
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCCC pincer Ru complex-catalyzed C–H vinylation/6π-E-cyclization of aldimines for constructing 4H-pyrido[1,2-a]pyrimidines†
Heng
Cai
a,
Yong-Qiang
Tu
 *ab,
Qiang
Niu
c,
Wen-Ping
Xie
b,
Bin
Wang
a,
Ka
Lu
a,
Zi-Hao
Li
b,
Fu-Min
Zhang
a and
Xiao-Ming
Zhang
a
*ab,
Qiang
Niu
c,
Wen-Ping
Xie
b,
Bin
Wang
a,
Ka
Lu
a,
Zi-Hao
Li
b,
Fu-Min
Zhang
a and
Xiao-Ming
Zhang
a
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: tuyq@lzu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: tuyq@sjtu.edu.cn
cNational Enterprise Technology Center, Inner Mongolia Erdos Electric Power and Metallurgy Group Co., Ltd, Ordos, 016064, Inner Mongolia, China
First published on 3rd September 2024
Abstract
An unusual cascade C–H activation, vinylation and 6π-electrocyclization of 2-pyridyl aldimines with vinyl bromides/triflates was achieved using catalysis with a unique CCC pincer NHC–Ru(III) complex (Cat B). This reaction was found to enable a rapid and diverse synthesis of polycyclic 4H-pyrido[1,2-a]pyrimidine derivatives in mostly good to high yields, and with a broad substrate scope. A mechanistic study suggested the formation of a semi-opened Ru(III) intermediate chelating/activating the aldimine, and the occurrence of single-electron transfer (SET) to generate a vinyl radical, followed by vinylation and then an intramolecular 6π-electrocyclization of 1N,3N-hexatrene to form the product. This protocol provides a convenient approach for preparing and seeking new drug candidates.
Introduction
Organic N-heteropolycycles often play substantial roles in pharmaceuticals, bioactive molecules, and functional materials.1 The 4H-pyrido[1,2-a]pyrimidine derivatives represent a large class of versatile N-heterocyclic molecules. At least ten members have been used as clinical drugs, having antidepressant,2 anticancer,3 antiallergic,4 antioxidant,5 quorum sensing inhibition,6 and other functions.7 Additionally, some other derivatives show utility in other fields, such as photocatalysis (Scheme 1a).8 Over the past decades, however, only a few methods for constructing these N-heteropolycycles have been developed, and commonly involve the catalytic cyclization of multiple components or use of substrates premade in multiple steps (Scheme 1b).9 Some of the approaches also show limitations: poor diversity or functionality of generated products, requirement of pre-installation and post-removal of activating/directing groups (DGs), etc.10 Thus alternative development of a more efficient strategy is necessary for constructing diverse derivatives of this N-heterocyclic framework to meet the demands of multiple research fields.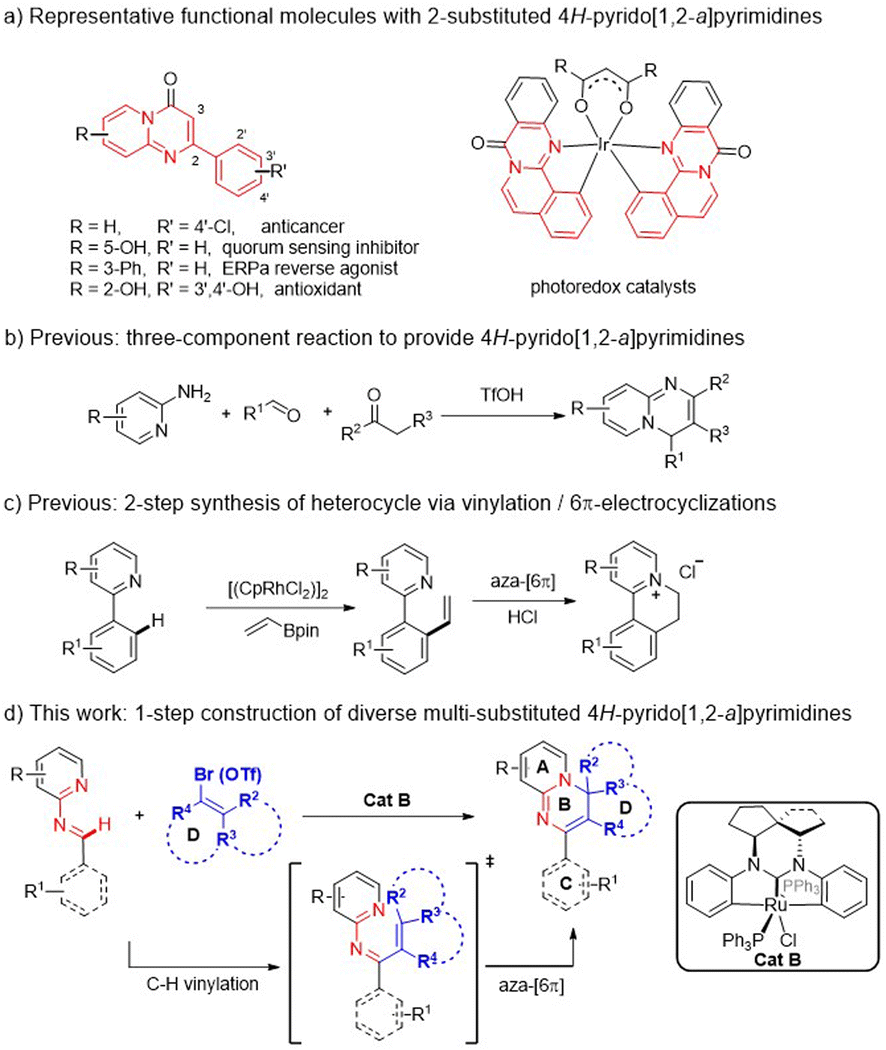 | ||
| Scheme 1 Features of multi-substituted 4H-pyrido[1,2-a]-pyrimidines. | ||
The transition-metal-catalyzed C–H vinylation (Heck reaction) and annulation of arenes with vinyl species is recognized as a useful approach in organic synthesis.11 To the best of our knowledge, there have hardly been any effective approaches reported for C–H vinylation and aza-cyclization to construct these N-heteropolycycles, except for the few examples using the simple vinyl partner (Scheme 1c).12 To effectively access diverse multi-substituted 4H-pyrido[1,2-a]pyrimidine derivatives, we hope to find a suitable catalyst system to investigate a more effective cascade strategy involving C–H activation, vinylation and aza-cyclization from aldimine and unactivated multi-substituted vinyl species (Scheme 1d). Here the aldimine substrates can be readily assembled from commercial aldehyde and amine materials, and importantly the N-atom of the pyridine moiety can serve as both a transient guide for C–H vinylation and the reaction site for 6π-electrocyclization, avoiding the use of an extra DG. In this approach, however, there may exist some challenges that would need to be overcome: an undesired C–H vinylation may take place competitively at aromatic ring C under direction of the N-atom of the aldimine;13 and if the vinyl partners are unactivated and multi-functionalized for achieving product diversity, such as more rings B/D, a sufficiently powerful catalyst system would be required. Given our successful development of transition-metal (Ir, Ru) NHC pincer catalysts, which have proven to robustly enable C–H activation/functionalization,14 here we set out to evaluate this series of catalysts to achieve our goal of being able to synthesize diverse polycyclic 4H-pyrido[1,2-a]pyrimidine derivatives.
Results and discussion
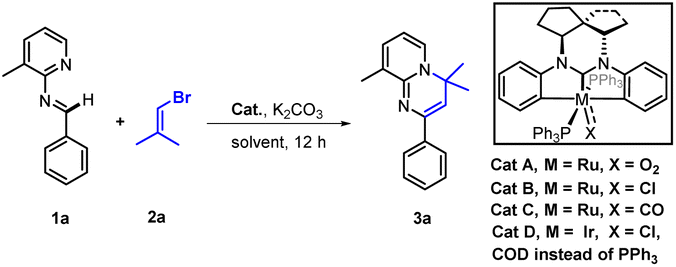
We began our study with the preparation of representative spirocycle-fused NHC pincer Ru and Ir complexes, Cat A–D, according to methods we reported.14a,d Then we tried to realize a model cascade reaction by screening the transition-metal catalysts and reaction conditions with N-(3-methylpyridin-2-yl)-1-phenylmethanimine (1a) as a substrate and the unactivated 1-bromo-2-methylpropene (2a) as its vinylation partner. Initially using the conditions described in Table 1 (see ESI† for details), to our delight we obtained the desired product 3a, albeit in 40% yield, with Cat A, and a little better 43% yield with Cat B. In contrast, using Cat C or D failed to give product 3a (entries 1–2 vs. 3–4). Fortunately, we did not detect the undesired product of C–H vinylation at aromatic ring C in this model reaction. This selectivity could be explained by the results of density functional theory (DFT) calculations, which indicated that the desired activation of the aldimine C–H (energy barrier ΔG = 28.0 kcal mol−1) was easier than that (ΔG = 37.3 kcal mol−1) of the undesired reaction (see ESI, Fig. S8†). Since Cat A and B gave the positive results (entries 1–2), additional commercial Ru-catalysts were examined, but no reaction was observed (entries 5–9). A control experiment without any catalyst resulted in no product. Subsequently we turned our attention to investigating the effect of solvent (entries 10–12), and found that 1,4-dioxane gave the better result (entry 12). Finally, the best 96% yield of 3a was obtained by raising the reaction temperature to 130 °C, and the loading of Cat B to 5 mol% (entry 13).|
|
|||
|---|---|---|---|
| Entry | Cat | Solvent | Yieldb/% |
| a General conditions: 1a (0.1 mmol), 2a (0.2 mmol), Cat (3 mol%), K2CO3 (0.15 mmol) and solvent (1 mL), 110 °C for 12 h under argon. b Yields determined from 1H NMR results. c 130 °C. d Cat B (5 mol%). | |||
| 1 | A | Toluene | 40 |
| 2 | B | Toluene | 43 |
| 3 | C | Toluene | NR |
| 4 | D | Toluene | NR |
| 5 | Ru(bpy)3Cl2 | Toluene | NR |
| 6 | Cp(PPh3)2RuCl | Toluene | NR |
| 7 | RuCl3 | Toluene | NR |
| 8 | Ru(acac)3 | Toluene | NR |
| 9 | RuO2 | Toluene | NR |
| 10 | B | DMSO | NR |
| 11 | B | MeCN | 12 |
| 12c | B | 1,4-Dioxane | 89 |
| 13 | B | 1,4-Dioxane | 96 |
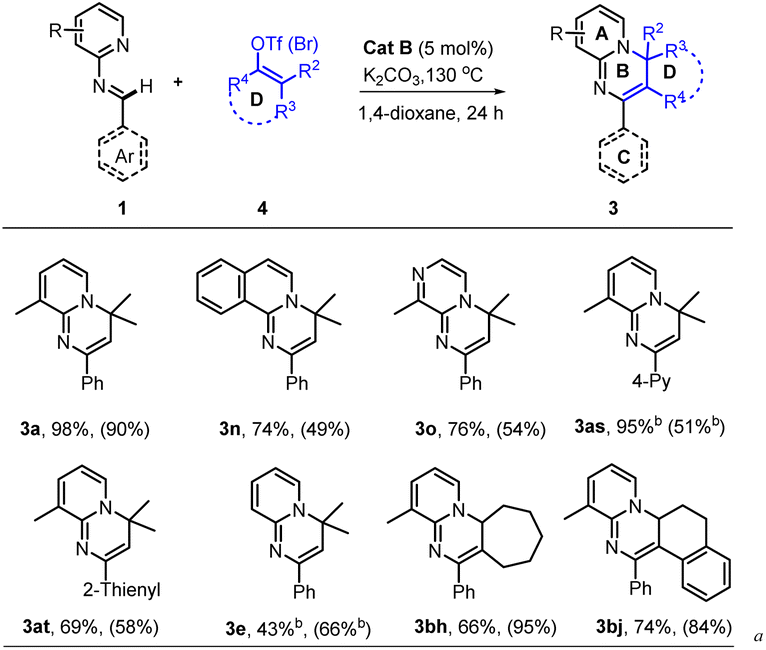
With the optimal conditions (Table 1, entry 13) in hand, we then set out to expand the substrate 1 scope first by testing various substituents on ring A. The experimental results indicated that substitution with C3-alkyls, Ph, CF3, and H gave medium to high yields of desired products 3a–3e (Table 2), with the C3–Et substitution giving the best yield (3b). C4- (3f–3i) and C5-substitutions (3j, 3k) gave on average lower yields of products than did the C-3 substitutions, with examples (3f, 3g, 3j) bearing electron-donating groups (EDGs) giving higher yields than those (3h, 3i, 3k) bearing electron-withdrawing groups (EWGs). The lower yield on average with EWGs was possibly due to a resulting decreased ability of the N-atom on ring A to coordinate with the Ru-catalyst. When a substituent (Me, OMe or F) was placed at the C6 position of the pyridine moiety, no reaction was observed, possibly due to steric hindrance from the neighboring C6 substituent. Furthermore, a 3,4-dimethyl substituent on ring A gave a high 91% yield of 3m, whereas fusing benzene to ring A in one experiment and replacing A with 3-methyl-pyrazine in another experiment gave only modest yields of 3n and 3o, respectively. In short, substitution with an EDG at C3, C4 or C5 of ring A could favor this reaction, while any substitution at C6 disfavored it.
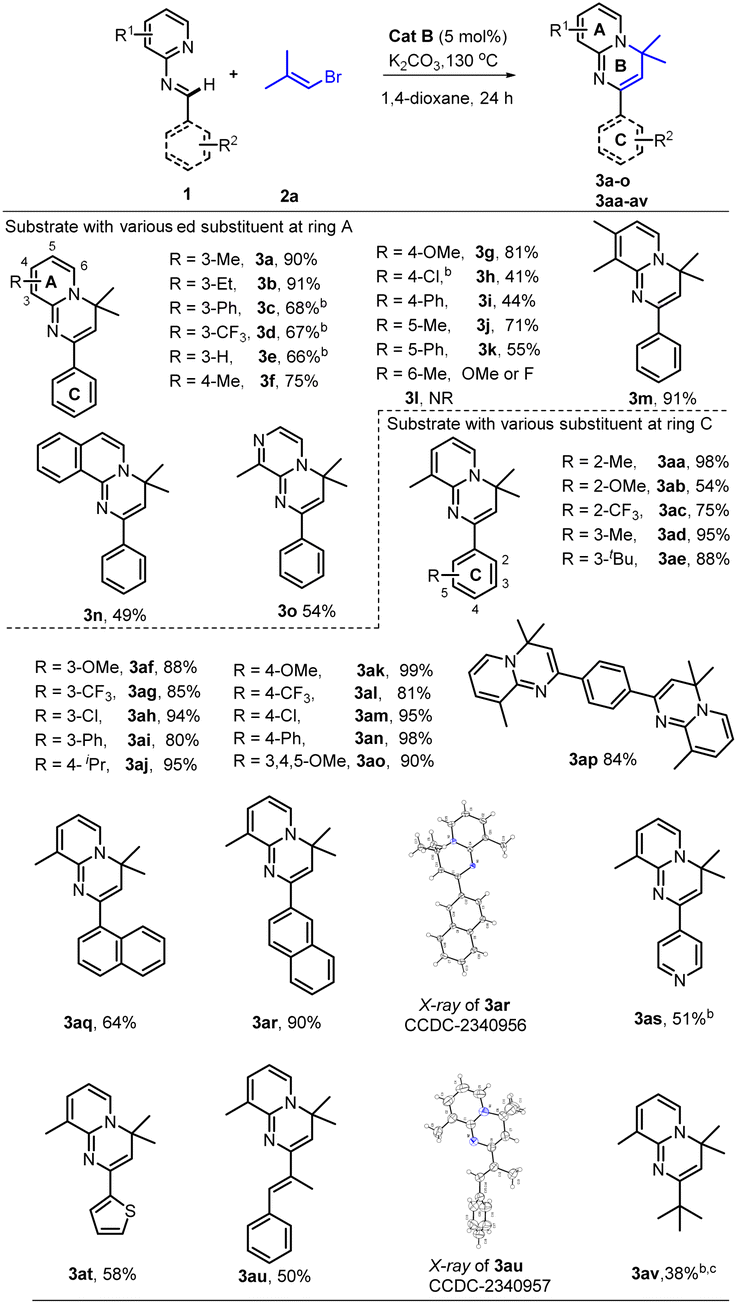
Subsequently we examined the substrate scope with 3-methyl-pyridyl fixed as ring A and testing various substitutions at C2–C4 of ring C. The experimental results demonstrated wide tolerance of the reaction to EDG (Me, Et, iPr, or OMe), neutral phenyl, and EWG (Cl, CF3) substituents here, generating mostly good to excellent yields of products (3aa, 3ad–3ao)—except for 3ab and 3ac, which gave products with only moderate yields. Further replacing ring C with 1,4-disubstituted phenyl, α- or β-naphthalene, and 4-pyridyl or 2-thio-phenyl or phenylalkenyl, was still effective for this reaction, generating moderate (3aq, 3as–3au) to good (3ap) and high (3ar) yields of desired products. Additionally, even a non-aromatic tertiary-butyl aldimine could be effective with this catalytic system, generating the corresponding product 3av, albeit with a relatively low yield of 38%.
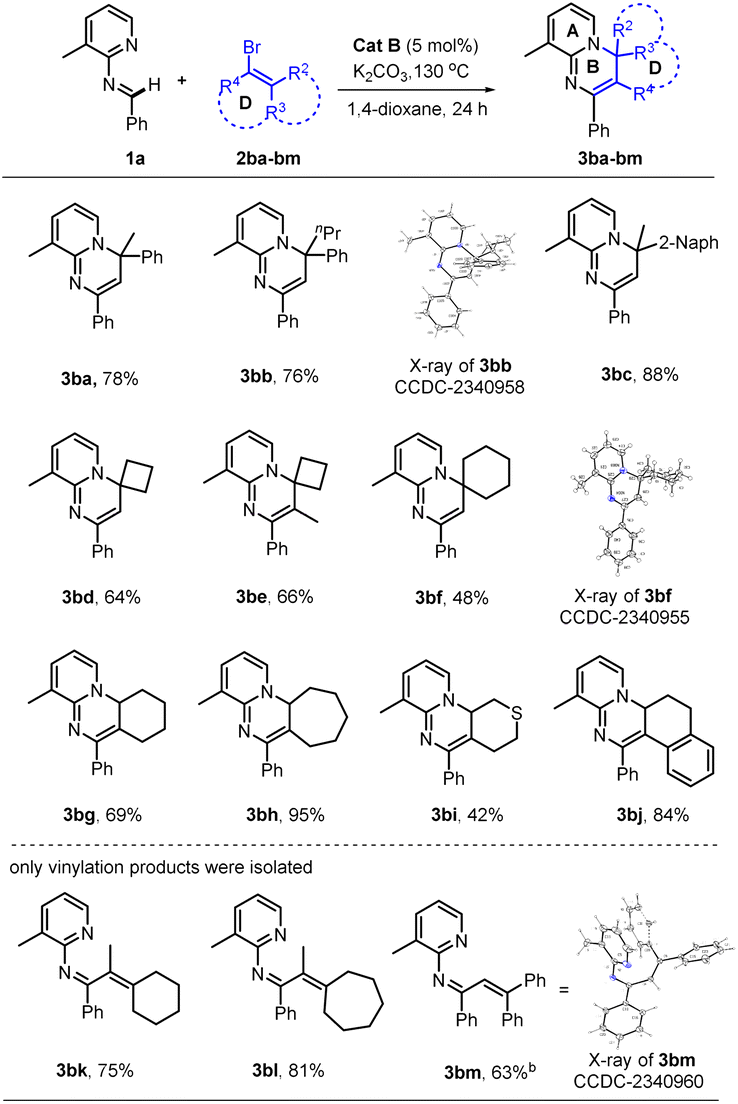
Of particular note, this catalyst system showed a robust ability to activate many unactivated vinyl partners 2, enabling construction of diverse versions of ring B, even those bearing a spiro- or fused-ring D in many cases. As indicated in Table 3, this catalytic reaction could tolerate both acyclic and even cyclic vinyl species, smoothly generating tetra- (3ba, 3bb, 3bd–3bi) and pentacyclic (3bc and 3bj) products with up to five rings in medium to good yields. However, when the vinyl species carried tri- or large 2,2-disubstituents, only the vinylation products were isolated, without further 6π-electrocyclization observed, possibly due to their greater steric hindrance at the terminus of the 1,3-diazo-1,3,5-triene moiety (3bk–3bm). This observation also confirmed that the reaction process underwent a 6π-electrocyclization of a diazo-triene intermediate instead of a azo-[4 + 2] D–A type cyclization of aldimides 1 with vinyl species 2 followed by dehydrobromination.
As the vinyl trifluoromethyl sulphonate (triflate, OTf) reagent is usually easier to prepare from ketone or aldehyde precursors, but less active than the bromide, we also examined the performance of the catalyst with some vinyl triflates 4. As indicated in Table 4, the reaction efficiencies of several examples were examined, and a significant substrate dependence was found. When 1a with 3-Me at ring A, 1n with benzene-fused ring A, 1o with 3-Me-pyrazine as ring A, 1as with 4-pyridine as ring C, and 1at with 2-thiophene as ring C, were subjected to the cascade reaction separately with 2,2-dimethyl vinyl triflates, they all gave the higher yields of products (3a, 3n, 3o, 3as and 3at) than they respectively did with the corresponding bromide. While 1e was used to reaction, a reverse result was obtained (3e). Importantly, when cyclic vinyl triflates (4bh and 4bj) were examined with 1a, moderate yields were observed but lower than those with corresponding vinyl bromides. This result provided valuable information for seeking a reaction yield as high as possible, namely by screening vinyl partners, though a clear structure-efficiency relationship has not yet been identified.
In order to obtain insight into the mechanism of this reaction, several supporting experiments and DFT calculations were conducted. Initially, inspection of the model reaction of 1a with 2a under catalysis of 20 mol% Cat B with in situ high-resolution mass spectroscopy (HRMS) after one hour revealed the presence of the bromide Cat E, and Ru-fragments I, III, IV and VI (see ESI, Fig. S2†), whose structures are shown in Fig. 1. The molecular peak of Cat B was not detected in this case. A repeated experiment of this model reaction with 5 mol% Cat E instead of Cat B led to 3a with a yield of 95% (Scheme 2a). This result indicated that Cat E participated in the catalytic cycle in the standard conditions with 2.0 equivalents of vinyl bromide 2. We nevertheless still used Cat B instead of Cat E as the catalyst in this work, since the former was easier to prepare. When the deuterium-labeled-1a (D-1a) was subjected to the model reaction with 20 mol% Cat B or Cat E separately, two fragments with Ru-species D-IV and 2D-IV were indicated by the HRMS data (see ESI, Fig. S3 and S5†) in each case, suggesting that a D-atom of D-1a was transferred onto the phenyl(s) of Ru-species IV. These results showed that reaction process underwent a semi-opening of Ru-species I and then re-cyclization after trapping aldimine substrate (I → III) via Ru intermediate II. Subsequently, a radical-trapping experiment with 2.0 equivalents of TEMPO was carried out, and found to form the TEMPO-linked vinyl product with a trace of 3a formed, detected both using TLC and HRMS (Scheme 2b). This result showed that the C–Br bond cleavage of 2a generated the vinyl radical, which was proposed based on the DFT calculations (ΔG = 33.2 kcal mol−1, see ESI, Fig. S9†) to be the rate-determining step of the whole reaction. Finally, a kinetic isotope effect (KIE) experiment revealed a primary KIE of 1.05 (Scheme 2c), suggesting that C–H cleavage of aldimine was not the rate-determining step in this transformation.15
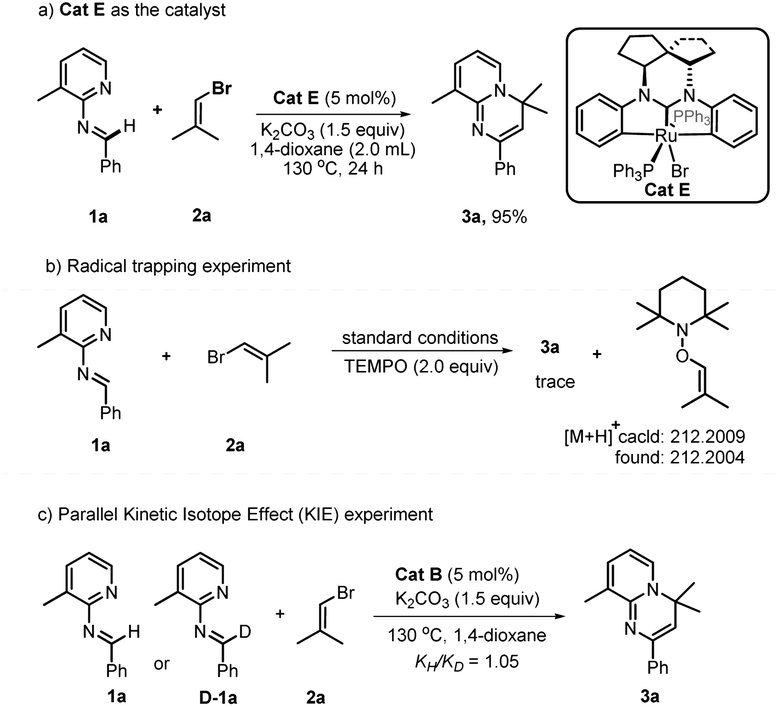 | ||
| Scheme 2 Mechanistic experiments. | ||
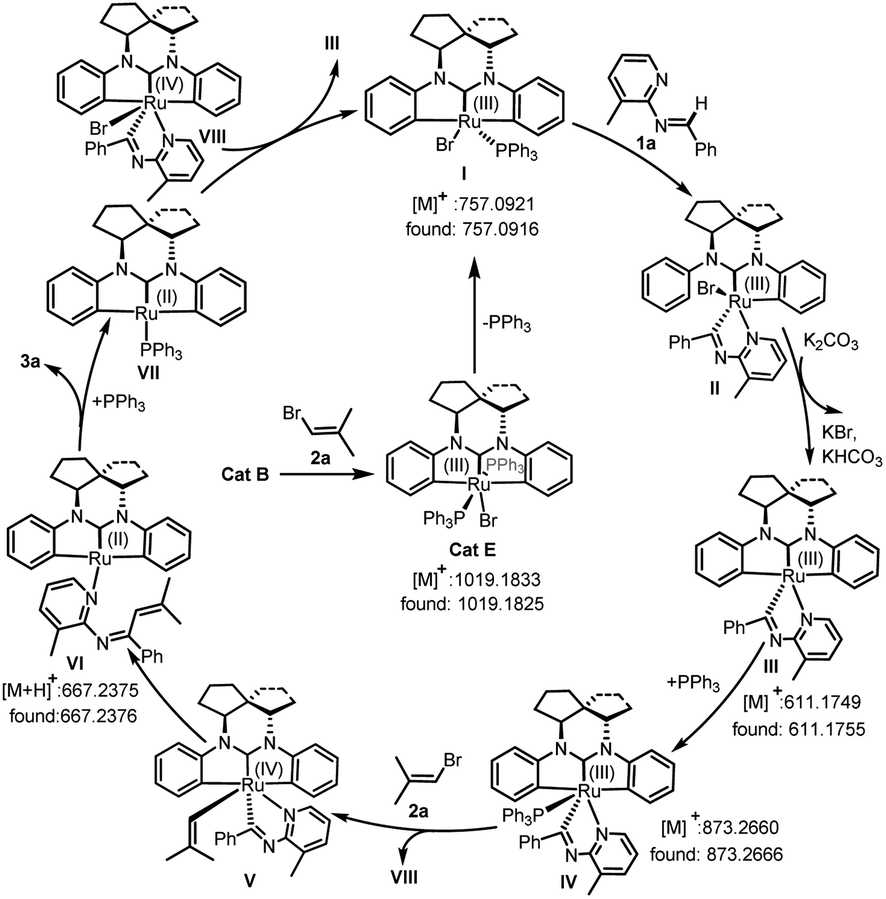 | ||
| Fig. 1 Proposed reaction mechanism. | ||
On the basis of the acquired evidence together with the related literature,14,16 a plausible mechanism was proposed (Fig. 1). Initially, according to this mechanism, Cat B was in situ transferred to Cat E, which released a PPh3 ligand to form the Ru-species I. Then the central Ru of I complexed substrate 1a, which was accompanied with a Ru–Ph bond breaking and transfer of the aldimine H to Ph to form the semi-opened Ru(III) species II. After releasing the second PPh3 ligand, eliminating HBr under assistance of K2CO3 and re-formation of a Ru–Ph bond, Ru-species III was formed. Complexation of III with PPh3 formed the Ru intermediate IV, which combined with the SET-generated vinyl and Br radicals to form Ru(IV)-species V and VIII, respectively. Intramolecular reductive vinylation in V gave the Ru(II)-chelated 1N,3N-hexatriene VI, which generated 3a and Ru(II)-species VII after 6π-electrocyclization/PPh3-ligand exchange. Subsequently, the Ru(II) species VII received a Br from VIII to regenerate species I and III for repeating the catalytic cycle.
Conclusions
In conclusion, we have developed a CCC pincer NHC–Ru(III)-catalyzed cascade C–H vinylation/6π-electrocyclization of pyridyl aldimines with a series of unactivated vinyl bromides/triflates. This procedure displays advantages of broad substrate scope and multiple product functionality. A range of N-heteropolycycles have been produced efficiently, which we believe will find good potential utility in medicinal chemistry. Preliminary mechanistic studies indicated that this reaction proceeds through a radical pathway and a semi-opened Ru(III) intermediate. This success displays again one more powerful reactivity of our catalyst series, and inspires us to further the catalytic research and its applications.Data availability
All data supporting the findings of this study are available within the article and its ESI† file.Author contributions
H. Cai performed all of the experiments and prepared the ESI.† B. Wang prepared materials and catalysts for this reaction. K. Lu and Z.-H. Li performed the DFT calculations. Q. Niu, W.-P. Xie, F.-M. Zhang and X.-M. Zhang discussed the results and commented on the manuscript. Y.-Q. Tu wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank prof. W.-X. Zhang (Peking University) for his helpful discussion of the reaction mechanism. We also acknowledge the “National Key R&D Program of China” (2023YFA1506400, 2023YFA1506403); NSFC (92256303); Shanghai Science and Technology Committee (19JC1430100) and Shanghai Jiao Tong University (WH410111001) for financial support.Notes and references
- (a) T. Kametani, C. V. Loc, T. Higa, M. Koizumi, M. Ihara and K. Fukumoto, J. Am. Chem. Soc., 1977, 99, 2306–2309 CrossRef CAS; (b) A. Witt and J. Bergman, Curr. Org. Chem., 2003, 7, 659–677 CrossRef CAS; (c) T. Xu and H. Alper, Org. Lett., 2015, 17, 1569–1572 CrossRef CAS PubMed; (d) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (e) T. Sena, K. Neog, S. Sarma, P. Manna, H. P. D. Boruah, P. Gogoi and A. K. Singha, Bioorg. Med. Chem., 2018, 26, 4942–4951 CrossRef; (f) K. P. Rakesh, H. K. Kumara, H. M. Manukumar and D. C. Gowad, Bioorg. Chem., 2019, 87, 252–264 CrossRef CAS; (g) U. H. F. Bunz and J. Freudenberg, Acc. Chem. Res., 2019, 52, 1575–1587 CrossRef CAS; (h) J. Zhang, X. Wang, D. Chen, Y. Kang, Y. Ma and M. Szostak, J. Org. Chem., 2020, 85, 3192–3201 CrossRef CAS PubMed; (i) L. Lv and C.-J. Li, Chem. Sci., 2021, 12, 2870–2875 RSC; (j) T. Wesp, T. Bruckhoff, J. Petry, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2022, 28, e202200129 CrossRef CAS PubMed; (k) Y. Zhang, S. Ling, P. Li, Z. Chen and X. F. Wu, Org. Lett., 2022, 24, 8864–8869 CrossRef CAS; (l) Z. Luo, J. Jiang, L. Zou, X. Zhou, J. Liu, Z. Ke, F. Chen, H. Jiang and W. Zeng, Sci. China Chem., 2024, 67, 374–382 CrossRef CAS; (m) Y. Gao, H. Li, S. Yang, Y. Huo, Q. Chen, X. Li, Z. Wang and X.-Q. Hu, Sci. China Chem., 2024, 67, 595–603 CrossRef CAS.
- (a) M. V. Mey, A. D. Windhorst, R. P. Klok, J. D. M. Herscheid, L. E. Kennis, F. Bischoff, M. Bakker, X. Langlois, L. Heylen, M. Jurzakb and J. E. Leysen, Med. Chem., 2006, 14, 4526–4534 Search PubMed; (b) L. E. J. Kennis, F. P. Bischoff, C. J. Mertens, C. J. Love, F. A. F. V. Keybus, S. Pieters, M. Braeken, A. A. H. P. Megens and J. E. Leysen, Bioorg. Med. Chem., 2000, 10, 71–74 CrossRef CAS PubMed.
- G. Priyadarshani, S. Amrutkar, A. Nayak, U. C. Banerjee, C. N. Kundu and S. K. Guchhait, Eur. J. Med. Chem., 2016, 122, 43–54 CrossRef CAS.
- Y. Yanagihara, H. Kasai, T. Kawashima and T. Shida, Jpn. J. Pharmacol., 1988, 48, 91–101 CrossRef CAS.
- C. L. Motta, S. Sartini, L. Mugnaini, F. Simorini, S. Taliani, S. Salerno, A. M. Marini, F. D. Settimo, A. Lavecchia, E. Novellino, M. Cantore, P. Failli and M. Ciuffi, J. Med. Chem., 2007, 50, 4917–4927 CrossRef PubMed.
- S. Hiroaki and J. Igarashi, WO Pat., WO2009063901, 2009 Search PubMed.
- (a) F. Awouters, J. Vermeire, F. Smeyers, P. Vermote, R. Beek and C. J. E. Niemegeers, Drug Dev. Res., 1986, 8, 95–102 CrossRef CAS; (b) P. Molina, E. Aller, A. Lorenzo, P. López-Cremades, I. Rioja, A. Ubeda, M. C. Terencio and M. J. Alcaraz, J. Med. Chem., 2001, 44, 1011–1014 CrossRef CAS; (c) D. G. Shulman, L. Amdahl, C. Washington and A. Graves, Clin. Therapeut., 2003, 25, 1096–1106 CrossRef CAS; (d) R. J. Perner, C. H. Lee, M. Jiang, Y.-G. Gu, S. DiDomenico, E. K. Bayburt, K. M. Alexander, K. L. Kohlhaas, M. F. Jarvis, E. L. Kowaluk and S. S. Bhagwat, Bioorg. Med. Chem. Lett., 2005, 15, 2803–2807 CrossRef CAS PubMed; (e) C. L. Motta, S. Sartini, L. Mugnaini, F. Simorini, S. Taliani, S. Salerno, A. M. Marini, F. D. Settimo, A. Lavecchia, E. Novellino, M. Cantore, P. Failli and M. Ciuffi, J. Med. Chem., 2007, 50, 4917–4927 CrossRef; (f) M. Donghi, O. D. Kinzel and V. Summa, Bioorg. Med. Chem. Lett., 2009, 19, 1930–1934 CrossRef CAS; (g) J. S. Debenham, C. B. Madsen-Duggan, J. Wang, X. Tong, J. Lao, T. M. Fong, M.-T. Schaeffer, J. C. Xiao, C. C. R.-R. Huang, C.-P. Shen, D. S. Stribling, L. P. Shearman, A. M. Strack, D. E. MacIntyre, J. J. Hale and T. F. Walsh, Bioorg. Med. Chem. Lett., 2009, 19, 2591–2594 CrossRef CAS; (h) L. Peng, X. Gao, L. Duan, X. Ren, D. Wu and K. Ding, J. Med. Chem., 2011, 54, 7729–7733 CrossRef CAS PubMed; (i) P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS.
- (a) S. Philipp, B. Esther and J. Dominik, WO Pat., WO2011044988, 2011 Search PubMed; (b) T. Hisato, W. Osamu, H. Hiromitsu, M. Makoto and T. Seiji, JP Pat., JP2000156288, 2000 Search PubMed.
- K. Yang, J. Xiang, G. Bao, Q. Dang and X. Bai, ACS Comb. Sci., 2013, 15, 519–524 CrossRef CAS PubMed.
- (a) Y. Xie, T. Chen, S. Fu, H. Jiang and W. Zeng, Chem. Commun., 2015, 51, 9377–9380 RSC; (b) Y.-F. Liang, R. Steinbock, A. Münch, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2018, 130, 5482–5486 CrossRef; (c) R. T. Bhawale, A. S. Chillal and U. A. Kshirsagar, J. Heterocycl. Chem., 2023, 60, 1356–1373 CrossRef CAS.
- (a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2964 CrossRef CAS PubMed; (b) S. Jagtap, Catalysts, 2017, 7, 267–319 CrossRef; (c) D. Paul, S. Das, S. Saha, H. Sharma and K. R. Goswami, Eur. J. Org Chem., 2021, 14, 2057–2076 CrossRef; (d) R.-X. Liang and Y.-X. Jia, Acc. Chem. Res., 2022, 55, 734–745 CrossRef CAS; (e) J. Cai, B. Sun, S. Yu, H. Zhang and W. Zhang, Int. J. Mol. Sci., 2023, 24, 8252–8280 CrossRef CAS PubMed.
- X. Jiang, Z. Zeng, Y. Hua, B. Xu, Y. Shen, J. Xiong, H. Qiu, Y. Wu, T. Hu and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 15585–15594 CrossRef CAS PubMed.
- (a) B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, ACS Catal., 2011, 1, 1221–1224 CrossRef; (b) H. Wang, S. Yu, Z. Qi and X. Li, Org. Lett., 2015, 17, 2812–2815 CrossRef CAS; (c) Y.-F. Liang, X. Wang, Y. Yuan, Y. Liang, X. Li and N. Jiao, ACS Catal., 2015, 5, 6148–6152 CrossRef CAS; (d) C. Ma, C.-Q. Zhao, L.-P. Li, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192 RSC; (e) J. Li, K. Korvorapun, S. D. Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Nat. Commun., 2017, 8, 15430–15437 CrossRef; (f) K. Korvorapun, N. Kaplaneris, T. Rogge, S. Warratz, A. C. f. Stückl and L. Ackermann, ACS Catal., 2018, 8, 886–892 CrossRef CAS; (g) M. T. Findlay, A. S. Hogg, J. J. Douglas and I. Larrosa, Green Chem., 2023, 25, 2394–2400 RSC; (h) X. Cong, Q. Zhuo, N. Hao, A. Mishra, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2024, 63, e202318203 CrossRef CAS PubMed; (i) X. Cong, N. Hao, A. Mishra, Q. Zhuo, K. An, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2024, 146, 10187–10198 CrossRef CAS PubMed.
- (a) Z.-B. Yan, K.-L. Dai, B.-M. Yang, Z.-H. Li, Y.-Q. Tu, F.-M. Zhang, X.-M. Zhang, M. Peng, Q.-L. Chen and Z.-R. Jing, Sci. China Chem., 2020, 63, 1761–1766 CrossRef CAS; (b) Z.-B. Yan, M. Peng, Q.-L. Chen, K. Lu, Y.-Q. Tu, K.-L. Dai, F.-M. Zhang and X.-M. Zhang, Chem. Sci., 2021, 12, 9748–9753 RSC; (c) K.-L. Dai, Q.-L. Chen, W.-P. Xie, K. Lu, Z.-B. Yan, M. Peng, C.-K. Li, Y.-Q. Tu and T.-M. Ding, Angew. Chem., Int. Ed., 2022, 61, e202206446 CrossRef CAS; (d) H. Cai, Y.-Q. Tu, K. Lu, Q.-L. Chen, F.-M. Zhang, X.-M. Zhang, Y.-J. Pan and Z.-B. Yan, Sci. China Chem., 2023, 66, 2791–2796 CrossRef CAS; (e) W.-F. Wang, K. Lu, P.-R. Liu, H.-H. Zeng, L.-M. Yang, A.-J. Ma, Y.-Q. Tu and J.-B. Peng, ACS Catal., 2024, 14, 5156–5166 CrossRef CAS.
- M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef.
- (a) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (b) Z. Shi, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 4892–4896 CrossRef CAS; (c) J. H. Kim, S. Greßies and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5577–5581 CrossRef CAS; (d) H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386–10390 CrossRef CAS PubMed; (e) Y.-F. Liang, V. Mgller, W. Liu, A. Mgnch, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 9415–9419 CrossRef CAS; (f) H. Wang, M. M. Lorion and L. Ackermann, ACS Catal., 2017, 7, 3430–3433 CrossRef CAS; (g) K. Korvorapun, R. C. Samanta, T. Rogge and L. Ackermann, Synthesis, 2021, 53, 2911–2934 CrossRef CAS; (h) M. T. Findlay, P. Domingo-Legarda, G. McArthur, A. Yen and I. Larrosa, Chem. Sci., 2022, 13, 3335–3362 RSC; (i) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS; (j) S. Kamio, M. Nakamoto, T. Yamagishi, M. Oestreich and H. Yoshida, Chem. Commun., 2024, 60, 6379–6382 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2340955, 2340956, 2340957, 2340958, 2340960 and 2340961. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05067c |
| This journal is © The Royal Society of Chemistry 2024 |