
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking flavin photoacid catalysis through electrophotochemistry†
Samuel
Gary
a,
Jack
Woolley
 b,
Sofia
Goia
c and
Steven
Bloom
*a
b,
Sofia
Goia
c and
Steven
Bloom
*a
aDepartment of Medicinal Chemistry, University of Kansas, Lawrence, 66045, USA. E-mail: spbloom@ku.edu
bDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
cForensic Centre for Digital Scanning and 3D Printing, WMG, University of Warwick, Coventry, CV4 7AL, UK
First published on 18th June 2024
Abstract
Molecular flavins are one of the most versatile photocatalysts. They can coordinate single and multiple electron transfer processes, gift hydrogen atoms, form reversible covalent linkages that support group transfer mechanisms, and impart photonic energy to ground state molecules, priming them for downstream reactions. But one mechanism that has not featured extensively is the ability of flavins to act as photoacids. Herein, we disclose our proof-of-concept studies showing that electrophotochemistry can transform fully oxidized flavin quinones to super-oxidized flavinium photoacids that successfully guide proton-transfer and deliver acid-catalyzed products. We also show that these species can adopt a second mechanism wherein they react with water to release hydroxyl radicals that facilitate hydrogen-atom abstraction and sp3C–H functionalization protocols. Together, this unprecedented bimodal reactivity enables electro-generated flavinium salts to affect synthetic chemistries previously unknown to flavins, greatly expanding their versatility as catalysts.
Introduction
Flavins are Nature's multi-tools, choreographing a myriad of chemical reactions. Flavins catalyze single-1–3 and multiple-electron transfer processes,4,5 group transfer chemistries,6 and other redox fluctuations7 that orchestrate complex bimolecular synthesis and cellular crosstalk. Flavins also have rich photochemistry.1 Depending on their redox state—quinone, semiquinone, or hydroquinone—flavins serve as powerful photo-oxidants and -reductants and can even confer their newly acquired photonic energy to ground state molecules, activating them for high-barrier transformations.8,9 Many of these pathways have been successfully leveraged for modern organic synthesis with visible light.10–12 A less explored pathway involves the use of flavins as photoacids. Protonation of the flavin quinone affords a flavinium salt, FL+ox. In the excited state (λmax = 394 nm), these species become ‘strong’ acids. Rarely do organic photoacids exceed pKa < 0.13–18 Schulman determined a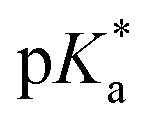 for protonated riboflavin (RF+ox) of −6.2.19 Liu et al. theorized that bis-protonation of the flavin quinone gives rise to an intermediate, FLH+ox_N1/N5, with even greater acid-like character in the ground state, pKa values of 0 and −10.1 at N1 and N5, respectively.20 Photoexcitation increases the acidity of N1 to −4.9 and shifts the basicity of N5 to +1.7, Fig. 1. Unlocking the unique photoacid reactivity of FL+ox has been a significant challenge. In the excited state, FL+ox is a strong oxidant, and promotes competitive single-electron transfer (SET) and hydrogen-atom transfer (HAT) mechanisms.21–26 Strong acids are typically used to generate FL+ox from their precursor quinones but cause competing background reactions with many acid-sensitive substrates.27 Mager et al. showed that flavin quinones are rapidly converted to FL+ox by anodic oxidation at +1.9 V vs. SCE in MeCN solvent, Fig. 1.28 The advantage of this electrochemical approach is that many unproductive free radicals are eliminated by reaction at the cathode or anode, suppressing background SET and HAT processes. It also precludes the use of strong acids as additives. Hence, the combination of electrochemistry and photochemistry, a.k.a., electrophotochemistry (EPC), could offer a superior route to access FL+ox, enabling organic chemists to probe its photoacid reactivity for the first time, Fig. 1. These studies would further complement previous work showing that the combination of electricity and flavins can entice new reaction discovery29 and that intermediates generated through EPC can facilitate synthetic disconnections and mechanisms not possible with prior art.30–34
for protonated riboflavin (RF+ox) of −6.2.19 Liu et al. theorized that bis-protonation of the flavin quinone gives rise to an intermediate, FLH+ox_N1/N5, with even greater acid-like character in the ground state, pKa values of 0 and −10.1 at N1 and N5, respectively.20 Photoexcitation increases the acidity of N1 to −4.9 and shifts the basicity of N5 to +1.7, Fig. 1. Unlocking the unique photoacid reactivity of FL+ox has been a significant challenge. In the excited state, FL+ox is a strong oxidant, and promotes competitive single-electron transfer (SET) and hydrogen-atom transfer (HAT) mechanisms.21–26 Strong acids are typically used to generate FL+ox from their precursor quinones but cause competing background reactions with many acid-sensitive substrates.27 Mager et al. showed that flavin quinones are rapidly converted to FL+ox by anodic oxidation at +1.9 V vs. SCE in MeCN solvent, Fig. 1.28 The advantage of this electrochemical approach is that many unproductive free radicals are eliminated by reaction at the cathode or anode, suppressing background SET and HAT processes. It also precludes the use of strong acids as additives. Hence, the combination of electrochemistry and photochemistry, a.k.a., electrophotochemistry (EPC), could offer a superior route to access FL+ox, enabling organic chemists to probe its photoacid reactivity for the first time, Fig. 1. These studies would further complement previous work showing that the combination of electricity and flavins can entice new reaction discovery29 and that intermediates generated through EPC can facilitate synthetic disconnections and mechanisms not possible with prior art.30–34
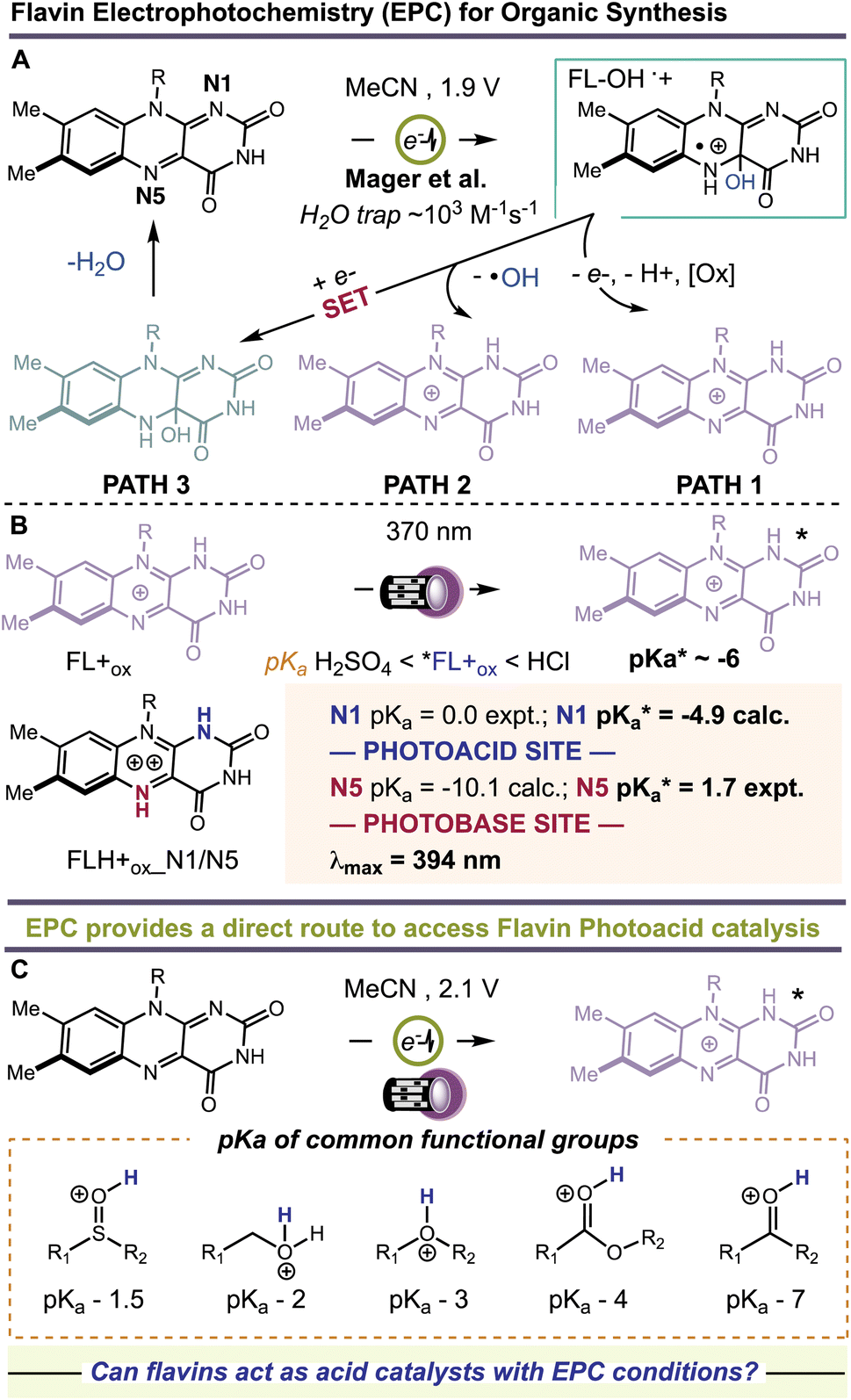 | ||
| Fig. 1 (A) Electrochemical generation of 4a-hydroxy flavins and decomposition to flavinium salts. (B) Photoacid and photobase behavior of flaviniums. (C) Electrophotochemistry route to flavin photoacid catalysis. | ||
Results and discussion
Sclareolide (1) is an ideal substrate to probe the competency of *FL+ox as a photoacid. Functionalization of sclareolide has been demonstrated with a swathe of chemical platforms that include HAT,35–37 SET,38 enzymatic,39–41 base-mediated,42,43 and transition metal-catalyzed processes,44,45Fig. 2. Most of these technologies replace a single C–H bond with a new functional group. No extent method directly replaces the key skeletal atoms of sclareolide, i.e., carbon and oxygen, with new atomic structures; although some methods do facilitate O → N ‘swapping’ by first opening the ‘C’ ring lactone to a hydroxamate or dioxazolone, and then recyclizing the ring.46,47 A more straightforward path could be achieved through photoacid catalysis. The carbonyl oxygen of the ‘C’ ring lactone can be protonated with a suitable acid, pKa CH3(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OH+)OMe = −3.9.48 Protonation induces C–O bond heterolysis and 3° carbocation formation. The carbocation can be trapped with a suitable nucleophile, such as solvent MeCN, to establish a new C–N bond. Closure of the ‘C’-ring through a Mumm rearrangement affords an imide product, excising the ‘O’ atom from the former lactone and introducing a ‘N’ atom in its place.49,50 If *FL+ox is a sufficient photoacid, we should obtain the imide product in lieu of C–H functionalized products. For this pilot reaction we selected riboflavin tetrabutyrate (RFTB) as a catalyst (10 mol%). RFTB (λAbs = 440 nm, λEm = 519 nm) undergoes irreversible electrooxidation at +1.9 V vs. SCE to form the flavinium RFTB+ox (λAbs = 396 nm, λEm = 512 nm), Fig. 1A. The four large butyrate groups give RFTB exceptional solubility in MeCN and shield the photoexcited state from nucleophilic attack, extending its lifetime in solution. The
OH+)OMe = −3.9.48 Protonation induces C–O bond heterolysis and 3° carbocation formation. The carbocation can be trapped with a suitable nucleophile, such as solvent MeCN, to establish a new C–N bond. Closure of the ‘C’-ring through a Mumm rearrangement affords an imide product, excising the ‘O’ atom from the former lactone and introducing a ‘N’ atom in its place.49,50 If *FL+ox is a sufficient photoacid, we should obtain the imide product in lieu of C–H functionalized products. For this pilot reaction we selected riboflavin tetrabutyrate (RFTB) as a catalyst (10 mol%). RFTB (λAbs = 440 nm, λEm = 519 nm) undergoes irreversible electrooxidation at +1.9 V vs. SCE to form the flavinium RFTB+ox (λAbs = 396 nm, λEm = 512 nm), Fig. 1A. The four large butyrate groups give RFTB exceptional solubility in MeCN and shield the photoexcited state from nucleophilic attack, extending its lifetime in solution. The 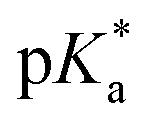 of RFTB+ox is expected to be in line with other protonated flavins, −5 to −8.19 We elected to use an IKA ElectraSyn 2.0 Pro system with an undivided cell, a pair of graphite electrodes, and MeCN solvent. LiClO4 was included as an electrolyte and AcOH was added as a cathodic oxidant. An alternating current of +2.1 V vs. SCE was applied for 16 h and irradiation was provided by two 370 nm Kessil lights (40 W each). From this reaction, we observed a trace amount of sclareolide-imide product (2). Increasing the loading of RFTB to 50 mol% improved the isolated yield to 17%. The imide product retained the original ‘C’ ring stereochemistry, yielding a single stereoisomer by X-ray crystallography (see ESI, page S105†). No C–H functionalization products were evident. A large fraction of the remaining mass balance is unreacted sclareolide, which was not consumed even with longer reaction times (>4 days). The imide product could not be formed in MeCN through bulk electrolysis at +4.0 V vs. SCE, direct photolysis at 370 nm, or treatment with stoichiometric acids such as H2SO4 and HClO4. All conditions led to epimerization and/or carbon skeleton rearrangements, as determined by crude 1H NMR of the reaction mixtures.51,52 Epimerization with acids like H2SO4 and HClO4 is consistent with reversible proton transfer to the lactone. Proton transfer from *FL+ox is irreversible (pKa of ground state FL+ox < 0), obviating epimerization, and allowing the intermediate carbocation that results from C–O bond heterolysis to be trapped by solvent acetonitrile. Ammonolysis with NH3 in MeOH also failed to produce the imide product, affording only the ‘C’ ring-opened amide alcohol.53 Alternative photoacid catalysts, i.e. eosin Y, pyranine, Schreiner's thiourea, and 7-bromonaphthalen-2-ol, were likewise unable to furnish imide 2. One-step formation of an imide 2 was only observed with flavin photoacid catalysis.
of RFTB+ox is expected to be in line with other protonated flavins, −5 to −8.19 We elected to use an IKA ElectraSyn 2.0 Pro system with an undivided cell, a pair of graphite electrodes, and MeCN solvent. LiClO4 was included as an electrolyte and AcOH was added as a cathodic oxidant. An alternating current of +2.1 V vs. SCE was applied for 16 h and irradiation was provided by two 370 nm Kessil lights (40 W each). From this reaction, we observed a trace amount of sclareolide-imide product (2). Increasing the loading of RFTB to 50 mol% improved the isolated yield to 17%. The imide product retained the original ‘C’ ring stereochemistry, yielding a single stereoisomer by X-ray crystallography (see ESI, page S105†). No C–H functionalization products were evident. A large fraction of the remaining mass balance is unreacted sclareolide, which was not consumed even with longer reaction times (>4 days). The imide product could not be formed in MeCN through bulk electrolysis at +4.0 V vs. SCE, direct photolysis at 370 nm, or treatment with stoichiometric acids such as H2SO4 and HClO4. All conditions led to epimerization and/or carbon skeleton rearrangements, as determined by crude 1H NMR of the reaction mixtures.51,52 Epimerization with acids like H2SO4 and HClO4 is consistent with reversible proton transfer to the lactone. Proton transfer from *FL+ox is irreversible (pKa of ground state FL+ox < 0), obviating epimerization, and allowing the intermediate carbocation that results from C–O bond heterolysis to be trapped by solvent acetonitrile. Ammonolysis with NH3 in MeOH also failed to produce the imide product, affording only the ‘C’ ring-opened amide alcohol.53 Alternative photoacid catalysts, i.e. eosin Y, pyranine, Schreiner's thiourea, and 7-bromonaphthalen-2-ol, were likewise unable to furnish imide 2. One-step formation of an imide 2 was only observed with flavin photoacid catalysis.
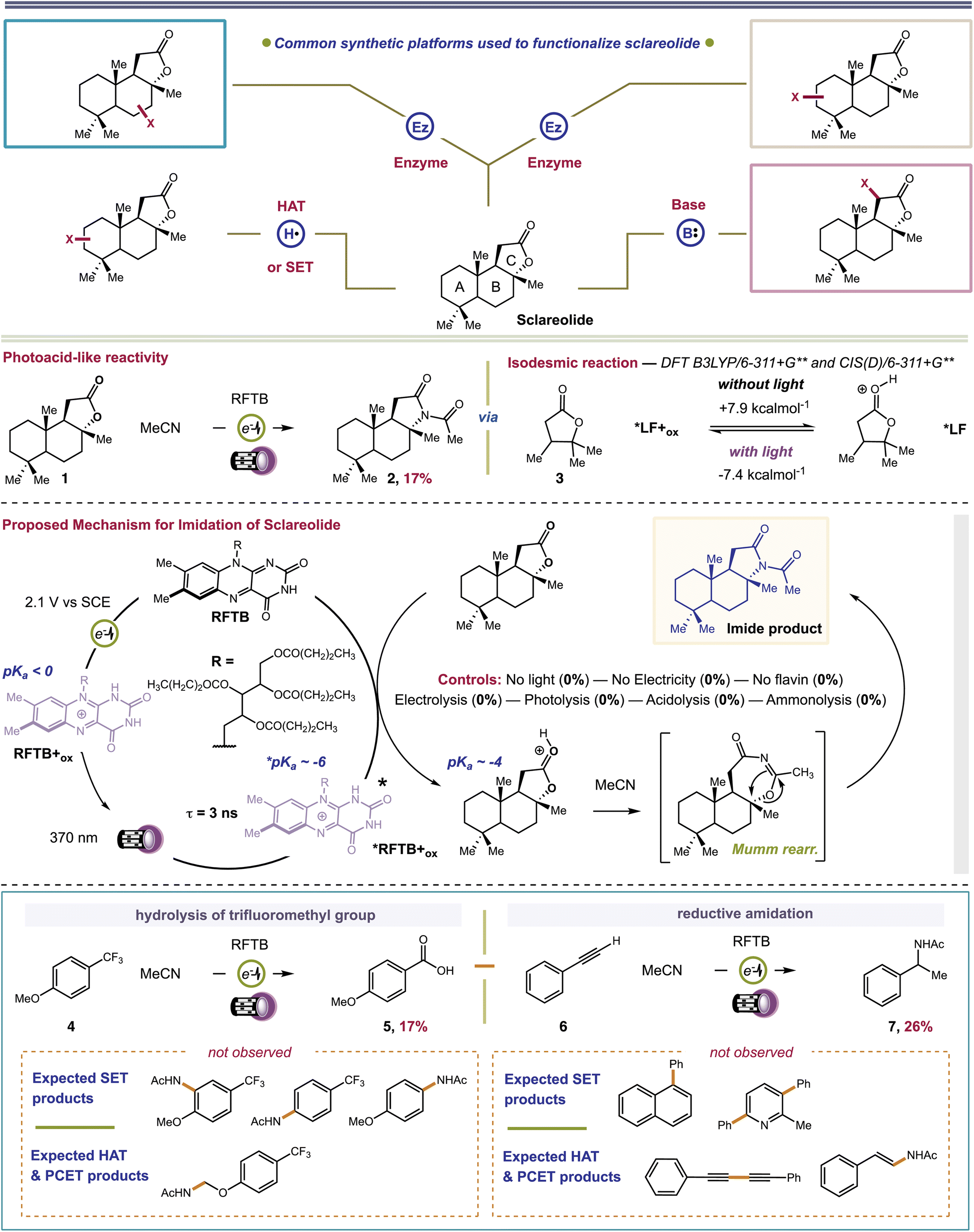 | ||
| Fig. 2 Top panel: Established platforms and regioselectivities observed for functionalization of sclareolide. Second panel: Observed imidation of sclareolide under flavin photoelectrocatalysis conditions and isodesmic calculations for excited-state proton transfer using a computational surrogate for sclareolide. Third panel: Proposed mechanism for formation of imido-sclareolide 2. Bottom panel: Substrates used to distinguish photoacid reactivity from alternate mechanisms. | ||
We attempted to optimize the reaction by testing alternative organic acids, electrodes, and electrolytes, but none of these changes offered any improvement. We also surveyed different flavin photocatalysts, e.g. lumiflavin (R = Me), riboflavin, and riboflavin 5′-monophosphate. Only lumiflavin afforded imide 2 (29% yield), but its cost is prohibitive at the high catalyst loadings needed in our reaction. Based on these results, we performed four sets of mechanistic experiments to gain deeper insight into our reaction. First, we carried out transient spectroelectrochemistry experiments to determine the excited state lifetime of RFTB+ox. Passing a current of +1.7 V vs. SCE through an MeCN solution of RFTB with irradiation at 390 nm produced *RFTB+ox, which decayed over ∼3 ns by fluorescence at ∼512 nm (see ESI, page S44†). A 3 ns lifetime is comparable to riboflavin tetraacetate in 10 mM HClO4, τ = 2.4 ns, which has a relatively low fluorescence quantum yield, ΦF ∼ 0.03.24 The fact that *RFTB+ox exhibits a nanosecond lifetime means that it is sufficiently long-lived to engage in bimolecular photocatalysis. One important observation made during these experiments was that extended exposure to light and anodic current converted small amounts of RFTB to several side products. By LC-MS, we tentatively assigned these products as oxygenated and amidated materials, and flavin derivatives having one less butyrate group attached (ESI, page S39†). These same byproducts were detected in our crude reaction mixture with sclareolide. While these products may only form in low amounts, their appearance suggests that at least some of our catalyst degrades over time and this could hinder the efficiency of our reaction. It also provides experimental evidence that some reactive intermediates, such as free radicals derived from MeCN (amidated catalyst) or water (oxygenated catalyst), may still exist in our reaction, and their open-shell chemistries could become more apparent when substrates with non-basic functional groups are employed; a case that we will address later in this manuscript. Second, we performed computations to study the thermochemistry of proton transfer. We used lactone 3 as a computational surrogate for the ‘C’ ring of sclareolide. Ground state proton transfer from a simplified flavin, lumiflavin+ox (LF+ox; R = CH3), to 3 is endergonic by +7.9 kcal mol−1. It is conceivable that this barrier can be overcome by visible light excitation of LF+ox. Absorption of a single photon of 370 nm light provides +77 kcal mol−1 driving force. In the first singlet excited state, proton transfer becomes exergonic by −7.4 kcal mol−1. Our computational analysis shows that proton transfer is only favorable in the excited state, in agreement with the proposed role of RFTB+ox as a photoacid. It is also consistent with the fact that no imide product is formed in the absence of light. Third, we conducted Stern–Volmer quenching studies. We found that sclareolide quenched the excited state of RFTB+ox, generated by treatment of RFTB with HClO4. Sclareolide failed to quench neutral RFTB. However, the Stern–Volmer plot for RFTB+ox displayed non-linear quenching (ESI, page S15†). At high concentrations of sclareolide, we observed a negative curvature. This occurs when a decrease in the quenching constant is evident and can manifest with a change in the fluorescence spectra of the fluorophore.54 It was clear from our experiment that RFTB+ox fluorescence shifted from 510 to 505 nm. The absorption spectrum of RFTB+ox remained constant when subjected to increasing concentrations of sclareolide, ruling out a Stokes' shift due to a change in solvent polarity. A deviation in the fluorescence spectra is also associated with the existence of multiple fluorescent states, with at least two subpopulations of the fluorophore being available. In our case, RFTB+ox can exist in two tautomeric forms where the proton resides on either N1 or N5.19 These can interconvert upon absorption and fluorescence (phototropism), accompanied by a dramatic change in charge density, accounting for the observed negative curvature. But this finding does not distinguish between individual mechanisms, i.e., proton transfer, HAT, SET or PCET. To delineate these pathways, we proceeded to our fourth set of mechanistic experiments, that is, the extension of our method to other substrates. We chose two substrates where a photoacid product would be mechanistically distinct from PCET, HAT, SET, base-catalyzed or transition metal catalyzed processes. We initially examined 4-trifluoromethylanisole (4). Trifluoromethylarenes are hydrolyzed under strongly acidic conditions, producing benzoic acids.55–63 SET, HAT, or PCET mechanisms lead to SNAr of the methoxy-64–66 or trifluoromethyl-group,67 electrophilic aromatic substitution,68 or α-functionalization of the aryl ether,69–71Fig. 2. Benzoic acid 5 was isolated as the exclusive product from our reaction. We next examined phenylacetylene (6). Phenylacetylene undergoes aromatic dimerization and N-insertion chemistries when irradiated at 400 nm in the presence of strongly oxidizing (SET) photocatalysts in MeCN solvent.72 It can also react with electrophilic free radicals (HAT species) to give β-substituted styrenes or alkyne dimers.73,74 We observed none of these products. The only product we found was benzylic acetamide 7, which forms from styrene produced by partial reduction of the alkyne at the cathode. Indeed, the use of styrene as reactant also afforded 7, albeit with competitive styrene polymerization. Addition of the amide group to the benzylic carbon is consistent with formation of a carbocation intermediate, expected from a photoacid mechanism. Neither of the two products reported above could be formed by electrolysis, photolysis, acidolysis or ammonolysis. Collectively, our experiments provide evidence for the involvement of RFTB+ox as a photoacid and show that its catalytic efficiency may be compromised by low levels of radical intermediates that survive past the electrodes and covalently modify the catalyst.
We examined substrates that are known to react through acid-catalyzed mechanisms to give defined products. Our first efforts focused on the Pinacol rearrangement. Our approach converted pinacol (8) to the classical pinacolone product (9) with transposition of a methyl group. Next, we examined a Ritter reaction with tert-butyl acetate (10). Proton transfer from RFTB+ox causes heterolytic C–O bond fragmentation and the release of tBu+, which can be trapped by MeCN solvent to give acetamide 11 after hydrolysis. This reaction worked well, yielding tert-butyl acetamide 11 in 59% isolated yield. Finally, we examined a Meyer–Schuster reaction with propargyl alcohol 12 and carbonyl reduction with phthalic anhydride 14. For our Meyer–Schuster reaction, we obtained acetophenone (13) as the exclusive product. This species can be explained by acid-catalyzed [3,3]-sigmatropic rearrangement of the tertiary alcohol to give the α,β-unsaturated phenone followed by acid-catalyzed conjugate addition of water and a retro aldol. The acid-catalyzed reduction of a single carbonyl group in phthalic anhydride also proceeded smoothly to provide phthalide (15). Here, the flavin photoacid activates the carbonyl group through proton transfer and reducing equivalents of electrons are supplied by the cathode. This type of electrochemical reduction was demonstrated by Baran for a related phthalimide system.75 In all cases, we fully acknowledge that many of our isolated yields are ‘low to moderate’. We should mention that all isolations were complicated by co-elution of the desired products with the flavin catalyst. Multiple columns were required to fully remove the catalyst, lowering our isolated yields. Still, despite the lower yields obtained, this is the first time that a flavin has ever been shown to promote acid catalyzed reactions with any success. Moreover, control reactions show that none of the products in Fig. 3 can be made in the absence of either light or current.
 | ||
| Fig. 3 Extension of electrophotochemical flavin photoacid generation to alternative acid-catalyzed reactions. | ||
Earlier, we showed that in the absence of a suitably basic substrate, that our catalyst is covalently modified. This likely occurs by reaction with background free radical species. Mager et al. showed that electrophilic ˙OH can form during flavin electrooxidation, Fig. 1 – path 2. The ˙OH can abstract strong C–H bonds producing open shell C˙ intermediates. If this pathway becomes viable in the absence of non-basic substrates, then we should be able to observe C–H functionalized products in such cases. To test this, we examined cyclohexane as substrate. Cyclohexane contains no functional groups for our flavin photoacid to activate, and it is very unlikely that RFTB+ox is acidic enough to perform hydride abstraction or carbonium ion formation with alkanes, which requires acids with pKa < −20.76 Applying identical conditions as before, we isolated a small amount of N-cyclohexyl acetamide as the sole product from our reaction. A plausible mechanism involves C–H abstraction by ˙OH, electrooxidation of the cyclohexyl radical to a carbocation, trapping by MeCN, and hydrolysis of the transient nitrilium ion to give the final acetamide product. The only byproduct of the reaction is H2O, consistent with prior art by Lambert using electrophotochemical conditions.77,78 Mager showed that addition of H2O to FL+ox affords the 4a-hydroxy flavin, Fig. 1. This species is quite stable but can inevitably dehydrate to return the ground state flavin, which is then anodically re-oxidized. Because the final dehydration event dictates catalyst turnover, we wondered if we could accelerate this step. Glusac et al. showed that Lewis-acidic CeIV can encourage dehydration of 4a-hydroxy flavins by forming metal-hydroxides.79 We surveyed a number of Lewis acids for this task, finding a significant improvement with Sc(OTf)3. The cooperative effect between FL and Sc(OTf)3 is well established, and these two species are often combined to achieve new photocatalytic reactions.80–82 Before proceeding, we wished to validate the proposed free radical pathway. We ran a competitive Kinetic Isotope Effect (KIE) experiment with d12-cyclohexane and performed Stern–Volmer quenching studies with cyclohexane, *2ScRFTB and *RFTB+ox (ESI, pages S11–S14†). We obtained a KIE = 1.2, which is not typically consistent with Csp3–H HAT in the rate-determining step, especially by highly reactive radicals like ˙OH (k ∼ 1012 cm3 per molecule per s)83 or others that can be generated by electrophotochemical methods to activate C–H bonds.84–87 In our case, generation of hydroxyl radicals, which was experimentally validated using methylene blue quenching of our crude reaction mixture—see ESI, pages S40–S41†—from 4a-hydroxyflavin radical cation is expected to be a much slower process,88 and likely rate-determining, which better reflects our KIE value. We documented no quenching of any flavin species with cyclohexane, occluding the possibility of SET, proton transfer, or energy transfer.
We examined the scope and chemoselectivity of alkane amidation. For most alkanes, poor solubility was observed in MeCN solvent. Therefore, we included trichloroacetonitrile (TCA) as a cosolvent. TCA greatly improves solubility without hindering reaction efficiency or competing with MeCN in the solvent trapping step. We are aware that TCA can expel Cl˙ when irradiated with UV light and that Cl˙ can abstract hydrogen atoms from alkanes.89 In a control reaction with 370 nm or 254 nm lights and 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 MeCN/TCA solvent, no product was formed when RFTB was excluded, showing that Cl˙ does not engender product formation when TCA is used as a cosolvent. We found that cycloalkanes C5–C12 (compounds 16a–e, E0ox = 2.3–2.6 V vs. SCE)90 were converted to new acetamide products, isolated yields 18–37%. For alkanes with both 2° and 3° C–H bonds (compounds 17–20), we observed site-selectivities consistent with Ritter-type amidation procedures mediated by an electrophilic HAT species, like ˙OH.91–101 For instance, in methylcyclohexane, amidation at more sterically congested C2 is preferred over less crowded C4. The difference is that C2 has 2-fold more hydrogen atoms to choose from, mirroring C3. The fact that C3 has four hydrogen atoms and is sterically accessible makes it the most preferred site for kinetic C–H amidation. Our observed selectivity, C3 > C2 > C4 is reflective of some evolved P450 nitrene-transferases102 and metallaphotoredox platforms.103 This same effect can be seen for trans-decalin 20 and n-hexane 21. When the number of hydrogen atoms is equal, the methylene site that is most accessible is amidated. Thus, a general set of rules for predicting site-selectivity in alkane substrates is established. These regiochemical preferences should hold true regardless of the selected nitrile (nucleophile) or the experimental setup used to perform our reaction. To test this, we selected three commercial nitriles that replace acetonitrile as solvent and nucleophile, and a three-neck round bottom flask equipped with a DC power supply and graphite electrodes as a substitute for our standard IKA ElectraSyn 2.0 Pro electrochemical setup. For all experiments, norbornane was used as the test alkane and TCA was omitted due to improved solubility, Fig. 4B and D. All three nitriles fashioned a single amide product (compounds 23–25, yields 35–67%) that would be very expensive to procure otherwise. The site- and stereo-selectivities of the new amide products were identical to reactions performed with acetonitrile. In a three-neck flask, 23 was formed in 35% isolated yield. Despite its lower efficiency, this proof-of-principle experiment is evidence that our reaction can be performed in alternative setups, including those that are more conducive to large-scale batch reactions, without loss in product selectivity. Finally, we tested the capacity of our catalyst to upgrade off-the-shelf samples of common hydrocarbon fuels. The hydroxy radical formed by flavin electrooxidation is highly reactive and, therefore, can abstract a wide variety of C–H bonds (1°, 2°, and 3°) in different alkanes, improving feedstock throughput. This is harder to achieve with more selective HAT reagents that may only activate a select few hydrogen atoms, 2° and 3°. Our preliminary studies focused on gaseous propane (E0ox = 2.90 V vs. SCE) and kerosene (jet fuel), a primary distillate from crude oil that contains a heterogeneous mixture of C9–C16 alkanes and alkylarenes (Fig. 4D and E). Functionalization of C–H bonds in light alkanes (C1–C4) is intrinsically more difficult because of their higher bond-dissociation energies (BDE) and low polarities. Under our electrophotochemical conditions, N-isopropylacetamide (26) was forged in 6% LC-MS yield (Fig. 4D). For kerosene, our conditions produced a ∼12% NMR yield of amidated products, which includes ∼7 unique amides by 1H–13C HMBC. Thus, our system can harness petroleum side streams as veritable bench deposits to pan out value-added amide products.
:5 MeCN/TCA solvent, no product was formed when RFTB was excluded, showing that Cl˙ does not engender product formation when TCA is used as a cosolvent. We found that cycloalkanes C5–C12 (compounds 16a–e, E0ox = 2.3–2.6 V vs. SCE)90 were converted to new acetamide products, isolated yields 18–37%. For alkanes with both 2° and 3° C–H bonds (compounds 17–20), we observed site-selectivities consistent with Ritter-type amidation procedures mediated by an electrophilic HAT species, like ˙OH.91–101 For instance, in methylcyclohexane, amidation at more sterically congested C2 is preferred over less crowded C4. The difference is that C2 has 2-fold more hydrogen atoms to choose from, mirroring C3. The fact that C3 has four hydrogen atoms and is sterically accessible makes it the most preferred site for kinetic C–H amidation. Our observed selectivity, C3 > C2 > C4 is reflective of some evolved P450 nitrene-transferases102 and metallaphotoredox platforms.103 This same effect can be seen for trans-decalin 20 and n-hexane 21. When the number of hydrogen atoms is equal, the methylene site that is most accessible is amidated. Thus, a general set of rules for predicting site-selectivity in alkane substrates is established. These regiochemical preferences should hold true regardless of the selected nitrile (nucleophile) or the experimental setup used to perform our reaction. To test this, we selected three commercial nitriles that replace acetonitrile as solvent and nucleophile, and a three-neck round bottom flask equipped with a DC power supply and graphite electrodes as a substitute for our standard IKA ElectraSyn 2.0 Pro electrochemical setup. For all experiments, norbornane was used as the test alkane and TCA was omitted due to improved solubility, Fig. 4B and D. All three nitriles fashioned a single amide product (compounds 23–25, yields 35–67%) that would be very expensive to procure otherwise. The site- and stereo-selectivities of the new amide products were identical to reactions performed with acetonitrile. In a three-neck flask, 23 was formed in 35% isolated yield. Despite its lower efficiency, this proof-of-principle experiment is evidence that our reaction can be performed in alternative setups, including those that are more conducive to large-scale batch reactions, without loss in product selectivity. Finally, we tested the capacity of our catalyst to upgrade off-the-shelf samples of common hydrocarbon fuels. The hydroxy radical formed by flavin electrooxidation is highly reactive and, therefore, can abstract a wide variety of C–H bonds (1°, 2°, and 3°) in different alkanes, improving feedstock throughput. This is harder to achieve with more selective HAT reagents that may only activate a select few hydrogen atoms, 2° and 3°. Our preliminary studies focused on gaseous propane (E0ox = 2.90 V vs. SCE) and kerosene (jet fuel), a primary distillate from crude oil that contains a heterogeneous mixture of C9–C16 alkanes and alkylarenes (Fig. 4D and E). Functionalization of C–H bonds in light alkanes (C1–C4) is intrinsically more difficult because of their higher bond-dissociation energies (BDE) and low polarities. Under our electrophotochemical conditions, N-isopropylacetamide (26) was forged in 6% LC-MS yield (Fig. 4D). For kerosene, our conditions produced a ∼12% NMR yield of amidated products, which includes ∼7 unique amides by 1H–13C HMBC. Thus, our system can harness petroleum side streams as veritable bench deposits to pan out value-added amide products.
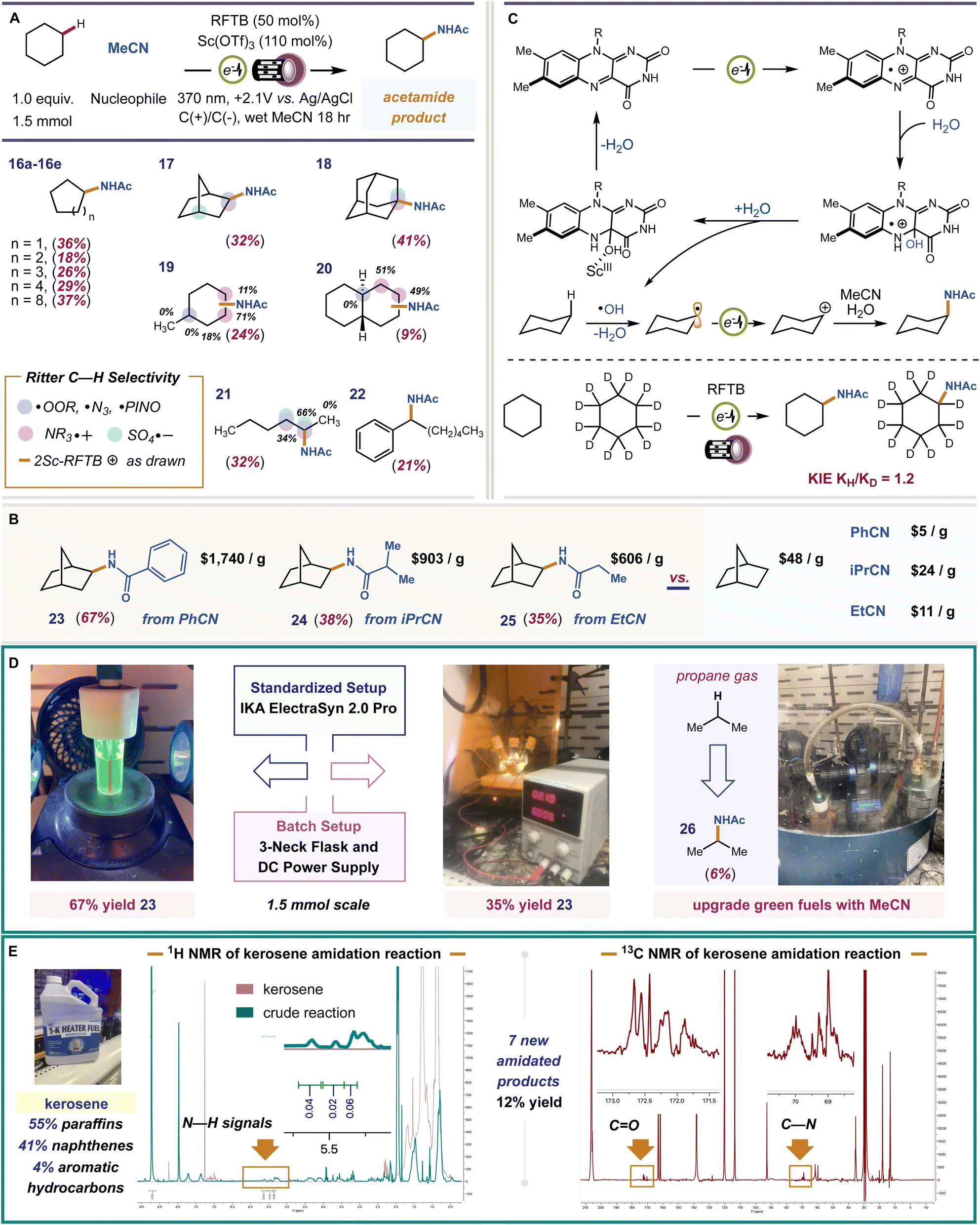 | ||
| Fig. 4 Scope of the reaction and application to hydrocarbon fuels. (A) Survey of alkane substrates and a comparison of observed site-selectivities to established amidation protocols (Ritter-type) that use different catalysts/mediators. % Regiochemical distribution for substrates with multiple products is indicated beside each site. (B) Scope of alternative nitrile nucleophiles with average prices of products and starting materials from commercial vendors. (C) Proposed mechanism of C–H amidation. (D) Results with gaseous propane and a comparison of reaction efficiencies between our standard IKA ElectraSyn 2.0 Pro setup and a three-neck flask electrochemical setup. (E) Experiments performed using commercial kerosene as C–H substrate. | ||
Conclusions
In summary, we show that electrophotochemistry provides a direct route to access flavin photoacids, allowing them to be harnessed in organic synthesis for the first time. We show that when substrates lacking a basic functional group are employed that ˙OH formation becomes more apparent, enabling C–H amidation protocols. Together, our results demonstrate the unique ability of flavins (FL+ox) to exhibit bimodal reactivity under electrophotochemical conditions, unveiling new mechanistic opportunities for using flavins as dynamic electrophotocatalysts.Data availability
Crystallographic data for compound 2 has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under accession number 2255574. All other data is present in the manuscript or ESI.†Author contributions
S. Ga. performed all chemical reactions. J. W. and S. Go. acquired all spectroelectrochemical spectra. S. B. supervised this work. The manuscript was written with input and approval from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in the Department of Medicinal Chemistry at the University of Kansas, and supported by the School of Pharmacy, University of Kansas Startup 2506031-099, and graduate teaching assistant support for S. Ga. This work was also supported by the National Institutes of Health Graduate Training at the Biology Chemistry Interface Grant T32 GM132061 from the National Institutes of General Medical Sciences (NIGMS) and NIGMS P20GM113117. Minor salary support for S. Ga. was provided by NIGMS R35GM147169. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Alonso Rodriguez Ugalde, Pei-Hsuan Chen, and Farjana Afroj for their help in reviewing the manuscript. We also thank Dr Travis Witte for assistance with collecting absorbance and fluorescence spectra, Dr Justin Douglas for help with the acquisition of 2D NMR spectra, Dr Scott Lovell and Dr Mohammad Rasel Mian for crystallography, and Dr Christopher Elles for helpful discussions. J. W. and S. Go. thank the Warwick Electrochemistry & Interfaces Group for their help with the non-aqueous spectroelectrochemical set-up. We thank Anjali John and Julie Macpherson (University of Warwick) for use of their electrochemical cell and for proofreading the manuscript.Notes and references
- V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, Synthetic applications of flavin photocatalysis: a review, RSC Adv., 2021, 11, 14251–14259 RSC.
- R. Lechner, S. Kümmel and B. König, Visible light flavin photo-oxidation of methylbenzenes, styrenes, and phenylacetic acid, Photochem. Photobiol. Sci., 2010, 10, 1367–1377 CrossRef PubMed.
- T. Hering, B. Mühldorf, R. Wolf and B. König, Halogenase-inspired oxidative chlorination using flavin photocatalysis, Angew. Chem., Int. Ed., 2016, 55, 5342–5345 CrossRef CAS PubMed.
- C. Smit, M. W. Fraaije and A. J. Minnaard, Reduction of carbon-carbon double bonds using organocatalytically generated diimide, J. Org. Chem., 2008, 73, 9482–9485 CrossRef CAS PubMed.
- J. Svoboda, H. Schmaderer and B. König, Thiourea-enhanced flavin photooxidation of benzyl alcohol, Chem.–Eur. J., 2008, 14, 1854–1865 CrossRef CAS PubMed.
- A. Rehpenn, A. Walter and G. Storch, Molecular flavin catalysts for C–H functionalisation and derivatisation of dehydroamino acids, Chem. Sci., 2022, 47, 14151–14156 RSC.
- Y. Imada and T. Naota, Flavins as organocatalysts for environmentally benign molecular transformations, Chem. Rec., 2007, 7, 354–361 CrossRef CAS PubMed.
- J. B. Metternich and R. Gilmour, A bio-inspired, catalytic E→Z isomerization of activated olefins, J. Am. Chem. Soc., 2015, 137, 11254–11257 CrossRef CAS PubMed.
- V. Mojr, E. Svobodová, K. Straková, T. Neveselý, J. Chudoba, H. Dvořáková and R. Cibulka, Tailoring flavins for visible light photocatalysis: organocatalytic [2+2] cycloadditions mediated by a flavin derivative and visible light, Chem. Commun., 2015, 51, 12036–12039 RSC.
- B. König, S. Kümmel, E. Svobodová and R. Cibulka, Flavin photocatalysis, Phys. Sci. Rev., 2018, 3, 20170168 Search PubMed.
- M. Jirásek, K. Straková, T. Nevesely, E. Svobodová, Z. Rottnerová and R. Cibulka, Flavin-mediated visible-light [2+2] photocycloaddition of nitrogen- and sulfur-containing dienes, Eur. J. Org Chem., 2016, 15, 2139–2146 Search PubMed.
- R. Cibulka and M. Fraaije, Flavin-Based Catalysis: Principles and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2021 Search PubMed.
- R. Simkovitch, S. Shomer, R. Gepshtein, M. E. Roth, D. Shabat and D. Huppert, Comparison of the rate of excited-state proton transfer from photoacids to alcohols and water, J. Photochem. Photobiol., A, 2014, 277, 90–101 CrossRef CAS.
- N. Sülzner, B. Geissler, A. Grandjean, G. Jung and P. Nuernberger, Excited-state proton transfer dynamics of a super-photoacid in acetone-water mixtures, ChemPhotoChem, 2022, 6, e202200041 CrossRef.
- I. Carmeli, D. Huppert, L. M. Tolbert and J. E. Haubrich, Ultrafast excited-state proton transfer from dicyano-naphthol, Chem. Phys. Lett., 1996, 260, 109–114 CrossRef CAS.
- Y.-J. Choi, H. Kim and O.-H. Kwon, Acid-base reaction of a super-photoacid with a cooperative amide hydrogen-bonded chain, Bull. Korean Chem. Soc., 2022, 43, 501–507 CrossRef CAS.
- J. Saway, Z. M. Salem and J. J. Badillo, Recent advances in photoacid catalysis for organic synthesis, Synthesis, 2021, 53, 489–497 CrossRef CAS.
- B. Finkler, C. Spies, M. Vester, F. Walte, K. Omlor, I. Riemann, M. Zimmer, F. Stracke, M. Gerhards and G. Jung, Highly photostable “super”-photoacids for ultrasensitive fluorescence spectroscopy, Photochem. Photobiol. Sci., 2014, 13, 548–562 CrossRef CAS PubMed.
- S. G. Schulman, pH dependence of fluorescence of riboflavin and related isoalloxazine derivatives, J. Pharm. Sci., 1971, 60, 628–631 CrossRef CAS PubMed.
- J. Wang and Y. Liu, Systematic theoretical study on the pH-dependent absorption and fluorescence spectra of flavins, Molecules, 2023, 28, 3315 CrossRef CAS PubMed.
- T. Hartman, M. Reisnerová, J. Chudoba, E. Svobodobá, N. Archipowa, R. J. Kutta and R. Cibulka, Photocatalytic oxidative [2+2] cycloelimination reactions with flavinium salts: mechanistic study and influence of the catalyst structure, ChemPlusChem, 2021, 86, 373–386 CrossRef CAS PubMed.
- A. Pokluda, Z. Anwar, V. Boguschová, I. Anusiewicz, P. Skurski, M. Sikorski and R. Cibulka, Robust photocatalytic method using ethylene-bridged flavinium salts for the aerobic oxidation of unactivated benzylic substrates, Adv. Synth. Catal., 2021, 363, 4371–4379 CrossRef CAS.
- T. Hartman and R. Cibulka, Photocatalytic systems with flavinium salts: from photolyase models to synthetic tool for cyclobutane ring opening, Org. Lett., 2016, 18, 3710–3713 CrossRef CAS PubMed.
- C. Pac, K. Miyake, Y. Masaki, S. Yanagida, T. Ohno and A. Yoshimura, Flavin-photosensitized monomerization of dimethylthymine cyclobutane dimer: remarkable effects of perchloric acid and participation of excited-singlet, triplet, and chain-reaction pathways, J. Am. Chem. Soc., 1992, 114, 10756–10762 CrossRef CAS.
- S. Fukuzumi and S. Kuroda, Photooxidation of benzyl alcohol derivatives by oxygen, catalyzed by protonated flavin analogues, Res. Chem. Intermed., 1999, 25, 789–811 CrossRef CAS.
- A. H. Tolba, F. Vávra, J. Chudoba and R. Cibulka, Tuning flavin-based photocatalytic systems for application in the mild chemoselective aerobic oxidation of benzylic substrates, Eur. J. Org Chem., 2019, 2020, 1579–1585 CrossRef.
- K. Miyake, Y. Masaki, I. Miyamoto, S. Yanagida, T. Ohno, A. Yoshimura and C. Pac, Flavin-photosensitized monomerization of dimethylthymine cyclobutane dimer in the presence of magnesium perchlorate, J. Photochem. Photobiol., 1993, 58, 631–636 CrossRef CAS PubMed.
- H. I. X. Mager, S.-C. Tu, Y.-H. Liu, Y. Deng and K. M. Kadish, Electrochemical superoxidation of flavins: generation of active precursors in luminescent model systems, Photochem. Photobiol., 1990, 52, 1049–1056 CrossRef CAS PubMed.
- W. Zhang, K. L. Carpenter and S. Lin, Electrochemistry broadens the scope of flavin photocatalysis: photoelectrocatalytic oxidation of unactivated alcohols, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, Electrophotocatalysis: Combining light and electricity to catalyze reactions, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed.
- L. Qian and M. Shi, Contemporary photoelectrochemical strategies and reactions in organic synthesis, Chem. Commun., 2023, 24, 3487–3506 RSC.
- J. P. Barham and B. König, Synthetic photoelectrochemistry, Angew. Chem., Int. Ed., 2019, 59, 11732–11747 CrossRef PubMed.
- J. Liu, L. Lu, D. Wood and S. Lin, New redox strategies in organic synthesis by means of electrochemistry and photochemistry, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed.
- F. Medici, V. Chiroli, L. Raimondi and M. Benaglia, Latest updates in electrophotochemical reactions, Tetrahedron Chem, 2024, 9, 100061 CrossRef CAS.
- G. Choi, G. S. Lee, B. Park, D. Kim and S. H. Hong, Direct C(sp3)–H trifluoromethylation of unactivated alkanes enable by multifunctional trifluoromethyl copper complexes, Angew. Chem., Int. Ed., 2020, 60, 5467–5474 CrossRef PubMed.
- Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, Scalable, electrochemical oxidation of unactivated C–H bonds, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed.
- R. K. Quinn, Z. A. Könst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. J. Alexanian, Site-selective aliphatic C–H chlorination using N-chloroamides enables a synthesis of chlorolissoclimide, J. Am. Chem. Soc., 2016, 138, 696–702 CrossRef CAS PubMed.
- S. Bloom, J. L. Knippel and T. Lectka, A photocatalyzed aliphatic fluorination, Chem. Sci., 2014, 5, 1175–1178 RSC.
- F. Li, H. Deng and H. Renata, Remote B-ring oxidation of sclareol with an engineered P450 facilitates divergent access to complex terpenoids, J. Am. Chem. Soc., 2022, 144, 7616–7621 CrossRef CAS PubMed.
- J. Li, F. Li, E. King-Smith and H. Renata, Merging chemoenzymatic and radical-based retrosynthetic logic for rapid and modular synthesis of oxidized meroterpenoids, Nat. Chem., 2020, 12, 173–179 CrossRef CAS PubMed.
- A. Ata, L. J. Conci, J. Betteridge, I. Orhan and B. Sener, Novel microbial transformations of sclareolide, Chem. Pharm. Bull., 2007, 55, 118–123 CrossRef CAS PubMed.
- Z. Li, X. Yao, X. Zhang, H. Mei and J. Han, Carboxylic acid O-H insertion reaction of β-ester diazos enabling synthesis of β-acyloxy esters, J. Org. Chem., 2022, 87, 15483–15491 CrossRef CAS PubMed.
- J. Guo, X. Xu, Q. Xing, Z. Gao, J. Gou and B. Yu, Furfuryl cation induced cascade formal [3+2] cycloaddition/double ring-opening/chlorination: an approach to chlorine-containing complex triazoles, Org. Lett., 2018, 20, 7410–7414 CrossRef CAS PubMed.
- N. D. Chiappini, J. B. C. Mack and J. Du Bois, Intermolecular C(sp3)–H amination of complex molecules, Angew. Chem., Int. Ed., 2018, 57, 4956–4959 CrossRef CAS PubMed.
- K. Chen, A. Eschenmoser and P. S. Baran, Strain release in C–H bond activation?, Angew. Chem., Int. Ed., 2009, 48, 9705–9708 CrossRef CAS PubMed.
- H. Jung, H. Keum, J. Kweon and S. Chang, Tuning triplet energy transfer of hydroxamates as the nitrene precursor for intramolecular C(sp3)–H amidation, J. Am. Chem. Soc., 2020, 142, 5811–5818 CrossRef CAS PubMed.
- H.-Y. Jung, S. Chang and S. Hong, Strategic approach to the metamorphosis of γ-lactones to NH γ-lactams via reductive cleavage and C–H amidation, Org. Lett., 2019, 21, 7099–7103 CrossRef CAS PubMed.
- A. Bagno and G. Scorrano, Acid-base properties of organic solvents, J. Am. Chem. Soc., 1988, 10, 4577–4582 CrossRef.
- R. Giri, I. Mosiagin, I. Franzoni, N. Y. Notel, S. Patra and D. Katayev, Photoredox activation of anhydrides for the solvent-controlled switchable synthesis of gem-difluoro compounds, Angew. Chem., Int. Ed., 2022, 61, e202209143 CrossRef CAS PubMed.
- J. Bolsakova and A. Jirgensons, The ritter reaction for the synthesis of heterocycles, Chem. Heterocycl. Compd., 2017, 53, 1167–1177 CrossRef CAS.
- A. Si, A. D. Landgraf, S. Geden, S. J. Sucheck and K. H. Rohde, Synthesis and evaluation of marine natural product-inspired meroterpenoids with selective activity toward dormant mycobacterium tuberculosis, ACS Omega, 2022, 7, 23487–23496 CrossRef CAS PubMed.
- H.-S. Wang, H.-J. Li, X. Nan, Y.-Y. Luo and Y.-C. Wu, Enantiospecific semisynthesis of puupehedione-type marine natural products, J. Org. Chem., 2017, 82, 12914–12919 CrossRef CAS PubMed.
- D. Li, S. Xhang, Z. Song, W. Li, F. Zhu, J. Zhang and S. Li, Synthesis and bio-inspired optimization of drimenal: discovery of chiral drimane fused oxazinones as promising antifungal and antibacterial candidates, Eur. J. Med. Chem., 2018, 143, 558–567 CrossRef CAS PubMed.
- J. Keizer, Nonlinear fluorescence quenching and the origin of positive curvature in Stern-Volmer plots, J. Am. Chem. Soc., 1983, 105, 1494–1498 CrossRef CAS.
- D. Herrera, D. Peral and C. J. Bayon, Preparation of carboxylic arylphosphines by hydrolysis of the trifluoromethyl group, RSC Adv., 2022, 12, 7103–7114 RSC.
- J. H. Simons and E. O. Ramler, Preparation and properties of some new trifluoromethyl compounds, J. Am. Chem. Soc., 1943, 65, 389–392 CrossRef CAS.
- H. Gilman and D. Blume, 5- and 7-trifluoromethylquinolines, J. Am. Chem. Soc., 1943, 65, 2467–2468 CrossRef CAS.
- G. M. Le Fave, Some reactions of the trifluoromethyl group in the benzotrifluoride series. I. Hydrolysis, J. Am. Chem. Soc., 1949, 71, 4148–4149 CrossRef CAS.
- G. M. Le Fave and P. G. Scheurer, Some reactions of the trifluoromethyl group in the benzotrifluoride series. II. Alcoholysis, J. Am. Chem. Soc., 1950, 72, 2464–2465 CrossRef CAS.
- P. G. Scheurer and G. M. Le Fave, Isophthalic and terephthalic acids, J. Am. Chem. Soc., 1950, 72, 3308–3309 CrossRef CAS.
- K. Wang, H. Li and J. Wen, Synthesis and mesomorphic properties of cholesteryl p-polyfluoroalkoxy-m-nitrobenzoate, J. Fluorine Chem., 2001, 109, 205–208 CrossRef CAS.
- Y. V. Zonov, V. M. Karpov and V. E. Platonov, Transformation of perfluorinated benzocycloalkenes and alkylbenzenes to their carbonyl derivatives under the action of CF3COOH/SbF5, J. Fluorine Chem., 2007, 128, 1058–1064 CrossRef CAS.
- A. Kethe, A. F. Tracy and D. A. Klumpp, Protolytic defluorination of trifluoromethyl-substituted arenes, Org. Biomol. Chem., 2011, 9, 4545–4549 RSC.
- X. Wu, W. Chen, N. Holmberg-Douglas, G. T. Bida, X. Tu, X. Ma, Z. Wu, D. A. Nicewicz and Z. Li, 11C-, 12C-, and 13C-cyanation of electron-rich arenes via organic photoredox catalysis, Chem, 2023, 9, 343–362 CAS.
- V. A. Pistritto, S. Liu and D. A. Nicewicz, Mechanistic investigations into amination of unactivated arenes via cation radical accelerated nucleophilic aromatic substitution, J. Am. Chem. Soc., 2022, 144, 15118–15131 CrossRef CAS PubMed.
- N. J. Venditto and D. A. Nicewicz, Cation radical-accelerated nucleophilic aromatic substitution for amination of alkoxyarenes, Org. Lett., 2020, 22, 4817–4822 CrossRef CAS PubMed.
- S. Wu, J. Žurauskas, M. Domański, P. S. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug, J. Hauer and J. P. Barham, Hole-mediated photoredox catalysis: tris(p-substituted)biarylaminium radical cations as tunable, precomplexing and potent photooxidants, Org. Chem. Front., 2021, 8, 1132–1142 RSC.
- T. Morofuji, G. Ikarashi and N. Kano, Photocatalytic C–H amination of aromatics overcoming redox potential limitations, Org. Lett., 2020, 22, 2822–2827 CrossRef CAS PubMed.
- Z.-Y. Dai, Z.-S. Nong, S. Song and P.-S. Wang, Asymmetric photocatalytic C(sp3)–H bond addition to α-substituted acrylates, Org. Lett., 2021, 23, 3157–3161 CrossRef CAS PubMed.
- S. M. Treacy and T. Rovis, Copper catalyzed C(sp3)–H bond alkylation via photoinduced ligand-to-metal charge transfer, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed.
- J. D. Bell, I. Robb and J. A. Murphy, Highly selection α-aryloxyalkyl C–H functionalization of aryl alkyl ethers, Chem. Sci., 2022, 13, 12921–12926 RSC.
- S. L. Mattes and S. Farid, Photosensitized electron-transfer reactions of phenylacetylene, J. Chem. Soc., Chem. Commun., 1980, 3, 126–128 RSC.
- A. Souibgui, M. ben Mosbah, R. ben Salem, Y. Moussaoui and A. Tlili, Hydrotrifluoromethylation of styrene and phenylacetylene derivates under visible-light photoredox conditions, Chemistry, 2022, 4, 1010–1015 CrossRef CAS.
- S. S. Zalesskiy, N. S. Shlapakov and V. P. Ananikov, Visible light mediated metal-free thiol-yne click reaction, Chem. Sci., 2016, 7, 6740–6745 RSC.
- Y. Kawamata, K. Hayashi, E. Carlson, S. Shaji, D. Waldmann, B. J. Simmons, J. T. Edwards, C. W. Zapf, M. Saito and P. S. Baran, Chemoselective electrosynthesis using rapid alternating polarity, J. Am. Chem. Soc., 2021, 143, 16580–16588 CrossRef CAS PubMed.
- G. A. Olah and R. H. Schlosberg, Chemistry in super acids. I. Hydrogen exchange and polycondensation of methane and alkanes in FSO3H-SbF5 (“magic-acid”) solution. Protonation of alkanes and the intermediacy of CH5+ and related hydrocarbon ions. The high chemical reactivity of “paraffins” in ionic solution reactions, J. Am. Chem. Soc., 1968, 90, 2726–2727 CrossRef CAS.
- T. Shen and T. H. Lambert, Electrophotocatalytic diamination of vicinal C–H bonds, Science, 2021, 371, 620–626 CrossRef CAS PubMed.
- T. Shen and T. H. Lambert, C–H Amination via Electrophotocatalytic Ritter-type Reaction, J. Am. Chem. Soc., 2021, 143, 8597–8602 CrossRef CAS PubMed.
- V. Sichula, Y. Hu, E. Mirzakulova, S. F. Manzer, S. Vyas, C. M. Hadad and K. D. Glusac, Mechanism of N(5)-ethyl-flavinium cation formation upon electrochemical oxidation of N(5)-ethyl-4a-hydroxyflavin pseudobase, J. Phys. Chem. B, 2010, 114, 9452–9461 CrossRef CAS PubMed.
- S. Fukuzumi, S. Kuroda and T. Tanaka, Flavin analog-metal ion complexes acting as efficient photocatalysts in the oxidation of p-methylbenzyl alcohol by oxygen under irradiation with visible light, J. Am. Chem. Soc., 1985, 107, 3020–3027 CrossRef CAS.
- D. Shen, F. Zhong, L. Li, H. Zhang, T. Ren, C. Sun, B. Wang, M. Guo, M. Chao and S. Fukuzumi, Aerobic oxyfunctionalization of alkynes by a bioinspired flavin–metal ion photocatalytic system, Org. Chem. Front., 2023, 10, 2653–2662 RSC.
- B. Mühldorf and R. Wolf, Photocatalytic benzylic C–H bond oxidation with a flavin scandium complex, Chem. Commun., 2015, 51, 8425–8428 RSC.
- E. S. C. Kwok and R. Atkinson, Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 1995, 29, 1685–1695 CrossRef CAS.
- P. Xu, P.-Y. Chen and H.-C. Xu, Scalable photoelectrochemical dehydrogenative cross-coupling of heteroarenes with aliphatic C–H bonds, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed.
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, Manganese-catalyzed oxidative azidation of C(sp3-H) bonds under electrophotocatalytic conditions, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed.
- L. Capaldo, L. L. Quadri, D. Merli and D. Ravelli, Photoelectrochemical cross-dehydrogenative coupling of benzothiazoles with strong aliphatic C–H bonds, Chem. Commun., 2021, 57, 4424–4427 RSC.
- H. Huang, Z. M. Strater and T. H. Lambert, Electrophotocatalytic C–H functionalization of ethers with high regioselectivity, J. Am. Chem. Soc., 2020, 142, 1698–1703 CrossRef CAS PubMed.
- H. I. X. Mager, D. Sazou, Y. H. Liu, S.-C. Tu and K. M. Kadish, Reversible one-electron generation of 4a,5-substituted flavin radical cations: Models for a postulated key intermediate in bacterial bioluminescence, J. Am. Chem. Soc., 1988, 110, 3759–3762 CrossRef CAS.
- Y. Toda, R. Matsuda, S. Gomyou and H. Suga, Use of trichloroacetonitrile as a hydrogen chloride generator for ring-opening reactions of aziridines, Org. Biomol. Chem., 2019, 17, 3825–3829 RSC.
- See ESI page S17† for a complete list of substrate oxidation potentials and S18–S30† for substrate cyclic voltammograms.
- Q. Michaudel, D. Thevenet and P. S. Baran, Intermolecular ritter-type C–H amination of unactivated sp3 carbons, J. Am. Chem. Soc., 2014, 134, 2547–2550 CrossRef PubMed.
- Y.-W. Zheng, R. Narobe, K. Danabauer, S. Yakubov and B. König, Copper(II)-photocatalyzed N–H alkylation with alkanes, ACS Catal., 2020, 10, 8582–8589 CrossRef CAS.
- V. Nair, T. D. Suja and K. A. Mohanan, A convenient protocol for C–H oxidation mediated by an azido radical culminating in ritter-type amidation, Tetrahedron Lett., 2005, 46, 3217–3219 CrossRef CAS.
- B. Tran, B. Li, M. Driess and J. F. Hartwig, Copper-catalyzed intermolecular amidation and imidation of unactivated alkanes, J. Am. Chem. Soc., 2014, 136, 2555–2563 CrossRef CAS PubMed.
- H. Lai, J. Xu, J. Lin, B. Su and D. Zha, Chemo-selective control of ritter-type reaction by coordinately unsaturated inorganic salt hydrates, Org. Chem. Front., 2022, 9, 1541–1549 RSC.
- L. Zhang, Y. Fu, Y. Shen, C. Liu, M. Sun, R. Cheng, W. Zhu, X. Qian, Y. Ma and J. Ye, Ritter-type amination of C(sp3)–H bonds enabled by electrochemistry with SO42−, Nat. Commun., 2022, 13, 4138 CrossRef CAS PubMed.
- K. Kiyokawa, K. Takemoto and S. Minakata, Ritter-type amination of C–H bonds at tertiary carbon centers using iodic acid as an oxidant, Chem. Commun., 2016, 52, 13082–13085 RSC.
- E. Kotani, S. Kobayashi, Y. Ishii and S. Tobinaga, A new combined oxidizing reagent system, Fe(CH3CN)63+–IO4−: oxidation of paraffin hydrocarbons, Chem. Pharm. Bull., 1985, 33, 4680–4684 CrossRef CAS.
- J. Lee, S. Jin, D. Kim, S. H. Hong and S. Chang, Cobalt-catalyzed intermolecular C–H amidation of unactivated alkanes, J. Am. Chem. Soc., 2021, 143, 5191–5200 CrossRef CAS PubMed.
- A. M. Fuentes, R. Gava, N. I. Saper, E. A. Romero, A. Cabellero, J. F. Hartwig and P. J. Pérez, Copper-catalyzed dehydrogenative amidation of light alkanes, Angew. Chem., Int. Ed., 2021, 60, 18467–18471 CrossRef PubMed.
- A. Bakhoda, Q. Jiang, Y. M. Badiei, J. A. Bertke, T. R. Cundari and T. H. Warren, Copper-catalyzed C(sp3)–H amidation: sterically driven primary and secondary C–H site selectivity, Angew. Chem., Int. Ed., 2019, 58, 3421–3425 CrossRef CAS PubMed.
- S. V. Athavale, S. Gao, A. Das, S. C. Mallojjala, E. Alfonz, Y. Long, J. S. Hirschi and F. H. Arnold, Enzymatic nitrogen insertion into unactivated C–H bonds, J. Am. Chem. Soc., 2022, 144, 19097–19105 CrossRef CAS PubMed.
- Q. Wang, S. Ni, L. Yu, Y. Pan and Y. Wang, Photoexcited direct amination/amidation of inert Csp3–H bonds via tungsten-nickel catalytic relay, ACS Catal., 2022, 12, 11071–11077 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, computational files, product characterization data and NMR spectra, and CV, UV/Vis, fluorescence, and transient absorption spectra. CCDC 2255574. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03054k |
| This journal is © The Royal Society of Chemistry 2024 |