
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organofunctionalized borotungstate polyoxometalates as tunable photocatalysts for oxidative dimerization of amines†
Nicole
Tsang
 ab,
Alexander J.
Kibler
ab,
Stephen P.
Argent
b,
Hon Wai
Lam
*ab,
Kieran D.
Jones
*ab and
Graham N.
Newton
*ab
ab,
Alexander J.
Kibler
ab,
Stephen P.
Argent
b,
Hon Wai
Lam
*ab,
Kieran D.
Jones
*ab and
Graham N.
Newton
*ab
aThe GSK Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Jubilee Campus, Triumph Road, Nottingham, NG7 2TU, UK. E-mail: hon.lam@nottingham.ac.uk; kieran.jones@nottingham.ac.uk; graham.newton@nottingham.ac.uk
bSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, UK
First published on 12th August 2024
Abstract
Organofunctionalized borotungstate Keggin polyoxometalates, (nBu4N)3H[HBW11O39(P(O)Ph)2] (PBW11), (nBu4N)3H[HBW11O39(As(O)Ph)2] (AsBW11), and (nBu4N)4[HBW11O39(PhSiOSiPh)] (SiBW11), were synthesized and structurally characterized. Cyclic voltammetry showed that the electronic properties of the clusters are dependent on the nature of the appended main group atoms (P, As, or Si). The first reduction potentials were found to shift positively with respect to that of the unmodified parent species (nBu4N)5[BW12O40], with PBW11 showing the largest shift at +100 mV. All clusters were evaluated as photocatalysts for the oxidative dimerization of amines where the organophosphonate hybrid PBW11 was found to be the most active. This study demonstrates how organofunctionalization of polyoxometalates may be used to tune and improve their performance as photocatalysts for organic reactions.
1 Introduction
Polyoxometalates (POMs) are anionic metal oxide clusters with tunable photo- and electrochemical properties.1 Typically, they can be reversibly interconverted between multiple redox states, allowing them to function as effective electron and proton reservoirs. This versatility renders POMs promising materials for applications such as energy storage,2–5 materials science,6,7 and catalysis.8–10 They have been shown to facilitate a range of organic reactions, including oxidations and reductions,11 hydrolysis,12 and Lewis acid processes,13,14 acting as both electrocatalysts15,16 and photocatalysts.17,18 The most prominent example of a POM photocatalyst in the literature is tetrabutylammonium decatungstate (TBADT), (nBu4N)4[W10O32], an isopolyoxometalate that has proven to be efficient for hydrogen atom transfer (HAT) chemistry.19,20 While other POM catalysts have been reported to operate through single electron transfer (SET)21 and proton-coupled electron transfer (PCET) reaction pathways,22 none have matched the reaction diversity and performance of TBADT.In contrast to isopolyoxometalates such as TBADT, in which the cluster anion comprises only metal ions and oxide ligands, heteropolyoxometalates contain templating anions (i.e. PO43−, SiO44−, or BO45−) that stabilize the clusters to the extent that metal vacancies can be introduced and lacunary POMs isolated. These metal vacancy sites on monolacunary polyoxotungstates can be filled by main group elements such as PV, AsV, SiIV, and SnIV, which can act as tethers to an effectively unlimited range of organic moieties yielding hybrid organic–inorganic POMs.23–25 The electrochemical and photochemical properties of these hybrid POMs depend on the nature of the metal addenda ions (commonly W or Mo), the templating anion, the counter-cations, the main group linker atoms, and the appended organic groups, which gives the synthetic chemist a plethora of options when designing functional POMs.26
The modification of heteropolyoxometalate structures, often by transition metal substitution,27–30 has been shown to improve POM performance as SET photocatalysts. The oxidation of amines by a photoexcited POM photocatalyst to yield aminium radical cations (Fig. 1), versatile reactive intermediates used in amination chemistry,31 followed by regeneration of the catalyst by an oxidant has served as a benchmarking reaction for these POM photocatalyts. While organofunctionalization has proven effective in enhancing the visible light activity of heteropolyoxometalates compared to their non-hybridized plenary anion counterparts,32–36 this strategy has yet to be demonstrated towards improved photocatalysis for organic synthesis. Hybridization with organophosphonates results in visible light active Wells–Dawson phosphotungstate photocatalysts for the photodegradation of the pollutant indigo dye,35 however, the stability of their reduced state means that these hybrid POMs are oxidized only slowly in air. Overall, this makes for an inefficient catalytic cycle, impacting their broader applicability as photocatalysis for organic synthesis. We hypothesized that modulation of the frontier orbital energies of POMs through organofunctionalization could lead to improved catalytic turnover, preferably using molecular oxygen as a sustainable oxidant, and application of the systems in organic reactions. Considering the limitations of existing phosphotungstate systems, hybrid POMs derived from underexplored heteropolytungstates may yield promising candidates for an aerobic-SET photocatalytic cycle.
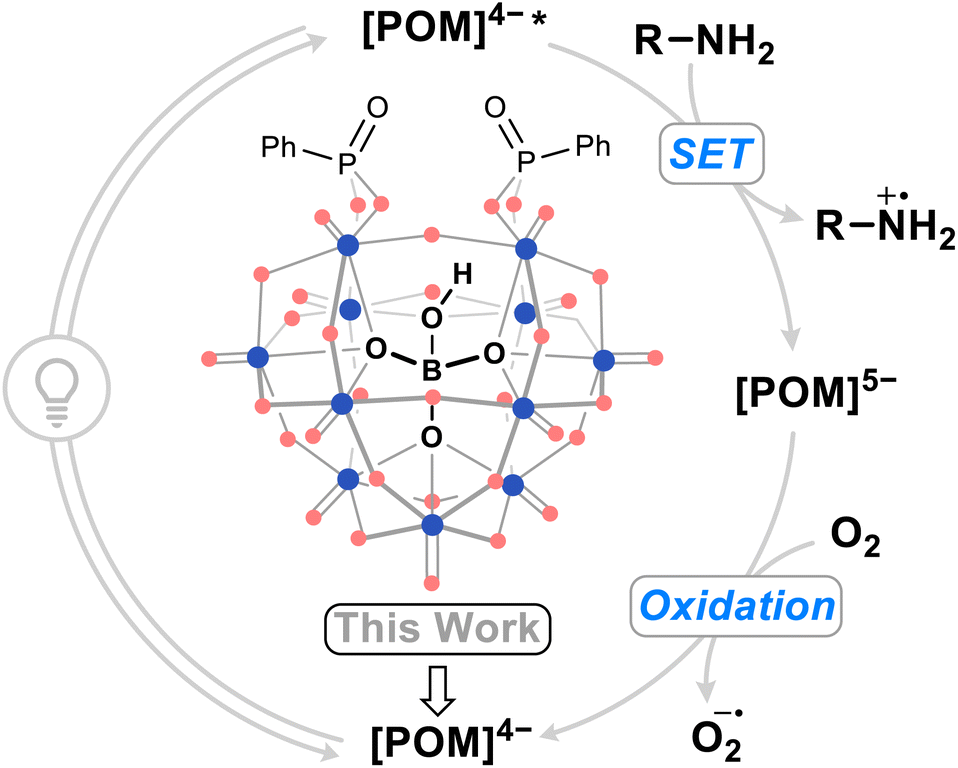 | ||
| Fig. 1 Summarized photocatalytic cycle of organic–inorganic hybrid borotungstate POM-mediated single electron transfer (SET) with amines. Blue dots = WVI, red dots = oxygen. | ||
To the best of our knowledge, only three examples of organofunctionalized borotungstates, all bearing organo(thio)phosphonate groups, are known in literature.37–39 These reports are inconsistent in their cluster charge assignments, and no electrochemical properties have been described. Herein, we report the synthesis and characterization of three organofunctionalized Keggin borotungstate hybrid POMs, namely (nBu4N)3H[HBW11O39(P(O)Ph)2] (PBW11), (nBu4N)3H[HBW11O39(As(O)Ph)2] (AsBW11), and (nBu4N)4[HBW11O39(PhSiOSiPh)] (SiBW11). Their structural characterization and electronic properties are discussed, with particular emphasis on the impact of hybridization on promoting photocatalytic behavior.
2 Results and discussion
2.1 Synthesis and characterization of hybrid borotungstates
Hybrid POMs (nBu4N)3H[HBW11O39(P(O)Ph)2] (PBW11), (nBu4N)3H[HBW11O39(As(O)Ph)2] (AsBW11), and (nBu4N)4[HBW11O39(PhSiOSiPh)] (SiBW11) were prepared by hybridization of the lacunary K8[HBW11O39] (KBW11) with phenylphosphonic dichloride, phenylarsonic acid, or trimethoxy(phenyl)silane, respectively (Fig. 2). Hybrid POMs PBW11, AsBW11, and SiBW11 were characterized by NMR, UV-Vis and IR spectroscopies and electrospray ionization mass spectrometry (see ESI† for details). These hybrid POMs exhibited an anionic charge of 4− instead of the expected 5− due to monoprotonation of the templating anion. This proton was observed as a distinctive singlet peak in the range 4.5–4.9 ppm in the 1H NMR spectra (400 MHz, CD3CN), corresponding to a B–O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) peak. By 11B NMR spectroscopy, the hybrid borotungstates
peak. By 11B NMR spectroscopy, the hybrid borotungstates ![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif) O4H is observed as a characteristic broad singlet (2.04–2.13 ppm, 128 MHz, CD3CN), due to the quadrupolar effect.40,41 Protonation of the templating anion is not observed in the analogous hybrid silicotungstate [SiW11O39(P(O)Ph)2]4− or phosphotungstate [PW11O39(P(O)Ph)2]3− and may be ascribed to the comparatively smaller size and greater charge density of the central boron atom.42
O4H is observed as a characteristic broad singlet (2.04–2.13 ppm, 128 MHz, CD3CN), due to the quadrupolar effect.40,41 Protonation of the templating anion is not observed in the analogous hybrid silicotungstate [SiW11O39(P(O)Ph)2]4− or phosphotungstate [PW11O39(P(O)Ph)2]3− and may be ascribed to the comparatively smaller size and greater charge density of the central boron atom.42
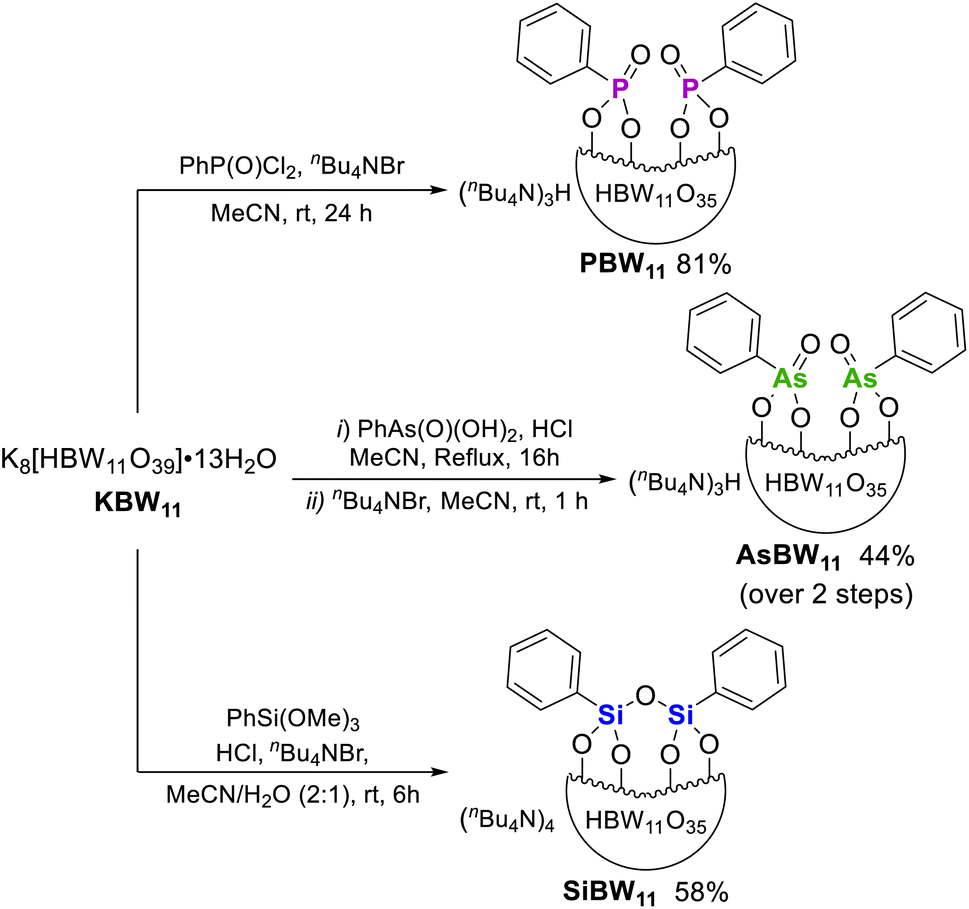 | ||
| Fig. 2 Synthesis of organic–inorganic hybrid borotungstates PBW11, AsBW11, and SiBW11 from the lacunary borotungstate. | ||
Crystals of K3H[HBW11O39(P(O)Ph)2] and K3H[HBW11O39(As(O)Ph)2] (Fig. 3) suitable for single crystal X-ray diffraction were prepared by the vapor diffusion method with MeCN and tert-butyl methyl ether (anti-solvent). The structures solve in monoclinic space groups P21 and P21/n, respectively (see ESI† for details). In both cases, three potassium cations per POM anion cluster were identified, supporting the assigned elemental compositions, and two phenylpnictogen groups were covalently bound to these clusters in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in a manner analogous to previously reported phenylpnictogen hybrid polyoxotungstates.23,35,37,43 Crystallographic support for the assignment and location of the templating boronate hydrogen atom for K3H[HBW11O39(As(O)Ph)2] was made by a hydrogen omit map calculated for all of the data which indicated an electron density peak of 0.49 e− Å−3 in the refined location of the hydrogen atom B–O (see ESI† for details). This marks the first crystallographic report supporting the assigned protonated hybrid borotungstate structure.
:1 ratio in a manner analogous to previously reported phenylpnictogen hybrid polyoxotungstates.23,35,37,43 Crystallographic support for the assignment and location of the templating boronate hydrogen atom for K3H[HBW11O39(As(O)Ph)2] was made by a hydrogen omit map calculated for all of the data which indicated an electron density peak of 0.49 e− Å−3 in the refined location of the hydrogen atom B–O (see ESI† for details). This marks the first crystallographic report supporting the assigned protonated hybrid borotungstate structure.
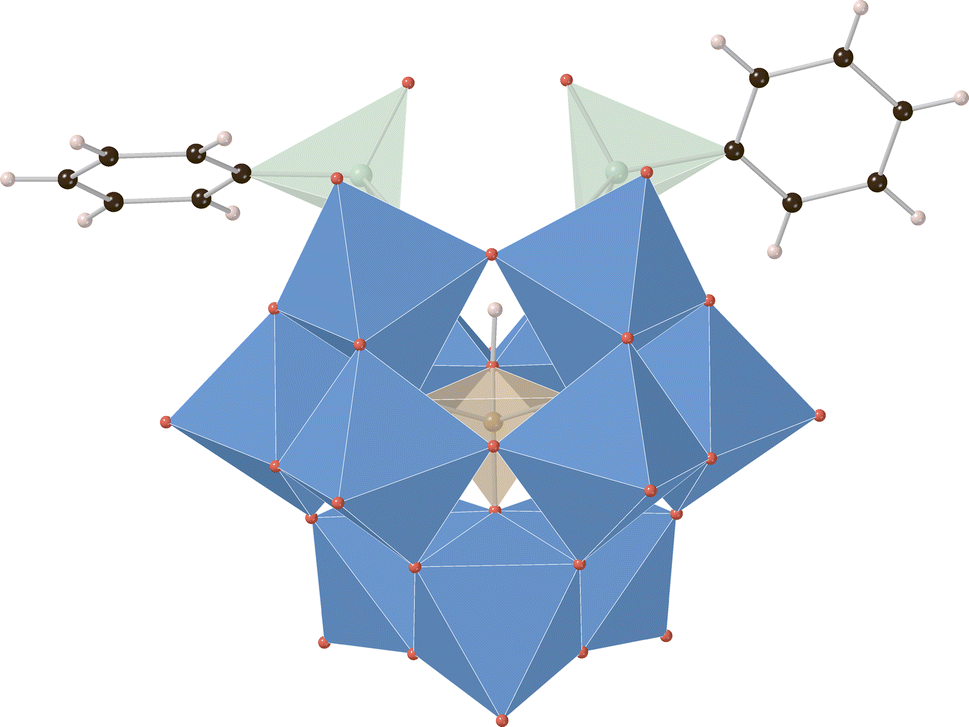 | ||
| Fig. 3 X-ray crystal structure of K3H[HBW11O39(As(O)Ph)2] with disordered elements, solvent, and potassium cations omitted for clarity. Color code: blue = W, red = O, black = carbon, green = As, orange = B, pink = H. | ||
The hydrolytic stability of a hybrid POM may be a useful indicator of the stability and suitability of the cluster as a photocatalyst, particularly under protic conditions. Previous studies have shown aryl phosphonate hybridized phospho-and silico-tungstate POMs to undergo hydrolytic cleavage of the phosphoryl group from the lacunary site within a period of 24 h.35,43,44 To date, the hydrolytic stability of hybrid borotungstates has not been investigated, and therefore compounds PBW11, AsBW11, and SiBW11 were dissolved in DMSO-d6:D2O (9:1) and monitored over time using 1H, 31P, and 11B NMR spectroscopies (Fig. S9–15†). The hydrolytic stability of organophosphonate hybrid PBW11 is similar to that reported for the isoionic silicotungstate PSiW11,43 with hydrolyzed phenyl phosphonate (12.97 ppm) accumulating slowly over a 24 h period as indicated by 31P NMR spectroscopy (162 MHz, 298 K). The starting material PBW11 (20.42 ppm) was still present as a major component even after 7 days. Conversely, organoarsonate hybrid AsBW11 was observed to undergo immediate hydrolysis upon the addition of D2O, evident through the disappearance of BO peak at 4.81 ppm and change in appearance of the aromatic region in the 1H NMR spectrum (400 MHz, 298 K). By 11B NMR spectroscopy (128 MHz, 298 K), the absence of the broad singlet at 1.83 ppm, indicative of AsBW11, and presence of a sharp singlet at 1.76 ppm suggests that following cleavage of the phenyl arsonate groups, the metastable lacunary [HBW11O39]8− reacts further to give the favored plenary Keggin species [BW12O40]5−. Finally, the organosilyl POM SiBW11 was found to have excellent hydrolytic stability over a month, with no apparent change in the aromatic region of the 1H NMR spectrum (400 MHz, 298 K) and unchanged 11B NMR spectroscopic (128 MHz, 298 K) data, suggesting little to no cleavage of the phenylsiloxane unit. Notably, the relative integration of the BO peak at 4.72 ppm in the 1H NMR spectrum decreased by ca. 65% over the first week; proton–deuterium exchange (B–OH/D) is proposed to have occurred without affecting the hybrid POM structure.
2.2 Electronic characterization
Cyclic voltammetry (CV) was performed in DMF [glassy carbon (d = 3 mm) working electrode, Pt wire counter electrode, and Ag+|Ag reference electrode, in 0.1 M nBu4NPF6 supporting electrolyte] to explore the redox properties of hybrid POMs PBW11, AsBW11, and SiBW11, with comparison to the parent plenary POM (nBu4N)5[BW12O40] (BW12) (Fig. 4, left). In all cases, quasi-reversible redox couples corresponding to one-electron WVI/WV processes were observed. The first reduction potentials of the hybrid POMs PBW11, AsBW11, and SiBW11 were shifted positively (by between 0.4 V and 1 V) compared to that of the unmodified BW12, attributed to the difference in cluster charge.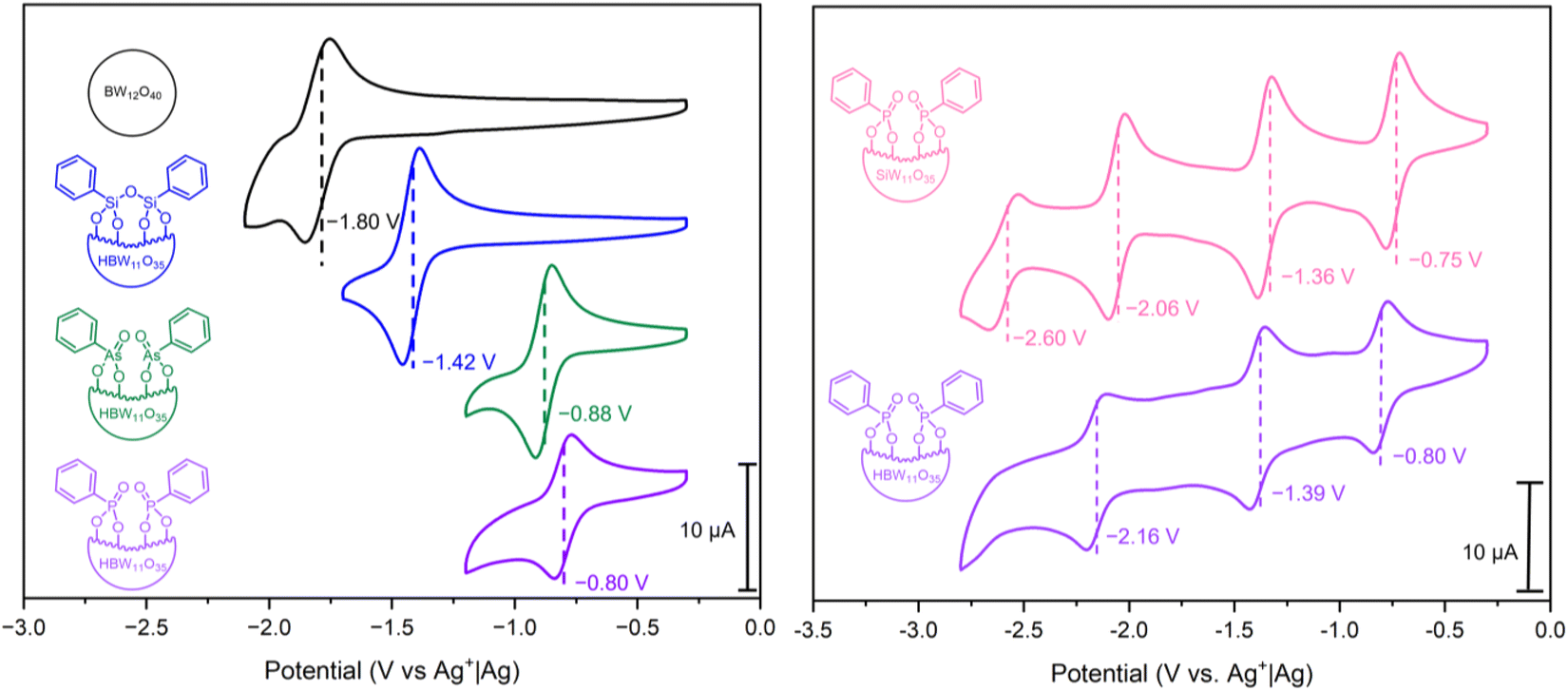 | ||
| Fig. 4 (Left) CV of 1 mM of compounds BW12, PBW11, AsBW11, and SiBW11 in anhydrous DMF with 0.1 M nBu4NPF6 supporting electrolyte. (Right) CV of 1 mM of compounds PBW11 and PSiW11 in anhydrous DMF with 0.1 M nBu4NPF6 supporting electrolyte. All CVs were recorded at 100 mV s−1 using a glassy carbon working electrode (d = 3 mm and A = 0.071 cm2), Pt wire counter electrode and a AgNO3|Ag non-aqueous reference electrode. The standard potentials are highlighted for the redox processes. | ||
Organophosphonate PBW11 exhibits the most positive first redox process (E1/2 = −0.80 V vs. Ag+|Ag), followed by the organoarsonate hybrid AsBW11 (E1/2 = −0.88 V vs. Ag+|Ag), and then the organosilyl hybrid SiBW11 (E1/2 = −1.42 V vs. Ag+|Ag). This trend in redox potential can be ascribed to the electron-withdrawing nature of the phosphonate and arsonate groups compared to the silyl anchoring group.23,45 Furthermore, the 80 mV difference between the phosphonate PBW11 and arsonate AsBW11 is likely due to the variance in bond lengths and hence differing frontier orbital overlap. A similar observation was made in analysis of the previously reported phenylpnictogen hybrid Wells–Dawson phosphotungstate variants.23
As an extension of this study, the previously reported organofunctionalized silicotungstate (nBu4N)3H[SiW11O39(P(O)Ph)2] (PSiW11) was synthesized and electronically characterized (see ESI† for details).43 Given the unique protonation of the central borate anion and subsequent charge of borotungstate hybrid POMs (4−), the silicotungstate hybrid can be described as an isoionic analogue of hybrid PBW11, with the only difference being the central templating anion. The CV of PSiW11 displayed four quasi-reversible redox processes (Fig. 4, right), with the first redox process being shifted +50 mV (E1/2 = −0.75 V vs. Ag+|Ag) compared to its isoionic analogue.42 Beyond the first redox potential, the voltammetry of PSiW11 and PBW11 share a similar voltametric profile with the next two reversible processes all falling within 100 mV of the corresponding processes in the voltammogram. Based on the relative reduction potentials, PSiW11 is the most readily reduced POM in the series. This is consistent with it having the lowest LUMO energy.
In the absorption spectra, a hyperchromic effect was observed for hybrids PBW11, AsBW11, and SiBW11 compared to the parent borotungstate POM BW12. A similar absorption profile was also observed for silicotungstate hybrid PSiW11. The borotungstate hybrids exhibit a higher absorbance in the UV region relative to the plenary POM, while hybrid SiBW11 has a lower absorbance relative to the other hybrids (Fig. S8†).
2.3 Photocatalytic oxidative dimerization of amines
The oxidative dimerization of amines serves as an effective reaction to benchmark the single electron transfer (SET) photocatalytic activity (Fig. 1) of our new borotungstate hybrid POMs (Table S6†).27,29,30,46,47 The plenary BW12, lacunary (nBu4N)8[HBW11O39] (BW11), hybrid PBW11, AsBW11, SiBW11 borotungstates, as well as the isoionic silicotungstate PSiW11 were tested as SET photocatalysts for the oxidative dimerization of benzyl amine. A solution of the amine 1a and POM (4 mol%) in MeCN was irradiated at 370 nm for 24 h (Fig. 5). The NMR yield of the target imine 2a was determined by 1H NMR spectroscopy, with 1,3-benzodioxole as the internal standard. Under aerobic conditions, the plenary (BW12) and lacunary (BW11) Keggin borotungstates gave imine 2a in low yields, 10% and 20% respectively. Under the same conditions, hybrids PBW11, AsBW11, and SiBW11 gave yields between 32% and 37%, indicating that organofunctionalization of Keggin borotungstates improves catalysis in this reaction. In comparison, the TBADT-catalyzed reaction gave the desired imine in 5% yield, accompanied by a multitude of side products attributed to competitive hydrogen atom transfer at the benzylic position and subsequent radical reactions with oxygen and imines leading to a series of hydroperoxides, amides, and various oligomers derived from 1a and 2a.19,20,48,49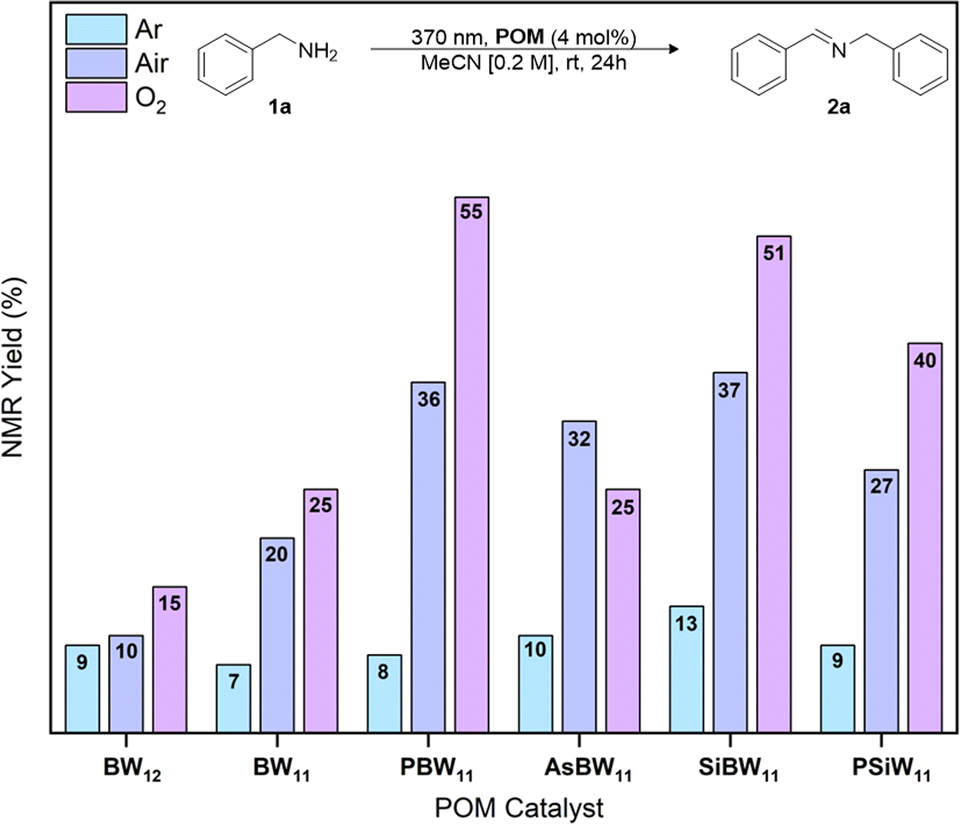 | ||
| Fig. 5 POM-mediated photooxidative dimerization of benzyl amine under anaerobic or aerobic conditions. NMR yield determined by 1H NMR spectroscopic analysis using 1,3-benzodioxole as an internal standard. | ||
Conducting the reaction in an anaerobic environment led to reduced yields for all POMs suggesting molecular oxygen is essential for catalytic turnover. With this in mind, we performed the same reactions under an oxygen atmosphere and observed a substantial increase in yield of the imine 2a when using PBW11 or SiBW11 (55% and 51%, respectively). The isoionic analogue PSiW11 followed a similar trend to the borotungstate PBW11, where an increase of yield from 27% to 40% was achieved with change of atmosphere (aerobic to O2, respectively). In comparison, these conditions did not improve the yield of 2a when AsBW11 was used, marking AsBW11 as the poorest performing catalyst of the hybrid POMs investigated, likely resulting from its limited hydrolytic and electrochemical stability (Fig. S6, S12 and 13†).
Based on these results, phenylphosphonate hybrid borotungstate PBW11 is the lead candidate as a SET photocatalyst for oxidative amine dimerization. The reaction parameters were further assessed using the Glorius robustness model and represented as a radar graph (Fig. 6).50 The percentage deviation in yield from the model reaction was determined, where negative integers represent a decrease in NMR yield, and positive integers signify an increase in NMR yield (Table S5†). In the absence of POM or light, no reaction occurred. The use of a longer wavelength of light (390 nm) was ineffective, as expected from the absorption spectrum of PBW11 (Fig. S8†). A 2 mol% catalyst loading was optimum, with a lower loading (0.5 mol%) resulting in a drop in yield (38%, −31% deviation from model), while increased loading did not significantly improve the yield. The reaction was robust towards concentration changes, and we therefore opted for a higher concentration of 0.4 M to reduce solvent waste. Changing the solvent from MeCN to pivalonitrile, DMF, or acetone was unfavorable, especially in the case of acetone which completely shut down reactivity. Finally, the optimal conditions were determined to be 24 h irradiation (370 nm – 43 W Kessil LED light) of benzylamine with POM catalyst PBW11 (2 mol%), dissolved in MeCN [0.4 M], under an oxygen atmosphere, which gave the desired imine product 2a in a moderate yield of 57%.
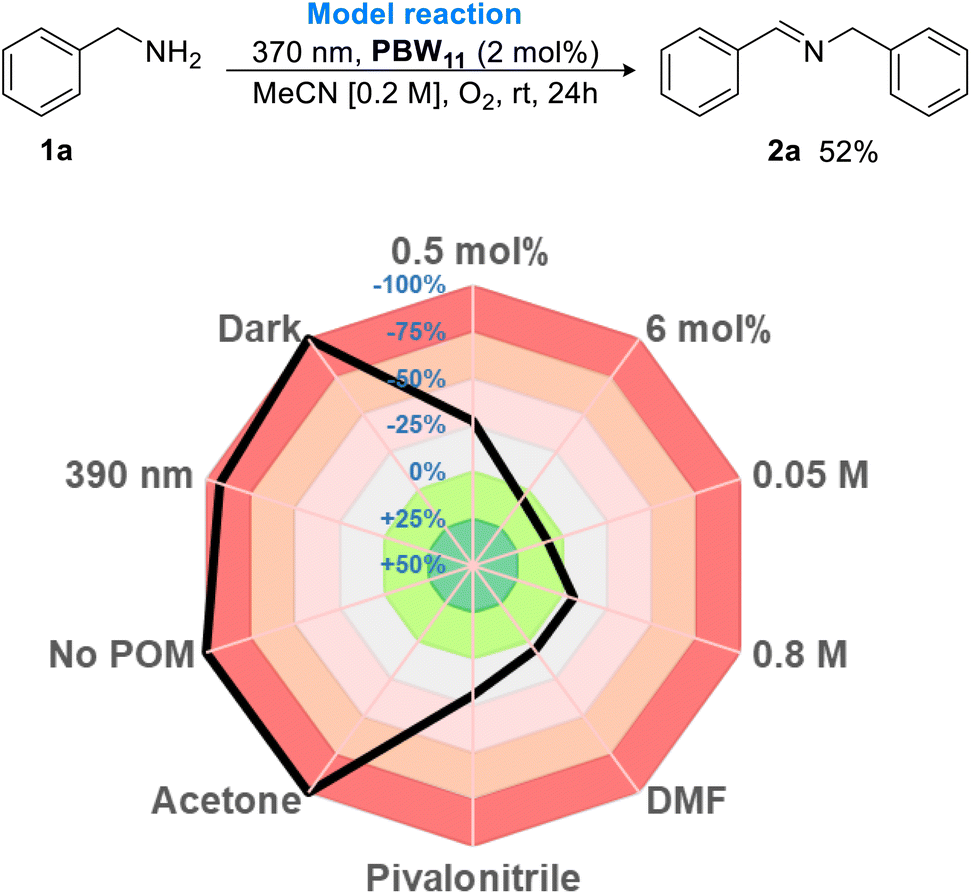 | ||
| Fig. 6 Optimization of PBW11 photocatalyzed oxidative dimerization of benzyl amine illustrated as a color-coded radar diagram. Yields determined by 1H NMR analysis using 1,3-benzodioxole as an internal standard and presented as a percentage deviation from the model reaction. | ||
With optimized conditions in hand, the scope of the borotungstate photocatalytic oxidative dimerization reaction of various amines was explored (Fig. 7). Cyclohexylmethylamine gave imine 2b in 8% yield, indicating the importance of an activated methylene unit for acceptable yields. Thus, a series of para-, meta-, and ortho-substituted benzyl amines bearing alkyl, methoxy, phenyl, as well as halogen-containing groups were evaluated and these gave the desired imines in moderate yields with little variance (44–62%), irrespective of substituent effects. Largely, these reactions proceeded cleanly with reasonable starting material recovery being observed for 2a, 2b, 2e–i and 2k (45%, 47%, 49%, 37%, 17%, 11%, 51%, 54%, respectively). Given the use of an energetic UVA light source with an atmosphere of oxygen, catalyst-free reactions were also conducted to assess radical-initiated autoxidation reaction pathways for each substrate.51,52 Generally, without the POM catalyst, oxidative dimerization of primary benzylic amines was insignificant (yields <10%), highlighting the efficacy of PBW11. However, anisole-, biphenyl-, and thiophene-bearing amines yielded secondary aldimines 2d, 2h, and 2l, respectively, in low to moderate yields (34–40%). Overall, PBW11 was determined to be a consistently effective photocatalyst for the oxidative dimerization of activated methylamines, albeit, giving moderate yields.
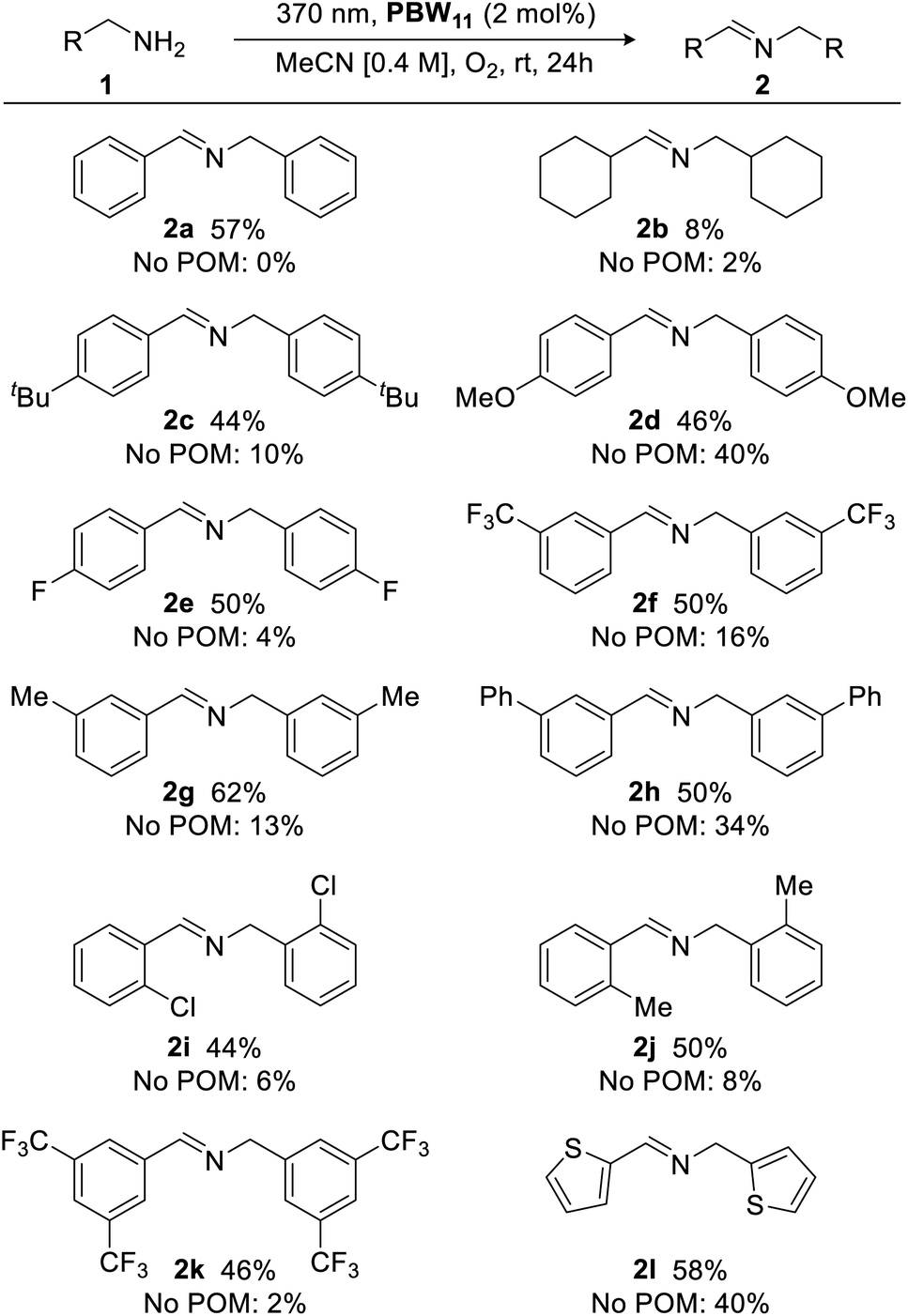 | ||
| Fig. 7 Photocatalytic oxidative dimerization of various amines. Yields determined by 1H NMR analysis using 1,3-benzodioxole as an internal standard. | ||
3 Conclusions
We have described the synthesis of three organofunctionalized Keggin borotungstate POMs bearing phenyl-substituted phosphonate (PBW11), arsonate (AsBW11) or siloxane (SiBW11) groups, each of which were structurally and electronically characterized, as well as investigated as photocatalysts for the oxidative dimerization of amines. p-Block organofunctionalization was found to dramatically influence the fundamental properties of borotungstate POMs with the phenylphosphonate hybrid determined to be the most effective catalyst in the series. PBW11 also outperformed the isoionic silicotungstate analogue PSiW11, indicating the importance of both the templating anion and linker group when considering hybrid heteropolyoxometalates as functional materials. Overall, this study has led to the development of the first generation of covalently modified organic–inorganic hybrid POMs capable of turning over photocatalytic single electron transfer chemistry for organic synthesis, a significant step forward in the field of hybrid POMs. It is also particularly interesting to note the improvement in catalytic performance of the organofunctionalized systems compared to the established photo-oxidation catalyst tetrabutylammonium decatungstate. The design strategy described in this study illustrates how POM-based catalysts can be tuned and optimized to drive specific organic reactions.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.Author contributions
GNN, KDJ and HWL conceptualized and supervised the project, contributed to data analysis and co-wrote the manuscript. NT performed experimental work, helped to develop the catalysis methodology and helped to write the manuscript. AJK contributed additional data analysis and experimental support. SPA managed the collection and analysis of single crystal data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) and Science Foundation Ireland (SFI) Centre for Doctoral Training in Sustainable Chemistry (grant number EP/S022236/1) and the EPSRC New Horizons scheme (grant number EP/V047124/1). We gratefully acknowledge Diamond Light Source for access to beamline I19 on award CY28766.Notes and references
- T. Yamase, Chem. Rev., 1998, 98, 307 CrossRef CAS PubMed.
- Y. Ji, L. Huang, J. Hu, C. Streb and Y. F. Song, Energy Environ. Sci., 2015, 8, 776 RSC.
- S. Herrmann, C. Ritchie and C. Streb, Dalton Trans., 2015, 44, 7092 RSC.
- Q. Li, L. Zhang, J. Dai, H. Tang, Q. Li, H. Xue and H. Pang, J. Chem. Eng., 2018, 351, 441 CrossRef CAS.
- C. L. Peake, A. J. Kibler, G. N. Newton and D. A. Walsh, ACS Appl. Energy Mater., 2021, 4, 8765 CrossRef CAS.
- J.-H. Kruse, M. Langer, I. Romanenko, I. Trentin, D. Hernández-Castillo, L. González, F. H. Schacher, C. Streb, J.-H. Kruse, I. Romanenko, F. H. Schacher, M. Langer, I. Trentin, C. Streb, D. Hernández-Castillo and L. González, Adv. Funct. Mater., 2022, 32, 2208428 CrossRef CAS.
- J. W. Jordan, J. M. Cameron, G. A. Lowe, G. A. Rance, K. L. Y. Fung, L. R. Johnson, D. A. Walsh, A. N. Khlobystov and G. N. Newton, Angew. Chem., Int. Ed., 2022, 61, e20211561 CrossRef PubMed.
- Y. Feng, F. Fu, L. Zeng, M. Zhao, X. Xin, J. Liang, M. Zhou, X. Fang, H. Lv and G. Y. Yang, Angew. Chem., Int. Ed., 2024, 63, e202317341 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed.
- R. Liu and C. Streb, Adv. Energy Mater., 2021, 11, 2101120 CrossRef CAS.
- M. Yamaguchi, K. Shioya, C. Li, K. Yonesato, K. Murata, K. Ishii, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2024, 146, 4549 CrossRef CAS PubMed.
- F. F. Martins, Á. Sánchez-González, J. Lanuza, H. N. Miras, X. Lopez, N. A. Bandeira and A. Gil, Chem.–Eur. J., 2021, 27, 8977 CrossRef CAS PubMed.
- Y. F. Liu, C. W. Hu and G. P. Yang, Chin. Chem. Lett., 2023, 34, 108097 CrossRef CAS.
- K. Suzuki, M. Sugawa, Y. Kikukawa, K. Kamata, K. Yamaguchi and N. Mizuno, Inorg. Chem., 2012, 51, 6953 CrossRef CAS PubMed.
- S. Kuramochi, J. M. Cameron, T. Fukui, K. D. Jones, S. P. Argent, S. Kusaka, R. Shiraishi, Y. Tamaki, T. Yatsu, T. Shiga, O. Ishitani, H. Oshio and G. N. Newton, Chem. Commun., 2023, 59, 10801 RSC.
- Y. Zhang, Y. Li, H. Guo, Y. Guo and R. Song, Mater. Chem. Front., 2024, 8, 732 RSC.
- C. Streb, K. Kastner and J. Tucher, Phys. Sci. Rev., 2019, 4, 20170177 Search PubMed.
- K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809 CrossRef CAS.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701 CrossRef CAS.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875 CrossRef CAS PubMed.
- K. Suzuki, N. Mizuno and K. Yamaguchi, J. Jpn. Pet. Inst., 2020, 63, 258 CrossRef CAS.
- A. A. Fertig and E. M. Matson, Inorg. Chem., 2022, 62, 24 Search PubMed.
- A. J. Kibler, N. Tsang, M. Winslow, S. P. Argent, H. W. Lam, D. Robinson and G. N. Newton, Inorg. Chem., 2023, 62, 3585 CrossRef CAS PubMed.
- A. J. Kibler and G. N. Newton, Polyhedron, 2018, 154, 1 CrossRef CAS.
- D. E. Salazar Marcano, M. A. Moussawi, A. V. Anyushin, S. Lentink, L. Van Meervelt, I. Ivanović-Burmazović and T. N. Parac-Vogt, Chem. Sci., 2022, 13, 2891 RSC.
- A. V Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382 RSC.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356 CrossRef CAS PubMed.
- Z. Shi, J. Li, Q. Han, X. Shi, C. Si, G. Niu, P. Ma and M. Li, Inorg. Chem., 2019, 58, 12529 CrossRef CAS PubMed.
- W. Chen, H. Li, J. Song, Y. Zhao, P. Ma, J. Niu and J. Wang, Inorg. Chem., 2022, 61, 2076 CrossRef CAS PubMed.
- S. Li, G. Li, P. Ji, J. Zhang, S. Liu, J. Zhang and X. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 43287 CrossRef CAS PubMed.
- J. M. Ganley, P. R. D. Murray and R. R. Knowles, ACS Catal., 2020, 10, 11712 CrossRef CAS PubMed.
- F. Odobel, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer, K. J. Elliott and A. Harriman, Chem.–Eur. J., 2009, 15, 3130 CrossRef CAS PubMed.
- S. Amthor, S. Knoll, M. Heiland, L. Zedler, C. Li, D. Nauroozi, W. Tobaschus, A. K. Mengele, M. Anjass, U. S. Schubert, B. Dietzek-Ivanšić, S. Rau and C. Streb, Nat. Chem., 2022, 14, 321 CrossRef CAS PubMed.
- A. Parrot, A. Bernard, A. Jacquart, S. A. Serapian, C. Bo, E. Derat, O. Oms, A. Dolbecq, A. Proust, R. Métivier, P. Mialane and G. Izzet, Angew. Chem., Int. Ed., 2017, 56, 4872 CrossRef CAS PubMed.
- S. Fujimoto, J. M. Cameron, R.-J. Wei, K. Kastner, D. Robinson, V. Sans, G. N. Newton and H. Oshio, Inorg. Chem., 2017, 56, 12169 CrossRef CAS PubMed.
- J. M. Cameron, D. J. Wales and G. N. Newton, Dalton Trans., 2018, 47, 5120 RSC.
- N. Andino, B. Artetxe, S. Reinoso, P. Vitoria, L. S. Felices, J. I. Martínez, F. L. Arbeloa and J. M. Gutiérrez-Zorrilla, Inorg., 2016, 4, 14 CrossRef.
- Z. G. Sun, Q. Liu and J. F. Liu, Polyhedron, 2000, 19, 125 CrossRef CAS.
- (a) Z. G. Sun, L. Y. Cui, Z. M. Zhu, X. H. Yu and X. F. Zheng, J. Coord. Chem., 2005, 58, 1321 CrossRef CAS; (b) Z. G. Sun, J. Liu, J. Ma and J. Liu, Met.-Based Drugs, 2002, 8, 257 CrossRef PubMed.
- A. Tézé, M. Michelon and G. Hervé, Inorg. Chem., 1997, 36, 505 CrossRef.
- R. I. Maksimovskaya and G. M. Maksimov, Inorg. Chem., 2011, 50, 4725 CrossRef CAS PubMed.
- I. M. Mbomekallé, X. López, J. M. Poblet, F. Sécheresse, B. Keita and L. Nadjo, Inorg. Chem., 2010, 49, 7001 CrossRef PubMed.
- G. S. Kim, K. S. Hagen and C. L. Hill, Inorg. Chem., 1992, 31, 5316 CrossRef CAS.
- S. S. Amin, K. D. Jones, A. J. Kibler, H. A. Damian, J. M. Cameron, K. S. Butler, S. P. Argent, M. Winslow, D. Robinson, N. J. Mitchell, H. W. Lam and G. N. Newton, Angew. Chem., Int. Ed., 2023, 62, e202302446 CrossRef CAS PubMed.
- M. Boujtita, J. Boixel, E. Blart, C. R. Mayer and F. Odobel, Polyhedron, 2008, 27, 688 CrossRef CAS.
- Y. Liu, J. Wang, K. Ji, S. Meng, Y. Luo, H. Li, P. Ma, J. Niu and J. Wang, J. Catal., 2022, 416, 149 CrossRef CAS.
- H. Li, W. Chen, Z. Yuan, Y. Jin, Y. Zhao, P. Ma, J. Niu and J. Wang, Inorg. Chem., 2022, 61, 9935 CrossRef CAS PubMed.
- V. I. Supranovich, V. V Levin and A. D. Dilman, Org. Lett., 2019, 21, 4271 CrossRef CAS PubMed.
- C. Gu, T. Yatabe, K. Yamaguchi and K. Suzuki, Chem. Commun., 2024, 60, 4906 RSC.
- L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572 CrossRef CAS PubMed.
- X. Guo, B. R. Shao, W. F. Jiang and L. Shi, J. Org. Chem., 2021, 86, 15743 CrossRef CAS PubMed.
- Y. Zhao, X. Hou, M. He, Y. Wang, S. Yang, W. Wang, M. Bao and X. Yu, Org. Lett., 2023, 25, 7344 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC Crystallographic data files and CheckCIFs for K3H[HBW11O39(P(O)Ph)2] and K3H[HBW11O39(As(O)Ph)2] can be accessed through the Cambridge Structural Database with deposition numbers CCDC 2358933 and CCDC 2358934 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03534h |
| This journal is © The Royal Society of Chemistry 2024 |