
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA structurally conserved helix enables leader-independent tyramine splicing of proteins†
Daniel
Richter
 ,
Alicia
Courvoisier-Clément
,
Anna Lisa
Vagstad
,
Sarolt
Magyari
and
Jörn
Piel
*
,
Alicia
Courvoisier-Clément
,
Anna Lisa
Vagstad
,
Sarolt
Magyari
and
Jörn
Piel
*
Institute of Microbiology, Eidgenössische Technische Hochschule (ETH) Zurich, Vladimir-Prelog-Weg 4, 8093 Zurich, Switzerland. E-mail: jpiel@ethz.ch
First published on 18th September 2024
Abstract
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are natural products that feature diverse modifications. They show a wide range of biological activities and are therefore of great interest for drug discovery and protein engineering. An unusual modification found in spliceotide RiPPs is the installation of β-amino acid residues with diverse side chains, generated by backbone excision of a tyramine moiety derived from tyrosine. We have previously shown that the modification can be adapted to protein engineering to greatly expand the set of amino acid residues and to introduce unique reaction centers for site-directed modification. To understand requirements for splicease–substrate interactions, we investigated the role of a RiPP recognition element (RRE) in spliceotide biosynthesis and provide evidence that it acts as an activator and enables leader-independent protein splicing. We leveraged this knowledge to engineer a simplified splicease system derived from Rheinheimera aquimaris B26 that processes splice tags introduced into proteins with high efficiency. This work expands the toolbox for peptide and protein engineering and contributes to an understanding of substrate recognition in RiPP biosynthesis.
Introduction
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a diverse class of natural products with antibiotic, anticancer, antifungal, antiviral, and other activities.1,2 RiPP biosynthesis involves a precursor that, with some exceptions, consists of an N-terminal leader and a C-terminal core section.2,3 Miscellaneous maturase enzymes install post-translational modifications (PTMs) in the core, a process that is commonly initiated by binding to the N-terminal leader. In many bacterial RiPPs, these interactions are mediated by an additional small, <100-residue peptide or domain called RiPP recognition element (RRE).4 RREs display high sequence divergence but in general share (partial) structural similarity to PqqD, which mediates the interaction between the precursor PqqA and the maturase PqqE in pyrroloquinoline quinone biosynthesis.5 Due to their presence in diverse RiPP biosynthetic gene clusters, RRE genes have been used as a bioinformatic tool for natural product discovery.6Spliceotides are a remarkable class of RiPPs that contain various α-keto-β-amino acid residues as a characteristic feature, in the following termed ketoamides.7 The ketoamide moiety is an important pharmacophore in natural and synthetic compounds of therapeutic interest, often in the context of protease inhibition.8 Examples of well-known (ribosome-independent) ketoamide compounds include the immunosuppressants rapamycin9 and FK506,10 the anticancer peptide aplidine,11 and HIV protease inhibitors such as the chloropeptins.12,13 In accordance, we found several tested spliceotides to exhibit potent protease–inhibitory activities.14 This property as well as the extraordinary introduction of diverse β-amino acids in gene-encoded products make the spliceotide modification an attractive reaction for drug development and synthetic biology.
The ketoamide residue is installed by a radical S-adenosyl methionine (rSAM) enzyme — the “X protein”, such as PlpX — that post-translationally excises a tyramine equivalent at XYG sequence motifs in precursors — the “A proteins” (Fig. 1a). In the initially discovered cyanobacterial spliceotide pathways,7 the X enzyme required an additional small protein — the “Y protein” — for activity. Y proteins share distant sequence similarity to PqqD and were therefore suspected to be an RRE, but their exact function was unknown. We showed that, contrary to conventional RiPP biosynthesis, PlpX does not require the leader for substrate modification.15 This enabled us to adapt the RiPP modification system to protein engineering: Introducing a core-derived splice tag at internal or terminal protein positions and co-production in vivo with the PlpXY splicease system provides access to the β-amino acid-containing proteins (Fig. 1b). This work was recently expanded by using the ketoamide group as a molecular handle for site-selective protein conjugation.16
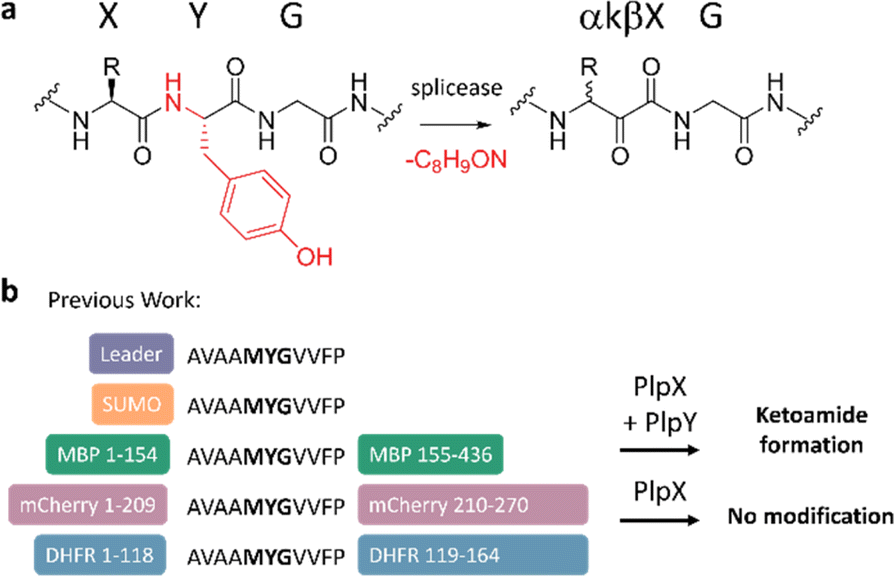 | ||
| Fig. 1 (a) Net splicing reaction. A tyramine equivalent (marked in red) is excised from the peptide backbone through enzymatic catalysis, forming an α-keto-β-amide (αkβX). (b) Leader-independent splicing of protein substrates by PlpX in the presence of PlpY. SUMO is small-ubiquitin-like modifier, MBP maltose binding protein, and DHFR dihydrofolate reductase. | ||
The observed leader-independence questioned our initial hypothesis that Y acts as an RRE to mediate binding between the X protein and the leader of the substrate. More recent work by our group has expanded the spliceotide family beyond the cyanobacterial phylum, revealing hundreds of pathways in diverse bacteria, which have been categorized into types based on the phylogeny of the rSAM splicease and the identity of the precursor protein.14 These all lack the Y protein that is present and essential for activity in the ternary cyanobacterial systems. The architectural simplicity and high sequence diversity makes these two-component pathways attractive candidates for protein engineering and library-based drug discovery, but an understanding of factors important for substrate recognition is required.
In this work, we present insights into the role of the Y protein in ternary splicease systems and into substrate requirements in binary pathways. This knowledge was then applied to eliminate the leader dependence of a binary system, providing a simplified, single-component splicease platform for protein modification with high splicing efficiency.
Results and discussion
PlpY and PcpY are the Y proteins of the initially reported type I cyanobacterial spliceotide pathways from Pleurocapsa spp. PCC 7319 and PCC 7327, respectively. To understand their roles, we tested a structural approach. Typical RREs share structural homology and only little sequence similarity.4 As we were not yet able to solve an experimental structure for a spliceotide protein, we resorted to computational predictions through Alphafold2.17,18 We predicted the ternary structure of PcpAXY and PlpAXY complexes and compared these to the type II binary RhaXA complex from Rheinheimera aquimaris B26, which does not contain a Y protein (Fig. 2a–c) and showed near-quantitative splicing in Escherichia coli co-production experiments.19 Several features stood out from the predictions: In the modeled structures, the XYG splice sites were positioned in the active center in close vicinity to the putative SAM binding pockets (Fig. 2e). This was surprising to us, as there is no experimental structure for these systems that the predictions could build on. Remarkably, for PcpA that contains two splice sites, AlphaFold provided structures with both sites modeled into the active center (Fig. S1†). The overall fold of the splicease was suggested to be a partial TIM barrel, typical for rSAM enzymes.20,21 Furthermore, the Y protein seems to bind both the splicease as well as the leader, in agreement with the function of RREs as mediators of maturase–leader interactions. We confirmed complex formation of PcpX, PcpY and PcpA by analytical high-performance size exclusion chromatography, where we detected a retention time shift for PcpXY incubated with PcpA, but not for PcpX alone with PcpA (Fig. 2e), indicating that PcpY mediates the interaction between PcpX and PcpA. Most importantly though, the Y protein of the modeled ternary complex binds with an α-helical region to a splicease site that is occupied in the binary Rha system by an α-helix of the RhaA leader. Not only were we able to see structural similarity of Y proteins to the N-terminal part of the RhaA leader, but similarity can also be detected in multiple sequence alignments of the Y proteins with regions homologous to the RhaA precursor and closely related spliceotide leader sequences (Fig. S2–S4†).14 Without the Y protein, PlpA3 and PcpA do not form these helices (Fig. S5†). As these features suggested shared functional roles of the splicease components, we wished to experimentally interrogate their relationship.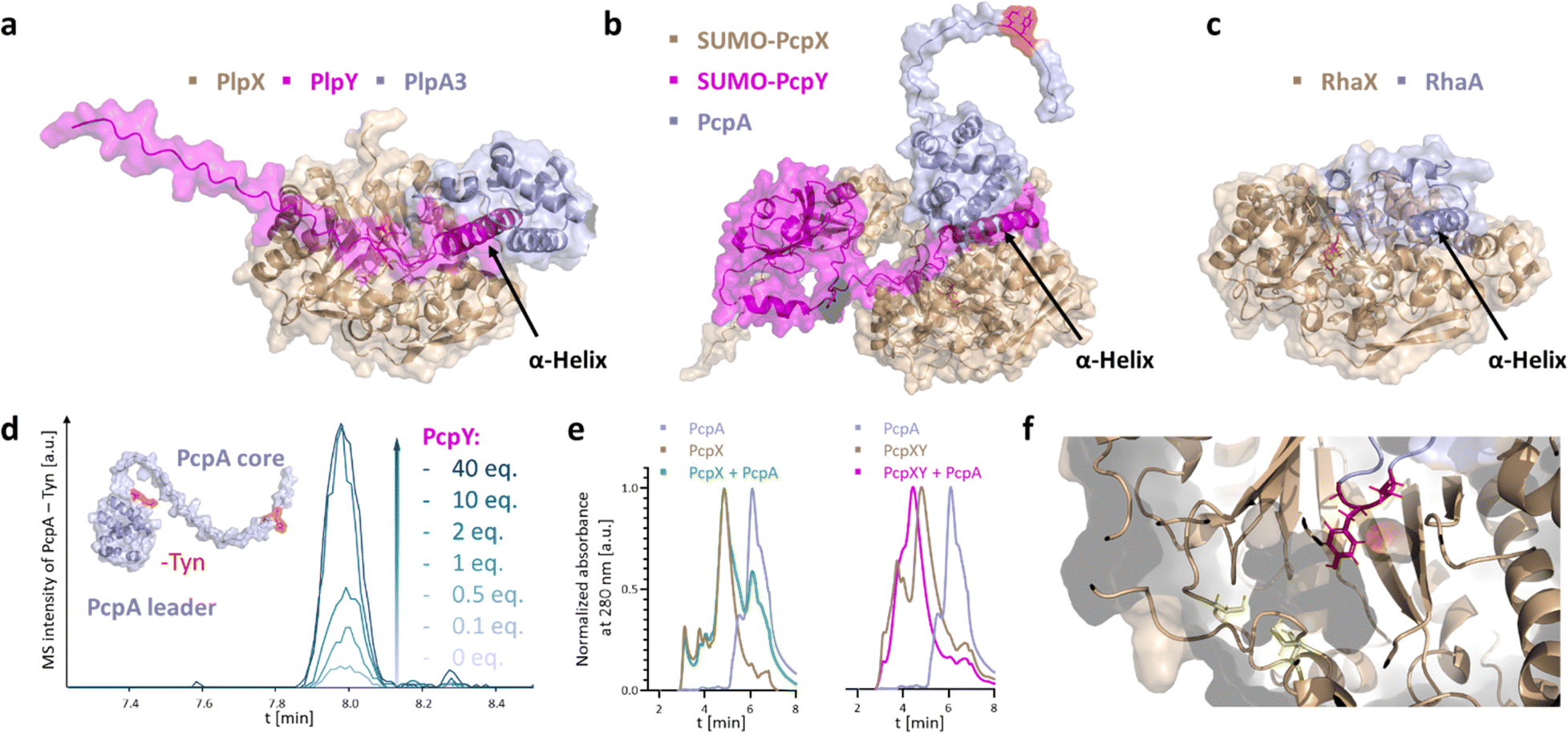 | ||
| Fig. 2 AlphaFold2 structure predictions of (a) PlpX (wheat), PlpY (magenta), and PlpA3 (light blue); (b) SUMO-PcpX (wheat), SUMO-PcpY (magenta), and PcpA (light blue); and (c) RhaX (wheat) and RhaA (light blue). Precursor and splicease are predicted to interact in all cases and the XYG site in the prediction is placed in the putative active site. PlpY and PcpY form an α-helical structure at a homologous splicease binding site as an α-helical structure of the RhaA leader sequence. (d) Extracted ion chromatograms (Trypsin and GluC fragment of PcpA with one excised tyramine from the N-terminal MYG site; m/z = 994.5303 [M + 3H]3+) of splicease reaction product for increasing relative concentrations of PcpY in in vitro reactions, showing PcpY is required for in vitro activity of PcpX towards PcpA. The red regions in the modeled PcpA structure are the two splice sites. (e) Analytical high-performance size exclusion chromatography data showing a retention time shift for PcpXY + PcpA but not PcpX + PcpA. (f) Structural model of PcpA and SUMO-PcpX with the N-terminal MYG site (red) placed in the putative active site near the putative iron-binding cysteines (yellow). | ||
We first investigated how the stoichiometry between PcpX and PcpY affects the activity of the splicease. PcpX and PcpY were purified as SUMO-tagged versions (for solubility reasons) and tested in in vitro reactions with PcpA, the native substrate. The iron–sulfur clusters of SUMO-PcpX were reconstituted with the cysteine desulfurase IscS before the reactions.22,23 Assays were carried out at room temperature for 3 h with E. coli FldA and Fpr as a redox regeneration system.22,23 Product formation was monitored by liquid chromatography coupled to mass spectrometry (LC-MS) after digestion of the precursor peptide with trypsin and GluC. The data showed that the addition of more than 1 equivalent of SUMO-PcpY in relation to the splicease increases conversion to the ketoamide product (Fig. 2d and S6†). As observed previously,7 we detected an additional peak with a mass that corresponds to the hydrate of the ketoamide. No further rate increase was observed above a 10-fold excess of PcpY. It cannot be excluded that the N-terminal SUMO tag causes an unbalanced stoichiometry by reducing affinity due to steric reasons. However, trials to study untagged PcpY failed, as the native protein was insoluble and aggregated upon purification. Nonetheless, this indicated to us that SUMO-PcpY does increase conversion to the product in a concentration-dependent manner well above equimolar concentrations to SUMO-PcpX.
To explore whether a ternary splice complex can be converted to a binary system, we tested if individual proteins can be fused to retain activity. Due to the higher enzymatic conversion in E. coli co-expressions, we performed in vivo experiments with PlpAXY instead of the pcp system. Natural RRE fusions exist in several RiPP biosynthetic pathways. For example, fusions were reported at the N-terminus of PqqD (in Methylobacterium extorquens AM1, oxidase fusion) or its C-terminus (Methylocystis sp. SC2, rSAM fusion).5 Considering the predicted structural similarity of the Y protein to a portion of the RhaA leader, we tested precursor variants in which the N-terminal part of the PlpA3 leader was exchanged against PlpY. Fusions were designed with the help of structure predictions, resulting in two variants, PlpYA3-33 and PlpYA3-45, which contain the sequence of PlpY in place of the first 33 or 45 amino acids of the PlpA3 leader sequence, respectively (Fig. 3a–c and S7–S9†). Both variants were tested by heterologous co-production in E. coli BL21(DE3) with PlpX and in the absence of PlpY. After purification by nickel-affinity chromatography and tryptic digestion, we detected modification on both fusion peptides, but not on the native substrate. This showed again that Y is needed, but that Y can also replace the leader sequence for activity.
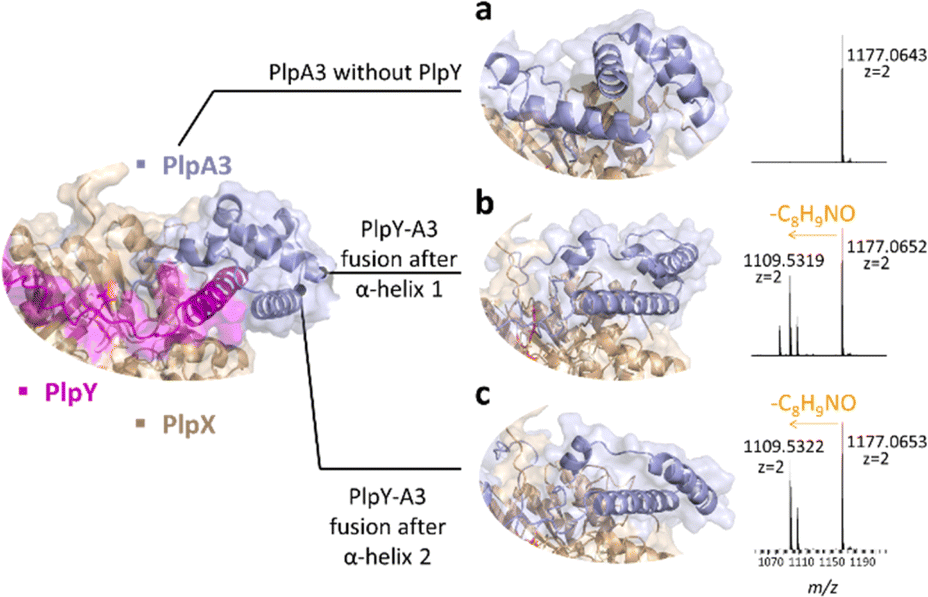 | ||
| Fig. 3 Fusion of PlpY to PlpA3. (a) AlphaFold prediction of the interface between PlpA3 (light blue) and PlpX (wheat) in the absence of PlpY (magenta) and mass spectrum showing lack of activity. (b) Structure prediction of PlpYA3-33 (light blue) and mass spectrum showing tyramine excision. (c) Structure prediction of PlpYA3-45 (light blue) and mass spectrum showing tyramine excision. | ||
Instead of fusing the PlpA3 precursor and PlpY, we next tested C-terminal fusions of PlpY to PlpX (Fig. 4). Fusions of RiPP leader sequences to the respective maturases have been used to accomplish leader-independent thiazoline, oxazoline, and thiazole formation, as well as lanthionine and microviridin/graspetide cyclization, termed constitutively active fusions (ConFusions).24–29 These fusions have also enabled the modification of proteins such as mCherry by RiPP heterocyclases.30 Fusions for leader-independent modification have been proposed as an alternative to chimeric leaders31 or leader peptide exchange32 for RiPP engineering. However, to our knowledge, RRE engineering strategies have not yet been employed although natural fusions of RREs and maturases are known.4,5,33 We therefore hoped that it was possible to design a similar system by fusing the RRE to the maturase. Structure predictions suggested that C-terminal fusions of Y proteins to the X proteins should bind the same interface on the X proteins without the need for a linker sequence (Fig. 4b, top).
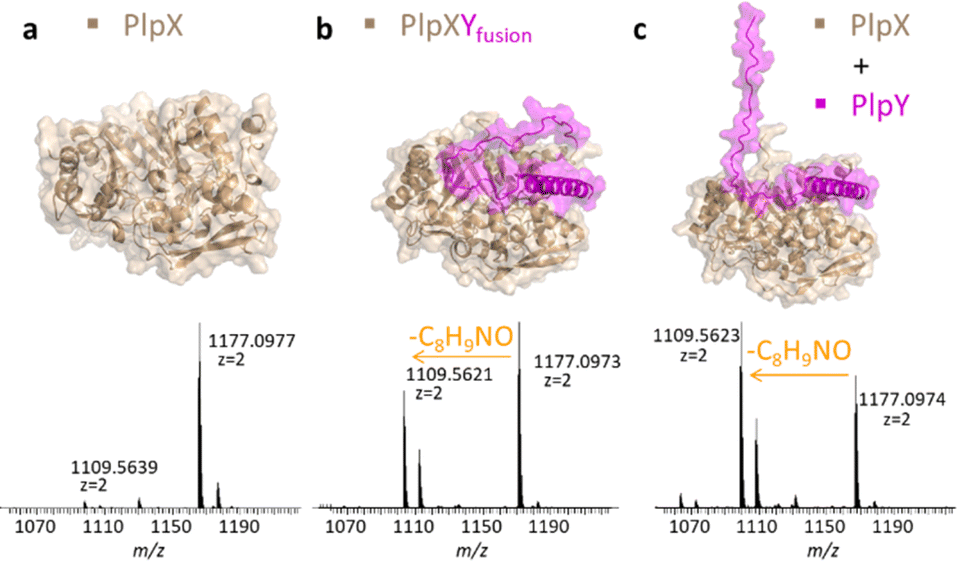 | ||
| Fig. 4 PlpXY fusion experiments. (a) Structure prediction of PlpX (wheat) and mass spectrum showing only traces of tyramine excision from the tryptic fragment of PlpA3. (b) Structure prediction of PlpXYfusion (wheat/magenta) and mass spectrum showing efficient tyramine excision from the tryptic fragment of PlpA3. (c) Structure prediction of PlpX (wheat) and PlpY (magenta) and mass spectrum showing efficient tyramine excision from the tryptic fragment of PlpA3. | ||
The fusion was termed PlpXYfusion for the plp system. We also produced SUMO-PcpXYfusion for the pcp system. When PlpXYfusion was tested by heterologous co-production in E. coli BL21(DE3) with the native PlpA3 substrate, we again saw modification of the substrate by LC-MS after nickel-affinity purification and tryptic digestion. The extent was similar to that of PlpX and PlpY co-production (Fig. 4a–c and S10†). In addition, we performed in vitro reactions for SUMO-PcpXYfusion with PcpA as a substrate, which showed comparable substrate modification as non-fused SUMO-PcpXY (Fig. S11†). Taken together with the leader-independence of PlpXY, this indicated that PlpY acts as an activator of PlpX.
Finally, we went back to our initial structure predictions that suggested similar binding of the Rheinheimera RhaA leader as compared to PlpY and PcpY. The structural and sequence similarities suggested the possibility that the RhaA leader is functionally analogous to the Y proteins. Asking whether the leader acts as an activator rather than a recognition element that guides the core peptide to the maturase, we fused the RhaA leader to the RhaX C-terminus similarly as for the PlpX-PlpY fusion (Fig. 5b and S12†). This fusion enzyme RhaXAleader was then tested for activity on the leaderless RhaA core in E. coli. Based on previously reported leader-independent modification of proteins by PlpXY,15 we introduced the RhaA core sequence as a splice tag into a diverse set of proteins (Fig. 5a): (1) small-ubiquitin-like modifier (SUMO) protein at either the N- or the C-terminus, (2) maltose binding protein (MBP) at the C-terminus or an internal loop at position 154, (3) dihydrofolate reductase (DHFR) at the N-terminus or an internal loop at position 118, and (4) mCherry at an internal loop at position 209.
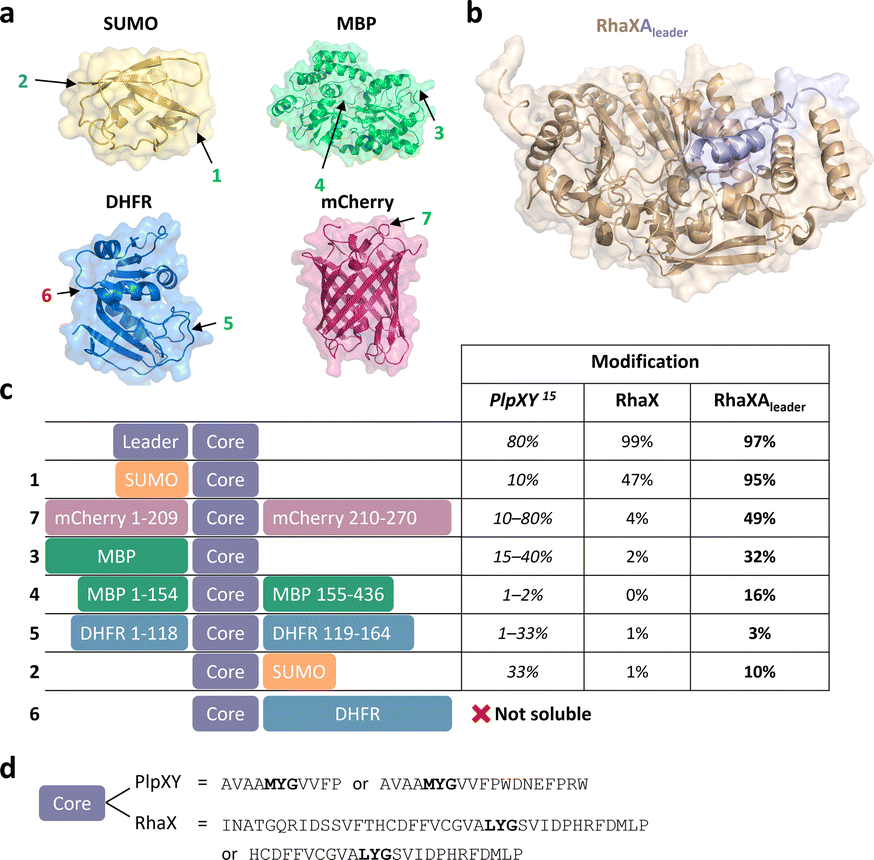 | ||
| Fig. 5 Engineering of a Y protein-independent protein splicing system. (a) Crystal structures of SUMO (yellow, PDB 4BKG), MBP (green, PDB 1MPD), DHFR (blue, PDB 1RF7), and mCherry (pink, PDB 2H5Q). Splice tag insertion sites are marked with an arrow and number and correspond to constructs in (c). (b) Alphafold2 prediction of the RhaXAleader (wheat/light blue) with the leader sequence of RhaA fused to the C-terminus of RhaX marked in light blue. (c) Constructs with the splice tag co-expressed either with PlpXY (data from Lakis et al.),15 RhaX or RhaXAleader. (d) Comparison of tag sequences between the plp and the rha systems. | ||
Gratifyingly, in all cases the fused RhaXAleader maturase modified the inserted cores with substantially higher conversion than RhaX and without requiring a substrate-fused leader (Fig. 5c). For comparison, the native RhaX splicease only converted ∼50% of the SUMO-RhaAcore protein (Fig. S13 and S14†) and only traces of mCherry209-RhaAcore (Fig. S15 and S16†) in a completely leader-independent manner, whereas the RhaXAleader splicease efficiently modified both substrates to yield the ketoamide-containing proteins. Interestingly, the modification rate for the SUMO-RhaAcore varied over time (Fig. S17†). RhaXAleader efficiently modified SUMO-RhaAcore after 4.5 h, 24 h, and 40 h of co-expression. However, RhaX showed efficient modification after 4.5 h but inefficient modification after 24 h, potentially suggesting low turnover or stability of the enzyme as limiting factors. Additionally, we achieved modification of the C-terminal-core fused MBP variant, the internal core MBP variant, the internal core DHFR variant and the N-terminal core SUMO variant (Fig. 5c and S18–S23†). For the N-terminally tagged DHFR variant we were unable to obtain soluble protein for the unmodified peptide (Fig. S21 and 23†). To investigate residues essential for recognition by the maturase, we performed truncation studies on the SUMO-RhaAcore protein. Truncated versions missing the internal T7 tag as well as the N-terminal 16 amino acids of the core were accepted by RhaXAleader (Fig. S24 and S25†), while a variant missing the N-terminal 24 amino acids of the core was not (Fig. S26†). Variants lacking the N-terminal 16 amino acid residues and C-terminal 4, 8, or 10 amino acids were not modified by the enzyme (Fig. S27–S29†). These results suggest a 25 amino acid core as a possible tag sequence. The engineered leader-independent modification by RhaXAleader is thus a simplified and binary protein splicing system that we obtained through structural analysis of the Y-proteins.
Conclusions
In this study we show that PlpY and PcpY act as activators of their respective enzymes PlpX and PcpX. Starting out from structure predictions, we realized that the Y proteins most likely bind the enzyme and the leader of substrates. We successfully introduced PlpY as a leader replacement into PlpA3 and showed efficient conversion with no additional PlpY required. By generating the fusion constructs PlpXYfusion and PcpXYfusion, we were able to combine the two proteins into single fully functional enzymes that modified substrates efficiently. Taken together with the previously reported leader-independence of PlpX, the data support PlpY to be an activator of PlpX that confers activity upon binding. These results are in agreement with insights from a recent study on lasso peptides that RREs confer activation functions in addition to binding.33 It will be worthwhile exploring whether the strategy of fusing enzymes and RREs can be used generally to facilitate engineering of RiPP pathways.Through a combination of structural predictions and rational protein engineering, we saw that the leader of the type II rha spliceotide biosynthetic gene cluster, which lacks a discrete Y protein, is predicted to bind the same site as the Y proteins of type I spliceotides through an α-helix. We leveraged our newly discovered knowledge of this Y-like structural element in the RhaA leader to develop the X-leader fusion RhaXAleader. The engineered fusion was able to modify leaderless protein substrates containing the RhaAcore sequence and much more efficiently than native RhaX. We were able to modify SUMO, MBP, DHFR, and mCherry at N-terminal, C-terminal, and internal loop regions to incorporate ketoamides. RhaXAleader in some cases provided higher conversion rates than previously characterized PlpXY for their cognate core-derived splice tags grafted into the same host proteins; although, it is important to note that we used a longer core sequence for RhaA relative to PlpA3. The remarkable ability of the binary Rha system to function with increased efficiency when the leader was relocated from the native precursor protein to the C-terminus of the RhaX splicease suggests that the native leader predominantly acts as an activator rather than merely a recognition element, a property that might also apply to other RREs.
With the proposed enzyme–leader fusions we aimed to apply protein splicing in a more efficient way and expanded splicing sequences. The knowledge gained here opens up a much larger toolbox of available splicease enzymes to install ketoamides in protein substrates and for drug development, with only a single protein required for modification. This approach expands the available enzymes for incorporation of non-canonical protein functionalities through RiPP maturases.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. R., A. C.-C., A. L. V., and J. P. designed the research and experiments. D. R. performed computational modelling, molecular cloning, in vivo and in vitro protein studies. A. C.-C. and M. S. performed molecular cloning and characterization of proteins tagged with the RhaA core. A. L. V. performed size-exclusion studies. D. R. and J. P. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank T. A. Scott and E. Lakis for plasmids. We thank A. O. Brachmann for assistance with Mass Spectrometry experiments. J. P. is grateful for funding from the European Research Council under the European Union's Horizon 2020 programme (742739, SynPlex), the Promedica Foundation (Project 1-001369-000), and the Swiss National Science Foundation (SNF project 205320_219638).References
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H. G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J. K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- M. A. Ortega and W. A. van der Donk, Cell Chem. Biol., 2016, 23, 31–44 CrossRef CAS PubMed.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS PubMed.
- J. A. Latham, A. T. Iavarone, I. Barr, P. V. Juthani and J. P. Klinman, J. Biol. Chem., 2015, 290, 12908–12918 CrossRef CAS PubMed.
- M. Kloosterman Alexander, E. Shelton Kyle, P. van Wezel Gilles, H. Medema Marnix and A. Mitchell Douglas, mSystems, 2020, 5, e00267 Search PubMed.
- B. I. Morinaka, E. Lakis, M. Verest, M. J. Helf, T. Scalvenzi, A. L. Vagstad, J. Sims, S. Sunagawa, M. Gugger and J. Piel, Science, 2018, 359, 779–782 CrossRef CAS PubMed.
- M. Robello, E. Barresi, E. Baglini, S. Salerno, S. Taliani and F. D. Settimo, J. Med. Chem., 2021, 64, 3508–3545 CrossRef CAS PubMed.
- C. Vézina, A. Kudelski and S. N. Sehgal, J. Antibiot., 1975, 28, 721–726 CrossRef PubMed.
- H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto and T. Taga, J. Am. Chem. Soc., 1987, 109, 5031–5033 CrossRef CAS.
- J. Lee, J. N. Currano, P. J. Carroll and M. M. Joullié, Nat. Prod. Rep., 2012, 29, 404–424 RSC.
- K. Matsuzaki, H. Ikeda, T. Ogino, A. Matsumoto, H. B. Woodruff, H. Tanaka and S. Omura, J. Antibiot., 1994, 47, 1173–1174 CrossRef CAS PubMed.
- H. Tanaka, K. Matsuzaki, H. Nakashima, T. Ogino, A. Matsumoto, H. Ikeda, H. B. Woodruff and S. Omura, J. Antibiot., 1997, 50, 58–65 CrossRef CAS PubMed.
- T. A. Scott, M. Verest, J. Farnung, C. C. Forneris, S. L. Robinson, X. Ji, F. Hubrich, C. Chepkirui, D. U. Richter, S. Huber, P. Rust, A. B. Streiff, Q. Zhang, J. W. Bode and J. Piel, Chem, 2022, 8, 2659–2677 CAS.
- E. Lakis, S. Magyari and J. Piel, Angew. Chem., Int. Ed, 2022, 61, e202202695 CrossRef CAS PubMed.
- D. Richter, E. Lakis and J. Piel, Nat. Chem., 2023, 15, 1422–1430 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Žídek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, Nucleic Acids Res., 2021, 50, D439–D444 CrossRef PubMed.
- C. Padhi, C. M. Field, C. C. Forneris, D. Olszewski, A. E. Fraley, I. Sandu, T. A. Scott, J. Farnung, H.-J. Ruscheweyh, A. N. Panda, A. Oxenius, U. F. Greber, J. W. Bode, S. Sunagawa, V. Raina, M. Suar and J. Piel, 2024, in revision.
- J. L. Vey and C. L. Drennan, Chem. Rev., 2011, 111, 2487–2506 CrossRef CAS PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS PubMed.
- A. L. Vagstad, E. Lakis, K.-S. Csizi, W. Walls, D. Richter, K. S. Lee, R. Stocker, M. Gugger, W. E. Broderick, J. B. Broderick, M. Reiher, and J. Piel, 2024, submitted.
- M. Kitagawa, T. Ara, M. Arifuzzaman, T. Ioka-Nakamichi, E. Inamoto, H. Toyonaga and H. Mori, DNA Res., 2005, 12, 291–299 CrossRef CAS PubMed.
- J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. Houssen, M. Jaspars and J. H. Naismith, Nat. Chem. Biol., 2015, 11, 558–563 CrossRef CAS PubMed.
- S. Gao, Y. Ge, A. F. Bent, U. Schwarz-Linek and J. H. Naismith, Biochemistry, 2018, 57, 5996–6002 CrossRef CAS PubMed.
- Y. Ge, C. M. Czekster, O. K. Miller, C. H. Botting, U. Schwarz-Linek and J. H. Naismith, Biochemistry, 2019, 58, 2125–2132 CrossRef CAS PubMed.
- T. J. Oman, P. J. Knerr, N. A. Bindman, J. E. Velásquez and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 6952–6955 CrossRef CAS PubMed.
- E. Oueis, B. Nardone, M. Jaspars, N. J. Westwood and J. H. Naismith, ChemistryOpen, 2017, 6, 11–14 CrossRef CAS PubMed.
- E. Reyna-González, B. Schmid, D. Petras, R. D. Süssmuth and E. Dittmann, Angew. Chem., Int. Ed., 2016, 55, 9398–9401 CrossRef PubMed.
- J. A. Walker, N. Hamlish, A. Tytla, D. D. Brauer, M. B. Francis and A. Schepartz, ACS Cent. Sci., 2022, 8, 473–482 CrossRef CAS PubMed.
- B. J. Burkhart, N. Kakkar, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, ACS Cent. Sci., 2017, 3, 629–638 CrossRef CAS PubMed.
- L. Franz and J. Koehnke, Chem. Commun., 2021, 57, 6372–6375 RSC.
- A. M. Kretsch, M. G. Gadgil, A. J. DiCaprio, S. E. Barrett, B. L. Kille, Y. Si, L. Zhu and D. A. Mitchell, Biochemistry, 2023, 62, 956–967 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03867c |
| This journal is © The Royal Society of Chemistry 2024 |