
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceViscoelasticity of globular protein-based biomolecular condensates†
Rachel S.
Fisher
 and
Allie C.
Obermeyer
*
and
Allie C.
Obermeyer
*
Department of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: aco2134@columbia.edu; Tel: +1-212-853-1315
First published on 15th November 2024
Abstract
The phase separation of biomolecules into biomolecular condensates has emerged as a ubiquitous cellular process. Understanding how intrinsically disordered protein sequence controls condensate formation and material properties has provided fundamental biological insights and led to the development of functional synthetic condensates. While these studies provide a valuable framework to understand subcellular organization via phase separation they have largely ignored the presence of folded domains and their impact on condensate properties. We set out to determine how the distribution of sticker interactions across a globular protein contributes to rheological properties of condensates and to what extent globular protein-containing condensates differ from those formed from two disordered components. We designed three variants of green fluorescent protein with different charge patterning and used dynamic light scattering microrheology to measure the viscoelastic spectrum of coacervates formed with poly-lysine over a timescale of 10−6 to 10 seconds, elucidating the response of protein condensates in this range for the first time. We further showed that the phase behavior and rheological characteristics of the condensates varied as a function of both protein charge distribution and polymer/protein ratio, behavior that was distinct to condensates formed with folded domains. Together, this work enhances our fundamental understanding of dynamic condensed biomaterials across biologically relevant length- and time-scales.
Introduction
Biomolecular condensates, membraneless mesoscale assemblies of proteins and nucleic acids, are increasingly understood to play critical roles in cellular regulation and organization.1 The formation of these condensed phases was initially claimed to be a result of liquid–liquid phase separation (LLPS) of the constituent biomolecules. However there is increasing evidence that biomolecular condensates are complex viscoelastic fluids with formation more readily described by coupled associative and segregative phase transitions (COAST), an umbrella term, that includes LLPS, but more broadly encompasses phase separation coupled to percolation and complex coacervation.2–8The emergent material properties of condensates are important to both the cellular function and their disfunction in the case of disease.9 Understanding how protein features contribute to the viscoelastic network of biomolecular condensates and consequently dictate emergent properties is therefore key. Multivalent interactions, or dynamic reversible crosslinks between species, are important drivers of condensate formation.1,10,11 The number of crosslinks, the spacing of crosslinks, and the strength of the molecular interactions forming these physical crosslinks are all key determinants of the underlying viscoelastic network properties.2,12,13 In recent years, intrinsically disordered proteins (IDPs) or disordered regions (IDRs) have emerged as important drivers of condensate formation, as they provide a flexible domain capable of forming a myriad of weak, transient interactions. A major and successful effort has been made to identify sequence level rules that govern the phase behavior of IDPs.11,14
The link between IDP sequence and condensate formation, has been leveraged to successfully design disordered sequences with sticker residues that form functional synthetic condensates.15 Strategic mutation of endogenous IDPs and design of de novo sequences have both been used to create cellular condensates that can recruit and release cargo.16 This enables the use of these engineered IDPs to create synthetic compartments that can control enzymatic activity, enhance reaction rates by co-localizing species,17,18 or disrupt disease pathways.19,20 Synthetic condensates also show great promise in protein delivery with several recent examples of condensates crossing membranes21 and entering living cells.22 While IDPs are important drivers of phase separation, for both endogenous proteins and many of the potential applications of condensates, the phase separating proteins also contain folded domains. In addition to the important biological function, such as catalytic activity, that these globular domains contribute these folded regions can impact phase behavior as well.
Despite the importance of folded domains, the same level of sequence to condensate material property understanding has yet to be established for proteins alone23 or in condensates.24 In the case of a globular protein condensate, the sticky points on the structured domain are not coupled to chain dynamics and cannot move independently. This may result in deviations from the traditional sticky Rouse type understanding that has been used to explain the behavior of polymer coacervates.25–29 While condensate function may depend on the rheological properties, the biological function, ranging from enzymatic activity to binding specificity, is clearly dependent on the activity of structured domains. Similarly, when designing condensates for applications in protein delivery or metabolic engineering nearly all will incorporate a folded protein. It is therefore crucial to shed light on the role folded domains play in both the physical and material properties of condensates.
With this work we asked the question, how do globular domains contribute to condensate material properties? We used model green fluorescent proteins (GFPs), engineered to be negatively charged, thus allowing them to form complex coacervates with poly-lysine. We investigated how charge distributed across a globular domain versus appended as an intrinsically disordered tag impacts phase separation propensity. Using passive video particle tracking and dynamic light scattering (DLS) microrheology we probed the viscoelasticity of these condensates across a timescale of seven orders of magnitude. We found globular protein-based condensates to have viscous behavior at long timescales with subtle differences in terminal viscosities, both between proteins and as a function of protein polymer stoichiometry. Compared to a poly-lysine and poly-aspartic acid polymer control, we found condensates that contained globular GFP to be significantly higher viscosity, interestingly, we found this to be solely due to differences in relaxation timescale. At intermediate timescales we observed a rubbery plateau due to formation of a range of sticky interactions between the oppositely charged macromolecules. We attributed differences in property to the number and distribution of the physical crosslinks formed. At short timescales we found behavior was governed by Zimm type scaling, indicative of the importance of hydrodynamic interactions at short times in the water swollen condensed phases. This work offers fundamental understanding of how globular domains contribute to protein condensates, and the viscoelastic behavior of condensates as a whole through the use of DLS-based microrheology.
Results and discussion
Distinct phase behavior of globular and tagged GFPs
To investigate the contribution of folded domains to condensate material properties we compared three negatively charged GFP variants (Fig. 1, ESI Table 1†) and their coacervation with a 30-mer of poly-L-lysine (polyK). The proteins each have an overall charge of approximately −12: in the case of iso-GFP this charge is distributed across the surface of the globular domain; for tag12-GFP the globular domain is net neutral and the excess negative charge is entirely localized to a C-terminal disordered, charged tag; for tag6-GFP the negative charge is split between the globular domain and a C-terminal tag (ESI Table 1†). For comparison, we also evaluated the phase behavior of this same polyK with a 30-mer of poly-L-aspartic acid (polyD). | ||
| Fig. 1 Schematic illustrating the complex coacervates evaluated herein. Coacervates between polyK (degree of polymerization (DP) = 30) and three proteins with increasing disordered peptide content were compared to a similar coacervate prepared with polyK and polyD (DP = 30). The protein structures show negative (red) and positive (blue) residue locations for iso-GFP, tag6-GFP and tag12-GFP (left to right). Polypeptide structures were predicted using PEP-FOLD3, while protein structures were predicted using AlphaFold. Optical microscopy images show the formation of liquid-like droplets at the optimal mixing ratio for phase separation. | ||
Initially, we explored the phase behavior of iso-, tag6- and tag12-GFP (40 µM) with polyK as a function of mixing ratio and salt concentration. As intrinsically disordered regions are known to promote condensate formation30 and IDPs with charged residues clustered in blocks as opposed to uniformly distributed have an increased propensity for phase separation we anticipated that the GFP derivatives with disordered tag regions would phase separate more readily. Increasing IDP length is also known to promote phase separation, consequently we expected tag12-GFP to exhibit the highest propensity for phase separation with the cationic peptide.
Phase separation was initially screened by monitoring the turbidity of solutions of the GFPs when mixed with increasing amounts of polyK (Fig. 2A). Unlike polyK/polyD coacervates, which had maximum turbidity when the polyions were present at the same ratio (ESI Fig. 1†), all three proteins were found to have maximum turbidity with excess polycation present. This was similarly observed for supercharged proteins including other GFP derivatives and has been attributed to a range of features of globular proteins.31,32 This includes the potential for induced charging of ionizable residues upon complexation with polyelectrolytes, the presence of regions of high charge density, or charge patches, as well as the globular structure resulting in local but not global charge neutrality.31,32 Interestingly, at low salt conditions maximum turbidity was closer to charge neutral conditions (charge fraction (f+) = 0.5 calculated as M+/(M+ + M−) where M+ and M− are the charge per polymer and protein, respectively) than at higher salt conditions, suggesting a potential role for salt influencing the protein charge state. The largest effect was observed for tag12-GFP indicating the localization of charged residues also played a role, likely because the density of charged residues at the terminus is most prone to induced charging.
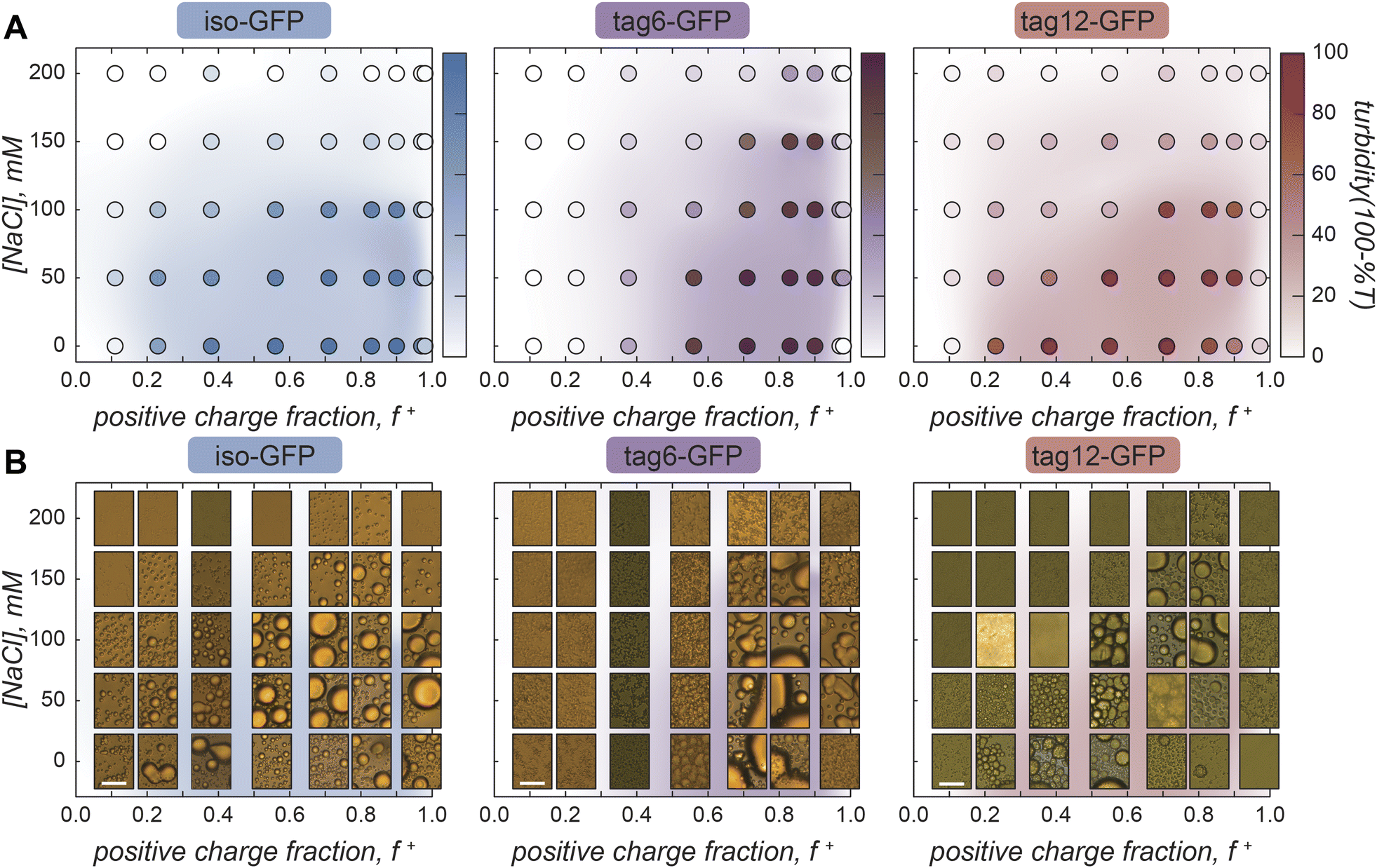 | ||
| Fig. 2 Phase diagrams with GFP and polyK. (A) Iso-GFP, tag6-GFP, and tag12-GFP turbidity as a function of sodium chloride concentration and charge fraction (f+) calculated as M+/(M+ + M−) where M+ and M− are the charge per polymer and protein, respectively. GFP 40 µM, Tris (10 mM, pH 7.4). (B) Brightfield images of GFP coacervates at varying sodium chloride concentrations and charge fraction. Scale bar 10 µm. | ||
Despite the increased charge blockiness of tag12-GFP, turbidity results suggest it does not have a greater phase separation propensity than iso-GFP (Fig. 2A). Upon imaging the samples however, the appearance of the condensed phases was starkly different (Fig. 2B). At peak turbidity conditions (f+ = 0.8) and salt concentrations below 150 mM NaCl, all three GFPs appear to form spherical liquid-like droplets, but with increasing amounts of salt tag-GFPs appear to form amorphous condensed material whereas iso-GFP still formed liquid-like material. These disparities in the condensed phases formed could result in different degrees of scattering and underrepresentation of solid-like material in turbidity measurements. Therefore, to corroborate maximum coacervate formation at f+ = 0.8, protein concentration in the dilute phase was also measured as a function of the mixing ratio of the protein and polymer. Similar behavior was observed, with protein concentration in the dilute phase minimized at this charge ratio (ESI Fig. 2†).
Striking differences between proteins can also be observed when comparing the impact of the positive charge fraction on the material state of the condensed phase. While iso-GFP formed liquid-like coacervates at nearly all conditions tested, tag6-GFP and tag12-GFP formed solid-like material when there was significant excess of either the protein or the polycation (e.g. low and high f+) (Fig. 2B, ESI Fig. 3–5†). This indicates that addition of a tag potentially increases the binding strength between the GFP and polyK, resulting in the formation of less dynamic, or kinetically arrested, condensates. PolyK/polyD coacervates appear liquid-like at all tested charge fractions (ESI Fig. 6†).
Viscosity varies between proteins and as a function of charge ratio
To quantify the effect protein charge distribution has on coacervate material properties, we used video particle tracking microrheology (VPT) to probe the viscosity of the GFP–polyK condensates. Fluorescent microspheres were embedded within condensates (Fig. 3A) and widefield microscopy was used to track their trajectories and ultimately report on condensate viscosity.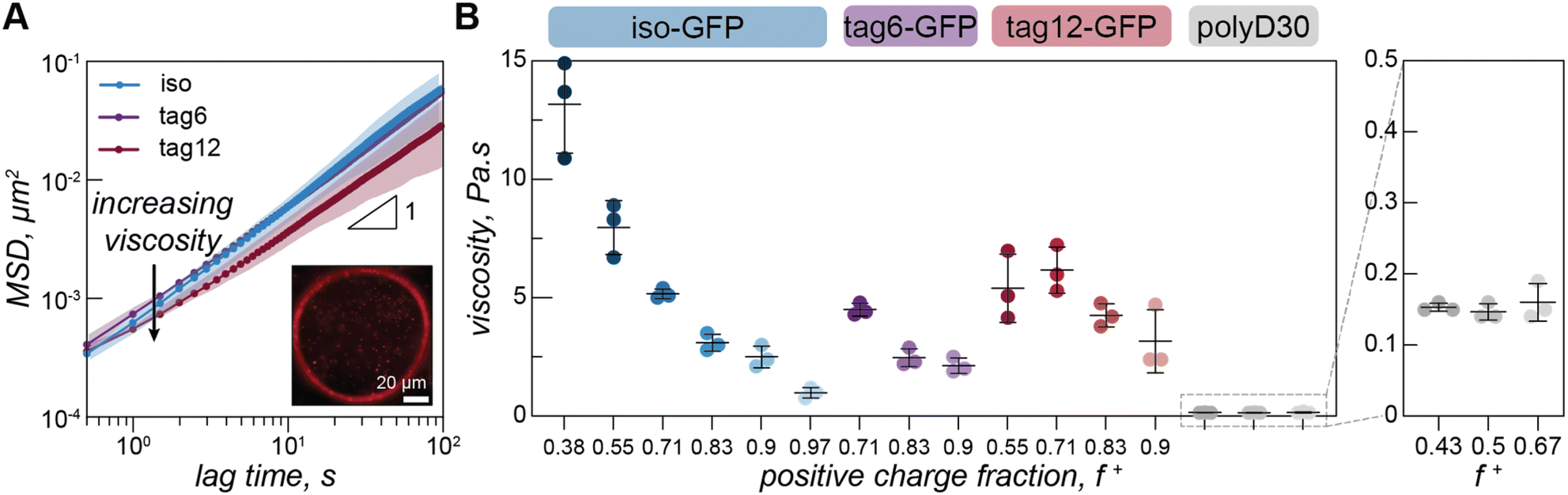 | ||
| Fig. 3 Video particle tracking in model condensates. (A) Mean squared displacement as a function of lag time for iso-GFP (blue), tag6-GFP (purple) and tag12-GFP (maroon) with polyK (f+ = 0.7) (Tris 10 mM, NaCl 100 mM). Plots depict average of three independent measurements. Shaded areas show standard deviation. Inset: single frame from particle tracking video of iso-GFP–polyK (f+ = 0.9) condensate with 500 nm beads embedded. Scale bar is 20 µm. (B) Viscosity for iso-GFP (blue), tag6-GFP (purple), and tag12-GFP (maroon) condensates when prepared at 100 mM NaCl with 40 µM protein and different polyK concentrations. Iso-GFP coacervates were prepared with 6 polyK concentrations: 10, 20, 40, 80, 148, 500 µM, corresponding to f+ = 0.38, 0.55, 0.71, 0.83, 0.9 and 0.97. Similarly, tag6-GFP coacervates were prepared with polyK at 40, 80 and 148 µM and tag12-GFP with polyK at 20, 40, 80 and 148 µM. Three separate measurements (data points), average (black line), and standard deviation (error bars), are shown. Enlarged portion of plot displays viscosity of polyK–polyD coacervates (grey), coacervates were prepared at 3 polyK concentrations 30, 40 and 80 µM, corresponding to charge ratios f+ = 0.43, 0.5, and 0.67. | ||
Due to the increased charge blockiness in the tagged GFP variants, we hypothesized that condensates containing these GFPs may have higher viscosities. However, we find that viscosities for all three proteins are only subtly different (Fig. 3B). At a positive charge ratio of 0.7, tag12-GFP, tag6-GFP, and iso-GFP have a viscosities of 6.2 ± 1.0 Pa s, 4.5 ± 0.3 Pa s, and 5.2 ± 0.2 Pa s respectively. Interestingly it is tag6-GFP, not iso-GFP, that has the lowest apparent viscosity.
Due to the differences between GFP phase diagrams, we hypothesized that viscosity differences could be due to location in phase space. We therefore measured the viscosity as a function of charge ratio. We varied charge ratio by keeping a constant GFP concentration and increasing the polyK concentration. Iso-GFP formed large coacervates amenable to microrheology measurements over the broadest range of conditions, consequently a broader range of concentrations were investigated. We find a general trend of decreasing viscosity with increasing polyK concentration for all three proteins. However, the viscosity of tag12-GFP coacervates appears to plateau or slightly decrease between a charge fraction of 0.55 and 0.7 and the overall change in viscosity across the measured range is less significant than that for iso-GFP.
Interestingly we do not observe similar charge ratio dependent behavior for coacervates formed from polyK/polyD (Fig. 3B). Here the viscosity appears to remain constant, an effect previously observed and attributed to one preferred polycation/polyanion ratio within the condensed phase, maintained by changes in the coacervate volume to compensate for initial condition changes.25,33 A change in coacervate viscosity can be induced by changing the polycation/polyanion ratio within the dense phase,34–36 suggesting that unlike in the polyK/polyD coacervates a compositional change in the condensed phase may take place in the GFP coacervates as a function of charge ratio. In support of this hypothesis, we estimated the ratio of polyanion (GFP or polyD) to polyK in the dense phase using two approaches (ESI Fig. 7 and 8†). Both confocal measurements of individual droplets and fluorescence measurements of resuspended coacervates indicated significant changes in the ratio of GFP to polyK as a function of charge fraction. In contrast, coacervates formed between two linear polyelectrolytes showed minimal changes in the composition as a function of charge ratio. We also found the viscosity of the polyK/polyD was significantly lower than that of the GFP containing coacervates. This could be attributed to the conditions probed being closer to the critical point for the polyK/polyD coacervates as well as potential differences in coacervate density, as has been shown for hybrid particle-linear polymer coacervates when compared to coacervates composed to two linear components.24,37
Condensates: a viscoelastic network
On the timescale of our particle tracking measurements all coacervates behaved as pure viscous fluids. To access the high frequency range required to probe rheological response corresponding to fast segmental chain dynamics and gain more detailed information about the fluid network, we turned to dynamic light scattering microrheology (DLSµR). This technique has previously been shown to reproduce oscillatory macrorheology results for synthetic polymer gels and been used to characterize precious biological fluids such as mucus.38–40 This approach is advantageous as proteins are liable to denature at elevated temperatures and the coacervate structure and density can vary significantly at elevated salt concentrations, preventing the use of temperature or salt-superpositioning as has been done for synthetic complex coacervates.25,27,41–43Comparing the mean squared displacement (MSD) determined from VPT microrheology to the MSD calculated from DLS for iso-GFP and polyK (40 µM) coacervates (Fig. 4A) highlights the different time regimes over which these techniques report. Both cover the 0.5 to ∼10 second time regime, and while there are some differences in the observed absolute value both scale as ∼1 and similar trends between samples were observed. While the slope of 1 found from VPT indicated beads were moving in a purely viscous fluid, at timescales faster than 0.5 seconds, the shortest time interval used for VPT, the MSD of the embedded beads measured by DLS can be seen to plateau, indicating non-Brownian behavior. The corresponding complex moduli (Fig. 4B) displays three distinct regions: a terminal relaxation or flow region (blue region), a rubbery plateau due to formation of reversible bonds (white region), and a Rouse-like transition region due to local monomer relaxation (green region). This is qualitatively similar to observations made in superposition experiments with polymer complex coacervates. Each of these regions corresponds to different potential interactions driving the observed viscoelastic behavior, shown in the schematic that represents the dominant interactions occurring within each timescale (Fig. 4C).
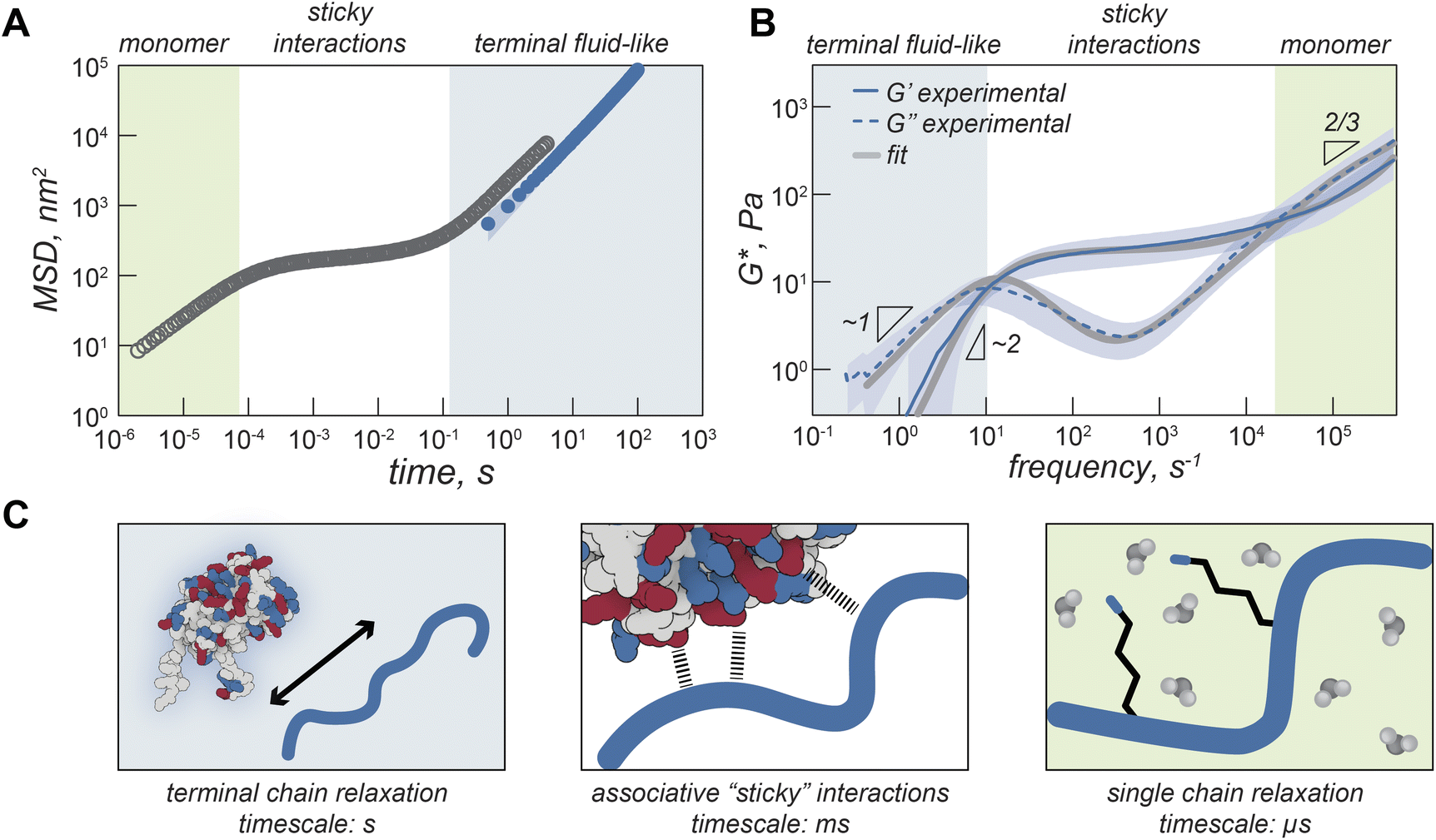 | ||
| Fig. 4 Viscoelasticity of GFP–polyK condensates. (A) Mean square displacement (MSD) of iso-GFP coacervates (polyK 40 µM) determined from DLS microrheology (grey) and MSD determined from video particle tracking microrheology (blue). Background colored regions qualitatively illustrate different timescales. (B) Complex modulus of iso-GFP coacervate from DLS microrheology. G′ solid line, G″ dashed line. Grey lines represent two-component Maxwell fit. (C) Schematic illustrating protein polymer behavior at different timescales. At longest time scales (blue background) proteins and polymers can flow. At intermediate times (white background) behavior is dominated by interactions between charged residues on GFP and polyK. At short timescales (green background) residue interactions with water dominate. | ||
At low frequencies, or the longest timescale probed, condensate behavior is dominated by the loss modulus (G″, indicated with a dashed line) indicating predominantly fluid behavior at long timescales where protein and polymer have time to disassociate. This region is shaded blue in Fig. 4A–C. In this region, we find that G′ scales as ω2 (slope = 1.9) and G″ scales as ω1 (slope = 0.97), which is consistent with the scaling expected for the viscoelastic response of unentangled ideal polymer chains. This is also consistent with the apparent liquid behavior we observed from video particle tracking microrheology as well as with previous work indicating condensates are Maxwell fluids.3,5,33
The characteristic relaxation time τrep, the reciprocal of the frequency at which the storage and loss modulus cross, represents the longest time taken for the biopolymers to disassociate from the network and form new crosslinks. For iso-GFP/polyK condensates we can see this occurs at ∼100 ms (Fig. 4B and Table 1). Interestingly we observe a much shorter relaxation time, ∼3 ms, for the polyK–polyD coacervates (Fig. 5F). At shorter timescales than τrep, we see elastic behavior begin to dominate and a marked plateau in the elastic modulus. This is characteristic of a network held together by sticky interactions, here physical crosslinks formed by electrostatic interactions between GFP and polyK.
| Protein | f + | NaCl, mM | G rep, Pa | G Rouse, Pa | G N, Pa | τ rep, ms | τ Rouse, µs | Viscosity, Pa s | Viscosity VPT, Pa s |
|---|---|---|---|---|---|---|---|---|---|
| Iso-GFP | 0.55 | 100 | 68 ± 9 | 430 ± 24 | 150 ± 39 | 48 ± 16 | 66 ± 8 | 6.7 ± 3 | 7.9 ± 1.1 |
| 0.71 | 100 | 9 ± 3 | 48 ± 17 | 25 ± 10 | 110 ± 60 | 47 ± 6 | 1.7 ± 0.6 | 5.2 ± 0.2 | |
| 0.90 | 100 | 3 ± 1 | 16 ± 5 | 8 ± 3 | 250 ± 210 | 58 ± 2 | 0.9 ± 0.4 | 2.5 ± 0.5 | |
| 0.71 | 50 | 50 ± 21 | 410 ± 95 | 230 ± 19 | 120 ± 70 | 41 ± 7 | 10.4 ± 3 | — | |
| Tag6-GFP | 0.71 | 100 | 11 ± 4 | 62 ± 14 | 29 ± 11 | 81 ± 48 | 42 ± 16 | 1.5 ± 0.4 | 4.5 ± 0.3 |
| 0.90 | 100 | 5 ± 1 | 28 ± 4 | 16 ± 2 | 91 ± 21 | 69 ± 4 | 0.9 ± 0.3 | 2.1 ± 0.3 | |
| 0.71 | 50 | 25 ± 7 | 150 ± 39 | 73 ± 21 | 120 ± 79 | 64 ± 15 | 5.6 ± 4 | — | |
| Tag12-GFP | 0.55 | 100 | 13 ± 2 | 90 ± 22 | 41 ± 7 | 140 ± 43 | 210 ± 18 | 3.6 ± 1.2 | 5.4 ± 1.4 |
| 0.71 | 100 | 11 ± 1 | 70 ± 24 | 33 ± 6 | 110 ± 27 | 70 ± 37 | 2.5 ± 0.8 | 6.2 ± 1 | |
| 0.71 | 50 | 26 ± 10 | 150 ± 60 | 71 ± 24 | 93 ± 49 | 68 ± 13 | 4.4 ± 1.6 | — | |
| PolyD | 0.5 | 100 | 19 ± 4 | — | 57 ± 10 | 3 ± 1 | — | 0.1 ± 0.02 | 0.15 ± 0.01 |
| 0.5 | 50 | 40 ± 9 | — | 109 ± 25 | 1 ± 0.1 | — | 0.1 ± 0.01 | — |
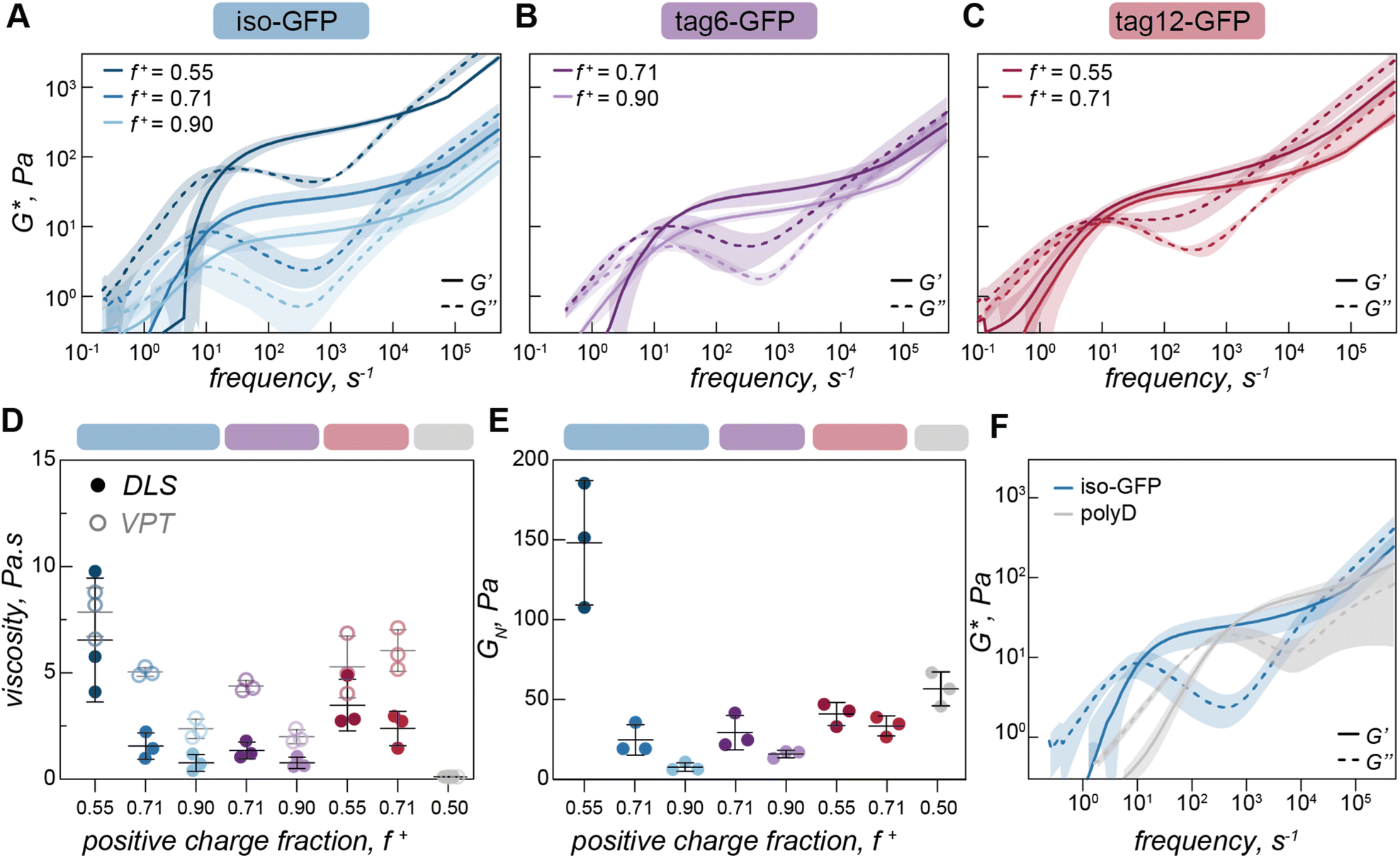 | ||
| Fig. 5 Condensate rheological properties as a function of mixing ratio. (A) Complex modulus of iso-GFP (40 µM) and polyK coacervates at different charge ratios (f+ = 0.55, 0.71, 0.90), (B) tag6-GFP (40 µM) and polyK at different charge ratios (f+ = 0.71, 0.90), (C) tag12-GFP (40 µM) and polyK at different charge ratios (f+ = 0.55, 0.71). (D) Viscosity (η0 = 2G0τ0) from DLS (filled circles) and VPT (empty circles) for iso-GFP (40 µM) and polyK coacervates at different charge ratios (f+ = 0.55, 0.71, 0.90) (blue); tag6-GFP (40 µM) and polyK at different charge ratios (f+ = 0.71, 0.90) (purple), tag12-GFP (40 µM) with polyK at different charge ratios (f+ = 0.55, 0.71) (red) and polyD30 (f+ = 0.50) (grey). (E) Plateau modulus (GN) of iso-GFP (40 µM) and polyK coacervates at charge ratios f+ = 0.55, 0.71, 0.90 (blue), tag12-GFP (40 µM) with polyK at charge ratios f+ = 0.71, 0.90 (purple), tag12-GFP (40 µM) with polyK at charge ratios f+ = 0.55, 0.71 (red) and polyD30 (f+ = 0.50) (grey). (F) Complex modulus of iso-GFP (blue) (40 µM) with polyK (40 µM, f+ = 0.71) in comparison to polyD (grey) (40 µM) with polyK (40 µM, f+ = 0.5). | ||
At the highest frequencies, above τRouse, we find that the elastic modulus scales as ω2/3. This region describes interactions occurring at the fastest time scales, typically monomer interactions or monomer solvent interactions for semidilute or dilute polymer solutions. Two of the most common methods to describe polymer behavior in this regime are the Rouse and Zimm models.40,44,45 Scaling of G′ as ω2/3 in the high frequency range is characteristic of the Zimm model in a Θ solvent, which differs from the original Rouse model (G′ ∼ ω½) by asserting the effect of hydrodynamic interactions between polymer monomers and solvent dominate over monomer–monomer interactions. A Zimm model describes well the behavior of dilute polymers where long-range interactions like hydrodynamics become increasingly important. This suggests that polyK is in the dilute solvent accessible environment. Due to this, the absence of a second rubbery plateau,41 and the short length of the polyK (N ≈ 30), these results indicate that there is no polymer chain entanglement. Interestingly as salt is decreased, we see a shift in modulus indicating increased material strength and scaling in the high frequency range as ω2/3 appears to slightly decrease (ESI Fig. 9† and Table 1). This suggests that Rouse-like monomer–monomer interactions may become increasingly important as the coacervate becomes less fluid with decreasing solution ionic strength.
A two component Maxwell model, with a slow relaxation originating from sticker interactions and a fast relaxation due to Rouse motion of the polymer between sticky bonds, describes the observed behavior well (ESI text, Fig. 4B, ESI Fig. 10†).
| G(t) = ∑Gslow/sticky(t) + ∑Gfast/Rouse(t) |
As expected, addition of further elements, accounting for the fact the proteins do not contain identical sticky interaction sites improves the fit, indicating that there are likely a range of relaxation processes associated with the different types of interactions, (ESI Fig. 10†).
Protein polymer charge ratio impacts material strength and relaxation time
Having observed changes in terminal viscosity as a function of charge ratio with VPT we wanted to interrogate this effect over a broader time range. Due to the larger volumes required for DLS rheology a subset of charge ratios were measured for the three GFP variants (f+ = 0.55, 0.71, and 0.90 for iso-GFP (Fig. 5A), f+ = 0.71 and 0.90 for tag6-GFP (Fig. 5B), and f+ = 0.55 and 0.71 for tag12-GFP (Fig. 5C)). Similar to the trend of decreasing viscosity with increasing polyK concentration observed from VPT, we see a decrease in both zero shear viscosity (Fig. 5D) and in the elastic modulus (Fig. 5E), indicating decreased material strength and increased fluidity.These changes in the complex modulus could be due to changes in sticker strength or fewer sticker interactions. If these coacervates were dominated by polymer behavior and governed by a sticky Rouse type mechanism, a decrease in material strength due to fewer or weaker interactions would be expected to occur alongside a corresponding increase in the relaxation frequency towards the higher frequency range, indicating that polymers within the condensate can relax more easily. Indeed, for polymer–polymer coacervates this relaxation does occur at a higher frequency indicating this network can relax more readily in comparison (Fig. 5F). However, the error associated with the crossover times in our measurements of protein coacervates is large in comparison to subtle changes in the crossover times, so it is unclear if this difference is statistically significant, preventing any conclusive findings of discrepancies with traditional sticky Rouse behavior.
The distinct viscoelastic behavior of protein coacervates indicates that the globular proteins are making a significant contribution to the material properties. Importantly, given the relatively fixed three dimensional structure of the globular proteins, each potential interaction site will be subtly different, which can result in the complex overlapping of multiple slightly different relaxation processes. In our model protein condensates, the sticky interaction sites largely correspond to different charged residues, or charge patches, on the protein surface, but we cannot exclude the ability of the protein to engage in other non-covalent interactions (e.g. cation–π, H-bonding) with polyK that may also contribute to the relaxation of the elastic modes of the coacervate. We hypothesize that as the polyK concentration increases, the relative proportion of GFP sticky points to polyK molecules decreases, thus increasing fluidity (ESI Fig. 7 and 8†). With excess polyK, we propose that the proportion of sticky interactions that contribute to the overall network and the elastic modulus decreases, while those that act as dangling ends increases. As in other systems, each time a sticky bond breaks, the most likely bond to form is between the same partners, but the characteristic relaxation time, or sticker interaction time, should be considered the timescale for an interaction to reform with a new partner, not simply the timescale for physical bond breaking. Therefore, the arrangement and physical proximity of neighboring interaction sites, which can be precisely controlled on globular protein surfaces, is key to material properties. For tag12-GFP the localization of charges to one region could increase the probability of one polyK forming multiple interactions with the same GFP, thus not contributing to the network elasticity. This results in a narrower range of conditions where viscoelastic fluids form and an increased propensity towards small clusters. For iso-GFP, as the charged patches are distributed across different regions of the protein surface, this potentially increases the probability that interactions on one GFP will be with multiple different polyK. This leads to viscoelastic fluids forming over a broad range of conditions. Additionally, the larger change in modulus for iso-GFP could be indicative of the presences of different charge patches across the protein surface, each with different strengths. In this case, when polyK is the limiting component only the dominant GFP charge patches interact, whereas when polyK is in excess less charged regions may also become potential sticky points. This could suggest that a spectrum of different clusters form impacting the material response.
Polymer–polymer condensates exhibit faster relaxation timescale
From VPT experiments we observed significantly lower viscosity for polyK–polyD, linear polymer complex coacervates (Fig. 3B, ESI Fig. 11†) than those containing a globular GFP. Using DLS-rheology we observe a similarly lower terminal viscosity (Fig. 5D). Interestingly, we find the plateau modulus to be of a similar magnitude to the globular protein condensates indicating interactions of a similar strength (Fig. 5E and F), likely the ionic interactions driving coacervate formation. The decrease in viscosity is therefore due to the faster timescale over which the physical, dynamic crosslinks form and relax in the linear polymer coacervates, not due to differences in interaction strength (Fig. 5F). We attribute this effect to the increased degrees of freedom available to a linear molecule which has segmental flexibility compared to a globular protein which has only translational and rotational degrees of freedom.Conclusions
The material properties of condensates are diverse, encompassing dynamic fluids, gel-like solids, glasses and aggregates.1,46,47 Changes in fluidity can impact biological function and transitions from dynamic to more solid like states are associated with pathologic response.46 Consequently, determining the emergent properties of condensates, such as their viscoelasticity, is of great importance. Condensates are composed of a range of biomolecules, the diffusion of these molecules, the interaction between different species (i.e. bond lifetime), and global properties of the condensates as a whole, all occur at different timescales. It is therefore imperative to understand material response at both long and short times, to capture all timescales relevant for function. Here we have determined the viscoelastic spectrum of model GFP and polyK condensates over a broad range of timescales, providing insight into these different regimes.Particle tracking rheology has been used to characterize a number of in vitro protein or peptide-based biomolecular condensate systems.48 This technique typically allows the MSD between 0.1 to 100 s to be measured. As with the condensates measured here, several display liquid-like behavior over this time regime,49–51 while for other systems features characteristic of viscoelastic fluids are observed.5,52 Alshareedah et al. showed for peptide–polyU condensates, a terminal region where G′ and G″ scaled as expected for a Maxwell fluid and a crossover frequency was observed and indicated the onset of network formation. They observed an increase in viscosity and corresponding increase in terminal relaxation time, as is expected for sticky Rouse type behavior. To capture faster timescales relevant for biological function, active microrheology performed using optical tweezers can be used to extend the observable frequency range.3,4 By measuring the rheological response of PGL-3 and FUS condensates as they age, Jawerth et al. ascertained these were aging Maxwell fluids.3 To our knowledge there have been no measurements of protein based coacervates across the frequency range shown in the present work.
Polyelectrolyte complex coacervates have been characterized over a much broader frequency/time domain than protein-based condensates, largely using time–temperature53 or time–salt superpositioning,25,42,43,54 creating master curves that resolve terminal, plateau, transition, and glassy regions. Due to the small range of salt concentrations these protein condensates form over, the sensitivity of protein conformation to salt and temperature, and the diverse range of interactions beyond electrostatic that contribute to the protein condensate interactome these superpositioning techniques may not be widely applicable to study protein coacervate systems.
The simplicity and ready availability of DLS instruments, combined with the wide frequency range measured makes this approach very well suited to the characterization of biomolecular condensates. Previously, condensate viscosities between ∼0.1 Pa s and 200 Pa s have been reported, this range is well accommodated by this technique, plus has the potential to measure much stiffer systems, with moduli up to 104.55 However, one downside compared to video particle tracking is the loss of spatial information. This means interesting heterogeneity across a sample could be obscured. Another advantage of video particle tracking is that by using protein tracers, the viscosity or viscoelastic response of condensates in cells can be probed directly.6,56 By combining DLS with video-particle tracking, this adds another technique to the host of methods currently used to provide detailed insights into condensate properties.
To design functional condensates, it is crucial to understand the underlying interactions that control material properties, including contributions from both disordered and globular domains. In the work presented here we demonstrate that condensates formed between polyK and a model anionic protein with charge differentially distributed between the globular domain and engineered disordered tags. We show that differences in charge distribution result in subtle differences in phase behavior and viscoelasticity. We further show that changes in charge fraction impact viscoelasticity, an effect we attribute to differences in composition that influence the number and types of interactions and how these interactions form a network. Critically, the behavior of condensates containing folded proteins differs significantly from those composed of two linear polypeptides. This work highlights the importance of globular domains when considering condensate properties, showing that the rules for disordered proteins and linear polypeptides do not necessarily directly translate. Many proteins known to drive phase separation in cellular organelles contain globular domains, their contribution should not be overlooked when considering multivalent interactions and therefore impact on viscoelasticity and other network properties. Looking forward we can begin to design condensates with tailored material properties by engineering both disordered and globular regions, thinking about how domains will behave independently and how they will interact together to control fluid networks.
Data availability
Data for this article, including turbidity, video particle tracking, and DLS microrheology, are available at dryad at https://doi.org/10.5061/dryad.4f4qrfjm9. Plasmids used in this article are available via Addgene.Author contributions
R. S. F. and A. C. O. designed the research and wrote the manuscript. R. S. F. performed research and analyzed the data.Conflicts of interest
The authors declare the following competing financial interest(s): A. C. O. is a co-founder of Werewool, a company that is engaged in the development of performance textiles that incorporate engineered proteins. R. S. F. declares no competing interests.Acknowledgements
R. S. F. and A. C. O. acknowledge the National Institutes of Health for funding under an award from the National Institute of General Medical Sciences (R35GM138378). Confocal imaging was performed with support from the Zuckerman Institute's Cellular Imaging platform. We also acknowledge Pam Cai for helpful discussions.Notes and references
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- R. V. Pappu, S. R. Cohen, F. Dar, M. Farag and M. Kar, Chem. Rev., 2023, 123(14), 8945–8987 CrossRef CAS PubMed.
- L. Jawerth, E. Fischer-Friedrich, S. Saha, J. Wang, T. Franzmann, X. Zhang, J. Sachweh, M. Ruer, M. Ijavi, S. Saha, J. Mahamid, A. A. Hyman and F. Jülicher, Science, 2020, 370, 1317–1323 CrossRef CAS PubMed.
- A. Ghosh, D. Kota and H.-X. Zhou, Nat. Commun., 2021, 12, 5995 CrossRef CAS PubMed.
- I. Alshareedah, M. M. Moosa, M. Pham, D. A. Potoyan and P. R. Banerjee, Nat. Commun., 2021, 12, 6620 CrossRef CAS PubMed.
- J. A. Riback, J. M. Eeftens, D. S. W. Lee, S. A. Quinodoz, A. Donlic, N. Orlovsky, L. Wiesner, L. Beckers, L. A. Becker, A. R. Strom, U. Rana, M. Tolbert, B. W. Purse, R. Kleiner, R. Kriwacki and C. P. Brangwynne, Mol. Cell, 2023, 83, 3095–3107 CrossRef CAS PubMed.
- I. Alshareedah, A. Singh, S. Yang, V. Ramachandran, A. Quinn, D. A. Potoyan and P. R. Banerjee, Sci. Adv., 2024, 10, eadi6539 CrossRef CAS PubMed.
- I. Alshareedah, W. M. Borcherds, S. R. Cohen, A. Singh, A. E. Posey, M. Farag, A. Bremer, G. W. Strout, D. T. Tomares, R. V. Pappu, T. Mittag and P. R. Banerjee, Nat. Phys., 2024, 20, 1482–1491 Search PubMed.
- R. Gallo, A. K. Rai, A. B. R. McIntyre, K. Meyer and L. Pelkmans, Dev. Cell, 2023, 58, 1880–1897 CrossRef CAS PubMed.
- P. Li, S. Banjade, H.-C. Cheng, S. Kim, B. Chen, L. Guo, M. Llaguno, J. V. Hollingsworth, D. S. King, S. F. Banani, P. S. Russo, Q.-X. Jiang, B. T. Nixon and M. K. Rosen, Nature, 2012, 483, 336–340 CrossRef CAS PubMed.
- J.-M. Choi, A. S. Holehouse and R. V. Pappu, Annu. Rev. Biophys., 2020, 49, 107–133 CrossRef CAS PubMed.
- Z. Zhang, Q. Chen and R. H. Colby, Soft Matter, 2018, 14, 2961–2977 RSC.
- F. Tanaka and S. F. Edwards, Macromolecules, 1992, 25, 1516–1523 CrossRef CAS.
- E. Gomes and J. Shorter, J. Biol. Chem., 2019, 294, 7115–7127 CrossRef CAS PubMed.
- Y. Dai, L. You and A. Chilkoti, Nat. Rev. Bioeng., 2023, 1–15 Search PubMed.
- B. S. Schuster, E. H. Reed, R. Parthasarathy, C. N. Jahnke, R. M. Caldwell, J. G. Bermudez, H. Ramage, M. C. Good and D. A. Hammer, Nat. Commun., 2018, 9, 2985 CrossRef PubMed.
- C. D. Reinkemeier, G. E. Girona and E. A. Lemke, Science, 2019, 363, eaaw2644 CrossRef CAS PubMed.
- E. M. Zhao, N. Suek, M. Z. Wilson, E. Dine, N. L. Pannucci, Z. Gitai, J. L. Avalos and J. E. Toettcher, Nat. Chem. Biol., 2019, 15, 589–597 CrossRef CAS PubMed.
- M. V. Garabedian, W. Wang, J. B. Dabdoub, M. Tong, R. M. Caldwell, W. Benman, B. S. Schuster, A. Deiters and M. C. Good, Nat. Chem. Biol., 2021, 17, 998–1007 CrossRef CAS PubMed.
- M. Yoshikawa, T. Yoshii, M. Ikuta and S. Tsukiji, J. Am. Chem. Soc., 2021, 143, 6434–6446 CrossRef CAS PubMed.
- T. Lu, X. Hu, M. H. I. van Haren, E. Spruijt and W. T. S. Huck, Small, 2023, 19, 2303138 CrossRef CAS PubMed.
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed.
- S. P. S. Deopa, S. S. Rajput, A. Kumar and S. Patil, J. Phys. Chem. Lett., 2022, 13, 9473–9479 CrossRef CAS PubMed.
- H.-X. Zhou, V. Nguemaha, K. Mazarakos and S. Qin, Trends Biochem. Sci., 2018, 43, 499–516 CrossRef CAS PubMed.
- E. Spruijt, M. A. C. Stuart and J. van der Gucht, Macromolecules, 2013, 46, 1633–1641 CrossRef CAS.
- S. Ali and V. M. Prabhu, Gels, 2018, 4, 11 CrossRef PubMed.
- R. G. Larson, Y. Liu and H. Li, J. Rheol., 2021, 65, 77–102 CrossRef CAS.
- Q. Wang and J. B. Schlenoff, Macromolecules, 2014, 47, 3108–3116 CrossRef CAS.
- K. Akkaoui, Z. A. Digby, C. Do and J. B. Schlenoff, Macromolecules, 2024, 57, 1169–1181 CrossRef CAS.
- C. P. Brangwynne, P. Tompa and R. V. Pappu, Nat. Phys., 2015, 11, 899–904 Search PubMed.
- A. C. Obermeyer, C. E. Mills, X.-H. Dong, R. J. Flores and B. D. Olsen, Soft Matter, 2016, 12, 3570–3581 RSC.
- R. A. Kapelner and A. C. Obermeyer, Chem. Sci., 2019, 10, 2700–2707 RSC.
- I. Alshareedah, G. M. Thurston and P. R. Banerjee, Biophys. J., 2021, 120, 1161–1169 CrossRef CAS PubMed.
- Y. Chen, M. Yang, S. A. Shaheen and J. B. Schlenoff, Macromolecules, 2021, 54, 7890–7899 CrossRef CAS.
- F. Weinbreck, R. H. W. Wientjes, H. Nieuwenhuijse, G. W. Robijn and C. G. de Kruif, J. Rheol., 2004, 48, 1215–1228 CrossRef CAS.
- D. S. Hwang, H. Zeng, A. Srivastava, D. V. Krogstad, M. Tirrell, J. N. Israelachvili and J. H. Waite, Soft Matter, 2010, 6, 3232–3236 RSC.
- A. M. Rumyantsev, O. V. Borisov and J. J. de Pablo, Macromolecules, 2023, 56, 1713–1730 CrossRef CAS PubMed.
- B. A. Krajina, C. Tropini, A. Zhu, P. DiGiacomo, J. L. Sonnenburg, S. C. Heilshorn and A. J. Spakowitz, ACS Cent. Sci., 2017, 3, 1294–1303 CrossRef CAS PubMed.
- T. G. Mason and D. A. Weitz, Phys. Rev. Lett., 1995, 74, 1250–1253 CrossRef CAS PubMed.
- P. C. Cai, B. A. Krajina, M. J. Kratochvil, L. Zou, A. Zhu, E. B. Burgener, P. L. Bollyky, C. E. Milla, M. J. Webber, A. J. Spakowitz and S. C. Heilshorn, Soft Matter, 2021, 17, 1929–1939 RSC.
- M. Yang, J. Shi and J. B. Schlenoff, Macromolecules, 2019, 52, 1930–1941 CrossRef CAS.
- K. Akkaoui, M. Yang, Z. A. Digby and J. B. Schlenoff, Macromolecules, 2020, 53, 4234–4246 CrossRef CAS.
- V. M. S. Syed and S. Srivastava, ACS Macro Lett., 2020, 9, 1067–1073 CrossRef CAS PubMed.
- M. Doi and S. F. Edwards, The Theory of Polymer Dynamics, Oxford University Press, Oxford, New York, 1988 Search PubMed.
- P. C. Cai, B. Su, L. Zou, M. J. Webber, S. C. Heilshorn and A. J. Spakowitz, ACS Cent. Sci., 2022, 8, 1318–1327 CrossRef CAS PubMed.
- A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef CAS PubMed.
- M. Kato, T. W. Han, S. Xie, K. Shi, X. Du, L. C. Wu, H. Mirzaei, E. J. Goldsmith, J. Longgood, J. Pei, N. V. Grishin, D. E. Frantz, J. W. Schneider, S. Chen, L. Li, M. R. Sawaya, D. Eisenberg, R. Tycko and S. L. McKnight, Cell, 2012, 149, 753–767 CrossRef CAS PubMed.
- D. Michieletto and M. Marenda, JACS Au, 2022, 2, 1506–1521 CrossRef CAS PubMed.
- N. Taylor, S. Elbaum-Garfinkle, N. Vaidya, H. Zhang, H. A. Stone and C. P. Brangwynne, Soft Matter, 2016, 12, 9142–9150 RSC.
- S. Elbaum-Garfinkle, Y. Kim, K. Szczepaniak, C. C.-H. Chen, C. R. Eckmann, S. Myong and C. P. Brangwynne, Proceedings of the National Academy of Sciences, 2015, 112, 7189–7194 Search PubMed.
- H. Zhang, S. Elbaum-Garfinkle, E. M. Langdon, N. Taylor, P. Occhipinti, A. A. Bridges, C. P. Brangwynne and A. S. Gladfelter, Mol. Cell, 2015, 60, 220–230 CrossRef CAS PubMed.
- M. Feric, N. Vaidya, T. S. Harmon, D. M. Mitrea, L. Zhu, T. M. Richardson, R. W. Kriwacki, R. V. Pappu and C. P. Brangwynne, Cell, 2016, 165, 1686–1697 CrossRef CAS PubMed.
- M. Yang, Z. A. Digby, Y. Chen and J. B. Schlenoff, Sci. Adv., 2022, 8, eabm4783 CrossRef CAS PubMed.
- D. Priftis, K. Megley, N. Laugel and M. Tirrell, J. Colloid Interface Sci., 2013, 398, 39–50 CrossRef CAS PubMed.
- M. Meleties, R. L. Martineau, M. K. Gupta and J. K. Montclare, ACS Biomater. Sci. Eng., 2022, 8, 2747–2763 CrossRef CAS PubMed.
- Y. Tseng, J. S. H. Lee, T. P. Kole, I. Jiang and D. Wirtz, J. Cell Sci., 2004, 117, 2159–2167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03564j |
| This journal is © The Royal Society of Chemistry 2024 |