
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatechol/o-benzoquinone exchange at gold(III)†
Félix
León
a,
Yago
García-Rodeja
b,
Sonia
Mallet-Ladeira
c,
Karinne
Miqueu
 *b,
György
Szalóki
*a and
Didier
Bourissou
*a
*b,
György
Szalóki
*a and
Didier
Bourissou
*a
aCNRS/Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA, UMR 5069), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: gyorgy.szaloki@univ-tlse3.fr; didier.bourissou@univ-tlse3.fr
bCNRS/Université de Pau et des Pays de l'Adour. E2S-UPPA, Institut des Sciences Analytiques et de Physico-Chimie pour l’Environnement et les Matériaux (IPREM, UMR 5254), Hélioparc, 2 Avenue du Président Angot, 64053 Pau Cedex 09, France. E-mail: karinne.miqueu@univ-pau.fr
cInstitut de Chimie de Toulouse (UAR 2599), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
First published on 18th September 2024
Abstract
Although gold(III) chemistry has tremendously progressed in the past 2 decades, gold(III) catecholate complexes remain extremely scarce and underdeveloped. Upon preparation and full characterization of P^C-cyclometalated gold(III) complexes, we serendipitously uncovered an intriguing catechol exchange process at gold(III). Electron-rich catecholates turned out to be readily displaced by electron-poor o-benzoquinones. DFT calculations revealed an original path for this transformation involving two consecutive Single Electron Transfer events between the catecholate and o-benzoquinone moieties while gold maintains its +III oxidation state. This catechol/o-benzoquinone exchange at gold(III) represents a new path for the exchange of X-type ligands at transition metals.
Introduction
Among the two stable oxidation states of gold in mononuclear complexes, Au(I) largely prevails over Au(III), but over the past 2 decades, research involving gold(III) complexes has gained considerable momentum. Spectacular progress has been achieved in the study, modulation and application of their optical and biological properties as well as of their chemical reactivity. As comprehensively reviewed recently by Rocchigiani and Bochmann,1 the variety of well-defined gold(III) complexes has been considerably expanded in terms of chemical structures and bonding situations.2 New reactivity paths have also been discovered and applications in homogeneous catalysis have emerged.N^C-cyclometalation is prevalent and pivotal in gold(III) chemistry.3–5 Nitrogen and carbon form strong bonds to gold. They are well-suited to the high oxidation state Au(III) to which cyclometalation brings stability. Our discovery of oxidative addition of C(sp2)–X bonds to Au(I) assisted by P-chelation6 has opened a straightforward and general entry to complementary P^C-cyclometalated Au(III) complexes. The soft character of phosphorus is somewhat compensated by the rigid naphthyl backbone, making complexes A (Fig. 1A) thermally stable and easy to work with. Nonetheless, they display rich reactivity.7 In particular, we could achieve and study migratory insertion8,9 and β-hydride elimination,10 two elementary steps with very little, if any, precedent in gold chemistry.11 P^C-cyclometalation also enabled us to access and analyze unusual coordination complexes such as π-alkene/alkyne/arene/allyl complexes,9,12–14 σ-CH/arene complexes15,16 and nucleophilic carbene complexes.17 In terms of catalysis, complexes A proved highly active in the intermolecular hydroarylation of alkynes,16,18 working under mild conditions with a broad scope of substrates.
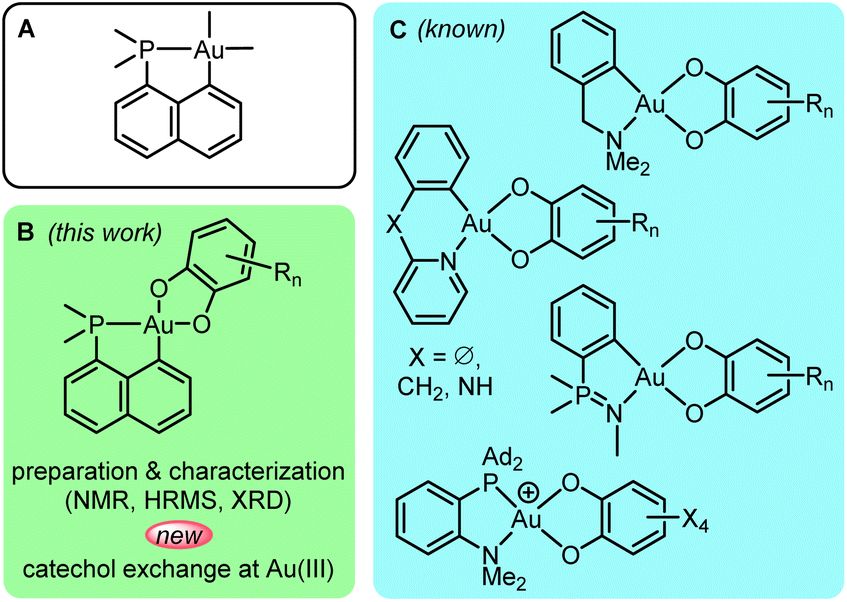 | ||
| Fig. 1 Known and new gold(III) catecholate complexes. | ||
In this work, we turned our attention to P^C-cyclometalated Au(III) catecholate complexes (Fig. 1B). Catecholates possess rich coordination chemistry and stand as archetypal redox-active ligands, with three possible oxidation states (the dianionic catecholate form, the radical monoanionic semiquinone form and the neutral o-quinone form). These redox features have been shown to impart unique properties to transition metal (TM) complexes.19 They expand reactivity beyond the common organometallic paths via ligand-based redox behaviour. While catecholate complexes of many transition metals (in particular molybdenum, rhenium, iron, ruthenium and copper) have been extensively studied, gold catecholate complexes remain chemical curiosities with only a few well-defined gold(III) species reported in the 2000–2010's (Fig. 1C).20–25 These complexes were classically prepared by reacting Au(III) dichloro or dibromo precursors with catechols under basic conditions. Some antitumor and antibacterial assays have been performed.20,21 A few punctual catalytic studies have also carried out (in the intermolecular hydroarylation of alkenes and intramolecular carboalkoxylation of alkynes).22–25 Of note, apart from one work,24 the reported gold(III) catecholate complexes are all supported by N^C-cyclometalated ligands, with amine, pyridine, iminophosphorane or oxazoline N donors. Our interest in ligand-enabled Au(I) to Au(III) oxidation26–35 prompted us to recently consider the use of chelating o-carboranyl diphosphine and hemilabile MeDalphos ligands to prepare Au(III) catecholate complexes. Accordingly, strongly oxidizing o-benzoquinones were found to react with Au(I) precursors to give new types of gold(III) catecholate complexes with P^P and P^N ligands.36 In addition, the transformation was found to be reversible upon addition of competing ligands such as chloride and alkenes. In this work, we became interested in preparing and investigating neutral P^C-cyclometalated Au(III) catecholate complexes. Thorough characterization of an unsymmetrically substituted complex led us to serendipitously uncover an unexpected and mechanistically original catechol/o-benzoquinone exchange process at gold(III). These results are presented and discussed in detail hereafter. Special attention has been given to the factors driving the catechol/o-quinone exchange, and to the way it operates. Experimental studies have been paralleled and completed by DFT calculations. The generality of the catechol/o-benzoquinone exchange has been substantiated by replacing the P^C-cyclometalated framework with a nido-carboranyl diphosphine ligand. While previously described cationic P^P and P^N-chelated Au(III) catecholate complexes were prone to reduction into Au(I) and o-quinone release,36 the neutral Au(III) complexes reported here maintain their oxidation state and undergo intra-ligand redox events.
Results and discussion
Our study began with the preparation of P^C cyclometalated Au(III) catecholate complexes. The diiodo Au(III) precursor 1 (which is readily accessible by P-chelation assisted oxidative addition)6 was reacted with sodium catecholates 2a–d in methanol at room temperature (Scheme 1). After 2 hours of vigorous stirring, complexes 3a–d were isolated in good yields (66–81%) by simple filtration, washing and drying under vacuum. They were unambiguously characterized by multi-nuclear NMR spectroscopy, mass spectrometry and X-ray diffraction analysis.37 The reduced state of the catechol ligands makes no doubt when referring to metrical parameters (Table S1†),37,38 which also supports the Au(III) assignment. Consistently, alternative forms of complexes 3a and 3d with the o-benzoquinone coordinated to a Au(I) fragment were predicted computationally to be >20 kcal mol−1 higher in energy than the Au(III) catecholate structures (Fig. S80†).37,39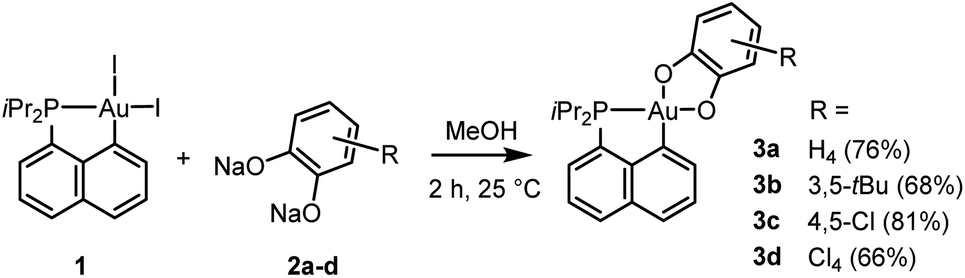 | ||
| Scheme 1 Synthesis of the P^C-cyclometalated gold(III) catecholate complexes 3a–d. | ||
Complex 3b deriving from the 3,5-di-tert-butyl-o-catecholate 2b deserves further comments. Due to its unsymmetrical substitution, it was obtained as a mixture of structural isomers 3b-1/3b-2 (in 84/16 ratio, according to 1H and 31P NMR spectroscopy, Fig. S9 and 10†).37 The isomer 3b-1 could be isolated (in 68% yield) from the mixture by removing 3b-2 with successive methanol washings. Single crystals of 3b-2 suitable for X-ray diffraction analysis could be obtained, so that the position of the tBu groups could be unambiguously determined (Fig. 2). Unexpectedly, upon characterization of the pure complex 3b-1, gradual isomerization into 3b-2 was observed in solution. After 2 hours at room temperature, an 80/20 mixture of 3b-1/3b-2 was obtained (Fig. S46†).37 The formation of isomeric mixtures of gold catecholates from unsymmetrically substituted catecholates has been already observed,20,23 but no sign of structural isomerization has been reported so far, to the best of our knowledge.40,41
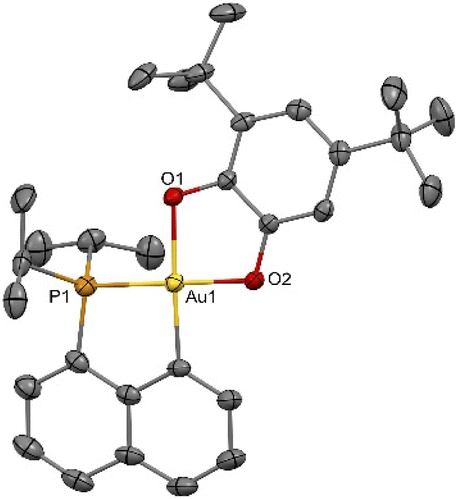 | ||
| Fig. 2 Molecular structure of the P^C-cyclometalated gold(III) catecholate complex 3b-2 (for sake of clarity, the hydrogen atoms and solvent molecules are omitted; ellipsoids shown at 50% probability). | ||
Besides being unprecedented, this transformation is also intriguing mechanistically. With such strongly chelating P^C and O^O ligands, a dissociation/recoordination path seems hardly conceivable. A 2-electron redox mechanism involving a gold(I) o-benzoquinone intermediate, as we encountered with the hemilabile P^N ligand MeDalphos,36 is also very unlikely. We thus hypothesized that some by-product or impurity may come into play. Apart from 3b-1 and 3b-2, no other species could be detected by NMR, but the presence of minute amounts of o-benzoquinone (which may result from oxidation of the sodium catecholate) could not be completely ruled out. With this idea in mind, we monitored the isomerization of 3b-1 in the absence and in the presence of 3,5-di-tert-butyl-o-benzoquinone 4b. Interestingly, the o-benzoquinone was found to accelerate the isomerization of 3b-1 (Scheme 2 and Fig. S41–46†).37
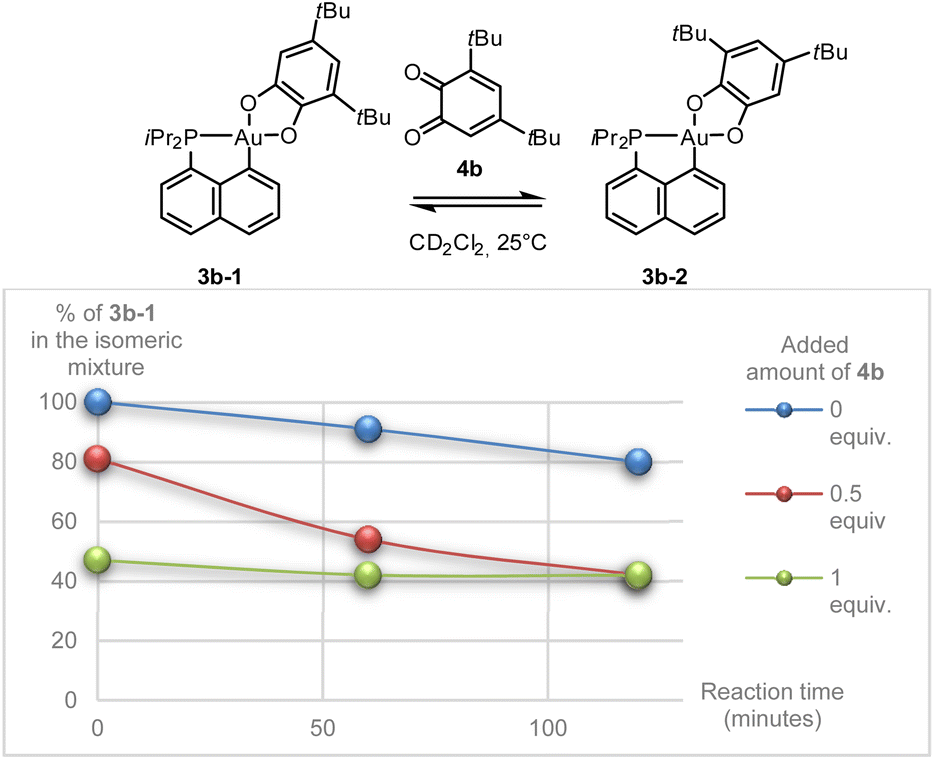 | ||
| Scheme 2 Isomerization of the gold catecholate complex 3b-1 in the presence of o-benzoquinone 4b, as observed by NMR spectroscopy. | ||
To probe the propensity of the P^C-cyclometalated Au(III) catecholate complexes to undergo catechol exchange in the presence of o-benzoquinones, complex 3b-1 was then reacted with other o-benzoquinones, namely the dichloro and tetrachloro o-benzoquinones 4c and 4d (Scheme 3). In both cases, 1H and 13C NMR monitoring revealed instantaneous and complete formation of the corresponding catechol complexes 3c and 3d with concomitant release of 3,5-di-tert-butyl-o-benzoquinone 4b (Fig. 3 and S51–55†).37 The scope of this catechol/o-benzoquinone exchange at gold was further expanded by reacting the parent P^C-cyclometalated Au(III) complex 3a with the tetrachloro o-benzoquinone 4d, resulting in quantitative formation of the tetrachloro-catecholate complex 3d along with the parent o-benzoquinone 4a (Fig. S47–50†).37 Thus, this catechol/o-benzoquinone exchange reaction at gold appears quite general and it seems electronically-driven, electron-rich catecholates being displaced by electron-poor o-quinones.
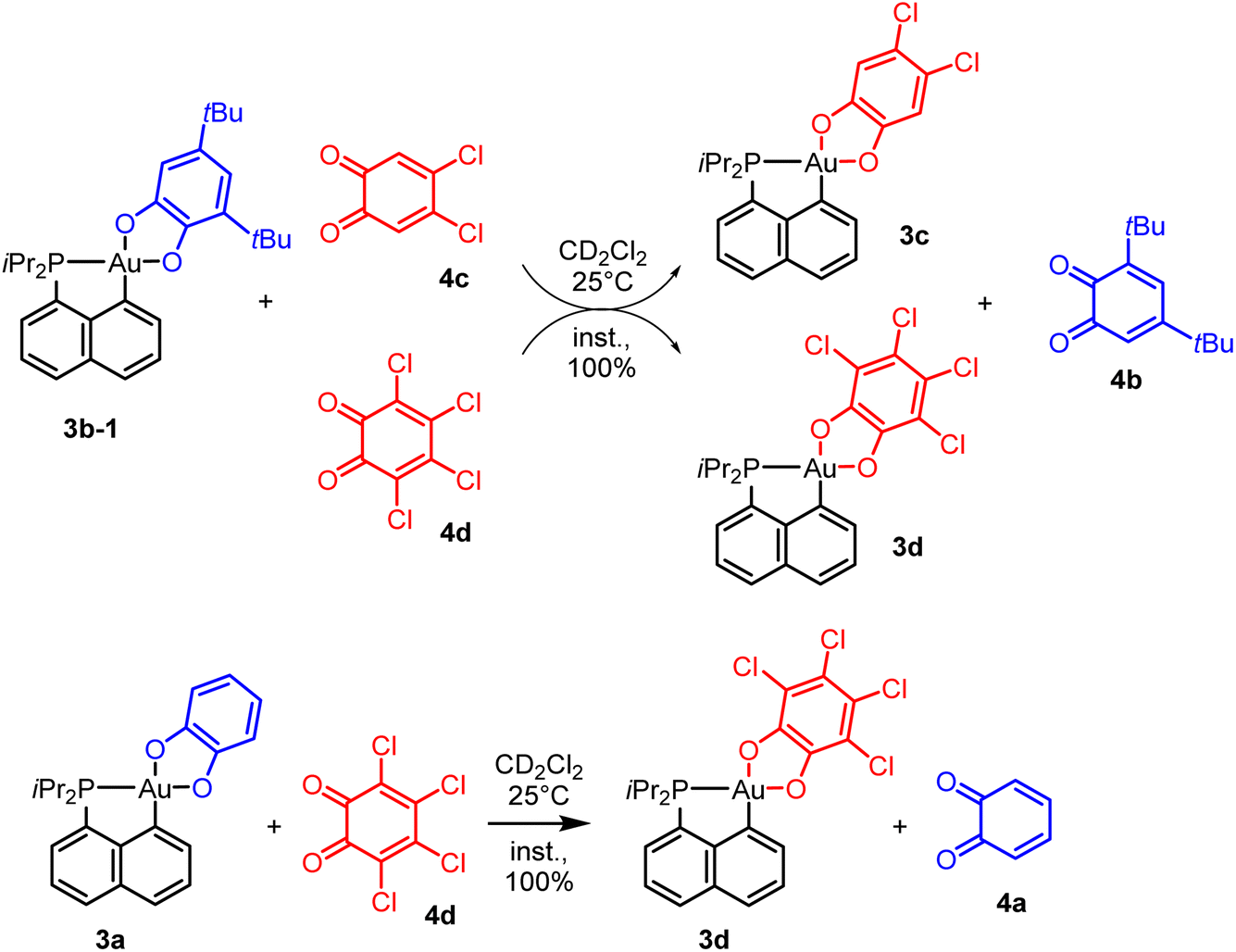 | ||
| Scheme 3 Exchange reactions between P^C cyclometalated catecholate Au(III) complexes and o-benzoquinones. | ||
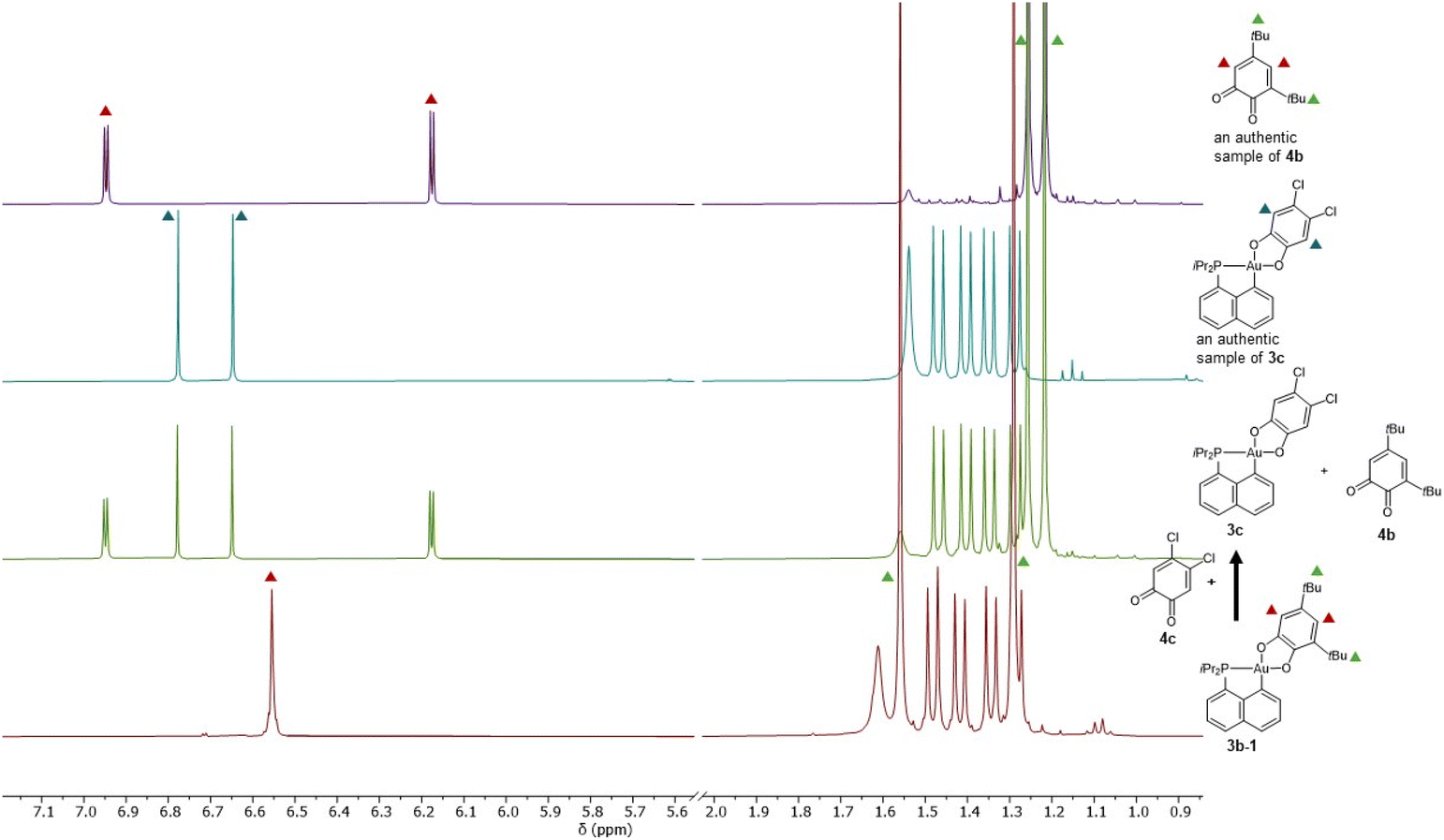 | ||
| Fig. 3 1H NMR monitoring of the reaction between 3b-1 and 4c. | ||
Efforts were then made to shed light on the mechanism of this original exchange reaction at gold. The reaction between the parent P^C-cyclometalated catecholate Au(III) complex 3a and the tetrachloro o-benzoquinone 4d was monitored by NMR spectroscopy at low temperature. The catechol/o-benzoquinone exchange occurred instantaneously and no intermediate could be detected even at – 80 °C (Fig. S64†).37 Considering the possible involvement of open-shell species as a result of Single Electron Transfer (SET), 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of the P^C-cyclometalated Au(III) catecholate complexes 3a, 3b and 3d with the respective o-benzoquinones 4a, 4b and 4d were also analysed by EPR spectroscopy, but no signal was observed at room temperature (Fig. S67–69†).37 On the other hand, EPR analysis of 3a and 4c at 150 K revealed a low intensity signal (Fig. S70†) that is tentatively assigned to a monoradical side-product, most likely a semi-quinone species, in free form or as a gold complex.42 The reaction between the parent P^C-cyclometalated catecholate Au(III) complex 3a and the dichloro o-benzoquinone 4c was also monitored at variable temperature by NMR and UV-vis spectroscopy.37 In this case, the catechol/o-benzoquinone exchange started at −80 °C but it was very slow. Gradual warm up of the solution revealed that the reaction was essentially complete after 15 min at −30 °C (Fig. S65†). During the course of the catechol/o-benzoquinone exchange, UV-vis spectroscopy revealed the transient appearance of a low-energy transition at λmax ∼745 nm attributable to a charge transfer/electron donor–acceptor complex (Fig. S66†).
:1 mixtures of the P^C-cyclometalated Au(III) catecholate complexes 3a, 3b and 3d with the respective o-benzoquinones 4a, 4b and 4d were also analysed by EPR spectroscopy, but no signal was observed at room temperature (Fig. S67–69†).37 On the other hand, EPR analysis of 3a and 4c at 150 K revealed a low intensity signal (Fig. S70†) that is tentatively assigned to a monoradical side-product, most likely a semi-quinone species, in free form or as a gold complex.42 The reaction between the parent P^C-cyclometalated catecholate Au(III) complex 3a and the dichloro o-benzoquinone 4c was also monitored at variable temperature by NMR and UV-vis spectroscopy.37 In this case, the catechol/o-benzoquinone exchange started at −80 °C but it was very slow. Gradual warm up of the solution revealed that the reaction was essentially complete after 15 min at −30 °C (Fig. S65†). During the course of the catechol/o-benzoquinone exchange, UV-vis spectroscopy revealed the transient appearance of a low-energy transition at λmax ∼745 nm attributable to a charge transfer/electron donor–acceptor complex (Fig. S66†).
DFT calculations were also performed, at the B3PW91-D3(BJ)/SDD + f(Au), 6-31G** (H, C, N, O, P, Cl) level of theory accounting for dispersion effects. The model reaction between (P^C)Au(O^O)–H4 and the 3,4,5,6-tetrachloro 1,2-benzoquinone was considered. Two paths involving first the displacement of the Au–O(catecholate) bond in trans or cis position to P by the incoming o-quinone were computed. Their energy profiles were found to be very similar, with activation barriers differing by only 0.5 to 1.2 kcal mol−1 (Fig. S71†).37
Only the path involving first the displacement of the Au–O bond in trans position to P is thus discussed in detail here (Fig. 4). The overall exchange reaction is thermodynamically favoured, with ΔG −8.1 kcal mol−1. To start with, the Au(III) catecholate complex and the 3,4,5,6-tetrachloro 1,2-benzoquinone form a charge transfer (CT) complex RC1 (ΔG −21.8 kcal mol−1), involving π–π interaction between the two O-substituted rings (with C⋯C distances of about 3 Å). It is likely that the absorption band we observed at λmax ∼745 nm during the low-temperature monitoring of the reaction 3a + 4c → 3c + 4a is due to such a CT complex. Then, two consecutive SET processes occur, corresponding to successive exchanges of the two Au–O bonds between the catecholate and the o-quinone moieties. The first SET corresponds to the exchange of the Au–O bond in trans position to P, to give the diradical intermediate INT (ΔG 7.3 kcal mol−1 from RC1). The second SET corresponds to Au–O bond exchange between the two semi-quinone moieties of INT to afford the charge transfer complex RC2, reciprocal to RC1 (i.e. between (P^C)Au(O^O)–Cl4 and the parent o-benzoquinone, ΔG −1.1 kcal mol−1 from RC1). The activation barriers for the two SET steps are very similar and easily accessible, at 12.7 and 13.0 kcal mol−1 (from RC1), in line with the experimental conditions. To refine the computational model, the solvent (CH2Cl2:DCM) was then taken into account using the universal solvation model based on solute electron density (SMD), upon optimization (Fig. 4, values into square brackets) or by single-point calculations (Fig. S72†).37 In both cases, very similar results were obtained compared with the gas phase. At the SMD(DCM)-B3PW91-D3(BJ) level, the exchange reaction is exergonic by −10.2 kcal mol−1, the charge transfer adduct RC1 sits 13.5 kcal mol−1 below the reactants and the activation barriers for the two SET steps amount to 10.5 and 11.7 kcal mol−1 (from RC1), respectively (Fig. 4). The path computed for the reaction 3a + 4d → 3d + 4a seems general, similar energy profiles were found for the exchange of the parent catechol by the parent and 4,5-dichloro o-benzoquinones (Fig. S79 and S81†).37
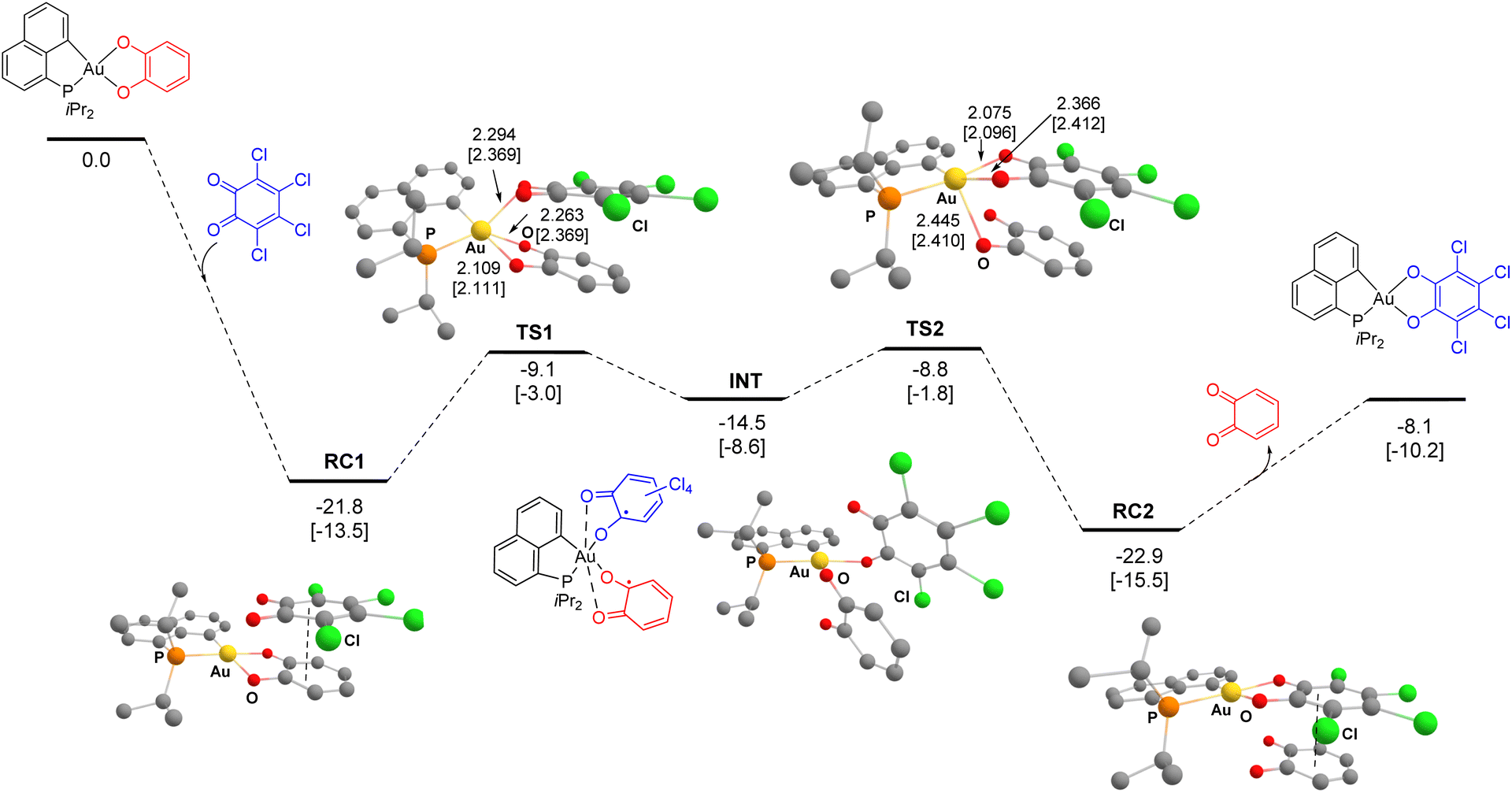 | ||
| Fig. 4 Energy profile (ΔG in kcal mol−1) of the catechol/o-benzoquinone exchange reaction at Au(III) computed at B3PW91-D3(BJ)/SDD + f(Au), 6-31G** (other atoms) level of theory. Path involving first the displacement of the Au–O bond in trans position to P. For comparison, the values computed at the SMD(DCM)-B3PW91-D3(BJ)/SDD + f(Au), 6-31G** (other atoms) level are reported into square brackets. Main Au–O distances in Å for the transition states of the two SET steps. Hydrogens omitted for clarity. | ||
The electronic structure and bonding situation of the two key intermediates, i.e. the charge transfer adduct RC1 and the Au(III) bis-semiquinone complex INT were then analysed in-depth. To this end, different approaches were employed: the Extended Transition State (ETS) method for Energy Decomposition Analysis (EDA) combined with the Natural Orbitals for Chemical Valence (NOCV) theory, the Electrostatic Potential Energy map (ESP) and the Atoms-in-Molecules analysis (AIM).
In line with a charge transfer adduct, the HOMO of RC1 was found to be mainly localized on the π-system of the H4-catecholate moiety (donor moiety) while the LUMO of RC1 is mainly localized on the π-system of the tetrachloro o-benzoquinone (acceptor moiety) (Fig. 5a). In the ETS-NOCV analysis, the predominant term of the ΔEorb contribution (ΔEorb(ρ1) = −27.4 kcal mol−1 compared to ΔEorb = −40.6 kcal mol−1, i.e. about 67.5%) corresponds to a π–π donor–acceptor interaction from the (P^C)Au(O^O)–H4 Au(III) complex to the (O^O)PhCl4o-benzoquinone, as visualized by the plot of the associated NOCV (Fig. 5b). The computed Hirshfeld charges indicate a substantial charge transfer of 0.42 electron from (P^C)Au(O^O)–H4 to (O^O)–Cl4. This CT is also clearly apparent from the ESP map (Fig. 5c). The ESP plot of the (P^C)Au(O^O)–H4 Au(III) complex alone shows a negative region (orange) delocalized on the ring of the (O^O)–H4 catecholate fragment whereas in the CT adduct RC1, this region becomes positive (blue), consistent with electron flow towards the tetrachloro o-quinone. The electronic structure of the Au(III) bis-semiquinone complex INT was also analysed in-depth. The two semi-quinone moieties are parallel and close to each other (the distance between the two centroids is ∼3.0 Å), suggesting strong π–π interaction. Antiferromagnetic coupling is clearly apparent from the frontier molecular orbitals (Fig. 6a). Indeed, the HOMO and LUMO of INT correspond, respectively, to the bonding and anti-bonding combinations of the SOMOs of the two semi-quinone moieties. The HOMO–LUMO energy gap ΔE is close to 2 eV. Such π-stacking interactions between delocalized radicals, commonly referred as “pancake bonding”,43–45 have some rare precedents with p- and o-semiquinones.46–50 Consistently, the Atom-In-Molecules (AIM) molecular graph of INT (Fig. 6b) shows three Bond Critical Points (BCP) corresponding to relatively short C⋯C contacts (from 2.706 to 3.121 Å), with electron densities ρ(r) ranging from 0.01 to 0.02 e. bohr−3 and bond indexes δ ranging from 0.04 to 0.09. Of note, no significant out-of-planarity distortion was noticed in the optimized geometry of INT. The triplet state of the Au(III) bis-semiquinone complex INTT was also computed (Fig. S75†).37 Here, the two semi-quinone moieties remain far away from each other (C⋯C distances > 3.8 Å) and essentially do not interact electronically. INTT sits only slightly above INT in energy (ΔG(S–T) 0.9 kcal mol−1) but the other stationary points of the triplet Potential Energy Surface (PES) lie significantly higher in energy (+10.3 kcal mol−1 for RC1T, +20.3 kcal mol−1 for RC2T, +5.9 kcal mol−1 for TS1T, +9.8 kcal mol−1 for TS2T, taking the corresponding stationary points of the singlet PES as origins). Accordingly, the catechol exchange/o-benzoquinone is likely to occur in the singlet state without spin-crossing.
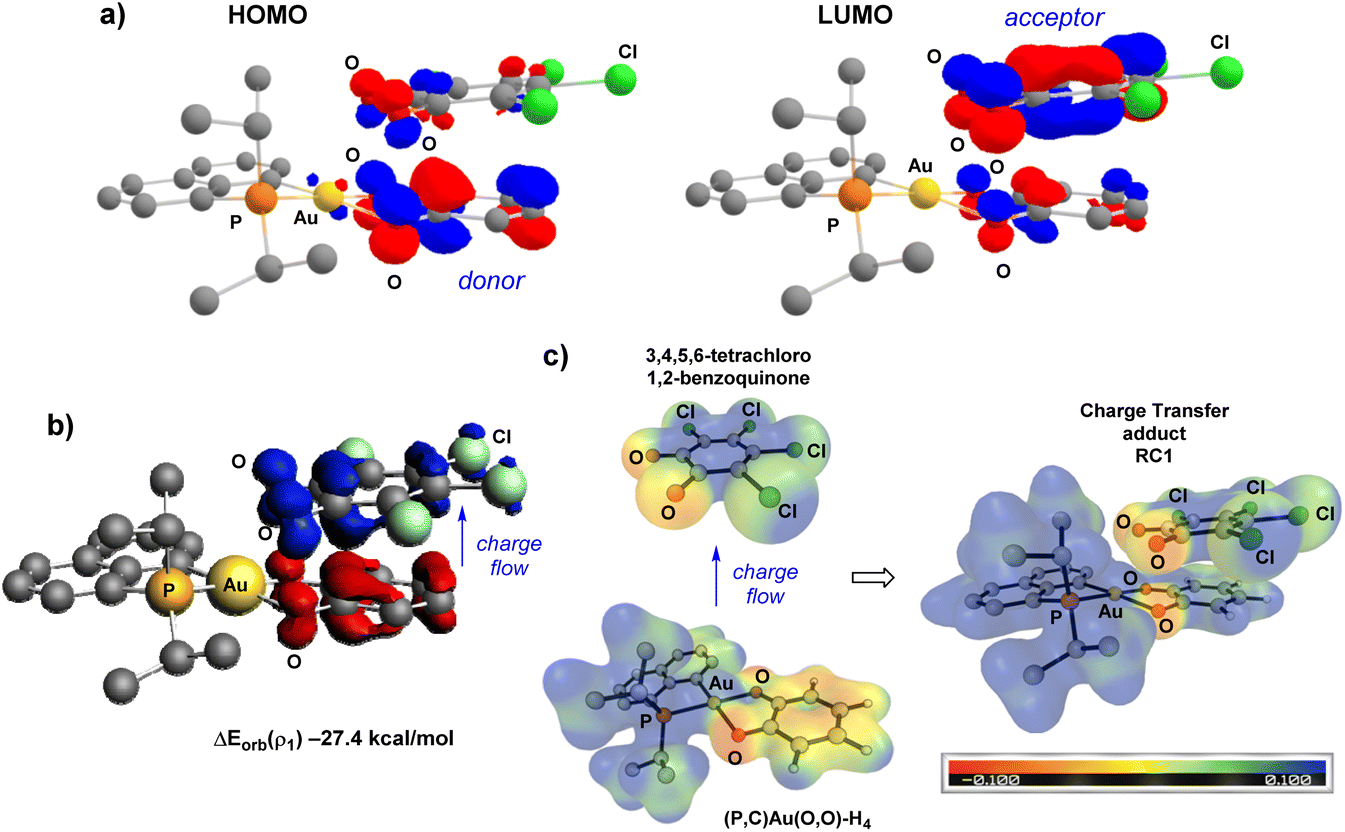 | ||
| Fig. 5 (a) Plot of the frontier molecular orbitals (cutoff: 0.05) for the charge transfer adduct RC1, computed at the B3PW91-D3(BJ)/SDD + f(Au), 6-31G** (other atoms) level. Hydrogens omitted for clarity. (b) Plot of the contour of deformation density contribution (Δρorb, 1) of the main pairwise orbital interaction between (P^C)Au(O^O)–H4 fragment and the 3,4,5,6-tetrachloro-1,2-benzoquinone, and associated orbital interaction energy contribution (ΔEorb, 1 in kcal mol−1), computed at ZORA-BP86-D3/TZ2P level of theory. The charge flow is red → blue (Δρ < 0 in red and Δρ > 0 in blue). The contour value for density is 0.001 a.u. (c) Electrostatic Potential (ESP) map for RC1 and the 2 fragments (P^C)Au(O^O)–H4 and 3,4,5,6-tetrachloro 1,2-benzoquinone in their ground-state structures, with an isosurface of 0.02. | ||
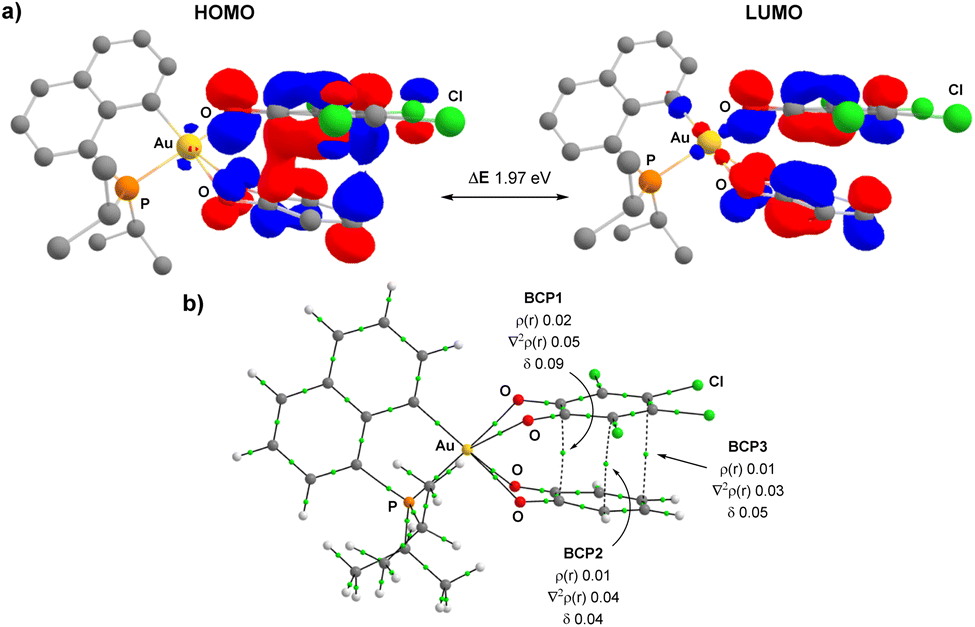 | ||
| Fig. 6 (a) Plot of the frontier molecular orbitals (cutoff: 0.04) for the Au(III) bis-semiquinone intermediate INT, computed at the B3PW91-D3(BJ)/SDD + f(Au), 6-31G** (other atoms) level. (b) Plot of the molecular graph for INT. For clarity, only the bond paths relative to the H4-semiquinone → Cl4-semiquinone interactions are shown. ρ(r): density at the main BCP in e bohr−3, ∇2ρ(r): Laplacian of density at the main BCP in e bohr−5 and δ: delocalization index. | ||
Next, we wondered whether this catechol/o-benzoquinone exchange reaction was restricted to cyclometalated complexes such as 3 or could also proceed with complexes featuring different ancillary ligands. To address this question, we turned to nido-carboranyl diphosphines which are anionic P^P chelating ligands51 In previous work on ligand-enabled oxidation of gold(I) complexes with o-quinones, we have prepared gold(III)-catecholate complexes deriving from a closo-carboranyl diphosphine and found them to slowly degrade by boron-decapping to give the corresponding nido-carboranyl diphosphine complexes36 Here, we developed an alternative preparative route, using the (nido-carboranyl diphosphine)AuCl2 precursor 6 (Scheme 4). The latter complex was readily and efficiently prepared by reacting the nido-carboranyl diphosphine 5 with (Me2S)AuCl followed by oxidation with PhICl2.37 Upon treatment with sodium catecholates 2a–d, the nido-carboranyl diphosphine gold(III)-catecholate complexes 7a–d were obtained in good yields (69–80%) and fully characterized.37
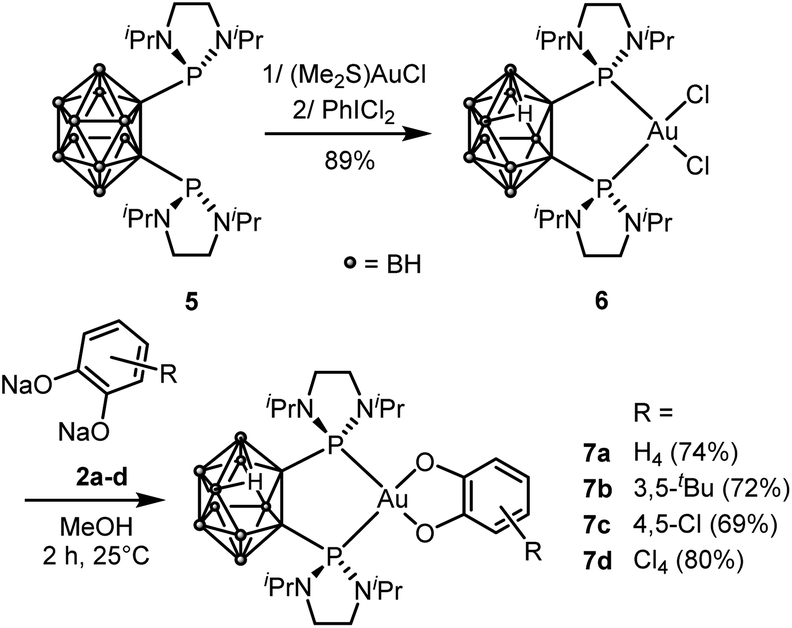 | ||
| Scheme 4 Synthesis of the P^P-chelated gold(III) catecholate complexes 7a–d. | ||
Then, the catechol/o-benzoquinone exchange was investigated. Gratifyingly, the nido-gold(III)-catecholate complexes 7a and 7b were found to react instantaneously and quantitatively with o-benzoquinones 4c and 4d (Scheme 5). The transformation is thus general and as for the P^C-cyclometalated complexes, electron-poor o-benzoquinones readily displace electron-rich catecholates at gold.
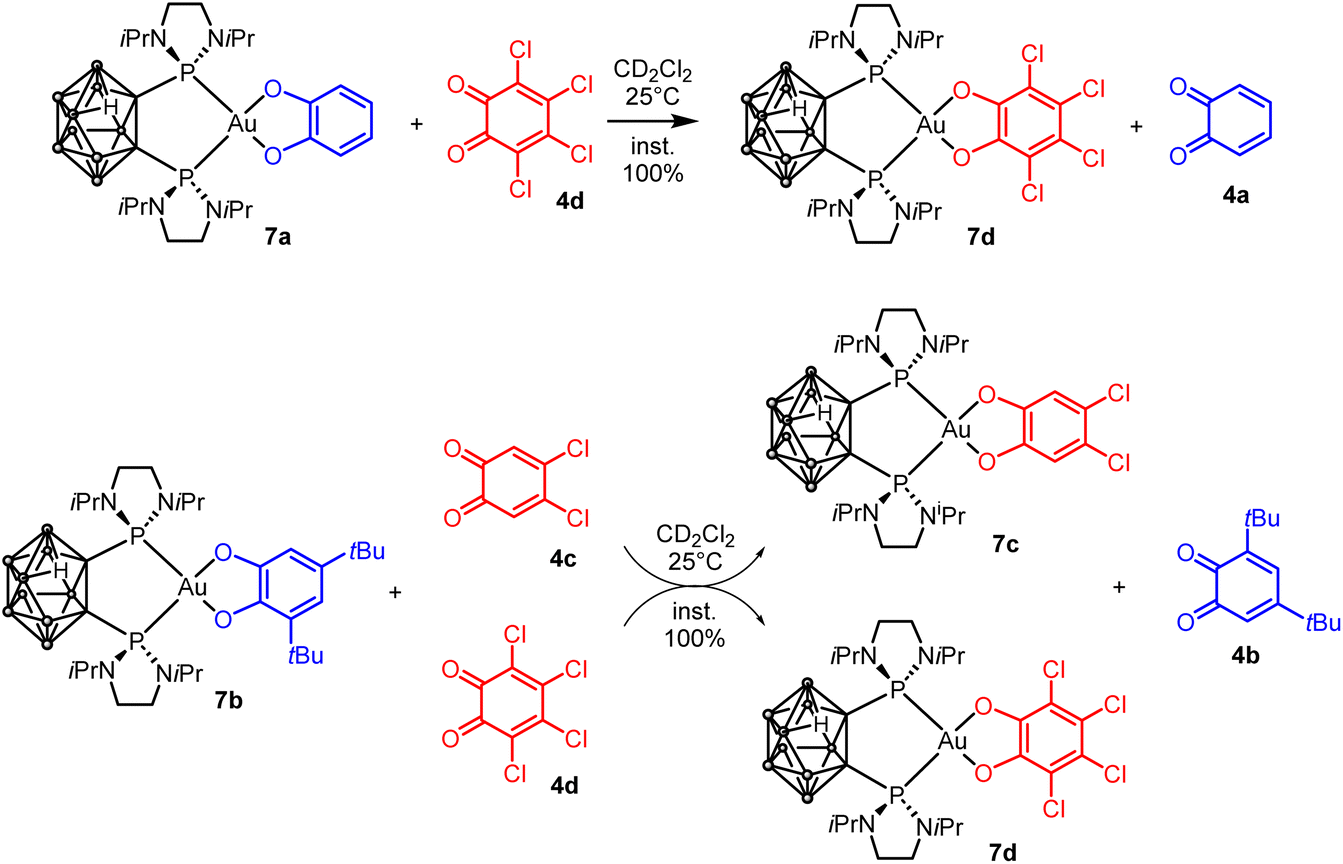 | ||
| Scheme 5 Exchange reactions between P^P-chelated catecholate Au(III) complexes and o-benzoquinones. | ||
Conclusion
In summary, neutral gold(III) catecholate complexes featuring P^C-cyclometalated and nido-carboranyl diphosphine ligands have been prepared and isolated. Full characterization by NMR spectroscopy and XRD analysis revealed an intriguing isomerization process for the unsymmetrical complex 3b.This turned to result from an original catechol exchange process at gold(III) upon reaction with external o-benzoquinones. The reaction is driven by electronic effects and elicits the displacement of electron-rich, easily oxidized catecholates by electron-poor, easily reduced o-benzoquinones. In this transformation, the gold center maintains its +III oxidation state. It acts as a redox-innocent host for the redox-active ligand and witnesses two consecutive SET events between the catecholate and o-benzoquinone moieties. The structure of the key bis-semiquinone Au(III) intermediate unveils how redox-active ligands may engage in intra-ligand SET, resulting ultimately in ligand exchange.
In a broader perspective, this catechol/o-benzoquinone exchange at gold(III) represents a new path for the exchange of X-type ligands at transition metals (Fig. 7a).11 Most common are the 2-electron routes involving oxidative addition/reductive elimination (Fig. 7b). σ-Complex Assisted Metathesis (σ-CAM) in which the transition metal oxidation state remains constant has also been recognized as a relevant path for X-type ligand exchange (Fig. 7c).52,53 Of note, a somewhat related catechol exchange has been recently evidenced by Greb et al. at silicon. Upon investigating bis(catecholato)silanes as Si(IV)-based Lewis acids, some dynamic covalent behaviour was authenticated. Here, monomeric and oligomeric structures interconvert via facile Si–O σ-bond metathesis.54,55 The double SET process we encountered in gold(III) catecholate complexes is likely to also be possible with other transition metals and even maybe with metalloids, in particular for square-planar compounds. Future work will aim to explore the generality of such ligand exchange and to develop the chemistry of gold complexes featuring redox-active ligands.56–59
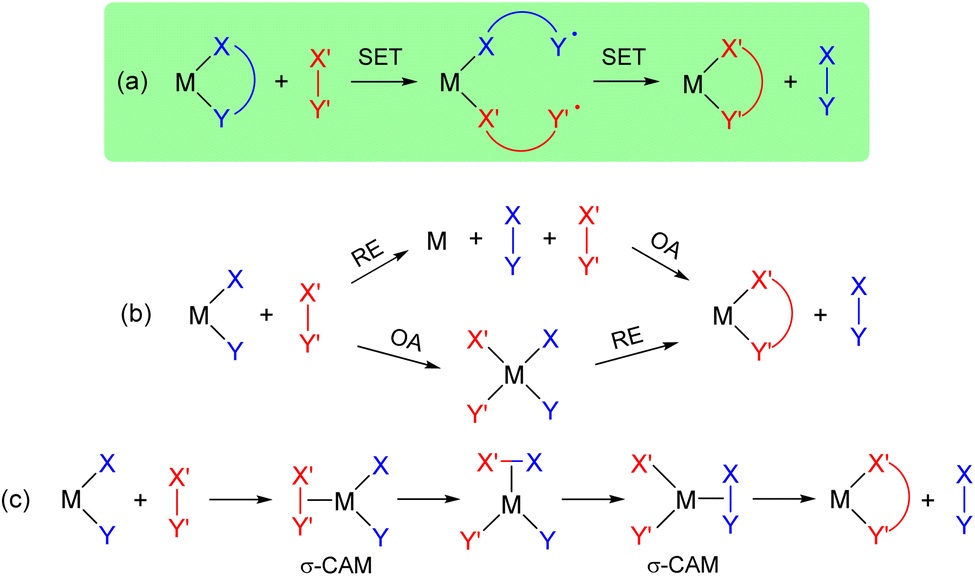 | ||
| Fig. 7 Schematic representation of the different paths for the exchange of X-type ligands at transition metals. SET: single electron transfer, OA: oxidative addition, RE: reductive elimination, σ-CAM: σ-complex assisted metathesis. | ||
The chemical stability of the P^C-cyclometalated Au(III) catecholate complexes towards reduction into Au(I) also make them worth considering as possible anticancer metallodrugs.60 The rigidity of the naphthyl backbone and the higher trans influence of phosphorus versus nitrogen may induce noticeable changes with respect to known N^C-cyclometalated species.
Data availability
The data that support the findings of this study are available in the ESI† of this article. Deposition Numbers 2332329–2332335 contain the supplementary crystallographic data for this paper.Author contributions
F. L. carried out the synthetic work, performed and interpreted the analytical characterizations. Y. G.-R. and K. M. carried out and analysed the computational studies. S. M-L. carried out the X-ray diffraction studies. K. M., G. S. and D. B. supervised the experimental and computational studies. D. B. conceived the project. All the authors contributed to the writing of the manuscript and validated it prior to submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Centre National de la Recherche Scientifique, the Université de Toulouse, the Agence Nationale de la Recherche (ANR-19-CE07-0037), the Foundation Del Duca de l’Institut de France (subvention to DB, post-doctoral fellowship to FLG) and European Union (ERC AdG grant to DB, Project 101097537 – Gold-Redox) is gratefully acknowledged. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. The help of Dr Dario Bassani (ISM, Bordeaux) and Dr Cédric Colomban (iSm2, Marseille) in conducting the low-temperature UV-vis experiments is highly appreciated. The NMR service of ICT, UAR 2599 (Marc Vedrenne) is acknowledged for assistance with the variable-temperature and monitoring NMR experiments. Thanks are due to Julien Babinot (LHFA) for preparation of some starting materials and some exploratory experiments on P^C-cyclometalated Au(III) complexes. The “Direction du Numérique” of the Université de Pau et des Pays de l’Adour and the Mésocentre de Calcul Intensif Aquitain (MCIA) are acknowledged for the support of computational facilities. This work was also granted access to the HPC resources of IDRIS under the allocation 2022/2023-[AD010800045R1/AD010800045R2] made by GENCI.Notes and references
- L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364 CrossRef CAS PubMed.
- R. P. Herrera and M. C. Gimeno, Chem. Rev., 2021, 121, 8311 CrossRef CAS PubMed.
- W. Henderson, Adv. Organomet. Chem., 2006, 54, 207 CrossRef CAS.
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994 CrossRef CAS PubMed.
- B. Bertrand, M. Bochmann, J. Fernandez-Cestau and L. Rocchigiani, in Pincer Compounds, Elsevier, 2018, p. 673 Search PubMed.
- J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778 CrossRef CAS PubMed.
- J. Monot, E. Marelli, B. Martin-Vaca and D. Bourissou, Chem. Soc. Rev., 2023, 52, 3543 RSC.
- F. Rekhroukh, R. Brousses, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 1266 CrossRef CAS PubMed.
- F. Rekhroukh, C. Blons, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Chem. Sci., 2017, 8, 4539 RSC.
- F. Rekhroukh, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 11920 CrossRef CAS PubMed.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022 CrossRef CAS PubMed.
- J. Rodriguez, M. S. M. Holmsen, Y. García-Rodeja, E. D. Sosa Carrizo, P. Lavedan, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2021, 143, 11568 CrossRef CAS PubMed.
- J. Rodriguez, G. Szalóki, E. D. Sosa Carrizo, N. Saffon-Merceron, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2020, 59, 1511 CrossRef CAS PubMed.
- M. S. M. Holmsen, D. Vesseur, Y. García-Rodeja, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2023, 62, e202305280 CrossRef PubMed.
- F. Rekhroukh, L. Estévez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2016, 55, 3414 CrossRef CAS PubMed.
- M. S. M. Holmsen, C. Blons, A. Amgoune, M. Regnacq, D. Lesage, E. D. Sosa Carrizo, P. Lavedan, Y. Gimbert, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2022, 144, 22722 CrossRef CAS PubMed.
- A. Pujol, M. Lafage, F. Rekhroukh, N. Saffon-Merceron, A. Amgoune, D. Bourissou, N. Nebra, M. Fustier-Boutignon and N. Mézailles, Angew. Chem., Int. Ed., 2017, 56, 12264 CrossRef CAS PubMed.
- C. Blons, S. Mallet-Ladeira, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 11732 CrossRef CAS PubMed.
- D. L. J. Broere, R. Plessius and J. I. Van Der Vlugt, Chem. Soc. Rev., 2015, 44, 6886 RSC.
- C. H. A. Goss, W. Henderson, A. L. Wilkins and C. Evans, J. Organomet. Chem., 2003, 679, 194 CrossRef CAS.
- K. J. Kilpin, W. Henderson and B. K. Nicholson, Inorg. Chim. Acta, 2009, 362, 3669 CrossRef CAS.
- K. J. Kilpin, B. P. Jarman, W. Henderson and B. K. Nicholson, Appl. Organomet. Chem., 2011, 25, 810 CrossRef CAS.
- T. S. Smith, J. R. Lane, M. R. Mucalo and W. Henderson, Transit. Met. Chem., 2016, 41, 581 CrossRef CAS.
- P. Byabartta and M. Laguna, Inorg. Chem. Commun., 2007, 10, 666 CrossRef CAS.
- J.-J. Jiang, J.-F. Cui, B. Yang, Y. Ning, N. C.-H. Lai and M.-K. Wong, Org. Lett., 2019, 21, 6289 CrossRef CAS PubMed.
- M. Joost, A. Zeineddine, L. Estévez, S. Mallet−Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed.
- M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236 CrossRef CAS PubMed.
- A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed.
- J. Rodriguez, A. Zeineddine, E. D. Sosa Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183 RSC.
- J. Rodriguez, A. Tabey, S. Mallet-Ladeira and D. Bourissou, Chem. Sci., 2021, 12, 7706 RSC.
- M. Navarro, A. Tabey, G. Szalóki, S. Mallet-Ladeira and D. Bourissou, Organometallics, 2021, 40, 1571 CrossRef CAS.
- M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069 RSC.
- B. Huang, M. Hu and F. D. Toste, Trends Chem., 2020, 2, 707 CrossRef CAS PubMed.
- P. Font and X. Ribas, Eur. J. Inorg. Chem., 2021, 2021, 2556 CrossRef CAS.
- T. McCallum, Org. Biomol. Chem., 2023, 21, 1629 RSC.
- G. Szalóki, J. Babinot, V. Martin-Diaconescu, S. Mallet-Ladeira, Y. García-Rodeja, K. Miqueu and D. Bourissou, Chem. Sci., 2022, 13, 10499 RSC.
- See ESI†.
- S. N. Brown, Inorg. Chem., 2012, 51, 1251 CrossRef CAS PubMed.
- For recent discussions about oxidation states, including the d10 M(I)/d8 M(III) descriptions of group 10 metal complexes, see: (a) E. A. Trifonova, I. F. Leach, W. B. de Haas, R. W. A. Havenith, M. Tromp and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2022, 62, e202215523 CrossRef PubMed; (b) I. F. Leach and J. E. M. N. Klein, ACS Cent. Sci., 2024, 10, 1406 CrossRef CAS PubMed.
- Ligand scrambling between symmetric bis(dithiolene) gold(III) complexes has been reported once, with the complexes in their neutral radical state, i.e. their one-electron oxidized form: R. Perochon, F. Barrière, O. Jeannin, L. Piekara-Sady and M. Fourmigué, Chem. Commun., 2021, 57, 1615 RSC.
- Rapid interconversion of cis and trans isomers of unsymmetrical bis(dithiolene) complexes has been reported for nickel(II) complexes in solution, but not for gold(III) complexes: A. Mizuno, H. Benjamin, Y. Shimizu, Y. Shuku, M. M. Matsushita, N. Robertson and K. Awaga, Adv. Funct. Mater., 2019, 29, 1904181 CrossRef CAS.
- M. Witwicki, A. Lewińska and A. Ozarowski, Phys. Chem. Chem. Phys., 2021, 23, 17408 RSC.
- K. E. Preuss, Polyhedron, 2014, 79, 1 CrossRef CAS.
- M. Kertesz, Chem.–Eur. J., 2019, 25, 400 CrossRef CAS PubMed.
- K. Molčanov, V. Milašinović and B. Kojić-Prodić, Cryst. Growth Des., 2019, 19, 5967 CrossRef.
- K. Molčanov, V. Milašinović, N. Ivić, V. Stilinović, D. Kolarić and B. Kojić-Prodić, CrystEngComm, 2019, 21, 6920 RSC.
- K. Molčanov, C. Jelsch, B. Landeros, J. Hernández-Trujillo, E. Wenger, V. Stilinović, B. Kojić-Prodić and E. C. Escudero-Adán, Cryst. Growth Des., 2019, 19, 391 CrossRef.
- T. Han, J. B. Petersen, Z.-H. Li, Y.-Q. Zhai, A. Kostopoulos, F. Ortu, E. J. L. McInnes, R. E. P. Winpenny and Y.-Z. Zheng, Inorg. Chem., 2020, 59, 7371 CrossRef CAS PubMed.
- V. Milašinović, A. Krawczuk, K. Molčanov and B. Kojić-Prodić, Cryst. Growth Des., 2020, 20, 5435 CrossRef.
- R. Maskey, H. Wadepohl and L. Greb, Angew. Chem., Int. Ed., 2019, 58, 3616 CrossRef CAS PubMed.
- F. Teixidor, C. Viñas, M. M. Abad, R. Kivekäs and R. Sillanpää, J. Organomet. Chem., 1996, 509, 139 CrossRef CAS.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578 CrossRef CAS PubMed.
- R. N. Perutz, S. Sabo-Etienne and A. S. Weller, Angew. Chem., Int. Ed., 2022, 61, e202111462 CrossRef CAS PubMed.
- R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717 CrossRef CAS PubMed.
- D. Hartmann, T. Thorwart, R. Müller, J. Thusek, J. Schwabedissen, A. Mix, J.-H. Lamm, B. Neumann, N. W. Mitzel and L. Greb, J. Am. Chem. Soc., 2021, 143, 18784 CrossRef CAS PubMed.
- V. Vreeken, D. L. J. Broere, A. C. H. Jans, M. Lankelma, J. N. H. Reek, M. A. Siegler and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2016, 55, 10042 CrossRef CAS PubMed.
- V. Vreeken, M. A. Siegler and J. I. van der Vlugt, Chem.–Eur. J., 2017, 23, 5585–5594 CrossRef CAS PubMed.
- P. Veit, C. Volkert, C. Förster, V. Ksenofontov, S. Schlicher, M. Bauer and K. Heinze, Chem. Commun., 2019, 55, 4615 RSC.
- M. P. Schrick, G. K. Ramollo, C.-M. S. Hirschbiegel, M. Fernandes, A. Lemmerer, C. Förster, D. I. Bezuidenhout and K. Heinze, Organometallics, 2024, 43, 69 CrossRef CAS.
- G. Moreno-Alcántar, P. Picchetti and A. Casini, Angew. Chem., Int. Ed., 2023, 62, e202218000 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2332329–2332335. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04374j |
| This journal is © The Royal Society of Chemistry 2024 |