
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of megapolipeptins by genome mining of a Burkholderiales bacteria collection†
Bruno S.
Paulo‡
 ab,
Michael J. J.
Recchia‡
c,
Sanghoon
Lee
c,
Claire H.
Fergusson
c,
Sean B.
Romanowski
ab,
Antonio
Hernandez
ab,
Nyssa
Krull
a,
Dennis Y.
Liu
c,
Hannah
Cavanagh
c,
Allyson
Bos
d,
Christopher A.
Gray
d,
Brian T.
Murphy
ab,
Roger G.
Linington
*c and
Alessandra S.
Eustaquio
*ab
ab,
Michael J. J.
Recchia‡
c,
Sanghoon
Lee
c,
Claire H.
Fergusson
c,
Sean B.
Romanowski
ab,
Antonio
Hernandez
ab,
Nyssa
Krull
a,
Dennis Y.
Liu
c,
Hannah
Cavanagh
c,
Allyson
Bos
d,
Christopher A.
Gray
d,
Brian T.
Murphy
ab,
Roger G.
Linington
*c and
Alessandra S.
Eustaquio
*ab
aDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, IL 60607, USA. E-mail: ase@uic.edu
bCenter for Biomolecular Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, IL 60607, USA
cDepartment of Chemistry, Simon Fraser University, Burnaby, BC V5H 1S6, Canada. E-mail: rliningt@sfu.ca
dDepartment of Biological Sciences, University of New Brunswick, Saint John, New Brunswick E2L 4L5, Canada
First published on 13th September 2024
Abstract
Burkholderiales bacteria have emerged as a promising source of structurally diverse natural products that are expected to play important ecological and industrial roles. This order ranks in the top three in terms of predicted natural product diversity from available genomes, warranting further genome sequencing efforts. However, a major hurdle in obtaining the predicted products is that biosynthetic genes are often ‘silent’ or poorly expressed. Here we report complementary strain isolation, genomics, metabolomics, and synthetic biology approaches to enable natural product discovery. First, we built a collection of 316 rhizosphere-derived Burkholderiales strains over the course of five years. We then selected 115 strains for sequencing using the mass spectrometry pipeline IDBac to avoid strain redundancy. After predicting and comparing the biosynthetic potential of each strain, a biosynthetic gene cluster that was silent in the native Paraburkholderia megapolitana and Paraburkholderia acidicola producers was cloned and activated by heterologous expression in a Burkholderia sp. host, yielding megapolipeptins A and B. Megapolipeptins are unusual polyketide, nonribosomal peptide, and polyunsaturated fatty acid hybrids that show low structural similarity to known natural products, highlighting the advantage of our Burkholderiales genomics-driven and synthetic biology-enabled pipeline to discover novel natural products.
Introduction
Bacterial metabolites have important ecological roles and are a major source of products for applications in medicine and agriculture. The bacterial order Streptomycetales (Actinomycetota phylum) has been the most widely explored for natural product and enzyme discovery.1 Yet, distinct taxonomy often implies distinct chemistry.2,3 In bacteria, the genes that encode the biosynthesis of natural products are usually co-localized forming biosynthetic gene clusters (BGCs). BGCs can be grouped into gene cluster families (GCFs) based on similarity. On average 74% of GCFs are unique to each phylum and diversity has been shown to drop at each taxonomic rank.2 Bacteria from the order Burkholderiales (Pseudomonodota phylum, previously named Proteobacteria)4 have emerged as an important source of natural products but they remain underexplored.5 Burkholderiales ranks top three (after two Actinomycetota orders) in terms of predicted natural product diversity based on available genomes.2 Furthermore, the predicted biosynthetic potential remains untapped within the available genomes, underscoring the need for continued sequencing efforts.2It has been recently estimated that only 3% of genome-predicted bacterial natural products have been isolated and structurally characterized.2 Obtaining the predicted products remains a bottleneck, in part because many BGCs are “silent”, that is they are not expressed in quantities practical enough to allow the detection and isolation of biosynthesized products.6 Approaches for targeted activation of BGCs include engineering of the native producer and heterologous expression in an optimized host strain.7 Heterologous expression has the potential to streamline the discovery process through standardization and automation. However, recent studies8–11 showed that the success rate of heterologous expression is still relatively low, varying from 11% to 32% when using model Escherichia coli and Streptomyces spp. as hosts. The choice of host strain can greatly impact success and product yields.12 For instance, a systematic analysis of host strains revealed a direct relationship between yields and the genetic identity of host and source DNA.13 Recently we have tested a Burkholderia sp. strain as an alternative host and demonstrated its ability to produce Burkholderiales natural products in titers that are two to three orders of magnitude higher than with E. coli.14
Here we report a pipeline to discover natural products from Burkholderiales that combines a suite of complementary approaches (Fig. 1). First, Burkholderiales bacteria were selectively isolated from environmental samples using methods we previously established.15,16 To select strains for genome sequencing, we performed matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) protein and metabolite analyses on cell material from bacteria colonies. The resulting data were processed using the bioinformatics pipeline IDBac to maximize strain and metabolite diversity while avoiding redundancy.17 Strain selection and sequencing was performed in two cycles. In cycle 1, a set of genomes was sequenced and analyzed for biosynthetic potential, which influenced strain selection and sequencing in the second round (Fig. 1A). The biosynthetic potential of each strain was predicted and compared (Fig. 1B). A BGC that was silent in the native producers was prioritized, cloned, and activated using heterologous expression in a Burkholderia sp. host strain18,19 (Fig. 1C). From this strain megapolipeptins A (1) and B (2) were isolated and structurally characterized (Fig. 1D). Megapolipeptins are unusual polyketide-nonribosomal peptides with varying polyunsaturated fatty acid components. They show low structural similarity to other known bacterial natural products (maximum Tanimoto similarity score of 0.58 (1) and 0.56 (2) compared to all entries in the Natural Product Atlas),1,20 highlighting the advantage of our Burkholderiales genomics-driven and synthetic biology-enabled pipeline to discover novel natural products.
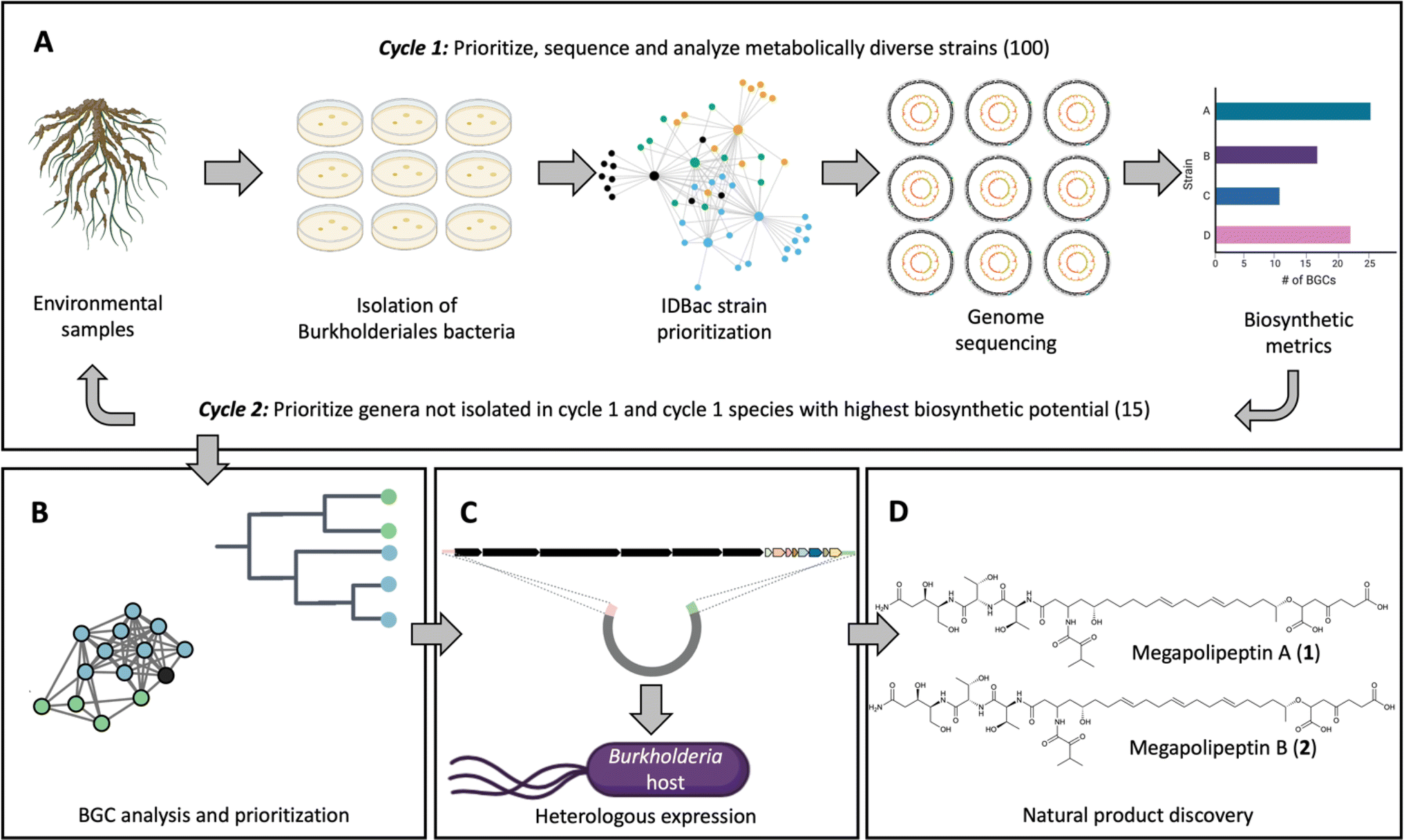 | ||
| Fig. 1 Overview of the approach used in this study. (A) Environmental samples (rhizosphere) were collected from British Columbia, Canada. Burkholderiales strains were then isolated from the rhizosphere of root samples using selective media.16 In the first cycle, 230 isolated strains were analyzed by MALDI-TOF MS/IDBac and 100 strains were selected for genome sequencing based on the analysis of metabolite association networks; the intent of this step was to avoid strain redundancy while maximizing metabolite diversity entering sequencing efforts (ESI Fig. S1†).17 One hundred draft genome sequences were obtained. Biosynthetic gene clusters (BGCs) were predicted using antiSMASH and the biosynthetic potential of strains was compared in terms of BGC numbers and biosynthetic class. Informed by the predicted biosynthetic potential, cycle 2 targeted any newly isolated genera not included in cycle 1 and species determined to be the most ‘talented’ in cycle 1, resulting in 15 additional strains sequenced from 86 analyzed. See ESI Tables S1–S3† for details on the strains sequenced. (B) Phylogenomic and gene cluster family (GCF) analyses were performed on a total of 115 strains to gain insight into GCF distribution and to prioritize BGCs for discovery. (C) A prioritized BGC with clade-specific distribution that was silent in the native strains was cloned and heterologously expressed in Burkholderia sp. FERM BP-3421. (D) Natural product isolation and structure elucidation yielded megapolipeptins A (1) and B (2). | ||
Results and discussion
Strain isolation, prioritization, and sequencing
We built a collection of Burkholderiales bacterial strains over the course of five years (2016–2021) using a method we previously developed.16 In brief, rhizosphere microbial communities were collected from plant samples in southern British Columbia and grown on solid agar selection media optimized for the growth of Burkholderiales strains. Individual colonies were identified by MALDI-TOF MS using a Bruker Biotyper and a custom mass spectral reference library15 and validated Burkholderiales strains archived as glycerol stocks. As the collection was being built, we used a two-cycle strategy to prioritize strains for genome sequencing. Strains were analyzed with IDBac, a technique that is based on MALDI-TOF MS analyses of protein and metabolite spectra obtained using cell mass from bacterial colonies.17 Strains were first grouped based on similarities within their protein spectra (2000–15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da), and further discriminated based on overlap of metabolites (200–2000 Da) observed in metabolite association networks. The intent of this process was to avoid redundancy while maximizing metabolite diversity heading into sequencing efforts. In the first prioritization cycle, we analyzed 230 isolated strains by IDBac and selected 100 for genome sequencing (Fig. 1A and S1†). Genomes were sequenced using Illumina technology, assembled, and analyzed for phylogeny and BGC diversity.
000 Da), and further discriminated based on overlap of metabolites (200–2000 Da) observed in metabolite association networks. The intent of this process was to avoid redundancy while maximizing metabolite diversity heading into sequencing efforts. In the first prioritization cycle, we analyzed 230 isolated strains by IDBac and selected 100 for genome sequencing (Fig. 1A and S1†). Genomes were sequenced using Illumina technology, assembled, and analyzed for phylogeny and BGC diversity.
In the second round of prioritization (Fig. 1A), 86 newly isolated strains were analyzed, and 15 strains were selected that either added phylogenetic diversity (new genus or species not included in cycle 1) or that were determined in the first round to be “talented” in terms of BGC number and diversity but that were underrepresented in the dataset. Thus, in the second round we included Herbasparillum, the only new genus we isolated that was not previously represented, and additional Paraburkholderia megapolitana and Paraburkholderia fungorum strains, which appeared “talented” in terms of BGC number and diversity but that were underrepresented (Fig. 2A and S2†). At the same time, we did not include new Caballeronia spp. because of the lower number of BGCs per strain observed in the first round (9.75 BGCs on average), nor did we include new P. sediminicola strains because they were already well represented in the first round.
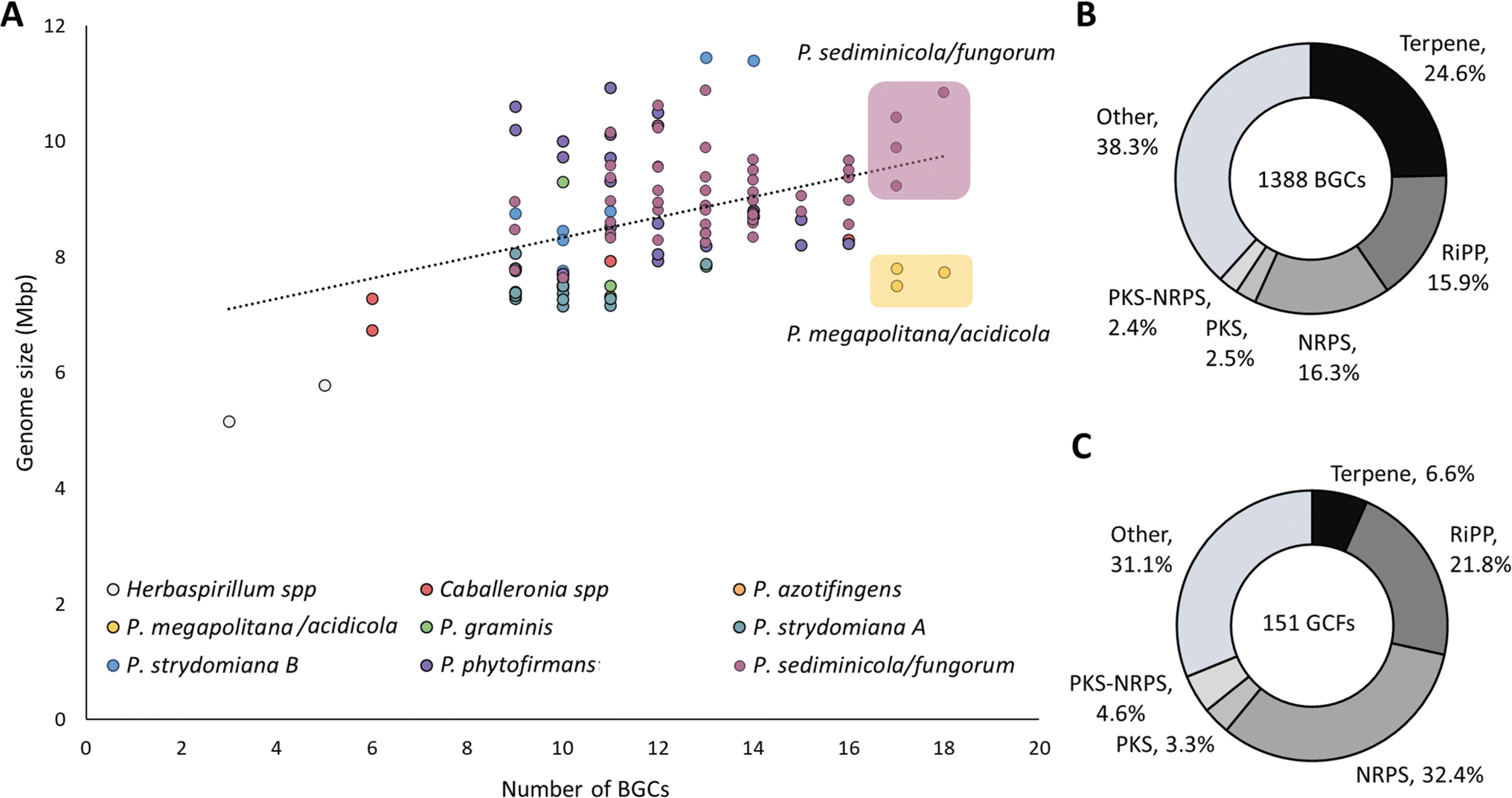 | ||
| Fig. 2 Genome library metrics. (A) Genome size by number of BGCs, color coded according to the clades attributed in Fig. 3. R2 = 0.18. The top 10% strains most prolific in terms of number of BGCs are highlighted in yellow (P. megapolitana/acidicola) and purple (P. sediminicola/fungorum). (B) Donut charts depicting the total number of either BGCs or (C) GCFs subdivided by biosynthetic class. | ||
In total, 115 draft genomes were obtained using short-read Illumina sequencing and Unicycler assembly (ESI Tables S1–S3†), resulting in 159 contigs on average (range of 33 to 780). Eight representative genomes were also sequenced using long-read Oxford Nanopore technology resulting in four complete genomes with circular replicons and four for which some contigs remained linear (ESI Table S4†).
Biosynthetic capacity
To detect BGCs, the genomes were analyzed with antiSMASH 6.0.21 A total of 1388 BGCs (after manual curation as described later) were identified and categorized into six groups, i.e., terpene, ribosomally synthesized and posttranslationally modified peptide (RiPP), nonribosomal peptide synthetase (NRPS), polyketide synthase (PKS), PKS-NRPS hybrid, and ‘other’ which included phosphonate, non-NRPS siderophore, redox-cofactor, ectoine, arylpolyene, phenazines, butyrolactones, furan and homoserine lactone (Fig. 2B, S2, and S3†).An average of ∼12 BGCs per strain were predicted in accordance with previous Burkholderiales studies.22 There was only weak association between number of BGCs and genome size (R2 = 0.18). Instead, the best predictor of biosynthetic capacity appeared to be phylogeny. Notably, the clade containing Paraburkholderia acidicola and Paraburkholderia megapolitana had the highest ratio of number of BGCs to genome size at 2.1 BGCs per Mbp followed by P. azotifigens at 1.6 and P. sediminicola/fungorum at 1.5 (Fig. 2A and ESI Table S5†).
In terms of biosynthetic class, terpene BGCs were the most abundant (341, 24.6%) and are present in every single strain in the collection. NRPS (226, 16.3%) and RiPP (220, 15.9%) are also well distributed amongst strains. PKS (35, 2.5%) and PKS-NRPS (34, 2.4%) are less abundant classes (Fig. 2B) with highest occurrence in Paraburkholderia megapolitana, Paraburkholderia fungorum, and some Paraburkholderia sediminicola strains (ESI Fig. S2†). In the ‘other’ category (532, 38.3%, ESI Fig. S3†), the main contributions came from homoserine lactones (134, 9.6%), phosphonates (115, 8.3%), arylpolyenes (112, 8.1%), betalactones (77, 5.5%), and redox cofactor (69, 5%) whereas minor groups included phenazines, non-NRPS siderophores, ectoines, butyrolactones, and furan (≤3, ≤0.2%).
As the same BGC may be present in many strains, total BGC count does not reflect natural product diversity. To explore the natural product diversity encoded in the strain library, we analyzed the 1388 BGCs using the biosynthetic gene similarity clustering and prospecting engine BiG-SCAPE23 which generates BGC similarity networks. Using a clustering threshold of 0.4 as previously reported to best represent similar natural products,2 151 gene cluster families (GCFs) were obtained, including 78 networks and 73 singleton BGCs. An analysis of biosynthetic class distribution (Fig. 2C) shows that there is more redundancy in the terpene space than could be determined from the total number of BGCs (341 BGCs [24.6%] grouping into 10 GCFs [6.6%]). In contrast, there is more diversity in the NRPS space (226 BGCs [16.3%] grouping into 49 GCFs [32.4%]). In the ‘other’ category (ESI Fig. S3†), redundancy is most apparent in arylpolyenes (112 BGCs [8.1%] grouping into 4 GCFs [2.6%]) and phosphonates (115 BGCs [8.3%], 4 GCFs [2.6%]).
Biosynthetic capacity based on phylogeny
Genomes were submitted to GenBank, and a species name was assigned based on Average Nucleotide Identity (ANI) equal to or greater than 96% to a previously deposited species. This led to 107 strains being assigned a species name and eight strains receiving a classification at the genus level only.We next performed a phylogenomic analysis of the 115 strains and observed three monophyletic groups representing three currently described genera, Herbaspirillum (2 strains), Caballeronia (4 strains), and Paraburkholderia (109 strains), with the latter having the largest representation in the collection (Fig. 3A and S4†). Moreover, Paraburkholderia strains also had the highest number of BGCs (Fig. 2A and S2†). To investigate correlations between BGC distribution and phylogeny, we subdivided the Paraburkholderia group into seven monophyletic groups as shown in Fig. 3A and S5.†
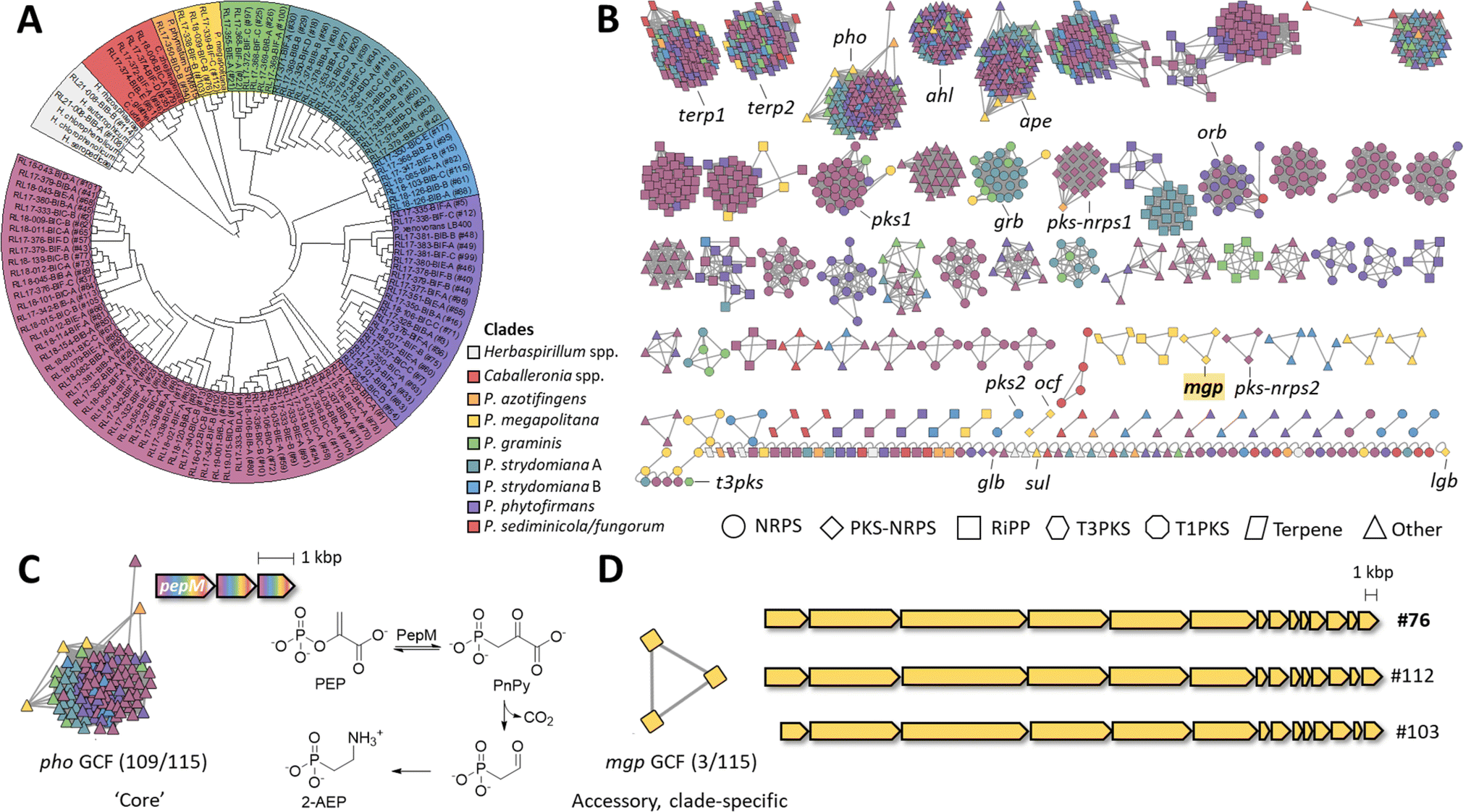 | ||
| Fig. 3 Phylogenomic analysis and BGC distribution. (A) Phylogenomic tree of Burkholderiales strains based on 49 genes within cluster of orthologous groups (ESI Table S6†). The tree was constructed using the neighbor-joining method. Select Paraburkholderia, Herbaspirillum and Caballeronia genomes available in public databases were included in addition to the 115 strains sequenced in this study which are shown with our internal strain numbering scheme. The Paraburkholderia clade was further subdivided into seven monophyletic groups as highlighted. See also ESI Fig. S4 and S5.† (B) BiG-SCAPE BGC Sequence Similarity Network within the 115 Burkholderiales genomes (distance cutoff = 0.4). A total of 1388 BGCs are displayed, color-coded according to the clades in panel A. Node shape indicates BGC class according to BiG-SCAPE classification. Known and orphan BGCs described in the text are highlighted. (C) Example of a widely distributed BGC that is part of the core genome of Paraburkholderia strains. BGCs in this family contain three core genes and varying gene neighborhoods. The core genes are predicted to encode the biosynthesis of 2-aminoethyl phosphonate (2-AEP) from phosphoenolpyruvate (PEP) via phosphonopyruvate (PnPy) and phosphonoacetaldehyde. (D) The clade-specific mgp GCF and BGC investigated in this work from genome #76 (ESI Table S1†). See ESI Table S7† for gene details. | ||
To explore the prevalence and potential novelty of BGCs from our collection, we included the reference BGCs from the Minimum Information about a Biosynthetic Gene cluster (MIBiG)24 database in the BiG-SCAPE23 analysis (ESI Fig. S6†). The large majority of the BGCs (1366 BGCs, 98.4%) did not associate with MIBiG nodes. The 1.6% that had an MIBiG counterpart included: (1) an NRPS-PKS similar to that encoding antifungal occidiofungin from Burkholderia pyrrocinia (ocf), which matched with two strains of P. megapolitana (RL18-039-BIC-B and RL17-339-BIF-C);25 (2) the antifungal lagriamide A (lga) encoded in strain P. acidicola RL17-338-BIF-B26 that we recently showed to encode a new compound lagriamide B (lgb BGC);27 (3) the nonribosomal peptide siderophore gramibactin (grb) encoded in 13 strains from the P. graminis and P. stydomiana clades;28 and (4) the β-lactam antibiotic sulfazecin (sul) encoded in P. acidicola RL17-338-BIF-B (ESI Fig. S6†).29 After manual curation, we further identified a glidobactin-like BCG (glb)30 in P. fungorum RL18-167-BIC-A and an ornibactin-like BGC (orb)31 in 21 strains.
Superclusters, which occur when two or more clusters closely co-localize and are treated as one entity, are a common issue with automated BGC prediction. The presence of superclusters in the direct antiSMASH output led to known PKS-NRPS BGCs ocf and lgb forming a network with terp2 (ESI Fig. S6†). We next manually curated PKS-NRPS BGCs that were likely part of superclusters to split the clusters and generate a new network. To facilitate visualization and extraction of networks by biosynthetic class, MIBiG nodes were removed to generate the sequence similarity network displayed in Fig. 3B.
Core and accessory BGCs
In pangenome analysis, core genes are those shared by all isolates of a particular group, whereas accessory genes are present in only some of the genomes.32 The top 5 most prevalent BGCs (Fig. 3B) belong to biosynthetic classes terpene (2 GCFs), phosphonate, N-acyl homoserine lactone, and aryl polyene (1 GCF each). The most prevalent BGCs were included in two terpene GCFs, one containing a squalene synthase (terp1) and another a phytoene synthase (terp2), which occurred in 113 and 111 of 115 strains, respectively. Next, an aryl polyene (ape) BGC was present in 111 strains. The ape BGC is widely distributed in Gram-negative bacteria and can be classified into three different subfamilies.33 The GCF present in our collection appears to belong to subfamily 1 which encodes a yellow pigment consisting of a 4-hydroxy-3-methylphenyl head group conjugated to hexenoic acid.33 Aryl polyenes have been shown to protect bacteria from reactive oxygen species.34 The prevalence of terpene and aryl polyene BGCs in our collection is consistent with a recent analysis showing these BGCs to be conserved in plant-associated microbiomes.35An N-acyl homoserine lactone BGC (ahl) was present in 109 strains. N-acyl homoserine lactones are involved in quorum sensing in bacteria and are known to regulate behaviors such as virulence and biofilm formation.36–39 Finally, phosphonates were also recurrent in our collection being present in 114 strains out of 115. The largest GCF (pho) contained 109 BGCs with three core genes (Fig. 3C). The first gene is common to most phosphonate pathways and is predicted to encode the phosphoenolpyruvate (PEP) mutase PepM, which reversibly converts PEP into phosphonopyruvate (PnPy). The operon contains two other genes predicted to encode a decarboxylase, and a transaminase, catalyzing decarboxylation of PnPy to the aldehyde and reductive amination, respectively, to yield 2-aminoethyl phosphonate (2-AEP). 2-AEP may be attached to structural components such as polysaccharides or lipids.40 The high prevalence of phosphonate BGCs in our collection agrees with prior reports. Based on the presence of pepM homologs, phosphonate biosynthesis was predicted to be encoded in ∼5% of bacterial genomes at large but in 94% of Burkholderia genomes.41
For the accessory BGCs, NRPSs (ESI Fig. S7†) are the most abundant and present in all strains except in Herbaspirillum rhizosphaerae RL21-008-BIB-B (#114, ESI Table S1†). Nearly all NRPS GCFs have a monophyletic distribution (Fig. 3B), except for siderophores gramibactin (grb) and ornibactin (orb) (ESI Fig. S7†). The clade-specific nature of NRPS clusters suggests vertical transmission of specialized functionalities with distinct nonribosomal peptides being produced by phylogenetically distinct strains. RiPPs follow the same tendency of clade-specific distribution with three exceptions (ESI Fig. S8†).
Type I PKS BGCs (ESI Fig. S9†) are less abundant (34/1388, 2.4%). The most prevalent BGC is an orphan, monomodular type I PKS present in 30 genomes, mainly from the P. sediminicola/P. fungorum clade (pks1, Fig. 3B and S9†). The remaining four BGCs fall into one GCF containing two members (pks2) and two singletons. Finally, one PKS belongs to type 3 (t3pks).
PKS-NRPS hybrid BGCs (ESI Fig. S10†) have low abundance as well (34/1388, 2.4%). They are present in P. sediminicola/fungorum, P. azotifingens, and P. megapolitana/acidicola clades. The largest GCF (23 BGCs, pks-nrps1) contains BGCs from P. sediminicola/fungorum and P. azotifingens clades. The highest diversity of PKS-NRPS BGCs comes from the P. megapolitana/acidicola clade, all three strains of which contain two PKS-NRPS BGCs that fall into the known ocf GCF, the known lgb27 singleton, and two orphan GCFs, pks-nrps2 and the mgp BGC studied here.
BGC prioritization, cloning and expression
We opted for prioritization based on biosynthetic class. All Burkholderiaceae natural products that have advanced in the drug discovery pipeline are hybrid polyketide nonribosomal peptides,5i.e., spliceostatins underwent pre-clinical development, rhizoxin entered clinical trials, and romidepsin is an approved anticancer agent.42–44 Thus, we decided to focus on PKS-NRPS BGCs. We were particularly intrigued by a BGC that is conserved in the P. megapolitana/acidicola clade and that also contains polyunsaturated fatty acid (PUFA) genes. Because assembly lines containing PKS, NRPS and PUFA gene clusters are uncommon, we expected to uncover new products. We named this BGC mgp for megapolipeptin (Fig. 3B and D and ESI Table S7†). Examples of such hybrid BGCs include those encoding polyamine antibiotics fabclavines from Xenorhabdus45 and zeamine from Serratia.46 However, the mgp BGC is distinct from these polyamine clusters.The mgp BGCs from P. megapolitana RL18-039-BIC-B (genome #76, Table S1†) and P. megapolitana RL17-339-BIF-C (#112) share 96.6% pairwise identity and 77% identity to the BGC found in P. acidicola RL17-338-BIF-B (#103), with the BGC from P. acidicola having a shorter mgpA PKS gene (Fig. 3D). Zheng et al.47 previously identified the BGC with the longer mgpA gene in P. megapolitana DSM 23488. Because the BGC was silent, the authors activated gene expression using promoter replacement in strain DSM 23488. Although mass spectrometry features could be detected, low yields precluded attempts at isolation and structure elucidation of any target molecules. We were likewise unable to detect the products of this BGC in the wild type strains from our collection. Thus, we cloned and expressed the BGC in Burkholderia sp. FERM BP-3421, a host we have been developing as an alternative synthetic biology chassis.18,48 Obtaining the product of the mgp BGC in sufficient quantity for characterization would allow the evaluation of the host's performance with a complex BGC and advance current knowledge of hybrid polyunsaturated fatty acid, polyketide, and nonribosomal peptide systems.
The mgp BGC is located on chromosome 2 of the three genomes (Fig. 4A, #76 shown) and it contains 14 open reading frames (ORFs) spanning 54 kbp (Fig. 3D). The mgp BGC from P. megapolitana RL18-039-BIC-B (#76) was cloned using a CRISPR-Cas9 based methodology.49 The obtained plasmid pBS001 was transferred into a spliceostatin-defective mutant (Δfr9A) of Burkholderia sp. FERM BP-3421,19 and exconjugants were confirmed by PCR (ESI Fig. S11†). Molecular networking was performed to identify molecular features present only in mutants harboring the mgp BGC but absent in strains containing the empty vector pBS003 (ESI Fig. S12–S16†). The analysis identified m/z 984.538 and m/z 958.522 (Fig. 4B) that were pursued for isolation. These features were the ones identified in strain DSM 23488 after promoter replacement, but quantities had not been enough for isolation.47
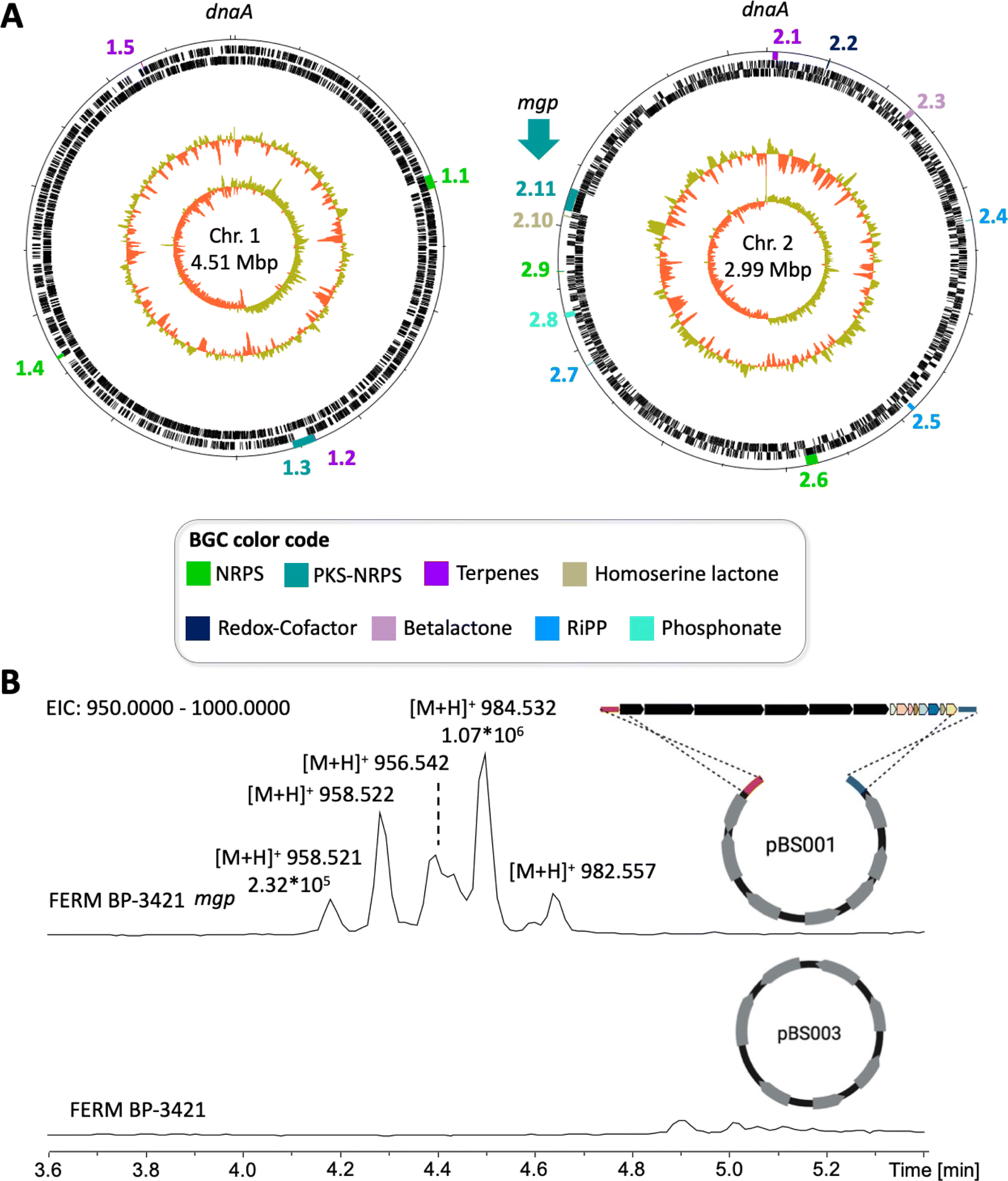 | ||
| Fig. 4 Heterologous expression of the mgp BGC from P. megapolitana RL18-039-BIC-B. (A) Genome map of P. megapolitana RL18-039-BIC-B with the two chromosomes oriented to replication gene dnaA. BGCs are color-coded according to biosynthetic class and numbered in clockwise order from the replication gene (lane 1 from the outside in). Predicted open reading frames (ORFs) on the leading and lagging strands are shown on lanes 2 and 3, respectively. A normalized and skewed plot of guanosine + cytosine (G + C) content (yellow/orange) is depicted in lanes 4 and 5, respectively. (B) Heterologous expression of mgp (BGC 2.11) in Burkholderia sp. FERM BP-3421 Δfr9A. LC-MS analysis of strains containing either the empty vector (pBS003, bottom trace) or the vector containing the mgp BGC (pBS001, top trace). Extracted ion chromatogram (EIC) of region m/z 950–1000. | ||
Isolation and structure elucidation of megapolipeptin A (1) and B (2)
Fermentation of Burkholderia sp. pBS001 (6.5 L) followed by liquid extraction with organic solvent (2:1 CH2Cl2/MeOH), solid-phase (C18) extraction, and mass-guided isolation by HPLC-MS, yielded 3.7 mg of 1 and 9.8 mg of 2 as amorphous, white solids (Fig. 5A). High-resolution mass spectrometry (HRMS) of 1 suggested a molecular formula of C45H75N5O17 based on the protonated cluster ion at [M + H]+m/z 958.52350 (calcd m/z 958.52307) (ESI Fig. S17†). Examination of the MS/MS spectrum revealed two sequential neutral losses consistent with threonine amino acid residues (Fig. 5B and S18†). This analysis, coupled with a comprehensive set of 1D and 2D nuclear magnetic resonance (NMR) spectra identified 1 as containing six isolated spin systems: 4-amino-3,5-dihydroxypentanamide (Ahpa), two threonine residues (1Thr and 2Thr), 3-amino-5,19-dihydroxyicosa-10,14-dienamide (Adhda), 2-hydroxy-4-oxoheptanedioic acid (Hoha), and a 2-methyl-propan-1-one (Mpo) moiety (ESI Fig. S19–25†). The assembly of these substructures was completed based on gHMBC correlations between the amide carbon of 1Thr-1 and Ahpa-NH, 1Thr-NH and 2Thr-1, 2Thr-NH and Adhda-1, Adhda-19 and Hoha-2 (ESI Fig. S26†).
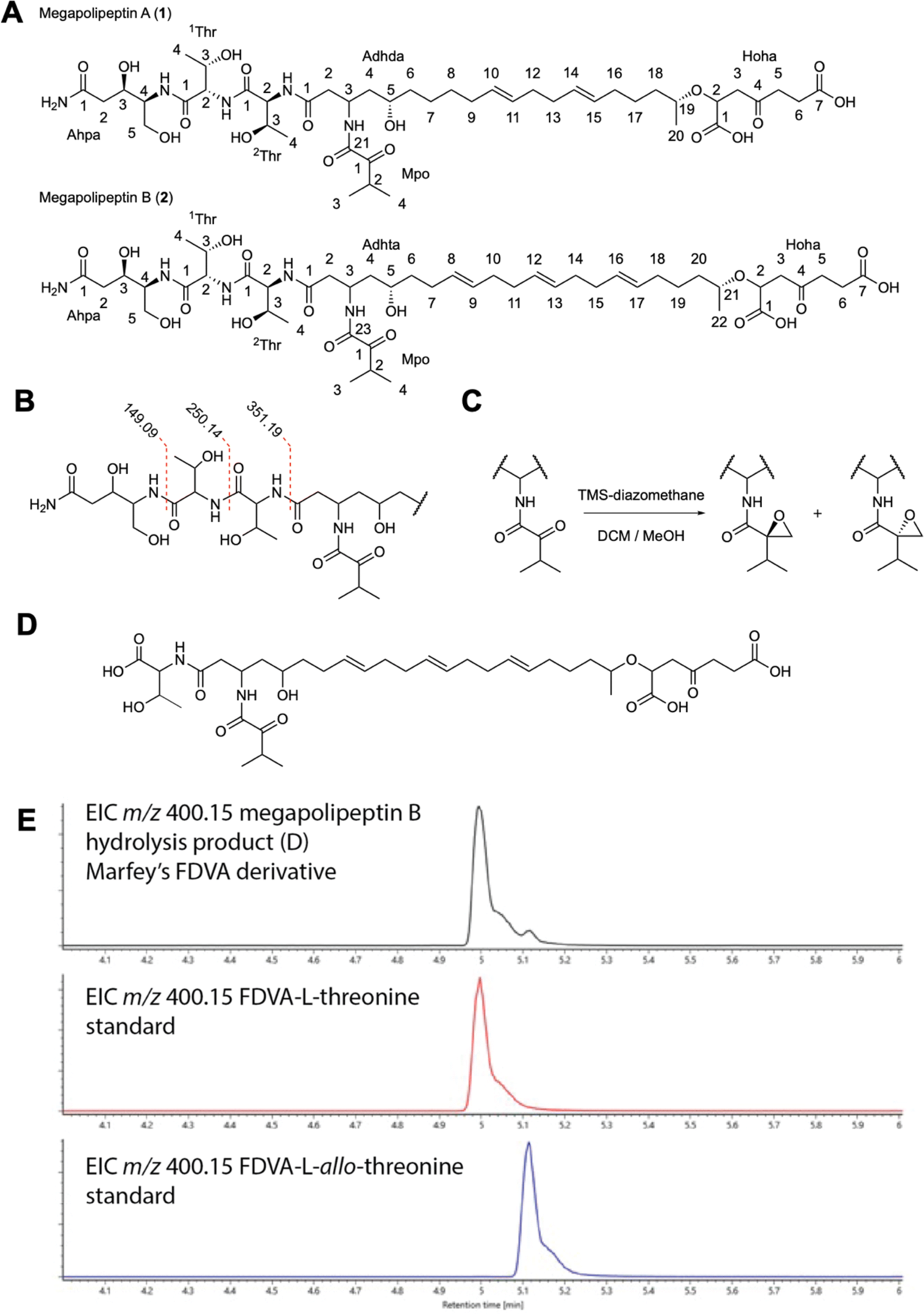 | ||
| Fig. 5 Structure elucidation of megapolipeptins from P. megapolitana RL18-039-BIC-B. (A) The structures of megapolipeptin A (1) and megapolipeptin B (2). (B) Key MS/MS fragments showing neutral losses of threonine amino acid residues on the peptidic terminus of 1 and 2. (C) Reaction scheme with TMS diazomethane in DCM/MeOH affording two diastereotopic epoxide-containing products via the Büchner–Curtius–Schlotterbeck reaction. (D) Structure of partial hydrolysis product used to determine configuration of threonine residues. (E) EIC traces of Marfey's derivative of threonine (m/z = 400.15) for partial hydrolysis product, and L-threonine and L-allo-threonine standards. | ||
The full planar structure could not be unambiguously determined from the NMR data as no correlations were observed between the Mpo moiety and the rest of the molecule. Possible connections included the carbonyl groups at Adhda-21, Hoha-1, or Hoha-7. To resolve this issue the molecule was treated with TMS diazomethane to convert the carboxylic acid groups to their corresponding methyl esters. LC-MS analysis of the methylated product showed two peaks, each with an increase in mass of 42 Da suggesting the addition of C3H6 which was unexpected given the presence of only two carboxylic acid moieties (ESI Fig. S27–29†). The derivatized products (3 and 4) were isolated and analyzed by NMR. The 1H-NMR and gHMBC spectra showed two methoxy signals correlating with Hoha-1 and Hoha-7. Closer inspection of the NMR data revealed that Mpo-1 was no longer present. Instead, the gCOSY spectrum revealed that this moiety had been converted to a 2-isopropyloxirane (Ipo), via a Buchner–Curtius–Schlotterbeck rearrangement between the ketone functional group of the Mpo subunit and TMS-diazomethane (Fig. 5C). The complete planar structures of 3 and 4 were determined using a full suite of 1D and 2D NMR experiments (ESI Fig. S30†). Based on these data, it was determined the Mpo moiety was attached to Adhda-21, completing the planar structure of 1 (Fig. 5A and ESI Table S8†).
HRMS and MS/MS fragmentation data of 2 displayed a protonated cluster ion at [M + H]+m/z 984.53821 (calcd m/z 984.53872) and neutral losses of adjacent threonine residues (1Thr and 2Thr) suggesting a molecular formula of C47H77N5O17 and a structural analog of 1 with a mass difference of 26 Da (ESI Fig. S31 and S32†). Examination of the 1H-NMR spectrum showed the presence of two additional vinylic methine protons along the hydrocarbon chain (Fig. 5A and S33†). The NMR data of 2 were comparable to 1 showing five identical spin systems: Ahpa, 1Thr, 2Thr, Hoha, and Mpo moieties. Further examination of the 2D NMR spectra revealed the final spin system as 3-amino-5,21-dihydroxydocosa-8,12,16-trienamide (Adhta) (Fig. 5A, ESI Table S9 and Fig. S33–38†). The position of the Mpo moiety was determined in similar fashion to 1 by treatment with TMS diazomethane to generate the 2-isopropyloxirane motif and identification of gHMBC correlations between Mpo-1 and Adhta-23, completing the planar structure of 2 (ESI Fig. S39 and S40†).
The configurational analysis of megapolipeptins A and B represents a significant analytical challenge. Megapolipeptin A contains 9 chiral centers and two double bonds, for a total of 2048 possible configurations. Because many of these centers are separated from one another by achiral regions it is not straightforward to directly relate their relative or absolute configurations. Instead, the absolute configurations of centers in each region must be determined independently. The absolute configurations of the threonine amino acid-derived stereocenters (1Thr and 2Thr) in 1 and 2 were examined using Marfey's analysis50 (ESI Fig. S41†) which revealed the presence of L-threonine and L-allo-threonine in both molecules. To determine the positions of each amino acid megapolipeptin B (2) was subjected to partial acid hydrolysis (1 N HCl, 110 °C, 30 minutes). UPLC-MS analysis of the hydrolysate revealed the presence of a product consistent with hydrolysis between the two threonine residues (Fig. 5D and S42†). HPLC purification, full acid hydrolysis, and Marfey's analysis of this product defined the configuration of the 2Thr residue as L-threonine (Fig. 5E), which by extension defined 1Thr as L-allo-threonine.
Traditionally, the configurations of disubstituted olefins are determined from the 3JHH coupling constant between the two olefinic signals (15–17 Hz = trans, ∼10 Hz = cis). However, in pseudosymmetrical systems such as the megapolipeptins the olefinic 1H signals are often highly overlapped. Fortunately, both the olefinic carbons (Adhda C10, C11, C14, C15) and the adjacent allylic carbons (Adhda C9, C12, C13, C16) possess diagnostic chemical shifts between cis and trans systems. In both 1 and 2 the olefinic carbons were all in the range 129.7 ± 0.7 ppm, indicative of an all-trans arrangement. This contrasts with cis olefins, where 13C shifts are ∼128.0 ppm.51 Further supporting evidence for the all-trans arrangement was provided by the allylic carbon chemical shifts centered around 32.0 ppm. In cis olefins these values center around ∼27.3 ppm.
Finally, as will be discussed in the following section on the proposed biosynthesis, the configurations of several centers could be inferred from the biosynthetic gene cluster. Analysis of the module responsible for the installation of the ketide-extended serine (Ahpa) indicated the installation of L-serine, followed by extension and reduction by the associated A-type ketoreductase (KR) to install a hydroxy group with L-orientation at position Ahpa-3. The KR in MgpA responsible for installing the hydroxyl group at C5 of the fatty acid (Adhda-5 (1) and Adhta-5 (2)) is also A type. The hydroxy groups at Adhda-19 (1) or Adhta-21 (2) are predicted to have D-orientation based on the B-type KR within MgpE. The configuration at Adhda-3 was not determined.
Bioactivity testing
Compounds 1 and 2 were tested for antimicrobial susceptibility against a panel of 17 bacterial pathogens (ESI Table S10†) in our previously developed BioMAP antibacterial profiling platform.52 No growth inhibition of pathogenic organisms was observed up to a maximum concentration of 128 μM (ESI Table S11†). Compounds 1 and 2 exhibited no antimycotic activity against Candida albicans or Saccharomyces cerevisiae in microbroth dilution assays up to a maximum concentration of 100 μM. Additionally, neither compound was active against Aspergillus niger or Purpureocillium lilacinum in qualitative filamentous screening assays up to a maximum concentration of 100 μM (ESI Tables S11 and S12†).Proposed biosynthesis
We propose the following working biosynthetic hypothesis, based on the elucidated structures and the content of the mgp BGC (Fig. 6A).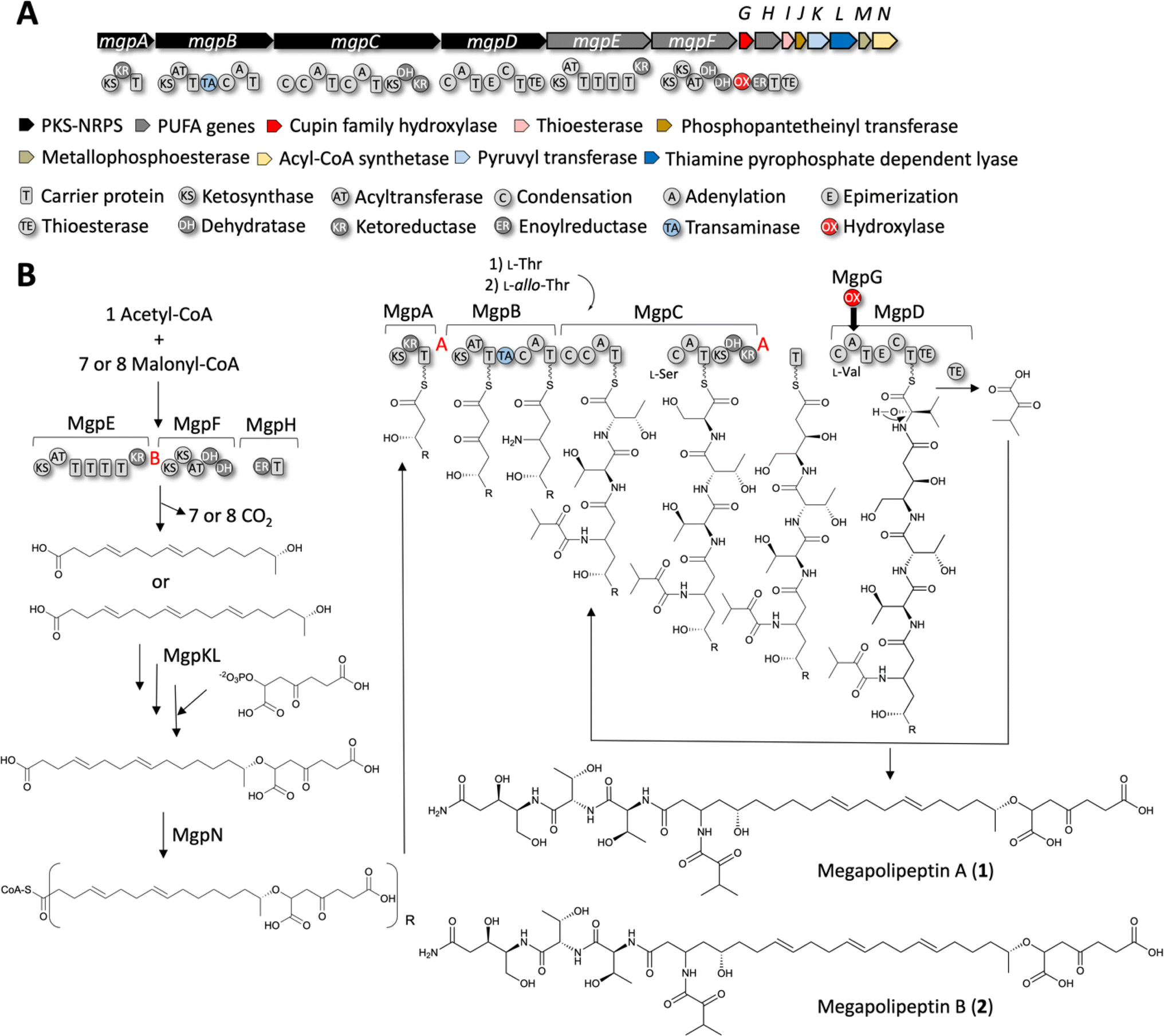 | ||
| Fig. 6 Biosynthetic proposal for mgp BGC from P. megapolitana RL18-039-BIC-B. (A) Megapolipeptin biosynthetic gene cluster from P. megapolitana RL18-039-BIC-A (genome #76). (B) Biosynthetic hypothesis based on gene/domain content and the observed structures. The configuration of chiral centers containing hydroxyl groups was predicted based on KR domain type (ESI Fig. S43†). The KR type is indicated with red A, B letters. | ||
Four proteins have been implicated in PUFA biosynthesis in bacteria, PfaA-PfaD, in addition to a phosphopantetheinyl transferase PfaE that may or may not be present in PUFA clusters.53,54 MgpE is homologous to PfaA that displays a KS-AT-(ACP)n-KR domain organization. MgpF appears to be a variation of PfaBC containing a KS-KS-AT-DH-DH domain organization, and MgpH encodes an ER domain, resembling PfaD. We propose MgpE, MgpF and MgpH catalyze biosynthesis of the fatty acid portion of megapolipeptins from acetyl-CoA and either 7 or 8 malonyl-CoA units leading to 16:2(6,10) or 18:3(4,8,12) unsaturated fatty acids, respectively. Based on the predicted B-type KR (ESI Fig. S43†) within MgpE, the hydroxyl group would possess the R configuration. Additionally, a 4-oxoheptanedioic moiety decorates the terminal ω-1 hydroxyl group. Biosynthesis of the potential precursor 1,5-dicarboxy-3-oxopentyl phosphate might be catalyzed by the putative pyruvyl transferase MgpK and thiamine pyrophosphate-dependent lyase MgpL.55 Alternatively, the 4-oxoheptanedioic moiety may derive from lipid peroxidation.56 Either way, the acyl-CoA synthetase MgpN would activate the fatty acid component for loading into MgpA (Fig. 6B).
MgpA is a PKS with a KS-KR-T organization that we propose extend the fatty acid chain with one malonate unit followed by reduction of the β-keto group to a hydroxyl. Based on the predicted A-type KR (ESI Fig. S43†), the hydroxyl group would possess the S configuration. MgpB and MgpC are hybrid PKS-NRPS enzymes containing unusual domain organization. MgpB (KS-AT-T-TA-C-A-T) would catalyze another C2 extension with malonate, followed by reductive amination of the β-carbonyl catalyzed by the transaminase (TA) domain as has been described for MycA in the biosynthesis of mycosubtilin.57 The A domain in this module is predicted by antiSMASH to load a proline, however seven residues are different from the expected proline code.58,59 We propose this A domain may load α-ketoisovaleric acid according to the structures of megapolipeptins, although the keto-acid code is also not conserved (ESI Table S13 and Fig. S44†).60
Based on sequence and phylogenetic analyses (ESI Fig. S45 and 46†), four of the six C domains clade with LCL domains, indicating that they process L-amino acids (the one within MgpB, the first and the third within MgpC and the first within MgpD). The first module in MgpC (C-C-A-T-C-A-T-KS-KR-DH) appears to catalyze the iterative addition of two L-threonine units (Fig. 6B), which agrees with the A domain code (ESI Table S13†). As determined by partial hydrolysis of megapolipeptin B followed by Marfey's analysis (Fig. 5D and E and S39†), L-threonine is incorporated first followed by L-allo-threonine. The second module in MgpC would add L-serine, followed by a ketide extension using malonate by an AT-less PKS module. The A-type KR in this domain would then reduce the β-carbonyl to a (S) hydroxyl group (Fig. 6B and S43†). The DH is predicted to be inactive as the catalytic histidine and aspartate residues are mutated (ESI Fig. S47†). Accordingly, no dehydration is expected, and the hydroxyl group is maintained in the final structure. The second C domain in MgpD (C-A-T-E-C-T-TE) clades with DCL domains in accordance with the presence of an epimerization domain in this module, suggesting that the L-valine selected by the A domain (ESI Table S13†) is epimerized to D-valine (Fig. 6B).
MgpD may be involved in terminal amide biosynthesis and perhaps in providing α-ketoisovaleric acid. Terminal amide biosynthesis has been described for myxothiazol and melithiazol (MtaG/MelG) from myxobacteria.61,62 MtaG/MelG display a C-A-MOX-A-T-TE domain organization where the A domain is split with a monooxygenase (MOX). Condensation with glycine followed by hydroxylation of the α-carbon catalyzed by MOX and dealkylation of the alcohol amide is proposed to yield the terminal amide, while the TE domain releases the α-ketoacid. Analogously, MgpD could add valine which followed by hydroxylation could result in the terminal amide and into α-ketoisovaleric acid to be condensed with the free amine product of MgpB (Fig. 6B). Although MgpD does not contain a MOX domain, MgpG encodes a standalone cupin-family hydroxylase63,64 that we propose may act in trans. Alternatively, MgpG could catalyze α-hydroxylation of the valine unit and dealkylation after product release from the NRPS by the TE domain. The second condensation domain of MgpC clades with starter C domains and may be the one responsible for capping of the free amine with the α-ketoisovaleric acid. All C domains possess the conserved catalytic motif HHxxxDG motif65,66 except for this second C domain in MgpC which contains the variation HHxxxDR (ESI Fig. S45†). All KS domains contain the catalytic triad of Cys-His-His essential for decarboxylative condensation and are thus predicted to be active (ESI Fig. S48†).
Finally, MgpI encodes a thioesterase that could have proof reading function67 as it is common for PKS-NRPS systems. MgpJ encodes a phosphopantetheinyl transferase that likely serves to activate PUFA, PKS and NRPS carrier proteins. The role of MgpM, a putative metallophosphoesterase is unclear.
Conclusion
In this study, we aimed to harness recent advancements in complementary technologies to establish a robust pipeline for natural product discovery from Burkholderiales bacteria. This approach is designed to be applicable not only to Burkholderiales but also to other bacteria (Fig. 1). The P. megapolitana/P. acidicola clade exhibited the highest ratio of number of BGCs to genome size and the highest number of BGCs per strain (Fig. 2A and ESI Table S5†). Despite these promising features, strains from this clade were rarely isolated from culture plates and consequently had low representation in the final library. To address this limitation, future isolation efforts should account for the slower growth characteristics of this clade.In terms of the biosynthetic capacity of our collection, terpene, and phosphonate BGCs are the most conserved (Fig. 3). NRPS and RiPP BGCs are also abundant but tend to show monophyletic distribution; thus, to find new NRPS and RiPP BGCs, taxonomic diversity is important. In contrast, PKS and PKS-NRPS gene cluster families are the rarest in the collection (Fig. 2B and C). The largest diversity of PKS-NRPS BGCs was found in the P. megapolitana/acidicola clade, where each strain contained two such BGCs, with the mgp BGC being conserved in all three strains in this clade. Because we did not detect potential products of the mgp BGC in the wild-type strains, we turned instead to heterologous expression in a Burkholderia sp. strain (Fig. 4) which resulted in the discovery of megapolipeptins A (1) and B (2) at 0.6 and 1.5 mg L−1 isolated yields, respectively (Fig. 5). This work expands recent genome mining efforts in P. megapolitana/acidicola.27,47,68
Megapolipeptins are bolaamphiphilic lipopeptides, that is, they exhibit a hydrophobic center and hydrophylic groups at each end of the molecule, such as the recently discovered bolagladins.69,70 We proposed biosynthetic hypotheses based on the structural features of megapolipeptins and the genetic information within the encoding BGC (Fig. 6), which serves as a starting point for future studies aimed at unraveling novel mechanisms of PKS-NRPS-PUFA biosynthesis. Despite choosing a gene cluster encoding an unusual enzyme combination for novelty, the isolated megapolipeptins 1 and 2 did not exhibit significant activity in the assays tested. Due to the low similarity of megapolipeptins to known compounds, it is difficult to predict their bioactivity. The top NP Atlas hits (ESI Table S14†) include herbicidal rotihibins (Tanimoto similarity score of 0.58) from Streptomyces scabis and siderophores crochelin (0.57) from Azotobacter chroococcum and megapolibactins (0.56) from Paraburkholderia megapolitana, none of which are bolaamphiphiles. Bolaamphiphile bolagladins showed antibacterial activity but display an even lower Tanimoto similarity score of 0.39. Future studies should aim at expanding the scope of tested assays beyond the ones conducted in this study. By doing so, we may uncover hidden aspects and functionalities of PKS-NRPS-PUFA products.
In summary, the low structural similarity of megapolipeptins to known natural products supports our Burkholderiales genomics-driven and synthetic biology-enabled pipeline for uncovering novel natural products from silent BGCs.
Experimental
General cultivation conditions
E. coli was routinely cultured in Lysogeny-Broth (LB) at 37 °C unless otherwise stated. Burkholderia sp. FERM BP-3421 and Burkholderiales strains isolated in this study were routinely cultured in LB at 30 °C unless otherwise stated. For plasmid selection, kanamycin at either 50 mg L−1 (E. coli) or 500 mg L−1 (Burkholderia) was used. For megapolipeptin analysis, a 20 μL aliquot of Burkholderia sp. FERM BP-3421 cryo stock containing either pBS001 or pBS003 was inoculated into 5 mL seed medium (10 g L−1 polypeptone, 5 g L−1 yeast extract, 5 g L−1 sodium chloride) and incubated for 48 h in an orbital-shaker at 220 rpm and 30 °C without antibiotics. An aliquot of the seed culture (1 mL) was transferred into 50 mL of 2S4G production medium (40 g L−1 glycerol, 20 g L−1 soytone, 2 g L−1 ammonium sulfate, 0.1 g L−1 magnesium sulfate heptahydrate, 2 g L−1 calcium carbonate) contained in 250 mL Erlenmeyer flasks and cultured at 25 °C, 220 rpm for five days. To induce gene expression, L-arabinose at 100 mM was used.Burkholderiales strain isolation
Rhizosphere microbial samples were collected by removing soil around the rootstock of each selected plant using a sterile scoopula and cutting a small (∼3 cm) section of root material with attached soil using sterile scissors, which was placed in a 15 mL centrifuge tube. Sterile 1× phosphate-buffered saline (PBS) solution was added, the tube was vortexed for 30 s, and then allowed to settle for 30 min. Next, 100 μl aliquots of the supernatant were spread onto agar plates containing six different selection media (PCAT, BIB, BIC, BID, BIE, BIF)15 using plastic spreaders and incubated for 5 to 7 days at 30 °C. Colonies of interest were selected using a sterile plastic loop and grown in LB liquid medium (10 mL) with shaking at 200 rpm overnight. For long-term storage, 500 μL of each culture was added to a sterile solution of 1:1 glycerol/water in cryo-microcentrifuge tubes and stored at −80 °C.
IDBac analyses and strain prioritization
See ESI† for sample preparation and analysis (ESI Table S15†). An example of strain prioritization is shown in ESI Fig. S1.†Genome sequencing and assembly
Genomic DNA was isolated as described under ESI† and submitted for short-read Illumina sequencing at the SeqCenter (Pittsburgh, PA). Obtained reads were quality controlled and adapter-trimmed using bcl2fastq (v. 2.20.0.422). Eight genomes were also sequenced with Oxford Nanopore for which reads quality control and adapter trimming was performed with porechop v0.2.3_seqan2.1.1. Genomes were assembled de novo using Unicycler (v 0.4.8)71 and statically recorded with QUAST (v 5.0.2).72 Annotation was carried out using a standard Prokka73 (v1.14.5) workflow. The annotation results were further evaluated by Rapid Annotations Subsystems Technology (RAST) server annotation. The assembly and annotation files were deposited in NCBI's GenBank. See ESI Tables S1–S3† for genome statistics, accession codes, and collection geolocation for all strains.Phylogenomic tree construction
The phylogenomic tree presented in Fig. 3A, S4 and S5† was created by using the ‘Insert Genome into SpeciesTree – v2.2.0’ app available at the KBase server74 and using a set of 49 core, universal genes (ESI Table S6†) defined by Cluster of Orthologous Genes (COG) families. In addition to genomes provided by the user, closely related, publicly available genomes were automatically included in the tree. The multiple sequence alignments (MSAs) were trimmed using GBLOCKS to remove poorly aligned sections, and the MSAs concatenated before the tree was constructed. SpeciesTree applied a heuristic variant of neighbor joining followed by a mix of nearest-neighbor interchanges and subtree-prune-regraft moves. For nucleotides sequences, SpeciesTree uses the Jukes-Cantor distance −0.75 × log(1–4/3d), where “d” is the proportion of positions that differ.Gene cluster analyses
All genomes were analyzed with antiSMASH v 6.0 (ref. 21) to predict BGCs. BiG-SCAPE23 was then used to cluster the identified BGCs into GCFs using .gbk files. The BiG-SCAPE analysis was supplemented with Pfam database version 32.0. The Singleton parameter in BiG-SCAPE was selected to ensure that BGCs with distances lower than the default cutoff distance 0.4 were included in the output data. The MIBiG flag (--mibig) in BiG-SCAPE was set to include the repository version MIBiG v 1.4 of annotated BGCs. The hybrids-off flag was selected to prevent hybrid BGC redundancy. The generated network files were recorded for raw distance cutoffs of 0.1–1.0 in increments of 0.1. Results of BiG-SCAPE were processed for visualization using Cytoscape version 3.9.1.75Cloning of the mgp BGC to yield pBS001
Cloning of the mgp BGC from Paraburkholderia megapolitana RL18-039-BIC-B (genome #76, Fig. 3D and ESI Table S7†) was performed by Terra Bioforge (Wisconsin, USA) using a CRISPR-Cas9 strategy,49 followed by isothermal DNA assembly with a linearized vector to yield pBS001 as described under ESI.† To be used as a negative control, the empty vector pBS003 was generated as described under ESI.†Heterologous expression of the mgp BGC into Burkholderia sp FERM BP-3421
Plasmids pBS001 and pBS003 were each transferred into Burkholderia sp. FERM BP-3421 via conjugation from E. coli S17-1/pACYC184_MBurI_MBurII as previously described48 and as detailed in ESI.† Obtained clones were confirmed by PCR as shown for pBS001 (ESI Fig. S11†).Comparative metabolite analysis, isolation, and structure elucidation of megapolipeptins
Burkholderia sp. containing either pBS001 or pBS003 were analyzed by LC-MS/MS as described under ESI (Fig. 4B and S12–16†). The structures of 1 and 2 were elucidated using HRMS, chemical derivatization, and 1D (1H and 13C) and 2D NMR (COSY, TOCSY, ROESY, phase sensitive HSQC, HMBC) experiments recorded in DMSO-d6 as described under ESI (ESI Tables S8 and S9 and Fig. S17–S42†).Data availability
The genome data that support the findings of this study are available in NCBI GenBank database under accession codes listed in ESI Table S1.† The NMR data for megapolipeptins A and B have been deposited in the Natural Products Magnetic Resonance Database (NP-MRD; https://www.np-mrd.org/) under accession numbers NP0332797 and NP0332798. Mass spectrometry data for MS/MS analysis of pure compounds has been deposited at the Global Natural Products Social molecular networking repository (https://gnps.ucsd.edu/) under accession numbers MSV000094914.Author contributions
ASE and RGL devised the project. CF isolated Burkholderiales strains from environmental samples, SBR, AH and NK analyzed strains using IDBac, BSP isolated genomic DNA and performed genome analyses. BSP performed heterologous expression and identification of candidate features. MR and SL isolated and structurally characterized megapolipeptins. DL, HC, and AB performed activity assays. CG mentored AB, BTM mentored AH and NK, RGL mentored MR, SL, CF, DL and HC, ASE mentored BSP and SBR. RGL and ASE obtained research funding. BSP, MR, RL and ASE wrote the paper draft. All authors commented on and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the International Patent Organism Depository of the National Institute of Technology and Evaluation (Japan) for strain FERM BP-3421, and David Mead and Robb Stankey (Terra Bioforge) for pBS001 construction. Financial support for this work was provided by the National Institute of General Medical Sciences (GM129344 to A. S. E. and R. G. L.), by the Office of the Director and the National Center for Complementary & Integrative Health (T32 AT007533 to S. B. R.), National Institutes of Health (NIH), and by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery program (R. G. L. and C. G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or NSERC.References
- J. A. Van Santen, et al., The Natural Products Atlas 2.0: A database of microbially-derived natural products, Nucleic Acids Res., 2022, 50, D1317–D1323 CrossRef CAS PubMed.
- A. Gavriilidou, et al., Compendium of specialized metabolite biosynthetic diversity encoded in bacterial genomes, Nat. Microbiol., 2022, 7, 726–735 CrossRef CAS PubMed.
- M. G. Chevrette and J. Handelsman, Needles in haystacks: Reevaluating old paradigms for the discovery of bacterial secondary metabolites, Nat. Prod. Rep., 2021, 38, 2083–2099 RSC.
- A. Oren and G. M. Garrity, Valid publication of the names of forty-two phyla of prokaryotes, Int. J. Syst. Evol. Microbiol., 2021, 71, 005056 Search PubMed.
- S. Kunakom and A. S. Eustáquio, Burkholderia as a source of natural products, J. Nat. Prod., 2019, 82, 2018–2037 CrossRef CAS PubMed.
- K. Scherlach and C. Hertweck, Mining and unearthing hidden biosynthetic potential, Nat. Commun., 2021, 12, 3864 CrossRef CAS PubMed.
- B. I. Adaikpoh, H. N. Fernandez and A. S. Eustáquio, Biotechnology approaches for natural product discovery, engineering, and production based on Burkholderia bacteria, Curr. Opin. Biotechnol., 2022, 77, 102782 CrossRef CAS PubMed.
- R. S. Ayikpoe, et al., A scalable platform to discover antimicrobials of ribosomal origin, Nat. Commun., 2022, 13, 6135 CrossRef CAS PubMed.
- N. Gummerlich, Y. Rebets, C. Paulus, J. Zapp and A. Luzhetskyy, Targeted genome mining—from compound discovery to biosynthetic pathway elucidation, Microorganisms, 2020, 8, 1–17 CrossRef PubMed.
- V. Libis, et al., Multiplexed mobilization and expression of biosynthetic gene clusters, Nat. Commun., 2022, 13, 5256 CrossRef CAS PubMed.
- B. Enghiad, et al., Cas12a-assisted precise targeted cloning using in vivo Cre-lox recombination, Nat. Commun., 2021, 12, 1171 CrossRef CAS PubMed.
- X. Xu, Y. Liu, G. Du, R. Ledesma-Amaro and L. Liu, Microbial chassis development for natural product biosynthesis, Trends Biotechnol., 2020, 38, 779–796 CrossRef CAS PubMed.
- G. Wang, et al., CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria, Nat. Microbiol., 2019, 4, 2498–2510 CrossRef PubMed.
- H. N. Fernandez, et al., High-yield lasso peptide production in a Burkholderia bacterial host by plasmid copy number engineering, ACS Synth. Biol., 2024, 13, 337–350 CrossRef CAS PubMed.
- C. H. Fergusson, J. M. F. Coloma, M. C. Valentine, F. P. J. Haeckl and R. G. Linington, Custom matrix-assisted laser desorption ionization-time of flight mass spectrometric database for identification of environmental isolates of the genus Burkholderia and related genera, Appl. Environ. Microbiol., 2020, 86, e354–e420 Search PubMed.
- F. P. J. Haeckl, et al., A selective genome-guided method for environmental Burkholderia isolation, J. Ind. Microbiol. Biotechnol., 2019, 46, 345–362 CrossRef CAS PubMed.
- C. M. Clark, M. S. Costa, L. M. Sanchez and B. T. Murphy, Coupling MALDI-TOF mass spectrometry protein and specialized metabolite analyses to rapidly discriminate bacterial function, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4981–4986 CrossRef CAS PubMed.
- S. Kunakom and A. S. Eustáquio, Heterologous production of lasso peptide capistruin in a Burkholderia Host, ACS Synth. Biol., 2020, 9, 241–248 CrossRef CAS PubMed.
- B. I. Adaikpoh, S. B. Romanowski and A. S. Eustáquio, Understanding autologous spliceostatin transcriptional regulation to derive parts for heterologous expression in a Burkholderia bacterial host, ACS Synth. Biol., 2023, 12, 1952–1960 CrossRef CAS PubMed.
- C. R. Pye, M. J. Bertin, R. S. Lokey, W. H. Gerwick and R. G. Linington, Retrospective analysis of natural products provides insights for future discovery trends, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5601–5606 CrossRef CAS PubMed.
- K. Blin, et al., AntiSMASH 6.0: Improving cluster detection and comparison capabilities, Nucleic Acids Res., 2021, 49, W29–W35 CrossRef CAS PubMed.
- A. J. Mullins and E. Mahenthiralingam, The hidden genomic diversity, specialized metabolite capacity, and revised taxonomy of Burkholderia sensu lato, Front. Microbiol., 2021, 12, 726847 CrossRef PubMed.
- J. C. Navarro-Muñoz, et al., A computational framework to explore large-scale biosynthetic diversity, Nat. Chem. Biol., 2020, 16, 60–68 CrossRef PubMed.
- B. R. Terlouw, et al., MIBiG 3.0: A community-driven effort to annotate experimentally validated biosynthetic gene clusters, Nucleic Acids Res., 2023, 51, D603–D610 CrossRef CAS PubMed.
- X. Q. Wang, et al., Occidiofungin is an important component responsible for the antifungal activity of Burkholderia pyrrocinia strain Lyc2, J. Appl. Microbiol., 2016, 120, 607–618 CrossRef CAS PubMed.
- L. V. Flórez, et al., An antifungal polyketide associated with horizontally acquired genes supports symbiont-mediated defense in Lagria villosa beetles, Nat. Commun., 2018, 9, 2478 CrossRef PubMed.
- C. H. Fergusson, et al., Discovery of a lagriamide polyketide by integrated genome mining, isotopic labeling, and untargeted metabolomics, Chem. Sci., 2024, 15, 8089–8096 RSC.
- R. Hermenau, et al., Gramibactin is a bacterial siderophore with a diazeniumdiolate ligand system, Nat. Chem. Biol., 2018, 14, 841–843 CrossRef CAS PubMed.
- R. Li, R. A. Oliver and C. A. Townsend, Identification and characterization of the sulfazecin monobactam biosynthetic gene cluster, Cell Chem. Biol., 2017, 24, 24–34 CrossRef CAS PubMed.
- B. Schellenberg, L. Bigler and R. Dudler, Identification of genes involved in the biosynthesis of the cytotoxic compound glidobactin from a soil bacterium, Environ. Microbiol., 2007, 9, 1640–1650 CrossRef CAS PubMed.
- K. Agnoli, C. A. Lowe, K. L. Farmer, S. I. Husnain and M. S. Thomas, The ornibactin biosynthesis and transport genes of Burkholderia cenocepacia are regulated by an extracytoplasmic function σ factor which is a part of the fur regulon, J. Bacteriol., 2006, 188, 3631–3644 CrossRef CAS PubMed.
- A. A. Golicz, P. E. Bayer, P. L. Bhalla, J. Batley and D. Edwards, Pangenomics comes of age: From bacteria to plant and animal applications, Trends Genet., 2020, 36, 132–145 CrossRef CAS PubMed.
- P. Cimermancic, et al., Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters, Cell, 2014, 158, 412–421 CrossRef CAS PubMed.
- T. A. Schöner, et al., Aryl polyenes, a highly abundant class of bacterial natural products, are functionally related to antioxidative carotenoids, ChemBioChem, 2016, 17, 247–253 CrossRef PubMed.
- A. Mukherjee, et al., Global analyses of biosynthetic gene clusters in phytobiomes reveal strong phylogenetic conservation of terpenes and aryl polyenes, mSystems, 2023, 8, e387–e423 Search PubMed.
- M. Givskov, et al., Two separate regulatory systems participate in control of swarming motility of Serratia liquefaciens MG1, J. Bacteriol., 1998, 180, 742–745 CrossRef CAS PubMed.
- K. Riedel, et al., N-acyl-l-homoserine lactone-mediated regulation of the lip secretion system in Serratia liquefaciens MG1, J. Bacteriol., 2001, 183, 1805–1809 CrossRef CAS PubMed.
- T. Burr, et al., Identification of the central quorum sensing regulator of virulence in the enteric phytopathogen, Erwinia carotovora: The VirR repressor, Mol. Microbiol., 2006, 59, 113–125 CrossRef CAS PubMed.
- L. Xu, et al., Role of the luxS quorum-sensing system in biofilm formation and virulence of Staphylococcus epidermidis, Infect. Immun., 2006, 74, 488–496 CrossRef CAS PubMed.
- K. S. Ju, J. R. Doroghazi and W. W. Metcalf, Genomics-enabled discovery of phosphonate natural products and their biosynthetic pathways, J. Ind. Microbiol. Biotechnol., 2014, 41, 345–356 CrossRef CAS PubMed.
- X. Yu, et al., Diversity and abundance of phosphonate biosynthetic genes in nature, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20759–20764 CrossRef CAS PubMed.
- A. W. Tolcher, et al., A phase I study of rhizoxin (NSC 332598) by 72-hour continuous intravenous infusion in patients with advanced solid tumors, Ann. Oncol., 2000, 11, 333–338 CrossRef CAS PubMed.
- N. El Omari, et al., Molecular mechanistic pathways underlying the anticancer therapeutic efficiency of romidepsin, Biomed. Pharmacother., 2023, 164, 114774 CrossRef CAS PubMed.
- S. Puthenveetil, et al., Natural product splicing inhibitors: A new class of antibody-drug conjugate (ADC) payloads, Bioconjugate Chem., 2016, 27, 1880–1888 CrossRef CAS PubMed.
- S. L. Wenski, et al., Fabclavine diversity in Xenorhabdus bacteria, Beilstein J. Org. Chem., 2020, 16, 956–965 CrossRef CAS PubMed.
- J. Masschelein, et al., A combination of polyunsaturated fatty acid, nonribosomal peptide and polyketide biosynthetic machinery is used to assemble the zeamine antibiotics, Chem. Sci., 2015, 6, 923–929 RSC.
- W. Zheng, et al., Establishment of recombineering genome editing system in Paraburkholderia megapolitana empowers activation of silent biosynthetic gene clusters, Microb. Biotechnol., 2020, 13, 397–405 CrossRef CAS PubMed.
- S. B. Romanowski, et al., Identification of the lipodepsipeptide selethramide encoded in a giant nonribosomal peptide synthetase from a Burkholderia bacterium, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2304668120 CrossRef CAS PubMed.
- J. W. Wang, et al., CRISPR/Cas9 nuclease cleavage combined with Gibson assembly for seamless cloning, Biotechniques, 2015, 58, 161–170 CrossRef CAS PubMed.
- P. Marfey, Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- J. Bus, I. Sies, L. K. Jie and S. F. Marcel, 13C-NMR of methyl, methylene and carbonyl carbon atoms of methyl alkenoates and alkynoates, Chem. Phys. Lipids, 1976, 17, 501–518 CrossRef CAS.
- W. R. Wong, A. G. Oliver and R. G. Linington, Development of antibiotic activity profile screening for the classification and discovery of natural product antibiotics, Chem. Biol., 2012, 19, 1483–1495 CrossRef CAS PubMed.
- I. M. Moi, et al., Polyunsaturated fatty acids in marine bacteria and strategies to enhance their production, Appl. Microbiol. Biotechnol., 2018, 102, 5811–5826 CrossRef CAS PubMed.
- C. N. Shulse and E. E. Allen, Widespread occurrence of secondary lipid biosynthesis potential in microbial lineages, PLoS One, 2011, 6, e20146 CrossRef CAS PubMed.
- M. Müller, G. A. Sprenger and M. Pohl, C-C bond formation using ThDP-dependent lyases, Curr. Opin. Chem. Biol., 2013, 17, 261–270 CrossRef.
- J. Guo, L. Hong, X. Z. West, H. Wang and R. G. Salomon, Bioactive 4-oxoheptanedioic monoamide derivatives of proteins and ethanolaminephospholipids: Products of docosahexaenoate oxidation, Chem. Res. Toxicol., 2016, 29, 1706–1719 Search PubMed.
- Z. D. Aron, P. C. Dorrestein, J. R. Blackball, N. L. Kelleher and C. T. Walsh, Characterization of a new tailoring domain in polyketide biogenesis: The amine transferase domain of MycA in the mycosubtilin gene cluster, J. Am. Chem. Soc., 2005, 127, 14986–14987 CrossRef CAS.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains, Chem. Biol., 2000, 7, 211–224 CrossRef CAS.
- D. A. Alonzo, C. Chiche-Lapierre, M. J. Tarry, J. Wang and T. M. Schmeing, Structural basis of keto acid utilization in nonribosomal depsipeptide synthesis, Nat. Chem. Biol., 2020, 16, 493–496 CrossRef CAS.
- B. Silakowski, et al., New lessons for combinatorial biosynthesis from myxobacteria, J. Biol. Chem., 1999, 274, 37391–37399 CrossRef CAS PubMed.
- S. Weinig, H.-J. Hecht, T. Mahmud and R. Müller, Melithiazol biosynthesis: Further insights into myxobacterial PKS/NRPS systems and evidence for a new subclass of methyl transferases, Chem. Biol., 2003, 10, 939–952 CrossRef CAS.
- N. Funa, M. Funabashi, E. Yoshimura and S. Horinouchi, A novel quinone-forming monooxygenase family involved in modification of aromatic polyketides, J. Biol. Chem., 2005, 280, 14514–14523 CrossRef CAS PubMed.
- L. M. Van Staalduinen, S. K. Novakowski and Z. Jia, Structure and functional analysis of YcfD, a novel 2-oxoglutarate/Fe2+-dependent oxygenase involved in translational regulation in Escherichia coli, J. Mol. Biol., 2014, 426, 1898–1910 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz, V. Bergendah and M. A. Marahiel, Peptide bond formation in nonribosomal peptide biosynthesis: Catalytic role of the condensation domain, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS PubMed.
- S. Dekimpe and J. Masschelein, Beyond peptide bond formation: The versatile role of condensation domains in natural product biosynthesis, Nat. Prod. Rep., 2021, 38, 1910–1937 RSC.
- F. Pourmasoumi, et al., Proof-reading thioesterase boosts activity of engineered nonribosomal peptide synthetase, ACS Chem. Biol., 2022, 17, 2382–2388 CrossRef CAS PubMed.
- R. Hermenau, et al., Genomics-driven discovery of NO-donating diazeniumdiolate siderophores in diverse plant-associated bacteria, Angew. Chem., 2019, 131, 13158–13163 CrossRef.
- B. Dose, et al., Food-poisoning bacteria employ a citrate synthase and a type II NRPS to synthesize bolaamphiphilic lipopeptide antibiotics, Angew. Chem., 2020, 59, 21535–21540 CrossRef CAS PubMed.
- Y. Dashti, et al., Discovery and biosynthesis of bolagladins: Unusual lipodepsipeptides from Burkholderia gladioli clinical isolates, Angew. Chem., 2020, 59, 21553–21561 CrossRef CAS PubMed.
- R. R. Wick, L. M. Judd, C. L. Gorrie and K. E. U. Holt, Resolving bacterial genome assemblies from short and long sequencing reads, PLoS Comput. Biol., 2017, 13, e1005595 CrossRef PubMed.
- A. Gurevich, V. Saveliev, N. Vyahhi and G. Tesler, QUAST: Quality assessment tool for genome assemblies, Bioinformatics, 2013, 29, 1072–1075 CrossRef CAS PubMed.
- T. Seemann, Prokka: Rapid prokaryotic genome annotation, Bioinformatics, 2014, 30, 2068–2069 CrossRef CAS PubMed.
- A. P. Arkin, et al., KBase: The United States department of energy systems biology knowledgebase, Nat. Biotechnol., 2018, 36, 566–569 CrossRef CAS PubMed.
- P. Shannon, et al., Cytoscape: A software environment for integrated models of biomolecular interaction networks, Gen. Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03594a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |