
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFour-step continuous-flow total synthesis of (−)-debromoflustramine B using a chiral heterogeneous Pd NP catalyst†
Junwen
Wang‡
a,
Feng
Liang‡
a,
Zhen
Dong
a,
Junrong
Huang
 a,
Yuxiang
Zhu
*d,
Hengzhi
You
*ab and
Fen-Er
Chen
*abc
a,
Yuxiang
Zhu
*d,
Hengzhi
You
*ab and
Fen-Er
Chen
*abc
aSchool of Science, Harbin Institute of Technology (Shenzhen), 518055, China. E-mail: youhengzhi@hit.edu.cn; rfchen@fudan.edu.cn
bGreen Pharmaceutical Engineering Research Center, Harbin Institute of Technology (Shenzhen), Shenzhen, 518055, China
cEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai, 200433, China
dSchool of Pharmaceutical Sciences (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Shenzhen, 518107, China. E-mail: zhuyx86@mail.sysu.edu.cn
First published on 9th September 2024
Abstract
Various prenylated indoline alkaloids with diverse biological activities, including (−)-debromoflustramine B with significant butyrylcholinesterase inhibitory activity, could be synthesized by dearomative prenylation reactions of tryptophan derivatives. However, previously reported dearomative prenylations were limited to batch reactions at the milligram scale, requiring multistep reactions and complex post-processing to obtain the desired natural products. The more efficient synthesis of alkaloids remains challenging, as does the recovery of expensive catalysts. Herein, we developed a chiral heterogeneous Pd nanoparticle (NP) catalyst supported on a polymer, which produces indoline alkaloids in high yields with excellent enantioselectivities. Additionally, the first gram-scale four-step continuous-flow total synthesis of (−)-debromoflustramine B was successfully achieved with this chiral Pd heterogeneous catalyst, requiring only a simple post-processing step.
Introduction
Dearomative prenylation reactions of tryptophan derivatives produce prenylated indoline alkaloids, featuring a skeleton of hexahydropyrrolo[2,3-b]indole with diverse biological activities (Fig. 1A).1–4 Well-known natural products with such a skeleton include flustramines isolated from the bryozoa Flustra foliacea and (−)-debromoflustramine B, which contain significant butyrylcholinesterase inhibitory activity,5–7 potentially offering curative effects for Alzheimer's disease.8 Therefore, the total synthesis of these compounds has received great attention in the field of pharmaceutical chemistry (Fig. 1B). In the past two decades, total synthesis of (−)-flustramine B and (−)-debromoflustramine B has been well developed using three different approaches: (i) asymmetric non-metal catalysis including iminium,9 bisguanidinium,10 chiral phosphoric acid (CPA),11 and phosphine12 catalyzed synthesis; (ii) asymmetric metal catalysis including Ru,13 Ir,14 and Pd15,16 catalyzed synthesis; (iii) chiral auxiliary-assisted synthesis, such as the use of chiral sulfinyl amides.17 Although numerous synthetic methods have been developed,18 these processes are still confined to multistep reactions at a small scale and require tedious post-processing. Furthermore, the recovery of expensive catalysts in the above syntheses remains challenging since homogeneous catalysts were employed.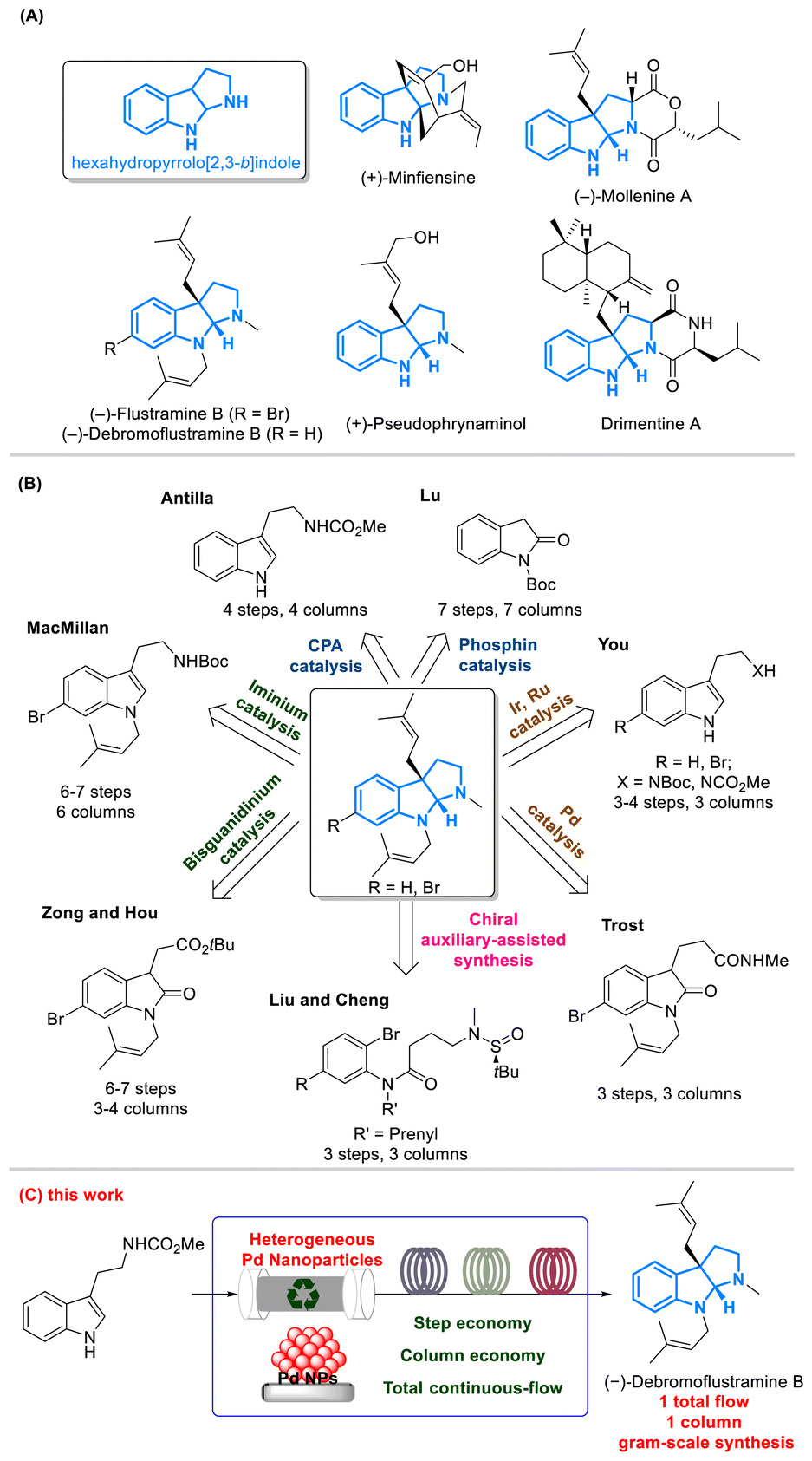 | ||
| Fig. 1 (A) Representative indoline alkaloids with a hexahydropyrrolo[2,3-b] indole skeleton; (B) reported asymmetric syntheses of (−)-debromoflustramine B and (−)-flustramine B; (C) Pd NP enabled multistep continuous-flow total synthesis of (−)-debromoflustramine B. | ||
Combining continuous-flow technology with heterogeneous catalysts provides tremendous advantages over conventional batch reactions in terms of reaction efficiency, scalability, safety, environmental friendliness19–23 and catalyst recovery.24–26 Besides these practical advantages, successful employment of heterogeneous catalysts in packed-bed reactors27,28 under flow processes further demonstrates their merits in line with the green chemistry concept and catalyst recovery. Additionally, the multistep continuous-flow total synthesis can achieve better efficiency by avoiding the tedious post-processing and the isolation of intermediates,29 realizing automated processes in synthetic chemistry. Therefore, developing a heterogeneous catalyst for the synthesis of prenylated indoline alkaloids under continuous-flow conditions holds significant importance in terms of step economy and column economy. This paves the way for feasible industrial-scale production, which benefits the ongoing research and the clinical application of these natural products.
Even though excellent pioneering studies on heterogeneous catalysts have been reported,30–40 cases employing multistep continuous-flow conditions for the synthesis of natural products remain a key challenge. Following our previous work on a chiral heterogeneous Cu NP catalyst,41 we have developed a chiral heterogeneous Pd NP catalyst supported on a polymer. This Pd NP catalyst exhibits excellent durability and maintains its activity compared to its homogeneous counterpart, leading to the production of various indoline alkaloids in high yields with excellent enantioselectivities. In addition, we have accomplished the first four-step continuous-flow total synthesis of (−)-debromoflustramine B on a gram scale (Fig. 1C). Importantly, there is no need for tedious post-processing or isolation of intermediates, as only one column chromatography step is required to obtain the optically pure product.
Results and discussion
Catalyst synthesis and catalyst characterization
The chiral heterogeneous Pd NP catalyst was prepared using a modified procedure described in our previous work.41 The catalyst synthesis began with the installation of a styrene substituent on the BINOL skeleton, leading to the production of the phosphoramidite monomer M1, which was then subjected to simple polymerization conditions to obtain the polymer. Lastly, the Pd NP catalyst was prepared by complexation of the polymer and a homogeneous palladium source (Fig. 2A). The Pd/ligand ratio was found to be approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as confirmed by ICP-OES analysis (Pd, 19.3%, 1.82 mmol g−1; P, 3.09%, 1 mmol g−1) of the Pd NP catalyst.
:1, as confirmed by ICP-OES analysis (Pd, 19.3%, 1.82 mmol g−1; P, 3.09%, 1 mmol g−1) of the Pd NP catalyst.
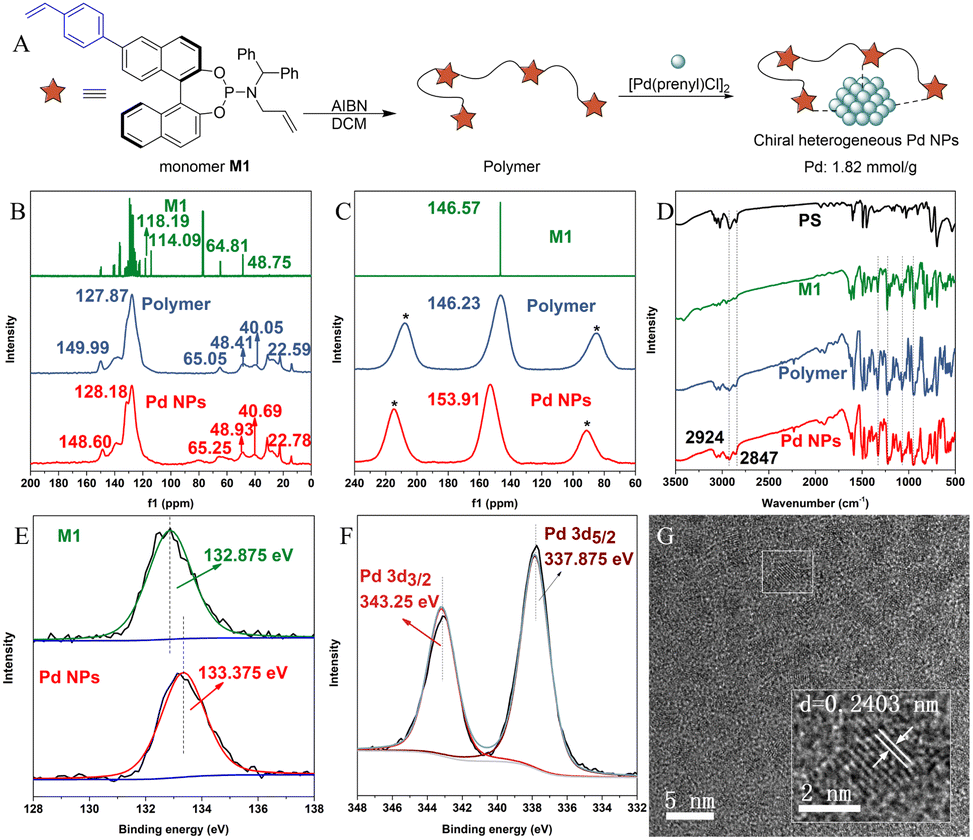 | ||
| Fig. 2 Catalyst synthesis and characterization. (A) Synthesis of the chiral Pd NP catalyst; (B) solid state 13C NMR spectra of M1, the polymer, and Pd NPs; (C) solid state 31P NMR spectra of M1, the polymer, and Pd NPs; (D) FT-IR spectra of PS, M1, the polymer and Pd NPs; (E) P 2p of XPS analysis for M1 and Pd NPs; (F) Rh 3d of XPS analysis for Pd NPs; (G) TEM of Pd NPs. | ||
To confirm and elucidate the interior structure of the heterogeneous Pd NP catalyst, comprehensive characterization studies were performed. The X-ray diffraction (XRD) patterns of the polymer (Fig. S1†) and Pd NPs (Fig. S2†) indicated that both are highly polymerized and amorphous. Connected peaks observed in solid-state 13C NMR spectra ranging from 127.87 to 149.99 ppm for the polymer and 128.18 to 148.60 ppm for the Pd NPs indicated the presence of aromatic rings in both. Similarly, peaks at 65.05/48.41 ppm for the polymer and 65.25/48.93 ppm for Pd NPs suggested the presence of an N-allyl substituent in both. Additionally, the characteristic peaks of the vinyl group in monomer M1 ranging from 110 to 120 ppm disappeared, and strong connected peaks from 22.59 to 40.05 ppm (polymer) and 22.78 to 40.69 ppm (Pd NPs) appeared, suggesting the successful polymerization of monomer M1 and immobilization of the chiral vinylbenzyl-substituted phosphoramidite ligands respectively (Fig. 2B). In solid-state 31P NMR spectra of the polymer and Pd NPs, peaks observed at 146.23 ppm and 153.91 ppm15 were similar to the peak in monomer M1, suggesting that the active centers (phosphorus) of chiral ligands remain stable after polymerization and coordination (Fig. 2C).
In FT-IR spectra, the characteristic absorption bands of polystyrene (PS) at 2924 and 2847 cm−1 were also presented in the polymer and Pd NPs. In addition, similar connected peaks ranging from 900 to 1330 cm−1 in monomer M1 were observed in the polymer and Pd NPs, indicating successful polymerization and coordination. This finding is consistent with the above conclusion (Fig. 2D). To better understand the coordination environment of the Pd NPs, X-ray photoelectron spectroscopy (XPS) analysis was conducted. The spectra showed the successful coordination of the palladium atom to the phosphorus atoms, as supported by the shift (0.5 eV) in binding energies between P 2p of monomer M1 and Pd NPs (Fig. 2E). Moreover, the metal species in Pd NPs was confirmed to be palladium using the spectra of Pd 3d (Fig. 2F).42 Energy dispersive X-ray spectroscopy (EDS) and transmission electron microscopy (TEM) of Pd NPs (Fig. S8†) were also performed to confirm the uniform distribution of the P, S, and Pd elements over the Pd NPs. Clear lattices were found uniformly in the image of TEM of Pd NPs (Fig. 2G), which contain a lattice spacing of 0.2403 nm (2.403 Å), similar to the average Pd–Pd distance43 (2.38 Å) in Pd NPs. In summary, the above characterization studies confirmed the successful and uniform immobilization of Pd atoms with the polymer and the desired coordination environment of the Pd atoms with the ligands.
Activity evaluation
This chiral heterogeneous Pd NP catalyst was first evaluated for enantioselective dearomative prenylation of tryptophan derivatives 1a under batch conditions at a 0.1 mmol scale (Table S1†). It was revealed that this Pd NP catalyst was applicable with excellent immobilization efficiency, enabling the production of desired alkaloid 3a in high yield (95%) with excellent enantioselectivity (91% ee).Substrate scope
With the optimized reaction conditions in hand, we further investigated the scope of the reaction. The Pd NP catalyst was confirmed to have excellent functional group tolerance to a series of tryptophan derivatives in enantioselective dearomative prenylation (Fig. 3). In the case of employing NHCO2Me as the nucleophile, both electron-deficient (Br-substituted) and electron-rich (OMe-substituted) tryptamines tolerated the system and gave products 3b and 3c in good yields (75–97%) with excellent enantioselectivities (89–90% ee). Substrates with different N-protecting groups, such as CO2Et, CO2iPr, tert-butoxycarbonyl (Boc), cyclopentoxycarbonyl and benzyloxycarbonyl (Cbz), also delivered excellent results (3d–3i, 74–99% yields with 90–93% ee). Moreover, using a flavoring agent and pharmaceutically useful L-menthol (–) menthyl oxycarbonyl as the N-protecting group yielded product 3j in 84% yield with 96% ee. Even the natural hormone melatonin and its derivate were readily transformed into the desired products 3k and 3l in excellent yields (88% yield for both) with excellent enantioselectivities (89–90% ee). Additionally, substrates possessing an alcohol nucleophile and a carbon (diester) nucleophile were able to produce the desired products 3m–3o with good yields and enantioselectivities (81–90% yields, 82–91% ee).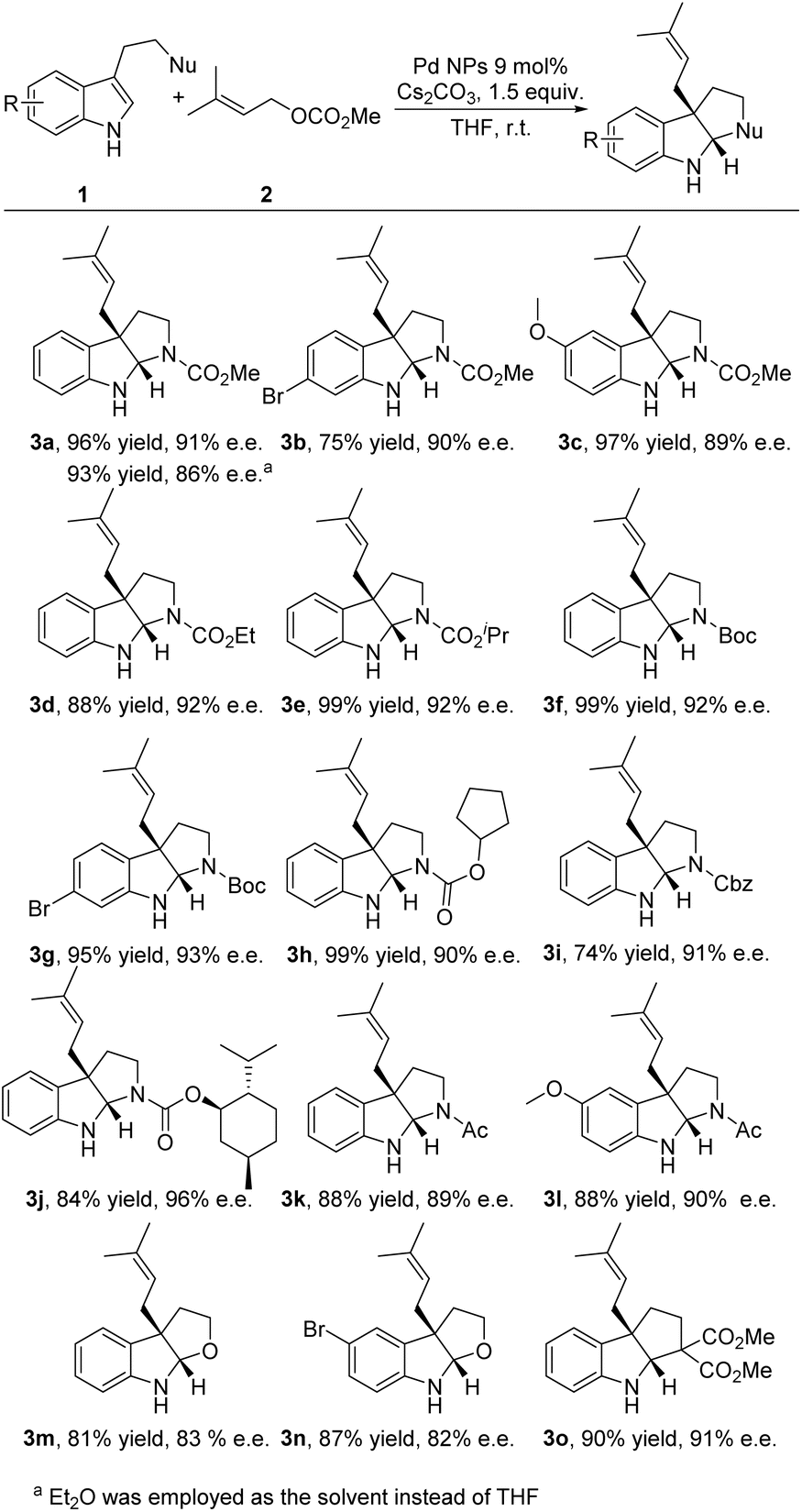 | ||
| Fig. 3 Scope of the Pd NP catalyzed dearomative prenylation. | ||
Multistep continuous-flow
The batch recycling experiment revealed Pd leaching during the catalyst recovery process. This occurred as the aqueous NH4Cl solution removed both Cs2CO3 and Pd from the catalyst (Tables S2 and S3†). Consequently, we believe that the flow process, which eliminates the need for post-processing, could be a more viable solution. To elucidate the versatility of the Pd NP catalyst and develop a more step- and column-economical approach for the synthesis of (−)-debromoflustramine B, a four-step continuous-flow process was developed, which required only one column chromatography step (Fig. 4). Although employing heterogeneous catalysts in packed-bed reactors provides numerous benefits, there are also problems such as system incompatibility in multistep natural product synthesis. To solve this problem, we improved the synthetic circuit by dividing it into four flow pathways. First, tryptophan derivatives 1a (0.6 M in Et2O,44 1 equiv.) and methyl prenyl carbonate 2 (0.9 M in Et2O,44 1.5 equiv.) were pumped into a column packed-bed reactor containing the chiral heterogeneous Pd NP catalyst and Cs2CO3 at the same velocity (15 μL min−1) in flow 1, leading to the production of prenylated indoline alkaloid 3a (Fig. 4), flow 1. Upon generation of 3a, it was premixed with tBuOK (0.45 M in THF, 1.5 equiv., 30 μL min−1) in an FEP tube for 5 min in flow 2 before being mixed in flow 3 with prenyl bromide (0.45 M in THF, 1.5 equiv., 30 μL min−1) to afford the precursor of (−)-debromoflustramine B. Subsequent reduction with LiAlH4 (1 M in THF, 10 equiv., 90 μL min−1) in flow 4 delivered the desired natural product (−)-debromoflustramine B on a gram scale (1.1 g). The product was obtained with an overall yield of 69% and complete retention of the enantiomeric purity (86% ee). It was worth noting that this process would produce solid blockage in the flow path. Therefore, a FEP tube with a large inner diameter was conducive to the process, and ultrasonic treatment was performed in the flow path to further prevent clogging. This four-step continuous-flow total synthesis method not only avoids multistep reactions and post-processing, but also enhances synthesis efficiency through improved step and column economy. Additionally, the catalyst can be easily recovered once the process is completed.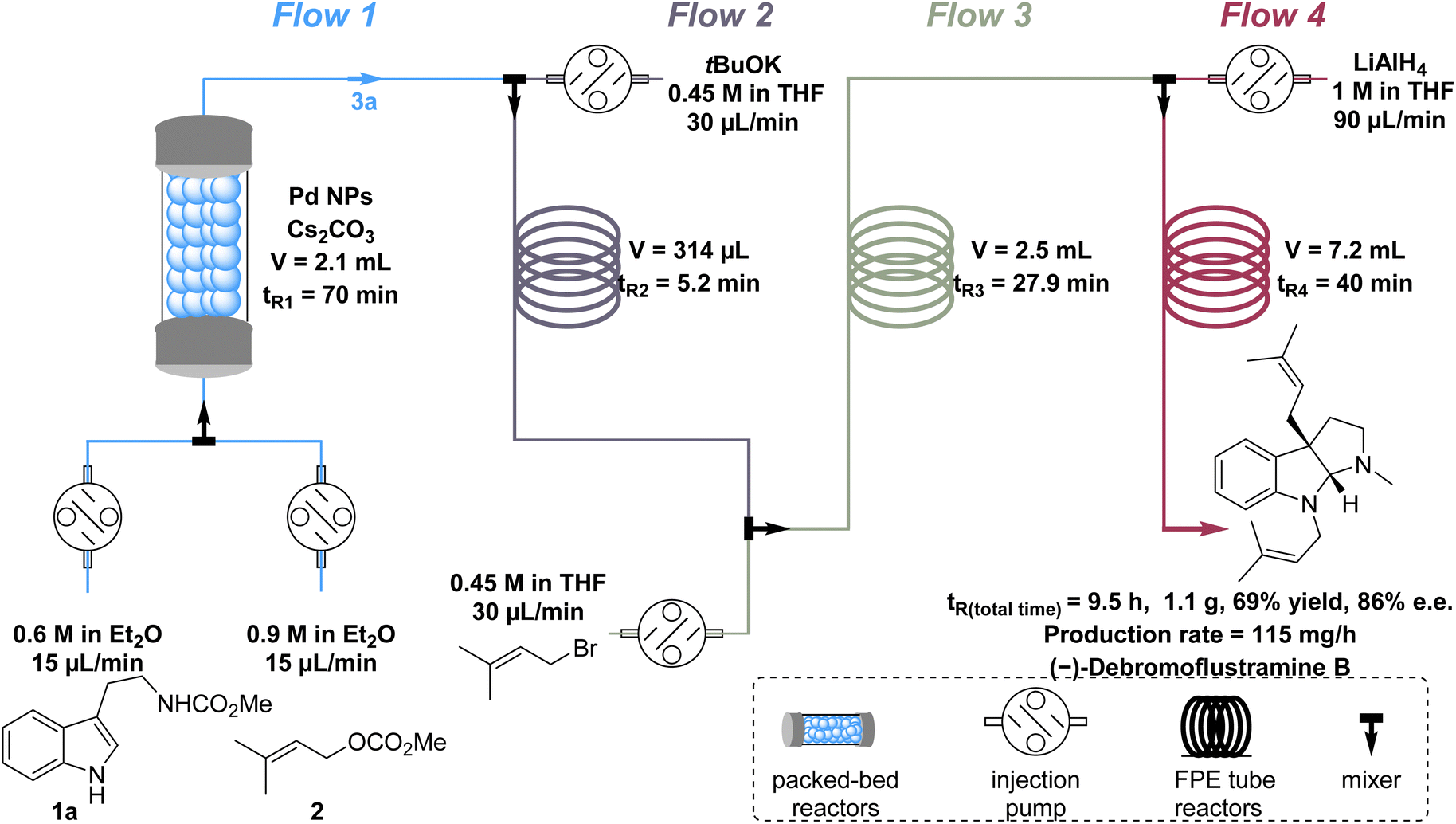 | ||
| Fig. 4 The four-step continuous-flow total synthesis of (−)-debromoflustramine B. | ||
Conclusions
In conclusion, we have elaborated the design and synthesis of an efficient polymer-supported chiral Pd NP catalyst for the enantioselective dearomative prenylation of tryptophan derivatives. The comprehensive characterization studies of the catalyst revealed that it is a highly polymerized matrix with well-distributed active catalytic centers. Meanwhile, the first four-step continuous-flow total synthesis of (−)-debromoflustramine B on a gram scale was successfully achieved, producing the desired product in 69% yield with 86% ee over 4 steps. Notably, the process requires only one multistep continuous-flow process and one column chromatography step, compared to the previously reported 3–7-step synthesis with at least 3 column chromatography steps. This work demonstrates the potential of developing an efficient heterogeneous catalyst for asymmetric catalysis, and its application in continuous flow systems offers a more efficient and industrially friendly solution for large-scale natural product synthesis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. W. and F. L. conducted the experiments and prepared the ESI;† Z. D. helped synthesize catalysts. J. H. contributed to the discussion. J. W. and Y. Z. designed the experiments and wrote the manuscript. H. Y. and F. C. directed the project. All the authors contributed to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Shenzhen Science and Technology Research Fund No. JSGG20201103153807021; GXWD20220811173736002; KCXFZ20230731094904009. We are also grateful to the Natural Science Foundation of Guangdong Province (No.2021A1515110366), the National Natural Science Foundation of China (No. 22302048; No. 82204231), the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (No. 24qnpy060) and Shenzhen Key Laboratory of Advanced Functional Carbon Materials Research and Comprehensive Application.Notes and references
- T. Lindel, N. Marsch and S. K. Adla, in Alkaloid Synthesis, ed. H.-J. Knölker, Springer, Berlin, Heidelberg, 2012, pp. 67–129 Search PubMed.
- S.-M. Li, Nat. Prod. Rep., 2009, 27, 57–78 RSC.
- R. M. Williams, E. M. Stocking and J. F. Sanz-Cervera, in Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, eds. F. J. Leeper and J. C. Vederas, Springer, Berlin, Heidelberg, 2000, pp. 97–173 Search PubMed.
- Y.-Z. Liu, H. Song, C. Zheng and S.-L. You, Nat. Synth., 2022, 1, 203–216 CrossRef.
- P. B. Holst, U. Anthoni, C. Christophersen and P. H. Nielsen, J. Nat. Prod., 1994, 57, 997–1000 CrossRef CAS.
- J. S. Carle and C. Christophersen, J. Am. Chem. Soc., 1979, 101, 4012–4013 CrossRef CAS.
- E. Rivera-Becerril, P. Joseph-Nathan, V. M. Pérez-Álvarez and M. S. Morales-Ríos, J. Med. Chem., 2008, 51, 5271–5284 CrossRef CAS PubMed.
- S. Zhou and G. Huang, Biomed. Pharmacother., 2022, 146, 112556 CrossRef CAS.
- J. F. Austin, S.-G. Kim, C. J. Sinz, W.-J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482–5487 CrossRef CAS.
- X. Zuo, Z. Wan, Z. Liao, C. Wang, C.-H. Tan, S. Huo and L. Zong, ACS Catal., 2023, 13, 15708–15714 CrossRef CAS.
- Z. Zhang and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 11778–11782 CrossRef CAS PubMed.
- X. Tang, H. Ni and Y. Lu, Org. Chem. Front., 2021, 8, 4485–4489 RSC.
- X. Zhang, Z.-P. Yang, C. Liu and S.-L. You, Chem. Sci., 2013, 4, 3239–3243 RSC.
- X. Zhang, L. Han and S.-L. You, Chem. Sci., 2014, 5, 1059–1063 RSC.
- H.-F. Tu, X. Zhang, C. Zheng, M. Zhu and S.-L. You, Nat. Catal., 2018, 1, 601–608 CrossRef CAS.
- B. M. Trost, S. Malhotra and W. H. Chan, J. Am. Chem. Soc., 2011, 133, 7328–7331 CrossRef CAS PubMed.
- X. Shen, T. Peng, F. Wang, S. Li, X. Lei, W. Yunxiao, F. Cheng and T. Liu, ChemistrySelect, 2021, 6, 5195–5197 CrossRef CAS.
- C. Zheng and S.-L. You, Nat. Prod. Rep., 2019, 36, 1589–1605 RSC.
- J. Liao, S. Zhang, Z. Wang, X. Song, D. Zhang, R. Kumar, J. Jin, P. Ren, H. You and F.-E. Chen, Green Synth. Catal., 2020, 1, 121–133 CrossRef.
- J. Britton and T. F. Jamison, Angew. Chem., Int. Ed., 2017, 56, 8823–8827 CrossRef CAS PubMed.
- C. Liu, J. Xie, W. Wu, M. Wang, W. Chen, S. B. Idres, J. Rong, L.-W. Deng, S. A. Khan and J. Wu, Nat. Chem., 2021, 13, 451–457 CrossRef CAS PubMed.
- B. Gutmann, J.-P. Roduit, D. Roberge and C. O. Kappe, Angew. Chem., Int. Ed., 2010, 49, 7101–7105 CrossRef CAS.
- T. Tsubogo, T. Ishiwata and S. Kobayashi, Angew. Chem., Int. Ed., 2013, 52, 6590–6604 CrossRef CAS.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- Y. Saito and S. Kobayashi, Angew. Chem., Int. Ed., 2021, 202112797 Search PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS.
- S. Cañellas, C. Ayats, A. H. Henseler and M. A. Pericàs, ACS Catal., 2017, 7, 1383–1391 CrossRef.
- J. Lai, M. Fianchini and M. A. Pericàs, ACS Catal., 2020, 10, 14971–14983 CrossRef CAS.
- T. Tsubogo, H. Oyamada and S. Kobayashi, Nature, 2015, 520, 329–332 CrossRef CAS.
- T. Kuremoto, T. Yasukawa and S. Kobayashi, Adv. Synth. Catal., 2019, 361, 3698–3703 CrossRef CAS.
- T. Kuremoto, R. Sadatsune, T. Yasukawa and S. Kobayashi, ACS Catal., 2021, 14026–14031 CrossRef CAS.
- Y. Saito and S. Kobayashi, Angew. Chem., Int. Ed., 2024, 63, e202313778 CrossRef CAS PubMed.
- Y. Otomaru, T. Senda and T. Hayashi, Org. Lett., 2004, 6, 3357–3359 CrossRef CAS.
- G. Shen, T. Osako, M. Nagaosa and Y. Uozumi, J. Org. Chem., 2018, 83, 7380–7387 CrossRef CAS.
- W.-Y. Huang, G.-Q. Wang, W.-H. Li, T.-T. Li, G.-J. Ji, S.-C. Ren, M. Jiang, L. Yan, H.-T. Tang, Y.-M. Pan and Y.-J. Ding, Chem, 2020, 6, 2300–2313 CAS.
- V. B. Saptal, V. Ruta, M. A. Bajada and G. Vilé, Angew. Chem., Int. Ed., 2023, 62, e202219306 CrossRef CAS PubMed.
- W.-H. Li, J. Yang, D. Wang and Y. Li, Chem, 2022, 8, 119–140 CAS.
- M. A. Bajada, J. Sanjosé-Orduna, G. D. Liberto, S. Tosoni, G. Pacchioni, T. Noël and G. Vilé, Chem. Soc. Rev., 2022, 51, 3898–3925 RSC.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, M. A. Ortuño, N. López, S. M. Collins, P. A. Midgley, S. Richard, G. Vilé and J. Pérez-Ramírez, Nat. Nanotechnol., 2018, 13, 702–707 CrossRef CAS PubMed.
- H. Ishitani, K. Kanai, W. Yoo, T. Yoshida and S. Kobayashi, Angew. Chem., Int. Ed., 2019, 58, 13313–13317 CrossRef CAS PubMed.
- J. Wang, J. Li, Y. Wang, S. He, H. You and F.-E. Chen, ACS Catal., 2022, 12, 9629–9637 CrossRef CAS.
- P. Brant, L. S. Benner and A. L. Balch, Inorg. Chem., 1979, 18, 3422–3427 CrossRef CAS.
- G. Liu, W. Zhou, Y. Ji, B. Chen, G. Fu, Q. Yun, S. Chen, Y. Lin, P.-F. Yin, X. Cui, J. Liu, F. Meng, Q. Zhang, L. Song, L. Gu and H. Zhang, J. Am. Chem. Soc., 2021, 143, 11262–11270 CrossRef CAS.
- Due to the gradual dissolution of Pd NPs in THF, Et2O was employed in the multistep continuous flow synthesis..
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03471f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |