
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphite conjugated nickel phthalocyanine for efficient CO2 electroreduction and Zn–CO2 batteries†
Jingwei
Han
a,
Qiang
Xu
a,
Fengkun
Tian
a,
Hai
Sun
a,
Yuanyuan
Qi
a,
Guodong
Zhang
 b,
Jun-Sheng
Qin
a and
Heng
Rao
*a
b,
Jun-Sheng
Qin
a and
Heng
Rao
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, International Center of Future Science, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China. E-mail: rao@jlu.edu.cn
bSchool of Chemistry and Chemical Engineering, Yangzhou University, Siwangting Road 180, Yangzhou, P. R. China
First published on 30th August 2024
Abstract
The linking chemistry between molecular catalysts and substrates is a crucial challenge for enhancing electrocatalytic performance. Herein, we elucidate the influence of various immobilization methods of amino-substituted Ni phthalocyanine catalysts on their electrocatalytic CO2 reduction reaction (eCO2RR) activity. A graphite-conjugated Ni phthalocyanine, Ni(NH2)8Pc-GC, demonstrates remarkable electrocatalytic performance both in H-type and flow cells. In situ infrared spectroscopy and theoretical calculations reveal that the graphite conjugation, through strong electronic coupling, increases the electron density of the active site, reduces the adsorption energy barrier of *COOH, and enhances the catalytic performance. As the cathode catalyst, Ni(NH2)8Pc-GC also displays remarkable charge–discharge cycle stability of over 50 hours in a Zn–CO2 battery. These findings underscore the significance of immobilization methods and highlight the potential for further advancements in eCO2RR.
Introduction
Utilizing renewable electricity to convert CO2, the primary greenhouse gas, into high-value-added fuels and chemicals presents substantial potential within the industrial landscape. Among the numerous electrocatalytic materials displaying high performance for CO2 conversion, stable transition metal porphyrin and phthalocyanine molecular complexes have garnered extensive attention for CO2 reduction.1,2 These complexes boast well-defined active sites and adjustable ligand structures, endowing them with distinct advantages in the realm of electrocatalytic CO2 reduction.3,4 However, the reported turnover numbers (TONs) and turnover frequencies (TOFs) for these molecular catalysts remain relatively low, which is far from the requirements of industrial application. Their catalytic selectivity and stability also encounter significant challenges. Therefore, designing and constructing transition metal-based porphyrin or phthalocyanine catalysts that can operate efficiently and reliably under industrial conditions is crucial for achieving the vision of carbon neutrality.Functional group modification strategies have been successfully employed to enhance the electrocatalytic performance of molecular catalysts. It has been observed that electron-withdrawing groups facilitate electron transfer by reducing electron density while simultaneously impeding proton transfer. Conversely, electron-donating groups promote proton transfer but increase the energy barrier of electron transfer.5 Therefore, it is imperative for functional group modification to strike a delicate balance between electron-withdrawing and electron-donating abilities. As a classical electron-donating group, the amino group (–NH2) offers a unique advantage due to its protonation potential, allowing for tunable electron density.6,7 For instance, Wang and colleagues reported that electron-donating amino substituents on the phthalocyanine ring significantly enhanced the stability of their catalytic system.8 Zheng and colleagues developed a donor–acceptor-modified copper porphyrin, wherein amino groups acted as electron donors to induce electron transfer to CuN4 sites, resulting in outstanding CO2-to-CH4 electroreduction performance.9 However, these molecular catalysts were directly dispersed and physically adsorbed onto the surface of the conductive carbon materials, which is easily deactivated and unsatisfied with the conditions for industrial applications.
Heterogenization of molecular catalysts is another enticing option, offering the potential for elevated current density and enhanced stability through the facilitation of charge transfer. Generally, this is realized through weak physical adsorption or electrostatic interactions that promote π–π stacking between catalysts and carbon supports.10 Unfortunately, the non-covalently attached catalysts may encounter challenges, chiefly pertaining to aggregation at high loadings or leaching issues.11,12 These complications are primarily attributed to the self-stacking effect and the frail connection between the molecular catalyst and its supporting substrate, as reflected by their poor long-term current stability.13,14 Therefore, covalent linkage employs diverse methods, including amide bonds,15,16 alkynyl linkages,17 diazo graftings,18 click reactions,19 and substitution reactions,20 to mitigate the prevalent predicament of self-stacking in molecule catalysts. Nevertheless, it is imperative to recognize that the electronic coupling resultant from such tethers may tend to be feeble, thereby introducing a higher barrier for electron transfer. This, in turn, restricts the rate of electron flow to the active site, curtailing overall catalytic efficiency.21–23 The aromatic linkages with highly delocalized electrons also significantly influence the electronic properties of the active site.24,25 Surendranath and colleagues demonstrated that graphite-conjugated catalysts (GCCs) greatly enhanced the oxygen reduction activity of molecular catalysts in alkaline media.26,27 Furthermore, GCCs containing Re complexes also exhibited efficient and selective reduction of CO2 to CO with quite low overpotential.28 The fundamental rationale behind this phenomenon lies in the fact that GCCs anchored via conductive pyrazine bonds exhibit the inner-sphere electron transfer (ISET) reaction, akin to metallic active sites.29,30 This strong electronic coupling significantly promotes the electron transfer between the active sites of the molecular catalyst and the carrier.
Herein, we engineer an amino substituted nickel phthalocyanine as transition-metal active sites for CO2 reduction on the edge of graphite planes. This was achieved by condensation of an organic 1, 2-diamine with ortho-quinone moieties native to graphitic carbon surfaces to form a conjugated pyrazine linkage. As a result, the eCO2RR performance of Ni(NH2)8Pc-GC (Ni(NH2)8Pc = (2,3,9,10,16,17,23,24-octaaminophthalocyaninato) nickel(II), GC = graphitic carbon) is dramatically improved compared to catalysts prepared via the non-conjugated covalent method or physical mixing method. Encouragingly, the graphite conjugated catalyst exhibits close to 100% CO selectivity over a wide potential window, in both H-type cell and flow cell setups. In the flow cell, Ni(NH2)8Pc-GC showcases turnover frequency for CO (TOFCO) exceeding 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and half-cell energy conversion efficiencies of 63% at an overpotential of 775 mV. Furthermore, we assemble a Zn–CO2 battery utilizing Ni(NH2)8Pc-GC as the cathode catalyst, which exhibits a peak power density of 0.73 mW cm−2 and an faradaic efficiency of CO (FECO) of 98% during the discharge process. This provides a promising approach to electric energy output and chemical conversion.
000 h−1 and half-cell energy conversion efficiencies of 63% at an overpotential of 775 mV. Furthermore, we assemble a Zn–CO2 battery utilizing Ni(NH2)8Pc-GC as the cathode catalyst, which exhibits a peak power density of 0.73 mW cm−2 and an faradaic efficiency of CO (FECO) of 98% during the discharge process. This provides a promising approach to electric energy output and chemical conversion.
Results and discussion
Synthesis and characterization of catalysts
The synthesis involved the condensation of o-phenylenediamine-containing phthalocyanine synthon, Ni(NH2)8Pc, with o-quinone moieties present on the edge planes of graphitic carbon using a modified literature procedure (Scheme 1).27,31 Specifically, after mixing the dimethyl sulfoxide (DMSO) solution of Ni(NH2)8Pc with the ethanol (EtOH) solution of graphite carbon in the absence of O2, the resulting mixture was reacted at 130 °C for 24 hours under argon protection. The crude product was then washed with DMSO and EtOH to eliminate any unsupported Ni(NH2)8Pc. Subsequently, the powder was treated with 0.1 M hydrochloric acid for 4 hours to hydrolyze the imine bonds formed on the surface. For comparison, we also prepared a nickel phthalocyanine modified graphitic carbon through non-conjugated amide bonding (Ni(NH2)4Pc-GC, Ni(NH2)4Pc = (tetraaminophthalocyaninato) nickel(II)). Initially, graphite carbon was treated with a mixed solution of thionyl chloride (SOCl2) and toluene, facilitating the conversion of carboxyl groups (–COOH) on its surface into acid chloride groups (–COOCl). Subsequently, the treated graphitic carbon and Ni(NH2)4Pc were dispersed in a DMSO solution and reacted under an argon atmosphere at 150 °C for 24 hours. Following the reaction, the product was washed with DMSO and EtOH, yielding Ni(NH2)4Pc-GC. The full synthesis details are described in the ESI.†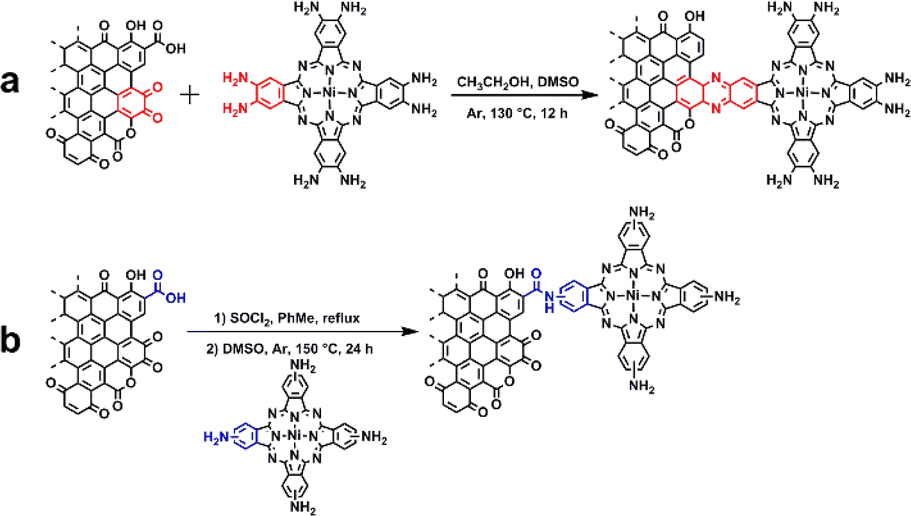 | ||
| Scheme 1 Synthesis of (a) Ni(NH2)8Pc-GC and (b) Ni(NH2)4Pc-GC. | ||
The prepared graphitic carbon catalysts were washed three times with DMSO to remove physisorbed active sites until the UV-visible spectra showed no absorption peaks corresponding to the Q-band of the nickel phthalocyanines (Fig. S1†). X-ray photoelectron spectroscopy (XPS) reveals the successful incorporation of phthalocyanine units onto the surface of the graphitic carbon substrate (Fig. 1a and b). In the case of Ni(NH2)8Pc-GC, the N 1s spectrum is deconvoluted into three distinct types of N species with a ratio of 4:10:1, corresponding to N–C (400.2 eV), N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (398.8 eV), and pyrazine N (399.8 eV). Regarding Ni(NH2)4Pc-GC, the peak centered at 397.5 eV is attributed to –NH–C. Notably, the Ni 2p3/2 spectrum of Ni(NH2)8Pc-GC exhibits a shift towards lower binding energy compared to that of Ni(NH2)4Pc-GC. This shift is attributed to the transfer of electrons from the graphitic carbon to the nickel phthalocyanine. These results substantiate the notion of enhanced electron transfer kinetics between conjugated active sites and carbon substrate, which also aligns with the subsequent DFT calculation results. Considered together, the XPS data support the formation of isolated nickel phthalocyanine active sites conjugated to the surface via pyrazine or non-conjugated to the surface via amide linkages. Furthermore, Raman spectra were employed to gain deeper insights into the structure and molecular interaction of the catalyst. As depicted in Fig. 1c, both Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC display similar Raman signals to graphitic carbon, indicating that the covalent immobilization process preserves the structural integrity of the graphitic carbon. The peak centered at ∼1330 cm−1 is attributed to the D band, the intensity of which is used to quantify defect concentration. The peak around 1590 cm−1 corresponds to the G band, reflecting the degree of graphitization.32,33 The ratio of the D to G peak intensity (ID/IG) of the molecule-modified carbon material exhibits a slight increase compared to the pristine graphitic carbon. This increase may arise from the destruction of the surface lattice of the carbon material after undergoing molecular covalent grafting.32,34 The Raman spectra and X-ray diffraction (XRD) patterns (Fig. S2†) of catalysts are dominated by graphitic carbon, which obscures the weaker nickel phthalocyanine characteristic signals. This observation is related to either a high dispersion or a low molecular loading of phthalocyanine on the graphitic carbon surface.35
C (398.8 eV), and pyrazine N (399.8 eV). Regarding Ni(NH2)4Pc-GC, the peak centered at 397.5 eV is attributed to –NH–C. Notably, the Ni 2p3/2 spectrum of Ni(NH2)8Pc-GC exhibits a shift towards lower binding energy compared to that of Ni(NH2)4Pc-GC. This shift is attributed to the transfer of electrons from the graphitic carbon to the nickel phthalocyanine. These results substantiate the notion of enhanced electron transfer kinetics between conjugated active sites and carbon substrate, which also aligns with the subsequent DFT calculation results. Considered together, the XPS data support the formation of isolated nickel phthalocyanine active sites conjugated to the surface via pyrazine or non-conjugated to the surface via amide linkages. Furthermore, Raman spectra were employed to gain deeper insights into the structure and molecular interaction of the catalyst. As depicted in Fig. 1c, both Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC display similar Raman signals to graphitic carbon, indicating that the covalent immobilization process preserves the structural integrity of the graphitic carbon. The peak centered at ∼1330 cm−1 is attributed to the D band, the intensity of which is used to quantify defect concentration. The peak around 1590 cm−1 corresponds to the G band, reflecting the degree of graphitization.32,33 The ratio of the D to G peak intensity (ID/IG) of the molecule-modified carbon material exhibits a slight increase compared to the pristine graphitic carbon. This increase may arise from the destruction of the surface lattice of the carbon material after undergoing molecular covalent grafting.32,34 The Raman spectra and X-ray diffraction (XRD) patterns (Fig. S2†) of catalysts are dominated by graphitic carbon, which obscures the weaker nickel phthalocyanine characteristic signals. This observation is related to either a high dispersion or a low molecular loading of phthalocyanine on the graphitic carbon surface.35
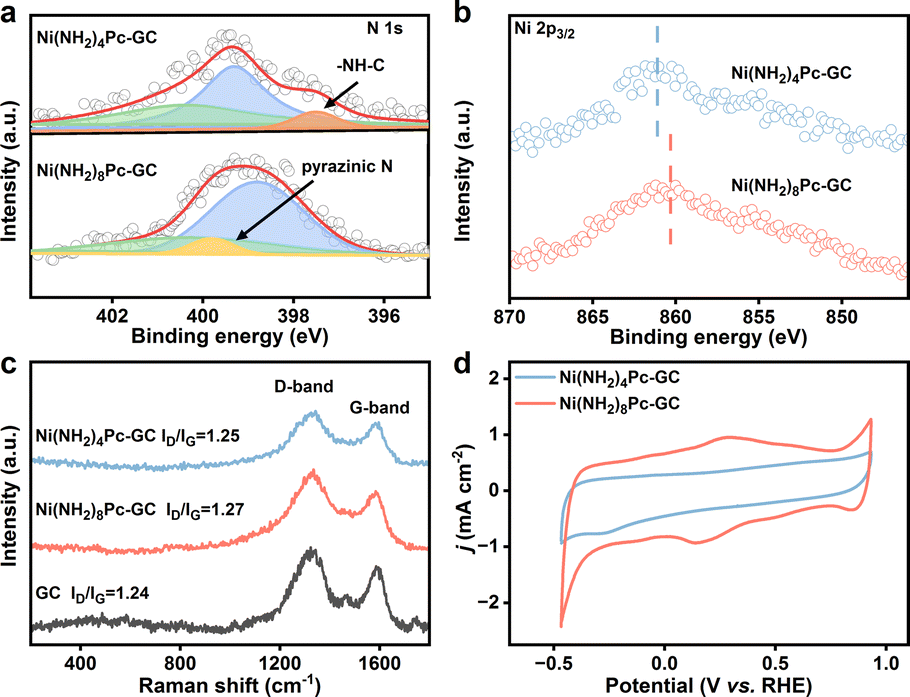 | ||
| Fig. 1 (a) N 1s XPS spectra of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC. (b) Ni 2p3/2 XPS spectra of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC. (c) Raman spectra of Ni(NH2)8Pc-GC, Ni(NH2)4Pc-GC and GC. (d) CV curves in Ar-saturated 0.5 M KHCO3 solution with a scan rate of 10 mV s−1 of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC. | ||
The concentration of the conjugated Ni centers was quantified by cyclic voltammetry (CV) and inductively coupled plasma optical emission spectroscopy (ICP-OES), with detailed methodologies available in the ESI.† Considering the instability of carbon materials and the activation process of the catalyst during potential cycling (Fig. S3†), we selected the stabilized fourth CV curve for integrating to determine the charge passed in the pyrazine wave. This approach enables an in situ and nondestructive measurement of the surface concentration of nickel active sites that are conjugated to the electrode. As shown in Fig. 1d, the broad redox waves centered at 0.15 V vs. RHE are attributed to the 2e−/2H+ reduction of the surface pyrazine units to dihydropyrazine.28 The results of ICP-OES revealed that the content of Ni in Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC was 0.16 wt% and 0.47 wt%, respectively. Following the method described in the literature,27 the Ni loading on the Ni(NH2)8Pc-GC electrode surface used in the subsequent study was 15.5 nmol cm−2. The Ni content in the electrode prepared with Ni(NH2)4Pc-GC was directly derived from the ICP-OES measurements, which was 39.7 nmol cm−2. Fig. S4, S5, and S6† show the transmission electron microscopy (TEM) images, scanning electron microscopy (SEM) images, and the corresponding element mappings, illustrating the uniform distribution of phthalocyanine molecules on the carbon surface.
Electrochemical reduction of CO2 in H-type cell
The electrocatalytic CO2 reduction performance of the Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC catalysts was initially evaluated in a three-electrode H-type cell with CO2-saturated KHCO3 solution. The linear sweep voltammetry (LSV) curves reveal that both catalysts exhibited higher current densities in the CO2-saturated 0.5 M KHCO3 electrolyte compared to the Ar-saturated electrolyte, indicating that the as-prepared catalysts are active for CO2RR (Fig. 2a). To more accurately assess the intrinsic activity of the catalysts, we also plotted the LSV curves normalized by the amount of substance in a CO2 atmosphere (Fig. S7†). The results indicate that Ni(NH2)8Pc-GC exhibits superior CO2RR activity compared to Ni(NH2)4Pc-GC. Encouraged by the results of the LSV test, a series of controlled potential experiments were performed within the potential range of −0.48 V to −0.93 V vs. RHE. Gaseous and liquid products were analyzed by gas chromatography (GC) and proton nuclear magnetic resonance spectroscopy (1H NMR), respectively. The results indicated that only two gaseous products, CO and H2, were generated at all applied potentials, with no liquid products detected. To investigate the origin of CO, we conducted differential electrochemical mass spectrometry (DEMS) analysis. Our findings reveal that the onset potential for the consumption of CO2 and the generation of CO is the same, indicating that CO is derived from dissolved CO2 in the electrolyte (Fig. 2b and S8†).36 Additionally, the results indicate that Ni(NH2)8Pc-GC exhibits a lower onset potential for CO2-to-CO conversion than Ni(NH2)4Pc-GC (Fig. S9†). The DEMS curves demonstrate a more negative onset potential than the LSV curves, which is attributed to the hysteresis effect of the instrument. Notably, the FECO for Ni(NH2)8Pc-GC achieves nearly 100% in the potential range of −0.68 to −0.88 V vs. RHE (Fig. 2c). The FECO of Ni(NH2)4Pc-GC is only approximately 92%. Moreover, the current density remains stable across all tested potentials (Fig. S10†). According to Fig. S11,† the CO partial current density (jCO) of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC follows a volcano-like trend. The maximum jCO for Ni(NH2)8Pc-GC is 17.6 mA cm−2 at −0.88 V vs. RHE, whereas Ni(NH2)4Pc-GC achieves 15.2 mA cm−2 at the same voltage of −0.88 V vs. RHE. The TOFCO provides further insights into the intrinsic activity of the catalysts. The maximum TOFCO of Ni(NH2)8Pc-GC reaches 6.3 s−1 at −0.88 V vs. RHE, which is 3.2 times that of Ni(NH2)4Pc-GC. For comparison, we also evaluated the eCO2RR performance of physically mixed catalysts (Ni(NH2)8Pc/GC and Ni(NH2)4Pc/GC). In this method, molecular catalysts are anchored on the surface of carbon supports by π–π stacking. The results presented in Fig. S12† indicate that both Ni(NH2)8Pc/GC and Ni(NH2)4Pc/GC exhibit activity for eCO2RR. The i–t curves of Ni(NH2)8Pc/GC and Ni(NH2)4Pc/GC remain steady across all tested potentials (Fig. S13†). However, compared to Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC, both FECO and jCO values are comparatively lower for Ni(NH2)8Pc/GC and Ni(NH2)4Pc/GC (Fig. S14 and S15†). The TOFCO values of Ni(NH2)8Pc/GC and Ni(NH2)4Pc/GC are found to be 1.2 s−1 and 0.8 s−1 at −0.88 V vs. RHE, respectively, as depicted in Fig. S16.† These values are significantly lower than that of Ni(NH2)8Pc-GC. In a word, Ni(NH2)8Pc-GC demonstrates extremely superior CO2RR activity compared with the recently reported Ni-based catalysts (Table S1†). We then investigated the long-term stability of the catalysts at −0.73 V vs. RHE, as revealed in Fig. 2d and S17.† Ni(NH2)8Pc-GC exhibits optimal stability, maintaining a FECO of over 95% in 10 hours. Importantly, the morphology of the catalysts remained intact after the long-term electrolysis (Fig. S18†). This signifies that covalent grafting significantly improves the stability of the catalyst, with the conjugated pyrazine linkage being even more effective than the non-conjugated amide linkage.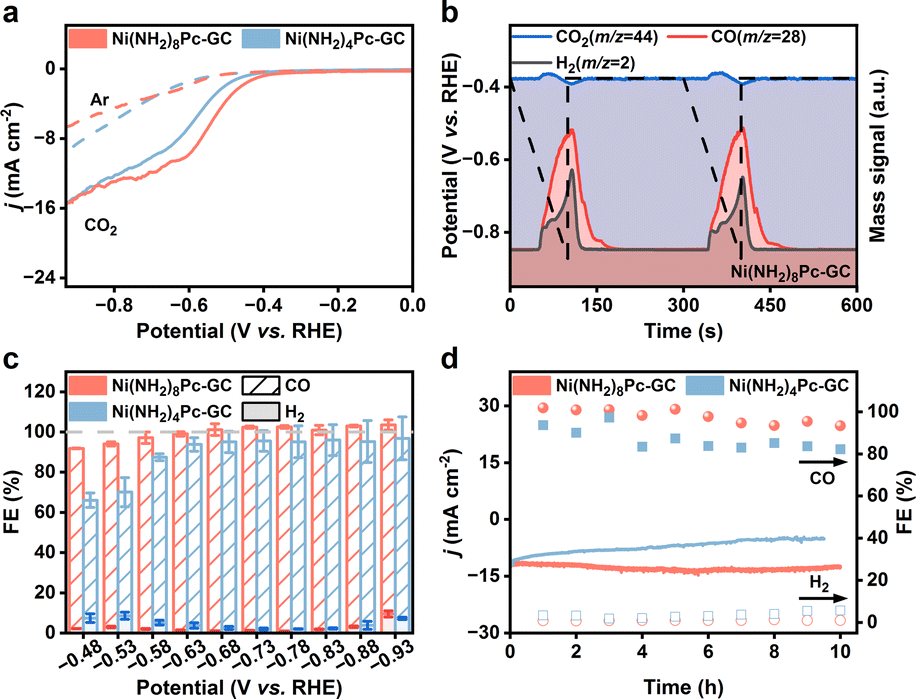 | ||
| Fig. 2 (a) LSV curves in Ar or CO2-saturated 0.5 M KHCO3 solution with a scan rate of 5 mV s−1 and 80% iR compensation (Rs = 7.4 Ω). (b) In situ DEMS spectra of Ni(NH2)8Pc-GC in a CO2-saturated 0.5 M KHCO3 electrolyte. The signals of CO2 (m/z = 44), CO (m/z = 28) and H2 (m/z = 2) were captured twice of LSV from −0.38 to −0.88 V vs. RHE. The dashed line represents the potential profile. (c) FEs of CO and H2 at different applied potentials. The error bars denote the standard deviations (SD) derived from three independent measurements. (d) Long-term stability tests. | ||
Electrokinetic studies were performed to explore the effect of different immobilization methods on the reaction mechanism. As illustrated in Fig. S19,† the Tafel slope for Ni(NH2)8Pc-GC, as fitted with the jCO, is 152 mV dec−1, which is lower than that observed for Ni(NH2)4Pc-GC (179 mV dec−1). In contrast, the Tafel slopes for the physically mixed Ni(NH2)8Pc/GC and Ni(NH2)4Pc/GC are much higher, reaching 194 mV dec−1 and 216 mV dec−1, respectively (Fig. S20†). The reported Tafel slopes for Ni-based catalysts generally fall within the 80–350 mV dec−1 range.33,35,37 The lower Tafel slope for Ni(NH2)8Pc-GC indicates faster reaction kinetics for CO2RR, enhancing CO formation.20,38 Additionally, the Nyquist plots of the catalysts were collected in the range of 10−1 to 105 Hz to compare the charge transfer resistance (Rct) at the catalyst interface. The results, summarized in Fig. S21 and S22,† reveal that the diameters of the semicircles in the high-frequency region for all catalysts are less than 5 Ω, signifying excellent conductivity of the catalysts. Notably, Ni(NH2)8Pc-GC exhibits the smallest Rct and the fastest electron transfer kinetics among all samples. To further understand the intrinsic catalytic activity of the electrocatalysts, the electrochemical active surface area (ECSA) evaluated by comparing the double-layer capacitance (Fig. S23†) was investigated. In the relationship between current density and scan rate (Fig. S24 and S25†), the capacitances for Ni(NH2)8Pc-GC, Ni(NH2)4Pc-GC, Ni(NH2)8Pc/GC, and Ni(NH2)4Pc/GC were calculated to be 21.85, 11.42, 36.98, and 16.94 mF cm−2, respectively. These data underscores that the active area is not the decisive factor affecting the catalytic activity, which is consistent with the results obtained from N2 adsorption–desorption curves (Fig. S26†).
Electrochemical reduction of CO2 in the flow cell
To further assess the potential of the Ni(NH2)8Pc-GC catalyst for industrial applications, we subsequently investigated its electrocatalytic performance in a flow cell setup. A gas diffusion electrode (GDE) was employed to directly transport CO2 to the catalyst surface, effectively overcoming the mass transfer limitation of CO2 in aqueous media. The composition of the flow cell and testing details are described in Fig. S27 and the ESI.† According to the LSV curves (Fig. 3a), the current densities are significantly higher compared to those in the H-type cell. Specifically, Ni(NH2)8Pc-GC achieves a current density of 400 mA cm−2 at −0.6 V vs. RHE, surpassing that of Ni(NH2)4Pc-GC (23 mA cm−2). Furthermore, Ni(NH2)8Pc-GC maintains a stable current density under all applied polarization potentials, reaching an industrial current density of 246 mA cm−2 at −1.03 V vs. RHE (Fig. 3b). The FECO of Ni(NH2)8Pc-GC approaches nearly 100% over a fairly wide potential range from −0.43 V to −1.03 V vs. RHE (Fig. 3c). In contrast, the maximum FECO and jCO for Ni(NH2)4Pc-GC are 95% at −0.43 V vs. RHE and 16.7 mA cm−2 at −0.58 V vs. RHE, respectively. (Fig. S28 and S29†). Long-term stability in the flow cell is also a crucial criterion for practical applicability. In the case of Ni(NH2)8Pc-GC, the constant-current electrolysis was performed at the current density of 100 mA cm−2. It demonstrated stability for at least 8 hours, with no significant increase in potential and an FECO close to 100%. However, after 10 hours of electrolysis, the FECO drops to 71.8% due to flooding and salting out (Fig. 3d).39,40 Replacing the cathode electrolyte with a fresh one restored the potential but could not be maintained for an extended duration, primarily because of the disruption of the gas–liquid balance on the catalyst's surface after prolonged electrolysis in a strong alkaline solution. However, the current density of Ni(NH2)4Pc-GC remains stable only for a short period. Its FECO declines rapidly after one hour of electrolysis (Fig. S30 and S31†). A more intuitive comparison of performance parameters at −0.88 V vs. RHE highlights the advantages of conjugate coupling in CO2RR, as shown in Fig. 3e. Remarkably, Ni(NH2)8Pc-GC achieves a high TOFCO of 342, 296 h−1, a single-pass CO2-to-CO conversion efficiency of 8.9%, a half-cell energy efficiency of 59% for CO production, and a maximum CO yield of 17.8 L min−1 m−2. The performance of Ni(NH2)8Pc-GC in the flow cell is truly exceptional, outperforming most of the recently reported catalysts (Fig. 3f and Table S2†).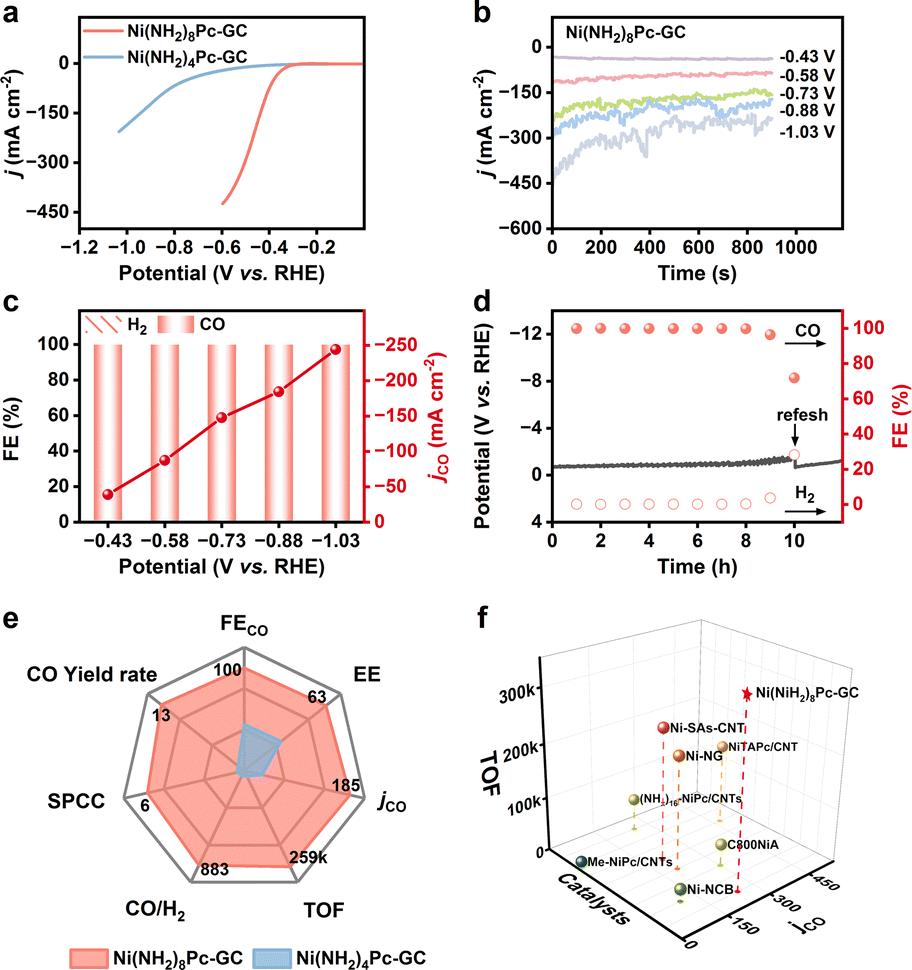 | ||
| Fig. 3 (a) LSV curves with a scan rate of 5 mV s−1 and 80% iR compensation (Rs = 2.0 Ω) of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC. (b) Total current densities at different applied potentials. (c) FEs and jCO of Ni(NH2)8Pc-GC. (d) Long-term chronopotentiometric stability with a current density of 100 mA cm−2 of Ni(NH2)8Pc-GC. (e) Performances comparison between Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC at −0.88 V vs. RHE. (f) Comparis of jCO and TOF for at −1.03 V vs. RHE and recently reported Ni-based catalysts. | ||
Electrocatalysis mechanism
In situ attenuated total reflectance surface-enhanced infrared spectroscopy (ATR-SEIRAS) was performed to investigate the transformation of intermediates at different potentials in a CO2-saturated solution of 0.5 M KHCO3. In the in situ ATR-SEIRAS analysis of Ni(NH2)8Pc-GC, the peak observed at 1640 cm−1 corresponds to the H–O–H bend of the adsorbed H2O molecules (Fig. 4a, b and S32†).41 Additionally, two infrared bands at 2321 and 2350 cm−1 represent the adsorbed CO2.42 The peaks at 1380 and 1545 cm−1 are assigned to the symmetric and asymmetric stretching vibrations of *COOH, respectively.43 As the reaction potential increases, the peak intensity of *COOH gradually increases. Furthermore, the signal observed at 2080 cm−1 is attributed to the Ni–CO vibration, which gradually broadens as the electrolysis potential becomes more negative.43 The presence of *CO and *COOH as crucial intermediates for CO generation aligns with subsequent theoretical calculations.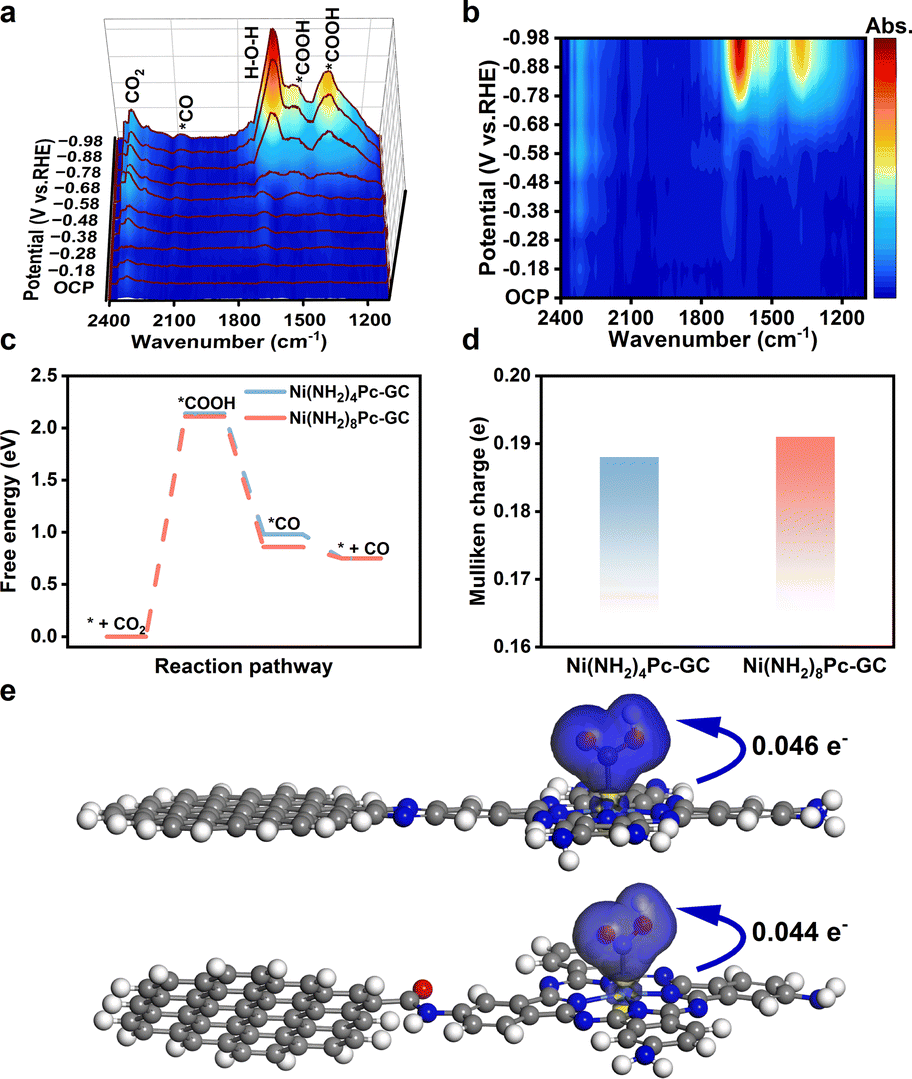 | ||
| Fig. 4 (a) In situ ATR-SEIRAS spectra of Ni(NH2)8Pc-GC at different potentials in CO2-saturated 0.5 M KHCO3. (b) The corresponding 2D colormap surface of (a). (c) Free energy diagrams of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC for CO2RR. (d) Mulliken charges of Ni atom in Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC. Different charge densities of (e) Ni(NH2)8Pc-GC (top) and Ni(NH2)4Pc-GC (bottom) when *COOH adsorbs on Ni site. Color-code atoms represent Ni (lightseagreen), C (gray), N (blue), O (red) and H (white). The isosurface value is 0.05 e Å−3. The color blue represents the charge increase and the color yellow represents the charge decrease. | ||
We also conducted density functional theory (DFT) calculations to deepen our comprehension of the thermodynamic reaction process. Taking into account the periodicity and edge effect of the carbon unit cell,26 we developed a model that incorporated amino-modified phthalocyanine coupled with a 14-ring polycyclic aromatic fragment (Fig. S33†). Based on the intermediates observed through in situ ATR-SEIRAS, this model allowed us to propose the steps required for the CO generation. Firstly, CO2 is adsorbed on the active site, and protons are acquired to form *COOH. Subsequently, the *COOH intermediate undergoes a proton-coupled electron transfer (PCET) reaction, resulting in the generation of the *CO intermediate. Finally, the *CO intermediate desorbs from the catalyst surface. The adsorption states of two critical intermediates on the catalysts Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC in the CO2-to-CO process are outlined in Fig. S34 and S35.† The free energy of the intermediate states on each model during the CO2RR process was calculated. The free energy diagram (Fig. 4c) confirms that the formation of *COOH on the Ni site is endothermic, while the generation of *CO is exothermic. The rate-determining step (RDS) is the formation of *COOH for both Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC, with energy barriers of 2.11 and 2.14 eV, respectively. Moreover, the energy barrier corresponding to Ni(NH2)8Pc-GC (0.86 eV) is significantly lower than that of the CO desorption model for Ni(NH2)4Pc-GC (0.98 eV). Altogether, the conjugated pyrazine linkage activates *COOH and promotes the desorption of *CO, giving rise to the favored synthesis of CO.44
The Mulliken charges of the Ni atom in Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC were analyzed (Fig. 4d). Ni(NH2)8Pc-GC exhibits a higher electron density at the Ni site than Ni(NH2)4Pc-GC, suggesting that the pyrazine linkage facilitates the transfer of electrons from the carbon support to the Ni site.35 The different charge densities were further examined to understand the impact of the electronic density of the Ni site on CO2 activation (Fig. 4e). It was observed that Ni(NH2)8Pc-GC converts more electrons (0.046 e−) to form *COOH, in contrast to Ni(NH2)4Pc-GC (0.044 e−), lowering the energy barrier for the reaction (* + CO2 + H+ + e− → *COOH).33 The presence of the pyrazine linkage facilitates enhanced electron transfer, which also improves the adsorption of *COOH and leads to enhanced activity.
Aqueous rechargeable Zn–CO2 battery
Considering the benefits of metal–CO2 batteries in promoting carbon recycling and affordability, significant attention has been paid to the development of aqueous rechargeable Zn–CO2 batteries.45,46 The principle of an aqueous rechargeable battery is displayed in Fig. 5a. In the experimental setup, a hydrophobic carbon paper with a catalyst loading of 0.5 mg cm−2 was used as the cathode, while a polished Zn foil served as the anode. The catholyte comprised 0.8 M KHCO3, and the anolyte was a mixture of 6 M KOH and 0.2 M Zn(CH3COO)2. In the discharge phase, CO2 is converted into value-added chemicals while generating electricity. Conversely, during the charging process, the cathode undergoes an oxygen evolution reaction (OER). The efficiency of the OER was tested to confirm the bifunctionality of the catalysts. The LSV curves were recorded in 0.8 M KHCO3 over a potential interval of 0.9–2.4 V vs. RHE using a three-electrode system. As displayed in Fig. S36,† Ni(NH2)8Pc-GC demonstrated a lower onset potential (1.81 V) and a higher current density of 51 mA cm−2 at 2.4 V vs. RHE compared to Ni(NH2)4Pc-GC (1.92 V, 39 mA cm−2 at 2.4 V vs. RHE). Furthermore, Ni(NH2)8Pc-GC requires an overpotential of 760 mV to achieve a current density of 10 mA cm−2, which is 110 mV lower than that for Ni(NH2)4Pc-GC. According to Fig. S37,† the Tafel slope for OER on Ni(NH2)8Pc-GC is calculated to be 265.4 mV dec−1, which is much lower than that of Ni(NH2)4Pc-GC, implying that the former achieves faster kinetics in a neutral electrolyte solution.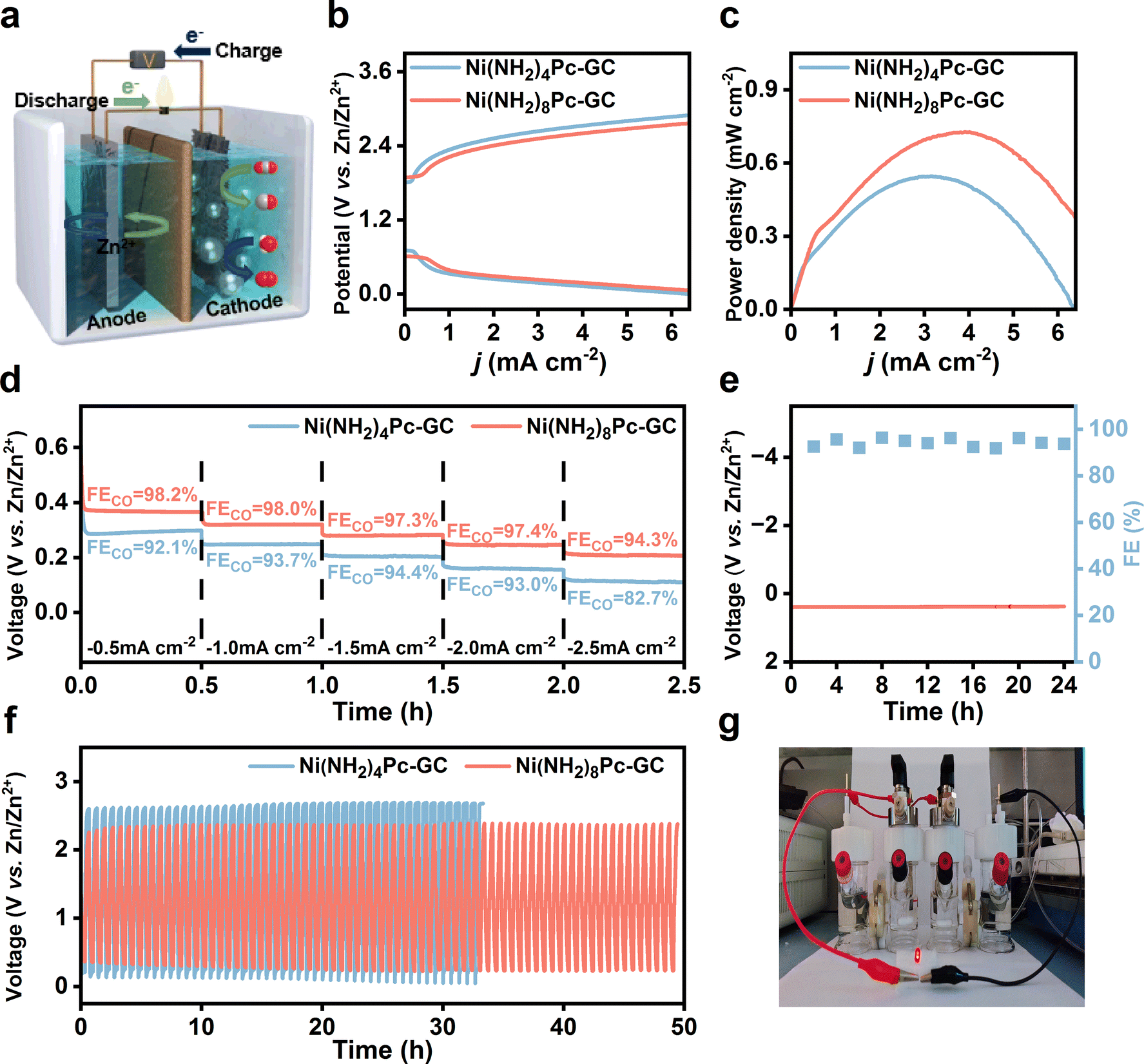 | ||
| Fig. 5 (a) Schematic diagram of aqueous rechargeable Zn–CO2 battery. (b) Charge–discharge polarization curves at a scan rate of 5 mV s−1, (c) power density curves, and (d) discharge curves and corresponding FECO values at different current densities of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC. (e) Prolonged discharge curve and FECO at −0.5 mA cm−2 with Ni(NH2)8Pc-GC cathode. (f) Galvanostatic discharge–charge cycling curves of Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC at ±0.5 mA cm−2. (g) The photograph shows an LED bulb (rated voltage: 1.6 V) powered by two ZCBs connected in series with Ni(NH2)8Pc-GC cathode. | ||
Based on the efficient CO2-to-CO and OER performance, an aqueous rechargeable Zn–CO2 battery was assembled to explore the potential of Ni(NH2)8Pc-GC in energy conversion and storage devices. The details of the battery assembly and possible charging and discharging processes were carefully recorded in the ESI.† As presented in Fig. S38 and S39,† the open circuit potential of the assembled battery reaches 1.6 V. When two batteries are connected in series, the open circuit potential reaches as high as 2.6 V. The charge and discharge polarization curves of the assembled Zn–CO2 battery were tested (Fig. 5b). It was observed that the charge–discharge gaps increased with higher current densities. Notably, the Ni(NH2)8Pc-GC displays a smaller gap, suggesting superior rechargeability for the battery.47–49Fig. 5c shows that the maximum power density of Ni(NH2)8Pc-GC is 0.73 mW cm−2 (at a loading of 0.5 mg cm−2), which is comparable to most values reported in the literature (Table S3†). Fig. 5d illustrates the relationship between discharge voltage and the faradaic efficiency of catalysts at different discharge current densities. At a constant discharge current density of 0.5–2.5 mA cm−2, both Ni(NH2)8Pc-GC and Ni(NH2)4Pc-GC consistently exhibit a FECO above 80%. At the same discharge current density, the discharge voltage of Ni(NH2)8Pc-GC is higher than that of Ni(NH2)4Pc-GC. At a discharge current density of 0.5 mA cm−2, Ni(NH2)8Pc-GC obtains a discharge voltage of 0.37 V and a maximum FECO of 98.2%. Therefore, we conducted a long-term discharge test and a galvanostatic charge–discharge cycle test at a current density of 0.5 mA cm−2 to assess the stability of the Zn–CO2 battery (Fig. 5e, f and S40†). During a 24 hour long-term discharge test of Ni(NH2)8Pc-GC, the discharge potential and FECO remain stable, with only a minor attenuation. The potential gap of Ni(NH2)8Pc-GC increases from 2 V to 2.17 V after 50 hours of charge–discharge cycling. However, the potential gap of Ni(NH2)4Pc-GC increases from 2.52 V to 2.72 V after only 32 hours of cycling, and its discharge potential decays to nearly 0 V. Frustratingly, ZnO precipitation in the anolyte and on the membrane is inevitable, which increases potential gap. Additionally, as a practical application demonstration, two batteries connected in series are shown to power a red LED bulb (1.6 V, 20 mA) (Fig. 5g). Therefore, aqueous rechargeable Zn–CO2 batteries are promising in alleviating energy and environmental challenges.
Conclusions
In conclusion, this study investigated the effect of different incorporation modes between amino-substituted Ni phthalocyanine and graphitic carbon surfaces for the electrocatalytic CO2RR. In both the H-cell and the flow cell, Ni(NH2)8Pc-GC consistently achieves a FECO of nearly 100% over a fairly wide potential range, demonstrating superior catalytic performance compared to Ni(NH2)4Pc-GC. Especially in the flow cell, Ni(NH2)8Pc-GC exhibits an exceptional TOF of 342, 296 h−1 at −1.03 V. Furthermore, in the rechargeable aqueous Zn–CO2 battery assembled with Ni(NH2)8Pc-GC, a FECO of 94.3% is obtained at a discharge current density of 2.5 mA cm−2, successfully demonstrating the utilization of CO2 fixing in the field of energy storage. Both experimental data and theoretical calculations consistently support the notion that graphite conjugated coupling accelerates the electron transfer kinetics between carbon supports and molecular catalysts, thereby increasing the electron density of Ni center. This study paves the way for the rational design of heterogeneous catalysts with enhanced CO2RR activity, providing a promising avenue for the utilization of CO2 in energy-related applications.Data availability
All experimental and computational procedures and associated data are provided in the ESI.†Author contributions
J. H. and H. R. conceived the project. J. H. prepared materials, performed measurements and analysed the data. Q. X. performed in situ ATR-SEIRAS analysis. F. T. performed in situ DEMS analysis. H. S., Y. Q., G. Z., and J. Q. helped with some of the experiments and characterization. All authors discussed the results and commented on the manuscript. J. H. and H. R. wrote the draft, revised and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Natural Science Foundation of Jilin Province (No. SKL202302017), the National Natural Science Foundation of China (No. 21901084, 21905106 and 22279041), the National Key Research and Development Program of China (No. 2022YFC2105800), the Specific Research Fund of the Innovation Platform for Academicians of Hainan Province, China (YSPTZX202321) and the 111 Project (B17020).References
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Metallophthalocyanine-based molecular materials as catalysts for electrochemical reactions, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS.
- G. F. Manbeck and E. Fujita, A review of iron and cobalt porphyrins, phthalocyanines and related complexes for electrochemical and photochemical reduction of carbon dioxide, J. Porphyrins Phthalocyanines, 2015, 19, 45–64 CrossRef CAS.
- K. Kosugi, M. Kondo and S. Masaoka, Quick and Easy Method to Dramatically Improve the Electrochemical CO2 Reduction Activity of an Iron Porphyrin Complex, Angew. Chem., Int. Ed., 2021, 60, 22070–22074 CrossRef CAS PubMed.
- Q. Chang, Y. Liu, J.-H. Lee, D. Ologunagba, S. Hwang, Z. Xie, S. Kattel, J. H. Lee and J. G. Chen, Metal-Coordinated Phthalocyanines as Platform Molecules for Understanding Isolated Metal Sites in the Electrochemical Reduction of CO2, J. Am. Chem. Soc., 2022, 144, 16131–16138 CrossRef CAS.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X.-a. Zhang and H. B. Kraatz, Enhanced Electrochemical Reduction of CO2 Catalyzed by Cobalt and Iron Amino Porphyrin Complexes, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- K. Chen, M. Cao, Y. Lin, J. Fu, H. Liao, Y. Zhou, H. Li, X. Qiu, J. Hu, X. Zheng, M. Shakouri, Q. Xiao, Y. Hu, J. Li, J. Liu, E. Cortés and M. Liu, Ligand Engineering in Nickel Phthalocyanine to Boost the Electrocatalytic Reduction of CO2, Adv. Funct. Mater., 2021, 32, 2111322 CrossRef.
- T. Zhang, W. Li, K. Huang, H. Guo, Z. Li, Y. Fang, R. M. Yadav, V. Shanov, P. M. Ajayan, L. Wang, C. Lian and J. Wu, Regulation of functional groups on graphene quantum dots directs selective CO2 to CH4 conversion, Nat. Commun., 2021, 12, 5265 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Domino electroreduction of CO2 to methanol on a molecular catalyst, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- P. Yu, X. Lv, Q. Wang, H. Huang, W. Weng, C. Peng, L. Zhang and G. Zheng, Promoting Electrocatalytic CO2 Reduction to CH4 by Copper Porphyrin with Donor–Acceptor Structures, Small, 2022, 19, 2205730 CrossRef PubMed.
- M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, Medium Effects Are as Important as Catalyst Design for Selectivity in Electrocatalytic Oxygen Reduction by Iron–Porphyrin Complexes, J. Am. Chem. Soc., 2015, 137, 4296–4299 CrossRef CAS.
- Y. Y. Birdja, R. E. Vos, T. A. Wezendonk, L. Jiang, F. Kapteijn and M. T. M. Koper, Effects of Substrate and Polymer Encapsulation on CO2 Electroreduction by Immobilized Indium(III) Protoporphyrin, ACS Catal., 2018, 8, 4420–4428 CrossRef CAS.
- W. W. Kramer and C. C. L. McCrory, Polymer coordination promotes selective CO2 reduction by cobalt phthalocyanine, Chem. Sci., 2016, 7, 2506–2515 RSC.
- A. Maurin and M. Robert, Noncovalent Immobilization of a Molecular Iron-Based Electrocatalyst on Carbon Electrodes for Selective, Efficient CO2-to-CO Conversion in Water, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS PubMed.
- M. Wang, L. Chen, T. C. Lau and M. Robert, A Hybrid Co Quaterpyridine Complex/Carbon Nanotube Catalytic Material for CO2 Reduction in Water, Angew. Chem., Int. Ed., 2018, 57, 7769–7773 CrossRef CAS PubMed.
- A. Maurin and M. Robert, Catalytic CO2-to-CO conversion in water by covalently functionalized carbon nanotubes with a molecular iron catalyst, Chem. Commun., 2016, 52, 12084–12087 RSC.
- S. Gu, A. N. Marianov and Y. Jiang, Covalent grafting of cobalt aminoporphyrin-based electrocatalyst onto carbon nanotubes for excellent activity in CO2 reduction, Appl. Catal., B, 2022, 300, 120750 CrossRef CAS.
- M. V. Sheridan, K. Lam and W. E. Geiger, An Anodic Method for Covalent Attachment of Molecules to Electrodes through an Ethynyl Linkage, J. Am. Chem. Soc., 2013, 135, 2939–2942 CrossRef CAS PubMed.
- A. N. Marianov and Y. Jiang, Covalent ligation of Co molecular catalyst to carbon cloth for efficient electroreduction of CO2 in water, Appl. Catal., B, 2019, 244, 881–888 CrossRef CAS.
- S. A. Yao, R. E. Ruther, L. Zhang, R. A. Franking, R. J. Hamers and J. F. Berry, Covalent Attachment of Catalyst Molecules to Conductive Diamond: CO2 Reduction Using “Smart” Electrodes, J. Am. Chem. Soc., 2012, 134, 15632–15635 CrossRef CAS.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y. F. Han, Covalently Grafting Cobalt Porphyrin onto Carbon Nanotubes for Efficient CO2 Electroreduction, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- T. Fukushima, W. Drisdell, J. Yano and Y. Surendranath, Graphite-Conjugated Pyrazines as Molecularly Tunable Heterogeneous Electrocatalysts, J. Am. Chem. Soc., 2015, 137, 10926–10929 CrossRef CAS PubMed.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Conjugated organic framework with three-dimensionally ordered stable structure and delocalized π clouds, Nat. Commun., 2013, 4, 3736 Search PubMed.
- M. Wang, M. Wang, H.-H. Lin, M. Ballabio, H. Zhong, M. Bonn, S. Zhou, T. Heine, E. Cánovas, R. Dong and X. Feng, High-Mobility Semiconducting Two-Dimensional Conjugated Covalent Organic Frameworks with p-Type Doping, J. Am. Chem. Soc., 2020, 142, 21622–21627 CrossRef CAS.
- Z. Meng, R. M. Stolz and K. A. Mirica, Two-Dimensional Chemiresistive Covalent Organic Framework with High Intrinsic Conductivity, J. Am. Chem. Soc., 2019, 141, 11929–11937 CrossRef CAS PubMed.
- Y. Zhang, X. Zhang, L. Jiao, Z. Meng and H.-L. Jiang, Conductive Covalent Organic Frameworks of Polymetallophthalocyanines as a Tunable Platform for Electrocatalysis, J. Am. Chem. Soc., 2023, 145, 24230–24239 CrossRef CAS PubMed.
- N. D. Ricke, A. T. Murray, J. J. Shepherd, M. G. Welborn, T. Fukushima, T. Van Voorhis and Y. Surendranath, Molecular-Level Insights into Oxygen Reduction Catalysis by Graphite-Conjugated Active Sites, ACS Catal., 2017, 7, 7680–7687 CrossRef CAS.
- C. J. Kaminsky, J. Wright and Y. Surendranath, Graphite-Conjugation Enhances Porphyrin Electrocatalysis, ACS Catal., 2019, 9, 3667–3671 CrossRef CAS.
- S. Oh, J. R. Gallagher, J. T. Miller and Y. Surendranath, Graphite-Conjugated Rhenium Catalysts for Carbon Dioxide Reduction, J. Am. Chem. Soc., 2016, 138, 1820–1823 CrossRef CAS.
- M. N. Jackson and Y. Surendranath, Molecular Control of Heterogeneous Electrocatalysis through Graphite Conjugation, Acc. Chem. Res., 2019, 52, 3432–3441 CrossRef CAS PubMed.
- M. N. Jackson, S. Oh, C. J. Kaminsky, S. B. Chu, G. Zhang, J. T. Miller and Y. Surendranath, Strong Electronic Coupling of Molecular Sites to Graphitic Electrodes via Pyrazine Conjugation, J. Am. Chem. Soc., 2018, 140, 1004–1010 CrossRef CAS PubMed.
- M. N. Jackson, M. L. Pegis and Y. Surendranath, Graphite-Conjugated Acids Reveal a Molecular Framework for Proton-Coupled Electron Transfer at Electrode Surfaces, ACS Cent. Sci., 2019, 5, 831–841 CrossRef CAS PubMed.
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Metallic tin quantum sheets confined in graphene toward high-efficiency carbon dioxide electroreduction, Nat. Commun., 2016, 7, 12697 CrossRef CAS PubMed.
- Z. Ma, X. Zhang, X. Han, D. Wu, H. Wang, Z. Gao, F. Xu and K. Jiang, Synergistic adsorption and activation of nickel phthalocyanine anchored onto ketjenblack for CO2 electrochemical reduction, Appl. Surf. Sci., 2021, 538, 148134 CrossRef CAS.
- J. Su, J.-J. Zhang, J. Chen, Y. Song, L. Huang, M. Zhu, B. I. Yakobson, B. Z. Tang and R. Ye, Building a stable cationic molecule/electrode interface for highly efficient and durable CO2 reduction at an industrially relevant current, Energy Environ. Sci., 2021, 14, 483–492 RSC.
- K. Chen, M. Cao, G. Ni, S. Chen, H. Liao, L. Zhu, H. Li, J. Fu, J. Hu, E. Cortés and M. Liu, Nickel polyphthalocyanine with electronic localization at the nickel site for enhanced CO2 reduction reaction, Appl. Catal., B, 2022, 306, 121093 CrossRef CAS.
- H. Kim, D. Shin, W. Yang, D. H. Won, H.-S. Oh, M. W. Chung, D. Jeong, S. H. Kim, K. H. Chae, J. Y. Ryu, J. Lee, S. J. Cho, J. Seo, H. Kim and C. H. Choi, Identification of Single-Atom Ni Site Active toward Electrochemical CO2 Conversion to CO, J. Am. Chem. Soc., 2021, 143, 925–933 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Molecular engineering of dispersed nickel phthalocyanines on carbon nanotubes for selective CO2 reduction, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- C. Hu, Y. Zhang, A. Hu, Y. Wang, X. Wei, K. Shen, L. Chen and Y. Li, Near- and Long-Range Electronic Modulation of Single Metal Sites to Boost CO2 Electrocatalytic Reduction, Adv. Mater., 2023, 35, 2209298 CrossRef CAS.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction, Adv. Mater., 2018, 30, 1803111 CrossRef.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Structural defects on converted bismuth oxide nanotubes enable highly active electrocatalysis of carbon dioxide reduction, Nat. Commun., 2019, 10, 2807 CrossRef PubMed.
- C. Wang, Y. Liu, H. Ren, Q. Guan, S. Chou and W. Li, Diminishing the Uncoordinated N Species in Co-N-C Catalysts toward Highly Efficient Electrochemical CO2 Reduction, ACS Catal., 2022, 12, 2513–2521 CrossRef CAS.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, Highly Selective CO2 Electroreduction to C2H4 Using a Metal–Organic Framework with Dual Active Sites, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- Y. L. Yang, Y. R. Wang, L. Z. Dong, Q. Li, L. Zhang, J. Zhou, S. N. Sun, H. M. Ding, Y. Chen, S. L. Li and Y. Q. Lan, A Honeycomb-Like Porous Crystalline Hetero-Electrocatalyst for Efficient Electrocatalytic CO2 Reduction, Adv. Mater., 2022, 34, 2206706 CrossRef CAS.
- M. Wang, Y. Yao, Y. Tian, Y. Yuan, L. Wang, F. Yang, J. Ren, X. Hu, F. Wu, S. Zhang, J. Wu and J. Lu, Atomically Dispersed Manganese on Carbon Substrate for Aqueous and Aprotic CO2 Electrochemical Reduction, Adv. Mater., 2023, 35, 2210658 CrossRef CAS.
- F. Wang, Y. Li, X. Xia, W. Cai, Q. Chen and M. Chen, Metal-CO2 Electrochemistry: From CO2 Recycling to Energy Storage, Adv. Energy Mater., 2021, 11, 2100667 CrossRef CAS.
- C. J. Fetrow, C. Carugati, X.-D. Zhou and S. Wei, Electrochemistry of metal-CO2 batteries: Opportunities and challenges, Energy Storage Mater., 2022, 45, 911–933 CrossRef.
- S. Liu, M. Jin, J. Sun, Y. Qin, S. Gao, Y. Chen, S. Zhang, J. Luo and X. Liu, Coordination environment engineering to boost electrocatalytic CO2 reduction performance by introducing boron into single-Fe-atomic catalyst, Chem. Eng. J., 2022, 437, 135294 CrossRef CAS.
- Y. Zhang, X. Wang, S. Zheng, B. Yang, Z. Li, J. Lu, Q. Zhang, N. M. Adli, L. Lei, G. Wu and Y. Hou, Hierarchical Cross-Linked Carbon Aerogels with Transition Metal-Nitrogen Sites for Highly Efficient Industrial-Level CO2 Electroreduction, Adv. Funct. Mater., 2021, 31, 2104377 CrossRef CAS.
- W. Zheng, Y. Wang, L. Shuai, X. Wang, F. He, C. Lei, Z. Li, B. Yang, L. Lei, C. Yuan, M. Qiu, Y. Hou and X. Feng, Highly Boosted Reaction Kinetics in Carbon Dioxide Electroreduction by Surface-Introduced Electronegative Dopants, Adv. Funct. Mater., 2021, 31, 2008146 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02682a |
| This journal is © The Royal Society of Chemistry 2024 |