
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free alkyne annulation enabling π-extension of boron-doped polycyclic aromatic hydrocarbons†
Mandala
Anitha
a,
To-Jen
Chin
a,
Guan-Cheng
Liu
a,
Chi-Tien
Hsieh
b,
Kuan-Hua
Wang
b,
Shu-Li
Li
a,
Mu-Jeng
Cheng
 b and
Jeffrey M.
Farrell
*ac
b and
Jeffrey M.
Farrell
*ac
aDepartment of Chemistry, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd., Taipei 10617, Taiwan. E-mail: farrell@ntu.edu.tw
bDepartment of Chemistry, National Cheng Kung University, Tainan 701, Taiwan
cCenter for Emerging Materials and Advanced Devices, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd., Taipei 10617, Taiwan
First published on 16th September 2024
Abstract
A C–H functionalizing annulation reaction of boron-doped polycyclic aromatic hydrocarbons (PAHs) with alkynes is described. This metal-free π-extension provides a new synthetic route to fusion atom B-doped polycyclic aromatic hydrocarbons (PAHs) that is demonstrated with the synthesis of a family of new, functionalized, structurally constrained 6a,15a-diborabenzo[tuv]naphtho[2,1-b]picenes. These annulation products exhibit deep LUMO energy levels, strong visible-range absorptions, and sterically accessible π-systems that can adopt herringbone or π-stacked solid-state structures based on choice of substituents. From regioselectivity and DFT calculations, we propose an annulation mechanism involving intramolecular electrophilic aromatic substitution of a zwitterionic intermediate.
Introduction
Molecular polycyclic aromatic hydrocarbon (PAH) substructures of graphene exhibit remarkable opto(electronic) properties and supramolecular behaviors that have led to their pivotal roles in modern functional organic materials.1 The replacement of PAH carbon atoms with one or more heteroatoms, or “heteroatom-doping,” has become an indispensable tool to modify optical and electronic properties while retaining PAH geometries.2 Substitution of PAH carbon atoms with neutral, three-coordinate boron renders them electron-deficient. Accordingly, B-doped PAHs are positioned to satisfy growing demands for electron-poor π-systems in applications including n-type semiconductor materials.3 Encouragingly, B-doped PAHs have been demonstrated in organic transistors,4 as non-fullerene acceptors in organic solar cells,5 as thermally activated delayed fluorescence emitters in organic light-emitting diodes,6 and for small molecule activations.7Synthetic challenges remain for B-doped PAHs. For example, “structurally constrained”8 B-doped PAHs with boron substitutions at fusion positions (shared by two or more of the fused rings) are rare, despite attractive characteristics (Fig. 1).9 These fusion B-doped PAHs are kinetically stabilized by structural constraint, so sterically bulky substituents are not needed to protect boron from nucleophilic attack.8 Consequently, ambient stability can be achieved while retaining unobstructed π-surfaces that could form π-stacked structures suited for charge-carrier transport. In addition, few existing syntheses produce B-doped PAHs bearing desirable “deep” LUMO energy levels, defined by Ingleson, Zysman-Colman, and co-workers as less than −3.0 eV.10 This property is highly dependent on π-core shape, size, and boron substitution pattern.5,11 Moreover, common co-dopants (e.g. N) can contribute electron pairs to the π-conjugated system, offsetting LUMO-lowering contributions of boron's empty p orbital.12 Finally, functionalized periphery substituents are often difficult to install due to chemical sensitivities typical of organoboranes and their syntheses.
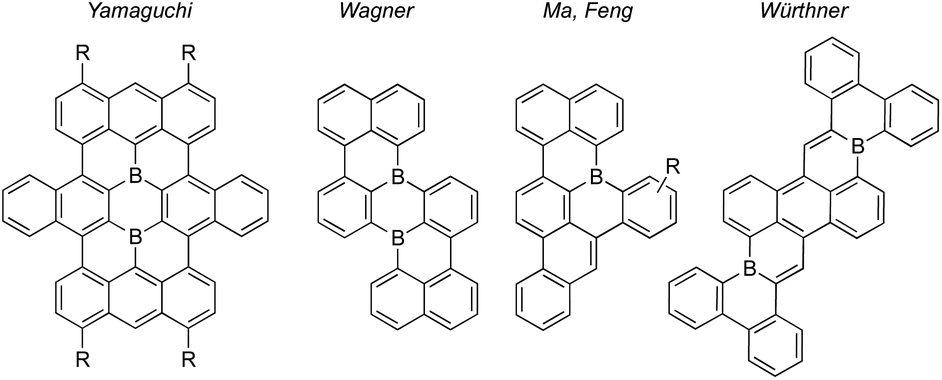 | ||
| Fig. 1 Examples of structurally constrained polybenzenoid fusion atom B-doped PAHs.9a,9d–f | ||
Strategies towards fusion B-doped PAHs include oxidative cyclodehydrogenation9a,11,13 or Ni-mediated ring–closing reactions9b,9d of properly substituted aryl boranes. Additionally, electrophilic borylation9c (sometimes followed by rearrangement)9e has proven useful in recent syntheses.9f,9h Nevertheless, the field lacks the extensive synthetic tools available for other classes of PAHs. Elsewhere, intermolecular annulative π-extension (or “APEX”)14 has become a valuable strategy for π-scaffold construction (Fig. 2), where the modular annulation of π-fragments allows elaboration of many products from a single polyaromatic precursor. Analogous π-extensions of B-doped π-scaffolds could alleviate the shortage of fusion B-doped PAHs, as annulations with different partners would sharply increase available structures. This could also allow B-doped PAHs bearing unprecedented shapes, sizes, substitution patterns, and functional groups to be realized, opening new avenues for the discovery and use of B-doped π-conjugated structures.
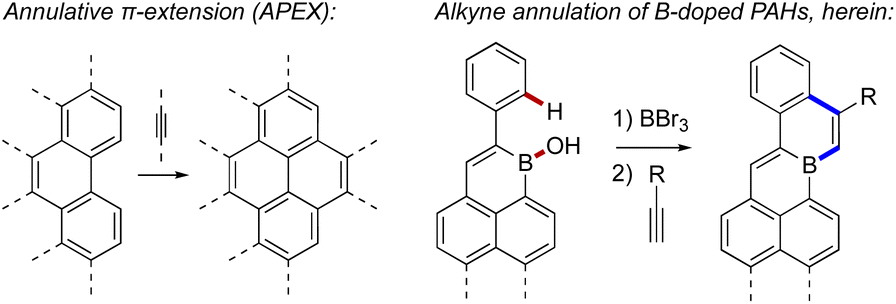 | ||
| Fig. 2 A schematic example of annulative π-extension14 (left) compared to the alkyne annulation of B-doped PAHs described herein (right). | ||
Herein we report an unanticipated metal-free, regioselective annulation reaction of a boron-doped PAH with a terminal alkyne. DFT calculations suggest this reactivity proceeds via intramolecular electrophilic aromatic substitution of a zwitterionic intermediate. The reaction was applied to the synthesis of a family of functionalized, fusion atom B-doped PAHs. Annulated products exhibited red-shifted absorption and emission spectra as well as substantially more positive reduction potentials compared to their precursors. DFT calculations corroborated low LUMO energy levels of the annulated products, and implied antiaromatic character of their boron-containing six-membered rings. Fusion B-doped PAHs synthesized herein are amongst the lowest-LUMO B-doped PAHs yet reported and exhibit solid-state packing arrangements that can be tailored by choice of alkyne annulation partner.
Results and discussion
We began our study by probing the reactivity of polycyclic borinic acid 1-hydroxy-2-phenyl-1-boraphenalene, 1.5 Excess BBr3 was reacted with 1 in CH2Cl2 at room temperature for five hours to effect –OH for –Br exchange. After removal of volatiles in vacuo, the residue was dissolved in CD2Cl2 and reacted with p-tolylacetylene at 70 °C in a sealed J Young NMR tube (Scheme 1). As the reactants were gradually consumed over 8 days, the formation of two major products could be observed by 1H NMR spectroscopy (Fig. S1 and S2†). One product was identified as the Markovnikov addition product of HBr to p-tolylacetylene, 1-(1-bromoethenyl)-4-methylbenzene. To our surprise, the other major reaction product could be identified as 3a, an annulation product with a bond formed between B and the alkyne terminal C, as well as a new C–C bond from an apparent C–H functionalization of the boraphenalene phenyl substituent (Scheme 1). Compound 3a crystalized from CD2Cl2 solution and its planarized structure could be confirmed by single-crystal X-ray crystallography. Further 1H NMR spectroscopy studies were conducted to optimize this reactivity (Table S1†). Rate of 3a formation could be increased by undertaking the reaction at 120 °C in C6D5Br. Noting that the overall transformation requires two equivalents of alkyne per annulation, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of alkyne:borane also improved conversion to 3a. Under these improved conditions, 3a could be isolated in 33% yield after recrystallization from 1:1 CH2Cl2:hexane. When BCl3 was employed in the –OH for halogen exchange step, the annulation was comparatively sluggish. As well, no formation of 3a was observed in the direct reaction between borinic acid 1 and p-tolylacetylene under the same conditions. We speculate that Lewis acid strength of the boron center is critical to the reaction. At 120 °C in C6D5Br for 180 h, diphenylacetylene and 1-phenyl-1-propyne each fail to react with 1 (after –OH for –Br exchange), indicating possible steric limitations of annulation reactivity. In attempts to sequester generated HBr with exogenous bases, reactions were undertaken using one equivalent each of alkyne and base (2,6-di-tert-butyl-4-methylpyridine, 2,6-dichloropyridine, 2,6-lutidine, or 2,2,6,6-tetramethylpiperidine) under otherwise optimized conditions (Table S1†). While inhibited formation of 1-(1-bromoethenyl)-4-methylbenzene was observed by 1H NMR spectroscopy for the latter two stronger bases, none of the basic additives improved conversions to 3a.
:1 stoichiometry of alkyne:borane also improved conversion to 3a. Under these improved conditions, 3a could be isolated in 33% yield after recrystallization from 1:1 CH2Cl2:hexane. When BCl3 was employed in the –OH for halogen exchange step, the annulation was comparatively sluggish. As well, no formation of 3a was observed in the direct reaction between borinic acid 1 and p-tolylacetylene under the same conditions. We speculate that Lewis acid strength of the boron center is critical to the reaction. At 120 °C in C6D5Br for 180 h, diphenylacetylene and 1-phenyl-1-propyne each fail to react with 1 (after –OH for –Br exchange), indicating possible steric limitations of annulation reactivity. In attempts to sequester generated HBr with exogenous bases, reactions were undertaken using one equivalent each of alkyne and base (2,6-di-tert-butyl-4-methylpyridine, 2,6-dichloropyridine, 2,6-lutidine, or 2,2,6,6-tetramethylpiperidine) under otherwise optimized conditions (Table S1†). While inhibited formation of 1-(1-bromoethenyl)-4-methylbenzene was observed by 1H NMR spectroscopy for the latter two stronger bases, none of the basic additives improved conversions to 3a.
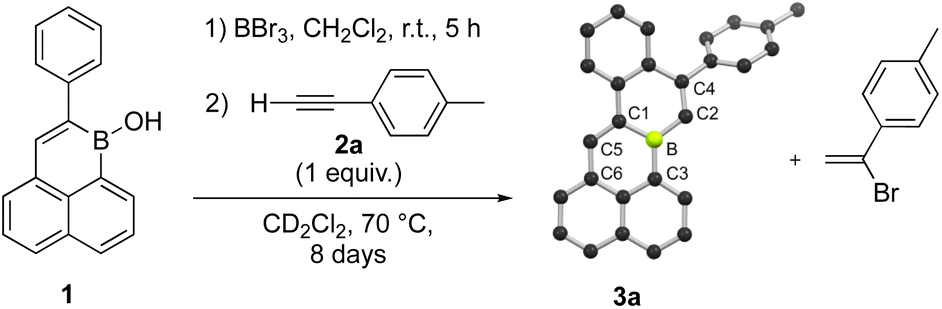 | ||
| Scheme 1 Formation of 3a by bromination of 1 followed by alkyne annulation. Compound 3a is illustrated by its solid-state structure. C: black, B: yellow-green, H atoms omitted for clarity. | ||
This annulation is markedly different to the reactions of alkynes with 5-membered boracyclic 9-halo-9-borofluorenes, for which alkyne insertion via 1,2-carboboration has been shown by Fukushima and co-workers.15 Other possible outcomes, such as 1,1-carboboration16 or haloboration,17 are apparently not favored under the given reaction conditions. The regioselectivity of the annulation of 1 after –OH for –Br exchange might suggest the involvement of a vinyl cation generated by attack of the boron center by the alkyne triple bond. Prerequisite resonance stabilization of such an intermediate18 (enroute to electrophilic aromatic substitution) would explain the observed positioning of the tolyl group in the major annulation product. Moreover, related intermediates, generated by the combination of alkynes and boranes, are implicated in the B(C6F5)3-mediated cascade syntheses of dibenzopentalenes of Yamaguchi, Erker, and co-workers,19 and in boron trihalide-mediated borylative cyclizations of Ingleson and co-workers.20 Indeed, Ingleson and co-workers have utilized the latter strategy to construct edge B-doped PAHs using combinations of well-designed alkynes and boron trihalides.21 However, unlike these examples which react alkynes with strong external Lewis acids, the present reaction between a brominated edge-B-doped PAH and an external alkyne must presumably rely on a much weaker Lewis acidic center for generation of a vinyl cation intermediate. The feasibility of this mechanistic proposal was not obvious to us, so we examined it using DFT calculations at the B3LYP-D3(BJ)/def2-TZVP//B3LYP-D3(BJ)/def2-SVP level (see ESI† for details). Indeed, an enthalpic local minimum was found for a vinyl cation-containing intermediate, generated from attack on boron by the alkyne triple bond (Fig. 3, 3a-2). The charge assignment on 3a-2 is supported by our Natural Population Analysis calculation; specifically, from reactants to 3a-2, the charge on the vinyl cation (C highlighted in red) increased by 0.38 e−, while the charge on boron decreased by 0.19 e−. The calculated pathway to form 3avia the ring closing from intermediate 3a-2 involves an intramolecular electrophilic aromatic substitution reaction, and its highest enthalpic barrier (7.0 kcal mol−1) is calculated for the C–C bond forming step of the annulation (Fig. 3, 3a-2 → 3a-3). In contrast, the enthalpy profile of the formation of an alternative regioisomer of 3a (wherein the positions of the alkyne substituents are reversed) possessed a higher enthalpic barrier (23.6 kcal mol−1) for product formation, and no enthalpic local minimum for a vinyl cation-containing intermediate (Fig. S71†).
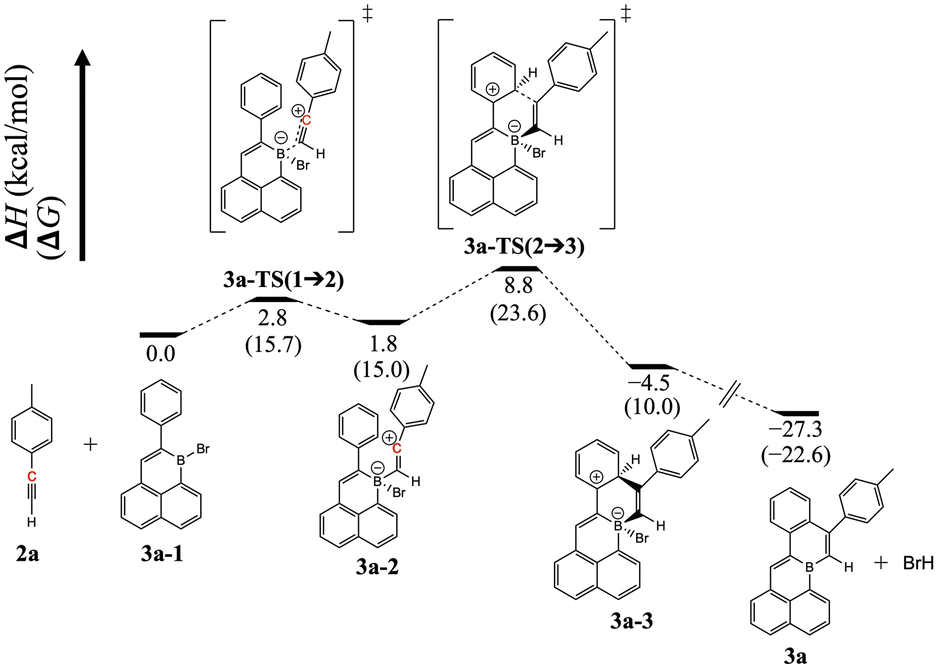 | ||
| Fig. 3 DFT-calculated enthalpy and Gibbs free energy profile of the formation of 3a. The structures were optimized in CPCM solvation model in CH2Cl2. | ||
We sought to apply this unusual intermolecular annulation to the synthesis of extended B-doped PAHs. As a target, we identified 6a,15a-diborabenzo[tuv]naphtho[2,1-b]picene as a B-doped core structure for which a potential precursor borinic acid, 4, has already been reported.5 Furthermore, DFT calculations suggest that 6a,15a-diborabenzo[tuv]naphtho[2,1-b]picenes should possess desirable low-LUMO energy levels and visible range absorptions (Table S4 and Fig. S62†). Thus, we conducted a series of experiments where 4 was reacted with BBr3 in CH2Cl2 at room temperature for 22 h and, after removal of volatiles in vacuo, further reacted with four equivalents of alkyne in C6H5Cl at 120 °C for 60 h. The resulting B-doped PAHs 5a–j were isolated in 3–24% yield as ambient-stable purple-to-black solids after solvent washing steps, filtration through celite in CHCl3 solution, and solvent removal in vacuo (Scheme 2). Aryl-, alkyl-, and alkenyl-substituted alkynes of varying electron-demand could be successfully employed in annulation reactions. Notably, thiophene, alkene, and ester functional groups are tolerated. No annulation products could be isolated from reactions with p-(trifluoromethyl)phenylacetylene or p-methoxyphenylacetylene, the former of which we speculate may be complicated by B-mediated fluoride abstraction.22 Assigned structures of poorly soluble 5a–j were confirmed using 1H NMR spectroscopy at elevated temperatures, solid-state 13C NMR spectroscopy, and high-resolution mass spectrometry. Solid samples of 6a,15a-diborabenzo[tuv]naphtho[2,1-b]picenes stored in ambient conditions for one week showed no indication of decomposition by 1H NMR spectroscopy.
 | ||
| Scheme 2 Annulative syntheses of extended fusion atom B-doped PAHs. | ||
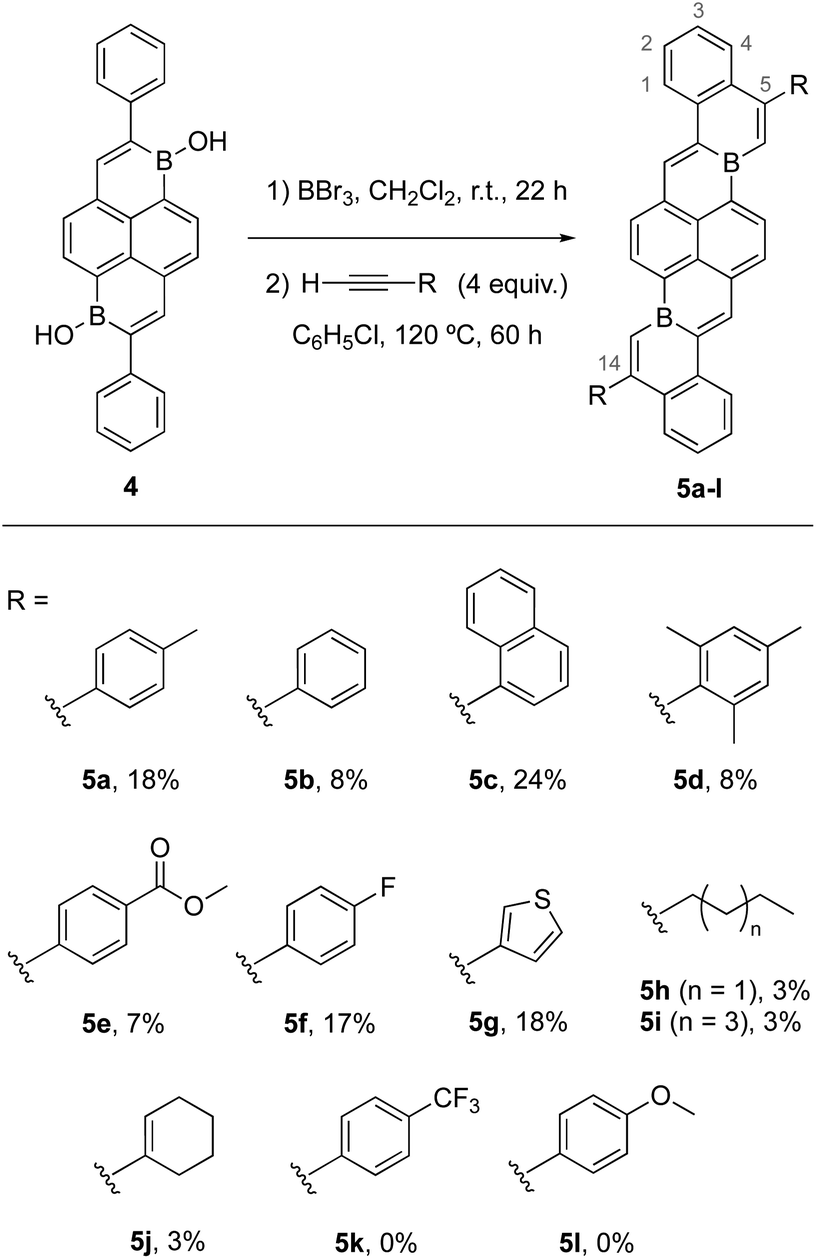
Crystals of 5a, 5c, 5d, 5f, and 5g suitable for X-ray crystallography could be obtained via sublimation (<10−6 torr, 390–420 °C). Solid-state structures confirmed anticipated polyaromatic structures with planar cove-free π-cores. Planarities about B centers are indicated by sums of C–B–C bond angles of 360° for each (e.g.Fig. 4a). The periphery C–B–C bond angles (e.g.Fig. 4a, C2–B–C3) are slightly enlarged from an ideal 120°, measuring 125°–126°. The B–C bonds of 5a, 5c, 5d, 5f, and 5g are 1.52–1.56 Å, slightly shorter than typical-B–C(sp2) single bonds (e.g. BPh3 B–C = 1.57–1.59 Å),23 but in line with B–C single bonds in other fusion atom B-doped systems.9a,9f In addition, C2–C4 and C1–C5 double bonds are slightly elongated compared to typical C–C double bonds, while C5–C6 bonds are slightly short for single bonds. Analogous bonding trends are observed for 3a (Scheme 1). These data suggest effective delocalization over B-doped π-surfaces, consistent with our DFT calculations that indicate highly delocalized frontier molecular orbitals (Fig. 4b, Table S4†). The delocalization of the LUMOs in combination with the structurally constrained environments of boron might temper the Lewis acidities of boron centers, contributing to the ambient stabilities of 5a–j. To understand the aromaticity of these π-systems, representative NICS24 analyses of 3a and 5a were performed (Fig. 4c and S74†). NICS(0) and NICS(1) calculations indicate that six-membered rings containing boron and a vinylic double bond are significantly less aromatic than carbon-only six-membered rings. These data are consistent with previous aromaticity assessments reported for B-doped PAHs.9f,9h
 | ||
| Fig. 4 (a) Solid-state structure of 5a determined by X-ray crystallography. C: black, B: yellow-green, H atoms omitted for clarity. (b) Frontier molecular orbitals of 5a from DFT calculations (isosurfaces = 0.07 Å−3). (c) NICS(1) values calculated for rings of 5a (B3LYP-D3(BJ)/def2-TZVP//B3LYP-D3(BJ)/def2-SVP). | ||
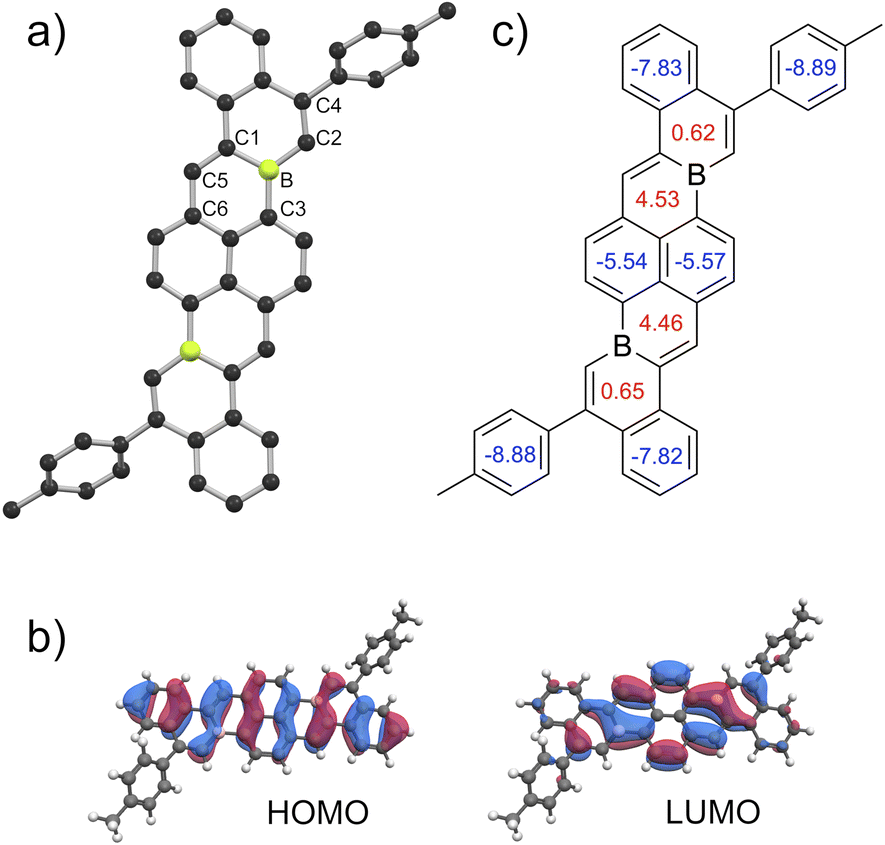
Solid-state packing structures of 5a, 5c, 5d, 5f, and 5g were dramatically influenced by substituent variations in the 5 and 14 position (Fig. 5a). The π-cores of mesityl-substituted 5d arrange into a herringbone structure with several close intermolecular C–H⋯π distances (2.6–3.1 Å) between B-doped π-cores. Conversely, π-cores of 3-thienyl-substituted 5g form continuous columnar π-stacks with significant overlap between π-systems and ∼3.4 Å interplanar distances (Fig. 5b). Interestingly, these stacks are composed of distinct trimeric sub-stacks of molecules of 5g in two different thienyl orientations, wherein the roll angle between molecules within each trimer is slightly reduced compared to the roll angle between trimers (Fig. 5c and S70).25 We note that C–H⋯π interactions between thienyl substituents of 5g are indicated by close proximities of ∼2.8 Å (Fig. 5c, dotted lines). The packings of 5c, 5f, and 5a show a continuum between the herringbone packing of 5d and the π-stacked packing of 5g (Fig. 5a).
 | ||
| Fig. 5 (a) Side-on views of solid-state packing structures of 5d, 5c, 5f, 5a, and 5g. (b) Alternate view of the π-stacked structure of 5g. (c) Diagram illustrating close contacts of thienyl groups of 5g and highlighting a representative trimeric sub-stack (red) of 5g molecules with two different 3-thienyl orientations (labelled with diamonds or squares). C: black, B: yellow-green, F: green S: yellow, H: grey, selected H atoms omitted for clarity. | ||
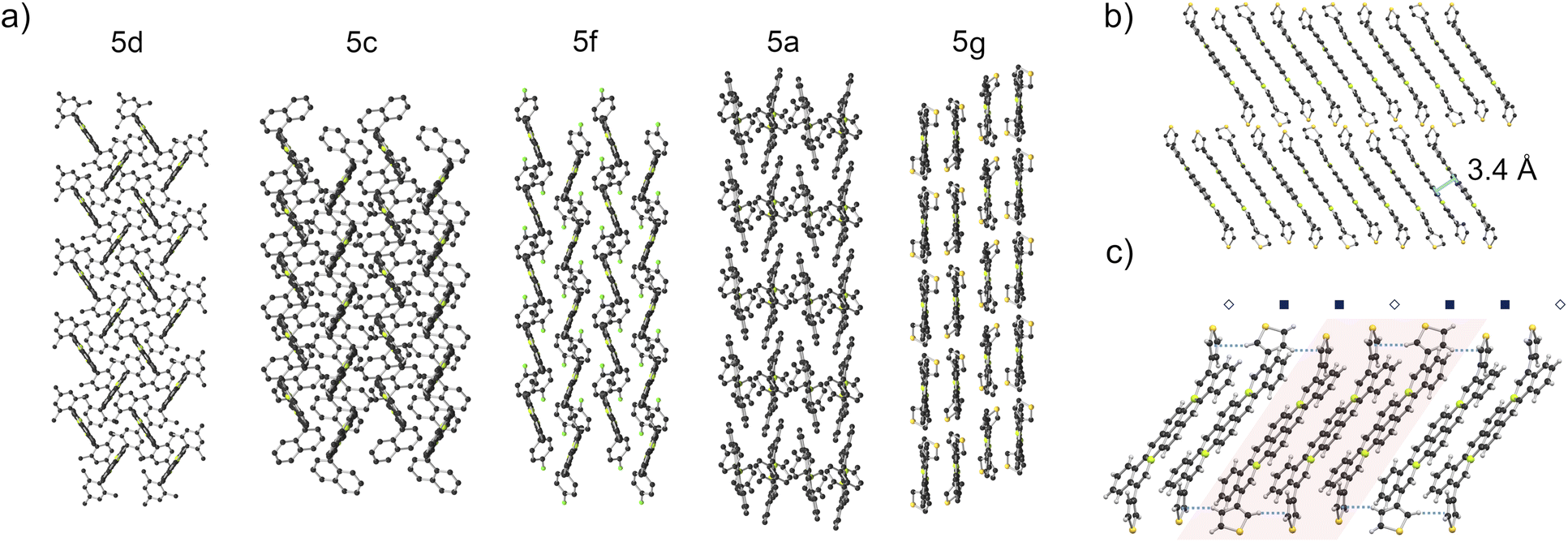
UV-Vis spectra of 5a–j in o-C6H4Cl2 (10−6–10−5 M, 298 K) show strong visible absorptions with lowest energy λabs between 546–563 nm and ε ranging 1.9–3.6 × 104 L mol−1 cm−1. For 3a the lowest energy λabs is 445 nm with ε = 8.3 × 103 L mol−1 cm−1 (Fig. 6a and Table 1). These lowest energy absorption maxima are all considerably red-shifted compared to respective precursors 4 (λabs = 425 nm) and 1 (λabs = 374 nm) in CHCl3,5 reflecting the extended conjugation of the annulated products and correlating well with TD-DFT predicted spectra (Fig. S72 and S73†). Fluorescence emission maxima of annulated compounds in o-C6H4Cl2 (10−6–10−5 M, 298 K) are also red-shifted compared to those of precursors 4 (λem = 561 nm) and 1 (λem = 489 nm) in CHCl3.5 Compounds 5a–j show emissions with λem ranging 601–616 nm and low quantum yields (Φ ≤ 0.03, Table S2 and Fig. S63†). These emissions correspond to apparent Stokes shifts between 1220–1760 cm−1. Compound 3a (8.5 × 10−6 M in o-C6H4Cl2, 298 K) exhibits broad emission (λem, = 587 nm) and a quantum yield of 0.14.
 | ||
| Fig. 6 (a) UV-Vis spectra of 3a and 5a–j (10−6–10−5 M in o-C6H4Cl2, 298 K). (b) Cyclic voltammogram of 5e (9.0 × 10−4 M in 0.1 M n-Bu4NPF6o-C6H4Cl2, 298 K). | ||
| λ abs [nm], (ε [M−1 cm−1]) | λ em [nm] | E 1/2 red 1 [V] | E 1/2 red 2 [V] | |
|---|---|---|---|---|
| a Optical measurements were performed at 10−6–10−5 M concentrations in o-C6H4Cl2 at 298 K. Cyclic voltammetry experiments were performed at 10−4–10−3 M concentrations in 0.1 M n-Bu4NPF6o-C6H4Cl2 at 298 K and referenced vs. Fc+/0 using ferrocene as an internal standard. All other experimental details are described in the ESI. | ||||
| 3a | 365 (4500), 445 (8300) | 587 | −1.62 | — |
| 5a | 429 (5700), 525 (17500), 561 (21800) |
608 | −1.01 | −1.33 |
| 5b | 431 (2600), 521 (14400), 559 (19400) |
603 | −0.98 | −1.29 |
| 5c | 432 (3600), 519 (16300), 556 (21900) |
607 | −0.97 | −1.29 |
| 5d | 430 (3900), 515 (21100), 553 (29000) |
610 | −1.00 | −1.49 |
| 5e | 433 (8500), 521 (23600), 558 (28800) |
607 | −0.95 | −1.24 |
| 5f | 431 (5500), 520 (19400), 559 (25000) |
616 | −0.97 | −1.26 |
| 5g | 430 (8700), 527 (28700), 563 (36200) |
616 | −0.99 | −1.30 |
| 5h | 425 (4700), 510 (19400), 546 (25400) |
604 | −1.05 | −1.36 |
| 5i | 426 (6500), 509 (23700), 546 (29400) |
601 | −1.08 | −1.39 |
| 5j | 427 (6400), 516 (16600), 551 (20300) |
607 | −1.02 | −1.33 |
Cyclic voltammetry was used to probe the electrochemical properties of 3a and 5a–j, (10−4–10−3 M in 0.1 M n-Bu4NPF6o-C6H4Cl2, 298 K, Table 1). A single reversible one-electron reduction was observed for 3a, and two reversible one-electron reductions were observed for each compound 5a–j (e.g.Fig. 6b and S52–S61†). The compounds studied exhibited +0.36–+0.52 V more positive first reduction potentials (E1/2 red 1) than their non-annulated precursors 1 and 4 (reported in CH2Cl2).5 Strikingly, first reduction potentials of 5a-j (−0.95–−1.08 V vs. Fc+/0) are amongst the most positive yet reported for boron-doped PAHs, and are all more positive (by +0.09–+0.22 V) than E1/2 red 1 reported for PC61BM in the same solvent.26 These E1/2 red 1 values correspond to very low estimated LUMO energy levels of −4.20–−4.07 eV.27 We note that 5a, 5c, 5d, 5f, and 5g each bear “deep LUMO”10 energy levels and exhibit similar strong visible range absorptions. However, they adopt solid-state packing motifs ranging from herringbone to continuous π-stacks. This embodies “heteroatom-doping” as a means to modify optical and electronic properties largely independently of molecular geometries and substituents, allowing the latter to be used for the control of solid-state packing.
Conclusions
We report a C–H-functionalizing annulation reaction of B-doped PAHs with alkynes. This reactivity complements existing B-doped PAH syntheses with a facile π-extension methodology for the synthesis of structurally constrained, fusion atom B-doped PAHs. We applied this synthetic strategy to ambient-stable, deep LUMO energy 6a,15a-diborabenzo[tuv]naphtho[2,1-b]picenes, whose solid-state packing can be directed via substituent effects. Annulation leads to red-shifted absorption and emission spectra, as well as significantly more positive reduction potentials, compared to corresponding B-doped PAH precursors. We anticipate that this new synthetic tool will aid the development and use of B-doped π-conjugated structures. Studies to this end are underway in our laboratory.Data availability
All experimental details and data supporting this article are available in the ESI.†Author contributions
A. M. and T.-J. C. performed the synthetic experiments. G.-C. L. measured optical and electronic properties. S.-L. L. performed solid-state NMR measurements. C.-T. H. and K.-H. W. performed DFT calculations. M.-J. C. directed DFT studies. J. M. F. supervised the research and wrote the manuscript. All authors edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support of the National Science and Technology Council of Taiwan (grant no. 110-2113-M-002-028-MY3) and the Yushan Young Scholar Program of the Ministry of Education of Taiwan. We acknowledge the mass spectrometry technical research services from Consortia of Key Technologies, National Taiwan University and the Instrumentation Center of National Taiwan Normal University. We also thank Mr Yi-Hung Liu (NTU) for X-ray crystal structure determination. We thank Prof. Pi-Tai Chou (NTU), Prof. Ken-Tsung Wong (NTU) for use of their laboratory's spectrometers and CVD equipment. We are also grateful to Prof. Jerry C. C. Chan (NTU) for assistance with solid-state NMR measurements.Notes and references
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef PubMed; (b) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491 CrossRef CAS; (c) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565 CrossRef CAS PubMed.
- (a) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413 CrossRef CAS; (b) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37 CrossRef CAS; (c) S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77 CrossRef CAS.
- (a) K. Matsuo, S. Saito and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 11984 CrossRef CAS; (b) T. Kushida, S. Shirai, N. Ando, T. Okamoto, H. Ishii, H. Matsui, M. Yamagishi, T. Uemura, J. Tsurumi, S. Watanabe, J. Takeya and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 14336 CrossRef CAS PubMed.
- J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096 CrossRef CAS PubMed.
- (a) T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235 CrossRef CAS; (b) C.-M. Hsieh, T.-L. Wu, J. Jayakumar, Y.-C. Wang, C.-L. Ko, W.-Y. Hung, T.-C. Lin, H.-H. Wu, K.-H. Lin, C.-H. Lin, S. Hsieh and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 23199 CrossRef CAS.
- (a) E. von Grotthuss, M. Diefenbach, M. Bolte, H. W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067 CrossRef CAS PubMed; (b) E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2018, 57, 16491 CrossRef CAS; (c) J. E. Barker, A. D. Obi, D. A. Dickie and R. J. Gilliard Jr, J. Am. Chem. Soc., 2023, 145, 2028 CrossRef CAS PubMed.
- (a) Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529 CrossRef CAS; (b) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291 CrossRef CAS PubMed.
- (a) C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206 CrossRef CAS; (b) K. Schickedanz, T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808 RSC; (c) F. Miyamoto, S. Nakatsuka, K. Yamada, K.-i. Nakayama and T. Hatakeyama, Org. Lett., 2015, 17, 6158 CrossRef CAS; (d) J. Radtke, K. Schickedanz, M. Bamberg, L. Menduti, D. Schollmeyer, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Sci., 2019, 10, 9017 RSC; (e) J.-J. Zhang, L. Yang, F. Liu, Y. Fu, J. Liu, A. A. Popov, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 25695 CrossRef CAS; (f) C. Mützel, J. M. Farrell, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2022, 61, e202115746 CrossRef PubMed; (g) W. Sun, J. Guo, Z. Fan, L. Yuan, K. Ye, C. Dou and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202209271 CrossRef CAS; (h) J. Guo, K. Zhang, Y. Wang, H. Wei, W. Xiao, K. Yang and Z. Zeng, Chem. Sci., 2023, 14, 4158 RSC.
- K. Yuan, A. K. Gupta, C. Si, M. Uzelac, E. Zysman-Colman and M. J. Ingleson, Org. Lett., 2023, 25, 5880 CrossRef CAS.
- Y. Liu, L. Yuan, J. Guo, W. Sun, Y. Wang and C. Dou, Angew. Chem., Int. Ed., 2023, 62, e202306911 CrossRef CAS.
- Nevertheless, heteroatom/B co-doped nanographenes have impressive applications, see: M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Chem. Soc. Rev., 2024, 53, 1624 RSC.
- H. Narita, H. Choi, M. Ito, N. Ando, S. Ogi and S. Yamaguchi, Chem. Sci., 2022, 13, 1484 RSC.
- (a) E. H. Fort, P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 16006 CrossRef CAS PubMed; (b) H. Ito, K. Ozaki and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 11144 CrossRef CAS PubMed.
- Y. Shoji, N. Tanaka, S. Muranaka, N. Shigeno, H. Sugiyama, K. Takenouchi, F. Hajjaj and T. Fukushima, Nat. Commun., 2016, 7, 12704 CrossRef.
- G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839 RSC.
- M. F. Lappert and B. Prokai, J. Organomet. Chem., 1964, 1, 384 CrossRef CAS.
- M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895 CrossRef CAS.
- C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992 CrossRef CAS PubMed.
- A. J. Warner, J. R. Lawson, V. Fasano and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 54, 11245 CrossRef CAS PubMed.
- (a) D. L. Crossley, R. J. Kahan, S. Endres, A. J. Warner, R. A. Smith, J. Cid, J. J. Dunsford, J. E. Jones, I. Vitorica-Yrezabal and M. J. Ingleson, Chem. Sci., 2017, 8, 7969 RSC; (b) R. J. Kahan, D. L. Crossley, J. Cid, J. E. Radcliffe and M. J. Ingleson, Angew. Chem., Int. Ed., 2018, 57, 8084 CrossRef CAS PubMed; (c) K. Yuan, R. J. Kahan, C. Si, A. Williams, S. Kirschner, M. Uzelac, E. Zysman-Colman and M. J. Ingleson, Chem. Sci., 2020, 11, 3258 RSC.
- T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS.
- F. Zettler, H. D. Hausen and H. Hess, J. Organomet. Chem., 1974, 72, 157 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318 CrossRef CAS PubMed.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532 CrossRef CAS.
- LUMO energy levels estimated using the equation: ELUMO = −e(E1/2 red) − 5.15 eV. Adapted from: C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 234163123416322341633234163423416352341636. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03781b |
| This journal is © The Royal Society of Chemistry 2024 |