molecule within polymorph-β adopts an assembly rule that optimally exploits its inherent helical conformation. In polymorph-β, the peptide molecules arrange themselves into a continuous straight α-helical column with head-to-tail interactions along the a- and b-axes, involving the acetyl groups and the carboxylic acid groups (Fig. 3d). The contiguous antiparallel helical columns further stack into a 2D lamellar structure through the zipper-type hydrophobic interaction, mediated by the linker region, benzene group, and hydrophobic side chains (Fig. 3e). This assembly is additionally reinforced by intramolecular C–H⋯π interactions between the benzene ring and the neighboring molecules (Fig. S4†). Subsequently, the adjacent 2D lamellas undergo 90-degree rotation stacking along the c-axis through hydrogen bonds facilitated by a hydrophilic interface and a solvent water molecule, ultimately resulting in the formation of block-shaped crystalline structures (Fig. 3f, g, and S4†).
The success in resolving distinct structures for CIHAAIa in two different morphologies encouraged us to explore the control of polymorphism by conformational transition. Temperature-dependent CD experiments of CIHAAIa revealed a conformational perturbation upon heating in an aqueous solution; the negative peak decreased significantly at 203 nm and 226 nm, and the positive peak increased significantly at 190 nm (Fig. 4a). This implied that the annealing temperature could potentially serve as a parameter for regulating conformational states, thereby influencing polymorphism. To confirm this hypothesis, we conducted a series of self-assembly experiments at varying heating temperatures, while maintaining a consistent annealing rate. The heating temperature was incrementally increased at intervals of 10 K, ranging from 333 K to 373 K. After each temperature interval, samples were collected for detailed characterization through SEM and Powder X-ray Diffraction (PXRD) analysis. The SEM results revealed the exclusive presence of rod-shaped crystals at 333 K and 343 K (Fig. 4b). Correspondingly, the PXRD results exhibited five distinct diffraction peaks at 10.8°, 14.2°, 18.9°, 21.7°, and 23.7°, aligning with the crystal faces (0, 1, 1), (1, 2, 1), (1, 3, 1), (2, 0, 3), and (2, 0, 4) in the simulated PXRD pattern of polymorph-α (Fig. 4c). Conversely, at 373 K, only tetragonal block-shaped crystals were observed (Fig. 4b), featuring three distinct diffraction peaks at 11.0°, 16.8°, and 22.4° in the PXRD results. These peaks corresponded to the crystal faces (0, 0, 8), (0, 0, 12), and (0, 0, 16) in the simulated PXRD pattern of polymorph-β (Fig. 4c). At 353 K and 363 K, both rod-shaped and block-shaped morphologies were observed (Fig. 4b), and all representative PXRD diffraction peaks characteristic of both polymorph-α and polymorph-β were detectable (Fig. 4c). These results indicated that CIHAAIa undergoes distinct stages of polymorph formation at specific temperatures. Taken together, this outcome underscores that the supramolecular assembly of CIHAAIa is a temperature-controlled process.
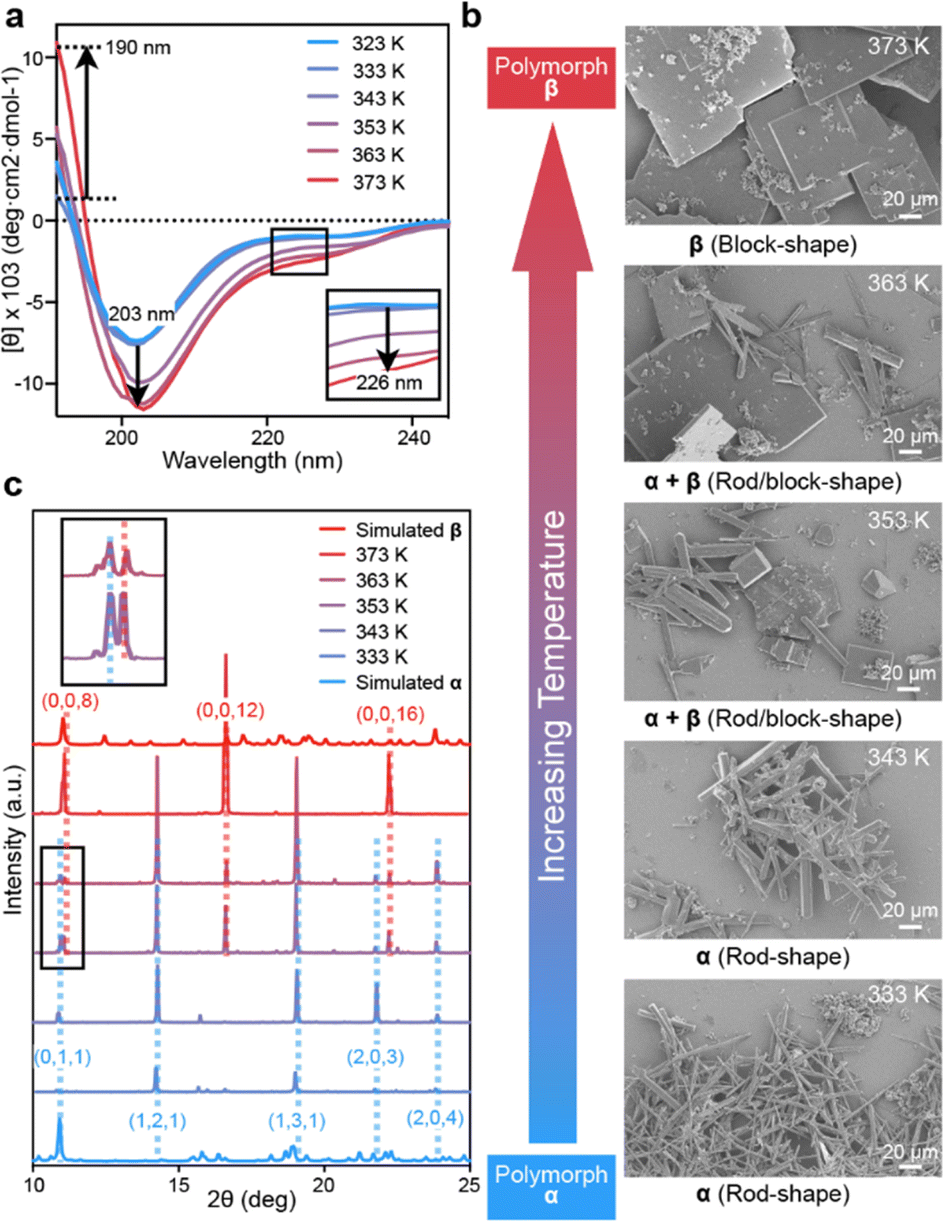 | ||
| Fig. 4 (a) CD spectra of CIHAAIa in ddH2O at variable temperatures. The arrows represent the increase of the peak at 190 nm and its decrease at 203 nm and 226 nm. The internal frame represented the magnified diagram around 226 nm. (b) SEM images of CIHAAIa crystals produced at different annealing temperatures from 5 mM solutions (scale bar: 20 μm). (c) PXRD patterns of CIHAAIa crystalline particles produced at different annealing temperatures from 5 mM solutions, and the simulated PXRD patterns of CIHAAIa by using the structures of conformer 1 (simulated α) and conformer 2 (simulated β). The collected crystalline particles were measured for PXRD patterns. The small number of observed diffraction peaks due to the strong preferred orientation in the PXRD patterns was ascribed to the rod shape and block shape of the crystalline particles. The representative PXRD diffraction peaks were marked by dashes, and the corresponding crystal faces were labeled (polymorph-α, blue; polymorph-β, red). | ||
After achieving precise temperature control over polymorphism, we next contemplated whether such polymorphism regulation was due to the effect of temperature on the conformation state of the peptide monomer as we expected. Therefore, we executed Density Functional Theory (DFT) calculations of CIHAAIa in terms of conformational transformation. Two calculated conformations, iL1, and iL2, were located (Fig. 5a), and both closely resembling the experimental conformations, conformer-1 and conformer-2, respectively (the detailed calculations and benchmark discussion are provided in Section 4.1 of the ESI†). Initially, with reference to iL1, the Gibbs free energy difference at room temperature for iL2 in the aqueous phase was 0.4 kcal mol−1 (Fig. 5b), indicating that the conformational transformation from iL1 to iL2 is thermodynamically prohibited as evidenced by the positive ΔG value. This finding aligns with the experimental result that the preferred polymorph is determined by conformer-1 at lower temperatures, which is polymorph-α. Furthermore, AIM analysis revealed the presence of CH⋯π attraction44–47 in iL1 (Fig. S5†), while no such interaction was observed in iL2, suggesting that the thermodynamically favored conformation relies on this specific CH⋯π interaction.
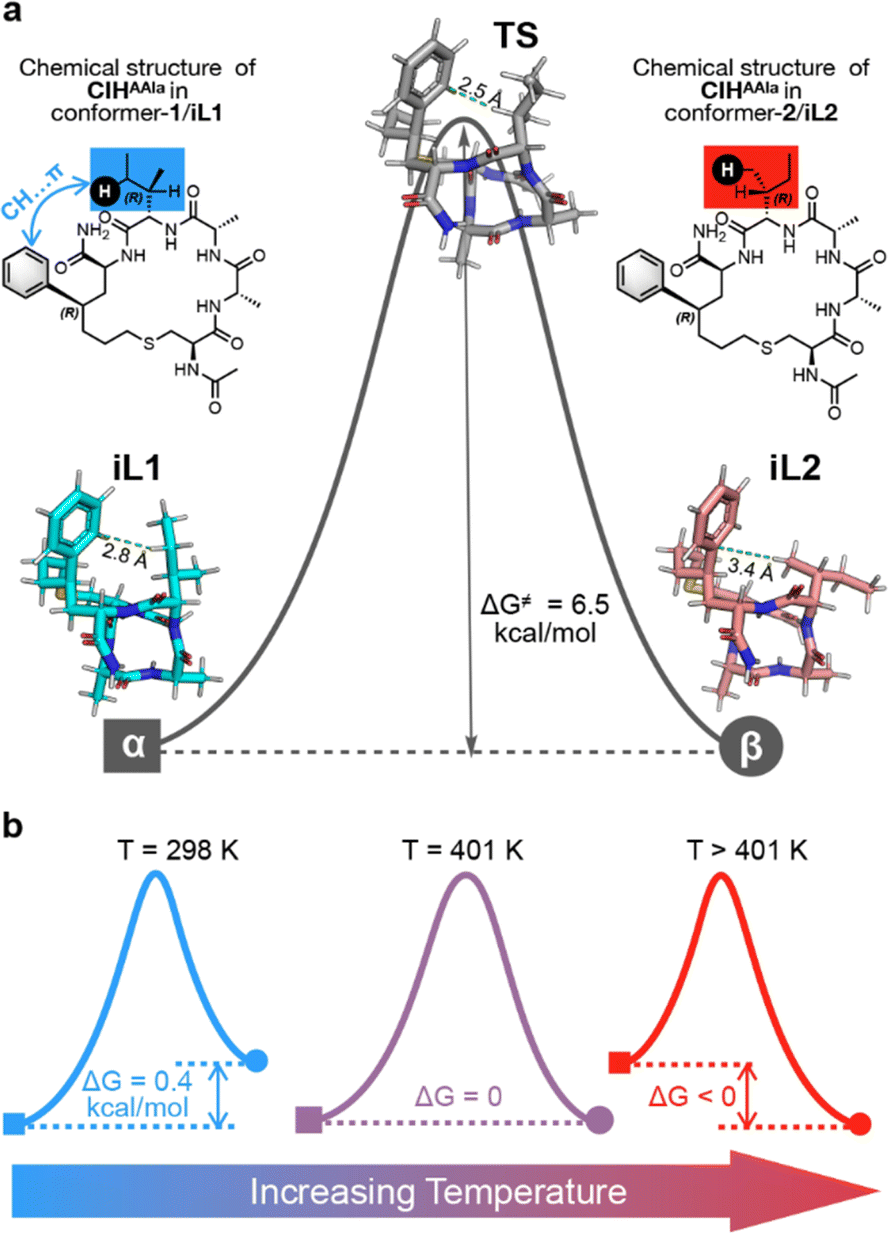 | ||
| Fig. 5 (a) The chemical structure of CIHAAIa (with/without CH⋯π interaction) and the calculated conformation of CIHAAIa (iL1, iL2, and TS). (b) Potential energy surface switching with increasing temperature. | ||
Notably, the activation Gibbs free energy difference associated with the transition state (TS) (Fig. 5a) for the conformational shift from iL1 to iL2 was determined to be merely 6.4 kcal mol−1 (Fig. 5a). This low kinetic barrier implies that conformational changes between iL1 and iL2 can readily occur once the reaction is thermodynamically permissible.48,49 Subsequent thermodynamic calculations, incorporating temperature correction, demonstrated that the energy gap between these two conformations gradually diminishes as the temperature increases (Fig. S6†). Beyond the critical temperature of approximately 401 K, where ΔG equals 0, the conformational transformation becomes thermodynamically permissible, resulting in the equilibrium coexistence of both conformations (Fig. 5b). This is consistent with experimental observations of the simultaneous appearance of the two polymorphic forms. Of note, the error between the calculated critical temperature and the practical temperature was accepted, which was attributed to the precision related to the DFT calculations that was less than 1 kcal mol−1. Finally, the sign of ΔG switched as the temperature continued to increase,50 consistent with the dominance of the conformer 2 and related polymorph-β at elevated temperatures (Fig. 5b).
The DFT calculations of CIHAAIa revealed that the CH⋯π interaction, responsive to temperature variations, could thermodynamically influence the peptide conformation. This underscores the significance of CH⋯π interaction in controlling polycrystalline forms. Of note, such CH⋯π interaction emanated from the Cγ1 atom of the fourth allo-isoleucine side chain, characterized by two C–H bonds and one C–C bond (Cγ1–Cδ1) (Fig. 6a). We hypothesized that altering the number of available C–H bonds on this Cγ1 atom could manipulate the possibility of CH⋯π interaction, consequently altering ΔG to regulate the conformation distribution and attain control over polymorphic assembly. Two distinct peptide design pathways arise from this hypothesis: (i) mutating the fourth allo-isoleucine to valine to increase the number of C–H bonds on the Cγ1 atom to three, thereby increasing the possibility of CH⋯π interaction; (ii) reducing the C–H bond on the Cγ atom proximal to the benzene ring to just one by the substitution of the fourth allo-isoleucine with the more hindered leucine (Fig. 6a).
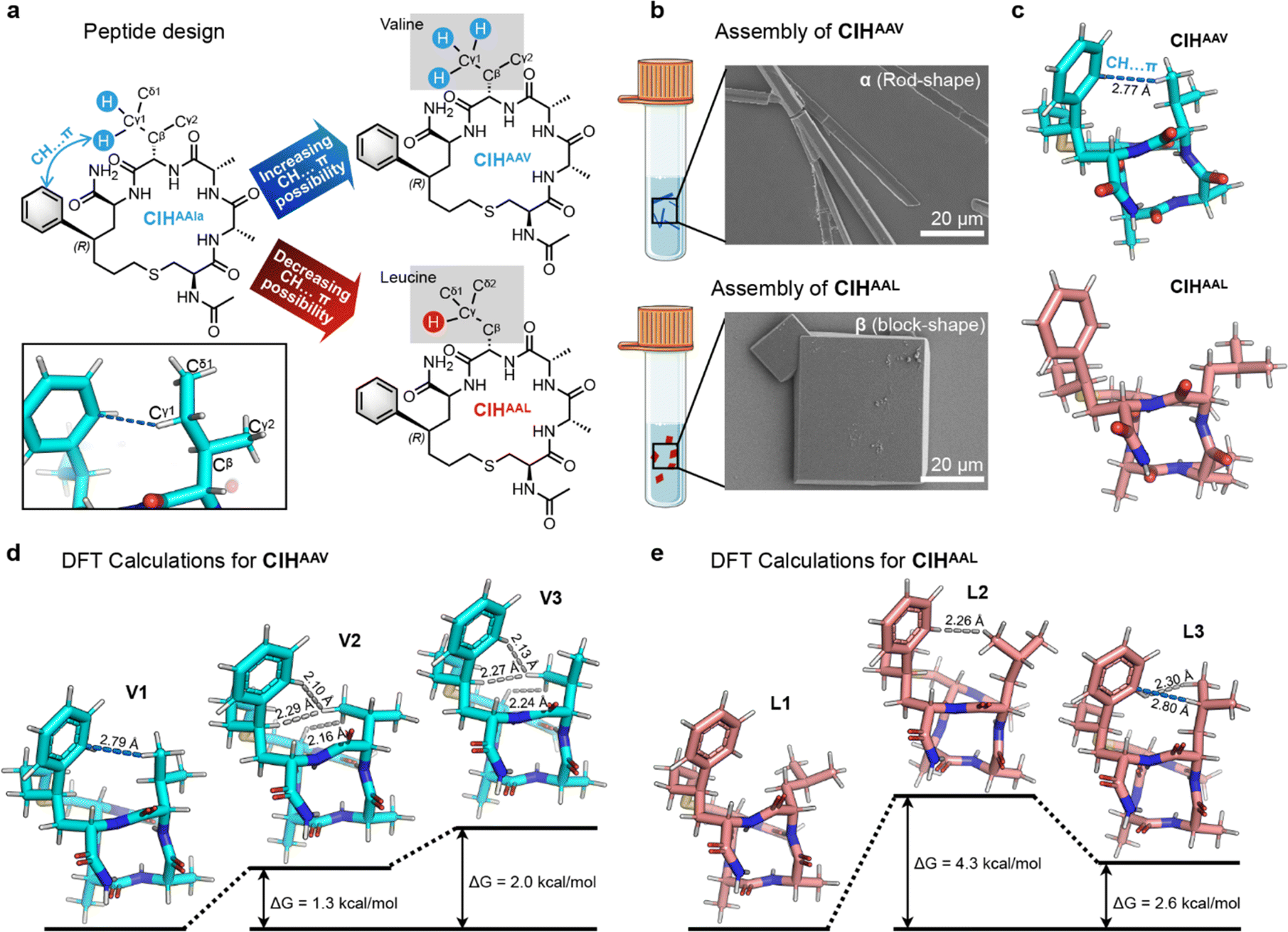 | ||
| Fig. 6 (a) Peptide design strategies of the sequence of CIHAAV and CIHAAL. (b) SEM images of CIHAAV and CIHAAL crystals (scale bar: 20 μm). (c) X-ray molecular structures of CIHAAV and CIHAAL. The CH⋯π interaction was labeled by blue dashes. (d) The DFT calculated structures and the related potential energy surfaces of CIHAAV. The CH⋯π interaction was labeled by blue dashes. The H⋯H steric hindrances were labeled by gray dashes. (e) The DFT calculated structures and the related potential energy surfaces of CIHAAL. The H⋯H steric hindrances were labeled by gray dashes. | ||
To validate our hypothesis, we synthesized CIHAAV and CIHAAL and conducted self-assembly experiments by the annealing method. SEM investigations revealed the exclusive formation of rod-shaped crystals for CIHAAV and block-shaped crystals for CIHAAL, respectively (Fig. 6b). SCXRD results indicated that both CIH peptides molecules exhibited helical conformations (Fig. 6c), consistent with CD analysis (Fig. S1†). Additionally, as anticipated, a 2.77 Å CH⋯π interaction was observed between the fourth valine side chain and benzene ring in CIHAAV, but no CH⋯π interaction was observed in CIHAAL (Fig. 6c). The single molecule X-ray structures of CIHAAV and CIHAAL were identical to the conformer-1 and conformer-2 of CIHAAIa, respectively (Fig. S7†). Furthermore, the space group, cell parameters, and molecular arrangements of the assemblies of CIHAAV and CIHAAL were consistent with polymorphs-α and -β of CIHAAIa, respectively (Table S2 and Fig. S8†). These results indicate that the modulation of CH⋯π interaction through amino acid substitution allows for the adjustment of CIH peptide conformation, consequently achieving uniformity in polymorphic assembly.
The temperature-dependent CD analysis revealed that the conformation of CIHAAV and CIHAAL remained unaltered with temperature variations (Fig. S9†). Moreover, the self-assembly form exhibited resilience to temperature changes, thereby indicating the inherent conformational stability of CIHAAV and CIHAAL. Therefore, the DFT calculations were employed to elucidate the thermodynamic stability of the conformations for CIHAAV and CIHAAL. For CIHAAV, three different conformations were located, which was definitely distinct from the isoleucine scenario. The preferred conformation V1 possessed an analogous CH⋯π interaction concerning the X-ray structure of CIHAAV (Fig. 6d). In addition, another two inferior conformations, which had no CH⋯π interaction, were less favored. In conformation V2, three severe H⋯H steric hindrances were induced within the valine side chain, the backbone of peptide, and the phenyl motif (Fig. 6d). In conformation V3, similar three serious H⋯H steric hindrances dominate the less favored consequence (Fig. 6d). These unfavorable elements in V1 and V2 contributed the higher Gibbs free energy with 1.3 and 2.0 kcal mol−1, underscoring that the reversal of the sign of ΔG with the less favored conformation demands higher temperature and less feasible under atmospheric conditions (Fig. 6d). Similarly, three distinct conformations were located for CIHAAL. As predicted, the preferred conformation L1 harbored less steric hindrance and no CH⋯π interaction was observed (Fig. 6e). The less favored conformation L2 was clearly understood that a dominant H⋯H repulsion was offered with 2.26 Å length, providing the higher 4.3 kcal mol−1 with Gibbs free energy (Fig. 6e). However, in another less favored conformation L3, besides an H⋯H steric hindrance with 2.3 Å was observed, a 2.8 Å CH⋯π interaction was also observed based on the DFT calculation (Fig. 6e). Nonetheless, the gap of the Gibbs free energy in terms of the preferred one was compensated to 2.6 kcal mol−1. Of note, the calculation related to the CIHAAL case was subtly different from the previous one (the detailed discussion is provided in Section 4.2 of the ESI†). Together, our DFT calculations are capable of providing molecular insight to reveal the essence of manipulating the polymorphism.
Conclusions
In summary, we have experimentally and computationally unraveled the conformational polymorphisms based on a chirality-induced helical (CIH) peptide system. The CIHAAIa assembled into two different molecular architectures with two molecular packing modalities (polymorphs-α and -β), induced by two distinct molecular conformations (conformer-1 and -2). DFT calculations revealed that the more compact conformer-1 possessed a characteristic C–H⋯π attraction. Of note, the ΔG between two conformations is only 0.4 kcal mol−1, underlying that only conformer-1 could exist at room temperature, and the other conformation without the C–H⋯π attraction can arise beyond critical temperature. The relative active Gibbs free energy is 6.4 kcal mol−1, underscoring that the transformation between these two conformations is a thermodynamically controlled and kinetically permitted process. In essence, the balance, that is the thermodynamic nature, between the two conformations enlightened us to manipulate the polymorphism with a finely tuned side chain in the CIH system utilizing the augmentation of ΔG. Subsequently, precise control of the self-assembly was provided by rational change of the side chain from allo-isoleucine to valine and leucine. The valine side chain was capable of stabilizing the CH⋯π interaction, while the leucine case offered the destabilization of the CH⋯π conformation with steric hindrance, leading to the other conformation preferred. We hope that the manipulation of the CIH molecular conformation to affect the polymorphism provided a pathway to understand the polymorphism in proteins because the dynamic conformational essence of proteins brought about the same local minimum point within the potential energy surface in nature.Data availability
This material is available free of charge via the Internet at https://pubs.acs.org/. CCDC 2292337, 2292338, 2292339, and 2292343. Materials, protocols, and data characterization for all experiments are provided in the ESI.†Author contributions
The idea for the work was conceived by Z. L. The synthesis and chemical analysis were performed by J. S., Z. T., Y. X. and J. L. The main hypothesis development and result analysis were undertaken by J. S., Z. T., and Z. L. The manuscript was written by J. S., Z. T., F. Y., Y. Y., and Z. L. with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Key R&D Program of China, No. 2021YFC2103900; the Natural Science Foundation of China grant 21977010; the National Center for Biological Medicine Technology Innovation, NCTIB2022HS01017; the Natural Science Foundation of Guangdong Province, 2022A1515010996; Shenzhen Science and Technology Program, RCJC20200714114433053, JCYJ20200109140406047, and JCYJ20220818095808019; the High-tech Zone Development Special Project of Shenzhen 29853M-KCJ-2023-002-07; the Tian Fu Jin Cheng Laboratory (Advanced Medical Center) Group Racing Project, TFJC2023010008; the Beijing National Laboratory of Molecular Science open grant BNLMS20160112 and Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions grant 2019SHIBS0004; the Shenzhen Fundamental Research Program Grant GXWD20201231165807007-20200827170132001 This work was supported by Proteomic Platform of Pingshan translational medicine center, Shenzhen Bay Laboratory.Notes and references
- N. M. Matsumoto, R. P. M. Lafleur, X. Lou, K. C. Shih, S. P. W. Wijnands, C. Guibert, J. van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, Polymorphism in Benzene-1,3,5-tricarboxamide Supramolecular Assemblies in Water: A Subtle Trade-off between Structure and Dynamics, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS.
- A. Karp, K. J. Edwards, M. Bruford, S. Funk, B. Vosman, M. Morgante, O. Seberg, A. Kremer, P. Boursot, P. Arctander, D. Tautz and G. M. Hewitt, Molecular technologies for biodiversity evaluation: opportunities and challenges, Nat. Biotechnol., 1997, 15, 625–628 CrossRef PubMed.
- X. Feng, S. Luo and B. Lu, Conformation Polymorphism of Polyglutamine Proteins, Trends Biochem. Sci., 2018, 43, 424–435 CrossRef PubMed.
- P. W. Hedrick, M. E. Ginevan and E. P. Ewing, Genetic Polymorphism in Heterogeneous Environments, Annu. Rev. Ecol. Syst., 1976, 7, 1–32 CrossRef.
- M. G. Bulmer, Protein polymorphism, Nature, 1971, 234, 410–411 CrossRef PubMed.
- P. Tompa and M. Fuxreiter, Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef.
- I. R. Gibbons, Cilia and flagella of eukaryotes, J. Cell Biol., 1981, 91, 107s–124s CrossRef PubMed.
- K. Namba and G. Stubbs, Structure of tobacco mosaic virus at 3.6 A resolution: implications for assembly, Science, 1986, 231, 1401–1406 CrossRef PubMed.
- B. K. Ganser, S. Li, V. Y. Klishko, J. T. Finch and W. I. Sundquist, Assembly and analysis of conical models for the HIV-1 core, Science, 1999, 283, 80–83 CrossRef CAS PubMed.
- C. Gao and G. Chen, Exploring and Controlling the Polymorphism in Supramolecular Assemblies of Carbohydrates and Proteins, Acc. Chem. Res., 2020, 53, 740–751 CrossRef CAS PubMed.
- J. M. Lehn, Toward complex matter: supramolecular chemistry and self-organization, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Molecular Self-Assembly and Supramolecular Chemistry of Cyclic Peptides, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed.
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Peptide Design and Self-assembly into Targeted Nanostructure and Functional Materials, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS.
- S. Bera, S. Mondal, S. Rencus-Lazar and E. Gazit, Organization of Amino Acids into Layered Supramolecular Secondary Structures, Acc. Chem. Res., 2018, 51, 2187–2197 CrossRef CAS PubMed.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Hierarchically oriented organization in supramolecular peptide crystals, Nat. Rev. Chem, 2019, 3, 567–588 CrossRef CAS.
- P. Sang and J. Cai, Unnatural helical peptidic foldamers as protein segment mimics, Chem. Soc. Rev., 2023, 52, 4843–4877 RSC.
- A. J. Metrano, N. C. Abascal, B. Q. Mercado, E. K. Paulson, A. E. Hurtley and S. J. Miller, Diversity of Secondary Structure in Catalytic Peptides with beta-Turn-Biased Sequences, J. Am. Chem. Soc., 2017, 139, 492–516 CrossRef CAS PubMed.
- F. Wang, O. Gnewou, A. Solemanifar, V. P. Conticello and E. H. Egelman, Cryo-EM of Helical Polymers, Chem. Rev., 2022, 122(17), 14055–14065 CrossRef CAS PubMed.
- J. Zhang, Y. Wang, B. J. Rodriguez, R. Yang, B. Yu, D. Mei, J. Li, K. Tao and E. Gazit, Microfabrication of peptide self-assemblies: inspired by nature towards applications, Chem. Soc. Rev., 2022, 51, 6936–6947 RSC.
- R. Chang, C. Yuan, P. Zhou, R. Xing and X. Yan, Peptide Self-assembly: From Ordered to Disordered, Acc. Chem. Res., 2024, 57, 289–301 CrossRef CAS PubMed.
- R. Xing, C. Yuan, W. Fan, X. Ren and X. Yan, Biomolecular glass with amino acid and peptide nanoarchitectonics, Sci. Adv., 2023, 9, eadd8105 CrossRef CAS.
- C. Yuan, Q. Li, R. Xing, J. Li and X. Yan, Peptide self-assembly through liquid-liquid phase separation, Chem, 2023, 9, 2425–2445 CAS.
- A. E. S. Van Driessche, N. Van Gerven, P. H. H. Bomans, R. R. M. Joosten, H. Friedrich, D. Gil-Carton, N. Sommerdijk and M. Sleutel, Molecular nucleation mechanisms and control strategies for crystal polymorph selection, Nature, 2018, 556, 89–94 CrossRef CAS.
- Z. Li, S. Chen, C. Gao, Z. Yang, K. C. Shih, Z. Kochovski, G. Yang, L. Gou, M. P. Nieh, M. Jiang, L. Zhang and G. Chen, Chemically Controlled Helical Polymorphism in Protein Tubes by Selective Modulation of Supramolecular Interactions, J. Am. Chem. Soc., 2019, 141, 19448–19457 CrossRef CAS PubMed.
- D. Thirumalai, G. Reddy and J. E. Straub, Role of water in protein aggregation and amyloid polymorphism, Acc. Chem. Res., 2012, 45, 83–92 CrossRef CAS PubMed.
- D. Eisenberg and M. Jucker, The Amyloid State of Proteins in Human Diseases, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- D. Li and C. Liu, Conformational strains of pathogenic amyloid proteins in neurodegenerative diseases, Nat. Rev. Neurosci., 2022, 23, 523–534 CrossRef CAS PubMed.
- X. Zottig, S. Al-Halifa, M. Babych, N. Quittot, D. Archambault and S. Bourgault, Guiding the Morphology of Amyloid Assemblies by Electrostatic Capping: from Polymorphic Twisted Fibrils to Uniform Nanorods, Small, 2019, 15, e1901806 CrossRef.
- J. Li and F. L. Deepak, In Situ Kinetic Observations on Crystal Nucleation and Growth, Chem. Rev., 2022, 122, 16911–16982 CrossRef CAS PubMed.
- K. W. Kurgan, A. F. Kleman, C. A. Bingman, D. F. Kreitler, B. Weisblum, K. T. Forest and S. H. Gellman, Retention of Native Quaternary Structure in Racemic Melittin Crystals, J. Am. Chem. Soc., 2019, 141, 7704–7708 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Supramolecular Assembly of Peptide Amphiphiles, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- L. K. Fong, M. J. Chalkley, S. K. Tan, M. Grabe and W. F. DeGrado, Elucidation of the molecular interactions that enable stable assembly and structural diversity in multicomponent immune receptors, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026318118 CrossRef CAS.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Peptide-Based Supramolecular Systems Chemistry, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS.
- J. Wang, K. Liu, R. Xing and X. Yan, Peptide self-assembly: thermodynamics and kinetics, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Energy landscapes and functions of supramolecular systems, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- X. Hu, M. Liao, H. Gong, L. Zhang, H. Cox, T. A. Waigh and J. R. Lu, Recent advances in short peptide self-assembly: from rational design to novel applications, Curr. Opin. Colloid Interface Sci., 2020, 45, 1–13 CrossRef CAS.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Trends in peptide drug discovery, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS.
- W. Jiang, S. Abdulkadir, X. Zhao, P. Sang, A. Tomatsidou, X. Zhang, Y. Chen, L. Calcul, X. Sun, F. Cheng, Y. Hu and J. Cai, Inhibition of Hypoxia-Inducible Transcription Factor (HIF-1alpha) Signaling with Sulfonyl-gamma-AApeptide Helices, J. Am. Chem. Soc., 2023, 145, 20009–20020 CrossRef CAS.
- K. L. Duncan and R. V. Ulijn, Short Peptides in Minimalistic Biocatalyst Design, Biocatalysis, 2015, 1, 67–81 Search PubMed.
- K. Hu, Y. Jiang, W. Xiong, H. Li, P. Y. Zhang, F. Yin, Q. Zhang, H. Geng, F. Jiang, Z. Li, X. Wang and Z. Li, Tuning peptide self-assembly by an in-tether chiral center, Sci. Adv., 2018, 4, eaar5907 CrossRef.
- Y. Jiang, W. Zhang, F. Yang, C. Wan, X. Cai, J. Liu, Q. Zhang, Z. Li and W. Han, Molecular design of stapled pentapeptides as building blocks of self-assembled coiled coil-like fibers, Sci. Adv., 2021, 7, eabd0492 CrossRef CAS PubMed.
- K. Hu, H. Geng, Q. Zhang, Q. Liu, M. Xie, C. Sun, W. Li, H. Lin, F. Jiang, T. Wang, Y. D. Wu and Z. Li, An In-tether Chiral Center Modulates the Helicity, Cell Permeability, and Target Binding Affinity of a Peptide, Angew Chem. Int. Ed. Engl., 2016, 55, 8013–8017 CrossRef CAS PubMed.
- Y. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, CH/πInteraction in the Crystal Structure of Organic Compounds. A Database Study, Bull. Chem. Soc. Jpn., 1998, 71, 1207–1213 CrossRef CAS.
- U. Koch and P. L. A. Popelier, Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density, J. Phys. Chem., 2002, 99, 9747–9754 CrossRef.
- P. L. A. Popelier, Characterization of a Dihydrogen Bond on the Basis of the Electron Density, J. Phys. Chem. A, 1998, 102, 1873–1878 CrossRef CAS.
- P. S. V. Kumar, V. Raghavendra and V. Subramanian, Bader's Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS.
- H. K. Ganguly, B. Majumder, S. Chattopadhyay, P. Chakrabarti and G. Basu, Direct Evidence for CH···π Interaction Mediated Stabilization of Pro-cisPro Bond in Peptides with Pro-Pro-Aromatic motifs, J. Am. Chem. Soc., 2012, 134, 4661–4669 CrossRef CAS PubMed.
- F. D. Suvire, L. N. Santagata, J. A. Bombasaro and R. D. Enriz, Dynamics of flexible cycloalkanes. Ab initio and DFT study of the conformational energy hypersurface of cyclononane, J. Comput. Chem., 2006, 27, 188–202 CrossRef CAS.
- D. B. Diaz, S. D. Appavoo, A. F. Bogdanchikova, Y. Lebedev, T. J. McTiernan, G. Dos Passos Gomes and A. K. Yudin, Illuminating the dark conformational space of macrocycles using dominant rotors, Nat. Chem., 2021, 13, 218–225 CrossRef CAS PubMed.
- D. F. Dourado, P. A. Fernandes and M. J. Ramos, Glutathione transferase classes alpha, pi, and mu: GSH activation mechanism, J. Phys. Chem. B, 2010, 114, 12972–12980 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2292337, 2292338, 2292339, and 2292343. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02545h |
| ‡ J. S., Z. T., and J. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |